
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review on noble-metal-free bifunctional heterogeneous catalysts for overall electrochemical water splitting
Ya
Yan
ab,
Bao Yu
Xia
c,
Bin
Zhao
*a and
Xin
Wang
*b
aSchool of Materials Science & Engineering, University of Shanghai for Science and Technology, 516 Jungong Road, Yangpu District, Shanghai 200093, China. E-mail: zhaobin@usst.edu.cn
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798 Singapore. E-mail: WangXin@ntu.edu.sg
cSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
First published on 17th October 2016
Abstract
Production of hydrogen by water splitting is an appealing solution for sustainable energy storage. Development of bifunctional catalysts that are active for both the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) is a key factor in enhancing electrochemical water splitting activity and simplifying the overall system design. Here, recent developments in HER–OER bifunctional catalysts are reviewed. Several main types of bifunctional water splitting catalysts such as cobalt-, nickel- and iron-based materials are discussed in detail. Particular attention is paid to their synthesis, bifunctional catalytic activity and stability, and strategies for activity enhancement. The current challenges faced are also concluded and future perspectives towards bifunctional water splitting electrocatalysts are proposed.
 Ya Yan | Dr Ya Yan is currently an Associate Professor of Special Appointment in the School of Materials Science & Engineering, University of Shanghai for Science and Technology. She received her bachelor's degree in chemical engineering from Northwest University (China) in 2010 and earned her Ph.D. degree under the supervision of Professor Xin Wang at Nanyang Technological University (NTU) in 2015. Her recent research interests focus on the development of highly efficient electrocatalysts and electrodes for water splitting and fuel cells. |
 Bao Yu Xia | Dr Bao Yu Xia received his Ph.D. degree in materials science and engineering from Shanghai Jiao Tong University (SJTU) in 2010. He then moved to NTU, Singapore, where he was a research fellow under the supervision of Prof. Xin Wang and Prof. Xiong Wen Lou. He is currently a full professor at Huazhong University of Science and Technology (HUST). His research interests are in the areas of nanostructured functional materials and their application in energy and the environment. |
 Bin Zhao | Dr Bin Zhao obtained his B.E. and M.E. from Dalian Jiaotong University and Dalian University of Technology, respectively; and completed his Ph.D. at Tsinghua University in 2004. As a postdoctoral fellow, he joined Prof. Y. Shimogaki's group at the University of Tokyo in 2004, and then Dr K. Hata's group at the National Institute of Advanced Industrial Science and Technology (Japan) in 2006. He joined the faculty of the University of Shanghai for Science and Technology in July 2009. His current research interests focus on low-dimensional carbon materials and their applications in energy storage and electrocatalytic energy conversion. |
 Xin Wang | Dr Xin Wang is currently a full professor in the School of Chemical and Biomedical Engineering, NTU. He received his bachelor's (1994) and master's (1997) degrees in chemical engineering from Zhejiang University and his Ph.D. degree (2002) in chemical engineering from Hong Kong University of Science and Technology. He has been working on electrocatalysis and electrochemical technologies for energy harvesting. His recent research focus includes electrocatalyst and electrode development for fuel cells, CO2 electroreduction, water electrosplitting, and electrochemical reactors with co-generation of electricity and valuable chemicals. |
1. Introduction
The global energy crisis, and its associated environmental issues, has motivated scientists to search for renewable energy resources that can replace fossil fuels.1 Driven by nature, available resources such as solar- or wind-derived electricity and water splitting technology provides an appealing way to produce hydrogen in a sustainable way.2,3 Water electrolysis produces hydrogen (H2) and oxygen (O2) from water by the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). At the anode, water is split into oxygen gas with accompanying protons and electrons. At the cathode, hydrogen gas evolves by recombination of the protons and electrons. By far, this energy conversion technology has attracted extensive attention due to its high energy conversion efficiency, negligible environment pollution, and potentially wide range of applications.4–6In practice, water electrolyzers require effective catalysts to facilitate both reactions. Currently, Pt-group metals are the most effective catalysts for HER while the benchmark catalysts for OER are Ir/Ru-based compounds. However, high cost and scarcity of these metals limit their widespread use. Enormous effort has been dedicated to the development of non-precious and highly efficient electrocatalysts for water splitting. Examples of these non-precious electrocatalysts include transition metal sulfides, carbides, and phosphides for HER;7–11 and transition metal phosphate, oxides, and hydroxides for OER.12–14 Ideally, catalysts for both HER and OER must operate in the same pH range so as to work together for water splitting. Furthermore, in order to minimize the overpotentials of the electrolysis system, water splitting is mostly conducted in either strongly acidic or basic solutions.15 However, this condition poses a challenge for most of non-noble metal water splitting catalysts as a highly active catalyst in acidic condition may be inactive or unstable in alkaline electrolyte. Therefore, integrating the merits of the HER and OER electrocatalysts to construct efficient bifunctional HER–OER heterostructures that possess binding force to both hydrogen- and oxygen-containing intermediates is of great benefit for improving the activity of overall water splitting. In addition, using a single bifunctional HER–OER catalyst simplifies the overall system design and lowers the cost.16 Alternatively, recent achievements in water splitting catalysts have made it possible to construct water electrolyzers using a bifunctional material that is active for catalyzing both the HER and OER reactions.16–18 This has encouraged the investigation of high efficiency bifunctional catalysts in the field of full water splitting.
Here, we summarized recent developments in bifunctional heterogeneous water splitting electrocatalysts with a focus on their design, characterization, and activity. Bifunctional HER–OER catalysts are mainly first-row transition metal-based compounds. They can be classified into three groups according to their composite elements: cobalt (Co) based catalysts, nickel (Ni) based materials, and iron (Fe) based materials. We have also reviewed some emerging metal oxides as well as carbon-based materials that have potential as bifunctional HER–OER catalysts. Based on the results discussed, the current challenges are briefly pointed out and several future opportunities are proposed.
2. Electrochemistry of water splitting
As shown in Fig. 1, a typical electrolyzer consists of three parts: an aqueous electrolyte, an anode, and a cathode. The bifunctional HER–OER catalysts are applied on both the anode and cathode to speed up the overall water splitting reaction. When an external voltage is applied between the two electrodes, the OER and HER occur at the anode and cathode, respectively.5 Depending on the electrolytes in which water splitting proceeds, the water splitting reaction can be described as shown below.19| Total reaction H2O → H2 + 1/2O2 | (1) |
In acidic solution
| Cathode 2H+ + 2e− → H2 | (2) |
| Anode H2O → 2H+ + 1/2O2 + 2e− | (3) |
In neutral or alkaline solution
| Cathode 2H2O + 2e− → H2 + 2OH− | (4) |
| Anode 2OH− → H2O + 1/2O2 + 2e− | (5) |
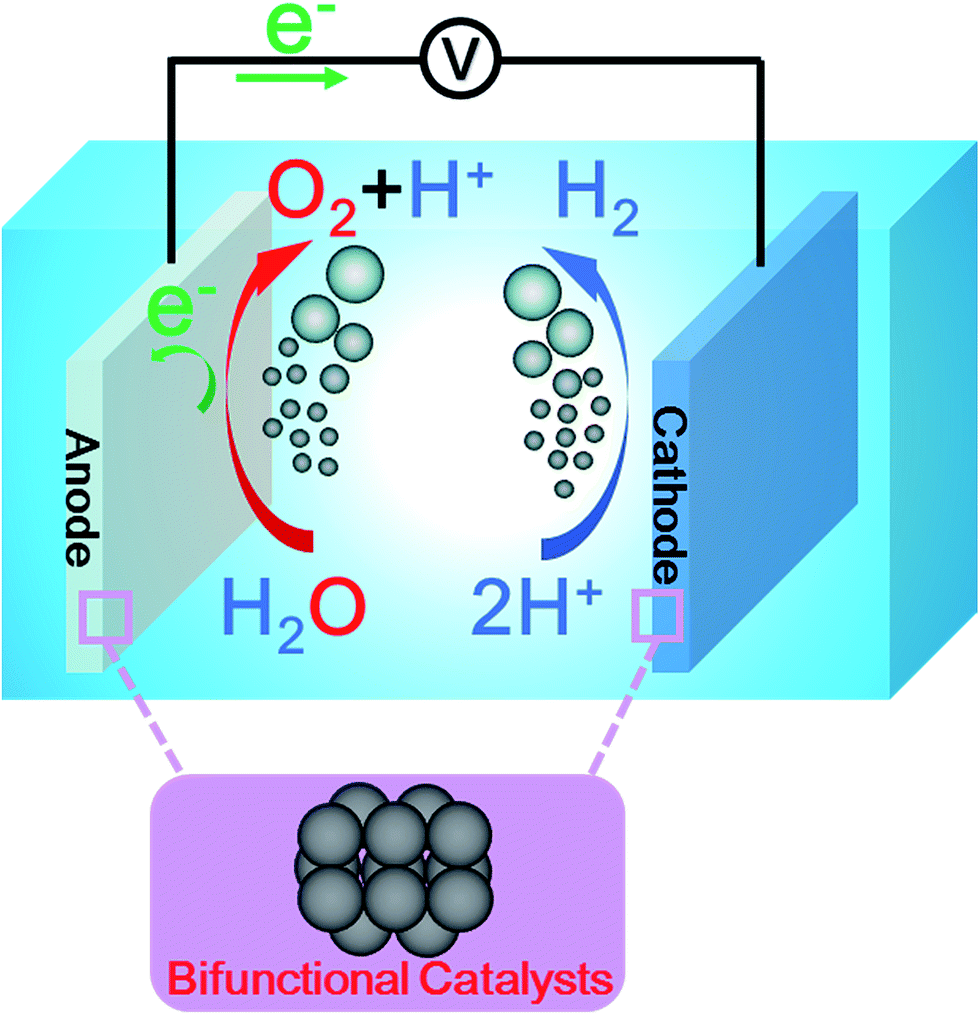 | ||
| Fig. 1 Schematic illustration of acidic water electrolyzer and its operating principle based on the electrochemical hydrogen and oxygen evolution reactions occurring on a bifunctionally active catalyst. | ||
Currently, the majority of reported bifunctional HER–OER catalysts work in alkaline electrolytes. Compared to acidic electrolytes, alkaline electrolytes allow for the replacement of precious metal catalysts (Pt, Ir, or Ru based) with inexpensive transition metal and/or functional carbon-based catalysts. In addition, alkaline electrolytes favor the electrocatalytic oxygen-evolving process, allowing a significantly faster rate than acidic electrolytes.20 The thermodynamic voltage of water splitting is known to be 1.23 V (25 °C and 1 atm) regardless of the reaction media.5 In order to carry out water splitting at a practical rate, we must apply extra voltages on the cell and the overall operational voltage (Vop) for water splitting can be described as:21
| Vop = 1.23 V + ηa + |ηc| + ηΩ | (6) |
2.1. Hydrogen evolution reaction
HER has been explored extensively for almost every transition metal.5 Generally, the HER process involves three steps in acidic and alkaline media, respectively:19Volmer reaction (electrochemical hydrogen adsorption)
| H+ + M + e → MHads (in acidic medium) | (7) |
| H2O + M + e → MHads + OH− (in alkaline medium) | (8) |
Heyrovsky reaction (electrochemical desorption)
| MHads + H+ + e → M + H2 (in acidic medium) | (9) |
| MHads + H2O + e → M + OH− + H2 (in alkaline medium) | (10) |
Tafel reaction (chemical desorption)
| 2MHads → 2M + H2 | (11) |
It is well known that the activity of the electrode material towards the HER depends on its electronic structure. As displayed in Fig. 2, Trasatti plotted the logarithm of the exchange current densities, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j0, on different metals against the metal–hydrogen (M–H) bond strength for the HER in acidic condition.21,22 This so-called volcano curve permits a quick comparison of the activities of different metals. However, the reported volcano plots for HER kinetics as a function of M–H bond energies (or metal properties) usually does not take into consideration other types of differences such as solution pH. In fact, a three-dimensional (3D) volcano plot should be plotted with pH in the third axis. However, reliable data for alkaline solution kinetics are far from sufficient for drawing such a plot. Therefore, future efforts to advance the volcano plot for the HER under basic conditions will be valuable for the overall alkaline electrolysis reaction.
j0, on different metals against the metal–hydrogen (M–H) bond strength for the HER in acidic condition.21,22 This so-called volcano curve permits a quick comparison of the activities of different metals. However, the reported volcano plots for HER kinetics as a function of M–H bond energies (or metal properties) usually does not take into consideration other types of differences such as solution pH. In fact, a three-dimensional (3D) volcano plot should be plotted with pH in the third axis. However, reliable data for alkaline solution kinetics are far from sufficient for drawing such a plot. Therefore, future efforts to advance the volcano plot for the HER under basic conditions will be valuable for the overall alkaline electrolysis reaction.
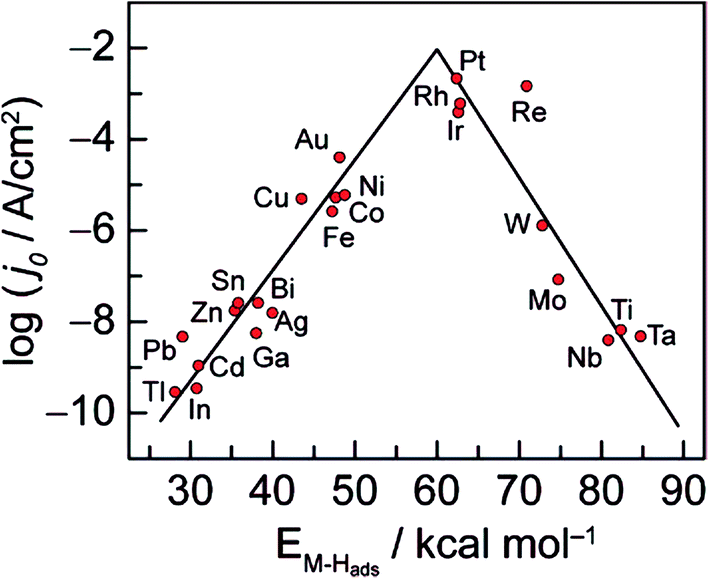 | ||
| Fig. 2 Volcano curve for the HER on metal electrodes in acidic media. The log of the exchange current density j0 is plotted versus the M–H bond energy for each metal surface. Reprinted with permission.21 Copyright 2010, American Chemical Society. | ||
2.2. Oxygen evolution reaction
Promotion of OER by transition metal oxides, hydroxides, and perovskites has been investigated extensively.23 OER mechanisms and pathways are relatively more complex compared to HER mechanisms. Oxygen is generally evolved from a metal oxide surface, but not from the bare metal, and the OER mechanism is different for oxides with different surface structures.24 Even oxides with identical compositions may give rise to different electro-kinetic profiles due to the differences in thickness and structures of their oxide layers and preparation methods. In spite of this, a general mechanism for the OER on oxides has been proposed as follows:21,25| M + H2O → MOH + H+ + e− | (12) |
| MOH → MO + H+ + e− or 2MOH → MO + M + H2O | (13) |
| 2MO → 2M + O2 or MO + H2O → M + O2 + 2H+ + 2e− | (14) |
Fig. 3 is the volcano plot showing the activity of O2 generation on first-row transition metal oxide surfaces against the transition enthalpy of the oxides in either acidic or basic conditions.21,25 As seen, noble metal oxides RuO2 and IrO2, are located at the top of the plot because of their low redox potential and good electrical conductivity. However, their high prices and limited HER activity make these precious-metal based oxides unsuitable for use as bifunctional catalysts in large scale water splitting. Conversely, cheaper and more sustainable bifunctional transition metal compound catalysts such as Co phosphates/phosphides and Ni–S composites have demonstrated high activity for both HER and OER with electrochemical stability.
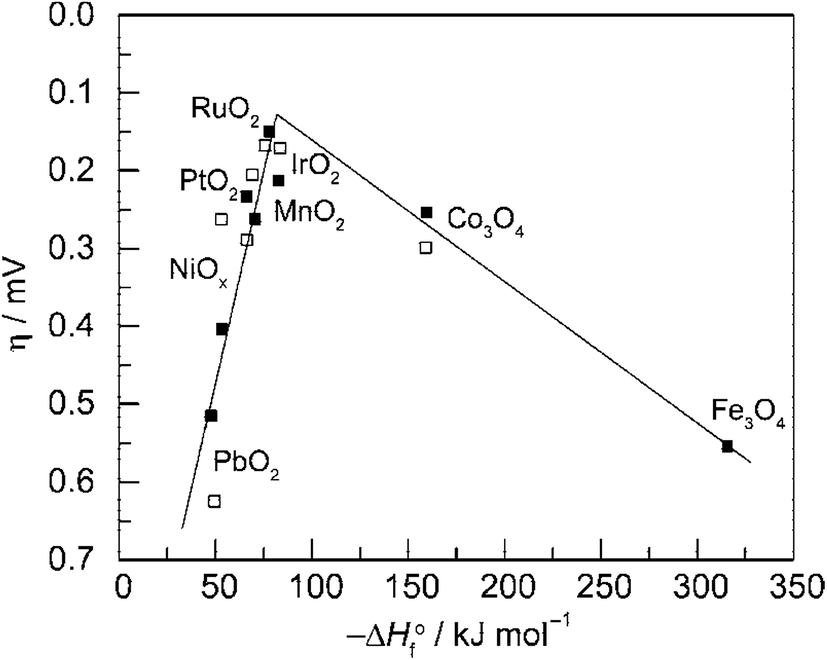 | ||
| Fig. 3 Volcano plot showing activity for O2 production on metal oxide surfaces versus the enthalpy of transition of the oxide in acidic (■) and basic (□) solutions. Overpotential measured relative to 0.1 mA cm−2 current density. Reprinted with permission.21 Copyright 2010, American Chemical Society. | ||
2.3. Evaluation of bifunctional electrocatalysts
An ideal HER–OER bifunctional electrocatalyst for full water splitting is one that is simultaneously cost-effective, active, and durable for both HER and OER in the same electrolyte. For water splitting, the dependence of steady-state current density on the anodic or cathodic overpotentials (ηa, ηc) can be depicted by Tafel plots. The overpotential is generally logarithmically related to the current density (j) and its linear portion is given as Tafel equation (η > 0.05 V):21,26| η = a + blog(j) | (15) |
Apart from the above discussed core parameters used for developing novel bifunctional HER–OER catalysts, there are still other properties that are important. Namely, an ideal bifunctional catalyst should possess a series of other advantages including:21 (i) tolerance for a wide pH range, (ii) the ability to work at moderate overpotentials with high current density, (iii) long-term stability (such as years to decades), (iv) composition with inexpensive earth-abundant materials, and (v) simple and economical preparation and fabrication methods. A single catalyst may never realistically hold all the qualities listed above. Therefore, choosing the appropriate catalysts for a particular application will be key for deciding development priorities of future water splitting electrolyzers. Most of time, a balance between efficiency for both HER and OER is the most attractive. For example, cost may surpass efficiency as the leading factor in some cases. Therefore, it is highly desirable for researchers to explore a wide range of bifunctional electrocatalysts with various properties to advance the development of the next generation of energy conversion technologies.
3. Synthesis of transition metal based bifunctional electrocatalysts for water splitting
First-row transition metal compounds such as Co-, Ni-, and Fe-based heterostructures have been extensively investigated as efficient bifunctional electrocatalysts for overall water splitting owing to their theoretically high electrocatalytic activities, earth abundance, and low cost. The majority of recently reported work belongs to this group of catalysts. These bifunctional catalysts demonstrate high HER–OER rates with some even surpassing the performance of benchmark noble metal catalysts. Most of the reported bifunctional catalysts are transition metal phosphides, but other compounds such as oxides, nitrides, and sulfides are also widely reported. Overall, these catalysts are mainly categorized by their metal composition, namely Co-based, Ni-based, and Fe-based compounds. The following sections will give a detailed discussion on their synthesis and overall water splitting performance.3.1. Co-based compounds
Cobalt has emerged as an attractive non-precious metal for its potential catalytic activity for water splitting in the past few decades. To date, enormous research effort has been devoted to developing Co-based inorganic compounds as heterogeneous bifunctional electrocatalysts.12,17,28–30 Among them, cobalt phosphates are one of the most investigated bifunctional electrocatalysts for water splitting.12,17,31,32 Several research groups have explored different approaches to engineer cobalt phosphates-based compounds for overall water splitting. In 2012, Cobo et al. first synthesized a Janus nanoparticulate H2–CoCat by the reductive electrodeposition from cobalt salts in a phosphate buffer (Fig. 4a).17 The obtained H2–CoCat consisted of metallic cobalt covered with the cobalt-oxo/hydroxo-phosphate layer and was used to catalyze HER. H2–CoCat could also be converted into the amorphous cobalt oxide film (O2–CoCat or CoPi) to catalyze O2 evolution on anodic equilibration (Fig. 4b). Remarkably, the switch between the two catalytic forms was fully reversible (Fig. 4c). Thus, the authors highlighted that the designed CoCat was a robust, bifunctional and switchable catalyst (Fig. 4d). Jiang et al. prepared a cobalt phosphorous-derived (Co–P) film via the potentiodynamic electrodeposition strategy for catalysis of overall water splitting.31 In their experiment, the as-prepared Co–P films demonstrated remarkable catalytic activity for both HER and OER in 1.0 M KOH. To achieve the current density of 10 mA cm−2, overpotentials of just −94 mV and 345 mV vs. RHE was required for HER and OER, respectively. Furthermore, their corresponding Tafel slopes were as small as 42 and 47 mV dec−1. Metallic cobalt and cobalt phosphide were the main components of the as-obtained and post-HER films, which partially transformed to cobalt oxide during OER. An electrolyzer constructed using Co–P films as both anode and cathode catalysts demonstrated superior activity and stability even when compared to the integrated Pt and IrO2 catalyst couple. | ||
| Fig. 4 (a) SEM images of electrodes modified by electrolysis at 1.0 V versus Ag/AgCl for 3 h versus RHE in phosphate buffer (KPi, 0.5 mmol l−1, pH 7). (b) SEM image of an H2–CoCat film on a FTO electrode equilibrated at 1.16 V versus Ag/AgCl for 90 min in a cobalt-free 0.5 mmol l−1 KPi, pH 7 electrolyte. (c) Charge passed through a FTO electrode (1 cm2) during controlled potential coulometry initially at 1.0 V versus Ag/AgCl (3 h, H2–CoCat deposition, black curve) in 0.5 mmol l−1 KPi, pH 7 electrolyte containing Co(NO3)2·6H2O (0.5 mmol l−1) and after transfer to a cobalt-free 0.5 mmol l−1 KPi, pH 7 electrolyte, with potential switching between oxidative (blue, 1.16 V versus Ag/AgCl) and reductive conditions (red, 1 V versus Ag/AgCl). (d) Specific rates of hydrogen (red) and oxygen (blue) evolution quantified through gas chromatography measurements during the same experiment. Panels (a–d) reprinted with permission.17 Copyright 2012, Nature Publishing Group. (e) Schematic of the fabrication process starting with a glass, silicon, ceramic, or stainless steel substrate. (f) Bifunctional water electrolysis tested by CV in 1 M KOH aqueous solution. Panels (e and f) reprinted with permission.32 Copyright 2015, Wiley-VCH. | ||
Given the successful examples of utilizing Co phosphate as water splitting catalysts, synthesizing porous Co phosphide/phosphate via nanostructuring may endow the material with unique properties, such as high surface area and rich mass transport channel. To this end, Tour's group designed a porous Co phosphide/phosphate-based thin film (PCPTF) by reacting anodized Co oxide porous films with phosphorus vapor (Fig. 4e).32 The obtained porous films could directly function as electrodes for catalyzing H2 and O2 evolution with superior activity having onset potentials of 35 and 220 mV vs. RHE and with Tafel slopes of 53 and 65 mV dec−1, respectively. The mixed phase film was able to directly produce H2 and O2 in a single electrolyzer through an alternating sequence (Fig. 4f).
Phosphide-based materials are an important class of compounds with superb electrocatalytic activities toward HER. In particular, Co-based phosphides have been widely studied as active HER catalysts.33 Recently some groups have begun to investigate its bifunctional activity for catalyzing both HER and OER.34–36 You et al. synthesized Co–P/NC nanopolyhedrons by a metal organic frame (MOF)-derived route for overall water splitting. The nanopolyhedrons were composed of CoP and Co2P nanoparticles embedded in N-doped carbon matrices (Fig. 5a).35 The as-prepared Co–P/NC possessed high specific surface areas (183 m2 g−1) and large mesopores. Importantly, Co–P/NC exhibited remarkable catalytic performance for both HER and OER. Co–P/NC afforded a current density of 10 mA cm−2 at the relevant overpotentials of −154 mV vs. RHE for the HER and 319 mV vs. RHE for the OER in 1.0 M KOH. Moreover, the Co–P/NC-based overall water electrolyzer yielded a large current density of 165 mA cm−2 at 2.0 V (Fig. 5b) along with superior durability (Fig. 5c). Similarly, using a sandwich-type MOF/GO as a template and precursor, Jiao's group developed layered CoP/rGO composites as electrocatalysts toward both the HER and OER.34 After annealing at 400 °C, the obtained nanocomposites (CoP/rGO-400) worked efficiently to afford both efficient H2 and O2 generation in a single alkaline electrolyzer. The excellent electrocatalytic activity and durability were ascribed to the synergistic effect between the CoP and rGO featured with porous nanostructures, high electrical conductivity, and high durability during the HER and OER.
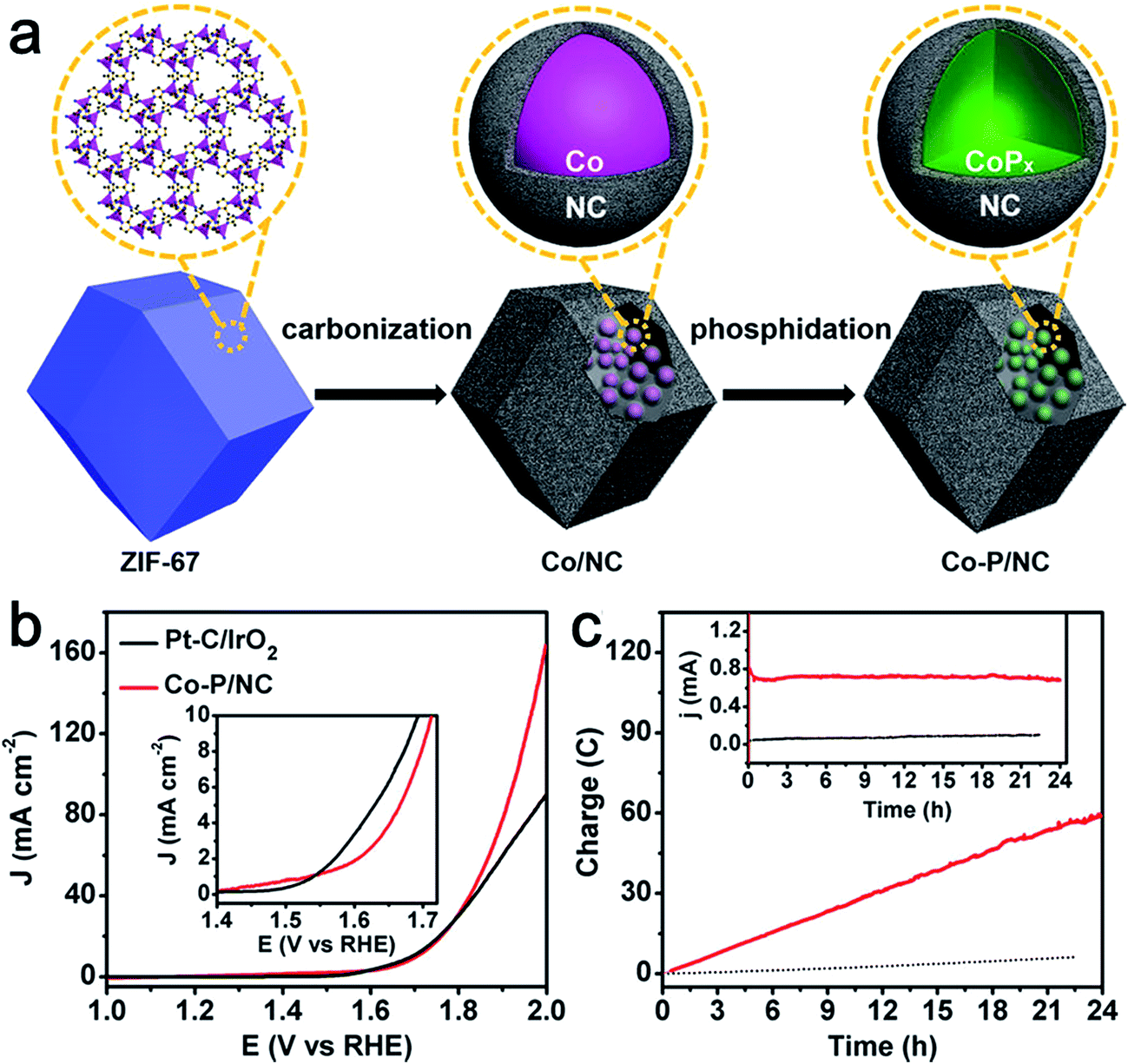 | ||
| Fig. 5 (a) Illustration of the two-step synthesis of CoP/NC nanopolyhedrons. (b) Polarization curves of Co–P/NC and Pt/IrO2 catalyst couples for overall water splitting in 1.0 M KOH. The mass loadings of Co–P/NC and Pt/IrO2 couples were 1.0 and 0.5 mg cm−2, respectively. Inset is the expanded region around the onsets of catalysis. (c) Long-term controlled potential electrolysis of Co–P/NC in 1.0 M KOH at η = 400 mV. Inset is the corresponding current change over time. Reprinted with permission.35 Copyright 2015, American Chemical Society. | ||
To further improve the conductivity of the Co phosphide and facilitate mass transfer at the electrode, Zhu et al. fabricated CoP mesoporous nanorod arrays supported on Ni foam.37 Featuring well-designed mesoporosity and a high electrochemical surface area, the synthesized materials could function as flexible bifunctional electrodes for both HER and OER. When placed in an alkaline electrolyzer, the flexible CoP nanorods array electrodes displayed exceptional activity (η10 mA cm−2 = 390 mV) and stability, which surpassed that of Pt and IrO2 electrodes. The catalyst's unique electrode configuration, consisting of an alighted nanorod array, was the main contributing factor behind its improved electric interconnection and enhanced mass transport.
Co–P based bifunctional catalysts, and the active components for HER and OER, are usually investigated by post-reaction characterization. However, in situ spectroscopic study is highly desirable for further understanding of real-time structural and composition evolution of the electrocatalysts in the process of electrolysis. This technique is still currently under investigation.
CoSe2 previously demonstrated high activity for catalyzing the generation of either H2 or O2 in water splitting.29,38,39 Further integration of other components into the CoSe2 system may endow it with bifunctional activity for overall water splitting. For example, Feng's group fabricated 3D coupled ternary compounds (EG/Co0.85Se/NiFe-LDH) by anchoring NiFe layered-double-hydroxide (NiFe-LDH) onto the Co0.85Se nanosheets vertically grown on the exfoliated graphene (EG) foil.40 The as-designed hierarchical composite exhibited superb OER performance with a potential of only 1.50 V vs. RHE to drive a current density of 150 mA cm−2. EG/Co0.85Se/NiFe-LDH was more efficient than other reported non-noble metal OER catalysts and the state-of-art Ir/C catalyst in alkaline conditions. Together with the excellent HER activity of EG/Co0.85Se/NiFe-LDH nanosheets, a water electrolyzer, with the ternary compounds as both anode and cathode, was built up and only needed a voltage of 1.71 V to reach a current density of 20 mA cm−2 in 1.0 M KOH. On the other hand, Li et al. integrated CoO domains with CoSe2 nanobelts on Ti (denoted as CoO/CoSe2).41 The catalyst was used as an active self-supported bifunctional electrocatalyst for both the HER and OER in neutral electrolyte.
Masa et al. reported an amorphous Co2B that was an exceptionally efficient electrocatalyst for the OER in alkaline conditions. Simultaneously, Co2B was also active for the HER.42 The annealed catalyst at 500 °C (Co2B-500) evolved O2 more efficiently than IrO2 and RuO2, and exhibited prominent stability during electrolysis tests (at 10 mA cm−2) for over 60 h. The B element introduced lattice strain in the metal crystal structure that then minimized the thermodynamic and kinetic barrier of the hydroxylation reaction and facilitated the formation of the OOH* intermediate for favoring the OER process. Complete water electrolysis tests using Co2B-500 as the anode and Co2B-500/NG as the cathode are illustrated in a two-electrode cell and vigorous and stable gas evolution was observed at moderate cell voltages.
Considering the chemical similarity between Co2B, CoP, and CoSe2, the distinctive electrochemical performance of these compounds indicates that other factors such as charge transfer capability, structural defects, and specific surface area may exert a strong influence on its electrochemical activity.
Superior electrical conductivity is extremely beneficial for the electrochemical activity of catalysts. Graphite carbon is often employed as a support to enhance the conductivity of the composite catalysts and the capability of tunable heteroatom-doping on carbon endows it with certain catalytic activity. The interplay between carbon and metal oxide nanoparticles can further modify the overall physicochemical and electronic structures to favor electron transfer at the interface.43 For instance, Jin et al. synthesized cobalt–cobalt oxide/N-doped carbon hybrids (CoOx@CN) for both the HER and OER through a one-pot thermal annealing method (Fig. 6a). The obtained CoOx@CN, which was composed of Co0, CoO, and Co3O4, exhibited low onset potential (85 mV), small charge-transfer resistance (41 Ω), and considerable stability for the HER in 1.0 M KOH.44 The excellent activity of CoOx@CN for the HER was attributed to the good electrical conductivity of N-doped carbon, synergistic effect between metallic Co and Co oxides, as well as the improved stability of carbon encapsulated Co nanoparticles. Moreover, the CoOx@CN hybrids showed high activity for the OER, yielding a current density of 10 mA cm−2 at an overpotential of only 0.26 V. Importantly, the catalytic activity of CoOx@CN for the OER was enhanced remarkably after carrying out the HER for 30 min. The authors attributed this to the improvement of the proportion of Co2+. Finally, an alkaline electrolyzer was designed by applying CoOx@CN as both cathode and anode electrocatalyst (Fig. 6b) with a voltage of only 1.55 V to drive a current density of ∼20 mA cm−2.
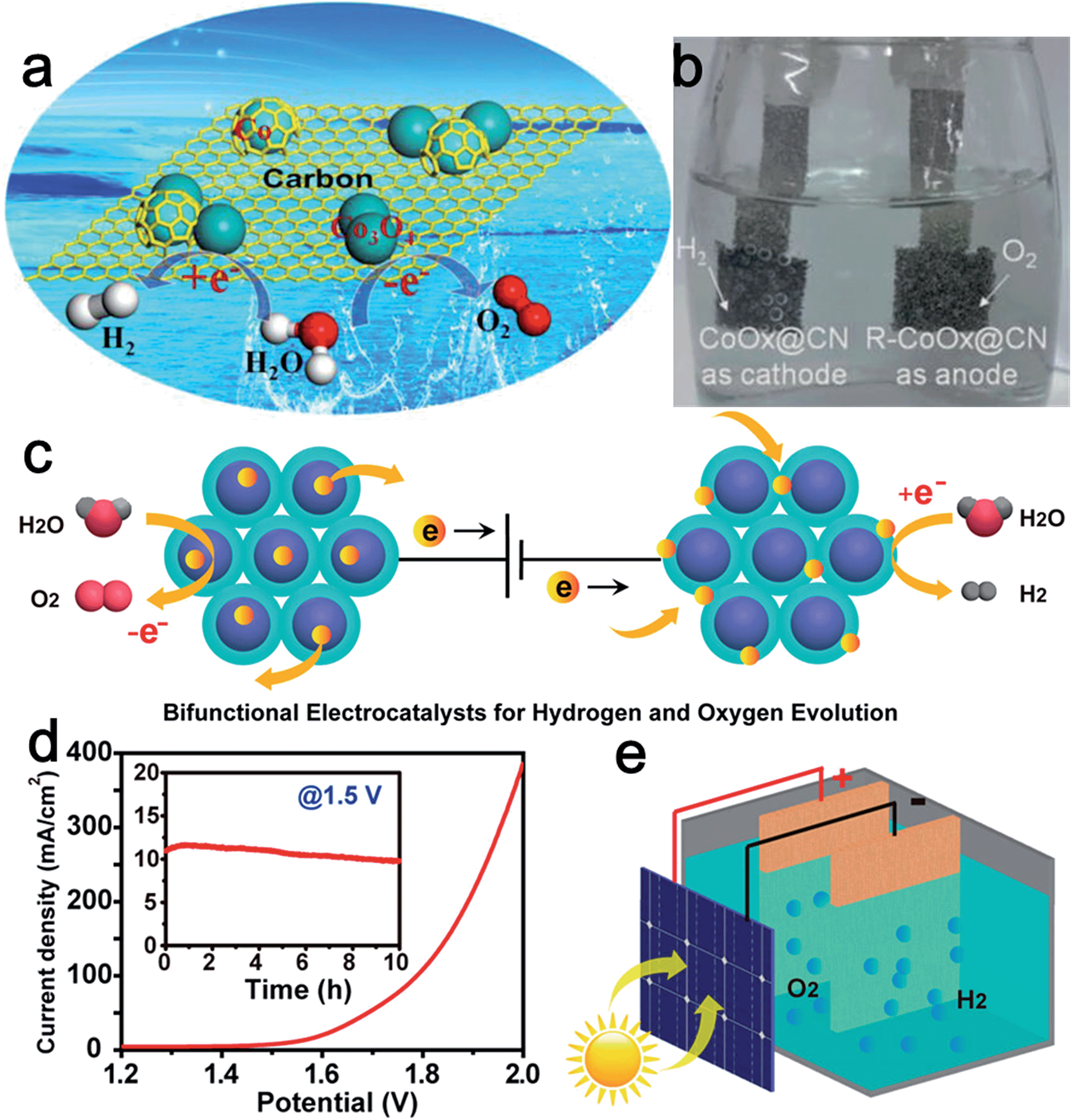 | ||
| Fig. 6 (a) Schematic illustration of the component of CoOx@CN, where cobalt oxide was mainly responsible for the OER while carbon-encapsulated Co nanoparticles were mainly responsible for the HER. (b) An optical photo showing the water electrolyzer with H2 and O2 bubbles generated. Panels (a and b) reproduced with permission.44 Copyright 2015, American Chemical Society. (c) Design of CoMnO@CN superlattices as a bifunctional OER and HER catalyst. The ordered CoMnO nanoparticles serve as the OER catalytic sites for water oxidation, and the nitrogen-doped carbon framework serves as the HER catalytic sites for water reduction. (d) Overall water splitting characteristics of the CoMnO@CN superlattice catalyst with a two-electrode configuration. Inset: chronoamperometric measurement at 1.5 V applied potential across the electrodes. (e) Schematic illustration of the solar water splitting cell without additional applied potential, using a commercial planar Si solar cell. Panels (c–e) reproduced with permission.48 Copyright 2015, American Chemical Society. | ||
Bimetallic oxides are expected to show higher intrinsic HER–OER activity than single metal oxides. Furthermore, the activity of bimetallic oxides can be easily optimized via controlled modulation of the valence and electronic states of either metal element.28,45,46 For example, Gao et al. recently reported the synthesis of Ni-incorporated hierarchical NiCo2O4 hollow microcuboids through a thermally driven conversion method. The hollow microcuboids proved to be highly active and stable electrode materials for both the HER and OER.47 NiCo2O4 electrodes also demonstrated exceptional catalytic performance for overall water splitting. The current density approached 10 mA cm−2 by applying 1.65 V across the two electrodes and the potential maintained very stable for at least 36 h. Furthermore, Zheng's group synthesized a highly ordered carbon-coated Co–Mn oxide nanoparticle superlattice (CoMnO@CN) as an effective bifunctional electrocatalyst for water splitting.48 Through this, the as-prepared catalyst could take advantage of the synergistic effects of bimetals, N-dopants, and carbon species. The uniform CoMnO nanoparticles served as an excellent OER reagent, with a smaller bandgap, stronger adsorption for OH−, and weaker adsorption for O2 than CoO and MnO. The authors emphasized that the surrounding N-doped carbon framework served as active HER catalytic sites, as well as providing efficient electron and ion transport toward the embedded CoMnO nanoparticles. In addition, the ordered superlattice structure with a carbon layer between adjacent CoMnO nanoparticles proved to be effective in increasing catalytic sites and preventing nanoparticles from aggregation or dissolution (Fig. 6c). Therefore, this CoMnO@CN superlattice structure could efficiently drive both OER and HER catalysis in the same alkaline electrolyte (Fig. 6d). When assembled with a silicon photovoltaic cell, this CoMnO@CN superlattice bifunctional catalyst enabled continuous solar water splitting for almost 5 days, corresponding to a solar-to-hydrogen conversion efficiency of nearly 8.0% (Fig. 6e).
Structure-wise, hierarchical 3D materials with hollow and mesoporous structures have been widely applied for electrochemical reactions. It is interesting to synthesize active electrocatalytic materials consisting of 1D nanostructures with hierarchical 3D porous structures and hollow interiors because the 1D nanowire/nanotube morphology, especially when directly attached to conducting substrates, ensure high surface area exposed, favorable conductivity, and fast charge transfer during electrolysis so as to enable excellent catalytic performance. 1D NiCo2S4 nanowire (NW) arrays directly grown on Ni foam (NF) were prepared by Sivanantham et al.49 The 3D NiCo2S4 NW/NF arrays showed remarkable bifunctional water splitting performance with prominent activity and stability. The 3D-Ni foam facilitated the directional growth of the NiCo2S4 NW, thus ensuing more exposure of active sites to take part in the HER and OER at the electrode–electrolyte interface. More recently, Zheng's group developed a homologous, all-nanowire based system (on Ti foil) of NiCo2O4 and Ni0.33Co0.67S2, as the OER and HER electrocatalysts, respectively.50 When assembled together, the NiCo2O4||Ni0.33Co0.67S2 nanowires represented a unique homologous, highly efficient system for overall water splitting. Meanwhile, Zheng's group also reported a homologous Co–Ni-based nanotube structure by employing the cation and anion exchange strategy.51 The CoNi(OH)x and NiNx nanotubes exhibited high current densities at low overpotentials and good stability for the OER and HER. Moreover, the assembled CoNi(OH)x||NiNx nanotube electrolyzer demonstrated promising full water splitting activity with long durability.
3.2. Ni-based compounds
In practice, Ni, particularly Ni foam, is usually used as the cathode material in commercial electrocatalytic electrolysis units in alkaline electrolytes. However as catalysts, Ni metals possess low efficiency and poor stability for water splitting. Thus, doping Ni metal or its oxides with other transition-metal and non-metal elements might offer some new solutions to this problem.52–54 In 2014, by incorporating Fe into Ni(OH)2, the Grätzel group reported a bifunctional earth-abundant catalyst, NiFe LDHs, which exhibited considerably high activity for both the HER and OER in alkaline electrolyte (Fig. 7a and b).16 Importantly, when assembled in a perovskite solar cells, the combination of the two yielded a value of 10 mA cm−2 with a 12.3% solar-to-hydrogen efficiency (Fig. 7c and d). | ||
| Fig. 7 (a) OER characteristics of different catalyst electrodes in a three-electrode configuration, scanned in the direction from positive to negative potential on the RHE scale. (b) HER characteristics of different catalyst electrodes in a three-electrode configuration, scanned in the direction from negative to positive potential. (c) Overall water splitting characteristics of different catalyst electrodes in a two-electrode configuration scanned from 2.0 V to 1.0 V. All scan rates were 1 mV s−1. (d) Schematic diagram of the water splitting device by combining the perovskite tandem cell with NiFe DLH/Ni foam electrodes. Reproduced with permission.16 Copyright 2014, American Association for the Advancement Science (AAAS). | ||
Additionally, combining Ni with non-metal elements has also received great attention. Ni-based phosphide and chalcogenide (including Ni sulfides and Ni selenides) electrocatalysts are promising catalysts for the full water splitting reaction. Hitherto, Ni phosphide materials were mainly used as effective cathodic electrocatalysts for hydrogen generation.55,56 However, pioneering work carried out by Hu group's showed that nickel phosphide (Ni2P) also exhibited high activity for oxygen evolution.57 N2P nanoparticles exhibited super OER activity (η10 mA cm−2 = 290 mV vs. RHE) and were remarkable stability in 1.0 M KOH. The active species for OER was a core–shell Ni2P/NiOx hybrid that was generated in situ during catalytic cycling (Fig. 8a–e). Subsequently, an alkaline water electrolyzer was constructed using Ni2P as catalyst for both HER and OER (Fig. 8f). Similarity, Lou and co-workers synthesized Ni–P porous nanoplates from Ni–Ni Prussian blue analogue precursors as active bifunctional electrocatalysts for both OER and HER.58 The authors emphasized that the surface nickel phosphides were partially oxidized to nickel oxides or hydroxides during the oxygen-evolving process. In situ formed oxidized nickel layers then functioned as the actual active sites for OER. It is noted that such an oxidation phenomenon is consistent with the results from Hu's group.
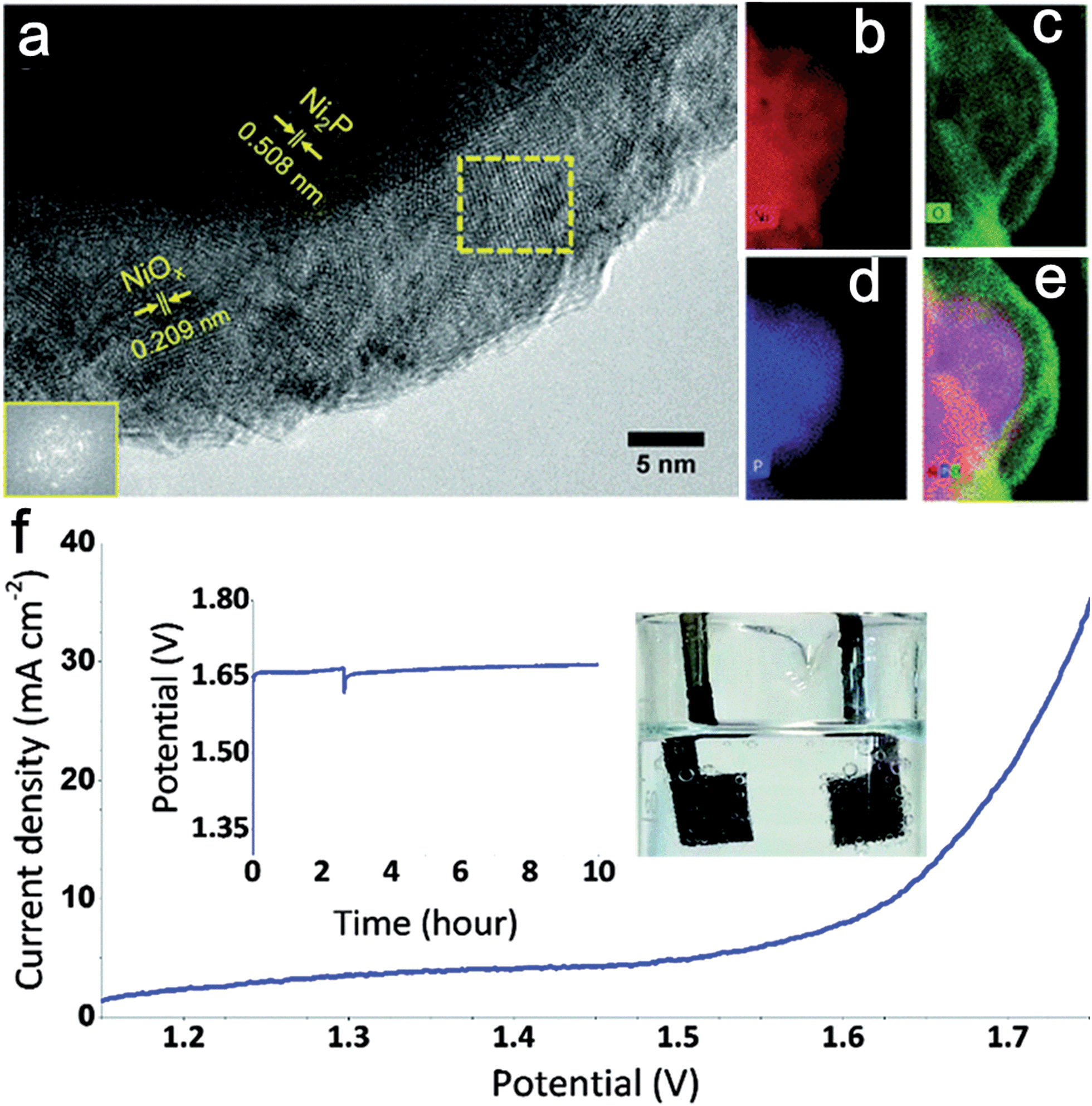 | ||
| Fig. 8 (a) HRTEM image of the Ni2P nanoparticles after electrochemical pretreatment at 1.5 V vs. RHE for one hour. Inset (lower left): FFT of the framed area (middle). (b) Nickel elemental mapping. (c) Oxygen elemental mapping. (d) Phosphorus elemental mapping. (e) Combined elemental mapping of Ni, O, and P. (f) Current–potential response of an alkaline electrolyzer using Ni2P as catalyst for both OER and HER. Support: Ni foam; loading of catalyst: 5 mg cm−2; electrolyte: 1 M KOH. At about 2.5 h, bubbles were mechanically removed from the electrode. Inset: galvanostatic electrolysis in 1 M KOH at a constant current density of 10 mA cm−2 over 10 hours. Photograph of the system showing the oxygen (left) and hydrogen (right) generation during water electrolysis. Reproduced with permission.57 Copyright 2015, Royal Society of Chemistry. | ||
Electrodes comprising of 3D catalysts generally exhibited higher electrocatalytic performances due to their larger number of active sites and improved electrical contact between the 3D electrode and catalysts. Ni foam is the most widely used conductive substrate for construction of 3D catalysts. Ni foam offers a long-range ordered porous framework, which not only facilitates charge transfer capabilities, but also enhances the mass transport rate.59 For example, Qiao and co-workers prepared 3D porous Ni/Ni8P3 and Ni/Ni9S8 electrodes via phosphorization or sulfurization of the acid-treated Ni-foam.60 The resulting materials could be directly used as free-standing bifunctional electrodes for full electrochemical water splitting in alkaline media. They also investigated the catalytic mechanism of Ni/Ni8P3 and Ni/Ni9S8 from the perspective of intermediate adsorption over the catalysts and the Ni–P/S bond length, and analyzed the reason for the relatively higher activity of Ni/Ni8P3 than Ni/Ni9S8. Finally, an advanced water electrolyzer by employing Ni/Ni8P3 as both anode and cathode was conducted in 1.0 M KOH solution; a cell voltage of just 1.61 V was required to deliver a 10 mA cm−2 water splitting current density. The excellent overall water splitting performance was attributed to its self-supported 3D architecture, strong physical contact, fast charge transport, and structure-induced electronic effect.
By using commercially available Ni foil as the Ni precursor, Shalom's group synthesized a self-supported highly ordered Ni5P4 sheets 3D electrode.61 The activity and stability of the catalyst towards the HER was exploited in both 0.5 M H2SO4 and 1.0 M KOH. The electrochemical performance of Ni5P4 for the OER in 1.0 M KOH was much higher than that of the pristine nickel or platinum. Importantly, when used as a bifunctional catalyst for overall water splitting in basic solution, Ni5P4 could achieve a current density of 10 mA cm−2 below 1.7 V. The authors attributed the excellent performance of Ni5P4 to its 3D hierarchical architecture, which facilitated the easy formation of catalytically active NiOOH on Ni5P4. It is believed that the formation of the Ni5P4/NiOOH heterojunction is important, which in turn altered the electronic properties resulting in lower OER overpotentials. In addition, the Shalom group also reported another approach towards encapsulating Mn-doped Ni materials into N-doped carbon as a high surface area electrocatalyst for the HER and OER.62 By thermal annealing in air, this highly porous material could be easily converted from a good catalyst of the HER to that of the OER. This simple treatment process also resulted in the formation of metal oxides on the surface and an increased surface area. Finally, a 70% overall water splitting efficiency was achieved.
Nickel chalcogenides have emerged as attractive HER catalysts.63–65 Recently, nickel sulfide also displayed potential activity toward the OER process and NiSOH was proposed to be the active species.66 Thereafter, Feng et al. reported the synthesis of stable, {![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 10} high-index faceted Ni3S2 nanosheet arrays supported on nickel foam (NF) as active bifunctional HER–OER catalysts.67 Thanks to its advantageous nanocatalytic features, as well as high index facets, the obtained Ni3S2/NF material exhibited efficient and ultra-stable electrocatalytic activity toward the HER (Fig. 9a) and OER (Fig. 9b). Theoretical calculations confirmed that the synergistic effect between the nanosheet array architecture and the {10} high-index facets accounted for its uniquely efficient catalytic ability for overall water splitting (Fig. 9c and d). Similarly, Tang et al. synthesized nickel foam supported NiSe nanowire film (NiSe/NF) for full water splitting.68 This was achieved by the hydrothermal reaction of NF with NaHSe. When employed as a 3D electrode for the OER, the NiSe/NF produced an anodic current density of 20 mA cm−2 at an overpotential of 270 mV vs. RHE and exhibited great stability in 1.0 M KOH. It was found the NiOOH layers formed on the NiSe surface served as the catalytic active species. This 3D electrode was also highly efficient for catalyzing hydrogen production in basic media. Therefore, an alkaline water electrolyzer consisting of this bifunctional electrode enabled a high activity with 10 mA cm−2 current at a cell voltage of 1.63 V.
10} high-index faceted Ni3S2 nanosheet arrays supported on nickel foam (NF) as active bifunctional HER–OER catalysts.67 Thanks to its advantageous nanocatalytic features, as well as high index facets, the obtained Ni3S2/NF material exhibited efficient and ultra-stable electrocatalytic activity toward the HER (Fig. 9a) and OER (Fig. 9b). Theoretical calculations confirmed that the synergistic effect between the nanosheet array architecture and the {10} high-index facets accounted for its uniquely efficient catalytic ability for overall water splitting (Fig. 9c and d). Similarly, Tang et al. synthesized nickel foam supported NiSe nanowire film (NiSe/NF) for full water splitting.68 This was achieved by the hydrothermal reaction of NF with NaHSe. When employed as a 3D electrode for the OER, the NiSe/NF produced an anodic current density of 20 mA cm−2 at an overpotential of 270 mV vs. RHE and exhibited great stability in 1.0 M KOH. It was found the NiOOH layers formed on the NiSe surface served as the catalytic active species. This 3D electrode was also highly efficient for catalyzing hydrogen production in basic media. Therefore, an alkaline water electrolyzer consisting of this bifunctional electrode enabled a high activity with 10 mA cm−2 current at a cell voltage of 1.63 V.
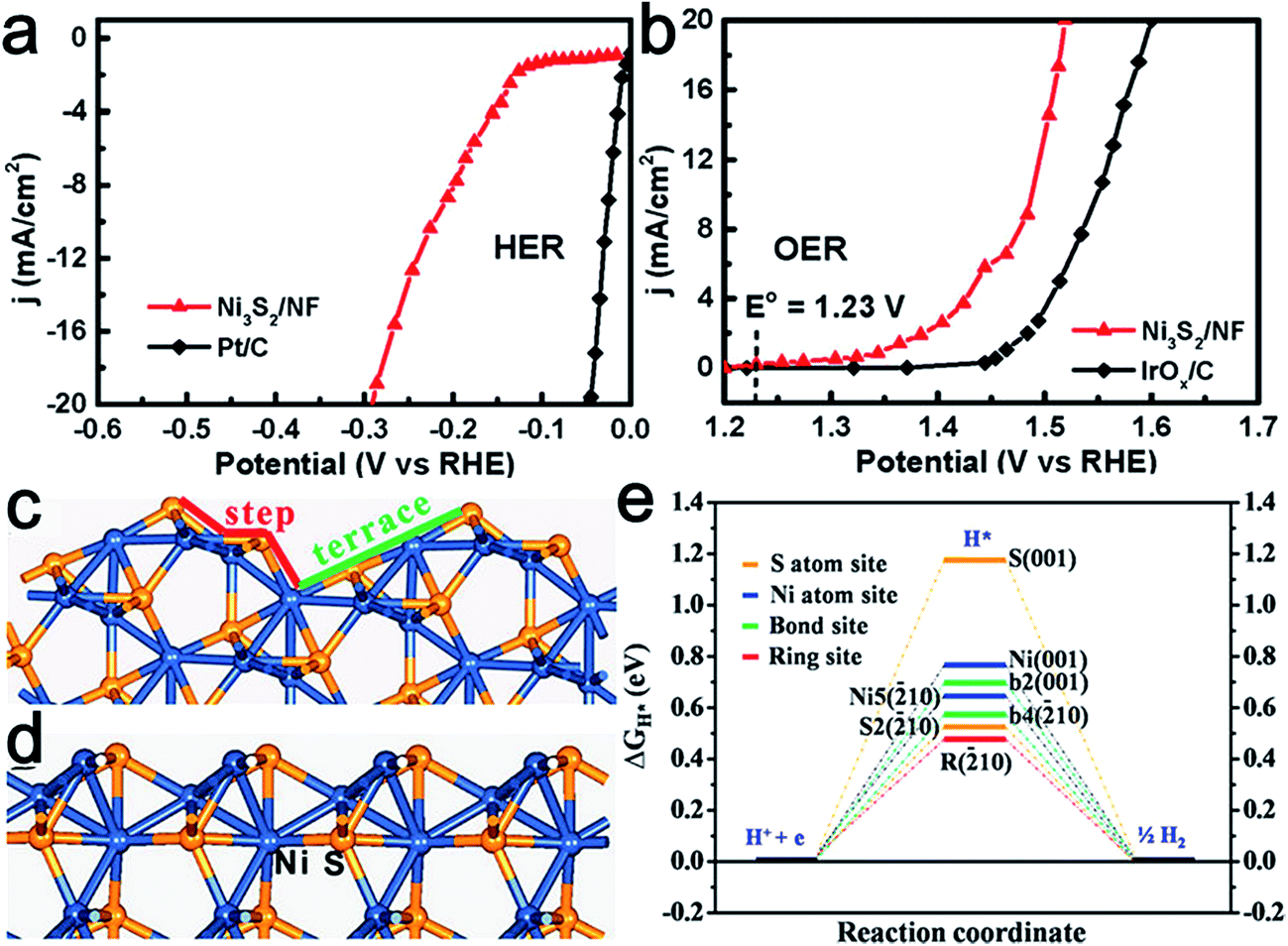 | ||
| Fig. 9 Steady-state current density as a function of applied voltage in alkaline media (pH 14) over Ni3S2/NF in (a) HER and (b) OER. Most stable terminations of (c) {10} and (d) (001) surfaces of Ni3S2. (e) Calculated free-energy diagram of HER over {10} and (001) surfaces at equilibrium potential. The blue and yellow spheres represent Ni and S atoms, respectively, of Ni3S2. Reproduced with permission.67 Copyright 2015, American Chemical Society. | ||
Increasing the surface area and active sites are also effective routes to lower the overpotential so as to enhance the electrocatalytic performance. For example, Feng's group presented the interface engineering of MoS2/Ni3S2 heterostructures on nickel foam as an advanced electrocatalyst for both the OER and HER in alkaline solution.69 An alkaline electrolyzer using the MoS2/Ni3S2 heterostructure as a bifunctional electrocatalysts was operated at a cell voltage of only 1.56 V. At this voltage, the heterostructure managed to achieve a current density of 10 mA cm−2, which was significantly lower than that of the state-of-the-art electrocatalysts for full water splitting. Together with density functional theory (DFT) calculations, they argued that the established interfaces between Ni3S2 and MoS2 and the in situ formed interfaces between NiO and MoS2 accelerated the simultaneous adsorption of hydrogen- and oxygen-containing intermediates, which consequently resulted in the improved overall water splitting efficiency.
Apart from nickel phosphides and chalcogenides, the overall water splitting activity of nickel nitrides-based materials have also been investigated. For example, Jia et al. synthesized Ni3FeN nanoparticles (Ni3FeN-NPs) with a particle size of 100 nm by thermal ammonolysis of ultrathin NiFe-LDH nanosheets. Ni3FeN-NPs was a highly active electrocatalyst for full water splitting.70 The enhanced performance of Ni3FeN-NPs originated from the metallic nature of the composites as well as its unique electronic structure, which in turn facilitated the charge transfer and H2O adsorption. Moreover, the nanosized particle effect was also thought to increase the accessibility of active sites for the electrocatalytic process. Besides, Zhang et al. reported the use of hierarchical TiN@Ni3N nanowire arrays to construct an efficient electrochemical water splitting device.71 The TiN@Ni3N nanowire arrays served as bifunctional catalysts for both the HER and OER in this device under alkaline conditions.
3.3. Fe-based compounds
Fe is more abundant than the other transition metals Co and Ni. As such, Fe is a highly attractive element in the design of low-cost catalysts. Fe-based electrocatalysts have been extensively investigated for either the HER or the OER,72–77 however, bifunctionally active Fe-based materials for water splitting have only been studied fairly recently. Martindale and Reisner recently reported an iron-only catalyst that was highly efficient for both water splitting half reactions in alkaline conditions. The obtained catalysts showed much higher activity than that reported from bifunctional Co and Ni catalysts.78 Interconversion between the Fe(0) phase under a cathodic bias and an iron oxide–hydroxide (FeOx) phase under an anodic bias resulted in the dual activity. Meanwhile, the authors also claimed that this interconversion of catalytic active species at the catalyst surface was reversible and switchable, which ensured the stability and durability of the electrolyzer system.Inspired by the bifunctional activity of Co and Ni phosphides for both the HER and OER, and the merits of 3D structured catalysts, Wang's group designed a flexible 3D electrode based on iron phosphide nanotubes (IPNT) toward full water splitting (Fig. 10a).79 The synthesized material was composed of FeP covered with iron oxide/phosphate species for catalyzing the H2 generation from both acidic and alkaline electrolytes with small onset overpotentials (35 and 31 mV vs. RHE, respectively). Remarkably, the in situ formed surface iron oxide/phosphate can be directly used to mediate O2 evolution at an onset overpotential of only 250 mV vs. RHE. Furthermore, the flexible iron phosphide nanotube electrode exhibited remarkable activity and durability for a constructed alkaline electrolyzer with 10 mA cm−2 current at a voltage of only 1.69 V (Fig. 10b).
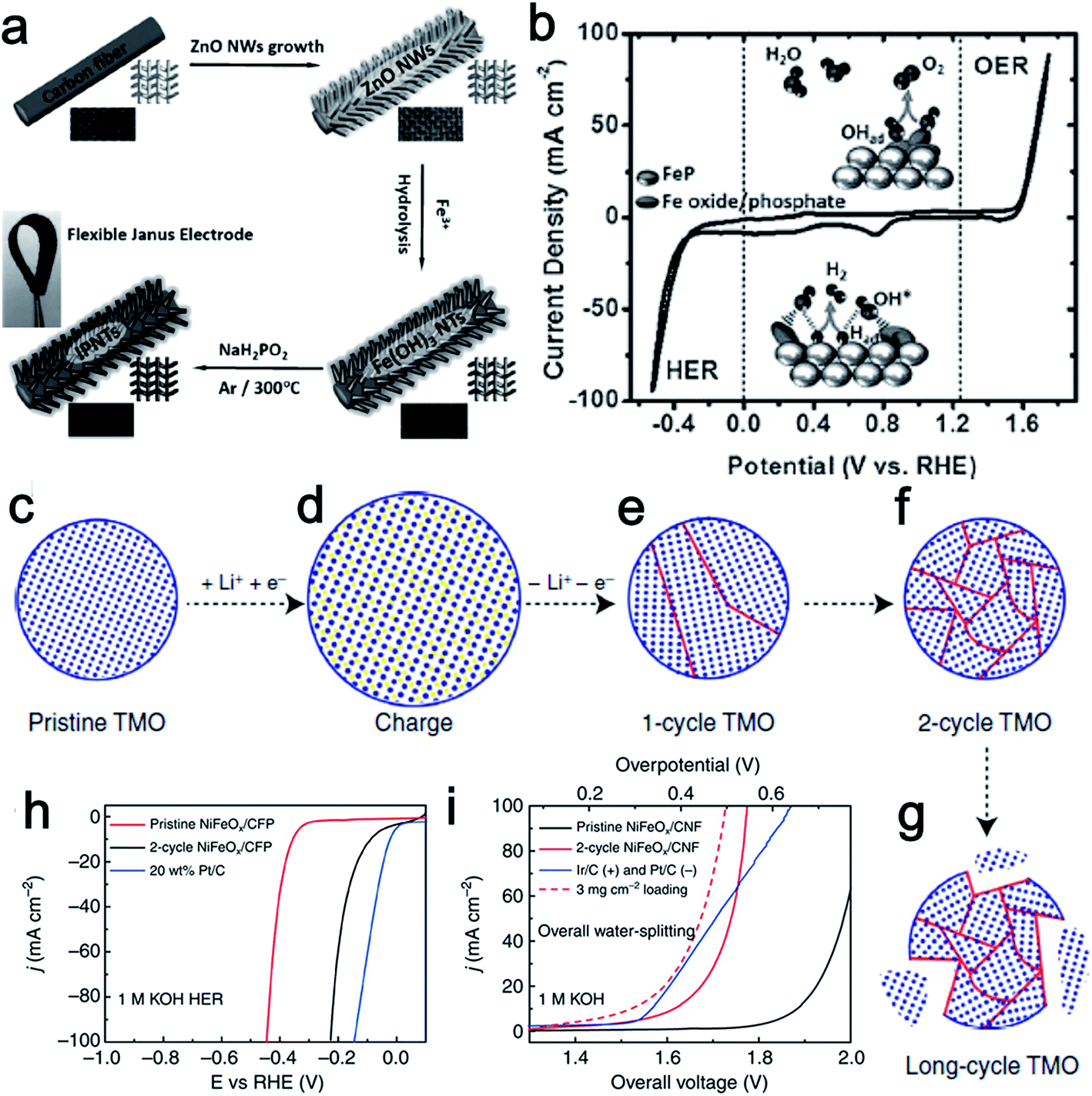 | ||
| Fig. 10 (a) Synthesis procedure of IPNT-based flexible bifunctional electrode. (b) CV curve of IPNTs in 1.0 M KOH with a scan rate of 50 mV s−1. Inset: proposed bifunctional mechanism of the catalyst for overall water splitting. Panels (a and b) reprinted with permission.79 Copyright 2015, Wiley-VCH. (c–g) Schematic of TMO morphology evolution under galvanostatic cycles. TMO particles gradually change from single crystalline to ultra-small interconnected crystalline NPs. Long-term battery cycling may result in the break-up of the particle. (h) The HER activity of 2-cycle NiFeOx/CFP is significantly improved from its pristine counterpart and close to the Pt/C benchmark. (i) Two-cycle NiFeOx/CFP as HER and OER bifunctional catalyst in 1 M KOH for overall water splitting. Ir/C and Pt/C as OER and HER benchmarks are tested side by side. Panels (c–i) reprinted with permission.18 Copyright 2015, Nature Publishing Group. | ||
As discussed above, a variety of methods have been used to prepare transition metal-based catalytic materials for overall water splitting. Yet, it is still highly desirable to develop more general and scalable methods to synthesize non-noble metal based bifunctional HER–OER catalysts. A good example of a general and scalable method was recently presented by Read et al. who synthesized transition metal phosphide electrodes by reacting commercial metal foils (Fe, Co, Ni, Cu, and NiFe) with various organophosphine sources.80 The resulting phosphide electrodes exhibited excellent electrocatalytic HER and OER performance that compared favorably with samples prepared using significantly more elaborate and costly procedures. Cui's group reported a lithium-induced conversion reaction to enhance the catalytic activity of various transition metal oxide (Fe, Co, Ni oxides, and their mixed oxides) nanoparticles (Fig. 10c–g).18 Due to the increased valency of the metal centers, the OER performance of those transition metal oxide improved dramatically through the electrochemically extracting lithium process. In addition, the increased density of electrochemically active sites and the covalent hybridization between the M-3d and O-2p states also made great contribution to the enhanced OER activity. The lithium-extracted ultra-small NiFeOx nanoparticles showed an especially remarkable performance, requiring only 1.51 V to achieve a current density of 10 mA cm−2 in a two-electrode configuration. Moreover, no obvious loss of the activity was observed even after 200 h of continuous electrolysis (Fig. 10h and i). These well-studied electrode materials enrich the family of highly efficient bifunctional catalysts for overall water splitting.
Summarizing the work reviewed above, it is not hard to find that the electrocatalytic performance of Co-, Ni-, and Fe-based bifunctional water splitting catalysts do not differ from each other too much; their bifunctional activities on full water splitting rely more on the structure and specific active surface area rather than on what kinds of metal are involved. This implies that the importance is on engineering the morphology and composition to maximize the density of catalytically active sites.
3.4. Other emerging compounds
In addition to the transition metal Co-, Ni-, and Fe-based bifunctional catalysts, other transition metal compounds such as Mo- and Cu-based oxides have also been proven to be bifunctionally active toward the HER and OER. For example, Jin et al. reported the preparation of porous MoO2 nanosheets via a simple wet-chemical route followed by annealing. Porous MoO2 nanosheets showed much higher dual activity for the HER and OER compared with bulk MoO2.81 They attributed the outstanding performance to the higher specific surface area and more catalytically active sites resulting from the porous nanostructure. Moreover, porous MoO2 required a cell voltage of only ∼1.53 V to reach a current density of 10 mA cm−2 and maintained its activity for almost 24 h in an alkaline electrolyzer. The water splitting configuration could even be driven by a 1.5 V AA battery at room temperature. Previously, Cu compounds were reported to exhibit biomimetic chemistry with O2, such as the reductive activation of O2 in enzymes and the laccase protein.82,83 Jahan et al. recently designed a Cu–MOF composite by incorporating GO with a copper-centered MOF as a multifunctional catalyst for not only the HER and OER, but also the ORR.84 It is noted that this Cu-based tri-functional electrocatalyst differed from other reported overall water splitting catalysts by its ability to work efficiently in an acidic electrolyzer. Most water splitting catalysts are usually active only under alkaline conditions. The excellent electrocatalytic performance of the GO–MOF hybrid was due to its special porous scaffold structure, enhanced charge transfer capability, and synergistic effect between the MOF and GO.Metal-free materials, particularly functional carbon materials such as heteroatom-doped graphene nanosheets and carbon nanotubes, have been extensively exploited as active oxygen or hydrogen catalysts.85–87 The catalytic properties of the obtained functional carbon catalysts can be significantly improved by doping the carbon-based materials with non-metal heteroatoms. In so doing, the electronic structure of the carbon species can be modified while the defects, which often serve as the catalytic active sites, are increased.88 However, the investigation of bifunctional activity on the same carbon surfaces for both the HER and OER is still rare; hence, exploring the application of functional carbon-based materials for full water splitting needs continued research efforts. In this respect, Lai et al. first reported the design of O, N, and P co-doped porous graphite nanocarbon as a bifunctional free-standing 3D electrode (ONPPGC/OCC) toward overall water splitting at all pH values.89 The constructed alkaline water electrolyzer exhibited especially excellent activity by employing ONPPGC/OCC for both the HER and OER catalyst, which could produce a current density of 10 mA cm−2 at a cell voltage of only 1.66 V with superior stability. The outstanding water splitting electrocatalytic performance of ONPPGC/OCC could be explained by the unique 3D porous structure, enhanced accessible active sites, improved transport properties, as well as the good electrical conductivity. Admittedly, this work shows an exciting novel avenue to explore the use of functional carbon materials for overall water splitting.
4. Conclusions and outlook
This review article has summarized recent research advances in the development of bifunctionally active HER–OER electrocatalysts for overall water splitting. The major goal of this research field is to replace expensive and rare noble metal-based electrocatalysts with low-cost and highly active nonprecious bifunctional ones. Table 1 lists a detailed comparison of the electrochemical performances of those bifunctional water splitting electrocatalysts. By far, enormous strides and many achievements have been made to advance these catalysts into practical application. First, the investigation of first row transition metal-based bifunctional heterogeneous catalysts toward both the HER and OER simplify the design of water splitting systems. This puts forwards a new way to solve the incompatibility of different catalysts for working in the same electrolyzer. Second, many efforts have been dedicated to construct self-supported catalysts on the conductive electrodes to avoid the use of carbon additives or polymer binder, ensuring good electronic contact between the water splitting catalysts with the support electrode. In addition, through intelligently designing the synergistic effect of nanostructured hybrid electrocatalysts, the performance of composite catalysts has been significantly maximized. Moreover, several general and scalable strategies have been proposed to prepared non-noble metal-based bifunctional electrocatalysts for overall water splitting, making potential directions for the scale-up of water photolysis/electrolysis technology for practical application.| Bifunctional catalyst | Electrolytes | Mass loading (mg cm−2) | η onset (mV) for HER, OER | η for HER, OER at corresponding j (mV@mA cm−2) | Tafel slope (mV dec−1) for HER, OER | Overall voltageb (V) | |||
|---|---|---|---|---|---|---|---|---|---|
| a All voltages presented in this table are shown versus the reversible hydrogen electrode (RHE). b The overall voltage is obtained from a two-electrode electrolyzer in basic conditions, except ONPPGC/OCC. | |||||||||
| Co-based materials | |||||||||
| CoCat film17 | 0.5 M KPi, pH 7 | — | 50 | — | 385@2 | — | 140 | — | — |
| Co–P film31 | 1.0 M KOH | 2.71 | — | — | 94@10 | 345@10 | 42 | 47 | 1.56onset |
| PCPTF32 | 0.5 M H2SO4 | 0.1 | 35 | — | 175@30 | — | 53 | — | — |
| 1.0 M KOH | — | 220 | 430@30 | 330@30 | — | 65 | |||
| Co–P/NC35 | 1.0 M KOH | 0.283 | — | 270 | 191@10 | 354@10 | 51 | 52 | 1.55onset |
| Co–P/rGO34 | 0.5 M H2SO4 | 0.28 | 13 | 105@10 | — | 50 | — | ||
| 1.0 M KOH | 150@10 | 340@10 | 38 | 66 | 1.70@j = 10 mA cm−2 | ||||
| CoP-MNA37 | 1.0 M KOH | 6.2 | — | 270 | 54@10 | 290@10 | 51 | 65 | 1.62@j = 10 mA cm−2 |
| CoP2/rGO36 | 1.0 M KOH | 0.285 | — | — | 88@10 | 300@10 | 50 | 96 | 1.56@j = 10 mA cm−2 |
| EG/Co0.85Se/NiFe-LDH40 | 1.0 M KOH | 4 | 240 | 240 | 260@10 | 270@150 | 160 | 57 | 1.67@j = 10 mA cm−2 |
| CoO/CoSe2 (ref. 41) | 0.5 M PBS | 1 | 150 | 320 | 337@10 | 510@10 | 131 | 137 | 2.18@j = 10 mA cm−2 |
| CoOx@CN44 | 1.0 M KOH | 1 | 85 | — | 232@10 | 260@10 | 115 | — | 1.55@j = 20 mA cm−2 |
| Co3O4 NCs30 | 1.0 M KOH | 0.35 | 50 | 290 | — | 350@16.5 | 116 | 101 | 1.91@j = 10 mA cm−2 |
| PNC/Co43 | 1.0 M KOH | 0.35 | 270 | — | 289@10 | 370@10 | 131 | 76 | 1.64@j = 10 mA cm−2 |
| NiCo2O4 (ref. 47) | 1.0 M NaOH | 1 | 50 | 230 | 110@10 | 290@10 | 49.7 | 53 | 1.65@j = 10 mA cm−2 |
| CoMnO@CN48 | 1.0 M KOH | 2.0 | — | 230 | 71@20 | 420@308 | — | — | 1.7@j = 54 mA cm−2 |
| NiCo2S4 NW/NF49 | 1.0 M KOH | — | — | — | 210@10 | 260@10 | 58.9 | 40.1 | 1.63@j = 10 mA cm−2 |
| NiCo2S4 NA/CC28 | 1.0 M KOH | 4 | 230 | 270 | 263@50 | 310@50 | 141 | 89 | 1.68@j = 10 mA cm−2 |
| CFP/NiCo2O4/Co0.57Ni0.43LMOs45 | 0.5 M H2SO4 | — | 29 | — | 52@10 | — | 34 | — | — |
| 0.1 M KOH | — | 90 | — | 340@10 | — | 63 | |||
| Co2B-500 (ref. 42) | 0.1 M KOH | 0.21 | — | — | 127@10 | 380@10 | 136.2 | 45 | — |
|
|||||||||
| Ni-based materials | |||||||||
| NiFe LDHs16 | 1.0 M NaOH | — | — | — | 210@10 | 240@10 | 1.7@j = 10 mA cm−2 | ||
| N2P57 | 1.0 M KOH | 0.14 | — | — | 220@10 | 290@10 | — | 59 | 1.63@j = 10 mA cm−2 |
| Ni5P4 (ref. 61) | 0.5 M H2SO4 | 3.5 | — | — | 140@10 | — | 40 | — | 1.7@j = 10 mA cm−2 |
| 1.0 M KOH | — | — | 150@10 | 290@10 | 53 | — | |||
| Ni–P58 | 0.5 M H2SO4 | 0.2 | — | — | 110@10 | — | 73 | — | — |
| 1.0 M KOH | — | — | — | 300@10 | — | 64 | |||
| Ni–P foam52 | 1.0 M KOH | — | — | — | 150@10 | 350@191 | — | 179.9 | 1.44@j = 5 mA cm−2 |
| Ni/Ni8P3 (ref. 60) | 1.0 M KOH | 10.58 | 70 | 260 | 130@10 | 270@30 | 58.5 | 73.2 | 1.61@j = 10 mA cm−2 |
| Ni/Ni9S8 (ref. 60) | 1.0 M KOH | 11.04 | — | — | 230@10 | 340@30 | 123.3 | 109.8 | |
| Ni3S2/NF67 | pH 14 | 1.6 | — | — | 223@10 | 260@10 | — | — | 1.76@j = 13 mA cm−2 |
| MoS2/Ni3S2 (ref. 69) | 1.0 M KOH | 7 | 50 | 175 | 110@10 | 218@10 | 83 | 88 | 1.56@j = 10 mA cm−2 |
| NiSe/NF68 | 1.0 M KOH | 2.8 | — | — | 96@10 | 270@20 | 120 | 64 | 1.63@j = 10 mA cm−2 |
| NiS/Ni foam65 | 1.0 M KOH | 43 | — | 320 | 158@20 | 335@50 | 83 | 89 | 1.64@j = 10 mA cm−2 |
| Mn1Ni1 (Mn0.1Ni1, 10 h)62 | 0.1 M KOH | 0.13 | 150 | — | 360@10 | — | — | — | — |
| — | 360 | — | 420@10 | ||||||
| Ni3FeN-NPs70 | 1.0 M KOH | 0.35 | — | — | 158@10 | 280@10 | 42 | 46 | — |
| TiN@Ni3N71 | 1.0 M KOH | 0.6 | 15 | 290 | 21@10 | 350@10 | 42.1 | 1.64@j = 10 mA cm−2 | |
|
|||||||||
| Fe-based materials | |||||||||
| Fe2P80 | 0.5 M H2SO4 | — | — | — | 191@10 | — | 55 | — | — |
| 1.0 M KOH | — | — | 300@10 | 390@10 | 126 | — | |||
| NiFeP80 | 0.5 M H2SO4 | — | — | — | 143@10 | — | 67 | — | — |
| 1.0 M KOH | — | — | 255@10 | 227@10 | 83 | — | |||
| FeP NTs79 | 0.5 M H2SO4 | 1.6 | 35 | — | 88@10 | — | 35.5 | — | 1.69@j = 10 mA cm−2 |
| 1.0 M KOH | 31 | 250 | 120@10 | 288@10 | 59.5 | 43 | |||
| FeOx78 | 0.1 M KOH | — | — | — | — | 400@10 | — | — | — |
| Ni2.5Co0.5Fe/NF54 | 0.1 M KOH | 0.25 | — | — | — | 275@10 | 93 | 105 | 1.62@j = 10 mA cm−2 |
| NiFeOx/CFP18 | 1.0 M KOH | 1.6 | 40 | 200 | 88@10 | 280@200 | 84.6 | 31.5 | 1.55@j = 10 mA cm−2 |
|
|||||||||
| Other emerging materials | |||||||||
| MoO2 (ref. 81) | 1.0 M KOH | 2.9 | 0 | 200 | 27@10 | 260@10 | 41 | 54 | 1.52@j = 10 mA cm−2 |
| Cu–MOF (8%GO)84 | 0.5 M H2SO4 | 0.226 | 87 | — | 209@30 | 110@2 | 84 | 65 | — |
| ONPPGC/OCC89 | 1.0 M KOH | 0.1 | — | — | 446@10 | 410@10 | 154 | 83 | 1.66@j = 10 mA cm−2 |
| 0.2 M PBS | — | — | 352@1 | 420@2 | 374 | 231 | 1.71@j = 2 mA cm−2 | ||
| 0.5 M H2SO4 | — | — | 386@10 | 470@10 | 109 | 200 | 1.75@j = 5 mA cm−2 | ||
Despite these advances, there are still many challenges that must be addressed before bifunctional water splitting electrocatalysts can find a wide range of practical uses for sustainable hydrogen production in a cost-effective and scalable way. (1) In the current stage of bifunctional catalyst development for water splitting, theoretical prediction and in situ spectroscopic study is lacking. These studies are necessary towards understanding the reaction mechanism and the real-time structural evolution of catalysts during electrolysis. Although DFT calculations provide a powerful tool to identify the active species and optimize the catalysts at the molecular level,90,91 more efforts are needed to pinpoint the exact catalytic/reaction mechanism on these composite catalysts. Whereas an in situ spectroscopic technique could be promising for that purpose, it is not well established and still under investigation. (2) To objectively evaluate the activity of HER–OER bifunctional electrocatalyst is not that straightforward. The activity of the electrocatalysts may be affected by many factors such as supporting substrates, operating conditions, and structure of the materials. In 2014, the Jaramillo group reported a benchmarking protocol for evaluating the activity, durability, and electrochemical active surface area of several electrocatalysts for the HER and OER by integrating them with a solar water splitting device in aqueous acidic or alkaline conditions.92 However, there is still more work to be done along this direction. (3) Currently, the majority of studies on HER–OER bifunctional catalysts is carried out in alkaline electrolyzers, this excludes their possible use in proton exchange membrane (PEM)-based electrolyzers.93 Consequently, acid-stable catalysts with high activity and stability are urgently needed, especially for anodic oxygen evolution.
Future research efforts with regard to bifunctional water splitting catalysts can be focused on the following aspects. (1) Understanding the specific mechanism of the HER/OER on the bifunctional electrocatalysts through theoretical and experimental studies, which may provide useful guidance towards the design of efficient bifunctional HER–OER catalysts via the modification of the crystal and electronic structures. The investigation into the mechanism that causes the degradation of the catalysts during cycling is also critical. (2) Exploitation and optimization of the catalytically active sites to maximize the potential of the electrocatalysts by engineering the morphology and composition of the catalyst. Engineering the morphology of bifunctional HER–OER catalysts into various nanostructures (such as nanorods, nanotubes, and nanowires) provides us with the means to improve the physical properties of the catalysts by increasing the number of exposed active sites and facilitating diffusion of reactants in the electrolyte.47,49–51 On the other hand, tuning the catalyst composition allows for modification of the electronic structure of active sites so as to optimize the intrinsic activity of the bifunctional catalysts.41,42,44,67 (3) Development of advanced functional carbon-based materials for catalyzing the water splitting reactions. Various heteroatom doped functional carbon materials featuring unique properties have demonstrated great potential for electrochemical water splitting reactions due to their controllable molecular structures, abundant active sites, and strong tolerance to acid/alkaline conditions.86 Therefore, functional carbon-based electrocatalysts are a promising new class of highly efficient HER–OER bifunctional catalysts. Finally, as solar energy is an abundant and renewable source of energy,21,86,94 it would be interesting to integrate these optimized bifunctional water splitting catalysts into solar cell devices for sustainable hydrogen production.5,16,95,96
Acknowledgements
This project is funded by the academic research fund AcRF tier 1 (M4011253 RG 7/14) and tier 2 (M4020246, ARC10/15), Ministry of Education, Singapore and The Program for Associate Professor of Special Appointment (Young Eastern Scholar) at Shanghai Institutions of Higher Learning (QD2016013). We also acknowledge financial support by the Natural Science Foundation of Shanghai (16ZR1423500).References
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- A. B. Laursen, S. Kegnaes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577–5591 CAS.
- Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693–1705 CrossRef CAS.
- W. F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896–8909 RSC.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1500985 CrossRef.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 CAS.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- E. A. Hernández-Pagán, N. M. Vargas-Barbosa, T. Wang, Y. Zhao, E. S. Smotkin and T. E. Mallouk, Energy Environ. Sci., 2012, 5, 7582–7589 Search PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- H. Wang, H. W. Lee, Y. Deng, Z. Lu, P. C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef CAS PubMed.
- A. Lasia, Handbook of fuel cells, 2010 Search PubMed.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- D. U. Lee, P. Xu, Z. P. Cano, A. G. Kashkooli, M. G. Park and Z. Chen, J. Mater. Chem. A, 2016, 4, 7107–7134 CAS.
- L. Jörissen, J. Power Sources, 2006, 155, 23–32 CrossRef.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- G. Burstein, Corros. Sci., 2005, 47, 2858–2870 CrossRef CAS.
- B. Conway, L. Bai and M. Sattar, Int. J. Hydrogen Energy, 1987, 12, 607–621 CrossRef CAS.
- D. Liu, Q. Lu, Y. Luo, X. Sun and A. M. Asiri, Nanoscale, 2015, 7, 15122–15126 RSC.
- D. Kong, H. Wang, Z. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed.
- S. Du, Z. Ren, J. Zhang, J. Wu, W. Xi, J. Zhu and H. Fu, Chem. Commun., 2015, 51, 8066–8069 RSC.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- Y. Yang, H. Fei, G. Ruan and J. M. Tour, Adv. Mater., 2015, 27, 3175–3180 CrossRef CAS PubMed.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- L. Jiao, Y.-X. Zhou and H.-L. Jiang, Chem. Sci., 2016, 7, 1690–1695 RSC.
- B. You, N. Jiang, M. Sheng, S. Gul, J. Yano and Y. Sun, Chem. Mater., 2015, 27, 7636–7642 CrossRef CAS.
- J. Wang, W. Yang and J. Liu, J. Mater. Chem. A, 2016, 4, 4686–4690 CAS.
- Y.-P. Zhu, Y.-P. Liu, T.-Z. Ren and Z.-Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337–7347 CrossRef CAS.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- M. R. Gao, J. X. Liang, Y. R. Zheng, Y. F. Xu, J. Jiang, Q. Gao, J. Li and S. H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 CAS.
- K. Li, J. Zhang, R. Wu, Y. Yu and B. Zhang, Adv. Sci., 2016, 3, 1500426 CrossRef.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- X. Li, Z. Niu, J. Jiang and L. Ai, J. Mater. Chem. A, 2016, 4, 3204–3209 CAS.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- J. Yin, P. Zhou, L. An, L. Huang, C. Shao, J. Wang, H. Liu and P. Xi, Nanoscale, 2016, 8, 1390–1400 RSC.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926–1929 CrossRef CAS PubMed.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290–6294 CrossRef CAS PubMed.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef CAS PubMed.
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 26, 4661–4672 CrossRef CAS.
- Z. Peng, D. S. Jia, A. M. Al-Enizi, A. A. Elzatahry and G. F. Zheng, Adv. Energy Mater., 2015, 5, 1402031 CrossRef.
- S. Li, Y. Wang, S. Peng, L. Zhang, A. M. Al-Enizi, H. Zhang, X. Sun and G. Zheng, Adv. Energy Mater., 2016, 6, 1501661 CrossRef.
- X. Wang, W. Li, D. Xiong and L. Liu, J. Mater. Chem. A, 2016, 4, 5639–5646 CAS.
- X. Liu, X. Wang, X. Yuan, W. Dong and F. Huang, J. Mater. Chem. A, 2016, 4, 167–172 CAS.
- X. Zhu, C. Tang, H.-F. Wang, B.-Q. Li, Q. Zhang, C. Li, C. Yang and F. Wei, J. Mater. Chem. A, 2016, 4, 7245–7250 CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- Z. Huang, Z. Chen, Z. Chen, C. Lv, H. Meng and C. Zhang, ACS Nano, 2014, 8, 8121–8129 CrossRef CAS PubMed.
- L. A. Stern, L. G. Feng, F. Song and X. L. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 CAS.
- X. Y. Yu, Y. Feng, B. Y. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 CAS.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- G.-F. Chen, T. Y. Ma, Z.-Q. Liu, N. Li, Y.-Z. Su, K. Davey and S.-Z. Qiao, Adv. Funct. Mater., 2016, 26, 3314–3323 CrossRef CAS.
- M. Ledendecker, S. Krick Calderon, C. Papp, H. P. Steinruck, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- M. Ledendecker, G. Clavel, M. Antonietti and M. Shalom, Adv. Funct. Mater., 2015, 25, 393–399 CrossRef CAS.
- H. Vandenborre, P. Vermeiren and R. Leysen, Electrochim. Acta, 1984, 29, 297–301 CrossRef CAS.
- N. Jiang, L. Bogoev, M. Popova, S. Gul, J. Yano and Y. Sun, J. Mater. Chem. A, 2014, 2, 19407–19414 CAS.
- W. Zhu, X. Yue, W. Zhang, S. Yu, Y. Zhang, J. Wang and J. Wang, Chem. Commun., 2016, 52, 1486–1489 RSC.
- W. Zhou, X.-J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 CAS.
- L. L. Feng, G. Yu, Y. Wu, G. D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef CAS PubMed.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS PubMed.
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef.
- Q. Zhang, Y. Wang, Y. Wang, A. M. Al-Enizi, A. A. Elzatahry and G. Zheng, J. Mater. Chem. A, 2016, 4, 5713–5718 CAS.
- Y. Yan, L. Thia, B. Y. Xia, X. M. Ge, Z. L. Liu, A. Fisher and X. Wang, Adv. Sci., 2015, 2, 1500120 CrossRef PubMed.
- P. Jiang, Q. Liu, Y. Liang, J. Tian, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 12855–12859 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 9476–9477 CrossRef CAS PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- B. C. M. Martindale and E. Reisner, Adv. Energy Mater., 2016, 6, 1502095 CrossRef.
- Y. Yan, B. Y. Xia, X. Ge, Z. Liu, A. Fisher and X. Wang, Chem.–Eur. J., 2015, 21, 18062–18067 CrossRef CAS PubMed.
- C. G. Read, J. F. Callejas, C. F. Holder and R. E. Schaak, ACS Appl. Mater. Interfaces, 2016, 8, 12798–12803 CAS.
- Y. Jin, H. Wang, J. Li, X. Yue, Y. Han, P. K. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785–3790 CrossRef CAS PubMed.
- S.-K. Lee, S. D. George, W. E. Antholine, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 6180–6193 CrossRef CAS PubMed.
- I. Bento, M. A. Carrondo and P. F. Lindley, J. Biol. Inorg. Chem., 2006, 11, 539–547 CrossRef CAS PubMed.
- M. Jahan, Z. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363–5372 CrossRef CAS.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- J. Lai, S. Li, F. Wu, M. Saqib, R. Luque and G. Xu, Energy Environ. Sci., 2016, 9, 1210–1214 CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- J. Gu, Y. Yan, J. L. Young, K. X. Steirer, N. R. Neale and J. A. Turner, Nat. Mater., 2016, 15, 456–460 CrossRef CAS PubMed.
- J. Gu, Y. Yan, J. W. Krizan, Q. D. Gibson, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Am. Chem. Soc., 2014, 136, 830–833 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |