
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates†
L.
Vereecken
 *ab,
A.
Novelli
a and
D.
Taraborrelli
a
*ab,
A.
Novelli
a and
D.
Taraborrelli
a
aForschungszentrum Jülich GmbH, Institute for Energy and Climate, IEK-8 Troposphere, 52428 Jülich, Germany. E-mail: L.Vereecken@fz-juelich.de
bMax Planck Institute for Chemistry, 55128 Mainz, Germany
First published on 28th November 2017
Abstract
Stabilized Criegee intermediates (SCI) are reactive oxygenated species formed in the ozonolysis of hydrocarbons. Their chemistry could influence the oxidative capacity of the atmosphere by affecting the HOx and NOx cycles, or by the formation of low-volatility oxygenates enhancing atmospheric aerosols known to have an important impact on climate. The concentration of SCI in the atmosphere has hitherto not been determined reliably, and very little is known about their speciation. Here we show that the concentration of biogenic SCI is strongly limited by their unimolecular decay, based on extensive theory-based structure–activity relationships (SARs) for the reaction rates for decomposition. Reaction with water vapor, H2O and (H2O)2 molecules, is the second most important loss process; SARs are also proposed for these reactions. For SCI derived from the most common biogenic VOCs, we find that unimolecular decay is responsible for just over half of the loss, with reaction with water vapor the main remaining loss process. Reactions with SO2, NO2, or acids have negligible impact on the atmospheric SCI concentration. The ambient SCI concentrations are further characterized by analysis of field data with speciated hydrocarbon information, and by implementation of the chemistry in a global chemistry model. The results show a highly complex SCI speciation, with an atmospheric peak SCI concentrations below 1 × 105 molecule cm−3, and annual average SCI concentrations less than 7 × 103 molecule cm−3. We find that SCI have only a negligible impact on the global gas phase H2SO4 formation or removal of oxygenates, though some contribution around the equatorial belt, and in select regions, cannot be excluded.
Introduction
Criegee intermediates (CI, carbonyl oxides as shown in Fig. 1) have the potential to be important oxidants in the atmosphere.1–4 Their main source is the ozonolysis of unsaturated volatile organic compounds (VOC), where the ozone molecule performs a cycloaddition on a double bond, forming the so-called primary ozonide (POZ) that dissociates to a CI and a carbonyl compound.1 The Criegee intermediates are formed with a wide range of internal energies, which affects their subsequent fate. Chemically activated CI with high energy content react promptly by unimolecular reactions, and are direct sources of acids and other oxygenated compounds, and of chain-propagating OH radicals (see Fig. 1). Depending on the size of the CI, its internal energy, and the bath gas properties and pressure, chemically activated CI can also dissipate their energy to the bath gas and become collisionally stabilized.5–7 Alternatively, the CI can be formed with less nascent internal energy; their longer lifetime allows them to attain a thermal energy distribution prior to reaction. Such CI with a thermal energy content, i.e. stabilized CI (SCI), can, in addition to a wide range of unimolecular reactions, undergo bimolecular reactions with a plethora of co-reactants,2–4 potentially affecting the oxidative capacity of the atmosphere and the formation of aerosols in a different manner than through their unimolecular reactions. A more detailed description of the complex ozonolysis reaction system is available in the ESI† (Section U.1.a). | ||
| Fig. 1 The ambient SCI concentration is determined by the balance between SCI formation in the ozonolysis of unsaturated organic compounds, against the dominant CI loss processes through unimolecular decay and reaction with water. A small fraction of the ambient SCI can react with SO2, acids, and other co-reactants forming low-volatility compounds that lead to more and larger aerosol formation in the atmosphere, in turn affecting climate and air quality. | ||
Terpenoids, i.e. C5H8, C10H16 and C15H24 biogenic hydrocarbons that make up the bulk of the non-methane VOCs emitted to the atmosphere,8 are the dominant parent molecules of SCI, through their ozonolysis reactions. Once formed, unimolecular decomposition or isomerisation, and reaction with water vapor (H2O and (H2O)2) are expected to be the dominant SCI loss processes. The ambient concentration of SCI is then determined by the relative rates of VOC ozonolysis, and these SCI losses (see Fig. 1). While the yield and speciation of the SCI formed in the ozonolysis are not well known, and depend strongly on the specific VOC, the total source strength is fairly well constrained by extensive literature data9 where ozonolysis is known to consume a sizable fraction of the 1000 Tg of organic matter yearly emitted to the atmosphere, though its contribution is highly region- and time dependent and ranges from near-negligible to >20%.8,10–12 The main uncertainty in quantifying the ambient SCI concentration lies in the loss processes, where only simple hydrogen- and methyl-substituted SCI have been investigated in detail,2–4 and only partial information exists on atmospherically more relevant SCI that are typically much larger, branched, and possibly hetero-substituted structures. The unimolecular and water reaction rate coefficients are highly dependent on the CI structure, making the analysis of CI chemistry in the atmosphere hitherto very speculative.
Several key questions remain as yet unanswered: what is the concentration of SCI in the atmosphere, what is their speciation, and how do these ambient SCI react and impact the atmosphere? Determining the concentration and speciation of ambient SCI requires a drastic increase in our knowledge on the loss processes, i.e. unimolecular decay and the reactions with water vapor, and this specifically for CI derived from biogenic VOCs. The atmospheric impact of the ambient SCI, on the other hand, lies partly in their ability to undergo fast reaction with many oxygenated species, thus forming larger, lower-volatility oxygenates and oligomers that can enhance aerosol growth (see Fig. 1). A central aspect of SCI chemistry comes from its ability to oxidize SO2 to SO3, which in turn forms H2SO4.13,14 The only other gas phase SO2 oxidation pathway, with OH radicals, has a low reaction rate,15 which limits the H2SO4 source strength. In contrast, the SCI + SO2 reactions are very fast,2,3,16 with rate coefficients approaching the collision limit, 2 to 16 × 10−11 cm3 molecule−1 s−1, and even moderate concentrations of SCI could strongly enhance H2SO4 formation. The extremely low vapor pressure of H2SO4 acid is a driving factor in the nucleation of secondary aerosols. Knowing the ambient SCI concentration is thus pivotal to assess, through formation of H2SO4 and of low-volatility oxygenates, the impact of SCI on aerosol formation, which in turn has a strong climate forcing potential as well as air quality impact. Several studies tried to estimate the SCI concentration and/or its impact on aerosol formation, by modeling or by examining related processes, but lack of data limits their reliability.17–22
In this work, we theoretically examine over 200 CI reactions, spanning a large range of substitutions: H-atoms, linear and branched aliphatic groups, β- and γ-unsaturated substituents, as well as oxo- and OH-substituents (see Fig. 2). These systematic calculations also examine group additivity trends, stereo-selectivity, kinetic isotope effects, chirality, and other aspects. From this, we obtain structure–activity relationships (SARs) for the temperature-dependent rate coefficients for the dominant unimolecular pathways, and for the reaction with H2O and (H2O)2, covering over 100 classes of CI most likely to be formed from terpenoids and other unsaturated compounds. Combined with field data and global modeling, we present a well-founded assessment of the ambient concentration of SCI in the atmosphere, and estimate its impact on SO2 oxidation. We also examine the reaction of SCI with formic acid, a key oxygenate in the atmosphere whose budget is not yet fully understood.23 While not discussed in detail at this time, our results will also help to identify which compounds are formed in the main CI loss processes, e.g. OH radicals, acids, hydroperoxides, etc., and enable a better assessment of the subsequent secondary chemistry, of the fate of chemically activated CI, and of the impact of CI on the oxidative capacity of the atmosphere.
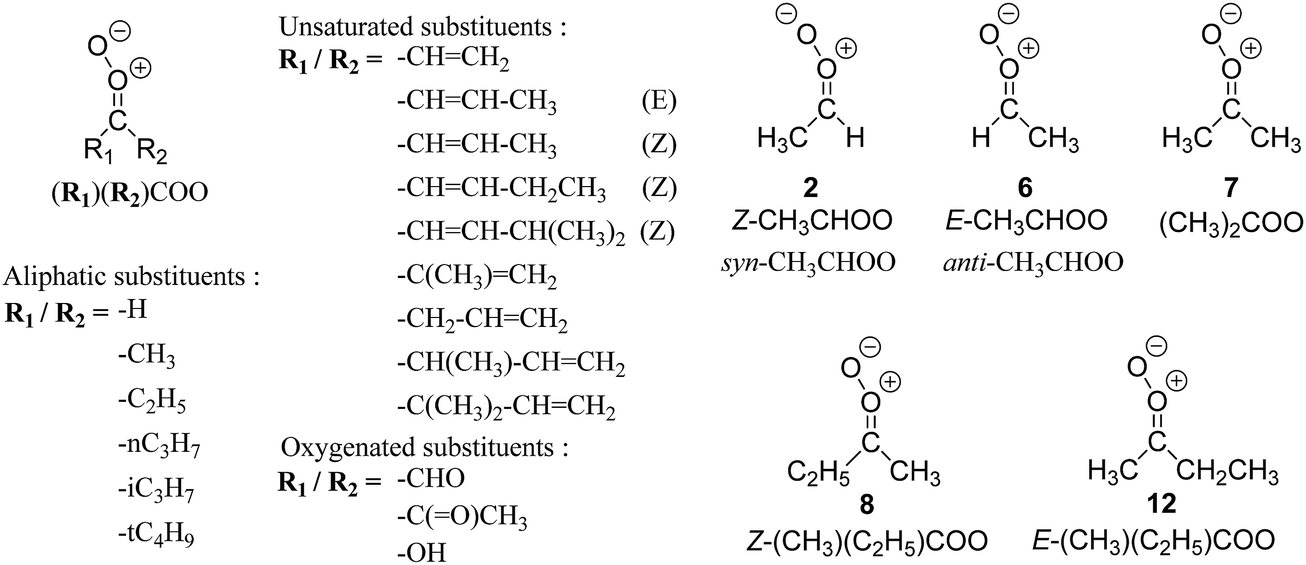 | ||
| Fig. 2 Stereospecific substitution of Criegee intermediates. The stereo-specific substituents R1 and R2 (defined in the top left) each lead to different reactivity. The list of R1 and R2 substituents studied in this work spans most atmospherically relevant compounds, covering aliphatic, (stereo-specific) unsaturated, and oxygenated moieties. Traditional syn/anti naming convention is superseded by the IUPAC Z/E stereodescriptors. Four examples (SCI 2, 6, 8, 12) are given on the right, selected out of the 69 SCI studied in this work (see ESI,† Section C); symmetric CI (e.g. SCI 7) do not need a stereo-descriptor. | ||
All results are discussed in the ESI,† which is arranged as a stand-alone reference document to accommodate the extensive data made available in this study. The main text here summarizes the key results, with links to the appropriate Sections in the ESI.†
Carbonyl oxide naming convention
The reactivity of CI is determined mainly by the substitution around the carbonyl oxide moiety, where the position of the substituents (see Fig. 2) relative to the oxide outer O-atom has a critical impact on reactivity. Earlier literature designated stereoisomers as syn- and anti-CI, but these terms are not accurate for CI that don't have a hydrogen atom substituent. Also, these terms have become overloaded with other meanings. This includes the expected reaction mechanism, where syn-CI undergo a 1,4-H-shift in the ‘vinyl hydroperoxide’ channel while anti-CI react by 1,3-ring closure in the ‘ester’ channel, or the presumed reaction products, where OH-forming CI are labeled syn-CI, and all others anti-CI.To allow a non-ambiguous description of the isomeric makeup of the CI, we propose here to adopt the Z/E IUPAC absolute stereodescriptors,24 already in use for double bonds, such as in olefines, to describe the position of the substituents across the CI double bond as depicted in Fig. 2. As discussed in the ESI† (Sections B and H), treating the carbonyl oxide moiety as containing a double bond is valid for thermalized SCI due to the strong zwitterionic wavefunction of the CI, and the concomitantly high barriers and negligible reaction rates for Z/E-stereo-isomerisation by (pseudo-)rotation. The relative priority of substituents is already well-established and familiar to most chemists due to their extensive use for olefins, so no particular problems are anticipated in adopting the Z/E stereodescriptors. In this text, we will use this notation throughout for absolute stereo-description, while the syn- and anti designators are only used to specify substituent positions relative to the carbonyl oxide outer O-atom.
Methodology
A detailed description, including additional references, benchmark results and error analysis, are presented in the ESI† (Sections F, U, and V), with only a very concise description here.The rovibrational characteristics of reactants and transition states were characterized at the M06-2X/aug-cc-pVTZ level of theory,25,26 with energies improved using CCSD(T)/aug-cc-pVTZ calculations,26,27 and empirical corrections based on experimental and higher-level theoretical literature data. The temperature-dependent rate coefficients were determined by canonical transition state theory (CTST), explicitly incorporating a full conformational analysis28,29 and asymmetric Eckart tunneling,30,31 yielding high-pressure Kooij kinetic expressions32 for k(T) for T between 250 and 450 K.
The SAR for unimolecular decay selects the dominant reactions for a set of CI classes, each defined by its specific type of syn- and anti-substituent; each class was found to have only a single contributing unimolecular decay process. The temperature-dependent rate coefficients are taken from the theoretical kinetic analysis described above. The SAR for SCI + H2O rate coefficients correlates the CTST k(T) based on our CCSD(T)//M06-2X data against experimental and high-level theoretical literature information, allowing ad hoc correction of the predicted rates to match the available data. The SAR for SCI reaction with (H2O)2 builds upon rate coefficient correlation against the H2O reaction. In all three SARs, rate coefficient estimates for SCI substituent combinations not explicitly studied was performed by extrapolating the reactivity trends in analogous reactions across the anti- and/or syn-substituents. The SARs are expected to be accurate within a factor 5, 10, and 40, respectively, for unimolecular decay, H2O- and (H2O)2 reactions, including error contributions from the quantum chemical calculations, the kinetic analysis, the literature reference data, and statistical errors (see ESI,† Sections F.3, R.4, S.6, and T.5).
SCI average concentrations from the field data at four locations,33–36 ranging from the pristine rain forest to a highly polluted mega-city, are obtained assuming steady state between speciated formation and loss processes. The production rate accounts for the average O3 and VOC concentrations, the site-specific ozonolysis rate coefficients, and the yield and speciation of the SCI. The loss rate for each SCI includes their unimolecular decomposition, reaction with water and water dimers, and with SO2, NO2, and organic acids. The analysis relies on direct experimental and theoretical studies where available, and estimates based on reaction trends and SARs otherwise.
For global modeling, we use the ECHAM/MESSy Atmospheric Chemistry (EMAC) model, which is a numerical chemistry and climate simulation system that includes sub-models describing tropospheric and middle atmosphere processes and their interaction with oceans, land and human influences.37 The core atmospheric model is the 5th generation European Centre Hamburg general circulation model (ECHAM5).38 VOC emissions8,39 and the Mainz Organics Mechanism (MOM) chemical model were already used by Lelieveld et al.,40 and comprise about 30 unsaturated organic compounds reacting with O3, spanning a wide range of terpenes, linear alkenes, and aromatics.
Chemical reactions of carbonyl oxides
a. Substituents of importance in atmospheric SCI
The most common substitution around the double bonds of atmospheric VOCs are H-atoms and aliphatic groups, where we find that the branching (i.e. primary, secondary,…) of the carbon atom connected to the carbonyl oxide moiety has the largest impact on SCI reactivity. The length of the carbon chain, on the other hand, was found to have only a minor influence on the CI reactivity. In this work, we thus focus on H, methyl, ethyl, iso-propyl, and tert-butyl substituents (see Fig. 2). Most of the terpenoids are poly-unsaturated compounds, where many show conjugated double bonds, or γ-unsaturated olefins (i.e. double bonds separated by a single aliphatic carbon). These additional unsaturated bonds, and their stereo-specific substitution, play an important role in the reactivity. Fig. 2 shows the range of unsaturated functionalities examined in this study; again, branching of the aliphatic substituents on these double bonds has a higher impact on reaction rates than the length of the chains. Multi-unsaturated compounds can also give rise to secondary CI, where a second double bond gets ozonized in the products of the first oxidation step of the parent VOC. This first oxidation can be initiated not only by O3, but also OH, NO3, Cl, photolysis, etc., leading to an extremely wide variety of compounds. However, typical oxidation processes in the atmosphere are known to yield predominantly carbonyl- and hydroxy-substituted oxygenates, so these are the hetero-substituents included in this work (see Fig. 2).b. Reactivity trends in SCI
The stability, and concomitantly the reactivity, of an SCI is determined mostly by the substituent in syn-position and its interaction with the oxide O-atom. The stereospecific reactivity of carbonyl oxides has been amply demonstrated for E- and Z-CH3CHOO,2–4,41–44 with rate coefficients for similar processes (e.g. the reaction with H2O) differing by up to several orders of magnitude for the stereoisomers due to their specific formation enthalpy. For the substituents examined in this work, the enthalpy differences between stereoisomers can be even larger than for CH3CHOO, resulting in even larger differences in rate coefficients between some stereoisomers. Methyl substituents, CH3, in syn-position lead to the most stable CI, while oxo-substituents, –C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–, in syn-position yield the most reactive CI with regard to similar reaction processes such as unimolecular 1,3-ring closure, or bimolecular reactions with water; H-atom and β-unsaturated substituents yield intermediate reactivity. We have summarized the stereoisomer relative energies in a group additivity analysis (see ESI,† Section H), which can help to quantify their relative energy, thus suggesting a relative reactivity.
O)–, in syn-position yield the most reactive CI with regard to similar reaction processes such as unimolecular 1,3-ring closure, or bimolecular reactions with water; H-atom and β-unsaturated substituents yield intermediate reactivity. We have summarized the stereoisomer relative energies in a group additivity analysis (see ESI,† Section H), which can help to quantify their relative energy, thus suggesting a relative reactivity.
Stereoisomerisation of SCI, either by internal rotation or by in-plane pseudo-rotation of the carbonyl oxide outer O-atom (reaction (1) in Fig. 3), is found to be very slow, in all cases slower than other available unimolecular and bimolecular reactions. Z- and E-isomers of asymmetric SCI thus act as separate atmospheric species and, given their often very different rate coefficients, their subsequent chemistry must be included separately in kinetic models. For conjugated CI, i.e. with a β-CC or β-CO group, we find that internal rotation along the central single bond (reaction (8) in Fig. 3) is, for practical purposes, always fast enough to equilibrate the s-trans, s-gauche, and/or s-cis isomers. These conformers will thus form a single reactant pool, greatly simplifying the treatment of such SCI in atmospheric kinetic models.
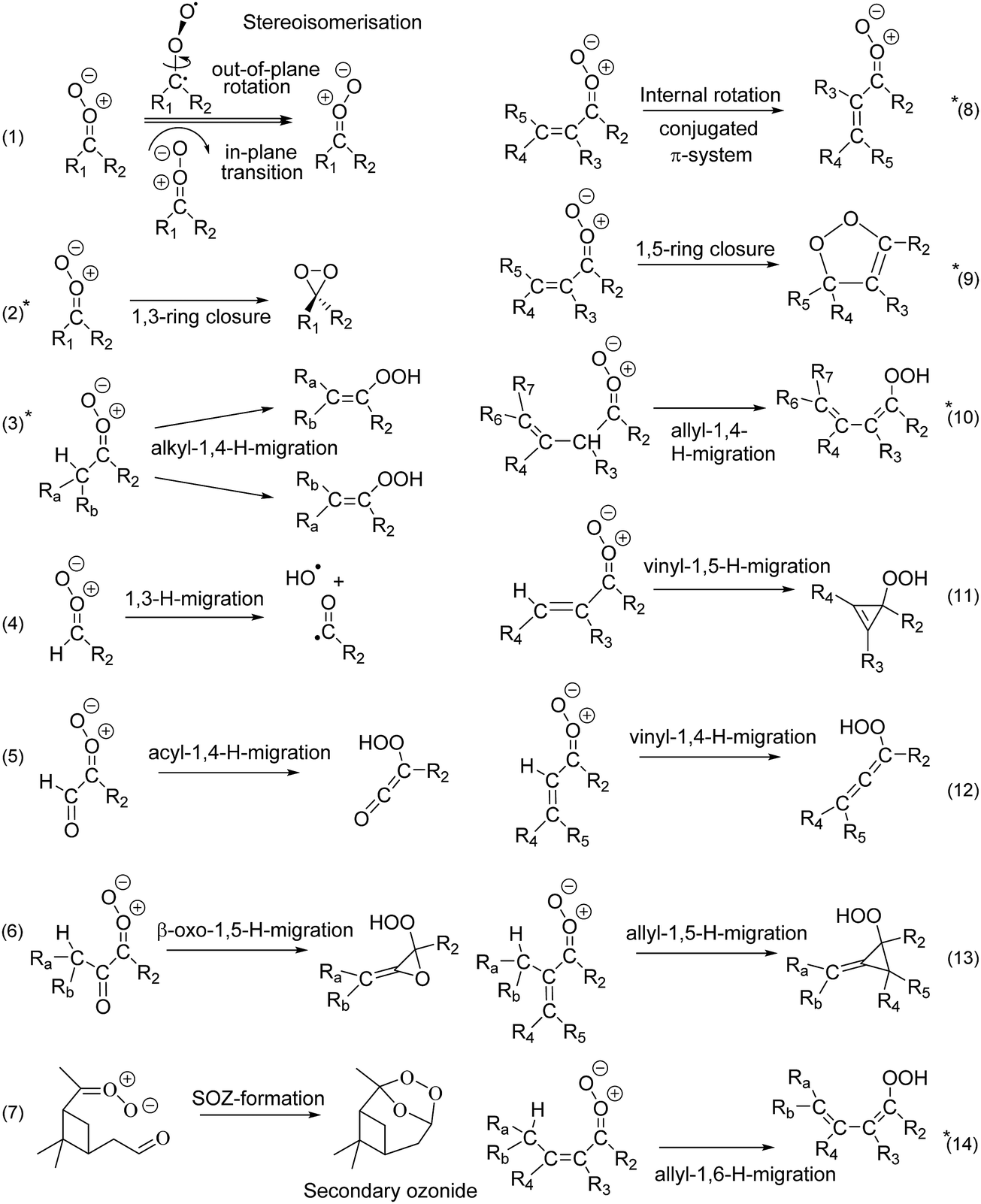 | ||
| Fig. 3 Unimolecular reaction classes implemented in this work; reactions marked with an asterisk (2, 3, 8, 9, 10, and 14) were found to be most important in the atmosphere. Rate coefficients for reactions (2), (3), (9), (10), and (14) are included in the SAR; reaction (8) is shown to be sufficiently fast to allow equilibration across the rotamers. See ESI† (Section D) for the full list of reaction mechanisms considered. | ||
c. SCI unimolecular decomposition
Carbonyl oxides have an extensive unimolecular chemistry,3 where we explicitly performed calculations for 14 reaction types, shown in Fig. 3, out of 24 reaction classes identified (see ESI,† Section D). Based on the ∼170 rate coefficients calculated in this work, we conclude that only six chemical reaction classes are relevant. Furthermore, for each SCI examined, a single reaction is always dominant, though which reaction this is it depends on the substitution pattern. We briefly describe these key reactions classes here, with extensive details and numerical data deferred to the ESI† (Sections G through Q).1,4-H-migration (reaction (3)) is the most important isomerisation channel if a suitably-placed, migratable H-atom is available. Lifetimes of the order of milliseconds are predicted here, in agreement with literature data,16,41,45–51 and allyl-resonance (reaction (10)) further increases the reaction rate. These reactions form vinyl hydroperoxides (VHP), where for asymmetric CI the E-VHP is by far the most dominant stereo-specific product. VHP are expected to dissociate, forming OH radicals with a vinoxy radical co-product,1 and H-migration is thus an important radical chain propagating step in the atmosphere. Contrary to the relative C–H bond strength, increasing substitution on the β-carbon slows down 1,4-H-migration due to unfavorable interaction of the additional alkyl groups with the carbonyl oxide moiety, negatively affecting both reaction barrier and entropy, and reducing rate coefficients by an order of magnitude from primary to tertiary H-atoms. For syn-β-unsaturated SCI, the 1,5-ring closure reaction (reaction (9)) becomes dominant,52,53 but if an H-atom is available in δ-position a very fast 1,6-H-migration (reaction (14)) becomes accessible, aided by the conjugated π-system. Both reactions yield SCI lifetimes of the order of microseconds. OH-substituted SCI have similarly short lifetimes, or even isomerize spontaneously to percarboxylic acids. The longest-lived SCI characterized have no migratable H-atom nor syn-β-unsaturation, and decay by 1,3-ring closure (reaction (2)) with unimolecular lifetimes up to an hour.
Some potentially important reactions were not studied in this work, as their rates are highly specific to the SCI molecular build. The most critical of these reactions are ring closure reactions where the carbonyl oxide moiety reacts with a hetero-substituted functionality elsewhere in the molecule. A well-known example of this type of reactions is the formation of secondary ozonides (SOZ) in a ring closure reaction with a carbonyl CO bond, but similar processes can occur with acids, alcohols, hydroperoxides, and other functionalities.
d. The reaction of SCI with water
The reaction with water is the main loss process for many of the atmospheric SCI with a longer unimolecular life-time. The rate coefficient depends very strongly on the CI stereo-specific substitution, and whether the coreactant is H2O or (H2O)2.2,3
We have performed calculations on 32 SCI + H2O reactions of importance in the atmosphere. The rate coefficients with H2O and (H2O)2 span over 10 orders of magnitude, as already predicted in the literature for a smaller subset of SCI.41–44,47,51,54–63 The SCI most reactive towards water were found to have an H-atom or oxo-substitutent in syn-position to the carbonyl oxide, and react with room temperature rate coefficients ≥ 10−11 cm3 molecule−1 s−1 towards (H2O)2, within an order of magnitude of the collision limit of ∼3 × 10−11 cm3 molecule−1 s−1, and with k(298 K) ≥ 10−14 cm3 molecule−1 s−1 towards H2O. For these SCI, the water reaction can dominate unimolecular decay. In contrast, the SCI least reactive to water have oxo- or vinyl stabilizing groups in the anti-position, combined with less-reactive alkyl substituents in syn-position; these have rate coefficients k(298 K) ≤ 10−16 cm3 molecule−1 s−1 towards both H2O and (H2O)2.
e. Other atmospheric SCI reactions
Stabilized carbonyl oxides can react with a wide range of coreactants; these are extensively reviewed in the literature,2–4 and an overview of the available literature is given in the ESI† (Section E). Most of these reactions are not directly relevant in the atmosphere, however, either due to low concentrations of the co-reactants (e.g. HO2, OH, RO2, H2SO4, other SCI,…), due to low rate coefficients (e.g. carbonyl compounds, O3, alkenes, NH3,…), or both. In addition to the unimolecular reactions and the reactions with H2O and (H2O)2, only the reactions with NO2, with acids, and with SO2, are considered to be of potential impact. An extensive literature overview of the data available for these reactions is given in the ESI† (Section E); only a brief summary is given here. None of these reactions appear to be overly sensitive to the SCI substitution pattern, simplifying the estimation of rate coefficients.The reactions of SCI with SO2 have high rate coefficients, ≥3 × 10−11 cm3 molecule−1 s−1, and furthermore the SCI + SO2 reaction could supplement the rather slow OH-initiated oxidation of SO2, enhancing the H2SO4 formation in the atmosphere and thus the nucleation and growth of particular matter. The reactions of NO2 with SCI have somewhat lower rate coefficient, ∼10−12 cm3 molecule−1 s−1; it was proposed that these reaction could affect the night-time NO3 concentration,19 though recent experimental and theoretical work suggests that the main product is not NO3 but a nitro-adduct, with a limited impact on the NOx cycle. Finally, the reactions of SCI with acids have very high rate coefficients, ≥5 × 10−11 cm3 molecule−1 s−1, equal to or even exceeding the collision limit, and forming lower-volatility oxygenates. The reactions are fast both with carboxylic acids and with inorganic acids; under dry conditions, it has been suggested that SCI could affect the atmospheric life time of e.g. formic acid.
In our analysis, none of these reactions have an important impact on the atmospheric concentration of SCI, though their impact on other atmospheric chemical cycles, such as H2SO4 formation, aerosol nucleation and growth, or acid lifetimes, is yet to be fully elucidated.
SARs for atmospheric SCI
The atmospheric SCI concentration is determined mostly by their unimolecular decay and reactions with water vapor. The absolute rate coefficients for these reactions are highly dependent on the SCI substitution patterns, affecting their relative competition, and thus the fate of the SCI in the atmosphere. Due to this substituent dependency, structure–activity relationships are essential to implement these reactions into a chemical kinetic model; thousands of VOCs are estimated to be emitted to the atmosphere,64 and studying each of these individually is impractical. To classify atmospheric SCI, we identified 18 syn-substituent classes, and 10 anti-substituent classes, which combined are able to describe a wide diversity of SCI expected to be formed in the atmosphere. Each SCI can be assigned to a class by matching its syn- and anti-substituent to the SAR substituent classes; the predicted rate coefficients for reaction can then be looked up in the SAR tables. A formal definition of the SAR substituent classes, and the full SAR lookup tables are available in the ESI† (Section R, and T), while a reduced version is given in Table 1. The ESI† also describes how to make predictions for substituents that are currently not included in the SARs, and how to treat cyclic SCI; additional examples of the SAR application are also available.|
|
|
|
|
|
|
|---|---|---|---|---|---|

|
1,4-H-migration
k uni = 478 k H2O = 7.5 × 10−18 k dimer = 1.8 × 10−14 |
1,4-H-migration
k uni = 433 k H2O = 9.1 × 10−18 k dimer = 2.1 × 10−14 |
1,4-H-migration
k uni = 611 k H2O = 4.3 × 10−18 k dimer = 1.1 × 10−14 |
1,4-H-migration
k uni = 373 k H2O = 3.5 × 10−17 k dimer = 6.8 × 10−14 |
1,4-H-migration
k uni = 136 k H2O = 6.7 × 10−19 k dimer = 2.1 × 10−15 |

|
1,4-H-migration
k uni = 689 k H2O = 1.5 × 10−17 k dimer = 3.2 × 10−14 |
1,4-H-migration
k uni = 534 k H2O = 3.1 × 10−17 k dimer = 6.0 × 10−14 |
1,4-H-migration
k uni = 257 k H2O = 3.5 × 10−17 k dimer = 6.7 × 10-14 |
1,4-H-migration
k uni = 12 k H2O = 7.8 × 10−17 k dimer = 1.4 × 10−13 |
1,4-H-migration
k uni = 205 k H2O = 1.5 × 10−18 k dimer = 4.3 × 10−15 |

|
1,4-H-migration
k uni = 14 k H2O = 9.8 × 10−18 k dimer = 1.9 × 10−14 |
1,4-H-migration
k uni = 13 k H2O = 1.3 × 10−17 k dimer = 2.8 × 10−14 |
1,4-H-migration
k uni = 19 k H2O = 1.6 × 10−17 k dimer = 2.8 × 10−14 |
1,4-H-migration
k uni = 1 k H2O = 3.2 × 10−17 k dimer = 5.9 × 10-14 |
1,4-H-migration
k uni = 6.7 k H2O = 9.2 × 10−18 k dimer = 2.1 × 10−14 |

|
1,3-Cyclisation
k uni = 0.17 k H2O = 1.5 × 10−17 k dimer = 3.0 × 10−14 |
1,3-Cyclisation
k uni = 0.15 k H2O = 2.0 × 10−17 k dimer = 3.9 × 10−14 |
1,3-Cyclisation
k uni = 1.1 k H2O = 2.4 × 10−17 k dimer = 4.5 × 10−14 |
1,3-Cyclisation
k uni = 0.075 k H2O = 5.2 × 10−17 k dimer = 9.4 × 10−14 |
1,3-Cyclisation
k uni = 1 × 10−2 k H2O = 1.0 × 10−18 k dimer = 2.8 × 10−15 |
|
1,3-Cyclisation
k uni = 53 k H2O = 2.3 × 10−14 k dimer = 2.7 × 10−11 |
1,3-Cyclisation
k uni = 74 k H2O = 1.6 × 10−14 k dimer = 1.7 × 10−11 |
1,3-Cyclisation
k uni = 102 k H2O = 3.5 × 10−14 k dimer = 3.8 × 10−11 |
1,3-Cyclisation
k uni = 111 k H2O = 4.5 × 10−14 k dimer = 4.7 × 10−11 |
1,3-Cyclisation
k uni = 0.3 k H2O = 8.7 × 10−16 k dimer = 1.4 × 10−12 |

|
Allyl-1,4-Hmig
k
uni = 14 k H2O = 4.1 × 10−18 k dimer = 2.9 × 10−14 |
Allyl-1,4-Hmig
k
uni = 10 k H2O = 2.9 × 10−17 k dimer = 5.4 × 10−14 |
Allyl-1,4-Hmig
k uni = 5250 k H2O = 3.3 × 10−17 k dimer = 6.1 × 10−14 |
Allyl-1,4-Hmig
k uni = 247 k H2O = 7.4 × 10−17 k dimer = 1.3 × 10−13 |
Allyl-1,4-Hmig
k uni = 4180 k H2O = 1.4 × 10−18 k dimer = 3.9 × 10−15 |

|
Allyl-1,6-Hmig
k uni = 1.4 × 107 k H2O = 1.8 × 10−18 k dimer = 4.8 × 10−15 |
Allyl-1,6-Hmig
k uni = 1.2 × 107 k H2O = 3.5 × 10−18 k dimer = 8.6 × 10−15 |
Allyl-1,6-Hmig
k uni = 1.4 × 107 k H2O = 4.0 × 10−18 k dimer = 9.7 × 10−15 |
Allyl-1,6-Hmig
k uni = 4.7 × 106 k H2O = 8.9 × 10−18 k dimer = 2.0 × 10−14 |
Allyl-1,6-Hmig
k uni = 4.8 × 106 k H2O = 1.2 × 10−19 k dimer = 4.3 × 10−16 |

|
1,5-Cyclisation
k
uni = 13 k H2O = 1.8 × 10−18 k dimer = 4.8 × 10−15 |
1,5-Cyclisation
k
uni = 13 k H2O = 3.5 × 10−18 k dimer = 8.6 × 10−15 |
1,5-Cyclisation
k
uni = 13 k H2O = 4.1 × 10−18 k dimer = 9.7 × 10−15 |
1,5-Cyclisation
k
uni = 13 k H2O = 8.9 × 10−18 k dimer = 2.1 × 10−14 |
1,5-Cyclisation
k uni = 14300 k H2O = 1.2 × 10−19 k dimer = 4.3 × 10−16 |

|
1,3-Cyclisation
k uni = 7.1 × 104 k H2O = 1.4 × 10−14 k dimer = 1.5 × 10−11 |
1,3-Cyclisation
k uni = 5.3 × 104 k H2O = 1.5 × 10−14 k dimer = 1.6 × 10−11 |
1,3-Cyclisation
k uni = 1.3 × 105 k H2O = 1.8 × 10−14 k dimer = 1.8 × 10−11 |
1,3-Cyclisation
k uni = 8.0 × 103 k H2O = 4.0 × 10−14 k dimer = 3.7 × 10−11 |
1,3-Cyclisation
k uni = 144 k H2O = 7.7 × 10−16 k dimer = 1.1 × 10−12 |



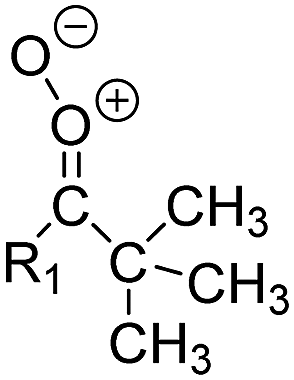

By analyzing the ∼170 unimolecular rate coefficients determined as described above, we found that a single unimolecular process is dominant in each SCI class, simplifying the implementation of SCI chemistry. For each CI substitution pattern, we thus select the reaction type with the highest rate coefficient (see ESI,† Section R). Not all substituent combinations were studied explicitly; substituent combinations for which no direct information is available are implemented using the reactivity trends available in the data, such that the SAR is broadly applicable. For each syn- and anti-substituent class combination, the SAR for unimolecular reactions thus provides the temperature-dependent rate coefficient, and identifies which reaction class is applicable. It should be stressed that the SAR includes only commonly found generic SCI reaction types (Fig. 3). For some specific compounds, potentially important pathways are not included; the most important example is secondary ozonide (SOZ) formation.
We also derive structure–activity relationships for both monomer and dimer water reactions as a function of the CI substitution (see ESI,† Sections S.5 and T.4), based on the available literature data, high-quality literature theoretical results, and additional calculations on 32 SCI + H2O reactions, matching the substituent classes already used for unimolecular SCI decay. These SARs likewise provide temperature-dependent rate coefficients.
The full SAR lookup tables are too extensive to include in the main paper. However, to illustrate some of the key reaction trends we include Table 1, showing a limited set of SCI substituent classes, with rate coefficients only at 298 K. An interesting feature is the impact of substituent branching and unsaturation on the H-migration rates, where higher branching can slow down the H-migration (e.g. iso-C3H7versus CH3 substituents), contrary to what might be expected based on C–H bond strength correlations. The underlying reason for this trend is the unfavorable interaction between the substituents, and between these and the carbonyl oxide moiety. Double bond conjugation can enhance H-migration (1,4-allyl-migration), or even enable H-migration across wider ranges (e.g. 1,6-allyl-migration). These H-migration reactions all form hydroperoxides, which are expected to predominantly dissociate to OH radicals and a vinoxy radical, affecting the atmosphere oxidative cycles. If no H-atoms suitable for migration are available, the reaction mechanism switches to ring closure reactions. The rate coefficients for reaction with H2O and (H2O)2 are seen to be correlated, i.e. they trace the reactivity of the SCI in a similar manner. The ratio of the rate coefficients varies significantly across the SCI substituent set, where for some SCI the unimolecular decay will be dominant, while for others the reaction with water vapor will be the main loss process. Table 3 shows the SCI classes that are most commonly formed in the atmosphere, and gives information on the relative importance of the loss processes. The sources and speciation of SCI in the atmosphere is discussed in more detail in the following section.
More extensive SARs are described formally in the ESI† (Sections R, S.5, and T.4), and are recommended for model development. These SARs are expected to be accurate within a factor 5, 10, and 40, respectively, for unimolecular decay, H2O- and (H2O)2 reactions, including error contributions from the quantum chemical calculations, the kinetic analysis, the literature reference data, and statistical errors (see ESI,† Sections F.3, R.4, S.6, and T.5), The ESI† also extensively tabulates rate coefficients for individual SCI, including the SCI formed from small alkenes, isoprene, α-pinene and β-pinene.
The atmospheric concentration and speciation of SCI
The VOCs emitted to the atmosphere span a wide variety of compounds. Ignoring methane, which does not react with ozone, the most dominant organic compounds are biogenically emitted compounds such as isoprene, monoterpenes, and sesquiterpenes, and a host of small alkenes which are mostly anthropogenic emissions. Hundreds of organic compounds have been identified in the atmosphere,64 of which a typical set of a few tens of compounds are found in the highest concentrations across many sites (see ESI,† Section U.1). To assess the concentration and speciation of SCI in the atmosphere, the rate coefficients for the main loss processes through unimolecular reaction and reaction with water vapor are needed across all relevant substitution patterns, and must be weighted across the speciated SCI and VOC contributions to quantify reaction fluxes across the many orders of magnitude difference in SCI reactivity. The substitution-, site-, and stereo-specificity prohibit easy generalization across multiple VOCs or even different CI formed from a single VOC. The modeling studies currently available in the literature17–22,54,65 did not have access to sufficiently detailed information, and of necessity adopted identical rate coefficients for very diverse families of SCI, resulting in large uncertainties and large variations across the SCI concentration predictions.Based on the rate coefficients obtained above for unimolecular reactions and reaction with H2O and (H2O)2, combined with site-specific ozonolysis rates from literature, and field measurement data33–36 on the relevant VOC, O3, NO2, SO2, carboxylic acid, and H2O concentrations, the speciated steady-state SCI concentration can then be estimated. We examined sites dominated by isoprene (Amazonian forest, Brazil) or monoterpenes (Sierra Nevada, USA), as well as a polluted urban environment (Mexico City) and rural near-free troposphere (Hohenpeissenberg, Germany). On each site, at least 100 different SCI are formed, with speciation varying strongly between the sites. Over half of these SCI are removed by unimolecular decay, while reaction with water is the only important remaining loss processes; SO2, carboxylic acids, or NO2 barely contribute to SCI loss. We discuss the main results here (see Table 2 and 3); details are given in the ESI† (Section U).
| Amazonia | Mexico city | Sierra Nevada | Hohenpeissenberg | |
|---|---|---|---|---|
| a The Mexico city region is not resolved at the grid size of the global model simulation. | ||||
| SCI total source strength (molecule cm−3 s−1) | 3.5 × 106 | 1.9 × 107 | 1.5 × 106 | 5.1 × 105 |
| SCI concentration (molecule cm−3) | 1.5 × 104 | 5.5 × 104 | 7.7 × 103 | 2.3 × 103 |
| Unimolecular decay | 54% | 53% | 58% | 53% |
| Reaction with water | 38% | 32% | 32% | 43% |
| Contribution SCI to total SO2 oxidation | ||||
| Field data (time series average) | 29% | 54% | 19% | 7% |
| Modeling (yearly average) | 62% | N/Aa | 10% | 5% |
| Gas phase lifetime of HCOOH to SCI | ||||
| Field data (time series average) | 3.5 days | 1 days | 7 days | 27 days |
| Modeling (yearly average) | 11 days | N/Aa | 70 days | 79 days |
The Amazonian forest shows a large contribution of isoprene-derived SCI, comprising 34% of the SCI source strength. Despite the dominance of isoprene in the VOC loading in this region, many other VOC contribute strongly, including 15% limonene-CI, and six other VOC contributing 45% combined. 60% of the SCI formed are lost by unimolecular decay, while >30% reacts with water. The steady state concentration is estimated at 1.5 × 104 molecule cm−3, where methyl vinyl ketone oxide (MVK oxide) formed directly from isoprene constitutes 42% of the ambient SCI, α-pinene-derived SCI 9%, and many other SCI each less than 5%. The smallest SCI, CH2OO, is formed from many compounds (see Table 3), but contributes negligibly to the ambient concentration owing to its fast reaction with (H2O)2. These predictions are fairly robust to uncertainties in the underlying rate coefficients and processes, e.g. neglecting all bimolecular SCI loss processes would only increase the non-CH2OO SCI concentration by a factor of ∼2, emphasizing the importance of unimolecular decay.
| SCI class smallest representative & number of VOC sources | Production rate (molecule cm−3 s−1) | k (298 K) | Example SCI | |
|---|---|---|---|---|
| Contribution in SCI production | Pseudo-first order (s−1) | |||
| Biogenic | Anthropogenic | |||
| CH2OO formed from 23 VOC |
1.0 × 106
19% |
3.8 × 106
20% |
k
uni = 0.3
k H2O = 260 k dimer = 280 |

|
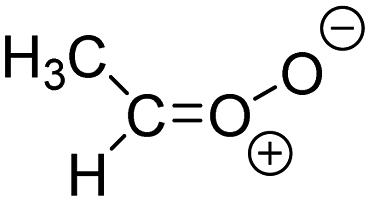
| Z-CH3CHOO formed from 6 VOC |
6.5 × 106
35% |
k
uni = 136
k H2O = 0.2 k dimer = 0.4 |
|
|
| E-CH3CHOO formed from 6 VOC |
1.6 × 106
10% |
k
uni = 53
k H2O = 6900 k dimer = 5400 |

|
|
| (CH3)2OO formed from 10 VOC |
5.0 × 105
9% |
5.2 × 106
28% |
k
uni = 478
k H2O = 2 k dimer = 4 |
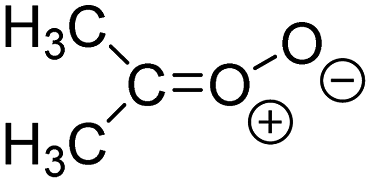
|
|
E-(CH3)(CHCH2)COO formed from 4 VOC |
4.7 × 105
9% |
k
uni = 50
k H2O = 0.02 k dimer = 0.06 |

|
|
| E-(C2H5)CHOO formed from 12 VOC |
4.7 × 105
9% |
6.0 × 105
3% |
k
uni = 74
k H2O = 4800 k dimer = 3400 |

|
| Z-(C2H5)CHOO formed from 14 VOC |
4.6 × 105
9% |
6.0 × 105
3% |
k
uni = 205
k H2O = 0.5 k dimer = 0.9 |

|
|
E-(CHCH2)CHOO formed from 10 VOC |
3.7 × 105
7% |
k
uni = 30
k H2O = 260 k dimer = 54 |

|
|
| E-(CH3)(C2H5)COO formed from 7 VOC |
3.0 × 105
6% |
k
uni = 433
k H2O = 3 k dimer = 4 |

|
|
| Z-(C2H5)(CH3)COO formed from 7 VOC |
3.0 × 105
6% |
k
uni = 689
k H2O = 5 k dimer = 6 |
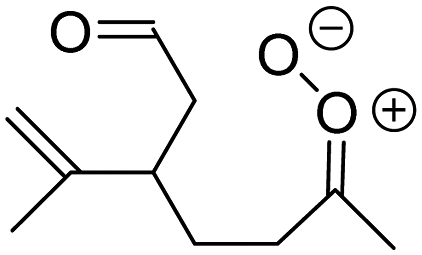
|
|
|
Z-(CHCH2)(CH3)COO formed from 4 VOC |
2.6 × 105
5% |
k
uni = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400
k H2O = 0.5 k dimer = 1 |

|
|
|
Z-(CH2–CHCH2)CHOO formed from 6 VOC |
2.6 × 105
5% |
k
uni = 4180
k H2O = 0.5 k dimer = 0.8 |

|
|
| Z-Nopinone-oxide formed from β-pinene |
2.4 × 105
4% |
k
uni = 2.0
k H2O = 130 k dimer = 120 |

|
|
| Z-Pinonaldehyde-K-oxide formed from α-pinene |
1.4 × 105
3% |
k
uni = 60
k H2O = 0.07 k dimer = 0.2 |

|
|
| E-Pinonaldehyde-K-oxide formed from α-pinene |
1.4 × 105
3% |
k
uni = 250
k H2O = 3 k dimer = 0.6 |
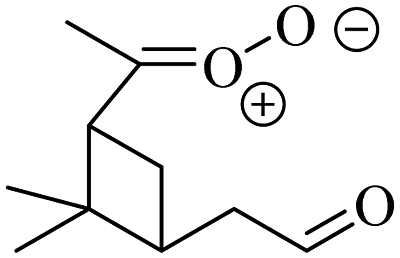
|
|
The SCI sources in the Sierra Nevada are more diffuse, with limonene, α- and β-pinene, δ-3-carene, MVK and myrcene jointly contributing 80% of the SCI source strength. 75% of the monoterpene SCI undergo rapid H-migration or ring closure reactions, with most other reacting quickly with water, resulting in a low predicted SCI concentration of 8 × 103 molecule cm−3. Some dominant longer-lived SCI are Z-nopinone oxide formed from β-pinene, contributing 19% to the ambient SCI concentration, with α-pinene-, limonene-, and MVK-derived SCI constituting an additional 33%.
The site at Hohenpeissenberg is characterized by low VOC and O3 concentration, with a predicted SCI concentration of only 2.3 × 103 molecule cm−3, in line with the recent field data analysis by Novelli et al.17 In contrast, the highly polluted air in Mexico City is predicted to have higher SCI concentration, ∼5.5 × 104 molecule cm−3, where the main contributors are anthropogenic emissions, i.e. C3 to C5 linear and branched alkenes.
While this analysis propagates the uncertainties on the rate coefficients estimates derived in this work, and relies on several additional assumptions, our uncertainty analysis shows that our predictions are not overly sensitive to any particular value due to the interaction of many competing processes; the overall uncertainty on the predicted concentrations is expected to be less than an order of magnitude.
The global distribution of SCI in the troposphere has also been assessed with the ECHAM/MESSy atmospheric chemistry model.37 The SCI formation and loss processes were implemented without affecting the overall chemistry, to prevent unbalancing the chemical system. Fig. 4 shows the global annual mean distribution of SCI concentrations at the Earth's surface, accounting for loss processes by unimolecular decay, water, SO2, NO2, and acids. Predicted peak SCI concentrations never exceed 105 molecule cm−3 (Fig. 4b), where the highest concentration are found in VOC-rich regions around the equator, or during biomass burning events like the intense fires in summer 2010 in Russia. Annual average concentrations are never larger than 7 × 103 molecule cm−3 (Fig. 4a), and in most parts of the world are ≤2 × 103 molecule cm−3, again in good agreement with earlier analysis by Novelli et al.17 The highest average concentrations are found in the forested regions in the equatorial belt. Similar to the field data, we find the modeled main loss processes of SCI at the Earth's surface are unimolecular decay and reactions with water (see Table 2), with negligible impact by SO2, NO2 and acids.
 | ||
| Fig. 4 Global annual distribution of SCI concentrations. (a) Concentration averaged over 1 simulation year. (b) Peak concentration predicted within that year. | ||
Impact of SCI on atmospheric chemistry
Due to the predicted low concentrations, the contribution of SCI to the gas phase oxidation of SO2 to SO3 and subsequently to H2SO4 is mostly small (Fig. 5a). The largest impact in absolute terms on a yearly basis is over the equatorial belt, where CI + SO2 is an important source of gas phase H2SO4; local hotspots are found in China, Eastern India and Myanmar. Relative to OH + SO2, significant production rates of H2SO4 from SCI are also predicted for regions impacted by biomass burning events, though the contribution is never overwhelming the OH-driven oxidation (see Table 2). These numbers are in stark contrast to early estimates that predicted up to 800% increase in SO2 oxidation in some regions, but are in line with more recent modeling estimates.18,22 Further studies are needed to investigate the subsequent impact of SCI-induced H2SO4 formation on the nucleation rates or aerosol chemistry in these regions. | ||
| Fig. 5 Impact of SCI in the atmosphere. (a) Contribution (fraction) of SCI in the gas phase formation of H2SO4, where the only other relevant contributing process is SO2 + OH. (b) Formic acid lifetime towards the gas phase loss processes with SCI. | ||
Fig. 5b shows the gas-phase sinks of HCOOH expressed as its lifetime towards CI. The traditional gas phase HCOOH sink due to reaction with OH leads typically to lifetimes in the range of 10 to 20 days in tropical regions. Over densely vegetated regions such as the Amazonian forest where HCOOH is most abundant, the reaction of formic acid with SCI is thus predicted to be the dominant gas phase sink, exceeding the loss by OH radicals. Near the surface and in clouds, however, wet and dry deposition will be dominant acid loss process, with typical HCOOH lifetimes of 2–4 days.23,66,67 Estimating the impact of SCI chemistry on formic acid concentration is thus not straightforward, and would require more extensive modeling.
The main products of SCI chemistry in the atmosphere are due to the unimolecular reactions, and reaction with water vapor, each contributing roughly half of the SCI loss. The impact of SCI in the atmosphere is thus mostly as source of oxygenated hydrocarbons. Unimolecular H-migration is an important reaction, forming (poly-unsaturated) vinylhydroperoxides (VHP), known to predominantly, and often promptly, eliminate an OH radical. The non-H-migration unimolecular SCI reactions, i.e. 1,3- and 1,5-ring closure reactions, lead to acids, ester, and cyclic peroxides. Little is known about the atmospheric chemistry of the latter, and further studies are needed to ascertain their role in the atmosphere. It can be expected, however, that these peroxides will at some point break the weak O–O bond, and decompose to smaller partially oxygenated compounds. The reaction with water and water dimers is known to form α-hydroxy-hydroperoxides. These compounds are already considered in chemical models, and are likely to partition to the aqueous phase. Gas phase decomposition, e.g. initiated by OH radicals, is again likely to lead to rupture of the weak peroxide bond, generating an OH radical. A detailed analysis of the overall impact of all these oxygenated compounds on the gas phase chemistry in the atmosphere, on aerosol nucleation, particulate matter growth and aging is outside the scope of this work.
Conclusions
From the theoretical data presented in this work, the SARs derived from it, and the application to field data and modeling, some important conclusions emerge. It is clear that the rate coefficients for unimolecular decay of SCI spans a much wider range than currently considered in models. In particular, unimolecular reactions are significantly more important for SCI derived from larger biogenic VOCs than for the smaller CI that have so far been studied most. Furthermore, several highly different unimolecular mechanisms are active, with distinct reaction products and hence differing subsequent chemistry. The H-migration mechanisms lead to (conjugated) vinyl hydroperoxides, and are likely to yield OH radicals in subsequent chemistry. In β-unsaturated CI, 1,5-ring closure yields cyclic peroxides, whose chemistry is not well-known, but it seems reasonable to assume they will decompose by O–O bond breaking and fragment to smaller oxygenates. Finally, 1,3-ring closure through the dioxirane channel is known to form acids and esters. For large SCI such as those formed from BVOC, hot acid decomposition is unlikely, and this pathway thus represents a termination of the radical reaction chain. The reaction of SCI with water vapor, the dominant loss process for about half of the SCI formed in the atmosphere, forms hydroxy-hydroperoxides, as already known from earlier studies. The large oxygenated compounds formed through these various channels could have a direct impact on SOA growth due to their lowered volatility.We conclude that it is important that SCI speciation is correctly incorporated in kinetic models. Our results show that one cannot generalize a limited set of rate coefficients or reaction mechanisms across all SCI, assign a single VOC as the main source of SCI, or claim that a small number of SCI species are representative of the entire SCI population. At the same time, we showed that for most of the SCI, unimolecular decay or reaction with water is overwhelmingly dominant, such that for those SCI species the same products are always formed, and the creation of the corresponding products can be lumped with the source ozonolysis reaction in a kinetic model without the need of explicit description of the intermediate SCI chemistry. While hundreds of different SCI can be formed from dozens of VOC at any one site, the largest part of the ozonolysis mass flux can thus be accommodated, and it is likely sufficient to model 20 to 30 specific SCI explicitly to represent the ambient population, and assess various aspects of SCI chemistry (e.g. oxidation of SO2) in the atmosphere. The total and speciated yield of SCI from the ozonolysis reactions remains poorly quantified; our uncertainty analysis, however, suggests that the ambient SCI concentration is not overly sensitive to this speciation.
Finally, we find that the ambient concentrations of SCI are low in most of the troposphere, always ≤1 × 105 molecule cm−3 even at peak concentration, which influences SO2 oxidation or the formic acid budget only around the equatorial belt or in specific regions such as megacities or biomass burning events.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. V. gratefully recognizes the support of the Max Planck Graduate Center with the Johannes Gutenberg-Universität Mainz (MPGC), Germany, for part of this research. We acknowledge the Jülich Supercomputer Centre for hosting the JuKinet computational cluster.References
- D. Johnson and G. Marston, The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- D. L. Osborn and C. A. Taatjes, The physical chemistry of Criegee intermediates in the gas phase, Int. Rev. Phys. Chem., 2015, 34, 309–360 CrossRef CAS.
- L. Vereecken and J. S. Francisco, Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- C. A. Taatjes, D. E. Shallcross and C. Percival, Research frontiers in the chemistry of Criegee intermediates and tropospheric ozonolysis, Phys. Chem. Chem. Phys., 2014, 16, 1704–1718 RSC.
- S. Hatakeyama, H. Kobayashi, Z. Y. Lin, H. Takagi and H. Akimoto, Mechanism for the reaction of peroxymethylene with sulfur dioxide, J. Phys. Chem., 1986, 90, 4131–4135 CrossRef CAS.
- N. M. Donahue, G. T. Drozd, S. A. Epstein, A. A. Presto and J. H. Kroll, Adventures in ozoneland: down the rabbit-hole, Phys. Chem. Chem. Phys., 2011, 13, 10848–10857 RSC.
- G. T. Drozd and N. M. Donahue, Pressure Dependence of Stabilized Criegee Intermediate Formation from a Sequence of Alkenes, J. Phys. Chem. A, 2011, 115, 4381–4387 CrossRef CAS PubMed.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef CAS.
- The mechanisms of atmospheric oxidation of the alkenes, ed. J. G. Calvert, R. Atkinson, J. A. Kerr, S. Madronich, G. K. Moortgat, T. J. Wallington and G. Yarwood, Oxford University Press, New York, 2000 Search PubMed.
- A. Geyer, B. Alicke, S. Konrad, T. Schmitz, J. Stutz and U. Platt, Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin, J. Geophys. Res.: Atmos., 2001, 106, 8013–8025 CrossRef CAS.
- L. Xue, R. Gu, T. Wang, X. Wang, S. Saunders, D. Blake, P. K. K. Louie, C. W. Y. Luk, I. Simpson, Z. Xu, Z. Wang, Y. Gao, S. Lee, A. Mellouki and W. Wang, Oxidative capacity and radical chemistry in the polluted atmosphere of Hong Kong and Pearl River Delta region: analysis of a severe photochemical smog episode, Atmos. Chem. Phys., 2016, 16, 9891–9903 CAS.
- S. S. Brown, W. P. Dube, J. Peischl, T. B. Ryerson, E. Atlas, C. Warneke, J. A. de Gouw, S. te, L. Hekkert, C. A. Brock, F. Flocke, M. Trainer, D. D. Parrish, F. C. Feshenfeld and A. R. Ravishankara, Budgets for nocturnal VOC oxidation by nitrate radicals aloft during the 2006 Texas Air Quality Study, J. Geophys. Res.: Atmos., 2011, 116, D24305 Search PubMed.
- R. L. Mauldin III, T. Berndt, M. Sipilä, P. Paasonen, T. Petäjä, S. Kim, T. Kurtén, F. Stratmann, V.-M. Kerminen and M. Kulmala, A new atmospherically relevant oxidant of sulphur dioxide, Nature, 2012, 488, 193–196 CrossRef PubMed.
- T. Berndt, T. Jokinen, M. Sipilä, R. L. Mauldin, H. Herrmann, F. Stratmann, H. Junninen and M. Kulmala, H2SO4 formation from the gas-phase reaction of stabilized Criegee Intermediates with SO2: Influence of water vapour content and temperature, Atmos. Environ., 2014, 89, 603–612 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, 1999 Search PubMed.
- R. Chhantyal-Pun, O. Welz, J. D. Savee, A. J. Eskola, E. P. F. Lee, L. Blacker, H. R. Hill, M. Ashcroft, M. A. H. H. Khan, G. C. Lloyd-Jones, L. A. Evans, B. Rotavera, H. Huang, D. L. Osborn, D. K. W. Mok, J. M. Dyke, D. E. Shallcross, C. J. Percival, A. J. Orr-Ewing and C. A. Taatjes, Direct Measurements of Unimolecular and Bimolecular Reaction Kinetics of the Criegee Intermediate (CH3)2COO, J. Phys. Chem. A, 2017, 121, 4–15 CrossRef CAS PubMed.
- A. Novelli, K. Hens, C. Tatum Ernest, M. Martinez, A. C. Nölscher, V. Sinha, P. Paasonen, T. Petäjä, M. Sipilä, T. Elste, C. Plass-Dülmer, G. J. Phillips, D. Kubistin, J. Williams, L. Vereecken, J. Lelieveld and H. Harder, Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 2017, 17, 7807–7826 CAS.
- C. Percival, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, D. O. Topping, D. Lowe, S. Utembe, A. Bacak, G. McFiggans, M. Cooke, A. T. Archibald, M. Jenkin, R. G. Derwent, I. Riipinen, D. Mok, E. P. F. Lee, J. Dyke, C. A. Taatjes and D. E. Shallcross, Regional and Global Impacts of Criegee Intermediates on Atmospheric Sulphuric Acid Concentrations and First Steps of Aerosol Formation, Faraday Discuss., 2013, 165, 45–73 RSC.
- D. Meidan, S. S. Brown and Y. Rudich, The potential role of Criegee intermediates in nighttime atmospheric chemistry. A modeling study, ACS Earth Space Chem., 2017, 1, 288–298 CrossRef CAS.
- M. Boy, D. Mogensen, S. Smolander, L. Zhou, T. Nieminen, P. Paasonen, C. Plass-Dülmer, M. Sipilä, T. Petäjä, L. Mauldin, H. Berresheim and M. Kulmala, Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations, Atmos. Chem. Phys., 2013, 13, 3865–3879 CrossRef.
- J. R. Pierce, M. J. Evans, C. E. Scott, S. D. D’Andrea, D. K. Farmer, E. Swietlicki and D. V. Spracklen, Weak global sensitivity of cloud condensation nuclei and the aerosol indirect effect to Criegee + SO2 chemistry, Atmos. Chem. Phys., 2013, 13, 3163–3176 CrossRef.
- M. A. H. Khan, W. C. Morris, M. Galloway, B. M. A. Shallcross, C. J. Percival and D. E. Shallcross, An Estimation of the Levels of Stabilized Criegee Intermediates in the UK Urban and Rural Atmosphere Using the Steady-State Approximation and the Potential Effects of These Intermediates on Tropospheric Oxidation Cycles, Int. J. Chem. Kinet., 2017, 49, 611–621 CrossRef CAS PubMed.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 2015, 15, 6283–6304 CAS.
- International Union of Pure and Applied Chemistry, ed. A. D. McNaught and A. Wilkinson, Compendium of chemical terminology: IUPAC recommendations, Blackwell Science, Oxford [England], Malden, MA, USA, 2nd edn, 1997 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- J. D. Watts, J. Gauss and R. J. Bartlett, Coupled-cluster methods with noniterative triple excitations for restricted open-shell Hartree–Fock and other general single determinant reference functions. Energies and analytical gradients, J. Chem. Phys., 1993, 98, 8718–8733 CrossRef CAS.
- L. Vereecken and J. Peeters, The 1,5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system, J. Chem. Phys., 2003, 119, 5159–5170 CrossRef CAS.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, Current Status of Transition-State Theory, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- C. Eckart, The penetration of a potential barrier by electrons, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- H. S. Johnston and J. Heicklen, Tunneling corrections for unsymmetrical Eckart potential energy barriers, J. Phys. Chem., 1962, 66, 532–533 CrossRef.
- D. M. Kooij, Über die Zersetzung des Gasförmigen Phosphorwasserstoffs, Z. Phys. Chem., 1893, 12, 155–161 Search PubMed.
- A. B. Jardine, K. J. Jardine, J. D. Fuentes, S. T. Martin, G. Martins, F. Durgante, V. Carneiro, N. Higuchi, A. O. Manzi and J. Q. Chambers, Highly reactive light-dependent monoterpenes in the Amazon, Geophys. Res. Lett., 2015, 42, 1576–1583 CrossRef CAS.
- E. Velasco, B. Lamb, H. Westberg, E. Allwine, G. Sosa, J. L. Arriaga-Colina, B. T. Jobson, M. L. Alexander, P. Prazeller, W. B. Knighton, T. M. Rogers, M. Grutter, S. C. Herndon, C. E. Kolb, M. Zavala, B. de Foy, R. Volkamer, L. T. Molina and M. J. Molina, Distribution, magnitudes, reactivities, ratios and diurnal patterns of volatile organic compounds in the Valley of Mexico during the MCMA 2002 & 2003 field campaigns, Atmos. Chem. Phys., 2007, 7, 329–353 CrossRef CAS.
- N. C. Bouvier-Brown, A. H. Goldstein, J. B. Gilman, W. C. Kuster and J. A. de Gouw, In-situ ambient quantification of monoterpenes, sesquiterpenes, and related oxygenated compounds during BEARPEX 2007: implications for gas- and particle-phase chemistry, Atmos. Chem. Phys., 2009, 9, 5505–5518 CrossRef CAS.
- G. M. Handisides, C. Plass-Dülmer, S. Gilge, H. Bingemer and H. Berresheim, Hohenpeissenberg Photochemical Experiment (HOPE 2000): Measurements and photostationary state calculations of OH and peroxy radicals, Atmos. Chem. Phys., 2003, 3, 1565–1588 CrossRef CAS.
- P. Jöckel, A. Kerkweg, A. Pozzer, R. Sander, H. Tost, H. Riede, A. Baumgaertner, S. Gromov and B. Kern, Development cycle 2 of the Modular Earth Submodel System (MESSy2), Geosci. Model Dev. Discuss., 2010, 3, 1423–1501 Search PubMed.
- E. Roeckner, R. Brokopf, M. Esch, M. Giorgetta, S. Hagemann, L. Kornblueh, E. Manzini, U. Schlese and U. Schulzweida, Sensitivity of simulated climate to horizontal and vertical resolution in the ECHAM5 atmosphere model, J. Clim., 2006, 19, 3771–3791 CrossRef.
- D. Cabrera-Perez, D. Taraborrelli, R. Sander and A. Pozzer, Global atmospheric budget of simple monocyclic aromatic compounds, Atmos. Chem. Phys., 2016, 16, 6931–6947 CAS.
- J. Lelieveld, S. Gromov, A. Pozzer and D. Taraborrelli, Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 2016, 16, 12477–12493 CAS.
- B. Long, J. L. Bao and D. G. Truhlar, Atmospheric Chemistry of Criegee Intermediates. Unimolecular Reactions and Reactions with Water, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS PubMed.
- J. M. Anglada and A. Solé, Impact of water dimer on the atmospheric reactivity of carbonyl oxides, Phys. Chem. Chem. Phys., 2016, 18, 17698–17712 RSC.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Direct Measurements of Conformer-Dependent Reactivity of the Criegee Intermediate CH3CHOO, Science, 2013, 340, 177–180 CrossRef CAS PubMed.
- L.-C. Lin, H.-T. Chang, C.-H. Chang, W. Chao, M. C. Smith, C.-H. Chang, J. J.-M. Lin and K. Takahashi, Competition between H2O and (H2O)2 reactions with CH2OO/CH3CHOO, Phys. Chem. Chem. Phys., 2016, 18, 4557–4568 RSC.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Communication: Real time observation of unimolecular decay of Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 144, 061102 CrossRef PubMed.
- Y. Fang, F. Liu, S. J. Klippenstein and M. I. Lester, Direct observation of unimolecular decay of CH3CH2CHOO Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 145, 044312 CrossRef PubMed.
- M. J. Newland, A. R. Rickard, M. S. Alam, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Kinetics of stabilised Criegee intermediates derived from alkene ozonolysis: reactions with SO2, H2O and decomposition under boundary layer conditions, Phys. Chem. Chem. Phys., 2015, 17, 4076–4088 RSC.
- M. C. Smith, W. Chao, K. Takahashi, K. A. Boering and J. J.-M. Lin, Unimolecular Decomposition Rate of the Criegee Intermediate (CH3)2COO Measured Directly with UV Absorption Spectroscopy, J. Phys. Chem. A, 2016, 120, 4789–4798 CrossRef CAS PubMed.
- J. D. Fenske, A. S. Hasson, A. W. Ho and S. E. Paulson, Measurement of Absolute Unimolecular and Bimolecular Rate Constants for CH3CHOO Generated by the trans-2-Butene Reaction with Ozone in the Gas Phase, J. Phys. Chem. A, 2000, 104, 9921–9932 CrossRef CAS.
- A. Novelli, L. Vereecken, J. Lelieveld and H. Harder, Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 2014, 16, 19941–19951 RSC.
- M. J. Newland, A. R. Rickard, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Atmospheric isoprene ozonolysis: impacts of stabilised Criegee intermediate reactions with SO2, H2O and dimethyl sulfide, Atmos. Chem. Phys., 2015, 15, 9521–9536 CrossRef CAS.
- K. T. Kuwata, L. C. Valin and A. D. Converse, Quantum Chemical and Master Equation Studies of the Methyl Vinyl Carbonyl Oxides Formed in Isoprene Ozonolysis, J. Phys. Chem. A, 2005, 109, 10710–10725 CrossRef CAS PubMed.
- K. T. Kuwata and L. C. Valin, Quantum chemical and RRKM/master equation studies of isoprene ozonolysis: Methacrolein and methacrolein oxide, Chem. Phys. Lett., 2008, 451, 186–191 CrossRef CAS.
- B. Ouyang, M. W. McLeod, R. L. Jones and W. J. Bloss, NO3 radical production from the reaction between the Criegee intermediate CH2OO and NO2, Phys. Chem. Chem. Phys., 2013, 15, 17070–17075 RSC.
- T. Berndt, R. Kaethner, J. Voigtländer, F. Stratmann, M. Pfeifle, P. Reichle, M. Sipilä, M. Kulmala and M. Olzmann, Kinetics of the unimolecular reaction of CH2OO and the bimolecular reactions with the water monomer, acetaldehyde and acetone at atmospheric conditions, Phys. Chem. Chem. Phys., 2015, 17, 19862–19873 RSC.
- L. Chen, W. Wang, L. Zhou, W. Wang, F. Liu, C. Li and J. Lu, Role of water clusters in the reaction of the simplest Criegee intermediate CH2OO with water vapour, Theor. Chem. Acc., 2016, 135, 252 CrossRef.
- J. M. Anglada, J. González and M. Torrent-Sucarrat, Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water, Phys. Chem. Chem. Phys., 2011, 13, 13034–13045 RSC.
- A. B. Ryzhkov and P. A. Ariya, A theoretical study of the reactions of parent and substituted Criegee intermediates with water and the water dimer, Phys. Chem. Chem. Phys., 2004, 6, 5042–5050 RSC.
- L. Sheps, A. M. Scully and K. Au, UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO, Phys. Chem. Chem. Phys., 2014, 16, 26701–26706 RSC.
- K. T. Kuwata, M. R. Hermes, M. J. Carlson and C. K. Zogg, Computational Studies of the Isomerization and Hydration Reactions of Acetaldehyde Oxide and Methyl Vinyl Carbonyl Oxide, J. Phys. Chem. A, 2010, 114, 9192–9204 CrossRef CAS PubMed.
- W. Chao, J.-T. Hsieh, C.-H. Chang and J. J.-M. Lin, Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor, Science, 2015, 347, 751–754 CrossRef CAS PubMed.
- T. R. Lewis, M. A. Blitz, D. E. Heard and P. W. Seakins, Direct evidence for a substantive reaction between the Criegee intermediate, CH2OO, and the water vapour dimer, Phys. Chem. Chem. Phys., 2015, 17, 4859–4863 RSC.
- M. C. Smith, C.-H. Chang, W. Chao, L.-C. Lin, K. Takahashi, K. A. Boering and J. J.-M. Lin, Strong Negative Temperature Dependence of the Simplest Criegee Intermediate CH2OO Reaction with Water Dimer, J. Phys. Chem. Lett., 2015, 6, 2708–2713 CrossRef CAS PubMed.
- A. H. Goldstein and I. E. Galbally, Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed.
- N. Sobanski, M. J. Tang, J. Thieser, G. Schuster, D. Poehler, H. Fischer, W. Song, C. Sauvage, J. Williams, J. Fachinger, F. Berkes, P. Hoor, U. Platt, J. Lelieveld and J. N. Crowley, Chemical and meteorological influences on the lifetime of NO3 at a semi-rural mountain site during PARADE, Atmos. Chem. Phys., 2016, 16, 4867–4883 CAS.
- A. Chebbi and P. Carlier, Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review, Atmos. Environ., 1996, 30, 4233–4249 CrossRef CAS.
- F. Paulot, D. Wunch, J. D. Crounse, G. C. Toon, D. B. Millet, P. F. DeCarlo, C. Vigouroux, N. M. Deutscher, G. González Abad, J. Notholt, T. Warneke, J. W. Hannigan, C. Warneke, J. A. de Gouw, E. J. Dunlea, M. De Mazière, D. W. T. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 2011, 11, 1989–2013 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Extensive tabulation and figures of theoretical kinetic results, discussion of quantum chemical and theoretical kinetic results, trend analysis, group additivity, structure–activity relationships, literature data, field data analysis, global modeling, and other information. Quantum chemical characteristics of all reactants and transition states characterized, at various levels of theory. See DOI: 10.1039/c7cp05541b |
| This journal is © the Owner Societies 2017 |