
Emerging investigator series: a 14-year depositional ice record of perfluoroalkyl substances in the High Arctic†
John J.
MacInnis
a,
Katherine
French
b,
Derek C. G.
Muir
b,
Christine
Spencer
b,
Alison
Criscitiello
 c,
Amila O.
De Silva
*b and
Cora J.
Young
*a
c,
Amila O.
De Silva
*b and
Cora J.
Young
*a
aDepartment of Chemistry, Memorial University, St. John's, NL, Canada A1B 3X7. E-mail: cora.young@mun.ca
bAquatic Contaminants Research Division, Environment and Climate Change Canada, Burlington, ON, Canada L7S 1A1. E-mail: amila.desilva@canada.ca
cDepartment of Geography, University of Calgary, Calgary, AB, Canada T2N 1N4
First published on 10th January 2017
Abstract
To improve understanding of long-range transport of perfluoroalkyl substances to the High Arctic, samples were collected from a snow pit on the Devon Ice Cap in spring 2008. Snow was analyzed for perfluoroalkyl acids (PFAAs), including perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs), as well as perfluorooctane sulfonamide (FOSA). PFAAs were detected in all samples dated from 1993 to 2007. PFAA fluxes ranged from <1 to hundreds of ng per m2 per year. Flux ratios of even–odd PFCA homologues were mostly between 0.5 and 2, corresponding to molar ratios expected from atmospheric oxidation of fluorotelomer compounds. Concentrations of perfluorobutanoic acid (PFBA) were much higher than other PFCAs, suggesting PFBA loading on the Devon Ice Cap is influenced by additional sources, such as the oxidation of heat transfer fluids. All PFCA fluxes increased with time, while PFSA fluxes generally decreased with time. No correlations were observed between PFAAs and the marine aerosol tracer, sodium. Perfluoro-4-ethylcyclohexanesulfonate (PFECHS) was detected for the first time in an atmospherically – derived sample, and its presence may be attributed to aircraft hydraulic system leakage. Observations of PFAAs from these samples provide further evidence that atmospheric oxidation of volatile precursors is an important source of PFAAs to the Arctic environment.
Environmental impactPerfluoroalkyl acids (PFAAs) are present in remote regions where no local sources exist. We present a suite of 19 PFAAs and perfluorooctane sulfonamide in samples from the Devon Ice Cap, Nunavut, Canada. These provide a 14-year record of atmospheric deposition. We detected perfluoro-4-ethylcyclohexanesulfonate (PFECHS) in the samples, which represents the first measurement of this compound in a remote location. High levels of perfluorobutanoic acid were also observed, which can be attributed to atmospheric oxidation of stratospheric ozone-depleting replacement compounds, such as hydrofluorocarbons. Evidence from homologue profiles on the ice cap and comparisons between the ice cap and ocean suggest that oxidation of volatile precursors, such as fluorotelomer compounds, are an important source of PFAAs to the ice cap. |
Introduction
Perfluoroalkyl acids (PFAAs) are found ubiquitously in the environment, including remote regions where local emissions are not expected. The prevalence of PFAAs in remote locations1 indicates these compounds can undergo long-range transport. Transport of PFAAs can be through the atmosphere, the ocean, or a combination of the two. In the atmosphere, PFAAs can be formed in the gas phase as oxidation products of volatile precursors, such as fluorotelomer alcohols2 (FTOHs) and perfluoroalkane sulfonamido substances (PASS), collectively known as perfluoroalkane sulfonamides (FASAs) and perfluoroalkane sulfonamido ethanols (FASEs).3 These precursors have sufficient atmospheric lifetimes to reach remote locations4–6 before subsequent oxidation to PFAAs. PFAAs are highly acidic organic compounds7 present primarily as anions in the aqueous phase under environmental conditions. Transport of PFAAs is feasible via ocean currents.8–10 Transport of persistent compounds to remote locations via ocean currents has been reported to take place on a scale of decades and is slow in comparison to atmospheric transport, which can occur on a scale of days.2 Considering PFAAs are surface-active compounds, they should be enriched in the surface microlayer (SML). The SML forms at the interface between the atmosphere and the ocean surface, has a thickness between 1–1000 μm, and is rich in organic compounds.11 During wave events, air bubbles containing particles enriched in organic compounds from the SML are formed and subsequently broken. Marine aerosols that contain SML components, as well as sea salts, are formed via this bubble bursting mechanism. This mechanism is poorly understood for PFAAs and has been examined in only two studies.12,13Elucidation of long-range transport mechanisms can be assisted through remote sample collection. Ice caps receive their contamination solely from atmospheric deposition because of their high elevation (e.g. Devon Ice Cap summit is 1800 m above sea level). PFAA atmospheric deposition to an ice cap in the Canadian High Arctic has been measured in a previous work.6 Long-chain PFAAs, including perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfluoroundecanoic acid (PFUnDA), and perfluorooctane sulfonic acid (PFOS) were detected in surface snow from multiple locations and a snow pit on the Devon Ice Cap. In this study, a deeper snow pit was sampled on the Devon Ice Cap in 2008. With increased analytical capabilities, it is possible to detect a greater number of analytes to further explore the mechanisms of long-range transport to the High Arctic. Herein, we discuss: (1) PFAA depositional and temporal trends; (2) transport mechanisms to the High Arctic; and (3) environmental implications.
Methods
Sample collection
Sampling procedures were described in detail previously.14 Briefly, samples were collected from the Devon Ice Cap, Devon Island, Nunavut (75° 20N, 82° 40W) in spring 2008. A 7 m snow pit was created near the highest point on the ice cap located a few km upwind from the nearest temporary research site. Samples were taken vertically using 4 L polypropylene (PP) bottles every 25 cm continuously along the face of the snow pit for a total of 28 samples. Samples designated for density and major ion chemistry were collected at 10 cm intervals along the face of the pit. Densities were determined by measuring the mass collected in the volume of a steel corer. Conductivity was measured using an Orion model 135 conductivity meter (Orion Research, Beverly, MA, USA). Major ions (SO42−, Cl− and Na+) were analyzed by the National Laboratory for Environmental Testing (Environment and Climate Change Canada, Canada Centre for Inland Waters, Burlington, ON, Canada; Fig. S1†). Duplicate samples, totalling approximately 16 L of snow, were collected at each depth. Products containing fluoropolymer coatings were strictly avoided at the sampling sites. Prior to sampling, the surface layer of the snowpit wall was removed using a stainless steel scraper. Samples were stored in new, unopened PP bottles and were kept frozen prior to analysis. Field blanks were taken from an unopened bottle of HPLC grade water and placed in PP bottles, which were transported to the sampling site, opened for 10 minutes and transported back to the laboratory with samples. Field blanks were compared to the water that was stored refrigerated in the laboratory in the same PP bottle, colloquially identified as a stay blank. A complete summary of PFAA concentrations in field, stay, and method blanks is provided in the ESI (Table S1†). Concentrations were not blank subtracted.Sample preparation and analysis
Detail of sample preparation is described comprehensively in Lescord et al.,15 with complementary information in the ESI (S1.1).† Targeted analytes were: perfluorobutanoic acid (PFBA), perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA), PFOA, PFNA, PFDA, PFUnDA, perfluorododecanoic acid (PFDoDA), perfluorotridecanoic acid (PFTrDA), perfluorotetradecanoic acid (PFTeDA), perfluorohexadecanoic acid (PFHxDA), perfluorooctadecanoic acid (PFOcDA), perfluorobutane sulfonic acid (PFBS), perfluorohexane sulfonic acid (PFHxS), perfluoroheptane sulfonic acid (PFHpS), perfluorooctane sulfonic acid (PFOS), perfluoro-4-ethylcyclohexanesulfonate (PFECHS), perfluorodecane sulfonic acid (PFDS) and perfluorooctane sulfonamide (FOSA). Samples were concentrated using a solid phase extraction method (S1.1†). Method blanks were used to validate the integrity of the extraction method, and isotopically labeled standards (Wellington Laboratories, Guelph, ON) were used to validate recovery and matrix effects (S1.1, Table S2†). The data presented herein were corrected on the basis of internal standard recovery in sample extracts (Tables S3 and S4†). Samples were analyzed using ultra performance liquid chromatography (Waters Acquity UPLC I) with tandem mass spectrometry (Waters Xevo® TQ-S, UPLC-MS/MS) detection operated in electrospray negative ionization mode. Samples were separated using a C18 column (Acquity UPLC ® BEH, 2.1 × 100 mm, 1.7 μm) with a water–methanol 0.1 mM ammonium acetate gradient method (Tables S5–S7 and Fig. S2–S5†). Interpretations of PFAA isomer signatures on the Devon Ice Cap was not possible at this time due to limitations in sample volume and speed of the chromatographic separation. The limit of detection (LOD) and limit of quantitation (LOQ) were defined as compounds having signal-to-noise (S/N) ratios of 3 and 10, respectively. S/N ratios were calculated by dividing analyte peak height by the standard deviation of the blank across compound-specific retention time windows. Values for LOD and LOQ can be found in the ESI (Table S8).† PFAA flux was calculated using measured concentrations and annual snow deposition, as reported in the ESI (S1.2).†Results and discussion
PFCA deposition to Devon Ice Cap
PFCAs from C4 (PFBA) to C14 (PFTeDA) were detected on the Devon Ice Cap, with concentrations ranging from pg L−1 to ng L−1 (Table S9†). These concentrations correspond to fluxes between <1 and hundreds of ng per m2 per year (Table S10†). The presence of PFHxDA and PFOcDA was sought, but not found. Fluxes of PFAAs and FOSA for the year 2007 are shown in Fig. 1. PFCA concentrations on the Devon Ice Cap were comparable to, but generally lower than, concentrations in precipitation collected at lower latitudes between 2006 and 2008.16 Concentrations were higher than those in another snow pit from the Devon Ice Cap collected in 2006 (Fig. S6†).6 The discrepancy between snow concentrations could be caused by differences in sampling location or analytical methods. When the 2006 samples were analyzed, the availability of isotopically labelled standards was limited. As a result, there were substantial analytical improvements in the current study, including use of recovery standards and recovery-correction, as well as isotopically labelled internal standards for each analyte. Differences in polybrominated diphenyl ether concentrations were also observed in samples collected at the same locations in 2006 and 2008.14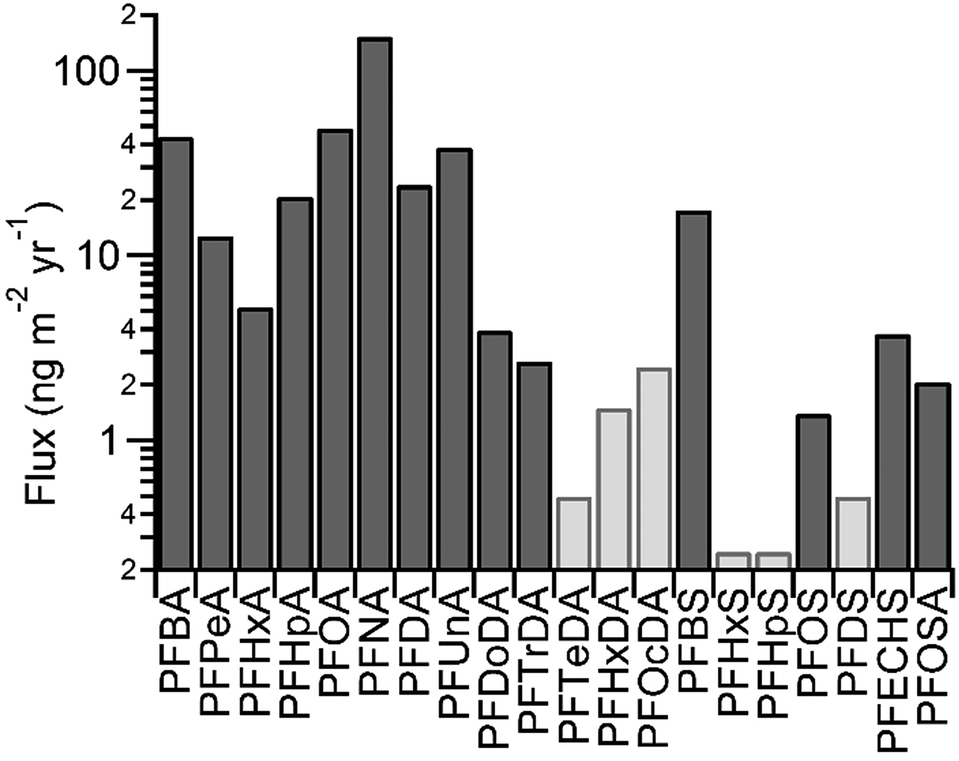 | ||
| Fig. 1 Flux of perfluoroalkyl substances for 2007. Where measurements were <LOD (light grey), LOD was used to calculate flux. | ||
Even–odd pairs are a useful metric to examine the role of fluorotelomer atmospheric oxidation in PFCA deposition. Smog chamber studies indicate that degradation of an X![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 fluorotelomer compound (i.e. containing a perfluoroalkyl moiety corresponding to F(CF2)X), will lead to comparable yields of two PFCAs, with X and X + 1 carbons.2 For example, atmospheric oxidation of the 8:2 FTOH leads to similar yields of PFOA and PFNA. It is expected that there will be some variability in the yields as a result of relative levels of NOx (NO + NO2) and peroxy radicals.17 Even–odd pairs of most PFCA homologues were deposited in similar amounts on the ice cap (Table S11, Fig. S7 and S1.3†) (Fig. 2). This was observed for PFOA–PFNA and PFDA–PFUnDA in previous samples from the Devon Ice Cap6 and for PFOA–PFNA from an ice core collected from a glacier on Svalbard.18 In this study, comparisons were made between observed concentrations of the even–odd pairs between C4 (PFBA) and C13 (PFTrDA). Correlations between pairs (C6–C13) were all statistically significant (two-tailed t-test), with correlation coefficients ranging from 0.60 to 0.80, and p-values all <0.0001 (Table S11†). Of the 58 flux measurement ratios for the 4 pairs from PFHxA to PFTrDA, 84% were within a factor of 2 and 98% of the measurement ratios were within a factor of 5. This is consistent with the expected ratios of even and odd PFCA homologues formed from gas phase atmospheric oxidation of fluorotelomer-derived compounds.2 This is further supported by the flux ratios of PFDA:PFUnDA and PFDoDA:PFTrDA observed at the Devon Ice Cap. These similar fluxes of even–odd homologues are likely the result of atmospheric oxidation of volatile precursors,19 ≥10:2 and ≥12:2 fluorotelomer compounds for PFDA, PFUnDA and PFDoDA, PFTrDA, respectively. Estimates of the quantities of long-chain PFCAs, from PFDA to PFTeDA present in the environment from direct and indirect sources vary substantially, but are generally thought to be dominated by indirect sources. This hypothesis has been corroborated by the presence of FTOHs20,21 and their oxidation products, fluorotelomer unsaturated carboxylic acids,21 in the Canadian Arctic. These observations suggest detection of long-chain PFCAs on the Devon Ice Cap is consistent with dominant contributions from indirect sources.
:2 fluorotelomer compound (i.e. containing a perfluoroalkyl moiety corresponding to F(CF2)X), will lead to comparable yields of two PFCAs, with X and X + 1 carbons.2 For example, atmospheric oxidation of the 8:2 FTOH leads to similar yields of PFOA and PFNA. It is expected that there will be some variability in the yields as a result of relative levels of NOx (NO + NO2) and peroxy radicals.17 Even–odd pairs of most PFCA homologues were deposited in similar amounts on the ice cap (Table S11, Fig. S7 and S1.3†) (Fig. 2). This was observed for PFOA–PFNA and PFDA–PFUnDA in previous samples from the Devon Ice Cap6 and for PFOA–PFNA from an ice core collected from a glacier on Svalbard.18 In this study, comparisons were made between observed concentrations of the even–odd pairs between C4 (PFBA) and C13 (PFTrDA). Correlations between pairs (C6–C13) were all statistically significant (two-tailed t-test), with correlation coefficients ranging from 0.60 to 0.80, and p-values all <0.0001 (Table S11†). Of the 58 flux measurement ratios for the 4 pairs from PFHxA to PFTrDA, 84% were within a factor of 2 and 98% of the measurement ratios were within a factor of 5. This is consistent with the expected ratios of even and odd PFCA homologues formed from gas phase atmospheric oxidation of fluorotelomer-derived compounds.2 This is further supported by the flux ratios of PFDA:PFUnDA and PFDoDA:PFTrDA observed at the Devon Ice Cap. These similar fluxes of even–odd homologues are likely the result of atmospheric oxidation of volatile precursors,19 ≥10:2 and ≥12:2 fluorotelomer compounds for PFDA, PFUnDA and PFDoDA, PFTrDA, respectively. Estimates of the quantities of long-chain PFCAs, from PFDA to PFTeDA present in the environment from direct and indirect sources vary substantially, but are generally thought to be dominated by indirect sources. This hypothesis has been corroborated by the presence of FTOHs20,21 and their oxidation products, fluorotelomer unsaturated carboxylic acids,21 in the Canadian Arctic. These observations suggest detection of long-chain PFCAs on the Devon Ice Cap is consistent with dominant contributions from indirect sources.
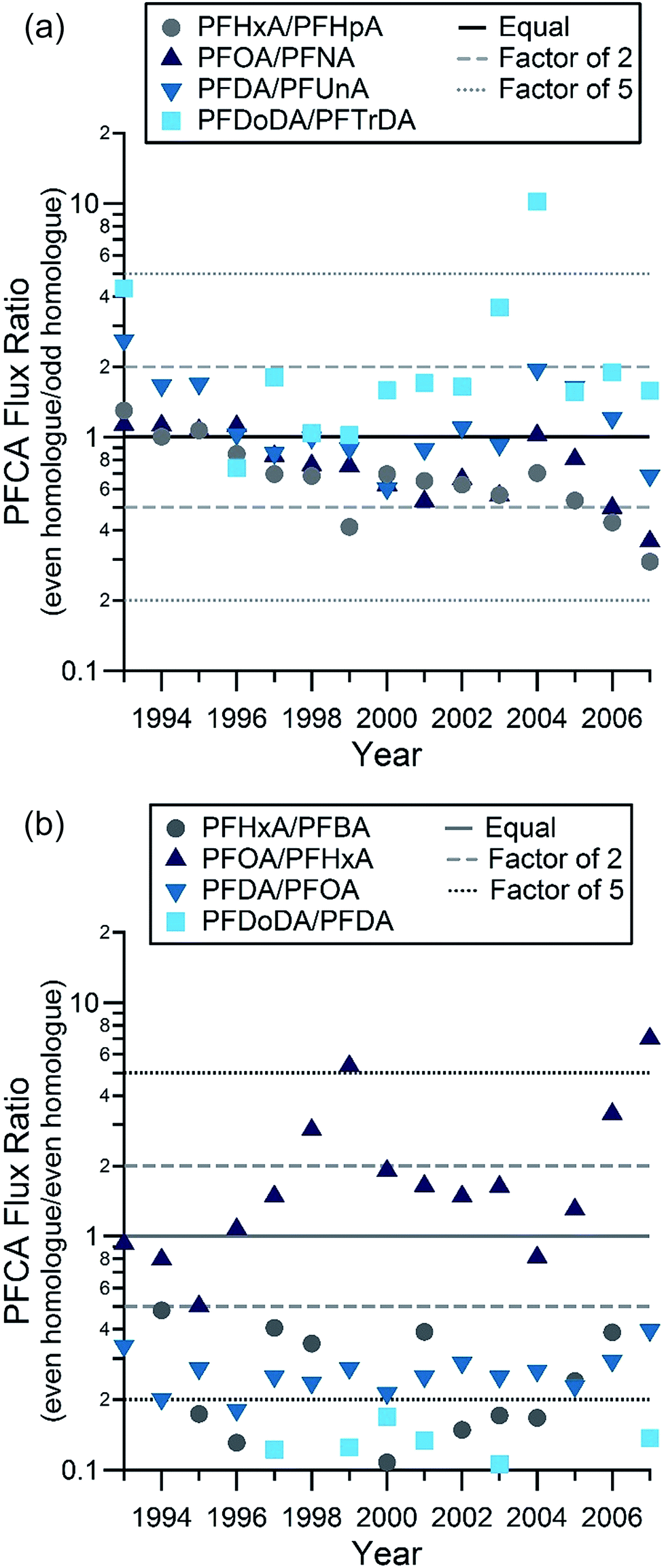 | ||
| Fig. 2 Ratio of even–odd (a) and even–even (b) PFCA homologue molar fluxes as a function of year. | ||
An additional source of PFCAs to the Devon Ice Cap could result from the atmospheric oxidation of PASS. Evidence from smog chamber experiments suggests the dominant oxidation products of N-methyl perfluorobutane sulfonamidoethanol (MeFBSE) and N-ethyl perfluorobutane sulfonamide (EtFBSA) in the presence of hydroxyl radicals are PFCAs.3,22 The yield was favourable under low NOx conditions, and is expected to be representative of the Arctic environment. It is possible the oxidation of FOSA or FASE could contribute to the observed flux of PFOA and shorter-chain PFCAs. However, no relationship was observed between PFOA and FOSA concentrations (R2 = 0.0006, p = 0.9010; Table S11†), suggesting PFOA deposition at the Devon Ice Cap is largely driven by other sources. Source apportionment is very challenging considering the number of volatile precursors and mechanisms involved in long-range atmospheric transport. It is difficult to assess the full extent of atmospheric oxidation of these species considering FOSA was the only PASS monitored in this work.
Notable exceptions to the patterns described above were relationships between PFBA and other PFCA homologues. No correlation was observed between PFBA and PFPeA (R2 = 0.20, p = 0.0217; Table S11†), while PFBA and PFHxA were weakly correlated (R2 = 0.30, p = 0.0016; Table S11†), suggesting a source other than atmospheric oxidation of fluorotelomer compounds. PASS oxidation could also lead to formation of PFBA. Because our measurements did not include any of these precursor compounds, this source cannot be excluded. However, we anticipate contributions of this source will be minor, given our observations for the long-chain PFCAs above and the lack of correlation observed between PFBA and PFBS (R2 = 0.008, p = 0.6587; Table S11†). Deposition of PFBA was much greater than PFPeA (mean of 6.0 ± 2.1 times larger) and most other homologues (Fig. 1). This observation is consistent with PFBA measurements from other studies, including Arctic char (Salvelinus alpinus) from Svalbard,23 glacial samples from Svalbard18 and Italy,24,25 snow from Tibet,25,26 and sea ice from the Arctic Ocean.27 This is particularly surprising in biota considering the greater bioaccumulation factor for PFPeA than PFBA.28 High concentrations of PFBA in previous studies have been attributed to direct emissions of PFBA25 or a shift in fluorotelomer production to short-chain homologues, such as the 4:2 FTOH.26 We argue that observations of high concentrations of PFBA and other short chain PFAAs29–33 in environmental samples are consistent with our knowledge of multiple non-fluorotelomer gas phase sources. Known sources of PFBA include hydrofluorocarbons (HFC), such as HFC-329 (CF3(CF2)3H),34 and hydrofluoroethers (HFE), such as HFE-7100 (C4F9OCH3) and HFE-7200 (C4F9OC2H5).35,36 Mixing ratios of several HFC compounds have been reported in the atmosphere, ranging from approximately 5–200 parts-per-trillion by volume (pptv).37 Levels of many HFCs,38 and likely HFEs, are increasing in the atmosphere, as these compounds are replacements for stratospheric ozone-depleting chlorofluorocarbons and hydrochlorofluorocarbons. Elevated concentrations of PFBA in relation to other PFCAs found on the Devon Ice Cap suggest formation is driven by the atmospheric oxidation of these other compounds. Mixing ratios of HFCs in the atmosphere are substantially higher39 than those of fluorotelomers and PASS, which are in the range of a few to a few hundred parts-per-quadrillion by volume (ppqv).40 Both the lack of correlation between PFBA and other PFCAs on the ice cap and the highly elevated concentrations of PFBA suggest an atmospheric source other than fluorotelomer and PASS, such as HFCs and/or HFEs. In contrast, our results suggest that PFPeA and all other long-chained PFCAs are formed predominantly from atmospheric oxidation of fluorotelomer compounds and PASS.
PFSA and FOSA deposition to Devon Ice Cap
C4 (PFBS), C8 (PFOS), C10 (PFDS), and cyclic C8 (PFECHS) PFSAs, along with FOSA, were detected on the Devon Ice Cap, with concentrations ranging from a few to hundreds of pg L−1. These correspond to fluxes between <1 and tens of ng per m2 per year. PFSAs and FOSA were detected at lower levels than PFCAs and with lower frequency. PFBS, PFOS, PFECHS, and FOSA were detected in most samples, while PFDS was detected in samples corresponding to four years of deposition. No measurements above the LOD were made for C6 (PFHxS) and C7 (PFHpS). Fluxes of PFSAs to the Devon Ice Cap from 2007 are shown in Fig. 1. Precipitation collected at lower latitudes between 2006 and 2008 contain similar concentrations of PFBS, PFOS, and FOSA.16 In general, concentrations of PFBS and FOSA were higher than PFOS. Elevated concentrations of PFBS and FOSA relative to PFOS were also observed in some snow samples collected from the surface of Arctic Ocean sea ice in 2010.27 Concentrations of PFOS were similar to those measured in another Devon Ice Cap snow pit in 2006, with the exception of elevated concentrations in the lowest 2 m of the older core (Fig. S8†). As described above, differences in concentration could be caused by different sample locations and analytical improvements.Both PFOS and PFBS can be produced through the atmospheric oxidation of commercially produced volatile precursors.3 However, no correlation was observed between measurements of PFOS and its potential semi-volatile precursor, FOSA (R2 = 0.01, p = 0.5886; Table S11†). The formation of PFBS was observed from oxidation of MeFBSE in one study,3 though this was not observed in a FASA oxidation study.22 Thus, it is possible that FOSA may not degrade to form PFOS. Precursors to PFSAs can also produce PFCAs with equal or fewer carbons. However, no correlation was observed between PFOS or FOSA and PFOA or short-chain PFCAs (R2 < 0.1, p > 0.1; Table S11†). Similarly, there was no correlation between PFBS and PFBA. From this data, it is difficult to allocate sources of these species to the Devon Ice Cap without complementary volatile precursor measurements and complete knowledge of precursor chemistry.
PFECHS was measured at low ng L−1 concentrations on the Devon Ice Cap, similar to observed levels of PFOS. PFECHS has been detected in surface waters in Lake Ontario,41 the Arctic,15 and in aquatic biota downstream from an airport.42 This is the first reported detection of PFECHS in an atmospherically derived sample. We note there are no volatile commercial products known to degrade in the environment to form PFECHS, suggesting deposition must be through direct sources.
Temporal trends
All detected PFCAs were observed to be constant or increasing with time. The temporal trends of fluxes of PFOS, PFBS, and FOSA are shown in Fig. 3. Differences were observed between the temporal trends of PFCA and PFOS fluxes from another snow pit on Devon Island collected in 2006.6 Fluxes of PFCAs calculated from samples collected from 2008 are generally higher (Fig. S9†). Calculated fluxes of PFOS are much higher in earlier years (1995–1999) of the 2006 samples (Fig. 3), while calculated fluxes from both sample sets are similar in later years (2001 onward). Differences between calculated fluxes are unsurprising, because sampling in 2006 accounted for only 30–40% of the vertical face of the snow pit surface, while sampling in 2008 collected the entire vertical face. Melting events within the snowpack may also bias the assignment of fluxes to the Devon Ice Cap. The surface of the Devon Ice Cap has been known to undergo seasonal melting due to increasing air temperature,43,44 which results in the formation of impermeable ice layers in the snowpack. Subsequent percolating meltwater within a snowpack will refreeze at the ice layer interface.44 Consequently, the accumulation of ice in the snowpack makes it challenging to deduce meaningful interpretations for the most recent years of deposition.45 This suggests that concentrations measured near the surface layer can result in inaccuracies in estimating PFCA deposition whereby overestimation occurs in certain years and underestimation in others. To our knowledge, only two studies have examined the elution behaviour of PFAAs and FOSA in the presence of percolating meltwater in which short-chain PFAAs eluted at a slower rate relative to long-chain PFAAs.46 The elution of PFAAs was found to be largely driven by water solubility within the snowpack, which may provide an explanation for some observations on the Devon Ice Cap; however, the percolation of meltwater in a snowpack is expected to refreeze within an annual year.47 Seasonal cycles of PFAA deposition would be inevitably biased, while annual interpretations of data, such as those presented here, should not be affected. Nevertheless, we have presented a three-year moving average to minimize impacts of melting (Fig. 3 and Fig. S9†).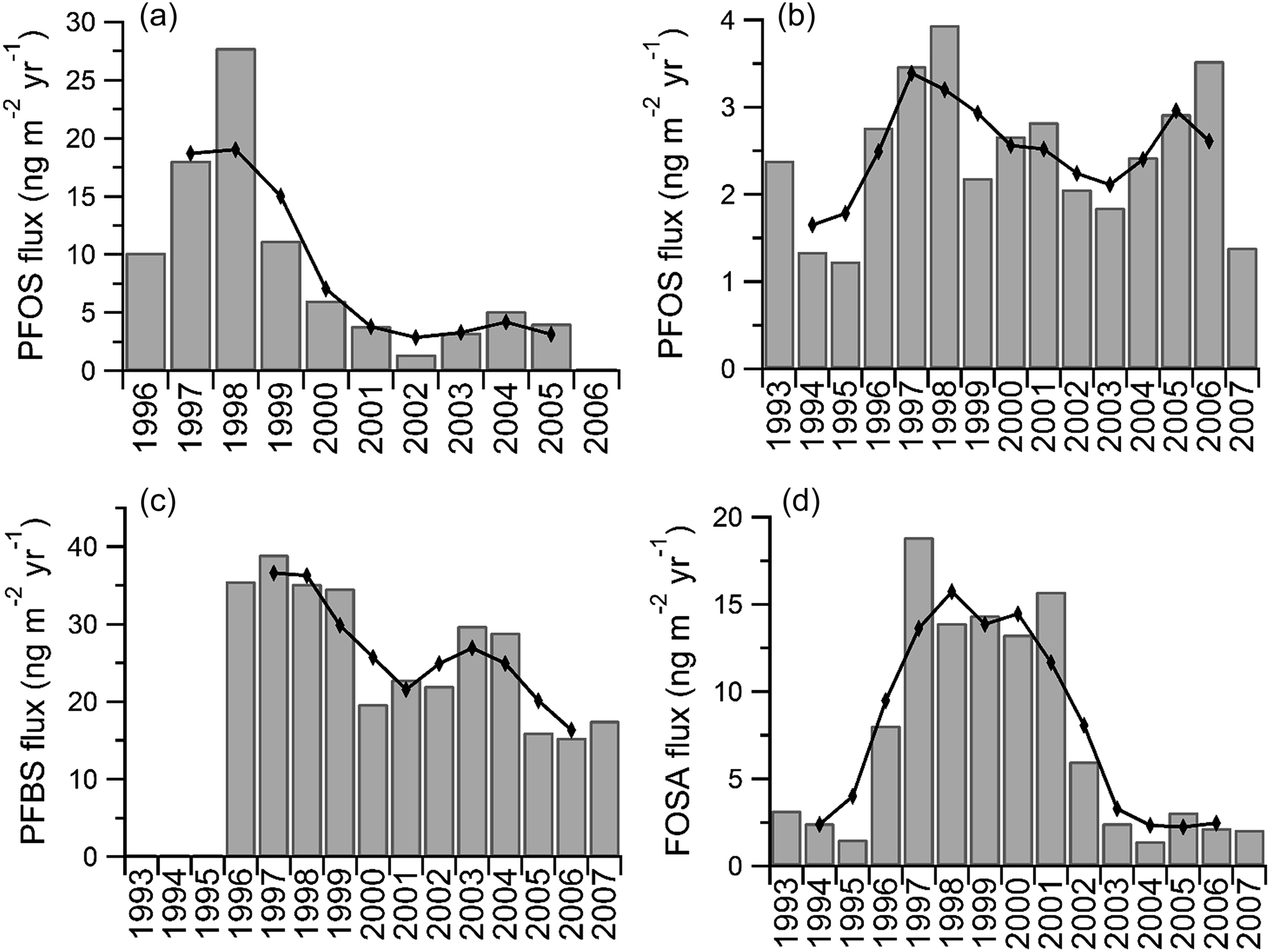 | ||
| Fig. 3 Annual deposition fluxes for the Devon Ice Cap (a) PFOS from Young et al.;6 (b) PFOS; (c) PFBS; (d) FOSA. Lines represent three-year moving average. | ||
In 2001, 3M, one of the largest global producers of perfluoroalkyl substances, voluntarily phased out the production of perfluorooctane sulfonyl fluoride (POSF) and all related products, including PFOS and its volatile precursors, such as FOSA.48 It was proposed that further production would use C4-based compounds derived from perfluorobutane sulfonyl fluoride (PBSF), which are believed to have lower bioaccumulation and toxicological effects.49 Reported production of PBSF in the United States has almost doubled in 2006 compared to production in 2002, while that of POSF decreased by more than two orders of magnitude between 2000 and 2002, and no production was reported for 2006.50 Production of PFOS has increased dramatically in Asia since 2001 and China is now the dominant producer of these compounds.51,52 To our knowledge, global emission data for other PFSAs and related volatile chemicals is unavailable and cannot be correlated to the temporal trends observed in the Devon Ice Cap.
In a previous study,6 PFOS was observed to increase on the ice cap up to 1999, after which it decreased (Fig. 3a). PFOS in these samples also indicates an increase up to 1998 followed by a decline until 2003 (Fig. 3b and S10†). Because transport through the atmosphere is fast, on the order of weeks, if atmospheric oxidation was the dominant source of PFOS to the ice cap, a fast change in response to the newly adopted syntheses might be observed after 2001. In the previous study, the decrease was thought to indicate a fast response to the production change. As described above, differences in sample collection may provide an explanation for this discrepancy between studies. It is also possible that melting effects in the ice cap prior to sampling could have created a false maximum.6 It is also germane to note that although both data sets suggest a decline in PFOS flux, when applying first order kinetics, neither data set indicated a statistically significant (p > 0.05, Table S12†) declining temporal trend in the period post-1998. Deposition of FOSA, which is also derived from POSF, increased in the 1990s, peaking around the year 2000, followed by a decrease up to 2007 (Fig. 3d). The observed behaviour of FOSA is likely in accordance with the voluntary phase-out of the C8-based chemistry in 2001. Considering FOSA is much less water-soluble than other PFAAs measured in this work, it should be less susceptible to percolating effects within a snowpack,46 and any observed trends should reflect changes in production. The overall decreasing trend of PFOS is also in general agreement. However, the flux of PFBS is inconsistent, showing a general decrease over the eight-year period. It has been previously demonstrated that the Devon Ice Cap receives air masses originating from Asian sources.53 The smaller-than-expected flux of PFBS to the Devon Ice Cap may be reflective of Asian air masses enriched in POSF-based product emissions.19 Atmospheric gas and particle measurements made in the Canadian Archipelago in 2005 have shown elevated levels of electrochemical fluorination (ECF) – derived POSF substances, MeFOSE and EtFOSE.20 These ECF intermediates are not produced using contemporary North American synthetic techniques (i.e. telomerization), which suggest ECF may be presently used in Asia. Recent studies have examined PFOA isomer profiles in blood serum54 and commercial products55 from China and have found fractions of branched isomers unique to ECF manufacturing processes. The presence of ECF-derived sulfonyl fluoride substances from Asian air masses in the Canadian Archipelago may provide an explanation for the discrepancy between reported emissions and PFAA profiles on the Devon Ice Cap if C4 related chemistries are not fully implemented in Asian manufacturing.
Direct transport of PFAAs to the Arctic
To examine the influence of long-range oceanic transport on PFCA deposition at the Devon Ice Cap, the concentrations of PFCAs and Na+, a tracer for marine aerosols, were compared.56 No correlation was observed between concentrations of Na+ and any PFAA or FOSA (R2 ≤ 0.1, p > 0.1; Table S11†). This suggests PFAA deposition on the Devon Ice Cap is not heavily influenced by transport of marine aerosols. A similar lack of correlation was seen by Kwok et al. between PFAAs and sea-salt derived sulfate in Svalbard.18 The influence of sea spray on the Devon Ice Cap could also be tested by comparing the ratio of Cl− to Na+ observed on the ice cap to expected ocean water ratios.57 The observed molar ratio of Cl− to Na+ on the Devon Ice Cap averages 0.3, while the expected ocean water ratio is 1.2 (Fig. S11†). However, ratios observed from 2006–2008 begin approaching the expected seawater ratio. These observations may be consistent with percolation events within the snowpack, or with decreasing sea ice coverage in the Canadian Archipelago.58 Melting of sea ice is expected to enhance flux of marine aerosol to the Devon Ice Cap.45 Another technique used to assess the influence of marine aerosol deposition of PFAAs to the ice cap is a comparison between ocean and ice cap homologue patterns. If marine aerosols were a major source of the PFAA contamination on the Devon Ice Cap, a similar homologue profile should be observed in both the ocean and the ice cap. Concentrations of PFCAs on the ice cap are generally higher than those in the ocean, though not for long-chain homologues (Fig. 4). Concentrations of PFOS and FOSA are higher than some ocean concentrations and lower than others. Concentrations of PFBS have not been widely measured in ocean water, so a comparison is not possible at this time. PFHxS was not detected at all on the ice cap, while it is measured in most ocean samples.59–61 In glacier samples from Svalbard, PFHxS was not detected, though it was detected in surface snow in coastal regions.18 According to reported usage, the synthetic precursor to PFHxS, perfluorohexane sulfonyl fluoride (PHxSF), has never been used for the production of commercial volatile PASS compounds.62,63 Thus, detection of PFHxS on the ice cap would confirm the role of direct sources. The absence of PFHxS in the Arctic deposition provides further evidence that marine aerosols are not a significant source of PFAAs to the ice cap. The differences in homologue profiles between the ice cap and the ocean cannot be accounted for by different surfactant properties. If differences in surfactant properties were leading to different homologue patterns, we would expect a greater enhancement of PFSAs relative to PFCAs on the ice cap because the former are stronger surfactants. In addition, we would expect longer-chain PFCAs to be preferentially enriched.13 Discrepancies between Cl−/Na+ ratios and homologue patterns provide further evidence that marine aerosols are not a dominant source of PFAAs to the Devon Ice Cap.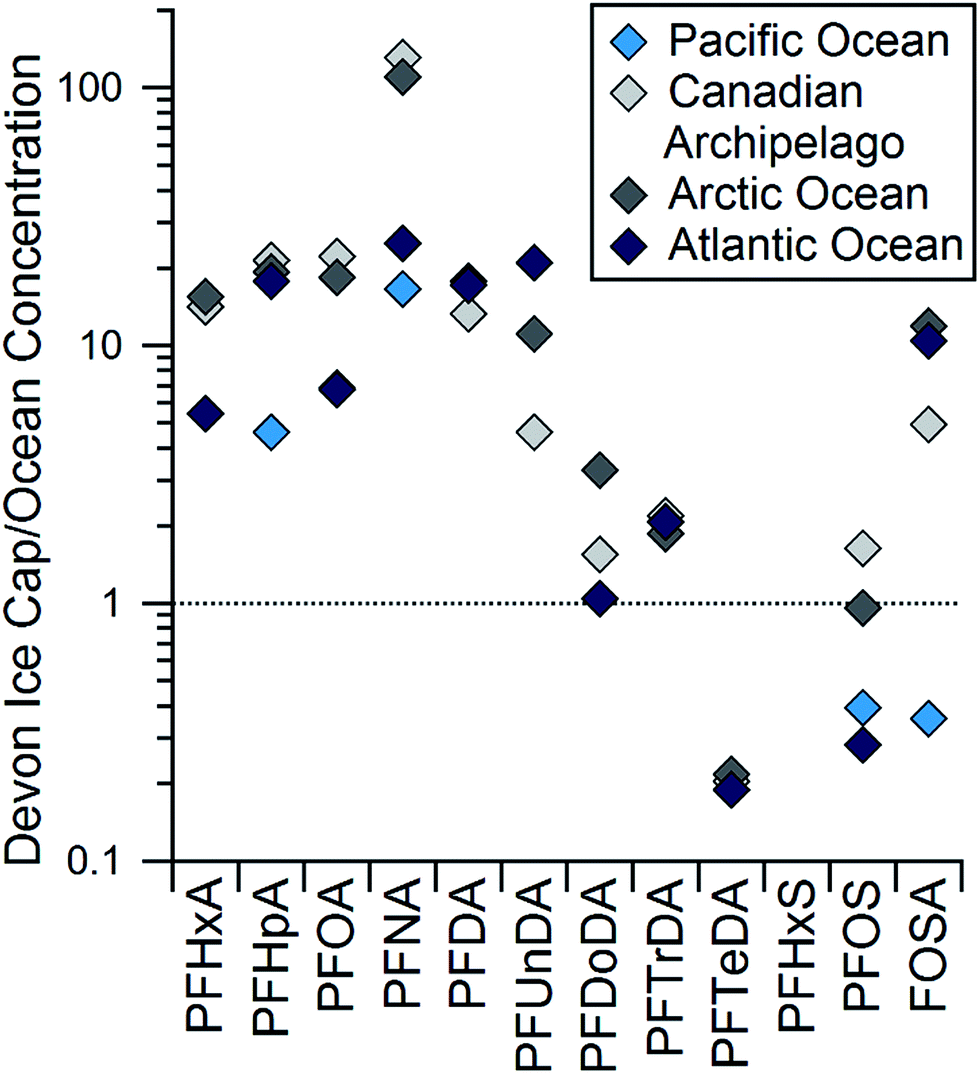 | ||
| Fig. 4 Ratios of observed concentrations in the Devon Ice Cap in 2004–2006 compared to levels in the Canadian Archipelago and Arctic Oceans collected in 2005 (ref. 59) and to the Atlantic59 and Pacific Oceans40 in the northern hemisphere. | ||
The presence of PFECHS on the Devon Ice Cap could be a result of long-range oceanic transport processes and marine aerosol formation. Although no marine measurements of PFECHS have been reported, there was no relationship between the concentration of sodium and PFECHS (R2 = 0.005, p = 0.7314; Table S11†), suggesting PFECHS deposition is likely dominated by another transport mechanism. The detection of PFECHS in freshwater Arctic lakes15 was attributed to contamination from a local airport, suggesting contemporary usage of the compound in the area. It is possible local inputs of PFECHS into freshwater Arctic lakes, followed by aerosol formation, could serve as a mechanism of transport for PFECHS to the Devon Ice Cap. It is also possible that PFECHS contamination on the Devon Ice Cap is a result of direct emission of the compound from aircraft during usage. PFECHS is used as an additive in phosphate ester-based hydraulic fluids to reduce erosion processes in commercial aircraft.64 In the event of water contamination, phosphate ester hydraulic fluids can undergo hydrolysis, leading to the production of acid products.64 The accumulation of acid in hydraulic fluid systems can promote metal erosion and subsequent leakage. Hydraulic system leakage in commercial aircrafts is an inherent problem in the aviation industry, despite recent advancements in the field.65,66 Considering hydraulic systems in aircraft are under pressure, a small leak could result in large emissions. The persistence of aircraft hydraulic fluid system leakage and absence of known volatile precursors of commercial relevance may provide an explanation for the presence of PFECHS in the Arctic environment. Regulatory documents in Canada67 list hydraulic fluid as a potential use for PFOS, suggesting detection of PFECHS on the ice cap indicates the possibility of another direct mechanism for PFOS long-range transport. Evidence of this may be substantiated by a report in 2001, which describes the use of Skydrol®, a PFOS-based hydraulic fluid employed in all commercial aircraft.68 Contemporary uses of PFOS as additives in aviation hydraulic fluids are permitted, according to the exemptions listed in the Stockholm Convention.69 The continued usage of these hydraulic fluids in commercial aircrafts could serve as direct emission sources of PFOS on the Devon Ice Cap. An industry report suggests PFECHS and PFOS would be present in different hydraulic fluids.68 Thus, lack of correlation between PFOS and PFECHS concentrations on the ice cap (R2 = 0.001, p = 0.8542; Table S11†) cannot be used to infer the validity of hydraulic fluid as a source of PFOS. It is challenging to interrogate aviation sources given their complexity; however, the presence of PFECHS and PFOS in an atmospherically derived sample could provide an explanation for the observations on the Devon Ice Cap.
Conclusions
There has been considerable discussion in the literature regarding long-range transport mechanisms of PFAAs. For certain homologues, such as PFOA70 and PFOS,10 both of which were produced directly in large quantities, it is clear that direct transport is the dominant mechanism to remote regions. For other homologues that were directly produced in lower quantities, the major transport pathways are less clear. Ice caps receive their contamination solely from atmospheric deposition and preserve a temporal record of that deposition. By sampling ice caps, more clarity can be brought to the role of the atmosphere in long-range transport of PFAAs. Samples collected from the Devon Ice Cap contained concentrations of PFAAs that were higher than those in the ocean for many homologues. The patterns and ratios of homologue concentrations, as well as the absence of a relationship to an ocean tracer, are consistent with a dominant indirect source, atmospheric oxidation of volatile precursors, for most PFAAs to the ice cap. The first observation of PFECHS in an Arctic atmospheric sample suggests the possibility of a new direct source to remote regions through leakage of airline hydraulic fluids.Acknowledgements
Funding for this project was provided by the Northern Contaminants Program, Environment Canada Chemical Management Plan, and Environment Canada Grants and Contributions. JJM thanks the Natural Science and Engineering Research Council of Canada for a postgraduate scholarship. The Authors thank Robert Di Lorenzo, Torsten Meyer for dating the ice cores, Alex Gardner for providing the Devon Ice Cap mass balance dataset, and the Environment and Climate Change Canada, Emergencies, Operational Analytical Laboratories and Research Support Division, for organizing and conducting sample collection.References
- C. M. Butt, U. Berger, R. Bossi and G. T. Tomy, Sci. Total Environ., 2010, 408, 2936–2965 CrossRef CAS PubMed.
- D. A. Ellis, J. W. Martin, A. O. De Silva, S. A. Mabury, M. D. Hurley, M. P. Sulbaek Andersen and T. J. Wallington, Environ. Sci. Technol., 2004, 38, 3316–3321 CrossRef CAS PubMed.
- J. C. D’eon, M. D. Hurley, T. J. Wallington and S. A. Mabury, Environ. Sci. Technol., 2006, 40, 1862–1868 CrossRef.
- D. A. Ellis, J. W. Martin, S. A. Mabury, M. D. Hurley, M. P. Sulbaek Andersen and T. J. Wallington, Environ. Sci. Technol., 2003, 37, 3816–3820 CrossRef CAS PubMed.
- M. D. Hurley, T. J. Wallington, M. P. Sulbaek Andersen, D. A. Ellis, J. W. Martin and S. A. Mabury, J. Phys. Chem. A, 2004, 108, 1973–1979 CrossRef CAS.
- C. J. Young, V. I. Furdui, J. Franklin, R. M. Koerner, D. C. G. Muir and S. A. Mabury, Environ. Sci. Technol., 2007, 41, 3455–3461 CrossRef CAS PubMed.
- J. Cheng, E. Psillakis, M. R. Hoffman and A. J. Colussi, J. Phys. Chem. A, 2009, 113, 8152–8156 CrossRef CAS PubMed.
- J. M. Armitage, M. Macleod and I. T. Cousins, Environ. Sci. Technol., 2009, 43, 1134–1140 CrossRef CAS PubMed.
- J. M. Armitage, M. Macleod and I. T. Cousins, Environ. Sci. Technol., 2009, 43, 5830–5836 CrossRef CAS PubMed.
- J. M. Armitage, U. Schenker, J. W. Martin, M. Macleod and I. T. Cousins, Environ. Sci. Technol., 2009, 43, 9274–9280 CrossRef CAS PubMed.
- O. Wurl, E. Wurl, L. Miller, K. Johnson and S. Vagle, Biogeosciences, 2011, 8, 121–135 CrossRef CAS.
- C. J. McMurdo, D. A. Ellis, E. Webster, J. Butler, R. D. Christensen and L. K. Reid, Environ. Sci. Technol., 2008, 42, 3969–3974 CrossRef CAS PubMed.
- M. Reth, U. Berger, D. Broman, I. T. Cousins, E. D. Nilsson and M. S. McLachlan, Environ. Chem., 2011, 8, 381–388 CAS.
- T. Meyer, D. C. G. Muir, C. Teixeira, X. Wang, T. Young and F. Wania, Environ. Sci. Technol., 2012, 46, 826–833 CrossRef CAS PubMed.
- G. L. Lescord, K. A. Kidd, A. O. De Silva, M. Williamson, C. Spencer, X. Wang and D. C. G. Muir, Environ. Sci. Technol., 2015, 49, 2694–2702 CrossRef CAS PubMed.
- K. Y. Kwok, S. Taniyasu, L. W. Y. Yeung, M. B. Murphy, P. K. S. Lam, Y. Horii, K. Kannan, G. Petrick, R. K. Sinha and N. Yamashita, Environ. Sci. Technol., 2010, 44, 7043–7049 CrossRef CAS PubMed.
- C. J. Young and S. A. Mabury, Rev. Environ. Contam. Toxicol., 2010, 208, 1–110 CAS.
- K. Y. Kwok, E. Yamazaki, N. Yamashita, S. Taniyasu, M. B. Murphy, Y. Horii, G. Petrick, R. Kallerborn, K. Kannan, K. Murano and P. K. S. Lam, Sci. Total Environ., 2013, 447, 46–55 CrossRef CAS PubMed.
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbuhler, Environ. Int., 2014, 70, 62–75 CrossRef CAS PubMed.
- M. Shoeib, T. Harner and P. Vlahos, Environ. Sci. Technol., 2006, 40, 7577–7583 CrossRef CAS PubMed.
- N. L. Stock, V. I. Furdui, D. C. G. Muir and S. A. Mabury, Environ. Sci. Technol., 2007, 41, 3529–3536 CrossRef CAS PubMed.
- J. W. Martin, D. A. Ellis, S. A. Mabury, M. D. Hurley and T. J. Wallington, Environ. Sci. Technol., 2006, 40, 864–872 CrossRef CAS PubMed.
- M. Garsjo, Perfluorinated alkylated substances (PFAS) in Arctic char (Salvelinus alpinus): A case study from Svalbard, Norwegian University of Life Sciences, 2013 Search PubMed.
- T. Kirchgeorg, A. Dreyer, P. Gabrielli, J. Gabrieli, L. G. Thompson, C. Barbante and R. Ebinghaus, Environ. Pollut., 2016, 218, 804–812 CrossRef CAS PubMed.
- T. Kirchgeorg, A. Dreyer, J. Gabrieli, N. Kehrwald, M. Sigl, M. Schwikowski, C. Boutron, A. Gambaro, C. Barbante and R. Ebinghaus, Environ. Pollut., 2013, 178, 367–374 CrossRef CAS PubMed.
- X. Wang, C. Halsall, G. Codling, Z. Xie, B. Xu, Z. Zhao, Y. Xue, R. Ebinghaus and K. C. Jones, Environ. Sci. Technol., 2014, 48, 173–181 CrossRef CAS PubMed.
- M. Cai, Z. Zhao, Z. Yin, L. Ahrens, P. Huang, M. Cai, H. Yang, J. He, R. Sturm, R. Ebinghaus and Z. Xie, Environ. Sci. Technol., 2012, 46, 661–668 CrossRef CAS PubMed.
- J. W. Martin, S. A. Mabury, K. R. Solomon and D. C. G. Muir, Environ. Toxicol. Chem., 2003, 22, 189–195 CrossRef CAS PubMed.
- S. Taniyasu, K. Kannan, L. W. Y. Yeung, K. Y. Kwok, P. K. S. Lam and N. Yamashita, Anal. Chim. Acta, 2008, 619, 221–230 CrossRef CAS PubMed.
- C. E. Wujcik, D. Zehavi and J. N. Seiber, Chemosphere, 1998, 36, 1233–1245 CrossRef CAS.
- M. Berg, S. R. Müller, J. Mühlemann, A. Wiedmer and R. P. Schwarzenbach, Environ. Sci. Technol., 2000, 34, 2675–2683 CrossRef CAS.
- B. F. Scott, C. A. Moody, C. Spencer, J. M. Small, D. C. G. Muir and S. A. Mabury, Environ. Sci. Technol., 2006, 40, 6405–6410 CrossRef CAS PubMed.
- B. F. Scott, C. Spencer, S. A. Mabury and D. C. G. Muir, Environ. Sci. Technol., 2006, 40, 7167–7174 CrossRef CAS PubMed.
- C. J. Young, M. D. Hurley, T. J. Wallington and S. A. Mabury, Chem. Phys. Lett., 2009, 473, 251–256 CrossRef CAS.
- T. J. Wallington, W. F. Schneider, J. Sehested, M. Bilde, J. Platz, O. J. Nielsen, L. K. Christensen, M. J. Molina, L. T. Molina and P. W. Wooldridge, J. Phys. Chem. A, 1997, 101, 8264–8274 CrossRef CAS.
- L. Chen, T. Uchimaru and S. Kutsuna, Chem. Phys. Lett., 2011, 514, 207–213 CrossRef CAS.
- G. Myhre, D. Shindell, F.-M. Bréon, W. Collins, J. Fuglestvedt, J. Huang, D. Koch, J.-F. Lamarque, D. Lee, B. Mendoza, T. Nakajima, A. Robock, G. Stephens, T. Takemura and H. Zhang, Clim. Chang. 2013 Phys. Sci. Basis. Contrib. Work. Gr. I to Fifth Assess. Rep. Intergov. Panel Clim. Chang., 2013, pp. 659–740 Search PubMed.
- M. Yao, B. Vollmer, M. K. Zhou, L. X. Henne, S. Reimann, S. Li, P. C. Wenger and A. Hill, Atmos. Chem. Phys., 2012, 12, 10181–10193 CrossRef.
- M. McFarland, S. Anderson, B. Miller, D. Fahey, B. Hall, L. Hu, C. Siso and J. Elkins, J. Phys. Chem. A, 2015, 119, 4439–4449 CrossRef PubMed.
- M. Cai, Z. Xie, A. Möller, Z. Yin, P. Huang, M. Cai, H. Yang, R. Sturm, J. He and R. Ebinghaus, Chemosphere, 2012, 87, 989–997 CrossRef CAS PubMed.
- A. O. De Silva, C. Spencer, B. F. Scott, S. Backus and D. C. G. Muir, Environ. Sci. Technol., 2011, 45, 8060–8066 CrossRef CAS PubMed.
- S. R. De Solla, A. O. De Silva and R. J. Letcher, Environ. Int., 2012, 39, 19–26 CrossRef CAS PubMed.
- M. Sharp, D. O. Burgess, J. G. Cogley, M. Ecclestone, C. Labine and G. J. Wolken, Geophys. Res. Lett., 2011, 38, L11501 CrossRef.
- P. Bezeau, M. Sharp, D. Burgess and G. Gascon, J. Glaciol., 2013, 59, 981–991 CrossRef.
- C. Kinnard, C. Zdanowicz, D. Fisher and C. Wake, Ann. Glaciol., 2006, 44, 383–390 CrossRef CAS.
- M. M. Plassmann, T. Meyer, Y. D. Lei, F. Wania, M. S. McLachlan and U. Berger, Environ. Sci. Technol., 2011, 45, 6872–6878 CrossRef CAS PubMed.
- R. M. Koerner, Ann. Glaciol., 2005, 42, 417–423 CrossRef.
- 3M Company, Phase-out plan for POSF-based products, US Environmental Protection Agency, St. Paul, MN, 2000 Search PubMed.
- T. Stahl, D. Mattern and H. Brunn, Environ. Sci. Eur., 2011, 23, 38 CrossRef.
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbuhler, Environ. Int., 2014, 69, 166–176 CrossRef CAS PubMed.
- S. Xie, T. Wang, S. Liu, K. C. Jones, A. J. Sweetman and Y. Lu, Environ. Int., 2013, 52, 1–8 CrossRef CAS PubMed.
- A. G. Paul, K. C. Jones and A. J. Sweetman, Environ. Sci. Technol., 2009, 43, 386–392 CrossRef CAS PubMed.
- X. Zhang, T. Meyer, D. C. G. Muir, C. Teixeira, X. Wang and F. Wania, Environ. Sci.: Processes Impacts, 2013, 15, 2304–2311 CAS.
- W. Jiang, Y. Zhang, L. Zhu and J. Deng, Environ. Int., 2014, 64, 40–47 CrossRef CAS PubMed.
- W. Jiang, Y. Zhang, L. Yang, X. Chu and L. Zhu, Chemosphere, 2015, 127, 180–187 CrossRef CAS PubMed.
- C. E. Whipkey, R. C. Capo, O. A. Chadwick and B. W. Stewart, Chem. Geol., 2000, 168, 37–48 CrossRef CAS.
- W. C. Keene, A. P. Pszenny, J. N. Galloway and M. E. Hawley, J. Geophys. Res., 1986, 91, 6647 CrossRef CAS.
- A. Tivy, S. E. L. Howell, B. Alt, S. McCourt, R. Chagnon, G. Crocker, T. Carrieres and J. J. Yackel, J. Geophys. Res., C: Oceans Atmos., 2011, 116, C06027 Search PubMed.
- J. P. Benskin, D. C. G. Muir, B. F. Scott, C. Spencer, A. O. De Silva, H. Kylin, J. W. Martin, A. Morris, R. Lohmann, G. Tomy, B. Rosenberg, S. Taniyasu and N. Yamashita, Environ. Sci. Technol., 2012, 46, 5815–5823 CrossRef CAS PubMed.
- L. Ahrens, Z. Xie and R. Ebinghaus, Chemosphere, 2010, 78, 1011–1016 CrossRef CAS PubMed.
- J. Sánchez-Avila, J. Meyer and S. Lacorte, Environ. Pollut., 2010, 158, 2833–2840 CrossRef PubMed.
- Persistent Organic Pollutants Review Committee, in Report of the Persistent Organic Pollutants Review Committee on the work of its sixth meeting, Geneva, Switzerland, 2010, pp. 5–38 Search PubMed.
- OECD, in Survey on the production, use and release of PFOS, PFAS, PFOA PFCA, their related substances and products/mixtures containing these substances, Paris, France, 2011, pp. 11–59 Search PubMed.
- Terrill D. Smith, United States Patent Office, 3,679,587, 1972.
- Leakage Prevention, Hydraul. Pneum., 2012, pp. 1–5 Search PubMed.
- The Challenges of Aircraft Hydraulic Design, Hydraul. Pneum., 1998, pp. 1–4 Search PubMed.
- Canada Environmental Protection Act: Prohibition of Certain Toxic Substances Regulations P.C. 2012-1714, 13 December, 2012, SOR/2012-285, Canada Gazette, Part I, vol. 147, pp. 19–67, 2013 January 2, ISSN 1494-6122 Search PubMed.
- P. Stifel, Boeing A&M Environmental Technotes: FLASHJET® Qualification Test Programs Expand Customer Applications, 2001, vol. 6 Search PubMed.
- Secretariat of the Stockhom Convention, Stockholm Convention on Persistent Organic Pollutants (POPs) as amended in 2009, 2009 Search PubMed.
- F. Wania, Environ. Sci. Technol., 2007, 41, 4529–4535 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: As described in text. See DOI: 10.1039/c6em00593d |
| This journal is © The Royal Society of Chemistry 2017 |