
Eukaryotic community diversity and spatial variation during drinking water production (by seawater desalination) and distribution in a full-scale network†
A.
Belila
a,
J.
El-Chakhtoura
*ab,
P. E.
Saikaly
a,
M. C. M.
van Loosdrecht
 b and
J. S.
Vrouwenvelder
abc
b and
J. S.
Vrouwenvelder
abc
aWater Desalination and Reuse Center (WDRC), Division of Biological and Environmental Science and Engineering (BESE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: Joline.Chakhtoura@kaust.edu.sa
bDepartment of Biotechnology, Faculty of Applied Sciences, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: J.ElChakhtoura@tudelft.nl
cWetsus, European Centre of Excellence for Sustainable Water Technology, Oostergoweg 9, 8911 MA Leeuwarden, The Netherlands
First published on 1st December 2016
Abstract
Eukaryotic microorganisms are naturally present in many water resources and can enter, grow and colonize water treatment and transport systems, including reservoirs, pipes and premise plumbing. In this study, we explored the eukaryotic microbial community structure in water during the (i) production of drinking water in a seawater desalination plant and (ii) transport of the drinking water in the distribution network. The desalination plant treatment involved pre-treatment (e.g. spruce filters), reverse osmosis (RO) membrane filtration and post-treatment steps (e.g. remineralization). 454 pyrosequencing analysis of the 18S rRNA gene revealed a highly diverse (35 phyla) and spatially variable eukaryotic community during water treatment and distribution. The desalination plant feed water contained a typical marine picoeukaryotic community dominated by Stramenopiles, Alveolates and Porifera. In the desalination plant Ascomycota was the most dominant phylum (15.5% relative abundance), followed by Alveolata (11.9%), unclassified fungi clade (10.9%) and Porifera (10.7%). In the drinking water distribution network, an uncultured fungi phylum was the major group (44.0%), followed by Chordata (17.0%), Ascomycota (11.0%) and Arthropoda (8.0%). Fungi constituted 40% of the total eukaryotic community in the treatment plant and the distribution network and their taxonomic composition was dominated by an uncultured fungi clade (55%). Comparing the plant effluent to the network samples, 84 OTUs (2.1%) formed the core eukaryotic community while 35 (8.4%) and 299 (71.5%) constituted unique OTUs in the produced water at the plant and combined tap water samples from the network, respectively. RO membrane filtration treatment significantly changed the water eukaryotic community composition and structure, highlighting the fact that (i) RO produced water is not sterile and (ii) the microbial community in the final tap water is influenced by the downstream distribution system. The study results raise questions concerning the source of the major eukaryotic community in the network and the emergence of fungi as a dominant group in the drinking water distribution system suggests that these microorganisms need special attention.
Water impactEukaryotic microorganisms constitute an important group in the drinking water system microbiome. They influence the bacterial ecology and can be opportunistic pathogens. This study highlights the taxonomic and phylogenetic diversity of the eukaryotic community during the (i) production of drinking water in a seawater desalination plant and (ii) transport of the produced drinking water in the distribution network. Results reveal the influence of different treatment processes and network distribution on the eukaryotic community structure, specifically the fungal community which emerged as a dominant group during reverse osmosis treatment and in the final household tap water. |
1. Introduction
From source to tap, different stages of drinking water production and transport systems offer unique habitats where complex microbial communities may thrive with their structure changing at each stage. These communities change dynamically with upstream microbial communities in e.g. feed water potentially seeding the downstream system, and thus influencing the final drinking water microbial quality. Eukaryotic microorganisms are present in drinking water production and distribution systems, where several studies worldwide have reported the presence of protozoa,1,2 algae,3,4 invertebrates5,6 and fungi.7–11 The presence of some microbial eukaryotes (i.e. free living protozoa) in drinking water distribution systems (DWDS) constitutes a public health concern either due to their potential pathogenicity such as Acanthamoeba, Candida and Aspergillus12–14 or by harboring a variety of pathogenic bacteria,15 including Legionella.16 Microbial eukaryotes may shelter potentially pathogenic microbes and provide disinfection protection, e.g. nematodes which were shown to be responsible for recurring high levels of total coliforms (i.e. Shigella spp.) in chlorinated drinking water storage reservoir pump stations.15 Also, amphipods, insect larvae and copepods isolated from a distribution system were found to contain putative opportunistic pathogens from various Aeromonas, Pseudomonas and Staphylococcus spp.17 Moreover, eukaryotes visibly occurring in drinking water will be perceived by consumers as indicative of low water quality and hygiene.Based on their main food source (bacteria, fungi and algae), eukaryotes potentially play an important role in shaping the in situ microbial composition of DWDS. However, little is known about microbial eukaryotic diversity in these engineered systems. Compared to 16S rRNA gene-based prokaryotic community analysis, limited data is available on the eukaryotic ecology of drinking water treatment plants (WTP) and drinking water distribution systems (DWDS). A few studies have assessed eukaryotic community diversity in the bulk water12,18,19 in comparison with biofilms.20–23 18S rRNA gene pyrosequencing has provided insight on the eukaryotic community structure of drinking water produced from surface water12,24 and groundwater.12
Reverse osmosis (RO) membrane-based seawater desalination has gained wide acceptance around the world as a solution for the alarming fresh water crisis in many regions. Compared with other WTPs, there is still scant information about the microbial ecology of desalination plants.25 The objectives of this study were to investigate the taxonomic and phylogenetic diversity of the eukaryotic community in water during the (i) production of drinking water in a seawater desalination plant and (ii) transport of the drinking water in the distribution network, including storage reservoirs, pipes and premise plumbing. The spatial variation of the eukaryotic community was characterized from the seawater (source) to several points of use in the drinking water distribution network.
2. Materials and methods
2.1. Desalination plant
The desalination plant is located at King Abdullah University of Science and Technology (KAUST) in the West of the Kingdom of Saudi Arabia. The facility was designed to provide all potable water needs for the campus as well as the residential areas. The plant has a drinking water production capacity of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m3 per day. The source is the Red Sea. The desalination plant consists of three parts: pre-treatment, reverse osmosis (RO) system and post-treatment. Pre-treatment involves the sequence of (i) open seawater intake, (ii) fine screen filtration, (iii) water chlorination at the intake point, and (iv) filtration through spruce multi-media filters (SMF) to remove particulate and colloidal matter (Fig. 1). The RO system involves the sequence of (i) anti-scalant dosage (1.5 mg L−1), (ii) cartridge filtration (10 μm pore size), (iii) sodium bisulphite (SBS) dosage and then the (iv) RO installation consisting of a seawater (SWRO) and brackish water RO (BWRO) pass. As for post-treatment, about 60% of the BWRO permeate is mixed with 40% of the SWRO permeate to meet the World Health Organization health-based guideline for Boron. Finally, the produced water is chlorinated to maintain 0.5–1.0 mg L−1 of residual chlorine. In addition, the following chemicals are dosed: (i) hydrated lime (43 mg L−1) for water conditioning to prevent pipe corrosion and (ii) CO2 (20 mg L−1) to adjust the pH to near a neutral value with positive Langelier saturation index (LSI). Subsequently, the produced drinking water is pumped into storage reservoirs (two vessels, each 10500 m3) feeding the drinking water distribution network. The hydraulic retention times in the break tank and the drinking water reservoirs were 4 h and 12.6 h, respectively.
000 m3 per day. The source is the Red Sea. The desalination plant consists of three parts: pre-treatment, reverse osmosis (RO) system and post-treatment. Pre-treatment involves the sequence of (i) open seawater intake, (ii) fine screen filtration, (iii) water chlorination at the intake point, and (iv) filtration through spruce multi-media filters (SMF) to remove particulate and colloidal matter (Fig. 1). The RO system involves the sequence of (i) anti-scalant dosage (1.5 mg L−1), (ii) cartridge filtration (10 μm pore size), (iii) sodium bisulphite (SBS) dosage and then the (iv) RO installation consisting of a seawater (SWRO) and brackish water RO (BWRO) pass. As for post-treatment, about 60% of the BWRO permeate is mixed with 40% of the SWRO permeate to meet the World Health Organization health-based guideline for Boron. Finally, the produced water is chlorinated to maintain 0.5–1.0 mg L−1 of residual chlorine. In addition, the following chemicals are dosed: (i) hydrated lime (43 mg L−1) for water conditioning to prevent pipe corrosion and (ii) CO2 (20 mg L−1) to adjust the pH to near a neutral value with positive Langelier saturation index (LSI). Subsequently, the produced drinking water is pumped into storage reservoirs (two vessels, each 10500 m3) feeding the drinking water distribution network. The hydraulic retention times in the break tank and the drinking water reservoirs were 4 h and 12.6 h, respectively.
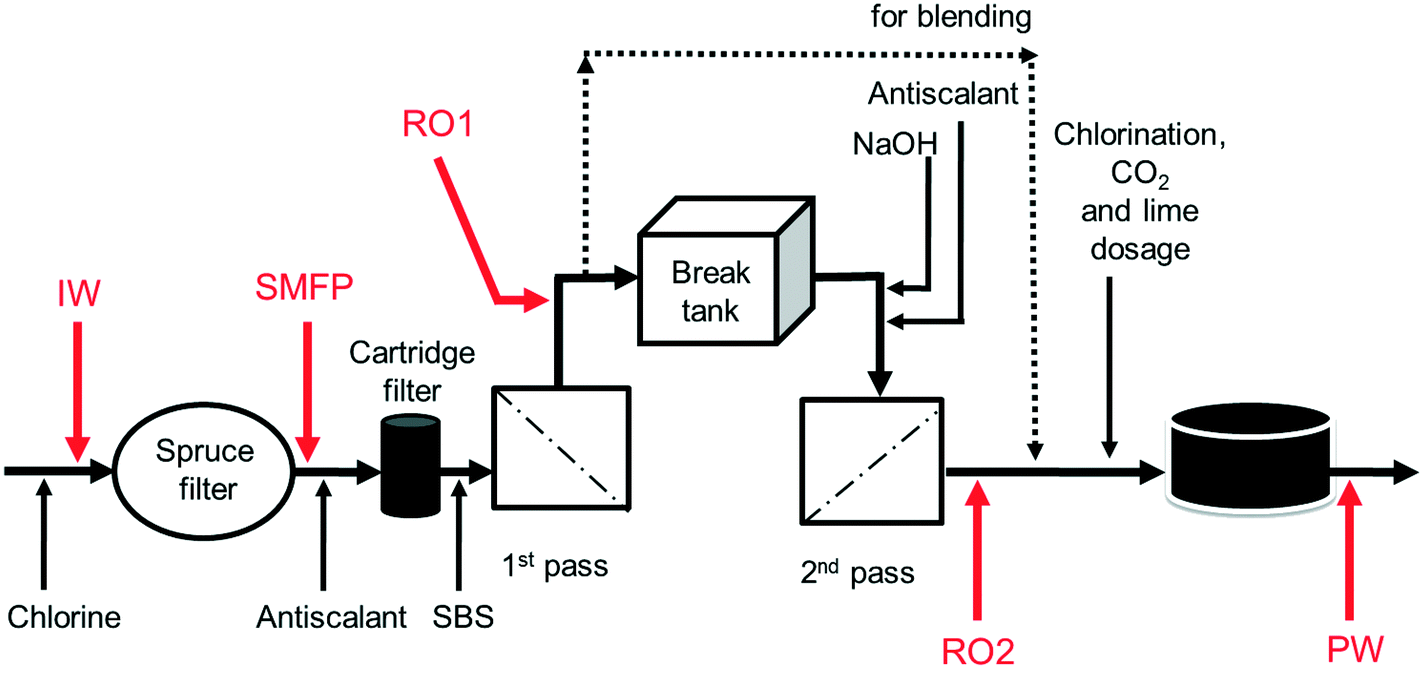 | ||
| Fig. 1 Schematic diagram of sampling points from KAUST desalination plant. IW: inlet water; SMFP: spruce media filter permeate; RO1: seawater reverse osmosis permeate; RO2: brackish water reverse osmosis permeate; PW: produced water. Dosed are chlorine, antiscalant, SBS (sodium bisulphite), NaOH, CO2 and lime. | ||
2.2. Distribution network
The full-scale desalination plant and the distribution network were taken into operation at the same time in 2009. The network piping material is only polyvinyl chloride (PVC). The total distribution network pipe length covered about 60 km and involved a confined area, meaning there was no mixing with drinking water from other sources. The piping network is periodically flushed with drinking water as part of routine maintenance.2.3. Water sampling and sample IDs
Ten water samples were collected on the same day (6 August 2012): five from the desalination plant and five from selected network taps, using 4 L sterile polyvinyl chloride (PVC) plastic bottles. The locations of sampling from the desalination plant were: the chlorinated raw seawater entering the spruce multimedia filters (IW), spruce media filter permeate (SMFP), seawater reverse osmosis permeate (RO1), brackish water reverse osmosis permeate (RO2) and the final chlorinated water (PW) (Fig. 1). The tap water sampling locations were situated at 1.5 (TP1), 2.5 (TP2), 3 (TP3), 4 (TP4) and 7.5 (TP5) km from the desalination plant (Fig. S1†), respectively. All tap water samples were collected after flushing the tap for 5 minutes, then the water samples were transported directly to the Water Desalination and Reuse Center laboratory at KAUST to analyze water quality parameters and then they were filtered through 0.2 μm pore-sized filters (Millipore) for subsequent molecular biology analyses.2.4. Water quality analysis
The water quality was analyzed through the measurement of pH (Cyberscan pH 6000, Eutech, USA), conductivity (CON 510, Oakton, USA), turbidity (2100Q, HACH, USA), total organic carbon (TOC) (TOC-VCPH Analyzer, Shimadzu, Japan) and residual chlorine concentration which was measured by the N,N-diethyl P-phenylenediamine (DPD) method using a pocket colorimeter TM II (HACH). All these analyses were done following standard methods for the examination of water and wastewater.262.5. DNA extraction
Water samples of 3 L from each location were filtered through polycarbonate membranes (pore size 0.2 μm, membrane diameter 45 mm, Millipore) which were rolled and placed into Cryo-vials (Nalgene, Nunc, Rochester, NY, USA) and frozen at −80 °C until DNA extraction. DNA was extracted from the membranes using a FastDNA spin kit for soil (FastDNA, MP Biomedical, Illkirch, France). The extracted DNA was concentrated in 50 μL DNAse-free water (extracted from 3 L water).2.6. PCR amplification and 18S rDNA gene pyrosequencing
The 18S rRNA gene fragments were amplified using primers Euk-A7F27 and Euk-570R27 following these PCR conditions: 94 °C for 3 minutes, followed by 30 cycles of 94 °C for 30 seconds; 53 °C for 40 seconds and 72 °C for 1 minute and a final elongation step at 72 °C for 5 minutes. After that, all amplicon products were mixed in equal concentrations and purified using Agencourt Ampure Beads (Agencourt Bioscience Corporation, MA, USA).Pyrosequencing was carried out at MR DNA Lab (Shallowater, TX, USA) on the Roche 454 FLX Titanium genome sequencer according to the manufacturer's instructions. Sequence data was processed at MR DNA Lab. In summary, sequences were depleted of barcodes and primers, then sequences <150 bp were removed, as well as sequences with ambiguous base calls and with homopolymer runs exceeding 6 bp. Sequences were denoised, operational taxonomic units (OTUs) generated and chimeras removed. OTUs were defined by clustering at 3% divergence (97% similarity). Final OTUs were taxonomically classified using BLASTn against a curated database derived from NCBI and RDP II.
2.7. Alpha and beta diversity measures
Rarified OTU tables were used to generate alpha and beta diversity metrics. Shannon diversity (H) and species richness estimator of Chao1 indices were generated for each sample using QIIME as a measure of alpha diversity. Spatial variation of eukaryotic community was analyzed with principal coordinate analysis (PCoA) in QIIME using unweighted UniFrac distance matrix as a measure of beta diversity. The Emperor visualization tool28 was used to visualize the PCoA graph. Shared and sample-specific OTUs were calculated using Venny online software29 (http://bioinfogp.cnb.csic.es/tools/venny/index.html).3. Results
3.1. Water parameters
During the seawater desalination process, 98.8% of the total organic carbon (TOC) was removed while the conductivity decreased by 99.6%. Crossing the spruce media filter, water pH remained stable (8.9), then increased to 10.1 after the second reverse osmosis stage due to the addition of NaOH and antiscalant necessary to enhance the removal of Boron. After treatment the pH of the produced water was corrected to 7.5 (±0.1) before distribution. No residual chlorine was detected during the desalination process but after chlorination, the free chlorine concentration detected was 0.60 mg L−1 in the produced drinking water. The water temperature during treatment was 25.6 °C. Minor variations were observed in the tap water parameters measured at various spots in the network (Table 1). The tap water pH ranged between 6.8 and 7.3, residual chlorine between 0.22 and 0.53 mg L−1; and turbidity ranged between 0.18 and 0.36 nephelometric turbidity units (NTU). All tap water measured parameters were in accordance with World Health Organization physicochemical quality standards for drinking water.30| TOC (mg L−1) | Temperature (°C) | pH | Conductivity (mS cm−1) | Free chlorine (mg L−1) | Turbidity (NTU) | Distribution distancea (km) | |
|---|---|---|---|---|---|---|---|
| Abbreviations: IW: inlet water, SMFP: spruce media filter permeate, RO1 and RO2: seawater and brackish water reverse osmosis permeates, PW: produced water, TP1–TP5: tap water samples. The values between parentheses represent the standard deviation of three measurements of the same water sample. a Distance between the produced water feeding the network and tap water sampling locations. | |||||||
| Desalination plant: samples during treatment | |||||||
| IW | 1.68(±0.06) | 25.6 | 8.9(±0.2) | 65.0(±0.3) | <0.02 | 2.10(±0.02) | — |
| SMFP | 1.10(±0.04) | 25.6 | 8.9(±0.2) | 61.3(±0.2) | <0.02 | 1.41(±0.03) | — |
| RO1 | 0.54(±0.05) | 25.6 | 7.7(±0.1) | 0.87(±0.02) | <0.02 | 0.48(±0.01) | — |
| RO2 | 0.04(±0.02) | 25.6 | 10.1(±0.3) | 0.30(±0.02) | <0.02 | 0.20(±0.00) | — |
| PW | 0.02(±0.02) | 25.6 | 7.5(±0.1) | 0.24(±0.01) | 0.6(±0.01) | 0.25(±0.00) | — |
| Distribution network: tap water samples | |||||||
| TP1 | 0.03 | 23.2 | 7.2 | 243 | 0.45 | 0.18 | 1.5 |
| TP2 | 0.02 | 25.1 | 6.8 | 283 | 0.42 | 0.18 | 2.5 |
| TP3 | 0.02 | 23.4 | 7.1 | 193 | 0.36 | 0.36 | 3.0 |
| TP4 | 0.02 | 24.4 | 7.0 | 252 | 0.53 | 0.20 | 4.0 |
| TP5 | 0.03 | 25.2 | 7.3 | 221 | 0.22 | 0.24 | 7.5 |
| Mean (SD) | 0.02(±0.01) | 24.2(±0.9) | 7.1(±0.2) | 238(±34) | 0.39(±0.12) | 0.23(±0.08) | — |
3.2. Alpha and beta diversity measures
Crossing the desalination plant, the Shannon diversity index decreased gradually from 6.54 (IW) to 2.39 (PW) (Table 2). The first reverse osmosis pass had the largest impact on the reduction of the Shannon diversity index (reduction from 5.40 to 2.49). After chemical treatment and the disinfection step by chlorination, the Shannon diversity index stabilized to around 2.39.| Number of raw reads | Average length (bp) | Observed OTUs | Richness estimate (Chao 1) | Shannon diversity index (H) | |
|---|---|---|---|---|---|
| Abbreviations: IW: inlet water, SMFP: spruce media filter permeate, RO1 and RO2: seawater and brackish water reverse osmosis permeates, PW: final produced chlorinated water before distribution, TP1–TP5: tap water samples. The operational taxonomic units (OTUs) were defined by clustering at 3% divergence. | |||||
| Desalination plant | |||||
| IW | 35307 |
461 | 478 | 510 | 6.54 |
| SMFP | 19205 |
456 | 440 | 414 | 5.40 |
| RO1 | 20199 |
460 | 30 | 28 | 2.49 |
| RO2 | 26670 |
480 | 178 | 82 | 3.34 |
| PW | 31413 |
472 | 119 | 50 | 2.39 |
| Distribution network | |||||
| TP1 | 12410 |
483 | 22 | 22 | 1.71 |
| TP2 | 9711 | 478 | 126 | 36 | 1.22 |
| TP3 | 10643 |
466 | 161 | 42 | 1.91 |
| TP4 | 28837 |
474 | 108 | 56 | 2.13 |
| TP5 | 35370 |
476 | 102 | 40 | 1.34 |
In the network, all measured tap water Shannon indices were lower than the produced water (PW) (Table 2). All tap water Shannon diversity indices were lower than 2 except for TP4 (2.13), which could be explained by the increase in the abundance of Blastocladiomycota and Amoebozoa in this tap water location (Fig. 2 and 3).
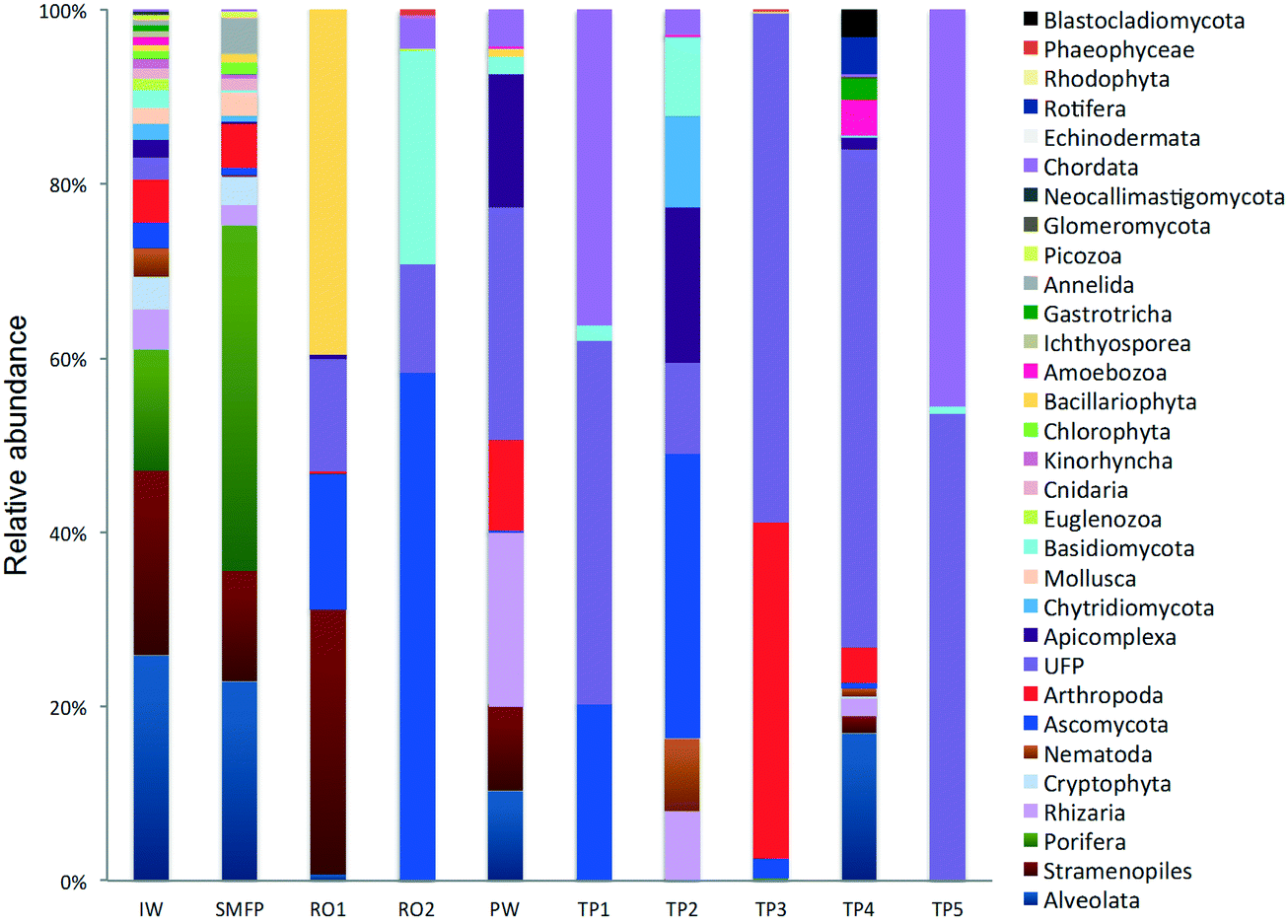 | ||
| Fig. 2 Relative abundance of the eukaryotic community (phyla level). IW: inlet water; SMFP: spruce media filter permeate; RO1: seawater reverse osmosis permeate; RO2: brackish water reverse osmosis permeate; PW: produced water; TP1–TP5: tap water samples; UFP: unclassified fungi phylum. | ||
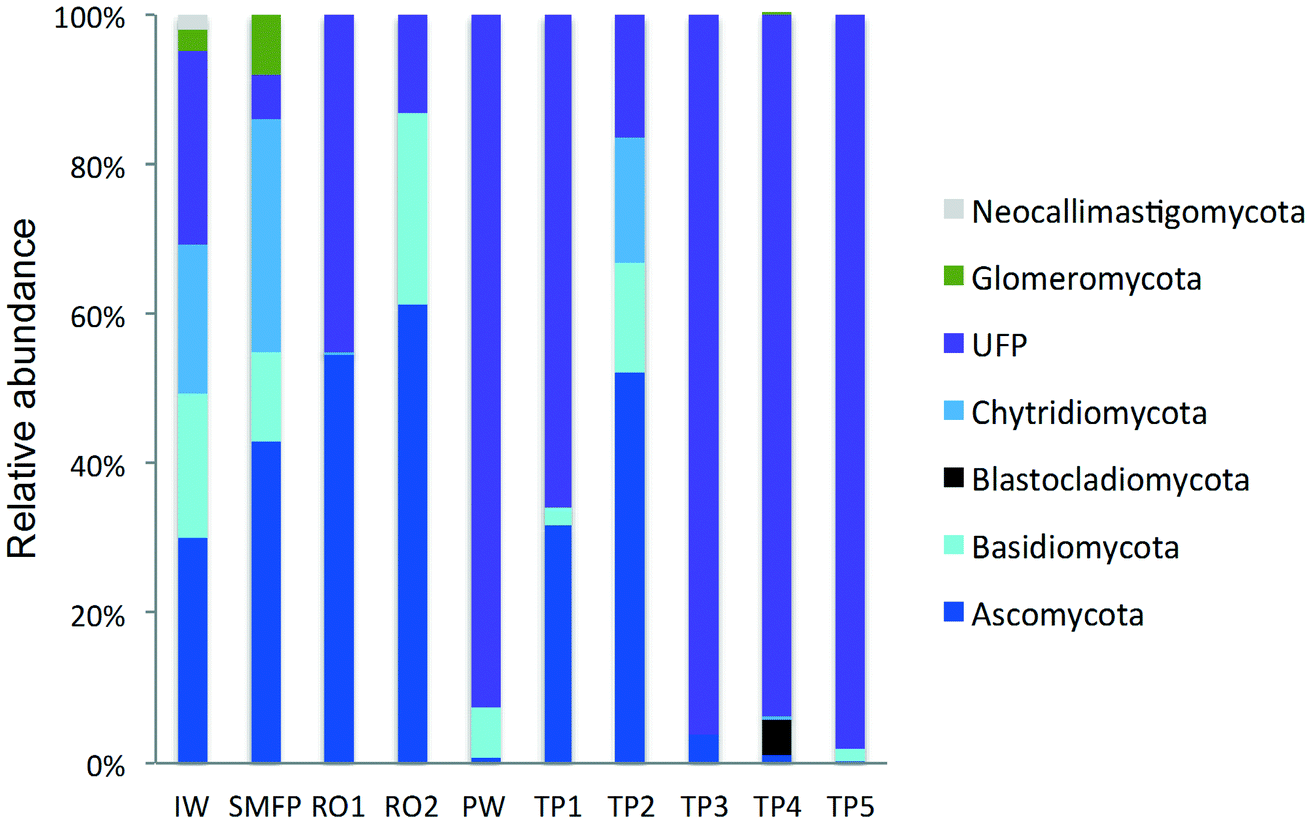 | ||
| Fig. 3 Variation in the fungal community composition (phyla level) during desalination and after water distribution. IW: inlet water; SMFP: spruce media filter permeate; RO1: seawater reverse osmosis permeate; RO2: brackish water reverse osmosis permeate; PW: produced water; TP1–TP5: tap water samples; UFP: unclassified fungi phylum. | ||
To estimate the variation of OTU richness during the water desalination process, nonparametric Chao 1 index was calculated for all samples. The highest OTU richness values (calculated at a cluster distance of 0.03) were detected in the inlet water (510) and spruce media filter permeate (414) (Table 2). Chao 1 index decreased by 93% after RO1 pass to reach 28 and then increased to 82 after RO2 pass and stabilized at 50 in PW. The Chao1 values calculated in the network for the tap water samples (i.e. TP1 to TP5) were lower than the produced water value except in TP4 where the Chao1 index was 56. The rarefaction curves did not reach a plateau for samples IW and SMFP as operational taxonomic units continued to emerge even after 5000 reads suggesting that there was additional diversity in those samples that was probably not captured by the pyrosequencing.
Based on unweighted UniFrac distance matrix, the PCoA plots showed divergent positions of the different water samples during the desalination process. A clear separation was found between (i) the pre-treatment water samples (IW and SMFP) and (ii) the post-treatment water samples (RO1, RO2 and PW). During pretreatment, a clear variation in the community structure was observed (Fig. 4). The most important changes in eukaryotic community structure were observed following the reverse osmosis trains 1 and 2 (RO1 and RO2, Fig. 4). The influence of water distribution on the eukaryotic community structure was relatively less significant than observed during the desalination process, indicating a relatively stable eukaryotic community in the distribution network. In fact, all tap water samples showed close or converging positions (Fig. 4). The main changes in the eukaryotic community structure in the drinking water distribution system (Fig. 4).
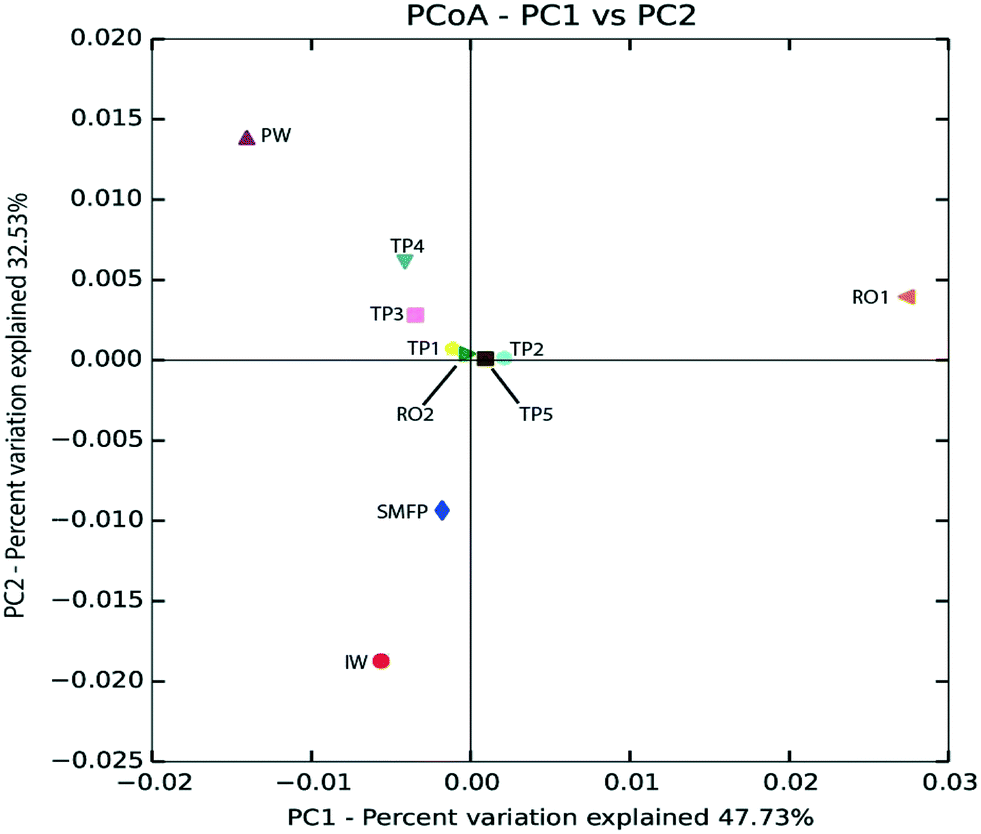 | ||
| Fig. 4 Principal coordinate analysis (PCoA) based on unweighted UniFrac distance matrix showing the phylogenetic link between eukaryotic communities in the desalination plant and after water distribution. IW: inlet water; SMFP: spruce media filter permeate; RO1: seawater reverse osmosis permeate; RO2: brackish water reverse osmosis permeate; PW: produced water; TP1–TP5: tap water samples. | ||
Aiming to evaluate the eukaryotic community response to the different treatment steps (e.g. filtration, chlorination, water distribution), the presence of core OTUs in the desalination plant and in the network distribution system was assessed applying Venn diagram analysis. “Core OTUs” implies the total of shared OTUs between the five sampling locations within the desalination plant and also the shared OTUs between the five sampling locations in the distribution network. Results showed the absence of a core community in the desalination plant. On the other hand, Venn diagram analysis revealed the presence of two distinct core OTUs (shared OTUs): in the desalination plant: the first during pre-treatment (IW + SMFP) and the second during the reverse osmosis treatment and post-treatment steps (Fig. S2†). During the pretreatment step, the core community was composed of 317 shared OTUs between the inlet water and spruce media filter permeate (Fig. S2A†) sampling sites, representing 52.7% of the total OTUs detected. Out of these 317 OTUs, 161 were identified as unique OTUs in the IW (26.8%) and 123 OTUs as unique OTUs in the SMFP (20.5%). Taxonomic assignment showed that 70 OTUs (22.0%) from the core community belonged to Grantessa sp., 22 OTUs to uncultured Stramenopiles and 19 OTUs to uncultured Dinoflagellate. During the reverse osmosis treatment and post-treatment steps (Fig. S2B†), the core community was composed of one unique OTU (0.4%) that was assigned to an uncultured fungus while 15 OTUs were shared between the RO1 and RO2 permeate. Remarkably, all 15 (5.5%) OTUs belonged to the kingdom of fungi: 14 were assigned to the fungal species Herpotrichiellaceae sp. and 1 OTU to Exophiala aquamarina. 13 unique OTUs were detected in RO1 (4.8%), 124 unique OTUs in RO2 (45.8%) and 78 unique OTUs were identified in the produced water (28.8%). The presence of similar organisms in the water before and after RO was not expected because of the strongly different environments: seawater and fresh water.
To better understand the effect of water distribution on the eukaryotic community structure we combined the OTUs of all the tap water samples and compared them to the core community of the produced water. The Venn diagram (Fig. S3†) showed that 84 OTUs (2.1%) formed the core community while 35 (8.4%) and 299 (71.5%) constituted unique OTUs in the PW and all combined tap water samples, respectively. Taxonomic analysis of the core community revealed that 75 OTUs (89.0%) forming the core community belonged to the kingdom of fungi. 64 of the 75 OTUs were assigned to an uncultured fungus with percentage of homology varying between 97.0% and 99.4%. 7 OTUs were assigned to Cochliobolus sp., 2 OTUs to an uncultured marine fungus and 2 OTUs to uncultured Climacodon. The rest of the OTUs forming the core community (9) were assigned to the kingdom Metazoa. 8 OTUs were assigned to the phylum Chordata and 1 OTU to the phylum Porifera.
51 shared OTUs were identified in both the inlet water and the tap water samples (TP1–TP5), implying that 13.1% of the OTUs detected in the network were supplied by the feed water. 94.1% of the shared OTUs between IW and TP1–TP5 were assigned to Grantessa sp. Additional analysis showed that 96 OTUs were shared between the tap water samples (TP1–TP5) and the reverse osmosis permeates (RO1 and RO2) which represented 25.0% of the OTUs detected in the drinking water distribution system. 88 of these shared OTUs were assigned to the fungi phylum and 39 of the fungal OTUs were assigned to an uncultured fungus. The rest of the fungal OTUs belonged to Ascomycota and Basidiomycota phyla. This result points on the role of reverse osmosis treatment in the growth of fungi in the drinking water distribution system. On the other hand, 64 OTUs were shared between IW, SMPF and the tap water samples (TP1–TP5) representing 16.6% of the total OTUs detected in the drinking water distribution system. 48 of the shared OTUs were assigned to Grantessa sp. and only one OTU was assigned to the phylum fungi. A varying number of shared OTUs was found when comparing the tap water samples to each other (Table S1†). The highest number of shared OTUs was observed between TP2 and TP3 (61 OTUs).
3.3. Taxonomic diversity of the eukaryotes in the desalination plant
18S rRNA gene reads were assigned to 8 supergroups namely: Picozoa, SAR supergroup (Stramenopiles, Alveolates and Rhizaria), Metazoa, Chromalveolata, Fungi, Euglenozoa, Archaeplastida, Amoebozoa, and Opisthokonta, based on the revised classification of Eukaryotes31 (Fig. 5). After trimming and chimera removal, a 97% similarity cut-off was used to delineate OTUs in downstream analyses. Following subsampling, a total of 1773 eukaryotic OTUs were acquired. 28 eukaryotic phyla were identified in the desalination plant dominated by Ascomycota (15.6%), followed by Alveolata (11.9%), uncultured fungi phylum (10.9%), Porifera (10.7%), Stramenopiles (10.5%), and Bacillariophyta (8.4%) whereas Annelida, Mollusca, Nematoda, Chytridiomycota were found to be minor groups (<1%) (Fig. 2). In the network, 20 eukaryotic phyla were identified in the tap water samples dominated by the uncultured fungi phylum (44.1%), followed by Chordata (17.1%), Ascomycota (11.1%) and Arthropoda (8.6%) while Rotifera, Amoebozoa, Blastocladiomycota and Nematoda were minor groups in the tap water samples (<1%) (Fig. 2). Throughout the full drinking water system, the unclassified fungi phylum and Ascomycota dominated the total eukaryotic community with a relative abundance of 27.5% and 13.3%, respectively.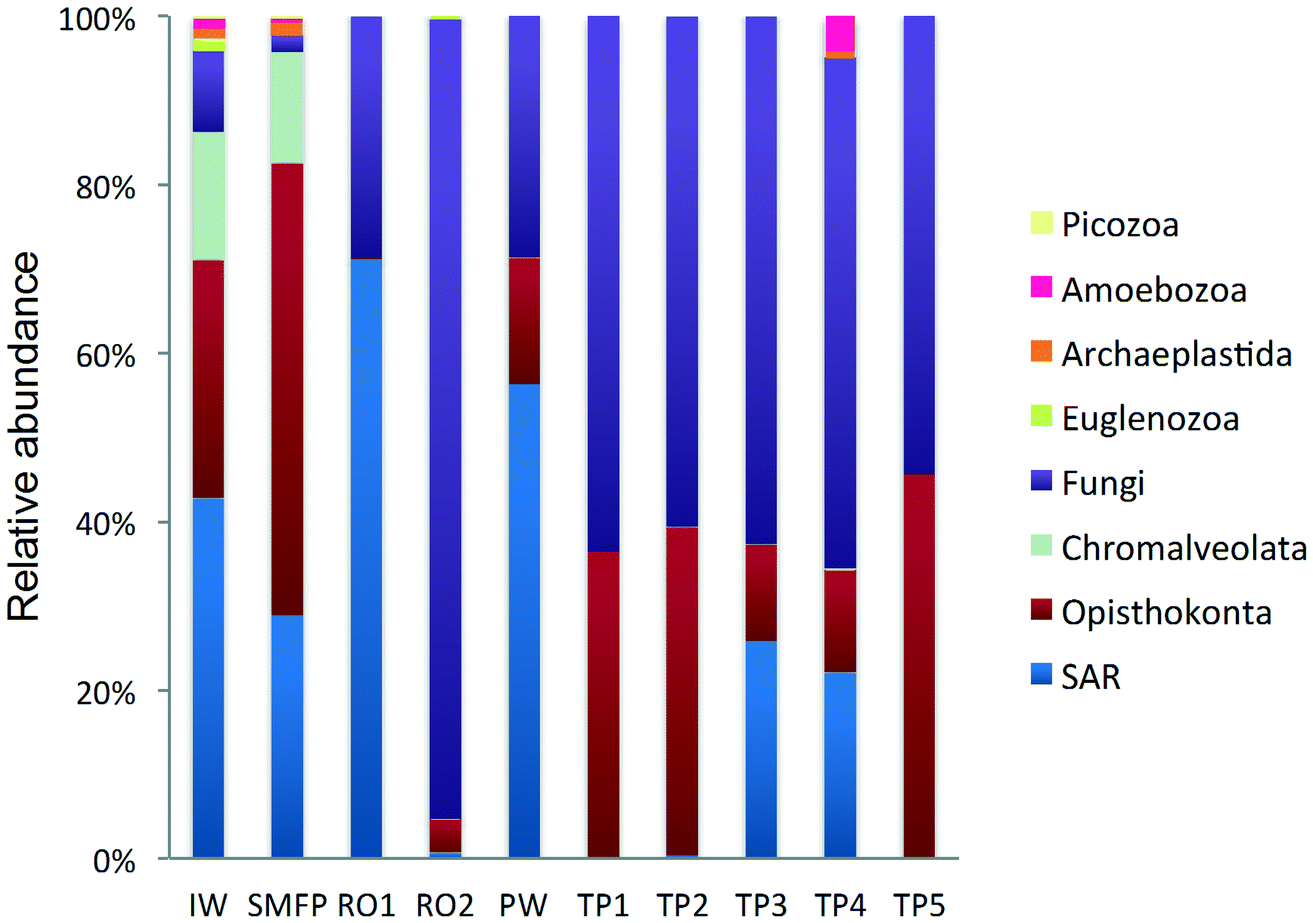 | ||
| Fig. 5 Relative abundance of the eukaryotic community (kingdom level) in KAUST water samples collected from IW: inlet water; SMFP: spruce media filter permeate; RO1: seawater reverse osmosis permeate; RO2: brackish water reverse osmosis permeate; PW: produced water; TP1–TP5: tap water samples; SAR: Stramenopiles, Alveolates and Rhizaria. | ||
In the inlet water feeding the multi-media filters, the eukaryotic community was dominated by the SAR and metazoan supergroups; only 9.7% of the sequences were assigned to fungi. The fungal community was represented by the Ascomycota, Basidiomycota, Chytridiomycota, Glomeromycota and Neocallimastigomycota phyla. After crossing the SMF, the eukaryotic community remained dominated by SAR and Metazoa while the relative abundance of fungi decreased to 2.1%. The same taxonomic composition of the fungal community was retrieved in the SMFP with the exception of Neocallimastigomycota which were absent within this permeate. Additionally an increase in the relative abundance of Ascomycota, Chytridiomycota and Glomeromycota was noted in the SMFP.
As for the rest of the eukaryotic community, Alveolata, Porifera, and Pinguiophyceae remained the major eukaryotic phyla in the filtered water (SMFP). The relative abundance of Porifera, represented by the class of Calcarea (Grantessa sp.), increased to 39.7% while the relative abundance of Stramenopiles, represented by the class of Dinophyceae, decreased to 12.7%. During this first filtration step, Chao 1 and Shannon diversity indices dropped by 19% and 17%, respectively.
After RO1 treatment in the desalination plant, the relative abundance of fungi increased to 28.5% (Fig. 5) and the fungal community was represented by the class of Eurotiomycetes (15.6%) (Fig. S4†) belonging to the phylum of Ascomycota and by an uncultured fungi clade (12.9%). Two dominant fungal species were identified; the Herpotrichiellaceae sp. lm294 and Exophiala aquamarina.
After RO2 treatment, the TOC concentration dropped to 4.1 μg L−1, water conductivity to 302 μS cm−1 and water turbidity to 0.2 NTU but alkaline conditions prevailed in this compartment (pH = 10.1) to enhance Boron removal. Fungi largely dominated in RO2 where they constituted 94.9% of the total eukaryotic community (Fig. 5). The taxonomic diversity of the fungi increased after RO2 with the detection of Basidiomycota fungi in addition to the fungal groups identified after RO1 treatment. After RO2 treatment, the Ascomycota were represented by the genera Aureobasidium, Dothidea, Cladosporium, Acephala, Penicillium and Exophiala while the Basidiomycota were represented by genera Bensingtonia, Cryptococcus, Trichosporon and Rhodotorula. The rest of the eukaryotic community (i.e. non-fungal community) was assigned to the metazoan phylum Chordata.
3.4. Taxonomic diversity of fungi in the desalination plant
To better understand the taxonomic diversity of the fungal community within the desalination plant, the fungal 18S rRNA gene sequences were analyzed separately from the rest of the eukaryotic community at the phylum (Fig. 3) and class levels (Fig. S4†). The fungal community (9.7%) identified in the inlet water of the desalination plant was of marine origin and was reported to play a diverse ecological role in marine ecosystems and has frequently been associated with parasitism of marine animals, plants and algae.33 Several Ascomycota and Chytridiomycota constitute parasitic agents of marine algae.34 Five fungal phyla were identified in the desalination plant: Ascomycota, Basidiomycota, Glomeromycota, Neocallimastigomycota and Chytridiomycota in addition to the unclassified fungi clade (Fig. 3). In the desalination plant, Ascomycota and the unclassified fungi clade had relatively equal abundance but in all tap water samples most of the sequences (69.4%) were assigned to the unclassified fungi clade (Fig. 3). Based on their relative abundance, Eurotiomycetes and Agaricomycetes were the most abundant classes of the Ascomycota and Basidiomycota groups in the desalination plant, respectively (Fig. S4†).3.5. Taxonomic diversity of fungi in the network
In the tap water samples, the fungal community was represented by five phyla: Ascomycota, Basidiomycota, Chytridiomycota, Blastocladiomycota and the unclassified fungi clade. The relative abundance and taxonomic composition of the fungal community varied within each tap water sample, as shown in Fig. 3 and S4:† Chytridiomycota were detected only in TP3, Basidiomycota were detected in TP1 and TP2, and Ascomycota in TP1, TP2 and TP3.At class level, Dothideomycetes and Tremellomycetes were the most abundant representatives of the Ascomycota and Basidiomycota groups, respectively. The Ascomycota in TP2 were represented by the class of Dothideomycetes and in TP3 by Dothideomycetes and Leotiomycetes. OTUs displaying high percentage of homology (≥97%) with known 18S rDNA sequences of the fungal species Cladosporium cladosporioides (Ascomycota, Dothideomycetes), shown to be linked with human infections7 (Table S2†) were identified within both tap water samples. Basidiomycota were represented by the class of Agaricomycetes in TP1 and TP5 and by Tremellomycetes in TP1. Chytridiomycota were represented by the class of Chytridiomycetes. At genus level, Ascomycota were represented by the dominant genus Cochliobolus, Basidiomycota were represented by genera Cryptococcus and Trichosporon, Chytridiomycota by genus Chytridium and Blastocladiomycota by the genus Catenaria.
3.6. Taxonomic diversity of non-fungal eukaryotes in the tap water samples
Sequences retrieved from the tap water samples were assigned to Metazoa, fungi, SAR and Amoebozoa groups. Remarkably, the fungi represented 60% of the total community and diverse metazoan groups were identified in tap water samples such as Arthropoda, Nematoda, Craniata, Calcarea, and Gastrotrichia. All eukaryotic groups detected in the different tap water samples were also identified in the produced water leaving the desalination plant (Fig. 2) suggesting that the eukaryotic community identified in the tap water samples originated from the plant water. Nevertheless, different eukaryotic communities were identified within each of the tap water samples. In TP1 and TP5, the Craniata class of the Chordata phylum was the unique eukaryotic group detected with Blarina sp. identified as the dominant species in both tap water samples. In addition, OTUs belonging to Nematoda and Arthropoda phyla (Table S2†), which are known to cause aesthetic problems in drinking water35 (such as insects), were detected in both tap water samples TP1 and TP5. In TP2, Arthropoda was represented by the class Maxillopoda, a diverse class of crustaceans where the most dominant taxonomic group was species Paramphiascella fulvofasciata. The taxonomic diversity of the eukaryotic community increased in TP3 and TP4. In TP3, the eukaryotic community was dominated by Coccidia (48%), an obligate intracellular parasite belonging to the phylum Apicomplexa and infecting the intestinal tracts of animals.36 In TP4, the eukaryotic community was the most diverse compared to the rest of the tap water samples, with representatives of the SAR supergroup; Arthropoda, Rotifera, Gastrotrichia and Amoebozoa present (Fig. 2). Unexpectedly, different eukaryotic communities were identified within each of the five tap water samples in the network despite being supplied by the same drinking water leaving the desalination plant.4. Discussion
454 pyrosequencing revealed highly diverse and variable eukaryotic communities in the water during (i) production of drinking water in a seawater desalination plant and during (ii) transport of the drinking water in the distribution network, including storage reservoirs, pipes and premise plumbing.4.1. Variation of diversity indices and community structure during treatment
During the desalination process, each of the explored treatment compartments was characterized by a unique eukaryotic community composition and structure that changed during treatment. These changes illustrate the effect of each treatment step in shaping the eukaryotic community and in influencing the produced chlorinated drinking water composition. Chlorination was not capable of preventing eukaryotes.The unexpected increase in the eukaryotic community diversity after the second reverse osmosis pass could not be attributed to reduced RO2 membrane performance (e.g. conductivity and TOC of the RO2 permeate were very low, Table 1) but most probably to contamination and/or microbial regrowth (e.g. by maintenance). In fact, a small fraction of the living microorganisms can and does pass through reverse osmosis membranes due to minor imperfections, or they bypass the membrane entirely through tiny leaks in surrounding seals (such as fungi as shown previously by Valster et al.37). Moreover, the presence of diverse alkalophilic and alkalotolerant fungi38 that thrive in high pH environments (i.e. Acremonium and Sodiomyces genera from Ascomycota phylum) supports the fungal diversity detected in RO2 (with pH = 10.1).
4.2. Interpretation of the taxonomic composition of the eukaryotic community
The microbial eukaryotic community identified in the spruce media filter inlet water (IW) of the desalination plant was a typical diverse oligotrophic community of small eukaryotes with representatives from most major marine picoeukaryotic phyla such as SAR supergroup. A similar community was described in an oligotrophic Red Sea coastal site.39,40 Within this community, the Alveolates and the Stramenopiles constitute an important component of marine ecosystems and include phototrophic, heterotrophic and mixotrophic organisms which can represent the main primary producers in surface waters. Metazoa also constituted a significant proportion of the eukaryotic community in the explored WTP. They are naturally present in many drinking water sources and have been detected in chlorinated and unchlorinated drinking water samples.2,12,37 These eukaryotes can survive within distribution systems by deriving their food from organic matter in the water and biofilms. Metazoa are also commonly present in deposits on pipe and tank surfaces. The presence of Metazoa in drinking water has largely been regarded as an “aesthetic” problem, however they may have a beneficial effect by limiting the growth potential of bacteria and by reducing the total microbial biomass.41 Fungi constituted an important eukaryotic group in the desalination plant (32.8%) and in the network (60.4%). Fungi have been reported as common microbial constituents of drinking water distribution systems42 and their occurrence is described in drinking water sources,43 bottled mineral water44 and tap water.45,46 Fungal presence in water may contribute to the degradation of complex natural and anthropogenic substances due to their broad enzymatic capabilities.47 However, fungi may be linked to taste and odor problems, skin irritations and allergic reactions, as well as the increased occurrence of opportunistic systematic mycosis in immune-compromised patients.45Fungi have been poorly recovered from marine surface waters48 as reported in this study, only 9% of the total eukaryotic community in the feed water of the spruce filter media (IW) belonged to fungi. Similar results have been found in marine environments.48,49 Furthermore, a similar fungal community has been described in the bulk water of a drinking water production plant.18 Both molecular analyses50,51 and culture-based inventories52 have shown that fungi are non-diverse and poorly recovered from surface marine waters.53,54 Richards et al.48 have reported ecological, morphological, and trophical limitations for studying fungi in marine environments. The dominance of Ascomycota and Basidiomycota in the inlet water of the desalination plant (feed of spruce filter) is in agreement with previous results suggesting that both fungal phyla are the most recovered marine fungal lineages.48,54–56
4.3. Influence of the different treatment steps on eukaryotic community structure
The role of the spruce media filter as the main pre-treatment process has been described previously.57 It is an improved mechanism of the regular media filter to remove solid matter by mechanical retention and surface adsorption. The SMF allows excellent removal by extracting and retaining over 99.954% of particle counts. In this study the SMF influenced the community structure by reducing all the diversity indices.The increase in the relative abundance of fungi from 2% (in SMF permeate) to 95% in RO2 reflected the importance and influence of reverse osmosis as a key step in shaping the ecology of fungi in the desalination plant. Fungi and bacteria are considered the prime fouling microorganisms.58 Numerous fungi have been found to participate in early biofilm development on RO membranes such as Penicillium, Trichoderma, Mucor and Aspergillus sp.59 Moreover, the dispersion and colonization mechanisms could have facilitated the increase in the relative abundance of fungi in RO2. Fungi have been shown to be able to grow on and degrade reverse osmosis membrane materials such as acetate cellulose.60,61 The high water pH (10.1) of RO2 could have favored the growth of Ascomycota which have alkaliphilic traits.62 Kladwang et al.63 have also demonstrated the existence of fungi able to tolerate high pH (alkaline-tolerant fungi) such as marine Ascomycota and Basidiomycota.
The community structure in the desalination plant produced drinking water (PW) was not only influenced by the structure of the eukaryotic communities in RO2 and the blending water (RO1 + RO2) but also by the chemical treatment (lime dosage) and the disinfection step. Some eukaryotic groups of the PW were present in RO1 and others in RO2 but new eukaryotic groups were also detected in the PW such as Arthropoda, Rhizaria and Apicomplexa.
Concerning the fungal community, the chlorination step affected the taxonomic composition of the fungal community and their relative abundance which deceased to 28.7%. The relative abundance of Ascomycota decreased significantly from 61.3% to <1% and that of Basidiomycota from 25.6% to 6.9%. In contrast, an increase in the relative abundance of the unclassified fungi clade was observed after the relative decline in the free chlorine concentration in the network during the water distribution process (Fig. 3).
Previous studies have shown that chlorination,12,37 the nature of the disinfectant (chlorine or chloramines), its residual concentration, and water age (contact time) are important factors in shaping the eukaryotic community structure in drinking water network biofilms.20 Kinsey et al.64 showed that the disinfection efficiency of chlorination against fungi is variable between species: some groups show high resistance to disinfection and others are protected from inactivation by the water matrix components. The chlorine efficiency depends on water temperature and exposure time.65 Based on the high resistance of fungi to chlorine,66 it has been suggested that an initial free chlorine concentration of approximately 1 mg L−1 is sufficient for spore inactivation, providing sufficient residual chlorine in the system to assist in the prevention of microbial growth and biofilm development.64
4.4. Eukaryotic communities in the network tap water samples
In the drinking water samples taken at 5 tap locations in the same drinking water distribution network (Fig. S1,† TP1–TP5), different eukaryotic communities were identified within each of the tap water samples. These differences may reflect the existence of a variety of microhabitats defined by complex environmental conditions existing in the distribution network (reservoir, pipes and premise plumbing) that affect the eukaryotic community composition and structure.Both the number of shared eukaryotic groups and their relative abundance varied between the PW (drinking water feeding the network) and each of the tap water samples in the network (except Gastrotrich detected only in TP4). In addition, the taxonomic diversity of the fungal community in the network was higher than observed in the PW (Fig. S4†). Results showed that the core community shared between the PW and the all combined tap water samples was composed of 84 OTUs (28.1%). The core community between the PW and each of the tap water samples varied between 0 (TP1) and 68 OTUs (42.8%) (TP4) (Table S1†). No relationship was observed between the number of shared OTUs and the distance separating the tap water from the desalination plant, thus the changes did not depend on the distance separating the desalination plant effluent from the tap water sampling locations. In addition, 71.5% of the total OTUs detected in the network were unique and differed from the core community in the PW. This finding indicates that the PW was not the main source of the eukaryotic community identified in the network. This ‘unique’ community in the network could come from biofilms or sediments within the network and/or could have been introduced in the distribution system during installation of the network and premise plumbing pipes or even due to contamination from the environment (e.g. by network maintenance operations). On the other hand, these core community members may also exist in the PW samples at concentrations below the detection limit. Another reason could be the water age whereby the water sampled in the network does not correspond to the same water collected at the desalination plant effluent on the same day. It could be that the previous water from the treatment plant had these OTUs. The network is relatively small with relative high water consumption suggesting that residence time of the water in the network is relatively short.
Previous studies showed that fungi, Metazoa, SAR and Amoebozoa are the most prevalent eukaryotes in DWDSs. Metazoa were reported as the dominant bulk water eukaryotic group in Chinese,21 Norwegian23 and French (Paris) drinking water samples.12 In this study, Metazoa and fungi constituted major groups in the produced chlorinated drinking water samples produced by seawater desalination by RO. Similar results have also been found in chlorinated drinking water systems produced by a seawater desalination plant in a Caribbean DWDS. Amoebozoa constituted a minor group in the produced chlorinated drinking water in our study, similar to the results obtained by Otterholt and Charnock23 in Norwegian chlorinated drinking water however Amoebozoa constituted the second major group in finished chlorinated drinking water samples from three Parisian surface water treatment plants.12 This eukaryotic group has been reported – with fungi – as a major constituent of eukaryotic biofilms in DWDS21,22 where it preys on bacteria.67
The persistence of the emergent uncultured fungi clade observed in the drinking water distribution system was not expected in a chlorinated and nutrient-limiting environment. This unclassified clade formed a major eukaryotic group which likely represents a new marine fungal clade. Le Calvez et al.52 reported that marine environments host numerous uncultured deep branching fungal forms and a significant number of highly novel groups. Most molecular techniques do not discriminate between live, dead, and viable non-culturable (VBNC) states of microorganisms, limiting a proper interpretation of the genetic/phylogenetic diversity with respect to the ecology and function of these uncultured sequences. Thus, attempting to culture these unknown fungi may provide key insight on their ecological and physiological significance in such engineered ecosystems.
Pinto et al.68 found that the bacterial community structure of the drinking water microbiome in the network was governed by filtration process. Their study involved conventional water treatment (involving dual media rapid filters) for a mix of fresh ground and surface water. The study from Pinto et al.68 differed strongly in water source and water treatment and they investigated bacterial communities. Our study did not show that the seawater filtration by RO membranes governed the fungal community composition and structure in the network. Assessing the factors determining the growth of both bacteria and eukaryotes in the distribution networks would require addition research to unravel the influence of water source, treatment type and operation, and distribution network characteristics.
4.5. Accuracy of the applied sequencing method
While the pyrosequencing method allowed sensitive and deep analysis of the eukaryotic community, there are some limitations that should be taken into account when interpreting these results. First, the DNA-based molecular approach does not differentiate between viable and non-viable eukaryotic microbial organisms or fragmented cells nor the active or inactive physiological state of the cell. Secondly, the water filtration process used to concentrate the eukaryotes present in the water samples was shown69 to affect the community composition. In addition, the eukaryotic cell membrane structure and composition particularly for fungi could induce biases in DNA extraction and/or in the PCR reaction.70 Several methods have been used to extract eukaryotic DNA such as CTAB (cetyl-trimethyl-ammonium bromide) based method71 or phenol extraction,72 but commercial kits (i.e. MoBio, FastDNA) have proven their efficiency in studying eukaryotic communities.73,74 Despite 18S rRNA gene sequencing being an efficient and powerful tool for profiling the biodiversity of eukaryotic communities in natural aquatic (marine and fresh)75,76 and engineered ecosystems,19–21 its use as a unique molecular marker for the phylogenetic assignment of fungi is still controversial. In fact ITS sequences have been shown to be more taxonomically informative than 18S rRNA sequences in DNA-based studies.77 Although this high-throughput sequencing method has provided sufficient information for in-depth phylogenetic analyses of certain eukaryote groups, further investigation is needed concerning the use of 18S for phylogenetic assignment of fungi.78 The 18S rRNA gene has fewer hyper-variable domains in fungi and some regions of fungal 18S rRNA genes share high similarity with other eukaryotes.79 Moreover, several biases related to the specificity of 18S primers designed for fungi have been reported.80 With the emergence of fungi as a major microbial group in the assessed DWDS, the need for a second marker such as internal transcribed spacer (ITS) – proposed as the official primary barcoding marker for fungi81,82 in combination with a small subunit (SSU) marker – is necessary to achieve a better understanding of the taxonomic and phylogenetic composition of the fungal community in drinking water distribution systems83 and their implications on the drinking water quality. This can help determine the biological significance of the eukaryotic organisms detected within the drinking water system.4.6. Nature, growth and origin of fungi
In this study no discrimination could be made as to which fraction of the fungi was vegetative or spores. No odor-related problems have been reported, possibly suggesting the dominant presence of fungal spores.Fungi can grow on biodegradable organics originating from the membrane84 and other membrane module materials, e.g. the module seals. Microbial growth also could occur on the permeate side of the membrane.85 A membrane system is by definition not sterile because during the membrane module manufacturing and loading of the membrane elements into the membrane filtration installation microbial contamination can occur. Moreover, bacteria, fungi and other microorganisms can grow back into the membrane system. Most materials can leach out biodegradable compounds (e.g. plasticizers) and fungi can degrade polymers. It is possible that fungi in the network water originated from pipe sediments and biofilm. In summary, a variety of reasons may explain the presence of fungi in membrane systems and the produced water.
The microbial community shifts during the treatment process can be (i) generated by the selective pressure of treatment processes and also be (ii) caused by different contaminations at the individual treatment steps and (iii) caused by the combination thereof, with microbial population compositions independently developing, which might be a purely stochastic process. In the distribution network similar selective pressures and contaminations may play a role as well. In this study, no conclusive explanations for the occurrence of the respective fungal population shifts in the treatment plant and the distribution network can be given. Possibly, a similar study at another time period or at another location may yield different eukaryotic communities; temporal and spatial conditions affect microbial community dynamics.86,87
The presence of fungi is grossly underestimated in the drinking water world and this study revealed their presence very clearly. There is a need for follow-up studies to provide quantitative data on the presence of bacteria and fungi during drinking water production and distribution.
Conclusions
This study explored the taxonomic and phylogenetic diversity of the microbial eukaryotic community in water during the production of drinking water in a seawater desalination plant and during transport of the drinking water in the distribution network, including reservoirs, pipes and premise plumbing. Results revealed a highly diverse community containing fungi which constituted a dominant group in the water during the reverse osmosis treatment steps and in all tap water samples in the network. Moreover, 71.5% of the eukaryotic community in the network was not found in the produced drinking water supplying the network, implying a shift in the eukaryotic community composition during distribution in the network.Reverse osmosis membrane treatment significantly changed the water eukaryotic community composition and structure, highlighting the fact that (i) reverse osmosis produced water is not sterile and (ii) the microbial community in the tap water is influenced by the downstream distribution system. The emergence of fungi as a major group in the drinking water distribution system suggests that these microorganisms need special attention.
Abbreviations
| BWRO | Brackish water reverse osmosis |
| DWDS | Drinking water distribution system |
| IW | SMF inlet water |
| LSI | Langelier saturation index |
| NTU | Nephelometric turbidity unit |
| OTU | Operational taxonomic unit |
| PCoA | Principal coordinate analysis |
| PVC | Polyvinyl chloride |
| PW | Produced water |
| RO | Reverse osmosis |
| RO1 | Seawater reverse osmosis permeate |
| RO2 | Brackish water reverse osmosis permeate |
| SBS | Sodium bisulphite |
| SMF | Spruce multi-media filter |
| SMFP | Spruce media filter permeate |
| SWRO | Seawater reverse osmosis |
| WTP | Water treatment plant |
Acknowledgements
The research reported in this paper was supported by King Abdullah University of Science and Technology (KAUST), Saudi Arabia: Office of Competitive Research Funds (OCRF) under Award No. URF/1/1728-01-01 as well as Evides Waterbedrijf, The Netherlands. The authors would like to thank the KAUST desalination plant staff for their technical assistance.References
- J. F. Loret and G. Greub, Int. J. Hyg. Environ. Health, 2010, 213, 167–175 CrossRef CAS PubMed
.
- R. M. Valster, B. A. Wullings, G. Bakker, H. Smidt and D. van der Kooij, Appl. Environ. Microbiol., 2009, 75, 4736–4746 CrossRef CAS PubMed
- L. Heng, Y. L. Yanling, G. J. Weijia, L. Xing and G. B. Li, Desalination, 2008, 222, 74–80 CrossRef CAS
- S. J. Hoeger, B. C. Hitzfeld and D. R. Dietrich, Toxicol. Appl. Pharmacol., 2005, 203, 231–242 CrossRef CAS PubMed
- J. H. M. van Lieverloo, D. W. Bosboom, G. L. Bakker, A. J. Brouwer, R. Voogt and J. E. M. De Roos, Water Res., 2004, 38, 1101–1112 CrossRef CAS PubMed
- E. Wolmarans, H. H. du Preez, C. M. E. de Wet and S. N. Venter, Water Sci. Technol., 2005, 52, 171–175 CAS
- S. Hurtado-McCormick, L. Sanchez, J. Martinez, C. Calderon, D. Calvo, D. Narvaez, M. Lemus, H. Groot and M. R. Susa, Water Sci. Technol.: Water Supply, 2016, 16, 905–914 CrossRef
- G. Hageskal, N. Lima and I. Skaar, Mycol. Res., 2009, 113, 165–172 CrossRef PubMed
- L. A. Nagy and B. H. Olson, Can. J. Microbiol., 1982, 28, 667–671 CrossRef CAS PubMed
- V. J. Pereira, D. Fernandes, G. Carvalho, M. J. Benoliel, M. V. S. Romao and M. T. B. Crespo, Water Res., 2010, 44, 4850–4859 CrossRef CAS PubMed
- V. M. Siqueira, H. Oliveira, C. Santos, R. R. M. Paterson, N. B. Gusmão and N. Lima, Int. J. Environ. Res. Public Health, 2011, 8(2), 456–469 CrossRef CAS PubMed
- J. B. Poitelon, M. Joyeux, B. Welte, J. P. Duguet, J. Peplies and M. S. DuBow, Lett. Appl. Microbiol., 2009, 49, 589–595 CrossRef CAS PubMed
- F. Marciano-Cabral, M. Jamerson and E. S. Kaneshiro, J. Water Health, 2010, 8, 71–82 CrossRef CAS PubMed
- R. Y. Liu, J. G. Zhu, Z. S. Yu, D. Joshi, H. X. Zhang, W. F. Lin and M. Yang, J. Environ. Sci., 2014, 26, 865–874 CrossRef CAS
- E. Lupi, V. Ricci and D. Burrini, J. Water Supply: Res. Technol.--AQUA, 1995, 44, 212–218 Search PubMed
- M. W. Kuiper, B. A. Wullings, A. D. L. Akkermans, R. R. Beumer and D. van der Kooij, Appl. Environ. Microbiol., 2004, 70, 6826–6833 CrossRef CAS PubMed
- H. Y. Buse, J. Lu, I. T. Struewing and N. J. Ashbolt, Environ. Sci. Pollut. Res., 2013, 20, 6351–6366 CrossRef CAS PubMed
- W. Lin, Z. Yu, H. Zhang and I. P. Thompson, Water Res., 2014, 52, 218–230 CrossRef CAS PubMed
- H. Wang, S. Masters, M. A. Edwards, J. O. Falkinham and A. Pruden, Environ. Sci. Technol., 2014, 48, 1426–1435 CrossRef CAS PubMed
- H. Y. Buse, J. Lu, X. Lu, X. Mou and N. J. Ashbolt, FEMS Microbiol. Ecol., 2014, 88, 280–295 CrossRef CAS PubMed
- R. Liu, Z. Yu, H. Guo, M. Liu, H. Zhang and M. Yang, Sci.
Total Environ., 2012, 435–436, 124–131 CrossRef CAS PubMed
- R. M. Valster, B. A. Wullings and D. Van Der Kooij, Appl. Environ. Microbiol., 2010, 76, 7144–7153 CrossRef CAS PubMed
- E. Otterholt and C. Charnock, Water Res., 2011, 45, 2527–2538 CrossRef CAS PubMed
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, C. A. Lozupone, P. J. Turnbaugh, N. Fierer and R. Knight, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4516–4522 CrossRef CAS PubMed
- A. Belila, J. El-Chakhtoura, N. Otaibi, G. Muyzer, G. Gonzalez-Gil, P. E. Saikaly, M. C. M. van Loosdrecht and J. S. Vrouwenvelder, Water Res., 2016, 94, 62–72 CrossRef CAS PubMed
- APHA/ AWWA/ WEF, Standard Methods for the Examination of Water and Wastewater, American Public Health Association/ American Water Works Association/ Water Environment Federation, 1998 Search PubMed.
- R. Logares, S. Audic, S. Santini, M. C. Pernice, C. De Vargas and R. Massana, ISME J., 2012, 6, 1823–1833 CrossRef CAS PubMed
- Y. Vázquez-Baeza, M. Pirrung, A. Gonzalez and R. Knight, EMPeror: a tool for visualizing high-throughput microbial community data, GigaScience, 2013, 2, 16 CrossRef PubMed
-
J. C. Oliveros, http://bioinfogp.cnb.csic.es/tools/venny/index.html, 2007
- Guidelines for drinking-water quality, World Health Organization, 2011 Search PubMed.
- S. M. Adl, A. G. B. Simpson, C. E. Lane, J. Lukeš, D. Bass, S. S. Bowser, M. W. Brown, F. Burki, M. Dunthorn, V. Hampl, A. Heiss, M. Hoppenrath, E. Lara, L. L. Gall, D. H. Lynn, H. McManus, E. A. D. Mitchell, S. E. Mozley-Stanridge, L. W. Parfrey, J. Pawlowski, S. Rueckert, L. Shadwick, C. L. Schoch, A. Smirnov and F. W. Spiegel, J. Eukaryotic Microbiol., 2012, 59, 429–493 CrossRef PubMed
- A. Kamp, D. De Beer, J. L. Nitsch, G. Lavik and P. Stief, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5649–5654 CrossRef CAS PubMed
- T. G. Jephcott, C. Alves-de-Souza, F. H. Gleason, F. F. van Ogtrop, T. Sime-Ngando, S. A. Karpov and L. Guillou, Fungal Ecol., 2016, 19, 47–58 CrossRef
- G. C. Srivastava, Hydrobiologia, 1967, 30, 385–404 CrossRef
-
S. C. B. Christensen, Asellus aquaticus and other invertebrates in drinking water distribution systems – occurrence and influence on microbial water quality, PhD thesis, Technical University of Denmark, 2011 Search PubMed
- H. D. Chapman, Poult. Sci., 2014, 93, 501–511 CrossRef CAS PubMed
- R. M. Valster, B. A. Wullings, R. van den Berg and D. van der Kooij, Appl. Environ. Microbiol., 2011, 77, 7321–7328 CrossRef CAS PubMed
- A. A. Grum-Grzhimaylo, A. J. M. Debets, A. D. van Diepeningen, M. L. Georgieva and E. N. Bilanenko, Persoonia, 2013, 31, 147–158 CrossRef CAS PubMed
- F. Acosta, D. K. Ngugi and U. Stingl, Aquat. Biosyst., 2013, 9, 16, DOI:10.1186/2046-9063-9-16
- R. Massana, Annu. Rev. Microbiol., 2011, 65, 91–110 CrossRef CAS PubMed
- R. W. Sanders and S. A. Wickham, Mar. Microb. Food Webs, 1993, 7, 197–223 Search PubMed
- G. Hageskal, T. Vrålstad, A. K. Knutsen and I. Skaar, Mol. Ecol. Resour., 2008, 8, 1178–1188 CrossRef CAS PubMed
- V. J. Pereira, M. C. Basílio, D. Fernandes, M. Domingues, J. M. Paiva, M. J. Benoliel, M. T. Crespo and M. V. San Romão, Water Res., 2009, 43, 3813–3819 CrossRef CAS PubMed
- M. U. Yamaguchi, R. D. C. P. Rampazzo, S. F. Yamada-Ogatta, C. V. Nakamura, T. Ueda-Nakamura and B. P. Dias Filho, Braz. Arch. Biol. Technol., 2007, 50, 1–9 CrossRef CAS
- G. Hageskal, A. K. Knutsen, P. Gaustad, G. S. De Hoog and I. Skaar, Appl. Environ. Microbiol., 2006, 72, 7586–7593 CrossRef CAS PubMed
- G. Hageskal, P. Gaustad, B. T. Heier and I. Skaar, J. Appl. Microbiol., 2007, 102, 774–780 CrossRef CAS PubMed
- C. Junghanns, M. Moeder, G. Krauss, C. Martin and D. Schlosser, Microbiology, 2005, 151, 45–57 CrossRef CAS PubMed
- T. A. Richards, M. D. M. Jones, G. Leonard and D. Bass, Annu. Rev. Mar. Sci., 2012, 4, 495–522 CrossRef PubMed
- T. A. Richards and D. Bass, Curr. Opin. Microbiol., 2005, 8, 240–252 CrossRef CAS PubMed
- T. Kis-Papo, I. Grishkan, A. Oren, S. P. Wasser and E. Nevo, Mycol. Res., 2001, 105, 749–756 CrossRef
- G. Burgaud, T. Le Calvez, D. Arzur, P. Vandenkoornhuyse and G. Barbier, Environ. Microbiol., 2009, 11, 1588–1600 CrossRef PubMed
- T. Le Calvez, G. Burgaud, S. Mahé, G. Barbier and P. Vandenkoornhuyse, Appl. Environ. Microbiol., 2009, 75, 6415–6421 CrossRef CAS PubMed
- V. P. Edgcomb, D. Beaudoin, R. Gast, J. F. Biddle and A. Teske, Environ. Microbiol., 2011, 13, 172–183 CrossRef CAS PubMed
- S. J. Cathrine and C. Raghukumar, Mycol. Res., 2009, 113, 100–109 CrossRef PubMed
- R. Massana and C. Pedrós-Alió, Curr. Opin. Microbiol., 2008, 11, 213–218 CrossRef PubMed
- T. Stoeck, D. Bass, M. Nebel, R. Christen, M. D. M. Jones, H. W. Breiner and T. A. Richards, Mol. Ecol., 2010, 19, 21–31 CrossRef CAS PubMed
- R. M. Rachman, N. Ghaffour, F. Wali and G. L. Amy, Desalin. Water Treat., 2013, 51, 1091–1103 CrossRef CAS
- J. S. Baker and L. Y. Dudley, Desalination, 1998, 118, 81–90 CrossRef CAS
- V. J. Pereira, R. Marques, M. Marques, M. J. Benoliel and M. T. Barreto Crespo, Water Res., 2013, 47, 517–523 CrossRef CAS PubMed
- A. P. Murphy, C. D. Moody, R. L. Riley, S. W. Lin, B. Murugaverl and P. Rusin, J. Membr. Sci., 2001, 193, 111–121 CrossRef CAS
- L. C. W. Ho, D. D. Martin and W. C. Lindemann, Appl. Environ. Microbiol., 1983, 45, 418–427 CAS
- A. A. Grum-Grzhimaylo, M. L. Georgieva, S. A. Bondarenko, A. J. M. Debets and E. N. Bilanenko, Fungal Divers., 2016, 76, 27–74 CrossRef
- W. Kladwang, A. Bhumirattana and N. Hywel-Jones, Fungal Divers., 2003, 13, 69–84 Search PubMed
-
G. Kinsey, R. Paterson and J. Kelley, in Handbook of Water and Wastewater Microbiology, 2003, pp. 77–98, DOI:10.1016/B978-012470100-7/50006-6
-
R. R. M. Paterson and N. Lima, in Water Encyclopedia, John Wiley & Sons, Inc., 2005, DOI:10.1002/047147844X.wq1516
- R. P. J. Kelley, G. Kinsey, R. Pitchers and H. Rossmoore, Water Technology Conference Proceedings, 1997 Search PubMed.
- V. Delafont, A. Brouke, D. Bouchon, L. Moulin and Y. Héchard, Water Res., 2013, 47, 6958–6965 CrossRef CAS PubMed
- A. J. Pinto, C. Xi and L. Raskin, Environ. Sci. Technol., 2012, 46, 8851–8859 CrossRef CAS PubMed
- N. Sørensen, N. Daugbjerg and K. Richardson, Microb. Ecol., 2013, 65, 964–968 CrossRef PubMed
- C. Berney, J. Fahrni and J. Pawlowski, How many novel eukaryotic “kingdoms”? Pitfalls and limitations of environmental DNA surveys, BMC Biol., 2004, 2, 13 CrossRef PubMed
- A. Lanzén, A. Simachew, A. Gessesse, D. Chmolowska, I. Jonassen and L. Øvreås, Surprising Prokaryotic and Eukaryotic Diversity, Community Structure and Biogeography of Ethiopian Soda Lakes, PLoS One, 2013, 8(8), e72577 Search PubMed
- K. B. Heidelberg, W. C. Nelson, J. B. Holm, N. Eisenkolb, K. Andrade and J. B. Emerson, Characterization of eukaryotic microbial diversity in hypersaline Lake Tyrrell, Australia, Front. Microbiol., 2013, 4, 115 CrossRef PubMed
- C. Bachy, J. R. Dolan, P. López-García, P. Deschamps and D. Moreira, ISME J., 2013, 7, 244–255 CrossRef CAS PubMed
- P. A. Schmidt, M. Bálint, B. Greshake, C. Bandow, J. Römbke and I. Schmitt, Soil Biol. Biochem., 2013, 65, 128–132 CrossRef CAS
- A. Zhan, S. A. Bailey, D. D. Heath and H. J. Macisaac, Mol. Ecol. Resour., 2014, 14, 1049–1059 CAS
- H. M. Bik, D. L. Porazinska, S. Creer, J. G. Caporaso, R. Knight and W. K. Thomas, Trends Ecol. Evol., 2012, 27, 233–243 CrossRef PubMed
- I. C. Anderson and P. I. Parkin, J. Microbiol. Methods, 2007, 68, 248–253 CrossRef CAS PubMed
- S. Monchy, G. Sanciu, M. Jobard, S. Rasconi, M. Gerphagnon, M. Chabé, A. Cian, D. Meloni, N. Niquil, U. Christaki, E. Viscogliosi and T. Sime-Ngando, Environ. Microbiol., 2011, 13, 1433–1453 CrossRef PubMed
- C. L. Schoch, K. A. Seifert, S. Huhndorf, V. Robert, J. L. Spouge, C. A. Levesque, W. Chen, E. Bolchacova, K. Voigt, P. W. Crous, A. N. Miller, M. J. Wingfield, M. C. Aime, K. D. An, F. Y. Bai, R. W. Barreto, D. Begerow, M. J. Bergeron, M. Blackwell, T. Boekhout, M. Bogale, N. Boonyuen, A. R. Burgaz, B. Buyck, L. Cai, Q. Cai, G. Cardinali, P. Chaverri, B. J. Coppins, A. Crespo, P. Cubas, C. Cummings, U. Damm, Z. W. de Beer, G. S. de Hoog, R. Del-Prado, B. Dentinger, J. Diéguez-Uribeondo, P. K. Divakar, B. Douglas, M. Dueñas, T. A. Duong, U. Eberhardt, J. E. Edwards, M. S. Elshahed, K. Fliegerova, M. Furtado, M. A. García, Z. W. Ge, G. W. Griffith, K. Griffiths, J. Z. Groenewald, M. Groenewald, M. Grube, M. Gryzenhout, L. D. Guo, F. Hagen, S. Hambleton, R. C. Hamelin, K. Hansen, P. Harrold, G. Heller, C. Herrera, K. Hirayama, Y. Hirooka, H. M. Ho, K. Hoffmann, V. Hofstetter, F. Högnabba, P. M. Hollingsworth, S. B. Hong, K. Hosaka, J. Houbraken, K. Hughes, S. Huhtinen, K. D. Hyde, T. James, E. M. Johnson, J. E. Johnson, P. R. Johnston, E. B. G. Jones, L. J. Kelly, P. M. Kirk, D. G. Knapp, U. Kõljalg, G. M. Kovács, C. P. Kurtzman, S. Landvik, S. D. Leavitt, A. S. Liggenstoffer, K. Liimatainen, L. Lombard, J. J. Luangsa-Ard, H. T. Lumbsch, H. Maganti, S. S. N. Maharachchikumbura, M. P. Martin, T. W. May, A. R. McTaggart, A. S. Methven, W. Meyer, J. M. Moncalvo, S. Mongkolsamrit, L. G. Nagy, R. H. Nilsson, T. Niskanen, I. Nyilasi, G. Okada, I. Okane, I. Olariaga, J. Otte, T. Papp, D. Park, T. Petkovits, R. Pino-Bodas, W. Quaedvlieg, H. A. Raja, D. Redecker, T. L. Rintoul, C. Ruibal, J. M. Sarmiento-Ramírez, I. Schmitt, A. Schüßler, C. Shearer, K. Sotome, F. O. P. Stefani, S. Stenroos, B. Stielow, H. Stockinger, S. Suetrong, S. O. Suh, G. H. Sung, M. Suzuki, K. Tanaka, L. Tedersoo, M. T. Telleria, E. Tretter, W. A. Untereiner, H. Urbina, C. Vágvölgyi, A. Vialle, T. D. Vu, G. Walther, Q. M. Wang, Y. Wang, B. S. Weir, M. Weiß, M. M. White, J. Xu, R. Yahr, Z. L. Yang, A. Yurkov, J. C. Zamora, N. Zhang, W. Y. Zhuang and D. Schindel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6241–6246 CrossRef CAS PubMed
- J. Borneman and R. J. Hartin, Appl. Environ. Microbiol., 2000, 66, 4356–4360 CrossRef CAS PubMed
- E. Bellemain, T. Carlsen, C. Brochmann, E. Coissac, P. Taberlet and H. Kauserud, ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases, BMC Microbiol., 2010, 10, 189 CrossRef PubMed
- B. D. Lindahl, R. H. Nilsson, L. Tedersoo, K. Abarenkov, T. Carlsen, R. Kjøller, U. Kõljalg, T. Pennanen, S. Rosendahl, J. Stenlid and H. Kauserud, New Phytol., 2013, 199, 288–299 CrossRef CAS PubMed
- T. Větrovský, M. Kolařík, L. Žifčáková, T. Zelenka and P. Baldrian, Mol. Ecol. Resour., 2016, 16, 388–401 CrossRef PubMed
- U. Klun, J. Friedrich and A. Kržan, Polym. Degrad. Stab., 2003, 79, 99–104 CrossRef CAS
- S. K. Park and J. Y. Hu, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2010, 45, 968–977 CrossRef CAS PubMed
- E. I. Prest, D. G. Weissbrodt, F. Hammes, M. C. M. van Loosdrecht and J. S. Vrouwenvelder, PLoS One, 2016, 11, e0164445 CAS
- J. El-Chakhtoura, E. Prest, P. Saikaly, M. van Loosdrecht, F. Hammes and H. Vrouwenvelder, Water Res., 2015, 74, 180–190 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ew00265j |
| This journal is © The Royal Society of Chemistry 2017 |