
Utilization of LDH-based materials as potential adsorbents and photocatalysts for the decontamination of dyes wastewater: a review
Zhongzhu Yang
ab,
Fenghua Wang
*c,
Chang Zhang
*ab,
Guangming Zeng
ab,
Xiaofei Tan
ab,
Zhigang Yuab,
Yu Zhongab,
Hou Wangab and
Fang Cuiab
aCollege of Environmental Science and Engineering, Hunan University, Changsha 410082, China. E-mail: 952157786@qq.com; zhangchang@hnu.edu.cn; Fax: +86-731-88822312; Tel: +86-731-88822312
bKey Laboratory of Environmental Biology and Pollution Control (Hunan University), Ministry of Education, Changsha 410082, China
cInstitute of Physical Education, Xinjiang Normal University, Urumqi 830054, China
First published on 22nd August 2016
Abstract
Dye as a colored organic pollution has caused tremendous environmental problems. Removing dyes from effluents is of significant importance. Layered double hydroxides (LDHs), known as hydrotalcite-like compounds or ionic lamellar compounds, have attracted considerable attention recently due to the presence of large interlayer spaces, positively charged layers and solvation molecules. LDH-based materials are frequently used for environmental remediation especially for the elimination of dye compounds. This review compiles an extensive list of LDH-based low-cost sorbents and photocatalytic catalysts for dyes from the vast literature. Furthermore, performance, key factors and mechanisms involved in the processes are also included. Lastly, some major challenges together with prospects in this research field are discussed and highlighted. In conclusion, the application of LDH-based materials in the area of adsorption and catalysis science represents a useful and viable method, resulting in the outstanding capabilities in the removal of dyes from aqueous systems.
 Zhongzhu Yang | Zhongzhu Yang received his bachelor's degree of environmental engineering from Hunan University of Science and Technology. Now he is studying for a PhD degree at Hunan University. His research interests focus on clay-based materials, novel photocatalysts and adsorption materials for environmental applications. |
 Fenghua Wang | Fenghua Wang received her doctor's degree of analytical chemistry from Hunan University. Then she worked in Hunan Normal University as a post-doctor. Her research interests are in biochemistry and analytical chemistry. |
 Chang Zhang | Chang Zhang received his PhD degree at Hunan University. He ever worked as a as visiting scholar of Stanford University. He is an associate professor and doctoral supervisor in Hunan University now. His research interests are in remediation of water environment and water pollution control techniques. |
 Guangming Zeng | Guangming Zeng received his PhD from Wuhan University in 1988. Later, he worked at the College of Environmental Science and Engineering, Hunan University. Now he is a professor at the Hunan University and also the director of Environmental Science and Engineering. His research interests include biosensor and bioimaging, especially fluorescence imaging in biological system based on QDs. After years of efforts, he has been rewarded many prizes and cultivated hundreds of master degree candidate and doctoral candidate. |
 Zhigang Yu | Zhigang Yu received his Master's degree of environmental engineering from Hunan University. His research interest lies in pollutant removal from sediment and water, and also the synthesis of nanomaterials as adsorbents and photocatalysts in contaminated water. |
1. Introduction
Nowadays, the water pollution of organic dyes resulting from paper-made, dyeing, and other industries effluent has attracted global concern because of its significant impact on public health.1 Besides the obvious aesthetic consequences, dyestuff discharges increase the biotoxicity and carcinogenic effects.2,3 Furthermore, dyes are usually poorly biodegradable or recalcitrant to environmental conditions because of their complex structure and xenobiotic properties. Hence, color removal from dye effluents is one of the several major environmental concerns. Several methods have been developed for the remediation of wastewater containing dyes, including coagulation,4 biological treatment,5 ion exchange,6 filtration,7 adsorption8 or photocatalysis.9 Among these approaches, adsorption and photocatalysis have been widely used and proved to be reliable and capable of removing dyestuffs from wastewater.10–15Adsorption offers flexibility in design and operation, in many cases it will generate high-quality treated effluent. In addition, owing to the reversible nature of most adsorption processes, the adsorbents can be regenerated by suitable desorption processes for multiple use. Adsorption is hence recognized as the most versatile process used in lesser developing countries and is currently being used extensively for the removal of organic pollutants from the aqueous media.16,17 Recently, many conventional sorbents have been used to remove dyes from wastewater, such as activated carbon,18 bentonite,19 and metal oxides,20 fly ash21 and coal.22 However the main obstacles of these adsorbent materials are the relatively low adsorption capacity and the difficulty to reuse. The search for better efficiency and less-cost adsorbents seems unceasing.23 At the meantime, photocatalysis which utilizes renewable solar energy to activate the chemical reactions via oxidation and reduction is a sustainable technology to provide solution for environmental issue. This photocatalysis system has attracted great interests from science community as the most promising way to solve the environmental problems, especially getting rid of residual dyes pollutants from wastewater stream.24,25 This technique is based on the band theory and the n-type semiconductor is used as the photosensitized material, which is mainly metal oxide or metal sulfide. TiO2, for example, is one of the most widely used photocatalyst, based on its good properties like chemically inert, non-toxic to organisms, well-resourced, high energy gap.26,27 But the industrial application is constricted by its low specific surface area, low surface adsorption rate and difficulty in recycling.28 Developing recyclable semiconductor material which has a high quantum yield and visible light absorption rate is of great importance for the photocatalysis field.
Layered double hydroxides (LDHs), which are referred to anionic clays in comparison with cationic clays and also as hydrotalcite-like compounds (HT) are an important class of ionic lamellar solids. Layered double hydroxides (LDHs) with the general formula [M1−x2+Mx3+(OH)2(An−)x/n]x+·mH2O, where M2+ and M3+ are divalent (e.g., Mg2+, Co2+, Ni2+, Zn2+, Cu2+) and trivalent cations (e.g., Al3+, Fe3+, Ga3+), respectively; x, ranging from 0.20 to 0.33, stands for the molar fraction of M3+ in the metallic ions; and An− is interlayer gallery anion (e.g., CO32−, Cl−, NO3−, SO42−). The excessive positive charges are balanced by intercalated hydrated anions in the inter-lamellar domain. The cation composition of the hydroxide layers, their charge density given by a M2+/M3+ molar ratio, as well as interlayer anion composition can be tailored during LDH synthesis.29–31 The structure of LDHs and a typical octahedral unit are shown in Fig. 1. By heating to 450–500 °C, LDHs can be converted into mixed metal oxides (MMOs), which exhibit fine dispersion of metal cations and high surface area. An important property of MMOs is the so-called ‘‘memory effect’’, that is the calcined anionic clays can reconstruct their original layered structure after adsorption of various anions.32,33
 | ||
| Fig. 1 The structure of a layered double hydroxide, with interlayer SO42− anions. | ||
A set of characterization was performed to get better insight into the structural properties of LDH based materials. Typical XRD patterns of LDHs and CLDHs both before and after sorption experiments are shown in Fig. 2. A series of sharp (00l) peaks indexed as (003), (006), and (009) appearing as symmetric lines at low 2θ angle correspond to the basal spacing, indicating the presence of an ordered stacking sequence. The basal spacing of LDHs can be calculated based on the XRD results. After the adsorption, the XRD patterns of the recovered LDHs (Fig. 2b) are almost unchanged compared with the original LDHs (Fig. 2a), but their basal spacing may be different. The interlayer spacing is always dependent on the size and orientation of the charge-balancing anion. The XRD pattern of CLDHs (Fig. 2d) shows that layered structure of the original LDHs is completely destroyed. The similar peak positions of the synthesized LDH and CLDH imply that the layered structure of LDH can be maintained after dyes sorption by CLDHs.34 The morphology of the as-prepared adsorbent was observed by SEM and TEM. Fig. 3 shows the typical SEM image and TEM image of the LDHs. SEM image proves that the pure LDH has a hexagon structure and overlapping crystals.35 As illustrated in Fig. 3, TEM image shows that the LDHs are smooth, well-shaped in hexagonal form, and overlapping crystals.36
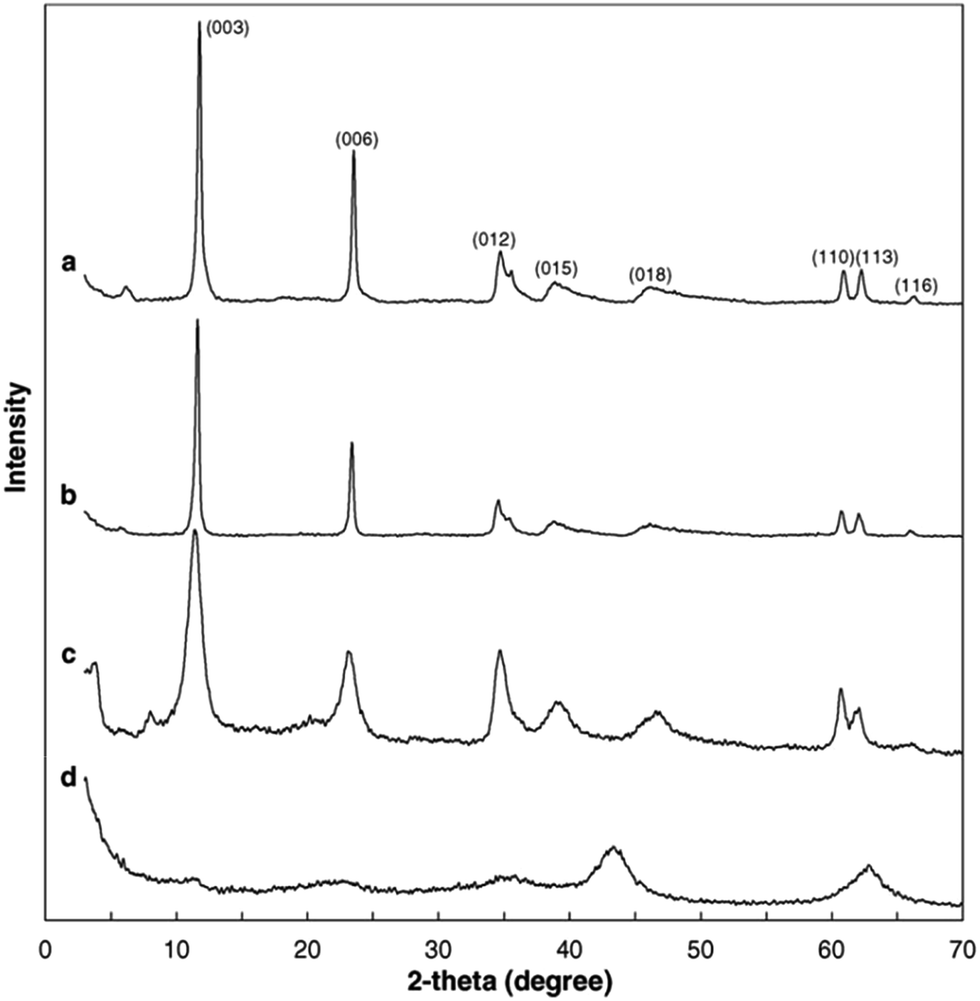 | ||
| Fig. 2 X-ray diffractograms of layered double hydroxides (LDHs) and calcined LDHs (CLDHs) before and after Brilliant Blue R (BBR) sorption. (a) LDHs; (b) LDHs after BBR sorption; (c) CLDHs after BBR sorption; (d) CLDHs (adapted from ref. 34 with Copyright from 2005 Elsevier). | ||
 | ||
| Fig. 3 SEM images (a) and TEM images (b) of Zn–Al LDH (image (a) adapted from ref. 35 with Copyright from 2014 Elsevier and image (b) adapted form ref. 44 with copyright from 2016 Elsevier). | ||
LDHs have been used as adsorbents or anion-exchangers for the removal of various anionic species in aqueous solutions, because these compounds present properties such as a layered structure, high porosity, high surface area, and interlayer anion mobility.37 Our previous research has demonstrated that the Fe–Al LDH is capable for the reduction of bromate in solution.38 Besides the excellent efficiency the field of adsorption, the photo-decomposition performance of dyes was found on LDHs composed of various metals such as Ni, Zn, and Cr. It resulted from the well distribution of metals in the hierarchical structure, because that doping or composition would enhance the dye sorption inside the particle and also high light absorption in the spectral range.11,28,39 Indeed, increasing interest has been diverted to evaluating the ability of LDHs to remove dyes from aqueous solutions by the process of adsorption and photocatalytic degradation. This is because LDHs have exhibited a great potential to efficiently remove harmful dyes due to their superior characteristics as mentioned in the preceding discussion.
Although the existence of reviews highlights dyes removal from aqueous solution by adsorption40,41 and the application of LDH-based materials in the area of environmental remediation,31,42,43 there is no review article describes the use of LDH materials in the removal of dyes. This review introduces briefly the use of LDH materials as adsorbents and catalysts in the decontaminant of dyes. We addressed the complexity of the many factors that influence the adsorption process as well as mechanisms. Besides we also examine various LDHs based materials used as photocatalysis that are capable of degrading dyes, and describe their performance, characteristics, advantages and limitations. Finally, challenges and outlook are put forward to facilitate exciting developments in this promising area in the future.
2. Adsorption of dyes on LDHs
The high anion exchange capacity and large surface area of LDHs, their flexible interlayer region that is accessible to various ionic and non-ionic compounds are important characteristics promising their better removal performance of contaminants from aqueous system. Thus far, various dyes including anionic dyes, cationic dyes, and non-ionic dyes are investigated. Fig. 4 shows a process flow diagram of LDH preparation from coprecipitation method and batch adsorption process for dye removal. The summaries of LDHs that have been used to treat various dyes along with their adsorption capacity of each are presented in Tables 1 and 2. It was clearly that dye sorption was influenced by pH, temperature and various functional groups presented on LDHs. Here we have outlined the above mentioned factors that influenced the dyes removal process.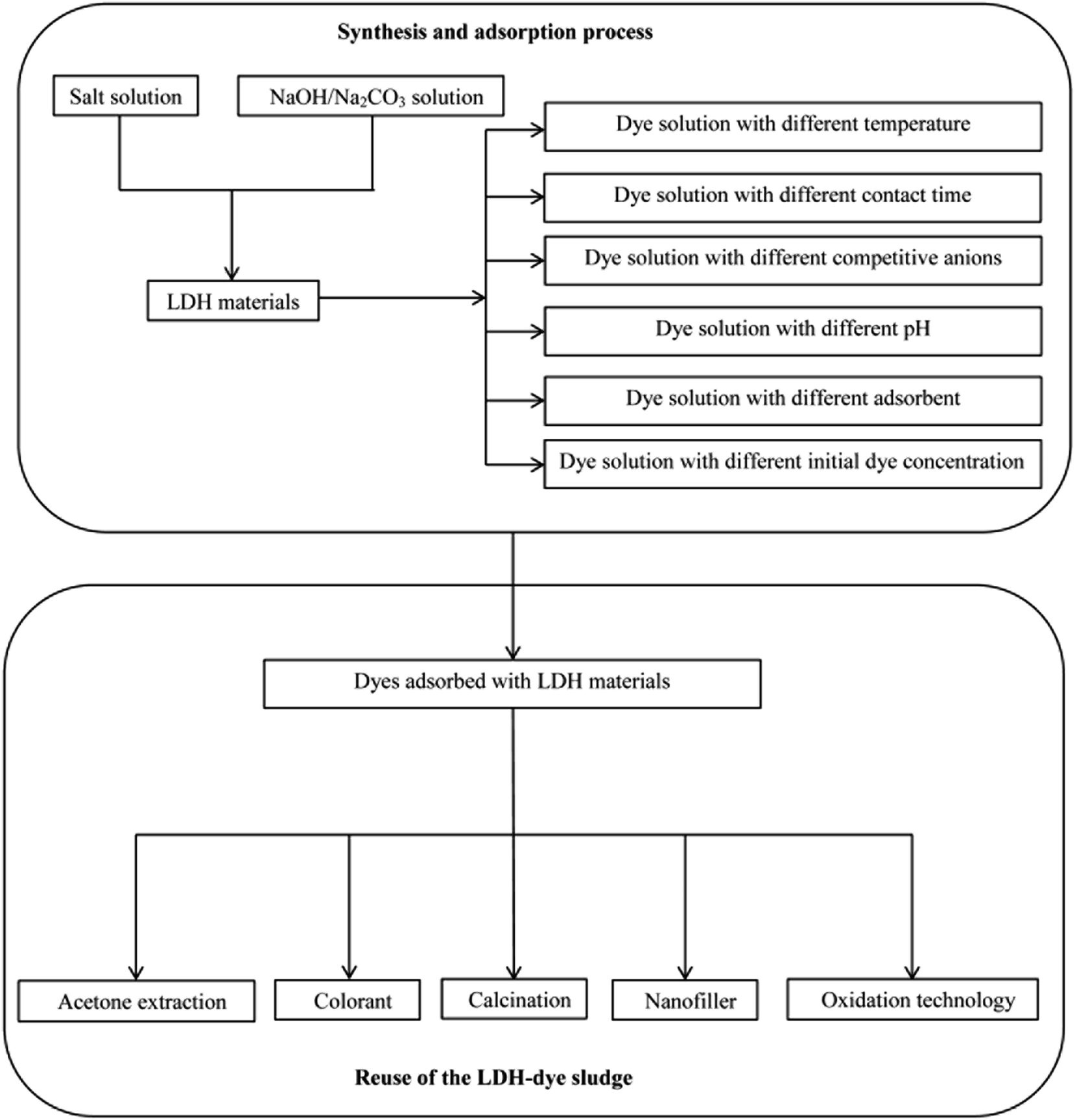 | ||
| Fig. 4 Schematic diagram of the system for various dyes removal and reuse of the LDH-dye sludge. | ||
| LDHs | Synthesis | Dyes | Optimum pH | Nature of adsorption | Adsorption capacity (mg g−1) | Kinetic model | Isotherm | Ref. |
|---|---|---|---|---|---|---|---|---|
| Mg–Al LDH | Coprecipitation | Congo red | — | Endothermic | 111.111 | 2nd | L | 45 |
| Mg–Al LDH | Hydrothermal | Methyl orange | 4 | — | 329.27 | 2nd | F | 10 |
| Mg–Al LDH | Coprecipitation | RR, CR, AR 1 | — | — | 59.49, 37.16, 108 | 2nd | L | 46 |
| Mg–Al LDH | Coprecipitation | MO, OII, OG | — | Exothermic | 1800, 1189, 769 | 2nd | — | 47 |
| Mg–Al LDH | Colloidal deposition | Methylene blue | — | 185 | 2nd | L | 48 | |
| Mg–AL LDH | Coprecipitation | Brilliant red K-2BP | 4 | Endothermic | 657.5 | 2nd | L | 49 |
| Mg–AL LDH | — | Acid blue 113 | — | — | 47 | 2nd | L | 50 |
| Mg–AL LDH | Coprecipitation | Benzopurpurin 4B | — | Endothermic | 153.88 | — | — | 51 |
| Mg–Al LDH | Coprecipitation | Acid blue 9 | — | — | 59.5 | — | L | 52 |
| Mg–Al LDH | Coprecipitation | Acid green 68:1 | — | — | 99.1 | 2nd | L | 53 |
| Mg–Al LDH | Coprecipitation | Reactive red | 2–10 | — | 59.49 | 2nd | L | 54 |
| Mg–Al LDH | Coprecipitation | Congo red | 4 | — | 37.16 | 2nd | L | 54 |
| Mg–Al LDH | Coprecipitation | Acid red 1 | 9 | — | 108 | 2nd | L | 54 |
| Mg2–Al LDH | Coprecipitation | Orange II | — | Endothermic | 1265 | — | L | 55 |
| Mg–AL LDH | Coprecipitation | Green bezanyl-F2B | 5–9 | — | 52.08 | 2nd | L | 56 |
| Mg–Fe LDH | Coprecipitation | Yellow GX | — | Endothermic | 53.76 | 2nd | L | 57 |
| Mg–Ni–Al LDH | Coprecipitation | Methyl orange | 6–9 | Endothermic | 118.5 | 2nd | L | 58 |
| Ca–Al LDH | Coprecipitation | Sunset yellow FCF | 4 | — | 398.41 | — | L | 59 |
| Ni–Fe LDH | Hydrothermal | Evans blue | — | — | — | — | — | 60 |
| Ni–Al LDH | Coprecipitation | Brilliant red X-3B | — | Exothermic | 48 | 2nd | L | 61 |
| Ni–Zn–Cr LDH | Accelerated carbonation | Acid scarlet GR | 5 | — | 122 | I-P | L | 62 |
| Cu–Al LDH | Coprecipitation | Methyl violet 2B | — | — | 361 | 2nd | L | 63 |
| Zn–Al LDH | Stepwise growth | Methyl orange | — | — | 523 | — | — | 64 |
| Zn2–Al LDH | Coprecipitation | Acid red 97 | — | — | 299.5 | 2nd | — | 65 |
| Zn2–Al LDH | Coprecipitation | Evans blue | — | Endothermic | 491.93 | — | L | 66 |
| Zn2–Al LDH | Coprecipitation | Chicago blue sky | — | Endothermic | 501.36 | — | L | 66 |
| Zn2–Al LDH | Coprecipitation | Niagara blue | — | Endothermic | 536.13 | — | L | 66 |
| Zn3–Al LDH | Coprecipitation | Methyl orange | — | — | — | — | F | 67 |
| Zn3–Al LDH | Coprecipitation | Fast green | — | — | 51.24 | — | F | 67 |
| Zn–Mg–Al LDH | Coprecipitation | Methyl orange | 3 | — | 883.24 | 2nd | L | 68 |
| HB–Zn–Al LDH | Coprecipitation | Methylene blue | 4 | — | 30.87 | 2nd | L | 69 |
| Fe3O4–Zn–Cr LDH | Hydrothermal | Methyl orange | 6.4–7.3 | — | 240.16 | 2nd | — | 70 |
| Fe3O4–Zn–Cr LDH | Hydrothermal | Methyl orange | — | — | 528 | — | L | 71 |
| DA–Mg–Al LDH | Coprecipitation | Methyl orange | 3 | Exothermic | — | 2nd | — | 72 |
| ILs–Mg–Al LDH | Coprecipitation | Reactive orange 5 | — | Endothermic | 300.9 | 2nd | F | 73 |
| DGLN–Mg–Al LDH | Coprecipitation | Victorial blue B | — | — | 1064 | — | L | 74 |
| DGLN–Mg–Al LDH | Coprecipitation | Weak acidic green GS | — | — | 131 | — | L | 74 |
| DGLN–Mg–Al LDH | Coprecipitation | Ethyl violet | — | — | 415 | — | L | 74 |
| SDS–Mg–Al LDH | Self assembly | Disperse violet 28 | — | — | — | 2nd | Linear | 75 |
| SDBS–Mg–AL LDH | Coprecipitation | Safranin | 5–8 | — | 40.5 | 2nd | L | 76 |
| SDS–Mg–AL LDH | Coprecipitation | Safranin | 5–8 | — | 83.3 | 2nd | L | 76 |
| SDS–Mg–AL LDH | Calcination rehydration | Green bezanyl-F2B | 5–9 | — | 188.68 | 2nd | L | 56 |
| SDS–Mg–Al LDH | Coprecipitation | Direct blue G-RB | 5–10 | Endothermic | 707.76 | 2nd | L | 77 |
| SDS–Mg–Al LDH | Coprecipitation | Yellow 4 GL | 5–10 | Endothermic | 392.88 | 2nd | L | 77 |
| SDS–Mg–Al LDH | Coprecipitation | Acid red GR | 5–10 | Endothermic | 137.33 | 2nd | F | 77 |
| SDS–Mg–Al LDH | Coprecipitation | Disperse red 3B | 5–10 | Endothermic | 249.24 | 2nd | L | 77 |
| SDS–Mg–Al LDH | Coprecipitation | Basic blue | 5–10 | Endothermic | 165.11 | 2nd | L | 77 |
| LDHs | Synthesis | Dyes | Optimum pH | Nature of adsorption | Adsorption capacity (mg g−1) | Kinetic model | Isotherm | Ref. |
|---|---|---|---|---|---|---|---|---|
| C–Mg–Fe LDH | Coprecipitation | Methyl orange | — | Exothermic | 194.9 | 2nd | L | 78 |
| C–Mg–Fe LDH | Coprecipitation | Acid brown 14 | 4–11 | — | 370.0 | 2nd | L | 33 |
| C–Mg–Fe LDH | Coprecipitation | Orange G | 3–13 | Endothermic | 378.8 | 2nd | L | 79 |
| C–Mg–AL LDH | Coprecipitation | C.I. acid blue 9 | — | — | 194.48 | — | L | 80 |
| C–Mg–Al LDH | Opal inverse | Orange II | — | — | 1550.5 | — | L | 81 |
| C–Mg–AL LDH | — | Acid blue 113 | — | — | 2544 | Avrami's | L | 50 |
| C–Mg–Al LDH | Coprecipitation | Acid blue 29 | — | — | 36 | — | L | 82 |
| C–Mg–AL LDH | Coprecipitation | Indigo carmine | 5–9 | — | 1343 | — | F | 32 |
| C–Mg–AL LDH | Coprecipitation | Remazol red 3BS | 6 | Endothermic | 134.4 | 2nd | L | 83 |
| C–Mg–Al LDH | Coprecipitation | Acid green 68:1 | 3–10 | — | 154.8 | 2nd | L | 53 |
| C–Mg–Al LDH | Coprecipitation | Brilliant blue R | 3.5–13 | — | 615 | 2nd | F | 34 |
| C–Mg–AL LDH | Coprecipitation | Benzopurpurin 4B | — | Endothermic | 417.36 | — | — | 51 |
| C–Mg–Al LDH | Coprecipitation | Acid red G | 10 | Exothermic | 93.1 | — | — | 84 |
| C–Mg–Al LDH | Coprecipitation | Acid orange 10 | 4 | Endothermic | 665 | 2nd | L | 85 |
| C–Mg–Ni–Al LDH | Coprecipitation | Methyl orange | 6–9 | Endothermic | 375.4 | 2nd | L | 58 |
| C–Ni–Al LDH | Coprecipitation | Remazol brilliant violet | 6 | — | 150 | — | — | 86 |
| C–Zn–Al LDH | Hydrothermal | Methyl orange | — | — | 2nd | L | 87 | |
| C–Zn–AL LDH | Coprecipitation | Methyl orange | 6 | Endothermic | 181.9 | 2nd | F | 88 |
| C–MnOx–Zn–Al LDH | Intercalation reduction | Methyl orange | — | Endothermic | 617.28 | 2nd | R–P | 89 |
| C–Au–Zn–Al LDH | Coprecipitation | Methyl orange | — | — | 627.51 | — | L | 90 |
| C–GO–Ni–Al LDH | Hydrothermal | Methyl orange | — | — | 210.8 | 2nd | R–P | 91 |
2.1. Adsorption isotherms and capacities
Adsorption equilibrium is one of the crucial pieces of information needed for the proper analysis and the isotherm indicates how the molecules distribute between the liquid and solid phase when reaching the equilibrium state. Besides, isotherms help to provide information about the optimum use of adsorbents.92 A number of empirical models have been employed to analyze experimental data and describe how adsorbates interact with adsorbents. The Langmuir and Freundlich isotherms are the most frequently used to examine the adsorption date of dyes adsorption onto LDHs. The Langmuir model is based on the assumptions that all the adsorption sites are equivalent, and the adsorption occurs in a monolayer form without any interactions between adsorbed molecules,93 while the Freundlich isotherm assumes the heterogeneous surfaces and is known to be satisfactory for low concentrations.94The adsorption capacities of calcined LDHs for various dyes in aqueous system are also summarized in Table 2. It is evident that the calcined sample exhibited better removal efficiency compared with its parent hydrotalcite. At a similar BBR equilibrium concentration of 100 mg L−1, sorption capacity of calcined LDHs was more than 10 times greater than that of LDHs.34 In another adsorption experiment, calcined and non-calcined LDHs were used as adsorbents to remove azo dye acid green 68:1 in an aqueous solution. The results indicated that calcined Mg–Al LDH possesses greater adsorption capacity (154.8 mg g−1) than non-calcined Mg–Al LDH (99.1 mg g−1). The larger sorption capacity of CLDH than that of LDH can be attributed to their property of structural reconstruction, higher surface area, the presence of stronger basic sites and anion exchange.33,53
2.2. Adsorption kinetics
The calculated kinetic parameters can be of a great practical value for technological applications. Many studies used kinetic models to study the fast changing kinetic data to estimate the sorption rates and to determine the possible controlling mechanism and the potential rate-limiting steps.97 In most experiments the amounts of sorbed dyes increased rapidly within the initial time and remained almost unchanged after complete equilibrium. Among the models to study the adsorption of dyes using LDHs materials: (1) the pseudo-first-order kinetic model; (2) the pseudo-second-order kinetic model; and (3) the intra-particle diffusion model are the three most widely used. The assumption of the pseudo-first-order model was physical adsorption and the solute uptake rate with time is directly proportional to ratio of the solute concentration and the amount of solid, while the pseudo-second-order kinetic model is based on the assumption that the rate-limiting step may be chemical adsorption or chemisorption involving valence forces through sharing or exchange of electrons between adsorbent and adsorbate.98Besides the surface adsorption, intraparticle diffusion or pore diffusion plays a key role, and therefore the rate-limiting step may be involved in film and/or pore diffusion. To determine if the rate limiting step is intraparticle diffusion or not, some researchers applied the Weber–Morris model diffusion law. In the removal of CR by Mg–Fe LDH, the effect of intra particle diffusion resistance on adsorption was evaluated and results showed that intra particle diffusion had a significant role in the adsorption.95
It is noted that pseudo-second-order model well fitted to the kinetic data for all the studied process rather than pseudo-first order (Tables 1 and 2). This demonstrates that the rate-limiting step in the adsorption involves chemisorption due to valence forces through the sharing or exchange of electrons between sorbent and sorbate, complexation, coordination, and/or chelation.100 The mechanism of dyes sorption by LDHs is complex and probably involves a combination of external mass transfer and intraparticle diffusion through the macropores/micropores of LDHs.
2.3. Factors affecting adsorption of dyes on LDHs
Optimisation of aqueous conditions will greatly help the development of industrial-scale dye removal treatment process. There are seven factors that influence the dyes removal, as mentioned in the following sector.Apart from the endothermic adsorption process, several studies reported on adsorption process are of exothermic in nature. For example, adsorption of MO, OII and OG on fresh Mg–Al LDH, and direct calorimetry measurements of the enthalpy change accompanying the dye retention by LDH were carried out to demonstrate the exothermic character of the overall mechanism.47 The other example was the uptakes of MO onto DA–Mg–Al LDH decreased with an increase of temperature, which reflected that LDH may be unstable at high temperature.72 Tong et al.84 studied the effect of temperature on adsorption of ARG onto calcined Mg–Al LDH from 298 K to 338 K and found the adsorption slightly decreased with increasing temperature, which indicated that the lower temperature favored the adsorption of ARG and it was an exothermic process.
 | ||
| Fig. 5 Effect of adsorbent dosages on the removal of AB 14 by Mg–Al LDH and calcined Mg–Al LDH (adapted from ref. 33 with Copyright from 2013 Elsevier). | ||
The percentage removal of MO increased from 39% to 96% when the adsorbent dosage increased from 0.2 to 10 g L−1. This was probably due to the low adsorbent dosage that causes the dispersion of LDHs grains in aqueous solution, therefore all types of sites on the adsorbent surface were entirely exposed which will facilitate the accessibility of MO molecules to a large number of sites. In this case, the adsorption on the surface was saturated quickly, showing a high uptake capacity. Lower adsorption capacity of the calcined Mg–Al–Ni LDHs was, however, obtained with the higher adsorbent dose. Because at higher particle concentrations the availability of sites with higher energy decreased with a larger fraction of sites with lower energy becoming occupied, leading to a lower adsorption capacity. Besides, higher adsorbent amount enhanced the probability of collision between solid particles and therefore created particle aggregation, causing a decrease in the total surface area and an increase in diffusion path length, both of which contribute to the decrease in the adsorption capacity.58 However, the OG amount adsorbed by both LDH and CLDH increased with increasing sorbent dose until 1.4 g L−1, from this dose the quantities of dye adsorbed on both materials were still unchanged. This may be explained by the good dispersion of materials particles on OG solutions, where the adsorbed and exchanged sites of materials were probably more open.79 These controversial findings highlight the needs for a comprehensive investigation of the material property in order for cost-effective application.
The surface charge of LDH was positive when pH < pHPZC and negative when pH > pHPZC. Theoretically, at pH < point of zero charge (PZC), the surface gets positively charged, which enhances the adsorption of the negatively charged dye anions through electrostatic forces of attraction. At pH > pHPZC, the surface of LDHs particles gets negatively charged, which favors the adsorption of cationic dye.103 Therefore, at a solution pH lower than pHPZC, hydrogen ions were in the solution making the surface of the adsorbent with more positive charges, which promoted the electrostatic attraction between the negatively charged anion of the dye and the surface of the adsorbent. In contrast, the surface of the LDH may acquire negative charges at a solution pH higher than pHPZC. The competitive effects of OH− ions and the electrostatic repulsion between the anionic dye molecules and the negatively charged active adsorption sites on the LDH would result in a decrease in the adsorption capacity.10,104 For example, the adsorption of Sunset Yellow FCF food dye was favored at pH values near 4.0. Between pH 4.0 and 10.0, the adsorption of dye by Ca–Al LDH decreased with increasing solution pH. The authors reported that the decreased adsorption was due to the deprotonation and competition with the OH−.59 The effect of the pH value on the adsorption of MO on the uncalcined and calcined Mg–Ni–Al LDH were exampled. There was a decrease in MO adsorption when the equilibrium solution pH was less than 4 and greater than 10, which was partially caused by the dissolution of LDHs.58 Similar explanation was given for the adsorption of MO onto Zn–Al calcinated LDH, which the percentage of adsorption reached the maximum at pH 6.88
However, a few sorption cases were found to be independent of pH, this is probably because of the buffering properties of uncalcinated LDHs.76,77,105 At low pH values, the hydroxyl groups in the LDH interlayers can neutralize the hydrogen ions in the solution, while at high pH values, the surface is deprotonated, and thus may have buffering effects on the hydrogen groups. These conditions also happened in the calcinated LDH that the effect of initial pH over a wide pH range on the adsorption were minimal.32–34,79 Effect of pH on BBR sorption at a fixed initial concentration and CLDH dosage is shown in Fig. 6. It may be concluded that the effect of initial pH over a wide pH range (3.5–13.0) on anionic dye removal by CLDHs is minimal. The reason was probably due to the reconstruction of the LDH structure with the intercalation of dye molecular into the CLDH33 or OH− release34 in aqueous solutions. It should be noted that, however, the effect of pH on both cationic and anionic dye sorption by many other adsorbents, for example, fly ash and acid-activated bentonite was pronounced.106,107 This is an advantage of CLDH in relation to other types of adsorptive materials that usually show a large dependency on pH for adsorption.
 | ||
| Fig. 6 Effect of initial pH on Brilliant Blue R (BBR) sorption by calcined layered double hydroxides (CLDHs) and on equilibrium pH (adapted from ref. 34 with Copyright from 2005 Elsevier). | ||
The relationship between the initial and final pH for MO adsorption on Mg–Al LDH was checked. At low initial pH (pHi), the final pH values (pHf) were higher than pHi values, which was due to the protonation and/or the dissolution of LDH.10 Fig. 6 also shows the difference between initial and final pH values. It can be seen that when initial pH values were lower than 10.0, final pH values can be enhanced and finally stabilized within a narrow alkaline range of 10.6–10.8. This is environmentally meaningful for precipitation or co-precipitation of some co-existing metal cations like Ca2+, Pb2+ and Cd2+.34 The increase of the final pH value was observed and this is due to the proton consumption during the reconstruction process. Thus, the pH increase probably produces an increase of the negative surface charges and the interaction between the anion dye and the surface charges decrease.85 At very low pH values, there are the possibility of dissolution and/or leaching of the CLDH. The dissolved material causes a fast increase in pH medium and consequently a decrease of the dissolution rate. Thus, the remaining adsorbent material is then responsible for the high adsorption capacity.53,69,95 It should be mentioned that the decrease in uptake capacity with decreasing pH may be due to the structure of layered materials with hydroxide sheets was partial destroyed.84,85,104 The discrepancies on the reported in lower pH influencing on adsorption may be due to the different dye molecule forms, dissociation constant67,95 and different origins of the LDH samples.50
 | ||
| Fig. 7 Effect of material component on MO removal of Au/ZnAl LDO nanocomposites. (a) Different Zn content with constant Au content of 1% and (b) different Au content with constant Zn content of 87% (adapted from ref. 90 with Copyright from 2013 Elsevier). | ||
For a given choice of metal cations and interlayer anions, the best crystalline LDH phase was generally obtained with an M2+/M3+ ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.108 In the removal of OG, various Mg/Fe molar ratios of 2–5 were prepared. As seen from XRD, the influence of Mg/Fe molar ratios was apparent and the good crystallinity was obtained when Mg/Fe molar ratio equals 3.79 However, in the adsorption of AB 9, AY 23, and AR 37 onto Mg–Al LDH of variable Mg/Al molar ratio, the optimum molar ratio was reported to be 4.109 The adsorption abilities of calcined and uncalcined Mg–Al LDH with different Mg/Al molar ratios were investigated to adsorb AB 9. The optimum adsorption of AB 9 was achieved by LDHs with Mg/Al molar ratios varying from 3.1 to 4.4. The highest pore volumes were measured for materials with the best dye affinity at Mg/Al molar ratios of 3.1 and 4.4. When Mg/Al molar ratio was above 4.4, the surface area started to decrease and so did the adsorptive capacity.52 In the case of CLDH, initial dye affinity tests indicated particularly strong adsorption for materials with Mg/Al ratios of 3.1 to 7.6. At low initial Mg/Al, there would be less MgO in the CLDH product but with more Mg(Al)O mixed oxide phase that can be formed after calcination. This therefore can lead to less basic sites that may be responsible to less adsorption of AB 9.80
:1.108 In the removal of OG, various Mg/Fe molar ratios of 2–5 were prepared. As seen from XRD, the influence of Mg/Fe molar ratios was apparent and the good crystallinity was obtained when Mg/Fe molar ratio equals 3.79 However, in the adsorption of AB 9, AY 23, and AR 37 onto Mg–Al LDH of variable Mg/Al molar ratio, the optimum molar ratio was reported to be 4.109 The adsorption abilities of calcined and uncalcined Mg–Al LDH with different Mg/Al molar ratios were investigated to adsorb AB 9. The optimum adsorption of AB 9 was achieved by LDHs with Mg/Al molar ratios varying from 3.1 to 4.4. The highest pore volumes were measured for materials with the best dye affinity at Mg/Al molar ratios of 3.1 and 4.4. When Mg/Al molar ratio was above 4.4, the surface area started to decrease and so did the adsorptive capacity.52 In the case of CLDH, initial dye affinity tests indicated particularly strong adsorption for materials with Mg/Al ratios of 3.1 to 7.6. At low initial Mg/Al, there would be less MgO in the CLDH product but with more Mg(Al)O mixed oxide phase that can be formed after calcination. This therefore can lead to less basic sites that may be responsible to less adsorption of AB 9.80
These observations collectively demonstrate that the nature and content percent of precursor metals have a significant impact on the adsorption due to the influence on crystallinity of the samples.
Considering the superior adsorption capacity of nanomaterials, a few nanomaterials based LDHs have been prepared to check the performance. Zhang et al.90 synthesized Au/ZnAl–LDO nanocomposites through a facile calcination process of HAuCl4 intercalated Zn–Al LDH nanocomposites. AuNPs with small size were well dispersed in calcined Zn–Al LDO matrix. The presence of AuNPs with smaller size, not only had the adsorption ability for MO, but could fabricate lot of porous channel in LDO matrix for the adsorption, resulting in a great high adsorption property. The percentage removal of MO from the solution was 98.99% compared to 48.18% without Au loading. In another study, ZnO nanorods decorated calcined Mg–Al LDH was prepared via a homogeneous precipitation process, the sample exhibited a good sorption capacity of 242 mg g−1 for ARG.110 The same study group synthesized ZnO nanoparticles immobilized on flaky layered double hydroxides as enhanced adsorption for removal of ARG, which resulted in specific surface area of FLDHs (249 m2 g−1) and a short adsorption equilibrium time.111
Using magnetic LDH nanoparticles has aroused great interests for the removal of dyes from aqueous solutions. The magnetic adsorbent with adsorbed dye can be easily separated magnetically and recycled after catalytic regeneration with advanced oxidation technology. Several researchers have investigated the use of magnetic LDH nanocomposites in the removal of dyes molecular. Yang et al.91 synthesized reduced graphene oxide, zero-valent nickel, and Ni–Al mixed metal oxides (rGO/Ni/MMO) by calcining graphene oxide (GO)/layered double hydroxide (LDH) for the purpose of removing MB and found it to be very effective (210.8 mg g−1) compared to GO/MMO at equilibrium 72.6 mg g−1. The high adsorption ability of the hybrid may benefit from the hybrid with less carboxyl groups and small-sized nanoparticles.112–114 Taking advantage of unique magnetic response and large surface area of magnetic Fe3O4. Chen et al.71 prepared the novel magnetic Fe3O4/Zn–Cr LDH and applied it to remove MO. The material had an excellent adsorption capacity (528 mg g−1), which resulted from the higher surface area (114 m2 g−1). The same research group subsequently synthesized the same material from electroplating wastewater and studied the adsorption of MO from heavy metal wastewater. The maximum capacity of MO was found to be 240.16 mg g−1, at the same time 99% of heavy metal ions can be effectively removed into precipitates. In addition, this material with adsorbed dye can be easily separated by a magnetic field (Fig. 8).70
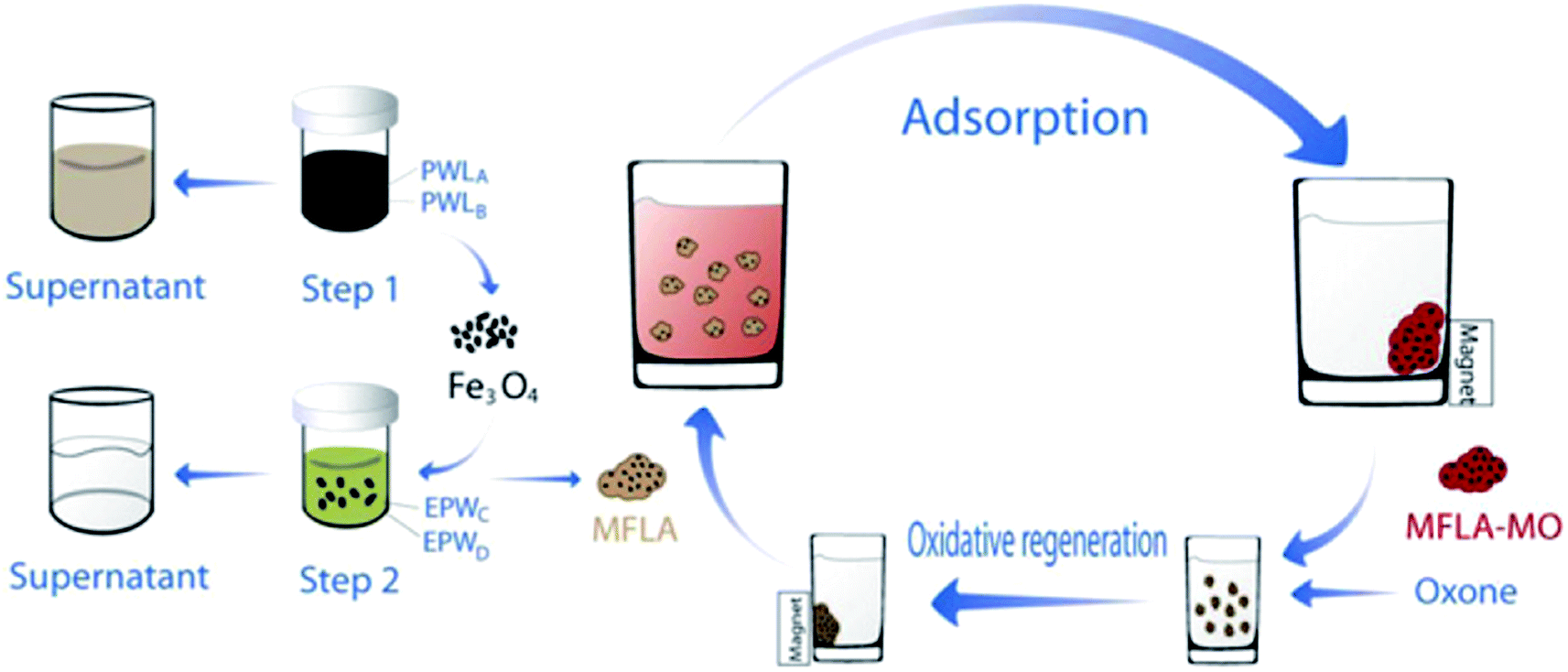 | ||
| Fig. 8 Synthesis route of MFLA and their application for removal of MO with the help of an external magnetic field (MFLA: magnetic Fe3O4/Zn–Cr LDH adsorbent, PWL: pickling waste liquor, EPW: electroplating wastewater. Adapted from ref. 70 with Copyright from 2012 Elsevier). | ||
From the above literature study, it is clear that modified LDHs achieve a higher dye removal ratio as compared to the virgin LDHs. This is because of the larger surface area and functional groups generated by the modification. The surface and functional groups act as active sites which are easily accessible to the dye molecules.
The corresponding pore size distributions of the uncalcined and calcined Mg–Ni–Al LDH and the experimental results of MO adsorptions at various initial concentrations with contact time are shown in Fig. 9. The calculated percentage of MO removal decreased from 95% to 86.8% with initial MO concentration increasing from 20 to 100 mg L−1. The calcined LDH performed a better adsorption percent than the uncalcined LDH because the pore volume of the Mg–Ni–Al LDH increased by heating. The difference between equilibrium time obtained with Mg–Ni–Al LDH and calcined Mg–Ni–Al LDH could be explained by the fact that MO anions adsorption onto calcined Mg–Ni–Al occurs by surface and ion exchange phenomena by reconstruction.58 For a given adsorbent dose the total numbers of available adsorption sites are fixed, thereby the same amount of dyes could be adsorbed. The removal percentage of AC 97 onto Zn2–Al LDH decreased with an increase in initial dye concentration. That resulted from the ratio of initial mole numbers of AC 97 to the available surface area was high at a higher initial concentration.65
 | ||
| Fig. 9 The pore size distribution curves (a) and effect of time and initial concentration and (b) in the adsorption of MO on the two samples (adapted from ref. 58 with Copyright from 2012 Elsevier). | ||
The effects of competing anions SO42−, Cl− and CO32− on BBR sorption by CLDHs were invested, the effects of SO42− and Cl− on the process were minimal although the molar ratios of Cl− or SO42− to BBR were up to 12. The adsorption even slightly increased with the increment of molar ratios from 2 to 12, which was probably caused by a salting out effect. Evidently, the dyes are not “pushed out” from the sorbent by inorganic salts, which is important feature for potential applications to real wastewaters.106 Unlike the performance of Cl− and SO42−, the presence of CO32− suppressed the sorption, due to the fact that the previously existing interlayer CO32− ions in LDHs are difficult to be exchanged by other anions.34,115 However, for the case of CO32−, the CLDH still demonstrated a high sorption capacity for AB 14 even at a high concentration of CO32−.33 In the case of MO adsorption on calcined Zn–Al LDH, the influence of competing anions was reported to follow the order PO43− > CO32− > SO42− > NO3− > Cl−, which confirmed that the divalent anions had more effect on MO adsorption compared to the monovalent anions.88 In another study of MO adsorption by uncalcined and calcined Mg–Ni–Al LDH. The results showed that adsorption of MO decreased with increasing NaCl concentration. With the increasing ionic strength, the adsorption capacity decreases due to the competing Cl− anions with MO for surface adsorption.58
In general, it could be concluded that the anions of higher valence have a more significant interfering effect than the monovalent anions in the dyes adsorption by calcined LDHs. Among the anions, PO43− and CO32− appear to be the most competitive anions in the adsorption. Unfortunately, large questions remain as to how competing species regulate the adsorption and existed in the hydroxides, compelling evidence in this regard is currently lacking.
2.4. Mechanism of sorption
Adsorption are defined as the accumulation of a substance or material at an interface between the solid surface and the bathing solution.116 Mechanism studies of dyes adsorbed by LDHs have been carried out with either the assistance of the characterization of the structures of the LDHs before and after adsorption such as FTIR, XRD, TEM and SEM, or the postulation from comprehensive experimental observations on sorption kinetic and sorption isotherm. Generally, the dyes may be removed by LDHs via (1) surface adsorption (2) interlayer anion exchange (3) reconstruction/memory effect of calcined LDH precursors (4) electrostatic interactions.53,69,117It has been found that many organic compounds have a strong affinity to the surface of clay minerals. The surface adsorption involves the adhesion of dyes to the LDHs surface. Clearly, the parameter of sorbent surface area plays a significant role in this mechanism.82 The prepared ZnO–LDHs with different mass ratios have different specific areas and the one having a larger surface area shows a stronger adsorption capacity.110 For the adsorption of AB 9, the powder XRD analysis showed no obvious change in the d(003) spacing. Thus, at low concentration surface adsorption of the dye occurred.52
An anion exchange mechanism also plays an important role. The level of exchange depends on the substituent anions, M2+/M3+ ratio and the anions to be intercalated.53,97 Some ligands were intercalated to form organic–inorganic composite adsorbents, the intercalated sodium alginate helps in widening the interlayer space of the hydrotalcite-like anionic clays, making them accessible for intercalation of orange II dye.118 The adsorption of MO and GR onto Ca–Al LDH was studied by X-ray diffraction (XRD), infrared spectroscopy (MIR), scanning electron microscope (SEM), and near-infrared spectroscopy (NIR). All results indicated that MO ion was intercalated into Ca–Al LDH interlayers, and acidic scarlet GR was only adsorbed upon Ca–Al LDH surfaces.119 The mechanism of these interaction models is displayed in Fig. 10. Anion exchange may also happen on the surface. The surface carbonates or hydrogen carbonates are easily replaced by anionic dyes, and anion exchange can still be achieved even if the original interlayer anion is carbonate an ion that can be difficult to exchange from the LDH interlayer region.52 In the adsorption of AB 14 dye on Mg–Fe LDH, the result showed that the interlayer distance changed a little, meaning the exchange occurred mainly on the outer planner surfaces and at the edges of inter-layers in LDH containing CO32−.33
 | ||
| Fig. 10 The models illustration of MO and GR interacted with Ca–Al LDH (adapted from ref. 119 with copyright from 2012 Elsevier). | ||
Electrostatic interactions happened between the groups of the dye and the ligands intercalated into interlayers and this usually used for the removal of cationic dyes. Researchers synthesised heteropoly blue intercalated layered double hydroxide. The intercalation of large cluster anion [PW10Mo2O40]5− into LDH could induce the adsorption of cationic dye of MB onto HB–LDH, obviously. The HB–LDH shows much higher cationic dye adsorption capacity than pure LDH and the maximum adsorption capacity of MB was 30.87 mg g−1.69 Wei et al.74 prepared DGLN–LDHs hybrid to adsorb victorial blue B, through the electrostatic interactions between the DGLN and LDHs (Fig. 11), which was confirmed by the FTIR, resulting in the adsorption capacity of 1064 mg g−1.
 | ||
| Fig. 11 The proposed adsorption mechanism for cationic dyes with DGLH–LDH. | ||
Majority of studies related to the removal dyes by calcined LDH revealed predominant adsorption mechanism is reconstruct of the original layered structure in an aquatic environment, as expressed as follows:33
| [Mg1−xFex(OH)2](CO3)x/2 → Mg1−xFexO1+x/2 + (x/2)CO2 + H2O | (1) |
| Mg1−xFexO1+x/2 + (x/n)An− + (x)AB 14 + (1 + x/2)H2O → Mg1−xFex(OH)2(AB 14)x + xOH− | (2) |
The adsorption mechanism is presumed that the CLDH reconstructed the layered structure after adsorption of AB 14, the anions of AB 14 intercalated into the interlayer of LDH by chemisorptions. Ni et al.88 studied the treatment of MO by calcined Zn–Al LDH and presumed a similar adsorption mechanism. Zn–Al LDO reconstructed the layered structure after MO adsorption, the most of interlayer anions are OH−, however, there are parts of MO ions and CO32− intercalated into the interlayer of LDH by chemisorption (Fig. 12). Also in the reconstruction process, OH− ions are simultaneously released. Consequently, final pH of the aquatic solution will be enhanced. This is environmentally meaningful for precipitation or co-precipitation of some co-existing metal cations like Ca2+, Pb2+ and Cd2+.120,121 The reconstruction also makes contribution to a much larger sorption capacity of CLDH than that of LDH resulting from higher surface area and the presence of stronger basic sites. The specific surface area of the Mg–Ni–Al LDH increased from 137.4 to 246.5 m2 and the pore volume from 0.451 to 0.590 cm by heating at 500 °C.58 Another advantage is the shorter equilibrium time. For the sorption of BBR, both film diffusion and pore diffusion were involved in ion exchanges in LDHs, however, structural reconstruction and BBR intercalation occurred almost simultaneously in CLDHs and the processes were obviously rapid.34 The reconstruction mechanism could also been affected by the exposure time. A surface adsorption mechanism is proposed to account for the adsorption of the dye onto the calcined Mg–Al layered double hydroxides. X-ray powder diffraction analysis indicated that given sufficient time it was possible to intercalate the dye within the layered double hydroxides' interlayer via a reconstruction mechanism.80
 | ||
| Fig. 12 The schematic illustration of MO adsorption by Zn–Al LDO. (a) The calcined layered double hydroxides. (b) The layered structure of recovered products is restructured by adsorption. (c) MO intercalated into LDH layered by chemisorption (adapted from ref. 88 with copyright from 2007 Elsevier). | ||
2.5. The disposal of LDH-dye sludge
After adsorption, how to deal with the adsorbent sludge is of great interest to both scientific research and practical application. There are two main solutions on the disposal of the resulting sludge: regeneration and reutilization, among which the regeneration is the most frequently used.Ulibarri et al.122 proposed that by combustion at 450–500 °C, adsorbed organic pollutants can be almost completely eliminated and LDH or CLDH sorbents can be regenerated into CLDH-like materials based on the “memory effect”. A lot of the regeneration tests were carried out by calcining the exhausted adsorbents. However, most of the adsorbent was feasible only within the first three cycles after thermal regeneration.79,88,95,111 In the removal of BBR, thermal regeneration of LDHs and CLDHs after sorption were feasible only within the first two cycles, after which the regenerated materials suffered from a large loss in their sorption capacities. The large loss resulted from progressively decreasing crystallinity of the LDHs in structural reconstruction after thermal regeneration, as well as by the incorporation of organic species remnant of the thermally decomposed dye. A similar conclusion was confirmed in the removal of AG 68:1 from aqueous solutions by calcined and uncalcined layered double hydroxides.53
A few LDH regeneration studies have been performed by sulfate radical-based oxidation technology70,91 and acetone extraction.69,76 Chen et al.70 recycled the adsorbent after catalytic regeneration with advanced oxidation technology and reported that the adsorption properties of were drastic reduced, due to the reduction of specific surface area. Bi et al.69 showed that after the first regeneration cycles by acetone extraction, the removal efficiency of MO decreased from 75% to 33%, which may resulted from an incomplete acetone extraction of the adsorbed dye.
As for the reutilization of the LDH adsorbent sludge, Xue et al.65 added the LDH adsorbent sludge into polypropylene (PP) and found that the Zn2Al–AC97 LDH could significantly improve the thermal stability and UV shielding ability of PP. In addition, the resulting sludge was reutilized as a colorant filled in polymer materials and it exhibited the resistance to bleeding and fire.74
3. The photocatalytic degradation of dyes by LDH materials
Photocatalytic degradation offers an attractive solution to organic contaminants by completely converting molecular to carbon dioxide, water, and mineral acids.24,110 A number of photocatalysts have shown high photocatalytic activity. However, taking into account the large band gap and economic cost, they are unsuitable for treating large amounts of wastewater. Incorporation of photocatalysts in LDHs is a promising strategy for wastewater treatment. The pollutants would not only be adsorbed by LDHs but also degraded, and this could greatly increase the wastewater treatment capacity.Seen from Table 3, in the context of degradation of dyes, three types of LDHs-based photocatalysts have been hitherto created: (1) direct photocatalysts (2) mixed metal oxide (MMO) materials derived from LDH single precursors (3) supports to photocatalytic guests.
| LDHs | Kinetics of degradation | Dyes | Light irradiation | Ref. |
|---|---|---|---|---|
| Mg–Al LDH | — | Methylene blue, methyl orange | Vis | 123 |
| Ni–Ti LDH | 1st | Methylene blue | Vis | 124 |
| Cu–Co–Cr LDH | — | Malachite green | Vis | 125 |
| Zn–Al LDH | 1st | Acridine orange | UV | 126 |
| Zn–Al LDH Mg–Al LDH | 1st | Methyl orange, fast green | UV | 67 |
| Zn/M LDH (M = Al, Fe, Ti, Fe/Ti) | 1st | Rhodamine B | Vis | 127 |
| Zn3–Al LDH | — | Methyl orange | UV | 51 |
| Zn–Ti LDH | — | Methylene blue | Vis | 128 |
| Zn–Cr LDH | — | Methylene blue, methyl orange | Vis | 123 |
| Zn–Cr LDH | 1st | Rhodamine B, rhodamine 6G | Vis | 129 |
| Zn–Fe LDH | — | Methyl violet, malachite green | Vis | 130 |
| Zn–Cd–Al LDH | — | Methylene blue | Vis | 131 |
| Pt–Zn–Ti LDH | 1st | Rhodamine B | Sunlight | 11 |
| C–Ni–Mg–Fe–Al LDH | — | Drimaren red, drimaren navy | UV | 132 |
| C–Ni–Ti LDH | 1st | Methylene blue | UV-Vis | 133 |
| C–Zn–Al LDH | — | Methyl orange | UV | 134 |
| C–Zn–Al LDH | 1st | Methylene blue, methyl orange | UV | 135 |
| C–Mg–Zn–In LDH | — | Methylene blue | Vis | 136 |
| CeO2/Zn–Ti LDH | — | Methyl orange, methylene blue | UV | 28 |
| C–Bi–Zn–Ti LDH | 1st | Indigo carmine | Vis | 137 |
| C–CNT/Co–Ni–Al–Zn LDH | — | C.I. acid red 14 | Vis | 138 |
| C-Legume/Zn–Al LDH | — | Polybenzenesulfonamido sulforhodamine B | UV | 139 |
| C–Zn–Al–Ti LDH | 1st | Methylene blue, rhodamine B | Vis | 140 |
| C–CG–Zn–Al LDH | — | Methylene blue, orange G | Vis | 141 |
| C–Ni–Zn–Al LDH | 1st | Orange G | Solar | 142 |
| TiO2/Mg–Al LDH | — | Methyl orange | UV | 143 |
| TiO2/Mg2–Al LDH | 1st | Acid orange 7 | UV | 144 |
| Cu2O/Mg–Al LDH | — | Methylene blue | Vis | 145 |
| ZnO/Mg–Al LDH | — | Acid red G | UV | 146 |
| Pd(II)–Bi2O3/Mg–Al LDH | 1st | Methylene blue | Vis | 147 |
| SnO2/Mg–Al LDH | — | Methylene blue | UV | 148 |
| Ag/AgBr/Co–Ni LDH | — | Methyl orange | UV/Vis | 149 |
| BiOBr/Co–Ni LDH | — | Methyl orange, rhodamine B | UV | 150 |
| TiO2/Cu–Mg–Al RLDH | — | Methylene blue | UV/Vis | 151 |
| Fe3O4/Zn–Cr LDH | 1st | Methylene blue, methyl orange | UV | 71 |
| CuPcTs/Zn–Al LDH | — | Methylene blue | Solar | 152 |
| MoO42−/WO42−–Zn–Y LDH | 1st | Rhodamine 6G | Vis | 153 |
| MxOy/Zn–Ti LDH (M = Fe, Sn, Ce) | 1st | Acid red 14 | Vis | 154 |
3.1. Direct photocatalysts
LDH was very much promising material for pollutant degradation in advanced oxidative process.129 With the properties that LDHs have two or three different metals and the ratio among these metals could be varied, LDHs can be regard as “doped semiconductor”. Doping or composition would result in a state of defect energy between the valence band and conduction band, which would provide a spring board for photon generated electrons (e−). And this photon generated electrons can activate valence band electrons transmit to conduction band by visible light with lower energy, which would make the absorption edge move to visible light region. Thus, it would reduce the band gap energy of materials, and also improve the photo-catalytic property.28 Some works aimed at revealing the role of pristine layered double hydroxide (LDH) materials in the elimination of organic pollutants from solution.M2+–Ti LDHs were synthesized and confirmed that the doping of Ti into LDHs could improve the catalytic ability of dyes.28,127,128 Zn–Ti LDH was synthesized and resulted in high visible light photocatalytic activity for MB, which resulted from the lower band gap, the hierarchical structure and high specific surface. The high specific surface area would provide strong adsorption ability toward target dyes and lead to the generation of photoinduced electron–hole pairs of active sites. A wide distribution of macropores is favorable for the transportation and diffusion of species.128,155,156 Xia et al.28 observed the photocatalytic degradation performance of MO and MB by three Ti-based layered double hydroxides, which followed the order: CeO2/Zn–Ti LDHs > Zn/Al–Ti/SB LDHs > Zn/Ti LDHs. The same group also checked the degradation performance of RB by a series of Zn/M–LDHs (M = Al, Fe, Ti, and Fe/Ti) and found that after three regeneration cycles, the percentage degradation rate was still close to 90%.127 These phenomenon were attributed to the basis of electronic structure property and textural parameters of LDH. Some other researchers also reported that surface areas, pore volume pore size distribution and crystallite size lead to the promotion of photo-induced degradations of dyes.67,128,130,134
Silva et al.157 developed a novel series of visible active Zn–M (M = Cr, Ti, Ce) LDH photocatalysts for oxygen generation from water and the result showed that it can be regarded as a “doped semiconductor” in which ‘M (III)’ used as dopant. These findings motivates researcher to further study the semiconductor properties of Zn–M LDH materials in the degradation of dyes. Zn–Cr LDH were prepared and used for the removal of MB, MO123 and xanthene dyes,129 the study demonstrated that Zn–Cr LDH was an efficient photocatalyst for degradation of organic pollutants, but the rapid charge recombination and low efficiency in electron/hole separation would suggest that photocatalytic activity would be greatly limited.123 A novel magnetic Fe3O4/Zn–Cr LDH composite was successfully fabricated and the results revealed a higher removal of dye on the composite, compared to that on Zn–Cr LDH under the same conditions.71 K. M. Parida130 synthesized Zn–Fe LDH with different intercalated anions and studied the photodegradation of MV and MG under solar light, the tentative mechanism may be photocatalytic degradation (mineralization):130
| Catalyst + hν → Catalyst (eCB− + hVB+) | (3) |
| Catalyst (hVB+) + H2O → Catalyst + H+ + OH˙ | (4) |
| Dye + OH˙ → Dye product (DP) | (5) |
| Catalyst (eCB−) + O2 → Catalyst + O2−˙ | (6) |
| O2−˙ + Dye → Dye product (DP) | (7) |
3.2. Mixed metal oxide (MMO) materials
After being calcined under high temperature, bimetallic oxide could be formed, the second metal could be high dispersed and doped into the LDH materials, and then the photocatalysis of these materials could be dramatically improved. In particular, the mixed oxides derived from Zn, Al containing LDHs without or with Fe, Sn and Ti, are successful photocatalysts for the degradation of organic compounds like MO, MB and phenols in aqueous media.140The Zn–Al LDH samples were calcined at different temperatures and found that the band gap energy decreased as the calcination temperature increased. The photocatalytic activity is highly improved when samples are calcined at 500 °C due to the large amounts of ZnO phase formed.134 In another study, ZnO/ZnAl2O4 composite photocatalyst was prepared by calcining Zn–Al LDH and also showed a high degradation efficiency of MB and MO. The calcined Ni–Ti LDH nanocomposite particles were photoresponsive in the UV-VI region and indeed exhibited good photocatalytic activity for MB.133 Zhao et al.139 reported the fabrication of biotemplated LDH film and MMO framework from the legume and demonstrated its effective and recyclable photocatalysis (recyclable for even 6 times) for the decomposition of two kinds of dyes due to its large specific surface area and wide pore size distribution.
LDHs containing tetravalent cations have been synthesized to explore the possibility for photocatalytic degradation of various dyes.125,132,136,140 The degradation performance of MB and RhB was found to increase slowly with the increase of Ti content in the calcined Zn–Al–Ti LDH catalyst. This increased activity may be attributed to resultant effects of decreased band gap energy and BET surface area.140 By the isomorphous replacement of Zn for Mg in coprecipitation system and the photocatalytic performance of Mg–Zn–In ternary layered materials have been invested by Li Huang.136 The research group found that the presence of Mg in the composite could improve the stability of the catalyst and doping zinc into the Mg–In system enlarged the surface area as well as the pore volume of calcined sample. The ternary Mg–Zn–In layered photocatalyst exhibits a considerably high activity in the degradation of MB resulted from the special structure of LDH. Both of the studies involved dye-sensitized process and the overall photocatalytic process schematically presented in Fig. 13.
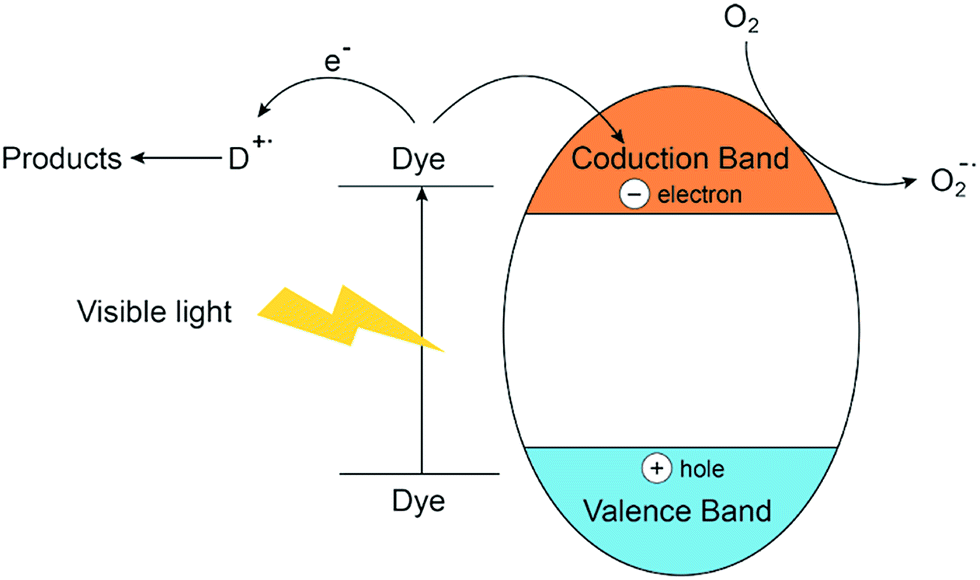 | ||
| Fig. 13 Electron transfer process from the excitation of dye in the visible region. | ||
Enrichment of reactants by adsorption is required for a highly efficient photocatalytic performance.158,159 After calcination, the large surface areas of the MMO produce a large amount of electron/hole pairs and ensure the adsorption of dye molecule to the active sites, therefore the catalytic efficiency is enhanced.160 The high dispersion of MMO nanocrystals and wide pore size distribution not only benefit the transfer of the light-generated charge carriers to the surface to react with dye molecules but also allow fast diffusion of reactants and products. In addition, the samples would be a relatively lower band gap energy after calcination. All these factors make the calcined LDHs showing better activities than the uncalcined LDHs.
3.3. Supports to photocatalytic guests
Semiconducting photocatalysts have attracted extensive attention due to their potential industrial applications.161 Semiconductor photocatalysts generates electrons and hole pairs (e−/h+) upon irradiation with light energy that can be utilized in initiating oxidation and reduction reactions, respectively. Nevertheless, the recombination of holes and electrons or agglomeration of the fine nanoparticles may lead to a decreased photocatalytic activity. Therefore, it is especially interesting to support semiconductor on suitable substrates for wastewater remediation.143 One of the attractive materials utilized to enhance photocatalytic activities of semiconductors-based materials is layered double hydroxides.135It was previously reported that the materials rich in OH groups, like LDHs, may favor the titania activity. The surface hydroxyl groups may be easily converted into HO˙ radicals, which are the primary species responsible for the dye degradation.162,163 Lu et al.151 fabricated supported TiO2 nanoparticles and proved TiO2/Cu–Mg–Al LDH sample has superior photocatalytic properties to the rehydrated single phase R-TiO2. The TiO2/LDH heterojunction nanostructure is proposed to contribute to the efficient spatial separation between the photogenerated electrons and holes, which can concomitantly improve the photocatalytic activity. The influence of initial Ti4+ location on the quality of the LDHs-type materials was studied and the Mg3Al0.5Ti0.5–LDH sample showed powerful photo-oxidative effect assigned to the segregation of small anatase nanocrystals on the highly hydroxylated layered surface.143 A new materials colloidal TiO2/LDH was prepared for photodegradation of AO7, a high surface area and the great adsorption of TiO2/LDHs particles facilitated electron injection so that a significant increase of the photodegradation rate of AO7 was obtained.144
Apart from TiO2/LDH composites, other hybrid photocatalysts have been researched for the decomposition of dyes such as NanoPt–LDH,11 CuPcTs–LDH,152 ZnO–FLDH, SnO2–LDH,148 Ag/AgBr/Co–LDH,149 MoO42−/WO42−–LDH.130 A new SnO2/Mg–Al LDH was obtained for MB degradation. The proposed mechanism was based on the shifting of flat band potential of SnO2 due to the interaction with Mg–Al LDH, this being energetically favourable to the formation of hydroxyl radicals responsible for methylene blue degradation.148 Another Ag/AgBr/Co–LDH nanomaterials have been developed as photocatalysts, the photoinduced holes in the VB of AgBr could easily induce formation of ·˙OH from OH groups of the LDH and these oxidative species would result in degradation of dyes, as shown in Fig. 14.149
 | ||
| Fig. 14 Schematic photocatalytic mechanism of Ag/AgBr/Co–Ni–NO3 LDH nanocomposites under visible-light illumination (adapted from ref. 149 with Copyright from 2013 Wiley). | ||
Above all, incorporation of photocatalysts into LDHs is a promising strategy for dye degradation. The pollutants would not only be adsorbed on the surface of LDHs, but also be degraded, and this can greatly increase the wastewater treatment capacity.
3.4. Kinetics of degradation
The kinetics of degradation dyes catalyzed by LDH materials were investigated based on the Langmuir–Hinshelwood model, which was widely used to describe the kinetics of photocatalytic reactions of dyes compounds in aqueous solutions.164 When the concentration of organic compounds is low and the adsorption is relatively weak, the equation can be simplified as follows:| r = KrKadC = KappC | (8) |
Setting eqn (1) under initial conditions of photocatalytic procedure, (t = 0, C = C0), eqn (2) is obtained:
 | (9) |
4. Conclusion and future perspectives
In summary, LDHs has been applied to construct different and promising sorbents and catalysts. By incorporated with nanoparticles or other substances, LDHs can offer their unique properties and possibly induce new performance. The use of LDHs in the removal of dyes results in markedly improved adsorption capacity and photocatalytic degradation efficiency over other conventional materials. A survey of the literature on the synthesis of LDHs reveals calcination, nanomaterials, inorganic oxides and surfactant have been used to enhance the adsorption performance. An analysis of adsorption mechanisms reveals that the surface adsorption, anion exchange and reconstruction are predominantly responsible for binding the organic pollutants. The main advantages of LDHs over the conventional anionic exchange resins include their higher anion exchange capacity for certain anion and their good thermal stability. LDHs and LDHs based materials (mixed metal oxide materials and supports to photocatalytic guest) are applied to degrade dyes and received a higher degradation percent as the result of extended light adsorption range and excellent charge separation–transportation capability.Although considerable progresses have been achieved in the field of adsorption and degradation of dyes by LDHs, the studies in wastewater remediation need to be further developed, at least, the following aspects should be considered:
(1) Most of the adsorption studies focused on the anionic dyes, only a few cases were on the removal of cationic dyes. Multi-purpose, economically feasible composites should be developed to remove not only for the anionic dyes but also for the cationic and nonionic dyes.
(2) An understanding of the adsorption process and photocatalytic degradation in the multi-dyes system should be totally understood. The mechanisms in both of the adsorption and photocatalytic degradation process should be explained by a variety of analytical tools.
(3) It is important to estimate the cost of any adsorption system or degradation system. However, LDHs have not been used at a commercial level to any extent, there is no data available to estimate the cost of production and application. An evaluation of the cost of LDHs between other low-cost sorbent should be encouraged and develop an economical and environment-friendly way to make use of the LDH-dye sludge.
(4) Finding the most appropriate element ratio, optimum surface area and calcination temperature to synthesize the LDHs for the particular dye to obtain a better removal percent. Since the release of the binding inorganic and organic glands would cause secondly pollution, using the environment-friendly modificators in the synthetic system is necessary.
(5) Attempts have been made to intercalate various dye molecules, in particular organic dyes inside layered inorganic lamella. Mg–Al LDH intercalated with Coomassie Brilliant Blue R anion was prepared to take up aromatic compounds from aqueous solutions.165 Evan's Blue incorporation into the Mg–Al LDH to form an organic–inorganic nanohybrid composite material was reported. Formation of such a material is useful, for example for controlled release purposes of dye for slow dyeing process.166 These developments of new organic–inorganic hybrid type materials open promising prospects for the reutilization of the LDH-dye sludge after adsorption.
(6) The development of new approach for preparation of novel based LDHs photocatalytic materials with improved activity and stability working in visible-light region is still highly valued. It is highly desirable to develop efficient visible-light driven photocatalysts that predominantly take advantage of solar energy and indoor illumination in the visible-to-infrared range.
(7) Designing and fabrication of an economically cheap treatment for the removal of dyes from diluted industrial effluent and invent a treatment system via combing the LDH adsorption with photocatalysis technology to improve the performance.
5. Abbreviations
| AB 9 | Acid blue 9 |
| AB 14 | Acid brown 14 |
| AB 113 | Acid blue 113 |
| AB 29 | Acid blue 29 |
| ARG | Acid red GR |
| AR 1 | Acid red 1 |
| AO 7 | Acid orange 7 |
| AG 68:1 | Acid green 68:1 |
| AY 23 | Acid yellow 23 |
| AR 37 | Acid red 37 |
| BBR | Brilliant blue R |
| CG | Carboxyl graphene |
| CLDH | Calcined LDH |
| CNT | Carbon nanotubes |
| CR | Congo red |
| DA | Dodecanoic acid |
| DGLN | Direct blending scarlet D-GLN |
| F | Freundlich |
| GB-F2B | Green bezanyl-F2B |
| GO | Graphene oxide |
| GR | Acidic scarlet GR |
| HB | Heteropoly blue |
| ILs | Hydroxyl ammonium ionic liquids |
| I-P | Intra-particle diffusion |
| L | Langmuir |
| LDO | Layered double oxides |
| MB | Methylene blue |
| MG | Malachite green |
| MMH | Mixed metal hydroxide |
| MO | Methyl orange |
| MV | Methyl violet |
| OG | Orange G |
| OII | Orange II |
| RBR K-2BP | Reactive brilliant red K-2BP |
| RhB | Rhodamine B |
| RhG | Rhodamine 6G |
| R–P | Redlich–Peterson |
| RR 3BS | Remazol red 3BS |
| RY | Reactive yellow 4 GL |
| SDS | Sodium dodecylsulfate |
| 1st | Pseudo-first-order |
| 2nd | Pseudo-second-order |
Acknowledgements
This study is supported by the project of Qing Hai Science & Technology Department (No. 2013-J-620). The project is financially supported by the National Natural Science Foundation of China (51179068, 51039001).References
- G. Zeng, M. Chen and Z. Zeng, Science, 2013, 340, 1403 CrossRef CAS PubMed.
- S. Li, Bioresour. Technol., 2010, 101, 2197–2202 CrossRef CAS PubMed.
- N. Bao, Y. Li, Z. Wei, G. Yin and J. Niu, J. Phys. Chem. C, 2011, 115, 5708–5719 CAS.
- J. Zhang, S. Chen, Y. Zhang, X. Quan, H. Zhao and Y. Zhang, J. Hazard. Mater., 2014, 274, 198–204 CrossRef CAS PubMed.
- M. Kornaros and G. Lyberatos, J. Hazard. Mater., 2006, 136, 95–102 CrossRef CAS PubMed.
- J. Labanda, J. Sabaté and J. Llorens, Chem. Eng. J., 2011, 166, 536–543 CrossRef CAS.
- Y. Zheng, G. Yao, Q. Cheng, S. Yu, M. Liu and C. Gao, Desalination, 2013, 328, 42–50 CrossRef CAS.
- J. L. Gong, B. Wang, G. M. Zeng, C. P. Yang, C. G. Niu, Q. Y. Niu, W. J. Zhou and Y. Liang, J. Hazard. Mater., 2009, 164, 1517–1522 CrossRef CAS PubMed.
- D. Rajamanickam and M. Shanthi, Spectrochim. Acta, Part A, 2014, 128, 100–108 CrossRef CAS PubMed.
- L. Ai, C. Zhang and L. Meng, J. Chem. Eng. Data, 2011, 56, 4217–4225 CrossRef CAS.
- Q. Fang and B. Chen, J. Mater. Chem. A, 2014, 2, 8941 CAS.
- S. Dong, J. Feng, Y. Li, L. Hu, M. Liu, Y. Wang, Y. Pi, J. Sun and J. Sun, Appl. Catal., B, 2014, 152–153, 413–424 CrossRef CAS.
- S. Dong, J. Sun, Y. Li, C. Yu, Y. Li and J. Sun, Appl. Catal., B, 2014, 144, 386–393 CrossRef CAS.
- S. Dong, Y. Cui, Y. Wang, Y. Li, L. Hu, J. Sun and J. Sun, Chem. Eng. J., 2014, 249, 102–110 CrossRef CAS.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- V. K. Gupta, R. Kumar, A. Nayak, T. A. Saleh and M. A. Barakat, Adv. Colloid Interface Sci., 2013, 193–194, 24–34 CrossRef CAS PubMed.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211–212, 317–331 CrossRef CAS PubMed.
- L. Yu and Y.-m. Luo, J. Environ. Chem. Ecotoxicol., 2014, 2, 220–229 CAS.
- M. Toor and B. Jin, Chem. Eng. J., 2012, 187, 79–88 CrossRef CAS.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- D. Sun, X. Zhang, Y. Wu and X. Liu, J. Hazard. Mater., 2010, 181, 335–342 CrossRef CAS PubMed.
- C. A. P. Almeida, A. dos Santos, S. Jaerger, N. A. Debacher and N. P. Hankins, Desalination, 2010, 264, 181–187 CrossRef CAS.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- V. K. Gupta, R. Jain, A. Mittal, M. Mathur and S. Sikarwar, J. Colloid Interface Sci., 2007, 309, 464–469 CrossRef CAS PubMed.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed.
- L. Ćurković, D. Ljubas, S. Šegota and I. Bačić, J. Alloys Compd., 2014, 604, 309–316 CrossRef.
- P. Muthirulan, C. N. Devi and M. M. Sundaram, Ceram. Int., 2014, 40, 5945–5957 CrossRef CAS.
- S. J. Xia, F. X. Liu, Z. M. Ni, W. Shi, J. L. Xue and P. P. Qian, Appl. Catal., B, 2014, 144, 570–579 CrossRef CAS.
- J. Wang, Y. Wei and J. Yu, Appl. Clay Sci., 2013, 72, 37–43 CrossRef CAS.
- F. Kovanda, E. Jindová, K. Lang, P. Kubát and Z. Sedláková, Appl. Clay Sci., 2010, 48, 260–270 CrossRef CAS.
- X. Liang, Y. Zang, Y. Xu, X. Tan, W. Hou, L. Wang and Y. Sun, Colloids Surf., A, 2013, 433, 122–131 CrossRef CAS.
- L. El Gaini, M. Lakraimi, E. Sebbar, A. Meghea and M. Bakasse, J. Hazard. Mater., 2009, 161, 627–632 CrossRef CAS PubMed.
- Y. Guo, Z. Zhu, Y. Qiu and J. Zhao, Chem. Eng. J., 2013, 219, 69–77 CrossRef CAS.
- M. X. Zhu, Y. P. Li, M. Xie and H. Z. Xin, J. Hazard. Mater., 2005, 120, 163–171 CrossRef CAS PubMed.
- L. Shao, Y. Yao, S. Quan, H. Wei, R. Wang and Z. Guo, Mater. Lett., 2014, 114, 111–114 CrossRef CAS.
- H. A. Z. E. Ghorbani and Z. Talleb, J. Iran. Chem. Soc., 2013, 1103–1112, DOI:10.1007/s13738-013-0255-z.
- C. Forano, T. Hibino, F. Leroux and C. Taviot-Guého, in Developments in Clay Science, ed. B. K. G. T. Faïza Bergaya and L. Gerhard, Elsevier, 2006, vol. 1, pp. 1021–1095 Search PubMed.
- Y. Zhong, Q. Yang, K. Luo, X. Wu, X. Li, Y. Liu, W. Tang, G. Zeng and B. Peng, J. Hazard. Mater., 2013, 250–251, 345–353 CrossRef CAS PubMed.
- C. G. Silva, Y. Bouizi, V. Fornés and H. García, J. Am. Chem. Soc., 2009, 131, 13833–13839 CrossRef PubMed.
- M. T. Yagub, T. K. Sen, S. Afroze and H. M. Ang, Adv. Colloid Interface Sci., 2014, 209, 172–184 CrossRef CAS PubMed.
- W. S. Wan Ngah, L. C. Teong and M. A. K. M. Hanafiah, Carbohydr. Polym., 2011, 83, 1446–1456 CrossRef CAS.
- K. H. Goh, T. T. Lim and Z. Dong, Water Res., 2008, 42, 1343–1368 CrossRef CAS PubMed.
- Z. P. Xu, J. Zhang, M. O. Adebajo, H. Zhang and C. Zhou, Appl. Clay Sci., 2011, 53, 139–150 CrossRef CAS.
- M. J. Barnabas, S. Parambadath, A. Mathew, S. S. Park, A. Vinu and C. S. Ha, J. Solid State Chem., 2016, 233, 133–142 CrossRef CAS.
- R. Lafi, K. Charradi, M. A. Djebbi, A. Ben Haj Amara and A. Hafiane, Adv. Powder Technol., 2016, 27, 232–237 CrossRef CAS.
- R. r. Shan, L. g. Yan, Y. m. Yang, K. Yang, S. j. Yu, H. q. Yu, B. c. Zhu and B. Du, J. Ind. Eng. Chem., 2015, 21, 561–568 CrossRef CAS.
- G. Darmograi, B. Prelot, G. Layrac, D. Tichit, G. Martin-Gassin, F. Salles and J. Zajac, J. Phys. Chem. C, 2015, 119, 23388–23397 CAS.
- M. Zhang, Q. Yao, C. Lu, Z. Li and W. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 20225–20233 CAS.
- Y. Li, B. Gao, T. Wu, B. Wang and X. Li, J. Hazard. Mater., 2009, 164, 1098–1104 CrossRef CAS PubMed.
- G. Bascialla and A. E. Regazzoni, Colloids Surf., A, 2008, 328, 34–39 CrossRef CAS.
- N. Drici Setti, N. Jouini and Z. Derriche, J. Phys. Chem. Solids, 2010, 71, 556–559 CrossRef CAS.
- A. R. Auxilio, P. C. Andrews, P. C. Junk, L. Spiccia, D. Neumann, W. Raverty and N. Vanderhoek, Polyhedron, 2007, 26, 3479–3490 CrossRef CAS.
- R. M. M. dos Santos, R. G. L. Gonçalves, V. R. L. Constantino, L. M. da Costa, L. H. M. da Silva, J. Tronto and F. G. Pinto, Appl. Clay Sci., 2013, 80–81, 189–195 CrossRef.
- R. r. Shan, L. g. Yan, Y. m. Yang, K. Yang, S. j. Yu, H. q. Yu, B. c. Zhu and B. Du, J. Ind. Eng. Chem., 2015, 21, 561–568 CrossRef CAS.
- M. Mustapha Bouhent, Z. Derriche, R. Denoyel, V. Prevot and C. Forano, J. Solid State Chem., 2011, 184, 1016–1024 CrossRef CAS.
- M. Bouraada, F. Belhalfaoui, M. S. Ouali and L. C. de Menorval, J. Hazard. Mater., 2009, 163, 463–467 CrossRef CAS PubMed.
- K. Nejati, Z. Rezvani, M. Mansurfar, A. Mirzaee and M. Mahkam, Z. Anorg. Allg. Chem., 2011, 637, 1573–1579 CrossRef CAS.
- H. Zaghouane-Boudiaf, M. Boutahala and L. Arab, Chem. Eng. J., 2012, 187, 142–149 CrossRef CAS.
- F. P. de Sá, B. N. Cunha and L. M. Nunes, Chem. Eng. J., 2013, 215–216, 122–127 CrossRef.
- F. B. Saiah, B. L. Su and N. Bettahar, J. Hazard. Mater., 2009, 165, 206–217 CrossRef CAS PubMed.
- C. Zhang, S. Yang, H. Chen, H. He and C. Sun, Appl. Surf. Sci., 2014, 301, 329–337 CrossRef CAS.
- C. L. Guoqing Zhang, J. Zhao, J. Zhi Zhou, G. Qian, J. Liu and Y. Wu, J. Water Sustainability, 2012, 2, 25–34 Search PubMed.
- A. Guzmán-Vargas, E. Lima, G. A. Uriostegui-Ortega, M. A. Oliver-Tolentino and E. E. Rodríguez, Appl. Surf. Sci., 2016, 363, 372–380 CrossRef.
- Y. Lin, Z. Zeng, J. Zhu, Y. Wei, S. Chen, X. Yuan and L. Liu, Mater. Lett., 2015, 156, 169–172 CrossRef CAS.
- T. Xue, Y. Gao, Z. Zhang, A. Umar, X. Yan, X. Zhang, Z. Guo and Q. Wang, J. Alloys Compd., 2014, 587, 99–104 CrossRef CAS.
- R. Marangoni, M. Bouhent, C. Taviot-Gueho, F. Wypych and F. Leroux, J. Colloid Interface Sci., 2009, 333, 120–127 CrossRef CAS PubMed.
- K. Morimoto, K. Tamura, N. Iyi, J. Ye and H. Yamada, J. Phys. Chem. Solids, 2011, 72, 1037–1045 CrossRef CAS.
- Y.-M. Zheng, N. Li and W.-D. Zhang, Colloids Surf., A, 2012, 415, 195–201 CrossRef CAS.
- B. Bi, L. Xu, B. Xu and X. Liu, Appl. Clay Sci., 2011, 54, 242–247 CrossRef CAS.
- D. Chen, Y. Li, J. Zhang, W. Li, J. Zhou, L. Shao and G. Qian, J. Hazard. Mater., 2012, 243, 152–160 CrossRef CAS PubMed.
- D. Chen, Y. Li, J. Zhang, J.-z. Zhou, Y. Guo and H. Liu, Chem. Eng. J., 2012, 185–186, 120–126 CrossRef CAS.
- L. Nong, C. Xiao and W. Jiang, Korean J. Chem. Eng., 2011, 28, 933–938 CrossRef CAS.
- Q. Zhou, F. Chen, W. Wu, R. Bu, W. Li and F. Yang, Chem. Eng. J., 2016, 285, 198–206 CrossRef CAS.
- Y. P. Wei, D. Q. Wei and H. W. Gao, Chem. Eng. J., 2011, 172, 872–878 CrossRef CAS.
- Y. Li, H. Y. Bi, Y. S. Jin and X. Q. Shi, RSC Adv., 2014, 4, 58307–58314 RSC.
- M. Bouraada, M. Lafjah, M. S. Ouali and L. C. de Menorval, J. Hazard. Mater., 2008, 153, 911–918 CrossRef CAS PubMed.
- P. Wu, T. Wu, W. He, L. Sun, Y. Li and D. Sun, Colloids Surf., A, 2013, 436, 726–731 CrossRef CAS.
- C. Peng, J. Dai, J. Yu and J. Yin, AIP Adv., 2015, 5, 057138 CrossRef.
- N. Benselka-Hadj Abdelkader, A. Bentouami, Z. Derriche, N. Bettahar and L. C. de Ménorval, Chem. Eng. J., 2011, 169, 231–238 CrossRef CAS.
- A. Auxilio, P. Andrews, P. Junk and L. Spiccia, Dyes Pigm., 2009, 81, 103–112 CrossRef CAS.
- E. Géraud, M. Bouhent, Z. Derriche, F. Leroux, V. Prévot and C. Forano, J. Phys. Chem. Solids, 2007, 68, 818–823 CrossRef.
- J. Orthman, H. Y. Zhu and G. Q. Lu, Sep. Purif. Technol., 2003, 31, 53–59 CrossRef CAS.
- D. D. Asouhidou, K. S. Triantafyllidis, N. K. Lazaridis and K. A. Matis, J. Chem. Technol. Biotechnol., 2012, 87, 575–582 CrossRef CAS.
- D. S. Tong, M. Liu, L. Li, C. X. Lin, W. H. Yu, Z. P. Xu and C. H. Zhou, Appl. Clay Sci., 2012, 70, 1–7 CrossRef CAS.
- R. Extremera, I. Pavlovic, M. R. Pérez and C. Barriga, Chem. Eng. J., 2012, 213, 392–400 CrossRef CAS.
- M. N. Pahalagedara, M. Samaraweera, S. Dharmarathna, C.-H. Kuo, L. R. Pahalagedara, J. A. Gascón and S. L. Suib, J. Phys. Chem. C, 2014, 118, 17801–17809 CAS.
- Z. Li, B. Yang, S. Zhang, B. Wang and B. Xue, J. Mater. Chem. A, 2014, 2, 10202 CAS.
- Z. M. Ni, S. J. Xia, L. G. Wang, F. F. Xing and G. X. Pan, J. Colloid Interface Sci., 2007, 316, 284–291 CrossRef CAS PubMed.
- Y. X. Zhang, X. D. Hao, T. Wang, Y. X. Meng and X. Han, Dalton Trans., 2014, 43, 6667–6676 RSC.
- Y. X. Zhang, X. D. Hao, M. Kuang, H. Zhao and Z. Q. Wen, Appl. Surf. Sci., 2013, 283, 505–512 CrossRef CAS.
- Z. Yang, S. Ji, W. Gao, C. Zhang, L. Ren, W. W. Tjiu, Z. Zhang, J. Pan and T. Liu, J. Colloid Interface Sci., 2013, 408, 25–32 CrossRef CAS PubMed.
- S. A. Dhahir, E. Abdul-Hussein and N. Faraj, Eur. Chem. Bull., 2013, 2, 866–872 CAS.
- Y. You, G. F. Vance and H. Zhao, Appl. Clay Sci., 2001, 20, 13–25 CrossRef CAS.
- Y. Yao, B. Gao, M. Inyang, A. R. Zimmerman, X. Cao, P. Pullammanappallil and L. Yang, J. Hazard. Mater., 2011, 190, 501–507 CrossRef CAS PubMed.
- I. M. Ahmed and M. S. Gasser, Appl. Surf. Sci., 2012, 259, 650–656 CrossRef CAS.
- X. Liang, Y. Zang, Y. Xu, X. Tan, W. Hou, L. Wang and Y. Sun, Colloids Surf., A, 2013, 433, 122–131 CrossRef CAS.
- N. K. Lazaridis, T. D. Karapantsios and D. Georgantas, Water Res., 2003, 37, 3023–3033 CrossRef CAS PubMed.
- L. Sun, S. Wan and W. Luo, Bioresour. Technol., 2013, 140, 406–413 CrossRef CAS PubMed.
- T. W. Panpan Wua, W. Hea, L. Suna, Y. Lia and D. Sun, Colloids Surf., A, 2013, 436, 726–731 CrossRef.
- F. Zhou, R. Shi and Y. Zhu, J. Mol. Catal. A: Chem., 2011, 340, 77–82 CrossRef CAS.
- M. A. M. Salleh, D. K. Mahmoud, W. A. W. A. Karim and A. Idris, Desalination, 2011, 280, 1–13 CrossRef CAS.
- R. Hao, X. Xiao, X. Zuo, J. Nan and W. Zhang, J. Hazard. Mater., 2012, 209–210, 137–145 CrossRef CAS PubMed.
- R. Liikanen, J. Yli-Kuivila, J. Tenhunen and R. Laukkanen, Desalination, 2006, 201, 58–70 CrossRef CAS.
- H. Abdolmohammad-Zadeh, E. Ghorbani and Z. Talleb, J. Iran. Chem. Soc., 2013, 10, 1103–1112 CrossRef CAS.
- S. Netpradit, P. Thiravetyan and S. Towprayoon, J. Colloid Interface Sci., 2004, 270, 255–261 CrossRef CAS PubMed.
- P. Janoš, H. Buchtová and M. Rýznarová, Water Res., 2003, 37, 4938–4944 CrossRef PubMed.
- A. S. Özcan and A. Özcan, J. Colloid Interface Sci., 2004, 276, 39–46 CrossRef PubMed.
- S. Wang and Z. H. Zhu, J. Hazard. Mater., 2006, 136, 946–952 CrossRef CAS PubMed.
- P. C. A. Anthony, R. Auxilio, P. C. Junk and L. Spiccia, Aust. J. Chem., 2010, 63, 83–91 CrossRef.
- S. Yuan, Y. Li, Q. Zhang and H. Wang, Colloids Surf., A, 2009, 348, 76–81 CrossRef CAS.
- Y. Zhi, Y. Li, Q. Zhang and H. Wang, Langmuir, 2010, 26, 15546–15553 CrossRef CAS PubMed.
- X.-L. Wu, L. Wang, C.-L. Chen, A.-W. Xu and X.-K. Wang, J. Mater. Chem., 2011, 21, 17353–17359 RSC.
- T. Wen, X. Wu, X. Tan, X. Wang and A. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 3304–3311 CAS.
- X. Yuan, Y. Wang, J. Wang, C. Zhou, Q. Tang and X. Rao, Chem. Eng. J., 2013, 221, 204–213 CrossRef CAS.
- J. Inacio, C. Taviot-Gueho, C. Forano and J. Besse, Appl. Clay Sci., 2001, 18, 255–264 CrossRef CAS.
- D. L. Sparks, Geoderma, 2001, 100, 303–319 CrossRef CAS.
- L. Xiao, W. Ma, M. Han and Z. Cheng, J. Hazard. Mater., 2011, 186, 690–698 CrossRef CAS PubMed.
- S. Mandal, V. S. Patil and S. Mayadevi, Microporous Mesoporous Mater., 2012, 158, 241–246 CrossRef CAS.
- P. Zhang, G. Qian, H. Shi, X. Ruan, J. Yang and R. L. Frost, J. Colloid Interface Sci., 2012, 365, 110–116 CrossRef CAS PubMed.
- T. Kameda, T. Yoshioka, T. Mitsuhashi, M. Uchida and A. Okuwaki, Water Res., 2003, 37, 4045–4050 CrossRef CAS PubMed.
- N. K. Lazaridis, Water, Air, Soil Pollut., 2003, 146, 127–139 CrossRef CAS.
- M. Ulibarri, I. Pavlovic, M. Hermosin and J. Cornejo, Appl. Clay Sci., 1995, 10, 131–145 CrossRef CAS.
- X. Liu, X. Zhao, Y. Zhu and F. Zhang, Appl. Catal., B, 2013, 140–141, 241–248 CrossRef CAS.
- P. Roy Chowdhury and K. G. Bhattacharyya, Dalton Trans., 2015, 44, 6809–6824 RSC.
- K. Parida, L. Mohapatra and N. Baliarsingh, J. Phys. Chem. C, 2012, 116, 22417–22424 CAS.
- R. Kun, M. Balázs and I. Dékány, Colloids Surf., A, 2005, 265, 155–162 CrossRef CAS.
- S. J. Xia, F. X. Liu, Z. M. Ni, J. L. Xue and P. P. Qian, J. Colloid Interface Sci., 2013, 405, 195–200 CrossRef CAS PubMed.
- M. Shao, J. Han, M. Wei, D. G. Evans and X. Duan, Chem. Eng. J., 2011, 168, 519–524 CrossRef CAS.
- L. Mohapatra and K. M. Parida, Sep. Purif. Technol., 2012, 91, 73–80 CrossRef CAS.
- K. M. Parida and L. Mohapatra, Chem. Eng. J., 2012, 179, 131–139 CrossRef CAS.
- X. Xu, R. Lu, X. Zhao, Y. Zhu, S. Xu and F. Zhang, Appl. Catal., B, 2012, 125, 11–20 CrossRef CAS.
- G. Carja, E. Husanu, C. Gherasim and H. Iovu, Appl. Catal., B, 2011, 107, 253–259 CrossRef CAS.
- J. H. Xin Shu and D. Chen, J. Phys. Chem. C, 2008, 112, 4151–4158 Search PubMed.
- E. M. Seftel, E. Popovici, M. Mertens, K. D. Witte, G. V. Tendeloo, P. Cool and E. F. Vansant, Microporous Mesoporous Mater., 2008, 113, 296–304 CrossRef CAS.
- R. Huo, Y. Kuang, Z. Zhao, F. Zhang and S. Xu, J. Colloid Interface Sci., 2013, 407, 17–21 CrossRef CAS PubMed.
- L. Huang, S. Chu, J. Wang, F. Kong, L. Luo, Y. Wang and Z. Zou, Catal. Today, 2013, 212, 81–88 CrossRef CAS.
- B. Benalioua, M. Mansour, A. Bentouami, B. Boury and H. Elandaloussi el, J. Hazard. Mater., 2015, 288, 158–167 CrossRef CAS PubMed.
- F. Khodam, Z. Rezvani and A. R. Amani-Ghadim, RSC Adv., 2015, 5, 19675–19685 RSC.
- Y. F. Zhao, M. Wei, J. Lu, Z. L. Wang and X. Duan, Nano, 2009, 3, 4009–4016 CAS.
- R. K. Sahu, B. S. Mohanta and N. N. Das, J. Phys. Chem. Solids, 2013, 74, 1263–1270 CrossRef CAS.
- Z. Huang, P. Wu, B. Gong, Y. Fang and N. Zhu, J. Mater. Chem. A, 2014, 2, 5534 CAS.
- X. Wang, P. Wu, Y. Lu, Z. Huang, N. Zhu, C. Lin and Z. Dang, Sep. Purif. Technol., 2014, 132, 195–205 CrossRef CAS.
- E. M. Seftel, M. Mertens and P. Cool, Appl. Catal., B, 2013, 134–135, 274–285 CrossRef CAS.
- Z. Bouberka, K. A. Benabbou, A. Khenifi and U. Maschke, J. Photochem. Photobiol., A, 2014, 275, 21–29 CrossRef CAS.
- Y. Zhou, W. Hu, J. Yu and F. Jiao, React. Kinet., Mech. Catal., 2015, 115, 581–596 CrossRef CAS.
- Y. L. Sujun Yuan, Q. Zhang and H. Wang, Res. Chem. Intermed., 2009, 35, 685–692 CrossRef.
- Y. Zhou, L. Shuai, X. Jiang, F. Jiao and J. Yu, Adv. Powder Technol., 2015, 26, 439–447 CrossRef CAS.
- E. Dvininov, M. Ignat, P. Barvinschi, M. A. Smithers and E. Popovici, J. Hazard. Mater., 2010, 177, 150–158 CrossRef CAS PubMed.
- H. Fan, J. Zhu, J. Sun, S. Zhang and S. Ai, Chemistry, 2013, 19, 2523–2530 CrossRef CAS PubMed.
- Y. Ao, D. Wang, P. Wang, C. Wang, J. Hou and J. Qian, RSC Adv., 2015, 5, 54613–54621 RSC.
- R. Lu, X. Xu, J. Chang, Y. Zhu, S. Xu and F. Zhang, Appl. Catal., B, 2012, 111–112, 389–396 CrossRef CAS.
- K. M. Parida, N. Baliarsingh, B. S. Patra and J. Das, J. Mol. Catal. A: Chem., 2007, 267, 202–208 CrossRef CAS.
- L. Mohapatra, K. Parida and M. Satpathy, J. Phys. Chem. C, 2012, 116, 13063–13070 CAS.
- S. j. Xia, X. b. Zhou, W. Shi, G. x. Pan and Z. m. Ni, J. Mol. Catal. A: Chem., 2014, 392, 270–277 CrossRef CAS.
- M. A. Carreon, S. Y. Choi, M. Mamak, N. Chopra and G. A. Ozin, J. Mater. Chem., 2007, 17, 82–89 RSC.
- S. Y. Chae, M. K. Park, S. K. Lee, T. Y. Kim, S. K. Kim and W. I. Lee, Chem. Mater., 2003, 15, 3326–3331 CrossRef CAS.
- C. u. G. Silva, Y. s. Bouizi, V. Fornés and H. García, J. Am. Chem. Soc., 2009, 131, 13833–13839 CrossRef PubMed.
- G. K. Zhang, X. M. Ding, F. S. He, X. Y. Yu, J. Zhou, Y. J. Hu and J. W. Xie, Langmuir, 2008, 24, 1026–1030 CrossRef CAS PubMed.
- C. Ooka, H. Yoshida, K. Suzuki and T. Hattori, Microporous Mesoporous Mater., 2004, 67, 143–150 CrossRef CAS.
- E. Dvininov, M. Ignat, P. Barvinschi, M. A. Smithers and E. Popovici, J. Hazard. Mater., 2010, 177, 150–158 CrossRef CAS PubMed.
- B. H. Lee, S. S. Hong, K. H. Lee and Y. D. Kim, J. Alloys Compd., 2004, 385, 264–268 CrossRef CAS.
- S. P. Paredes, M. A. Valenzuela, G. Fetter and S. O. Flores, J. Phys. Chem. Solids, 2011, 72, 914–919 CrossRef CAS.
- K. Lv, J. Yu, K. Deng, X. Li and M. Li, J. Phys. Chem. Solids, 2010, 71, 519–522 CrossRef CAS.
- H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432–22439 CrossRef CAS PubMed.
- T. Kameda, S. Sato and T. Yoshioka, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2012, 47, 2035–2039 CrossRef CAS PubMed.
- M. Z. b. Hussein, A. H. Yahaya and L. M. Ping, Dyes Pigm., 2004, 63, 135–140 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |