
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase-catalysed, one-step mechanochemical conversion of chitin and shrimp shells into low molecular weight chitosan†
Xi
Chen
a,
Huiying
Yang
a,
Ziyi
Zhong
b and
Ning
Yan
 *a
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Blk E5, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: ning.yan@nus.edu.sg
bInstitute of Chemical and Engineering Sciences, A*STAR, 1 Pesek Road, Jurong Island, Singapore 627833, Singapore
First published on 2nd March 2017
Abstract
A facile, solid-state mechanochemical method was developed for the synthesis of low molecular weight chitosan (LMWC) in one step from chitin and crude shrimp shell powders, in which chitin undergoes simultaneous deacetylation and depolymerisation in the presence of a base catalyst under mechanical milling. The method is advantageous over traditional multi-step methods, featuring enhanced efficiency and significantly reduced environmental impact. Compared to traditional approaches, the base usage is reduced to about 1/10, and the molecular weight of the obtained LMWC product is much more narrowly distributed, with a polydispersity value of only 1.1. The degree of deacetylation and the molecular weight can be adjusted by varying the ball milling parameters. The influences of different types of bases were investigated by solid-state NMR analysis and control experiments, pointing out the critical role of base in both depolymerisation and deacetylation. Base-catalysed mechanochemical transformation of chitin and shrimp shells provides a solvent-free way to effectively valorize shellfishery waste for valuable products.
Introduction
Various biomass resources represent an important renewable feedstock for chemicals and materials.1–4 Lignocellulosic materials and lipids have been widely investigated,5–10 and considerable attention has been drawn to other types of biomass in recent years.11,12 Chitin is the world's second most abundant biopolymer next to cellulose, widely existing in nature (crustacean shells, the skeletons of insects, fungi, etc.) with a global annual production of about 100 billion tons.13 Recently, the concept of shell biorefinery has been proposed,14,15 and is developing rapidly.16–26 In this concept, key components in crustacean shells such as chitin are fractionated via environmentally friendly protocols, following which, each fraction is further upgraded and utilized enabling an integrated process to produce value-added chemicals from shellfish waste.The use of chitin derivatives as functional polymers in material synthesis has a long history and still attracts considerable scientific interest.27–29 Chitosan, which is generally defined by a degree of deacetylation (DD) beyond 50%, is the most important chitin derivative with a number of applications and a global market of USD 1.5 billion in 2015.30–32 In particular, low molecular weight chitosan (LMWC) exhibits enhanced solubility and can be even dissolved in neutral water for direct use under physiological conditions.33 Additionally, it has more prominent antibacterial, antitumor, and immuno-enhancing activities than chitin and high molecular weight chitosan (HMWC), making it attractive in both biomedical and pharmaceutical applications.34–38 For instance, LMWC with an average molecular weight (MW) between 5 and 10 kDa is specifically suitable for DNA delivery showing more enhancement in biological activities than chitosan.39 LMWC is also superior to HMWC and chitin in material synthesis40–42 because of the remarkably improved reactivity in grafting, crosslinking, and other types of modifications. LMWC (2 to 6.5 kDa) was adopted as a pH-sensitive coating agent for tumor-specific drug delivery leading to the formation of smaller nanoparticles than using HMWC,43 while chitosan-alginate films using LMWC exhibited a reduced thickness and enhanced transparency compared to HMWC.
Several methods have been reported for the synthesis of LMWC from chitin, which always involve two separate steps including deacetylation followed by depolymerisation (see Fig. 1, top). Deacetylation is notorious for the use of a large amount of concentrated, corrosive basic solutions. A common protocol is to heat chitin in 40 wt% sodium hydroxide solution at 130 °C for 1 to 4 h, with a low chitin concentration (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 to 1:100 mass ratio).44 Environmental issues and elevated capital costs arise due to the handling for wastewater treatment, as well as special requirements for equipment and operation. Amended approaches such as microwave-coupled base treatment,45,46 multiple freeze–pump out-thaw methods,47 compression methods,48etc. have been proposed in the literature, but none of them are able to promote deacetylation and depolymerisation simultaneously. As such, a second step for depolymerisation using hydrochloric acid,49 hydrogen peroxide,50–52 nitrous acid,53 potassium persulfate,35 high-energy ultrasonication54 or plasma treatment55 is needed. Catalytic depolymerisation using enzymes56–60 has also been reported, but the procedure is costly and requires a long period of time.
:50 to 1:100 mass ratio).44 Environmental issues and elevated capital costs arise due to the handling for wastewater treatment, as well as special requirements for equipment and operation. Amended approaches such as microwave-coupled base treatment,45,46 multiple freeze–pump out-thaw methods,47 compression methods,48etc. have been proposed in the literature, but none of them are able to promote deacetylation and depolymerisation simultaneously. As such, a second step for depolymerisation using hydrochloric acid,49 hydrogen peroxide,50–52 nitrous acid,53 potassium persulfate,35 high-energy ultrasonication54 or plasma treatment55 is needed. Catalytic depolymerisation using enzymes56–60 has also been reported, but the procedure is costly and requires a long period of time.
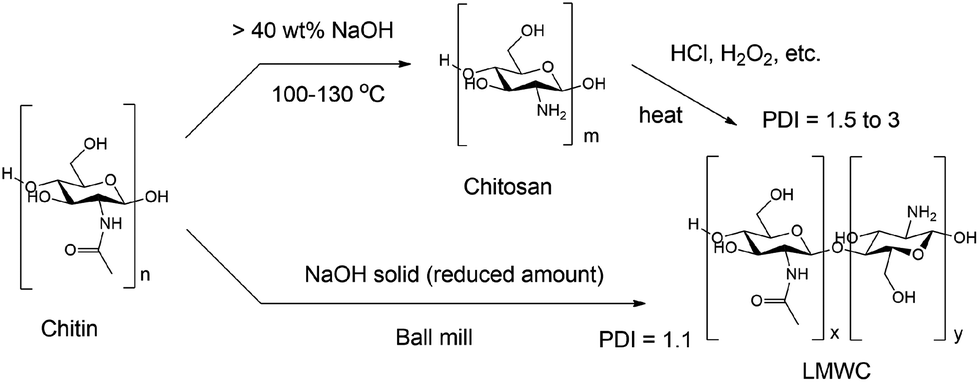 | ||
| Fig. 1 The reaction scheme of the traditional, two-step method (top) and the proposed one-step, mechanochemical method (bottom). Note that chitin and chitosan actually possess both NAG and glucosamine units. Only one representative unit is plotted because it is the dominant unit. The commercial chitin we use has a low DD value of 3.5% and the DD values of Chitosan-C and Chitosan-H are around 80%. | ||
Recently, mechanochemistry has been widely applied in chemical and material synthesis with the unique advantage of enabling efficient solid-state reactions and the ability to directly convert various recalcitrant feedstocks.61–74 A series of studies transforming biomass in a solvent-free way have been demonstrated. For example, cellulose was converted to water-soluble oligomers by both mineral acids and solid acids in a ball milling process,75,76 while lignin was decomposed similarly in the presence of a base.77 Yabushita et al.78 reported ball mill enhanced depolymerisation of acid-impregnated chitin into acetylated oligomers, and the subsequent transformation into N-acetyl glucosamine (NAG) in a separate step. Inspired by these studies, we envisaged the possibility of one-step conversion of chitin and raw shrimp shells into LMWC in the solid state by utilizing a synergistic mechanical force and base catalysis. The key assumption is that ball milling could significantly and simultaneously enhance base-catalysed depolymerisation and deacetylation of chitin.
We report here the studies along this line. LMWC was generated directly from chitin and shrimp shells in the presence of a base under ball milling conditions. Combined techniques including Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), gel permeation chromatography (GPC), matrix-assisted laser desorption ionization-time of flight (MALDI-TOF), electrospray ionisation mass spectrometry (ESI-MS) and solid-state nuclear magnetic resonance (NMR) analysis were used to fully characterize the products, and to understand the reaction pathways and roles of bases. Remarkably, raw shrimp shells were successfully transformed into LMWC with a high purity of ca. 90% using the protocol developed. The new approach is straightforward and solvent-free (Fig. 1, bottom) which obviates the generation of wastewater and the use of oxidants, acids, or other environment-unfriendly reagents. The base usage is sharply reduced to ca. 1/10 compared to traditional methods.
Experimental
Chemicals and materials
Chitin (92% purity, high molecular weight) was purchased from Wako Pure Chemical Industry. NaOH was from Schedelco. Acetic acid (HAc) was from Merck. Sodium borohydride (NaBH4) was from Alfa Aesar. A pullulan polysaccharide calibration kit was bought from Agilent Technologies. Barium hydroxide (Ba(OH)2·8H2O) was from Sinopharm Chemical Reagent Company. Boric acid was from Amresco. Commercial chitosan (denoted as Chitosan-C, >75% DD, >200 kDa, from shrimp shell), lithium hydroxide (LiOH), potassium hydroxide (KOH), calcium hydroxide (Ca(OH)2), sodium acetate (NaAc), lithium chloride (LiCl), deuterium water (D2O), NAG, methanol, dimethylacetamide (DMA) and other chemicals were purchased from Sigma Aldrich. Chitosan-H denotes the homemade chitosan from chitin, which is obtained by suspending 1 g of chitin in 20 mL of 45 wt% NaOH solution, which was stirred and heated at 120 °C for 3 h. The mixture was centrifuged, and Chitosan-H was obtained as a light-yellow solid after washing and drying.General procedure for the mechanochemical process
The ball milling machine is a Pulverisette P7 Premium Line (Fritsch) with a chamber (made of zirconium oxide (ZrO2)) volume of 45 mL and the balls (ZrO2) have a diameter of 5 mm. In a typical procedure of mechanochemical conversion of chitin, 0.4 g chitin and an equivalent amount of a base were loaded together without pre-mixing into the chamber of the ball mill reactor with a certain number of balls and the reactor was capped tightly. Note that 100 balls were used in most of the experiments if not specifically stated. With the setting of ball milling time and speed, the ball milling machine was operated. The ball milling time was counted by cycles and one cycle includes 10 min of milling time and 5 min of rest. After ball milling, the solid products were passed through an alumina grid and collected.Two methods have been used for the separation of the LMWC products from the bases. When the base was NaOH, the ball milled solids were washed with methanol repeatedly until the pH value was neutral and then oven-dried at 70 °C overnight. In the study of various bases (some not soluble in methanol), dialysis was employed to separate the products by using a membrane pocket with a threshold of 3.5 kDa. The samples were dialysed for three days and the solution in the membrane pocket was freeze dried and collected. The product recovery rate was calculated by using the equation:
| Recovery% = Mproduct/Mfeedstock × 100% |
The solubility tests of the milled products were conducted as follows: after ball milling, the solids were collected through a grid. 300 mg of the solid mixture was taken and stirred in 50 mL water for 30 min, and then centrifuged. The solid residue was washed again using 50 mL water and the process was repeated twice. Thereafter, the undissolved solid was washed, oven dried and weighed. The calculation is as follows:
| Water-soluble product% = (1 − Mresidue/Mfeedstock) × 100% |
In the control experiment using cellobiose as a model compound, 0.4 g cellobiose was ball milled at 700 rpm for 5 min in the absence and presence of 0.1 g NaOH. The products were analysed by high-performance liquid chromatography (HPLC, a Shimadzu Nexera XR LC system) equipped with an Agilent Hi-Plex H column operating at 60 °C and a RID detector. The mobile phase was 0.005 M H2SO4 solution at a flow rate of 0.6 mL min−1. Gas chromatography-mass spectrometry (GC-MS) analysis was conducted on an Agilent 7890A GC system with a 7693 Autosampler and 5975C inert MSD with a triple-axis detector equipped with a HP-5 capillary column. Prior to GC-MS analysis, a silylation step was applied following a previous protocol.21
Analysis of the LMWC products
GPC analysis was carried out with a system equipped with a Waters 2410 refractive index detector, a Waters 515 HPLC pump and two Waters styragel columns (HT 3 and HT 4). The chitosan samples and products were measured following a previous protocol.19 XRD was performed on a Bruker D8 Advance Diffractometer with Cu Kα radiation at 40 kV. The scan range was from 5 to 40° without rotation. FTIR was conducted on a Thermo Scientific Nicolet iS50 FTIR spectrometer with both attenuated total reflection (ATR) mode and transmission mode. The DD values were calculated using the following equation:79| DD% = [1 − (A1320/A1420 − 0.3822)/3.133] × 100% |
1H NMR was performed on a Bruker ultrashield 400 plus spectrometer. The samples were dissolved in 1% HCl/D2O solution for analysis. The following equation was adopted to obtain the DD values:80
| DD% = (1 − 2HAc/H2 to 6) × 100% |
ESI-MS analysis was conducted on a Bruker MicroTOF-Q system. The MALDI-TOF mass spectra were obtained on a Bruker Daltonics Autoflex II TOF/TOF system. The sample was prepared by mixing 1 μL of 2 mg mL−1 product solution with 1 μL saturated 2,5-dihydroxybenzoic acid (2,5-DHB) matrix solution. Prior to 1H NMR and MS analysis, the solid products were first dissolved in 1% HAc and filtered, and then recovered by freeze drying. The solid-state NMR spectra were obtained at room temperature using a Bruker Ultrashield Avance 400WB with a MAS probe and 4 mm rotor at 5 kHz. The preparations of ball milled NAG samples with bases were conducted in a glove box to prevent the absorption of water.
Direct conversion of raw shrimp shells
The raw shrimp shell powder was prepared as follows: glass shrimps (Caridina heteropoda) were bought from a local supermarket. The shells on the back and tail were peeled off (no flesh residue) and air dried. Then, the shells were ground by using a blender to obtain crude shrimp shell powders. The contents of chitin, CaCO3 and proteins in the shell were quantified using a literature method,81 inductively coupled plasma atomic emission spectroscopy (ICP-OES, an iCAP 6000 series) and the Bradford method,82 respectively. In the Bradford method, shells were first boiled in 5% NaOH at 70 °C for 2 h and the solution was mixed with Bradford reagent. The values of the three were determined to be 28.3% of chitin, 23.0% of CaCO3, and 31.8% of proteins.The crude shrimp shell and NaOH catalyst were ball milled together at 700 rpm for 8 cycles. The solids were pasted to the balls and thus 10 mL of water was added into the chamber. The solids were washed down under low-speed ball milling at 250 rpm for 5 min. After separation from the balls, the suspension was centrifuged. The undissolved solid was washed and oven dried. The liquid solution was dialysed by using a membrane pocket with a threshold of 3.5 kDa, and then freeze dried. The content of CaCO3 in the products was determined by ICP-OES, while the contents of proteins were quantified by the Bradford method using bovine serum albumin (BSA) solutions as standards to plot the calibration curve.
Results and discussion
Mechanochemical conversion of chitin into LMWC products
In an initial test, we ball milled an equivalent mass of chitin and NaOH at the speed of 700 rpm for 8 cycles (denoted as NaOH-700-8). Each cycle includes 10 min of milling and 5 min of rest. To our delight, the light-yellow solid product obtained after the grinding became almost fully soluble in aqueous solution without the addition of any acids. Based on this, chemical transformation took place during the mechanical grinding process, in which insoluble chitin was converted to soluble compounds. A series of analytical techniques were subsequently employed to identify the structure of the product after thorough removal of NaOH by washing with methanol. The mass recovery rate of the product was beyond 90% indicating negligible mass loss in the post-treatment process. Chitin was also ball milled in the absence of NaOH following the same protocol for comparison (denoted as BM chitin).Infrared spectroscopy is an important tool to differentiate chitin and its derivatives. Commercially available chitin and chitosan (Chitosan-C) exhibit distinctively different FTIR spectra from 900 to 4000 cm−1 (see Fig. 2a). For example, the shape and the number of the peaks located between 2800 and 3500 cm−1, assignable to the vibrations of –NH, –OH and –CH groups, are different between the two. Moreover, the peak at 1115 cm−1 in the chitin spectrum is absent in that of Chitosan-C. The FTIR spectrum of NaOH-700-8 highly resembles that of Chitosan-C, with all fingerprint peaks fully matched, unambiguously indicating effective deacetylation in the mechanochemical process. In contrast, the FTIR spectrum of BM chitin remains largely the same as that of chitin suggesting negligible chemical transformations of functional groups. Hence, the FTIR analysis clearly demonstrates that chitosan-type products are only obtained in the presence of NaOH after ball milling. This is further confirmed by solid-state NMR spectra (see Fig. 2b), in which the peaks located at 25.8 and 176.8 ppm belonging to the two carbons on the acetamido group are much weaker in the spectra of NaOH-700-8 and Chitosan-C, compared with those of chitin and BM chitin.
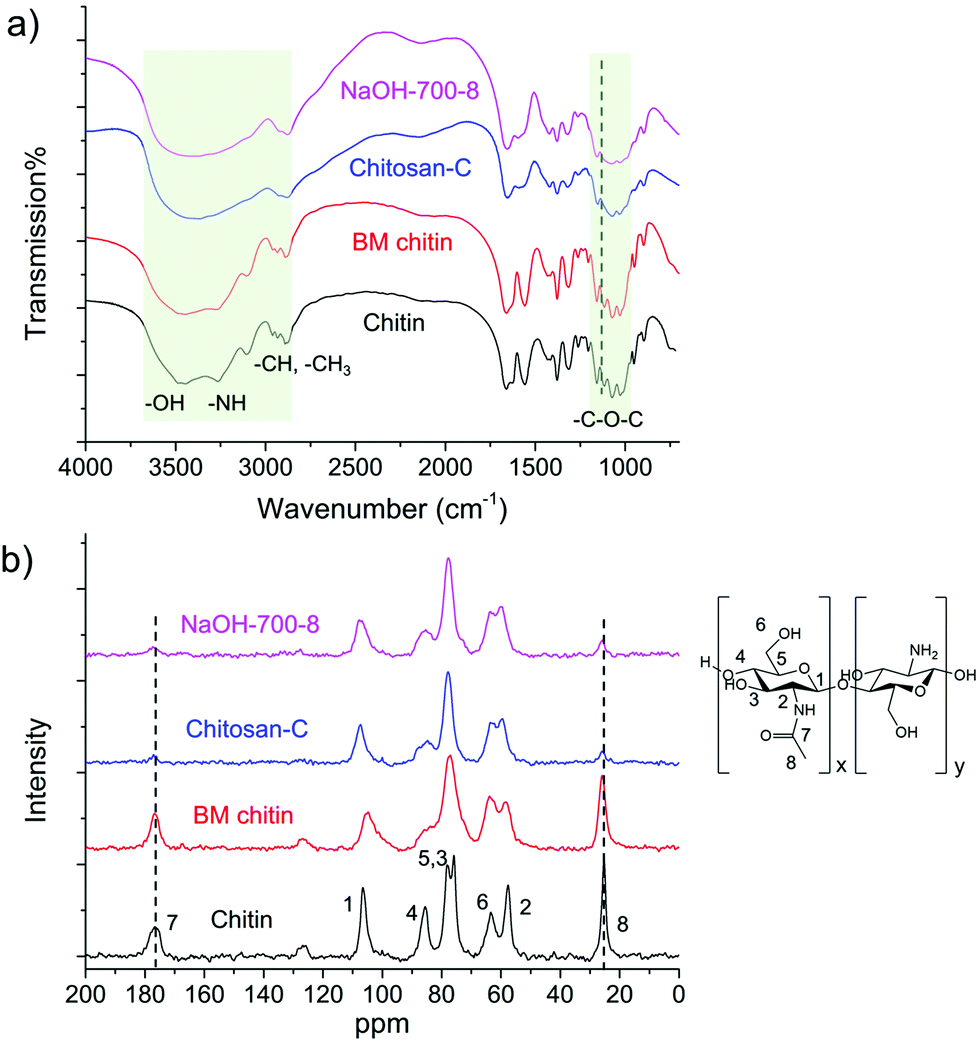 | ||
| Fig. 2 (a) FTIR and (b) solid-state NMR analysis of chitin, BM chitin, Chitosan-C and NaOH-700-8. | ||
NaOH-700-8 and control chitosan samples (Chitosan-C and Chitosan-H, the latter of which refers to homemade chitosan from the same chitin starting material by a traditional treatment using hot NaOH solution) were dissolved in acidic D2O and analyzed by liquid 1H NMR (see Fig. S1†). The position and relative intensities of the peaks for NaOH-700-8 were very similar to those of the chitosan control samples. The peak at 1.9 ppm is assigned to three protons on the acetamido group, while the peak at ca. 3.0 ppm is ascribed to the proton on the C2 position of the glucosamine unit. The group of peaks located from 3.3 to 3.8 ppm represents the non-anomeric protons on the ring skeleton,83 while the signal of the proton on the C1 position overlaps with the water signal. Note that the peaks of NaOH-700-8 are more resolved than Chitosan-C and Chitosan-H reflecting a decrease in MW (detailed in a later section).
The DD values of the samples were calculated from both the FTIR and 1H NMR results, as summarized in Table 1. The data derived from the two methods are in good agreement with deviations within 6%. The BM chitin has a DD value of merely 4%, similar to that of untreated chitin (3.5%), confirming that the mechanical force alone is not able to induce deacetylation. On the contrary, the DD value of NaOH-700-8 was 70% (based on FTIR) and 76% (based on NMR), comparable with those of Chitosan-C and Chitosan-H that were derived from the same chitin source. Based on the analysis, a highly-deacetylated chitosan product is formed after a simple ball milling process in the presence of NaOH.
| Entry | Sample | DD% by FTIR | DD% by NMR |
|---|---|---|---|
| 1 | Chitin | 3.5 | — |
| 2 | BM chitin | 4 | — |
| 3 | Chitosan-C | 78.9 | 82.4 |
| 4 | Chitosan-H | 78.6 | 78.7 |
| 5 | NaOH-700-8 | 70.0 | 76.4 |
| 6 | NaOH-700-2 | 39.1 | — |
| 7 | NaOH-700-4 | 53.1 | — |
| 8 | NaOH-700-12 | 83.3 | — |
| 9 | KOH-700-4 | 33.5 | — |
| 10 | LiOH-700-4 | 8.4 | — |
| 11 | Ba(OH)2-700-4 | 8.3 | — |
| 12 | Ca(OH)2-700-4 | 13.2 | — |
The MWs of NaOH-700-8, chitin, BM-chitin, Chitosan-C and Chitosan-H were examined by GPC, MALDI-TOF and ESI-MS techniques. In GPC analysis, BM chitin displayed a reduced MW of 79.7 kDa compared to that of chitin (362.4 kDa),19 due to the tensile stress exerted on the polymer chains during ball milling.84 The presence of a base substantially reduced the MW of NaOH-700-8 to only 8.5 kDa, which is two orders of magnitude lower than those of Chitosan-C (239.1 kDa) and Chitosan-H (149.3 kDa) (see Fig. 3a). Thus, despite the fact that the mechanical force itself is able to depolymerise chitin to a certain extent, it is the presence of a base that dramatically amplifies the effect affording low MW products. In addition, the peaks of Chitosan-C and Chitosan-H are also much broader than those of NaOH-700-8. The polydispersity (PDI) of NaOH-700-8 is only 1.1 by using the Breeze software indicating a very narrow MW distribution. For comparison, the LMWC obtained via H2O2 induced chitosan degradation had PDI values between 2 and 3.51 Even LMWC obtained via an enzymatic method, which is conceived to be very mild and selective, had a PDI value of 1.5.60
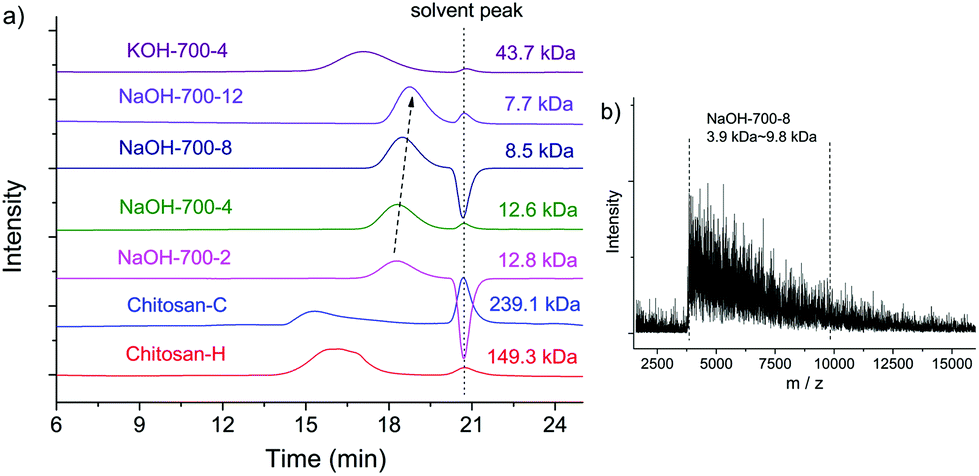 | ||
| Fig. 3 (a) GPC spectra of Chitosan-C, Chitosan-H and the products obtained with different milling times and bases; (b) MALDI-TOF spectrum of NaOH-700-8. | ||
In a typical MALDI-TOF spectrum of NaOH-700-8 (see Fig. 3b), peaks ranging from 3.9 kDa to 9.8 kDa were observed, which can be translated to ca. 24 to 60 monomer units in each chain. The average MW and the PDI were calculated to be 6.3 kDa and 1.1 respectively, consistent with the values from GPC analysis. ESI-MS was further employed for the analysis of MW. The chitosan chains seemed to be degraded in acidic solution during ESI-MS analysis and species were multiply-charged (up to the charge of M14+). Nevertheless, the signals show Gaussian-shaped peak clusters centered at around m/z 848.4. The distance between two adjacent clusters is m/z = 40 or 41 which is equal to the loss of an acetamido group with the addition of one or two protons (see Fig. S2†).
According to the XRD patterns (see Fig. S3†), chitin exhibited a crystal structure with a strong peak at 2θ ≈ 19.0° representing the (110) plane of the crystalline regions. The XRD patterns of Chitosan-C and Chitosan-H were almost identical with reduced crystallinity and the signal of the (110) plane shifted to 2θ ≈ 19.8°. In sharp contrast, the crystal structures for BM-chitin and NaOH-700-8 are fully amorphous indicating that the mechanical force effectively breaks the crystallinity of chitin, and plausibly makes subsequent chemical transformations easier to occur. Weak signals belonging to ZrO2 were observed in NaOH-700-8, which may be from stripping of the balls due to the presence of a strong base under friction.
All of the combined characterization provides compelling evidence that chitin has been transformed into a uniformly-distributed LMWC material during a simple ball milling process in the presence of NaOH. Ball milling destroys the high crystallinity of chitin and enables excellent contact between the chitin polymer chains and NaOH, leading to a deacetylated LMWC product with a narrow MW distribution.
The current method at the lab scale uses methanol in a large excess. In fact, the solubility of NaOH in methanol at room temperature is 23.8 g per 100 mL, so that the amount of methanol required for washing, in principle, is not large. Methanol is a low boiling point solvent which can be easily recycled and reused after distillation. For the manufacture of every kilogram of the product, a minimum of 4 L methanol will be needed for washing after which methanol should be distilled for the next-run use. The minimum energy consumption for methanol separation is 1.03 kWh to produce 1 kg product based on the enthalpy of vaporization at 25 °C, costing ca. 0.12 USD at the current electricity price in the USA.
The influences of ball milling parameters
The NaOH amount and ball milling parameters were varied and the yield of the water-soluble product was used as an indicator for the degree of deacetylation and depolymerisation. With the increase of the NaOH:chitin ratio (see Fig. S4†), the yield first increased to 43.8% when the ratio increased to 1.5, and then decreased to 28.2% with a higher ratio. A base is indeed necessary to promote LMWC formation but excessive NaOH is likely to cause severe water absorption thus inhibiting the mechanochemical process (water is detrimental as described in a later session). Ball milling parameters such as the speed, time and ball number have greater influences on the yield of the water-soluble product (see Fig. 4). When the ball number was increased from 50 to 140 (2 cycles of milling), the yield enhanced considerably from 21.6% to 96.4%. With a ball number of 100, the yield was always around 95% over 4 cycles of the milling time and above. As a result, a minimum of 4 cycles is required for high conversion. Milling speed is also a crucial factor, and the yields of the water-soluble product were 17.1%, 42.6% and 96.4%, respectively, at the speeds of 500, 600 and 700 rpm. From these results, it is understood that the extent of chitin transformation is not very sensitive to the amount of NaOH but is improved significantly with a reinforced mechanical power. In a previous study on chitin treatment by ball mill grinding,19 the crystallinity of chitin would be reduced accordingly with the increased ball number, time and speed, which resulted in enhanced chemical reactivity of chitin. Therefore, the conversion of chitin into water-soluble products was promoted here in a similar way. Note that the required NaOH:chitin weight ratio is not higher than 1 to achieve a full conversion. In a traditional method for chitin deacetylation, an NaOH solution of >40 wt% concentration and 20 to 30-fold weight is needed – this works out to be a NaOH:chitin ratio of around 10. Therefore, the new process can sharply decrease the usage of a base.
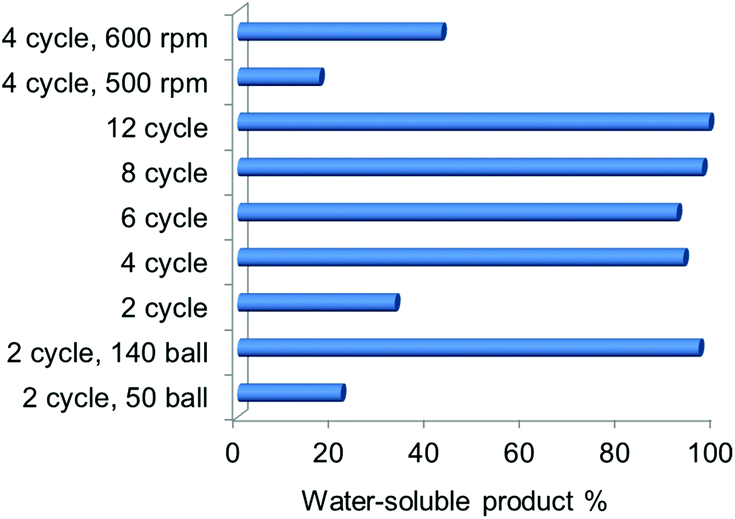 | ||
| Fig. 4 Yields of water-soluble products with different ball milling parameters including the ball numbers, milling time and speed. 0.4 g chitin was ball milled with an equivalent amount of NaOH. If not specifically mentioned, the number of balls used was 100 and the milling speed was 700 rpm. | ||
The influences of the ball milling time and speed on the DD and MW values of the products were also studied (Table 1 and Fig. 3). The DD values increased smoothly with a longer milling time (FTIR spectra shown in Fig. S5†): from 3.5% to 39.1% after 2 cycles, and further increased to 53.1%, 70.0%, and 83.3% after 4, 8, and 12 cycles, respectively, indicating that deacetylation occurs in the entire process of ball milling. On the other hand, the MW values sharply decreased to 12.8 kDa after 2 cycles of ball milling, and dropped further to 7.7 kDa after 12 cycles (ESI-MS analysis of NaOH-700-12 is also provided in Fig. S6†). To achieve a product with an even lower MW, a second round of ball milling is required. For example, the ball milling of purified NaOH-700-8 in the presence of an equivalent amount of NaOH for a 6 cycle milling time leads to a significant reduction of MW to only 1.2 kDa (see Fig. S7†). A long-time low-speed test was also conducted, in which chitin and NaOH were milled under 350 rpm for 24 cycles. The product retained the structure of chitin with negligible deacetylation (Fig. S8†). This highlights that a sufficient mechanical force is critical for the solid-state reaction between chitin and base regardless of the milling time. Under appropriate ball milling conditions, synergistic effects between the mechanical and chemical forces would lead to the effective formation of LMWC products, with tunable MW (1.2 to 13 kDa) and varied DD values (40% to 83%).
In a mechanical-assisted depolymerisation reaction, the role of water is two-sided. Water is a necessary reactant in the hydrolysis of glycosidic linkages, but excessive water impairs friction and mechanical impacts.75 Since chitin itself contains ca. 9 wt% of water, the depolymerisation can occur in a self-sustained manner. As expected, a monotonic increase of MW was observed when increasing the amount of water, reaching 67.4 kDa when chitin:water = 1:1 (see Fig. S9a†). The MW of the products was considerably influenced because of the reduced tensile stress which is a crucial factor for depolymerisation.84 Meanwhile, the DD values experienced an initial increase from 53.1% to 65.4% (chitin:water = 10:3) and then dropped to 34.8% (chitin:water = 1:1). Therefore, excessive water not only mitigates the mechanical force but also dilutes the base leading to reduced deacetylation. In addition, the chitin was dried in an oven at 105 °C for 24 h and used for a mechanochemical reaction at 700 rpm for 8 cycles. From GPC analysis (see Fig. S10†), it was found that the product had an almost identical MW to NaOH-700-8 (using native chitin), indicating that the amount of water originally in chitin did not inhibit depolymerisation. Nevertheless, starting from dried chitin, the DD value decreased significantly as observed by FTIR analysis (see Fig. S11†), showing that a certain amount of water is beneficial for deacetylation. As a result, it is not necessary to dry chitin prior to the mechanochemical process.
NaBH4 as an additive was also evaluated (see Fig. S9b†) because it is capable of the reductive cleavage of cyclic amides.85 Indeed, the DD values were enhanced with the addition of NaBH4, and reached a high value of 79.1% at a chitin:NaBH4 ratio of 4:3. The promotional effect of NaBH4 was not observed in conventional deacetylation86 and might be present only with the assistance of a mechanical force. However, the MWs increased from 12.6 kDa to 23.5 kDa, probably because NaBH4 inhibited depolymerisation.86 As a result, NaBH4 is beneficial for the process when products with high DD and relatively higher MW are required.
Reaction pathways
Transformation of chitin into LMWC comprises two major chemical reactions including depolymerisation and deacetylation. In a previous study, acid-impregnated chitin was ball milled into chitooligomers with negligible deacetylation, suggesting that the mechanical force mainly impacts the linkage of the polymer chains and would not affect the side chain.84 Our results highlight that the mechanical force, in fact, is able to promote deacetylation in the presence of a base. Base-catalysed cleavage of the acetamido group has a long-standing chemistry, and we find that the mechanochemical deacetylation occurs at a greatly reduced temperature.For the depolymerisation of chitin, a key question is whether it follows a hydrolytic pathway, or a free radical or oxidative pathway as proposed in the literature.51,87,88 Cellobiose—an analog of a chitin dimer bearing the same β-1→4 glycosidic bond—was therefore employed as a model compound, which was ball milled both in the absence and presence of NaOH. According to GC-MS analysis (see Fig. S12†) of the silanised products, cellobiose did not undergo any chemical changes in the absence of NaOH. In the presence of NaOH, however, glucose and a trace amount of fructose were detected. Indeed, 3.4% glucose and 0.6% fructose were determined by HPLC in the sample with NaOH, while none was found in the absence of a base (HPLC spectra shown in Fig. S13†). In both GC-MS and HPLC analysis, no oxidation products were detected. These control experiments suggest that the depolymerisation mainly follows a base-catalysed hydrolysis pathway.
ESI-MS was further employed to analyze the products, as it is more sensitive and informative than GC-MS and HPLC techniques. Again, no peaks of oxidation products were observed (see Fig. 5 and S14†). Condensation products were also detected in ball milled cellobiose in the absence of NaOH. For example, the signals at 264.1, 362.1 and 782.3 were assigned to [(C18H32O16) + H + Na]2+, [(C24H44O23) + Na + H]2+ and [(C12H22O11)2(C5H10O4) + H]+, respectively. This is in agreement with a previous study that short-chain oligomers such as glucose and cellobiose were prone to condensation into more stable longer-chain oligomers under a strong mechanical force.75 In contrast, the ESI-MS spectrum of ball milled cellobiose with NaOH is much cleaner. Only three major peaks were observed which were all ascribed to cellobiose and its assemblies. Similar results were obtained in the negative mode (see Fig. S14†). ESI-MS analysis reveals that NaOH not only promotes the hydrolysis of the glycosidic bond but also suppresses the side reactions leading to a more uniform product distribution.
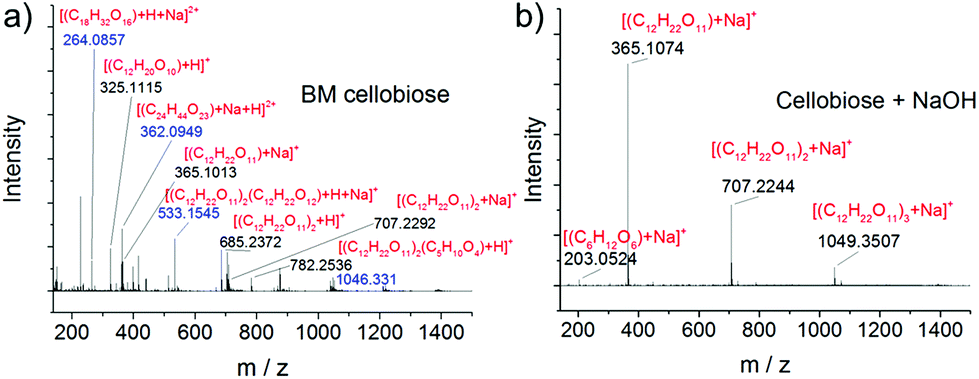 | ||
| Fig. 5 ESI-MS signals (positive mode) of (a) BM cellobiose and (b) ball milled cellobiose in the presence of NaOH. Conditions: 0.4 g cellobiose, none or 0.1 g NaOH, 700 rpm, 5 min. The blue color indicates the charge of the peak to be 2+. The proposed molecular formulae are shown in red. | ||
Role of a base
A series of bases including LiOH, NaOH, KOH, Ca(OH)2 and Ba(OH)2 were employed to study the role of the bases. Each base was ball milled with chitin (1:1 weight ratio) at 700 rpm for 4 cycles. Since most of these bases are not soluble in methanol, a different separation method using membrane dialysis (threshold of 3.5 kDa) was applied for base removal. Noteworthily, the recovery rate of LiOH-700-4 was the lowest (61.2%) whereas the recovery of the remaining samples lay in a narrow range of 80% (±5%). Therefore, lower MW products (<3.5 kDa) formed with LiOH, inferring a stronger ability of LiOH for chitin depolymerisation. On the other hand, the FTIR analysis (see Fig. S15†) showed that only NaOH and KOH are effective at promoting deacetylation of chitin to chitosan products with DD values of 53.1% and 33.5%, and MW values of 12.6 kDa and 43.7 kDa. LiOH, Ca(OH)2 and Ba(OH)2, in sharp contrast, hardly induce deacetylation as reflected by DD values of 8.4%, 13.2% and 8.3% respectively (see Table 1). From the above observations, NaOH is found to be most effective in regard of facilitating both depolymerisation and deacetylation. Note that the concentration of OH− for each base used is different, following the order: Ba(OH)2 < KOH < NaOH ≈ Ca(OH)2 < LiOH. However, the performances were not directly associated with the concentration of OH−. For example, LiOH showed negligible ability for chitin deacetylation.
Solid-state NMR analysis was employed to investigate the interactions between several bases and chitin monomer NAG. The mixtures after ball milling were directly subjected to NMR analysis without washing. As shown in Fig. 6, the signals of NAG were well-resolved and negligible peak broadening was found in BM NAG (physically milled NAG without any base). Upon the addition of bases, peak broadening was noticed indicating interactions between the substrate and the base. Meanwhile, no chemical shift was observed when NAG was ball milled with Ca(OH)2 but a significant down-field shift for almost all carbon atoms was identified in the presence of NaOH and LiOH. C1 and C4 peaks were shifted by 11.01 and 8.79 ppm after ball milling NAG with LiOH, and shifted by 11.26 and 8.78 ppm with NaOH (see Table S1†). The signals ascribed to other carbons of the pyranose ring also experienced similar shifts but to a lesser extent. Probably, this is due to the direct coordination of the cations of Li+ and Na+ with the oxygen atom of the –OH group on the C1 and C4 positions, which led to the remarkably decreased electron density of the carbon and thus prominent down-field shifts. This may explain the effectiveness of LiOH and NaOH in the depolymerisation of chitin, where the small sized Li+ and Na+ readily coordinate to the oxygens at the C1 and C4 positions. On the other hand, LiOH is not effective for deacetylation, consistent with previous papers,89,90 because it showed a weaker interaction with the acetamido side chain than NaOH or KOH. For these reasons NaOH is the best catalyst to prepare LMWC from chitin under ball milling conditions.
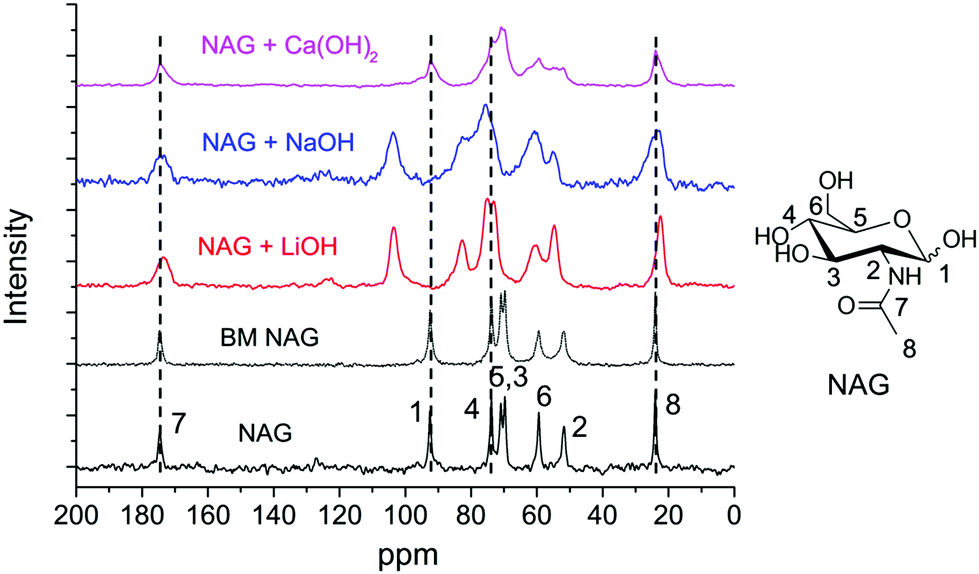 | ||
| Fig. 6 Solid-state NMR analysis of NAG without/with different bases before and after ball milling. Conditions: 0.4 g NAG, none or 0.2 g base, 600 rpm, 4 min. | ||
Solid-state NMR analysis was also conducted for ball milled chitin together with the bases. The signals were assigned as the final product rather than a chitin-base complex (see Fig. S16†). For instance, the signals of sodium acetate appeared in the spectrum of chitin milled with NaOH, as indicated by the two sharp and narrow peaks at 179.1 ppm and 27.7 ppm (partially overlapping with the signals of the acetamido group of chitin). Nevertheless, it again provides solid evidence that NaOH is superior for deacetylation since these peaks were absent for samples treated with other bases.
Direct transformation of shrimp shells
Commercial chitin is a premium product extracted from crab or shrimp shells via a series of processes. Direct synthesis of LMWC from waste shells ($0.1 kg−1) instead of using refined chitin is much more economically attractive. For this reason, we tested the transformation of shrimp shells (Caridina heteropoda) under NaOH catalysed ball milling conditions (8 cycles, 700 rpm). The starting material comprises of 28.3 wt% chitin, 23.0 wt% calcium carbonate (CaCO3), 31.8 wt% proteins and 13.1 wt% water. CaCO3 remained insoluble after ball milling and was removed simply by filtration after dispersing the products in water. Proteins in the shrimp shell underwent depolymerisation in the presence of a base and were removed via membrane dialysis, leaving a 20.9 wt% product (the images of the raw shrimp shell powders and the obtained final product are shown in Fig. 7a). In Fig. 7b, the FTIR spectrum of the product demonstrates the formation of chitosan type compounds. The DD value of the product was 80.5% and the MW was 7.9 kDa, proving the formation of the LMWC product from shrimp shells. On the other hand, the weak signals at 1536 cm−1 and 1395 cm−1 in the FTIR spectrum suggest the presence of residual CaCO3 and proteins. The content of protein residues in the final product was analysed by the Bradford method,82 in which a blue color is indicative of the presence of proteins. Fig. 7c shows the color changes of the water (control), the final product solution and the hydrolytes of shrimp shell (treated by NaOH solution) with the Bradford reagent. The intensive blue color suggests the rich content of proteins in the crude shrimp shells, whereas the color of our final product is similar to that of water indicating that only trace amounts of proteins were left (5.2 wt% compared with the total weight of product). ICP-OES revealed a CaCO3 content of 6.7 wt%. Altogether, the LMWC product was obtained from raw shrimp shell powders by a simple and one-step process with facile separation with a purity of around 90% higher than using a biological method.91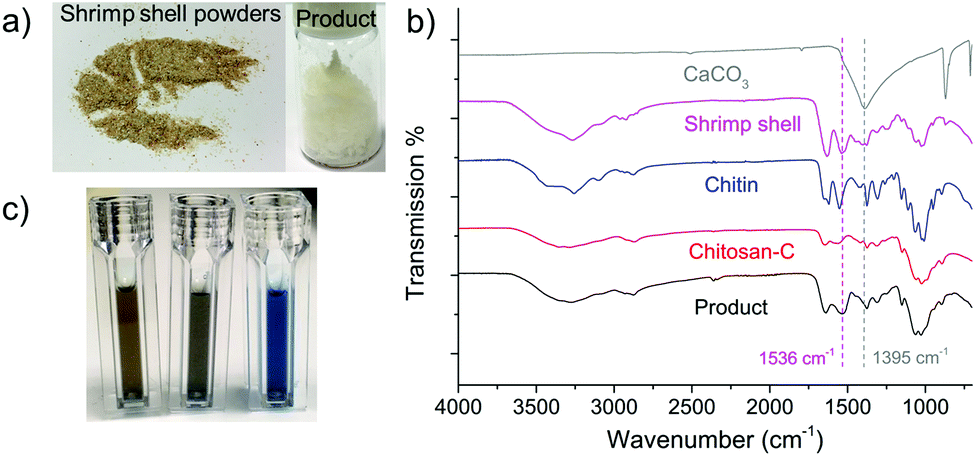 | ||
| Fig. 7 (a) The photos of the crude shrimp shell powders and the obtained LMWC product; (b) FTIR spectra of the product, Chitosan-C, chitin, shrimp shell powders and CaCO3; and (c) the images of the color changes of water (control), the product and shrimp shell powder hydrolyses with the Bradford reagent (in the order from left to right). Note that the FTIR signals in this figure were collected by using ATR mode, while the previous data were by using transmission mode. | ||
Conclusions
In this paper, a solvent-free mechanochemical method was developed to produce LMWC in one step from chitin and shrimp shells, which is efficient, cost saving and more environmentally friendly. The base usage is reduced to 1/10 in the new process. Moreover, the LMWC products exhibited a much narrower MW distribution than traditional methods, with tunable DD (40% to 83%) and MW (1 to 13 kDa) values. Synergistic effects of mechanical and chemical forces are crucial for the simultaneous depolymerisation and deacetylation. NaOH not only facilitates chitin transformation into LMWC but also inhibits side reactions under ball milling conditions. By using this simple process, the LMWC product with a purity of ca. 90% can be directly obtained from crude shrimp shell powders, putting forward a facile way to create value from the shell waste.Acknowledgements
We thank the National University of Singapore Young Investigator Award (WBS: R-279-000-464-133) and the A*STAR, PSF funding (WBS: R-279-000-403-305) for financial support. The authors thank Ms Lili Ong in ICES for her kind help in solid NMR measurement, and the undergraduate Mr Wen Yalong for his participation in this project.Notes and references
- J. H. Clark, Nat. Chem., 2009, 1, 12–13 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- C. Alvarez-Vasco, R. Ma, M. Quintero, M. Guo, S. Geleynse, K. K. Ramasamy, M. Wolcott and X. Zhang, Green Chem., 2016, 18, 5133–5141 RSC.
- S. Siankevich, Z. Fei, R. Scopelliti, P. G. Jessop, J. Zhang, N. Yan and P. J. Dyson, ChemSusChem, 2016, 9, 2089–2096 CrossRef CAS PubMed.
- S. Siankevich, Z. Fei, N. Yan and P. J. Dyson, Chimia, 2015, 69, 592–596 CrossRef CAS PubMed.
- J. Zhang and C. Zhao, ACS Catal., 2016, 6, 4512–4525 CrossRef CAS.
- Z. Luo, Y. Wang, M. He and C. Zhao, Green Chem., 2016, 18, 433–441 RSC.
- H. Li, S. Yang, A. Riisager, A. Pandey, R. S. Sangwan, S. Saravanamurugan and R. Luque, Green Chem., 2016, 18, 5701–5735 RSC.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- X. Chen and N. Yan, Catal. Surv. Asia, 2014, 1–13 Search PubMed.
- S.-K. Kim, Chitin, chitosan, oligosaccharides and their derivatives: biological activities and applications, CRC Press, 2010 Search PubMed.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- Y. Pierson, X. Chen, F. D. Bobbink, J. Zhang and N. Yan, ACS Sustainable Chem. Eng., 2014, 2, 2081–2089 CrossRef CAS.
- F. D. Bobbink, J. Zhang, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- X. Chen, Y. Gao, L. Wang, H. Chen and N. Yan, ChemPlusChem, 2015, 80, 1565–1572 CrossRef CAS.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073–20080 RSC.
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- Y. Wang, C. M. Pedersen, T. Deng, Y. Qiao and X. Hou, Bioresour. Technol., 2013, 143, 384–390 CrossRef CAS PubMed.
- Y. Ohmi, S. Nishimura and K. Ebitani, ChemSusChem, 2013, 6, 2259–2262 CrossRef CAS PubMed.
- M. Osada, K. Kikuta, K. Yoshida, K. Totani, M. Ogata and T. Usui, Green Chem., 2013, 15, 2960–2966 RSC.
- B. Duan, X. Zheng, Z. Xia, X. Fan, L. Guo, J. Liu, Y. Wang, Q. Ye and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 5152–5156 CrossRef CAS PubMed.
- R. Arun Kumar, A. Sivashanmugam, S. Deepthi, S. Iseki, K. P. Chennazhi, S. V. Nair and R. Jayakumar, ACS Appl. Mater. Interfaces, 2015, 7, 9399–9409 CAS.
- Y. Huang, M. Yao, X. Zheng, X. Liang, X. Su, Y. Zhang, A. Lu and L. Zhang, Biomacromolecules, 2015, 16, 3499–3507 CrossRef CAS PubMed.
- S. Ladet, L. David and A. Domard, Nature, 2008, 452, 76–79 CrossRef CAS PubMed.
- M. N. V. R. Kumar, R. A. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017–6084 CrossRef PubMed.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- S.-K. Kim and N. Rajapakse, Carbohydr. Polym., 2005, 62, 357–368 CrossRef CAS.
- S. Mansouri, P. Lavigne, K. Corsi, M. Benderdour, E. Beaumont and J. C. Fernandes, Eur. J. Pharm. Biopharm., 2004, 57, 1–8 CrossRef CAS PubMed.
- K. V. Harish Prashanth and R. N. Tharanathan, Biochim. Biophys. Acta, Gen. Subj., 2005, 1722, 22–29 CrossRef CAS PubMed.
- V. E. Tikhonov, E. A. Stepnova, V. G. Babak, I. A. Yamskov, J. Palma-Guerrero, H.-B. Jansson, L. V. Lopez-Llorca, J. Salinas, D. V. Gerasimenko and I. D. Avdienko, Carbohydr. Polym., 2006, 64, 66–72 CrossRef CAS.
- N. Duceppe and M. Tabrizian, Biomaterials, 2009, 30, 2625–2631 CrossRef CAS PubMed.
- N. Li, C. Zhuang, M. Wang, X. Sun, S. Nie and W. Pan, Int. J. Pharm., 2009, 379, 131–138 CrossRef CAS PubMed.
- S. Nimesh, M. M. Thibault, M. Lavertu and M. D. Buschmann, Mol. Pharm., 2010, 46, 182–196 CAS.
- Y.-H. Lin, F.-L. Mi, C.-T. Chen, W.-C. Chang, S.-F. Peng, H.-F. Liang and H.-W. Sung, Biomacromolecules, 2007, 8, 146–152 CrossRef CAS PubMed.
- G. Ke, W. Guan, C. Tang, W. Guan, D. Zeng and F. Deng, Biomacromolecules, 2007, 8, 322–326 CrossRef CAS PubMed.
- P.-J. Chien, F. Sheu and H.-R. Lin, Food Chem., 2007, 100, 1160–1164 CrossRef CAS.
- Z. Amoozgar, J. Park, Q. Lin and Y. Yeo, Mol. Pharm., 2012, 9, 1262–1270 CAS.
- C. M. d. Moura, J. M. d. Moura, N. M. Soares and L. A. d. A. Pinto, Chem. Eng. Process., 2011, 50, 351–355 CrossRef.
- T. Lertwattanaseri, N. Ichikawa, T. Mizoguchi, Y. Tanaka and S. Chirachanchai, Carbohydr. Res., 2009, 344, 331–335 CrossRef CAS PubMed.
- F. A. A. Sagheer, M. A. Al-Sughayer, S. Muslim and M. Z. Elsabee, Carbohydr. Polym., 2009, 77, 410–419 CrossRef.
- G. Lamarque, M. Cretenet, C. Viton and A. Domard, Biomacromolecules, 2005, 6, 1380–1388 CrossRef CAS PubMed.
- X. He, K. Li, R. Xing, S. Liu, L. Hu and P. Li, Egypt. J. Aquat. Res., 2016, 42, 75–81 CrossRef.
- J. C. Cabrera and P. Van Cutsem, Biochem. Eng. J., 2005, 25, 165–172 CrossRef CAS.
- C. Qin, Y. Du and L. Xiao, Polym. Degrad. Stab., 2002, 76, 211–218 CrossRef CAS.
- K. L. B. Chang, M.-C. Tai and F.-H. Cheng, J. Agric. Food Chem., 2001, 49, 4845–4851 CrossRef CAS PubMed.
- Y. Du, Y. Zhao, S. Dai and B. Yang, Innovative Food Sci. Emerging Technol., 2009, 10, 103–107 CrossRef CAS.
- K. Tømmeraas, K. M. Vårum, B. E. Christensen and O. Smidsrød, Carbohydr. Res., 2001, 333, 137–144 CrossRef.
- H. Liu, J. Bao, Y. Du, X. Zhou and J. F. Kennedy, Carbohydr. Polym., 2006, 64, 553–559 CrossRef CAS.
- I. Prasertsung, S. Damrongsakkul, C. Terashima, N. Saito and O. Takai, Carbohydr. Polym., 2012, 87, 2745–2749 CrossRef CAS.
- H. Zhang and S. H. Neau, Biomaterials, 2001, 22, 1653–1658 CrossRef CAS PubMed.
- H. Zhang, Y. Du, X. Yu, M. Mitsutomi and S.-i. Aiba, Carbohydr. Res., 1999, 320, 257–260 CrossRef CAS PubMed.
- H. Zhang and S. H. Neau, Biomaterials, 2002, 23, 2761–2766 CrossRef CAS PubMed.
- F. S. Kittur, A. B. V. Kumar and R. N. Tharanathan, Carbohydr. Res., 2003, 338, 1283–1290 CrossRef CAS PubMed.
- J. Li, Y. Du, J. Yang, T. Feng, A. Li and P. Chen, Polym. Degrad. Stab., 2005, 87, 441–448 CrossRef CAS.
- L. S. Ribeiro, J. J. M. Orfao and M. F. R. Pereira, Green Chem., 2015, 17, 2973–2980 RSC.
- V. Molinari, M. Antonietti and D. Esposito, Catal. Sci. Technol., 2014, 4, 3626–3630 CAS.
- S. Tabasso, D. Carnaroglio, E. Calcio Gaudino and G. Cravotto, Green Chem., 2015, 17, 684–693 RSC.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schüth, Energy Environ. Sci., 2013, 6, 92–96 CAS.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993–997 CrossRef CAS.
- L. Kratky and T. Jirout, Chem. Eng. Technol., 2011, 34, 391–399 CrossRef CAS.
- B. Içli, N. Christinat, J. Tönnemann, C. Schüttler, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2009, 131, 3154–3155 CrossRef PubMed.
- L. Konnert, A. Gauliard, F. Lamaty, J. Martinez and E. Colacino, ACS Sustainable Chem. Eng., 2013, 1, 1186–1191 CrossRef CAS.
- T. Rantanen, I. Schiffers and C. Bolm, Org. Process Res. Dev., 2007, 11, 592–597 CrossRef CAS.
- T. Kameda, M. Ono, G. Grause, T. Mizoguchi and T. Yoshioka, Ind. Eng. Chem. Res., 2008, 47, 8619–8624 CrossRef CAS.
- G. C. Paveglio, K. Longhi, D. N. Moreira, T. S. München, A. Z. Tier, I. M. Gindri, C. R. Bender, C. P. Frizzo, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, ACS Sustainable Chem. Eng., 2014, 2, 1895–1901 CrossRef CAS.
- A. Stolle, Ball milling towards green synthesis: applications, projects, challenges, Royal Society of Chemistry, 2014 Search PubMed.
- T. Szuppa, A. Stolle, B. Ondruschka and W. Hopfe, Green Chem., 2010, 12, 1288–1294 RSC.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS PubMed.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- T. Kleine, J. Buendia and C. Bolm, Green Chem., 2013, 15, 160–166 RSC.
- H. Kobayashi, M. Yabushita, T. Komanoya, K. Hara, I. Fujita and A. Fukuoka, ACS Catal., 2013, 3, 581–587 CrossRef CAS.
- J. Brugnerotto, J. Lizardi, F. M. Goycoolea, W. Argüelles-Monal, J. Desbrières and M. Rinaudo, Polymer, 2001, 42, 3569–3580 CrossRef CAS.
- A. Hirai, H. Odani and A. Nakajima, Polym. Bull., 1991, 26, 87–94 CrossRef CAS.
- F. Shahidi and J. Synowiecki, J. Agric. Food Chem., 1991, 39, 1527–1532 CrossRef CAS.
- N. J. Kruger, The protein protocols handbook, 2009, pp. 17–24 Search PubMed.
- M. R. Kasaai, Carbohydr. Polym., 2010, 79, 801–810 CrossRef CAS.
- M. Yabushita, H. Kobayashi, K. Kuroki, S. Ito and A. Fukuoka, ChemSusChem, 2015, 8, 3760–3763 CrossRef CAS PubMed.
- J. Hubert, J. Wijnberg and W. N. Speckamp, Tetrahedron, 1975, 31, 1437–1441 CrossRef CAS.
- A. Tolaimate, J. Desbrieres, M. Rhazi and A. Alagui, Polymer, 2003, 44, 7939–7952 CrossRef CAS.
- F. Tian, Y. Liu, K. Hu and B. Zhao, J. Mater. Sci., 2003, 38, 4709–4712 CrossRef CAS.
- S.-C. Hsu, T.-M. Don and W.-Y. Chiu, Polym. Degrad. Stab., 2002, 75, 73–83 CrossRef CAS.
- Y. Fang, B. Duan, A. Lu, M. Liu, H. Liu, X. Xu and L. Zhang, Biomacromolecules, 2015, 16, 1410–1417 CrossRef CAS PubMed.
- J. Duan, X. Liang, Y. Cao, S. Wang and L. Zhang, Macromolecules, 2015, 48, 2706–2714 CrossRef CAS.
- F. Sedaghat, M. Yousefzadi, H. Toiserkani and S. Najafipour, Int. J. Biol. Macromol., 2016, 82, 279–283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc00089h |
| This journal is © The Royal Society of Chemistry 2017 |