
Hidden complexity of synergistic roles of Dopa and lysine for strong wet adhesion†
Ying
Li‡
 a,
Chao
Liang‡
bc,
Ling
Gao
d,
Shiyu
Li
e,
Yizhe
Zhang
e,
Jiang
Zhang
b and
Yi
Cao
*b
a,
Chao
Liang‡
bc,
Ling
Gao
d,
Shiyu
Li
e,
Yizhe
Zhang
e,
Jiang
Zhang
b and
Yi
Cao
*b
aJiangsu Engineering Technology Research Centre of Environmental Cleaning Materials, Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Jiangsu Joint Laboratory of Atmospheric Pollution Control, Collaborative Innovation Center of Atmospheric Environment and Equipment Technology, School of Environmental Science and Engineering, Nanjing University of Information Science & Technology, 219 Ningliu Road, Nanjing, Jiangsu 210044, P. R. China
bCollaborative Innovation Center of Advanced Microstructures, National Laboratory of Solid State Microstructure, Department of Physics, Nanjing University, Nanjing 210093, P. R. China. E-mail: caoyi@nju.edu.cn
cDepartment of Chemistry and Biology, College of Science, National University of Defense Technology, Changsha 410073, P. R. China
dAnimal, Plant and Food Inspection Center, Jiangsu Entry-Exit Inspection and Quarantine Bureau of People's Republic of China, 210001, P. R. China
eReading Academy, Nanjing University of Information Science & Technology, 219 Ningliu Road, Nanjing, Jiangsu 210044, P. R. China
First published on 24th October 2017
Abstract
Dopa and lysine are widely found in mussel foot proteins and are suggested to play synergistic roles in wet adhesion; yet, the detailed molecular mechanism remains unclear. Here, using PEG conjugated dipeptides as the model system, we found that the neighboring lysine can significantly enhance surface binding of Dopa through three distinct mechanisms: (1) displacing surface water and ions to increase the effective binding sites; (2) being directly involved in cooperative surface binding in a sequence dependent manner; (3) enhancing cohesion by Michael addition to oxidized species or forming cation–π interactions. This study may be helpful for rational design of biomimetic strong adhesives for biomedical applications.
 Yi Cao | Yi Cao received his bachelor's degree in 2001 and Master's degree in 2004 from Nanjing University. He then obtained his PhD in 2009 from the University of British Columbia. After a one-year postdoc at the same place, he started his independent career at the Department of Physics, Nanjing University as a full professor. He was the recipient of the 2014 IUPAP Young Scientist Prize in Biological Physics for his contributions to the study of nanomechanics and mechanical unfolding dynamics of proteins using single molecule atomic force microscopy. His current research focuses on the application of single molecule methods and molecular engineering to designing biomaterials with tailored mechanical properties. |
Mussels are able to form strong and tough attachments to rock surfaces in the sea to prevent them from being dislodged by the tide. This is achieved by secreting a variety of mussel foot proteins (Mfps) to form the advanced and sophisticated holdfast byssus.1 Biochemical studies revealed that the adhesive proteins of mussels are rich in a special catecholic residue, 3,4-dihydroxyphenylalanine (Dopa), which is produced through post-translational modification of tyrosine.2 Inspired by this design, various biomimetic polymers bearing catecholic functional groups have been synthesized to provide strong wet adhesion, surface coating, and anchoring properties,3 despite the fact that the adhesion mechanisms are not fully understood. Extensive studies suggest that Dopa plays multiple roles in surface binding: it can form bidentate hydrogen bonds and metal chelation bonds with mineral surfaces to promote adhesion.4 It can also form catechol-Fe3+ coordination and oxidative coupling (e.g. Dopa–quinone coupling and quinone–amine coupling) to enhance cohesion.5
Recent studies show that the adhesion strength of natural Mfps does not simply correlate with the amount of Dopa. For example, although mfp-1 and mfp-3 have similar Dopa contents, they show distinct adhesive properties.6 The local chemical environments provided by the primary sequence of Mfps may also have an important contribution to the adhesive performance of Mfps. In particular, it has been found that Dopa residues are often present in the vicinity of positively charged lysine residues in many Mfps. (See Table 1 for statistic analysis of different Mfp sequences.) Therefore, they may function cooperatively for wet adhesion. This hypothesis has been confirmed by surface force apparatus (SFA) measurements.7 It was found that neighboring lysine residues are critical for dewetting mineral surfaces, thus promoting catechol binding to the underneath metal oxides. Parallel to the SFA studies, single molecule atomic force microscopy (AFM) experiments suggested that the positively charge lysine residues may be directly involved in surface binding by forming ionic bonds with negatively charged mineral surfaces.8 More interestingly, it was found that the order of lysine and Dopa in the primary sequence plays an important role in the synergistic binding. The binding strength of Dopa–lysine is significantly higher than that of lysine–Dopa when the peptides are pulled from the N-termini. These studies provide clear evidence that Dopa and lysine have multiple synergistic effects for strong wet adhesion. However, these studies are conducted at the nanoscale based on small molecules and relatively flat surfaces. It remains unexplored whether the synergistic effects of Dopa–lysine persist at a bulk level in synthetic polymers and rough surfaces.
| Protein | Species | Accession no. | M w (kDa) | pI | Y (%) | K (%) | YK or KY (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| The molecular weights (Mw), isoelectric points (pI) and amino acid compositions were calculated by an online ExPASy ProtParam tool, using signal peptide eliminated mature mfp sequences retrieved from GenBank database. Y denotes 3,4-dihydroxyphelanine (Dopa) while K is the standard abbreviation of lysine. YK or KY (%) refers to the percentage of those dopa residues that align in close vicinity to lysine. It should be noted that all the parameters were calculated without taking any post-translational modifications into consideration, and the content of Y was obtained by assuming that all the tyrosines in mfps were modified into Dopa. Me: Mytilus edulis; Mc: Mytilus californianus; Mg: Mytilus galloprovincialis. | ||||||||
| mfp-1 | Me | AY845258 | 62 | 10 | 18.7 | 20.4 | 48 | 9 |
| Mc | AY960601 | 83 | 10.1 | 19.2 | 20.8 | 47 | 10 | |
| mfp-2 | Me | AY845260 | 53 | 9.2 | 7.6 | 12.1 | 22 | 9 |
| Mc | KY627780 | 45 | 9.1 | 8.0 | 12.8 | 36 | 11 | |
| Mg | D43794 | 50 | 9.2 | 7.7 | 12.9 | 29 | 12 | |
| mfp-3 | Me | AF286136 | 6 | 10.4 | 20.8 | 4.2 | 20 | 13 |
| Mc | AY960607 | 5 | 10.2 | 23.8 | 16.7 | 40 | 14 | |
| Mg | AB049579 | 6 | 10.4 | 21.7 | 8.7 | 30 | 15 | |
| mfp-4 | Mc | DQ351535 | 88 | 10.2 | 2.3 | 5.5 | 0 | 16 |
| mfp-5 | Me | — | 9 | 9.9 | 27.4 | 20.5 | 55 | 17 |
| mfp-6 | Mc | DQ351537 | 12 | 8.9 | 22.2 | 8.1 | 23 | 18 |
To fully understand the synergistic effects between Dopa and lysine for wet adhesion, here we design Dopa and lysine containing polymers to measure the adhesion using standard lap shear experiments and the cohesion using rheology. We find that the presence of neighboring lysine residues can largely enhance surface adhesion, consistent with the “surface priming” mechanism revealed by SFA measurements. Moreover, we also confirm the sequence dependent “cooperative binding” mechanism of Dopa and lysine for adhesion performance discovered in single molecule AFM studies. Interestingly, the cohesive interactions, i.e. Fe3+–Dopa coordination and oxidative Dopa covalent cross-links, are also affected by the presence of neighboring positively charged residues. These studies provide new insights into the sequence dependent adhesion of Mfps and may inspire new biomimetic design of synthetic polymers decorated with Dopa motifs for strong wet adhesion.
The model peptides are depicted in Fig. 1A. We synthesized two model dipeptides (Lys–Dopa and Dopa–Lys) to reveal their sequence dependent synergistic effects for surface binding. We also synthesized two dipeptides without lysine residues (i.e. Gly–Dopa and Dopa–Gly) as the controls. The C-terminal carboxyl groups for all peptides except Dopa–Gly were blocked as esters to avoid the influence of negative charge on the binding. The lysine ε-amino group and catechol group of the peptides were protected with tert-butyloxocarbonyl (Boc) and acetonide (ac), respectively (Fig. 1B).
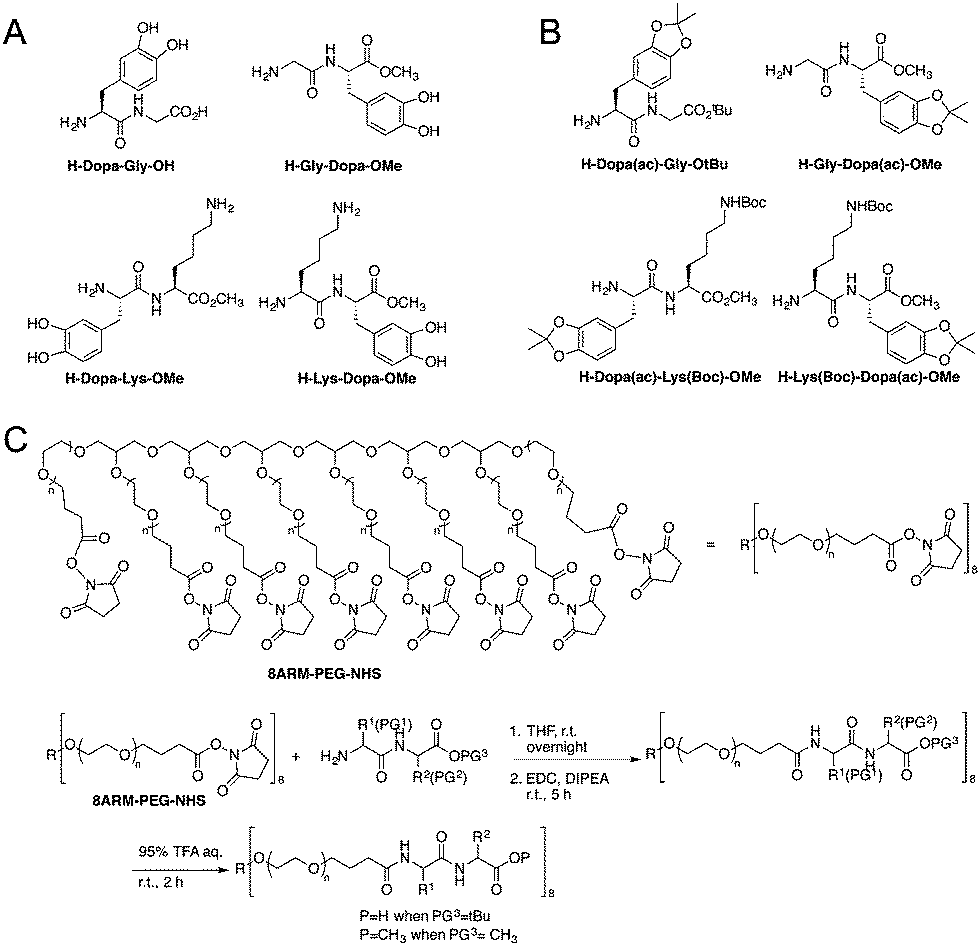 | ||
| Fig. 1 Structures of the four peptides used in this study and synthetic schemes for the peptide conjugated PEG polymers. (A) Structures of the peptides unattached to the polymer, (B) structure of peptide precursors for polymer derivatization, and (C) the reaction scheme for the polymer derivatization. | ||
To introduce the four peptides to an eight-armed poly(ethylene glycol) with terminal N-hydroxysuccinimide active ester (NHS) (8ARM-PEG-NHS, Mw: 20 kDa) respectively, the N-terminal amino group was deprotected by diethylamine to remove the fluorenylmethyloxocarbonyl (Fmoc) group from the precursor as shown in Scheme S1 (ESI†) and directly reacted with NHS on PEG (Fig. 1C). To ensure that all carboxylic acid reacted, equal amounts of a coupling reagent such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) were added to the reaction system to react for an additional 5 h. Then, the free catechol and lysine ε-amino group were release by TFA to yield the PEG–peptide conjugated polymers. The final products were dialyzed in degassed deionized water under argon protection to remove the small molecule byproducts and lyophilized before use. The detailed synthetic procedures can be found in the ESI† as shown in Schemes S1 and S2, along with the characterization.
To confirm that Dopa containing peptides were successfully conjugated to 8ARM-PEG, we measured the UV-vis spectra of all the PEG–peptide conjugates as well as the unreacted 8ARM-PEG-NHS. As shown in Fig. 2, all Dopa containing PEG–peptide complexes show characteristic absorption peaks of catechol groups at ∼280 nm. This peak is absent for 8ARM-PEG-NHS. Moreover, these are no additional peaks existing at wavelengths above 300 nm, indicating that Dopa has not been oxidized in the synthetic steps. Based on the absorbance of the polymer and the standard absorbance of catechol alone, the average conjugation ratios between peptide and PEG are estimated to be ∼8 for Dopa–Lys, 7.7 for Lys–Dopa, 6.4 for Gly–Dopa, and 7 for Dopa–Gly, respectively (the calibration curve based on dopamine at 280 nm is shown in Fig. S1, ESI†). Because the conjugation ratios are similar for all four peptides, the different adhesion and cohesion performance of the PEG–peptide complexes should solely result from the different sequences of the dipeptides.
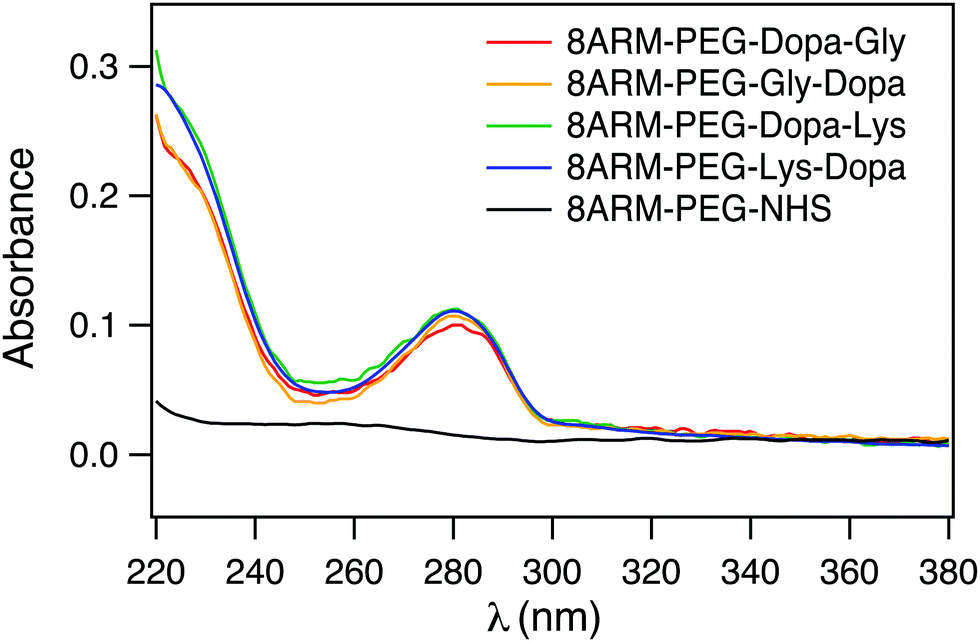 | ||
| Fig. 2 UV spectra of the peptide-conjugated PEG (1 mg mL−1 in ddH2O). | ||
Next, we measured the adhesion of the four peptides using standard lap shear experiments (Fig. 3A). Freshly prepared PEG–peptide solutions (30 μL, weight percentage of 10%, phosphate buffer, pH 8.0) were applied to the ends of a pair of glass slides to form a lap joint with an overlapping area of 25 mm × 20 mm. The samples were cured in an incubator at room temperature and 90% relative humidity for 24 h prior to the tearing test at a constant cross head speed of 1.0 mm min−1. The representative load-extension curves for the four samples are shown in Fig. 3B. Clearly, the four peptides showed distinct maximum separation forces, indicating different adhesion properties. After breaking the contact, the force did not return to zero, but remained at a high level, presumably due to the presence of friction between the two surfaces. The shear strengths measured based on multiple samples (≥6) are summarized in Fig. 3C. The PEG–peptide complexes with lysine showed significantly higher shear strength than that without lysine, presumably due to the displacement of hydrated salt ions by cationic lysine groups. Interestingly, 8ARM-PEG-Lys-Dopa showed much higher shear strength than 8ARM-PEG-Dopa-Lys, suggesting that the peptide sequence also plays an important role in Dopa–lysine synergistic binding. This finding is consistent with our previous single molecule AFM studies.8 We proposed that when being stretched from the N-terminus, Dopa–Lys and Lys–Dopa show distinct load distribution within the molecules, due to different lengths between anchoring points to the end surface binding groups (i.e. Dopa and lysine). The order of lysine and dopa within the polymer can have significant impacts on the binding strength. The lap shear experiments further confirmed this finding and provided a quantitative measure of the relative contributions between the two types of effects for synergistic adhesion (surface-priming and cooperative binding).
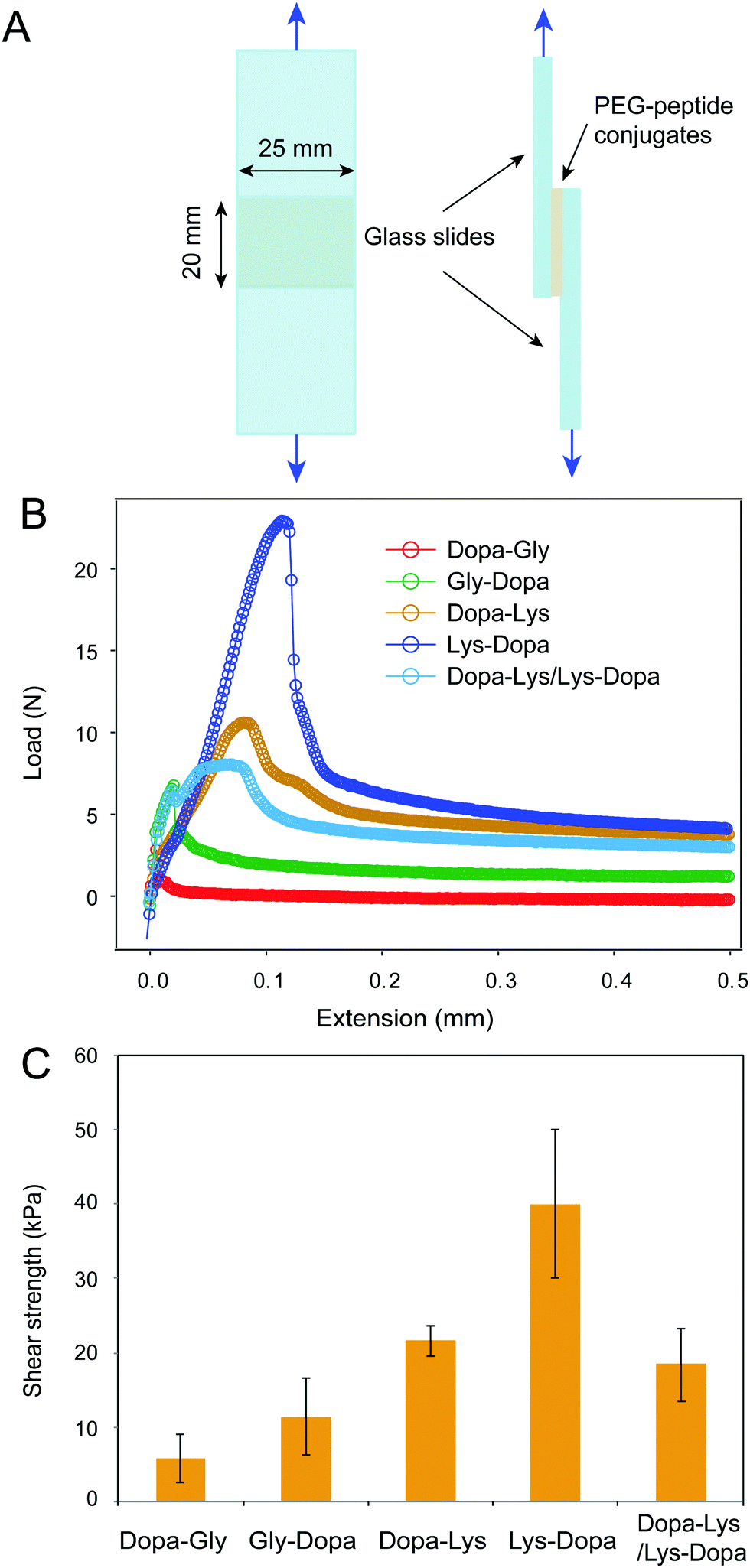 | ||
| Fig. 3 Lap shear analysis of the adhesion of peptide-conjugated PEG. (A) The scheme of the lap shear experiment, (B) representative lap shear curves for the peptide-conjugated PEG, and (C) summary of the shear strength for the peptide-conjugated PEG. For the Dopa–Lys/Lys–Dopa sample, the Dopa–Lys polymer construct was applied to one side and the Lys–Dopa on the other of the two glass slides. Error bars represent standard deviation. Number of samples: 6 for Dopa–Gly, 7 for Gly–Dopa, 7 for Dopa–Lys, 9 for Dopa–Lys and 7 for Dopa–Lys/Lys–Dopa. | ||
It was recently reported that cohesive cation–π interactions contribute strongly to interfacial adhesion.19 AFM images show that the height of the adhesive interlayer is about 300–400 nm for Dopa–Lys and Lys–Dopa in the lap shear experiments (Fig. S2, ESI†). The polymers remained on the surface were distributed inhomogeneously and the height was also not uniform, which may suggest a combined cohesion and adhesion failure. To further figure out whether the observed different adhesion strength between Dopa–Lys and Lys–Dopa was due to their different cohesive cation–π interactions instead of surface adhesion, we performed lap shear experiments with Dopa–Lys on one glass slide and Lys–Dopa on the other under the same experimental conditions. Because each slide was pre-covered by a single component, the separation of the two glass slides was mainly through the break of cohesive interactions, as confirmed by AFM imaging (Fig. S2, ESI†). The shear strength in this scenario was significantly lower than that for Lys–Dopa and slightly lower than that for Dopa–Lys (Fig. 3). Therefore, different cation–π interaction strength between Dopa–Lys and Lys–Dopa cannot fully explain the observed stronger adhesion for Dopa–Lys than that for Lys–Dopa. Note that, the failure mode in the hetero-surface experiments is distinct from that for the single component experiments (Fig. S2, ESI†). This may suggest that in the single component experiments, the cohesion is not only provided by cation–π interactions but also covalent cross-linking through oxidation.
We then set out to investigate how the intermolecular cohesion is affected by peptide sequences. We prepared hydrogels of the four PEG–peptide conjugates cross-linked by Fe3+ following the same procedure as reported in the literature.5d The PEG–peptide concentrations were fixed at 150 mg mL−1 and the Fe3+:Dopa ratios were fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Fe3+ can not only form chelation bonds with Dopa but also catalyze the oxidation of Dopa to form covalent cross-links. All four hydrogels were quickly formed within 2 min and in a blue color, but turned to dark brown when the hydrogels were aged for an additional 24 h, presumably due to the partial oxidation of Dopa (Fig. 4A and Fig. S2, ESI†). To reveal the intermolecular cross-linking mediated by Dopa in the four hydrogels, we measured their rheological properties (Fig. 4B). For covalent cross-links, the hydrogel network is expected to be permanent and the storage modulus (G′) is almost independent of frequency, while for dynamic cross-links, the G′ depends on the timescale comparable to the lifetime of the dynamic bonds. In the four hydrogels, the permanent covalent cross-links and transient Fe3+–Dopa coordination bonds coexist, as confirmed by the UV-vis spectroscopy (Fig. S4, ESI†). The G′ for Lys–Dopa and Dopa–Lys is significantly higher than that for Gly–Dopa and Dopa–Gly and is almost independent of angular frequency, indicating that lysine can also promote covalent cross-linking within the hydrogels. It is highly likely that the amino group of a lysine side chain participates in the cross-linking via Michael addition to the Dopa oxidized products (e.g. quinone). To fully confirm that lysine contributes to covalent cross-linking, NMR experiments should be performed in the future. The same G′ and G′′ for Lys–Dopa and Dopa–Lys suggest that the reactivity of the amino group of lysine is sequence independent even though the initial binding of Fe3+ to Dopa residues may still be sequence sensitive. Without lysine, the storage moduli (G′) for Gly–Dopa and Dopa–Gly are significantly lower. In particular, for Dopa–Gly, the G′ is the lowest and the hydrogel network is more dynamic with a frequency dependent G′, suggesting that Fe3+–Dopa coordination bonds are dominant to the covalent cross-links. The detailed molecular mechanism for different rheological behaviors for Gly–Dopa and Dopa–Gly is unknown and out of the scope of this work.
:1. Fe3+ can not only form chelation bonds with Dopa but also catalyze the oxidation of Dopa to form covalent cross-links. All four hydrogels were quickly formed within 2 min and in a blue color, but turned to dark brown when the hydrogels were aged for an additional 24 h, presumably due to the partial oxidation of Dopa (Fig. 4A and Fig. S2, ESI†). To reveal the intermolecular cross-linking mediated by Dopa in the four hydrogels, we measured their rheological properties (Fig. 4B). For covalent cross-links, the hydrogel network is expected to be permanent and the storage modulus (G′) is almost independent of frequency, while for dynamic cross-links, the G′ depends on the timescale comparable to the lifetime of the dynamic bonds. In the four hydrogels, the permanent covalent cross-links and transient Fe3+–Dopa coordination bonds coexist, as confirmed by the UV-vis spectroscopy (Fig. S4, ESI†). The G′ for Lys–Dopa and Dopa–Lys is significantly higher than that for Gly–Dopa and Dopa–Gly and is almost independent of angular frequency, indicating that lysine can also promote covalent cross-linking within the hydrogels. It is highly likely that the amino group of a lysine side chain participates in the cross-linking via Michael addition to the Dopa oxidized products (e.g. quinone). To fully confirm that lysine contributes to covalent cross-linking, NMR experiments should be performed in the future. The same G′ and G′′ for Lys–Dopa and Dopa–Lys suggest that the reactivity of the amino group of lysine is sequence independent even though the initial binding of Fe3+ to Dopa residues may still be sequence sensitive. Without lysine, the storage moduli (G′) for Gly–Dopa and Dopa–Gly are significantly lower. In particular, for Dopa–Gly, the G′ is the lowest and the hydrogel network is more dynamic with a frequency dependent G′, suggesting that Fe3+–Dopa coordination bonds are dominant to the covalent cross-links. The detailed molecular mechanism for different rheological behaviors for Gly–Dopa and Dopa–Gly is unknown and out of the scope of this work.
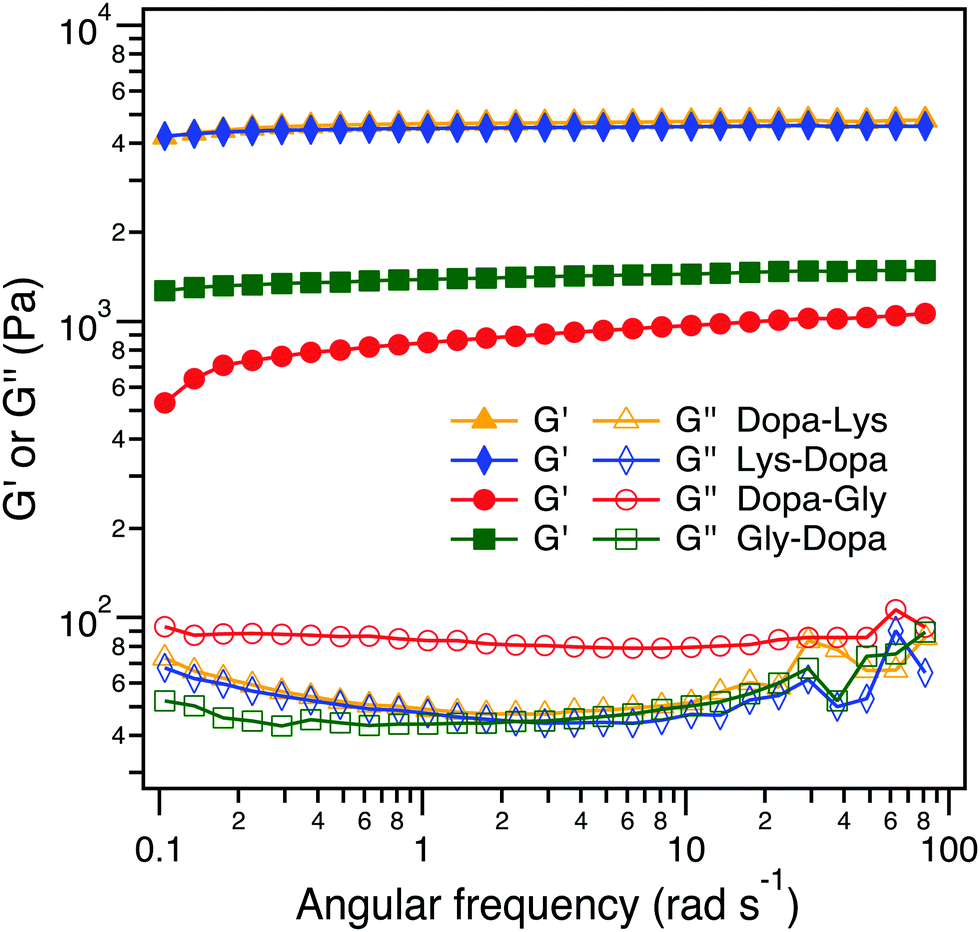 | ||
| Fig. 4 Rheological analysis of the peptide-conjugated PEG at the frequency sweep mode (10% strain at 20 °C). | ||
In summary, by designing a series of PEG–peptide conjugates, we investigate the synergistic effects of lysine and Dopa for wet surface adhesion in detail. Unlike natural Mfps, which contain multiple different interactions, our systems can be considered as a minimal model of such naturally occurring proteins and allow the effect of neighboring lysine residues on Dopa adhesion to be studied in a systematic way. We found that the adhesion of Dopa is indeed significantly enhanced with a neighboring lysine residue. Moreover, the order of Dopa and lysine is critical to such synergistic effects. These findings can be understood by the complex contributions of lysine to Dopa adhesion. It facilitates surface binding of Dopa by displacing surface bound water and ions; it directly binds to negatively charged surfaces in cooperation with Dopa binding; and it also promotes oxidative cross-linking of Dopa-containing polymers. These findings may enhance our current understanding of the design principle of natural adhesives and inspire new biomimetic efforts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (No. 21522402, 11304156, 11334004, 31170813, 81421091, and 91127026).Notes and references
- (a) J. H. Waite, Results Probl. Cell Differ., 1992, 19, 27–54 CrossRef CAS PubMed; (b) E. Bell and J. Gosline, J. Exp. Biol., 1996, 199, 1005–1017 Search PubMed.
- (a) B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed; (b) J. H. Waite, N. H. Andersen, S. Jewhurst and C. Sun, J. Adhes., 2005, 81, 297–317 CrossRef CAS; (c) R. Y. Floriolli, J. von Langen and J. H. Waite, Mar. Biotechnol., 2000, 2, 352–363 CAS; (d) B. K. Ahn, J. Am. Chem. Soc., 2017, 139, 10166–10171 CrossRef CAS PubMed.
- (a) B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed; (b) M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS PubMed; (c) H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed; (d) C. J. Kastrup, M. Nahrendorf, J. L. Figueiredo, H. Lee, S. Kambhampati, T. Lee, S. W. Cho, R. Gorbatov, Y. Iwamoto, T. T. Dang, P. Dutta, J. H. Yeon, H. Cheng, C. D. Pritchard, A. J. Vegas, C. D. Siegel, S. MacDougall, M. Okonkwo, A. Thai, J. R. Stone, A. J. Coury, R. Weissleder, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21444–21449 CrossRef CAS PubMed; (e) A. R. Statz, R. J. Meagher, A. E. Barron and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 7972–7973 CrossRef CAS PubMed; (f) J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS; (g) J. D. White and J. J. Wilker, Macromolecules, 2011, 44, 5085–5088 CrossRef CAS; (h) W. Wei, Y. Tan, N. R. Martinez Rodriguez, J. Yu, J. N. Israelachvili and J. H. Waite, Acta Biomater., 2014, 10, 1663–1670 CrossRef CAS PubMed; (i) N. R. Martinez Rodriguez, S. Das, Y. Kaufman, W. Wei, J. N. Israelachvili and J. H. Waite, Biomaterials, 2015, 51, 51–57 CrossRef CAS PubMed; (j) D. Wang, Y. Ha, J. Gu, Q. Li, L. Zhang and P. Yang, Adv. Mater., 2016, 28, 7414–7423 CrossRef CAS PubMed; (k) B. K. Ahn, S. Das, R. Linstadt, Y. Kaufman, N. R. Martinez-Rodriguez, R. Mirshafian, E. Kesselman, Y. Talmon, B. H. Lipshutz, J. N. Israelachvili and J. H. Waite, Nat. Commun., 2015, 6, 8663 CrossRef PubMed; (l) J. Zhou, Q. Xiong, J. Ma, J. Ren, P. B. Messersmith, P. Chen and H. Duan, ACS Nano, 2016, 10, 11066–11075 CrossRef CAS PubMed; (m) G. Fichman, L. Adler-Abramovich, S. Manohar, I. Mironi-Harpaz, T. Guterman, D. Seliktar, P. B. Messersmith and E. Gazit, ACS Nano, 2014, 8, 7220–7228 CrossRef CAS PubMed; (n) H. O. Ham, S. H. Park, J. W. Kurutz, I. G. Szleifer and P. B. Messersmith, J. Am. Chem. Soc., 2013, 135, 13015–13022 CrossRef CAS PubMed; (o) J. Su, F. Chen, V. L. Cryns and P. B. Messersmith, J. Am. Chem. Soc., 2011, 133, 11850–11853 CrossRef CAS PubMed; (p) H. Lee, Y. Lee, A. R. Statz, J. Rho, T. G. Park and P. B. Messersmith, Adv. Mater., 2008, 20, 1619–1623 CrossRef CAS PubMed; (q) X. M. Kang, W. H. Cai, S. Zhang and S. X. Cui, Polym. Chem., 2017, 8, 860–864 RSC.
- (a) H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed; (b) Y. Li, H. Liu, T. Wang, M. Qin, Y. Cao and W. Wang, ChemPhysChem, 2017, 18, 1466–1469 CrossRef CAS PubMed; (c) Y. Li, M. Qin, Y. Li, Y. Cao and W. Wang, Langmuir, 2014, 30, 4358–4366 CrossRef CAS PubMed; (d) Q. Lu, E. Danner, J. H. Waite, J. N. Israelachvili, H. Zeng and D. S. Hwang, J. R. Soc., Interface, 2013, 10, 20120759 CrossRef PubMed; (e) J. Yu, W. Wei, M. S. Menyo, A. Masic, J. H. Waite and J. N. Israelachvili, Biomacromolecules, 2013, 14, 1072–1077 CrossRef CAS PubMed.
- (a) M. J. Sever, J. T. Weisser, J. Monahan, S. Srinivasan and J. J. Wilker, Angew. Chem., Int. Ed., 2004, 43, 448–450 CrossRef CAS PubMed; (b) N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed; (c) D. E. Fullenkamp, D. G. Barrett, D. R. Miller, J. W. Kurutz and P. B. Messersmith, RSC Adv., 2014, 4, 25127–25134 RSC; (d) D. G. Barrett, D. E. Fullenkamp, L. He, N. Holten-Andersen, K. Y. Lee and P. B. Messersmith, Adv. Funct. Mater., 2013, 23, 1111–1119 CrossRef CAS PubMed; (e) Y. R. Li, J. Wen, M. Qin, Y. Cao, H. B. Ma and W. Wane, ACS Biomater. Sci. Eng., 2017, 3, 979–989 CrossRef CAS.
- Q. Lin, D. Gourdon, C. Sun, N. Holten-Andersen, T. H. Anderson, J. H. Waite and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3782–3786 CrossRef CAS PubMed.
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed.
- Y. R. Li, T. K. Wang, L. Xia, L. Wang, M. Qin, Y. Li, W. Wang and Y. Cao, J. Mater. Chem. B, 2017, 5, 4416–4420 RSC.
- US6987170B1, 2006.
- N. Holten-Andersen, H. Zhao and J. H. Waite, Biochemistry, 2009, 48, 2752–2759 CrossRef CAS PubMed.
- D. G. DeMartini, J. M. Errico, S. Sjoestroem, A. Fenster and J. H. Waite, J. R. Soc., Interface, 2017, 14 Search PubMed.
- K. Inoue, Y. Takeuchi, D. Miki and S. Odo, J. Biol. Chem., 1995, 270, 6698–6701 CrossRef CAS PubMed.
- S. C. Warner and J. H. Waite, Mar. Biol., 1999, 134, 729–734 CrossRef CAS.
- H. Zhao, N. B. Robertson, S. A. Jewhurst and J. H. Waite, J. Biol. Chem., 2006, 281, 11090–11096 CrossRef CAS PubMed.
- K. Inoue, Y. Takeuchi, D. Miki, S. Odo, S. Harayama and J. H. Waite, Eur. J. Biochem., 1996, 239, 172–176 CAS.
- H. Zhao and J. H. Waite, Biochemistry, 2006, 45, 14223–14231 CrossRef CAS PubMed.
- J. H. Waite and X. X. Qin, Biochemistry, 2001, 40, 2887–2893 CrossRef CAS PubMed.
- H. Zhao and J. H. Waite, J. Biol. Chem., 2006, 281, 26150–26158 CrossRef CAS PubMed.
- M. A. Gebbie, W. Wei, A. M. Schrader, T. R. Cristiani, H. A. Dobbs, M. Idso, B. F. Chmelka, J. H. Waite and J. N. Israelachvili, Nat. Chem., 2017, 9, 473–479 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00402h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |