
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient trifluoromethylation via the cyclopropanation of allenes and subsequent C–C bond cleavage†
Yang
Tang‡
a,
Qiong
Yu‡
b and
Shengming
Ma
 *ac
*ac
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China
bDepartment of Chemistry, East China Normal University, 3663 North Zhongshan Lu, Shanghai 200062, P. R. China
cLaboratory of Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P. R. China. E-mail: masm@sioc.ac.cn
First published on 23rd June 2017
Abstract
As we know, the incorporation of a trifluoromethyl group into organic molecules may significantly alter their physical and biological properties due to the high electronegativity, lipophilicity, and excellent metabolic stability of the trifluoromethyl substituent. Thus, an efficient method for the introduction of the trifluoromethyl group is of high current interest. On the other hand, vinylic cyclopropanes are a class of strained compounds capable of undergoing ring-opening reaction with other molecules. Here, CF3-substituted vinylic cyclopropanes have been highly selectively formed by a copper-catalyzed cyclic trifluoromethylation of (4,4-disubstituted-2,3-butadienyl)malonates with Togni's reagent II, in which the trifluoromethyl group was installed at the middle carbon of the allene unit by applying 1,10-phenanthroline as the ligand. Such unique cyclopropanes successfully bring the trifluoromethyl group to other useful organic skeletons by the selective cleavage of C–C bonds with an exclusive diastereoselectivity. Based on the mechanistic studies, an allene radical addition, oxidation, and allylic substitution pathway has been proposed.
Trifluoromethylated compounds have been widely used in all aspects of chemistry, such as materials, pharmaceuticals, agrochemicals, and fine chemicals due to the high electronegativity, lipophilicity, and excellent metabolic stability of the trifluoromethyl substituent.1–3 Of particular interest, compounds containing a 2,2,2-trifluoroethylcyclopropane unit have been identified as selective androgen receptor modulators,4a anti-inflammatory agents,4b,c immuno-modulators,4b and anti-tumor agents4c (Scheme 1). On the other hand, vinylcyclopropanes are the core structures of various pyrethroids such as pyrethrin,4d,e permethrin,4f,g cyhalothrin,4h and bifenthrin.4i Thus, we envisioned the structure of trifluoromethyl-substituted vinylcyclopropanes 2 and have been interested in developing methodologies for the efficient synthesis of this type of compound (Scheme 1). In addition, due to the high reactivity of the three-membered ring, such cyclopropanes 2 may also bring the trifluoromethyl group to other useful organic skeletons via the C–C bond cleavage reactions.5 To the best of our knowledge, there is no method for the construction of such CF3-substituted vinylic cyclopropanes and no report on their related reactivity study. We proposed a trifluoromethylative cyclization of allenes containing a malonate unit for the efficient synthesis of 2-type of compound (Scheme 1D).6–8 The challenge here would be the regioselectivity affording either the non-favored highly strained 3-membered products 2 or the most favored 5-membered products 3 as observed in the Pd-catalyzed cyclization of allenylmalonates with organic halides (Scheme 1C).9 Herein, we report our recent observation on the highly regioselective copper-catalyzed trifluoromethylation of (2,3-butadienyl)malonates using a hypervalent trifluoromethyl iodonium reagent, which allows for the exclusive formation of strained trifluoromethylated vinyl cyclopropanes 2.
 | ||
| Scheme 1 A and B) Some bioactive cyclopropanes containing a trifluoromethyl group; (C) Pd-catalyzed cyclization of allenyl malonates with organic halides; (D) concept of introducing a trifluoromethyl group via transition metal-catalyzed cyclopropanation. | ||
Our initial investigation started with the reaction of dimethyl 2-(buta-2,3-dienyl)malonate 1a with Togni's reagent II in the presence of 5 mol% of PdCl2 and 2 equiv. of K2CO3 in DCM at 50 °C, however, no trifluoromethylation product 2a was observed (Table 1, entry 1). Instead, a highly regioselective iodo-trifluoromethylation product 3a was detected albeit as a pair of Z/E stereoisomers.10 This clearly indicated that in this simplest case the trifluoromethyl group was directed to the terminal position of the allene unit in dimethyl 2-(buta-2,3-dien-1-yl)malonate 1a, excluding the possibility of forming the expected cyclized product 2a. Cu(I) or Cu(II) catalysts exhibited similar results, still affording low yields of 4a while the formation of 2a was not detected (Table 1, entries 2–4).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield of 4a (%) (Z/E)b | Recovery of 1ab (%) |
| a Reaction conditions: Unless otherwise specified, the reaction was carried out using 1a (0.2 mmol), Togni's reagent II (0.3 mmol), TBAI (0.3 mmol), K2CO3 (0.4 mmol), copper catalyst (0.03 mmol), and 2,2′-bipyridine (L1) (0.04 mmol) in 2 mL of CH2Cl2 under an argon atmosphere. b Determined by 19F NMR and 1H NMR spectroscopy using PhCF3 and CH2Br2 as the internal standards, respectively. c The reaction was carried out without bipyridine and TBAI. d The reaction was conducted on a 1 mmol scale of 1a with 2 equiv. of Togni's reagent II, K3PO4 (2 mmol) as the base, Cu(OAc)2 (10 mol%), and 1,10-phenanthroline (L2) (0.2 mmol) as the ligand; reaction time was 22 h. | |||
| 1 | PdCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
3 (2:1) |
48 |
| 2 | CuBr | 13 (1.5:1) |
26 |
| 3 | [Cu(CH3CN)4]PF6 | 20 (1.5:1) |
16 |
| 4 | Cu(OAc)2d |
33 (2.3:1) |
0 |
Thus, we envisioned to increase the steric hindrance of the terminal position of the allene unit in 1 for the possible direction of the trifluoromethyl group to the allene middle carbon atom to form a π-allylic metal species, which would be followed by nucleophilic substitution to possibly afford the cyclized products 2 or 3. When dimethyl 2-(buta-2,3-dien-1-yl)malonate 1a was replaced with dimethyl 2-(4-methyl-2,3-pentadienyl)malonate 1b, the reaction under the catalysis of Cu(OAc)2, CuCl2, CuF2·H2O and Cu(OTf)2 (Table 2, entries 1–4) did afford the designed trifluoromethylated vinylic cyclopropane 2b exclusively in moderate yields and the formation of the 5-membered ring 3b was not observed. Cu(OAc)2 gave the best results, affording 2b in 65% yield with 28% recovery of 1b (Table 2, entry 1). CuOAc could also catalyse the reaction with a slightly decreased yield (Table 2, entry 5). The reaction was even better with just 1.0 equiv. of TBAI (Table 2, entry 6). Rather unexpectedly, reducing the catalyst loading of Cu(OAc)2 from 15 mol% to 10 mol% improved the yield of 2b from 65% to 69% (Table 2, entry 7). When we further reduced the loadings of the catalyst and ligand, the yield dropped to 63% (Table 2, entry 8). By comparison, only 11% of product 2b was obtained in the absence of the copper complex (Table 2, entry 9).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield of 2bb (%) | Recovery of 1bb (%) |
| a Reaction conditions: Unless otherwise specified, the reaction was carried out using 1b (0.2 mmol), Togni's reagent II (0.3 mmol), TBAI (0.3 mmol), K2CO3 (0.4 mmol), copper catalyst (0.03 mmol), and 2,2′-bipyridine (L1) (0.04 mmol) in 2 mL of CH2Cl2 under an argon atmosphere. b Determined by 19F NMR and 1H NMR spectroscopy using PhCF3 and mesitylene or CH2Br2 as the internal standards. c The reaction was carried out using 1.0 equiv. of TBAI. d The reaction was carried out using 10 mol% of Cu(OAc)2. e The reaction was carried out with 5 mol% of Cu(OAc)2 and 10 mol% of L1. | |||
| 1 | Cu(OAc)2 | 65 | 28 |
| 2 | CuCl2 | 55 | 25 |
| 3 | CuF2·H2O | 35 | 24 |
| 4 | Cu(OTf)2 | 47 | 53 |
| 5 | CuOAc | 42 | 0 |
| 6 | Cu(OAc)2c |
65 | 15 |
| 7 | Cu(OAc)2c,d |
69 | 16 |
| 8 | Cu(OAc)2c,e |
63 | 14 |
| 9 | — | 11 | 51 |
|
|
||||
|---|---|---|---|---|
| Entry | L | Base | Yield of 2bb (%) | Recovery of 1bb (%) |
| a Reaction conditions: Unless otherwise specified, the reaction was carried out using 1b (0.2 mmol), Togni's reagent II (0.3 mmol), TBAI (0.2 mmol), base (0.4 mmol), Cu(OAc)2 (0.02 mmol), and ligand (0.04 mmol) in 2 mL of CH2Cl2 under an argon atmosphere. b Determined by 19F NMR and 1H NMR spectroscopy using PhCF3 and mesitylene or CH2Br2 as the internal standards. c The reaction was carried out with 2.5 equiv. of K3PO4. d The reaction was carried out with 2.0 equiv. of Togni's reagent II. e The reaction was carried out with 10 mol% of CuOAc instead of Cu(OAc)2. | ||||
| 1 | L1 | K2CO3 | 69 | 16 |
| 2 | L2 | K2CO3 | 77 | 13 |
| 3 | L3 | K2CO3 | 12 | 27 |
| 4 | L4 | K2CO3 | 42 | 19 |
| 5 | L5 | K2CO3 | 27 | 26 |
| 6 | — | K2CO3 | 10 | 34 |
| 7 | L2 | Na2CO3 | 63 | 17 |
| 8 | L2 | Cs2CO3 | 12 | 0 |
| 9 | L2 | K3PO4 | 82 | 9 |
| 10 | L2 | K3PO4c |
76 | 12 |
| 11 | L2 | K 3 PO 4 | 87 | 0 |
| 12 | L2 | — | 40 | 31 |
| 13d,e | L2 | K3PO4 | 42 | 0 |
With the optimized protocol in hand, we next turned to demonstrate the generality of this reaction. The results summarized in Table 4 show that this reaction indeed provides a straightforward entry to a series of trifluoromethylated vinyl cyclopropanes in moderate to good yields. Different substitutions at the terminal position of allene, such as methyl, ethyl, butyl, cyclobutyl, cyclopentyl and cyclohexyl could be compatible in this reaction, affording the corresponding trifluoromethylated vinyl cyclopropanes 2b–2j in moderate to good yields (Table 4, entries 1–9); the substrate with a sterically bulky isopropyl substituent also furnishes the corresponding product 2k in a moderate yield (Table 4, entry 10). Moreover, phenyl- and methyl-substituted malonate 1l afforded the corresponding product 2l in 66% yield with a ratio of 1:3 for the corresponding stereoisomers (Table 4, entry 11). To further demonstrate the potential of this reaction, we carried out this reaction on a gram scale under the standard conditions: when 1.0604 g of 1b was used, 1.1061 g of 2b was obtained in 79% yield.
|
|
||||
|---|---|---|---|---|
| Entry | R1, R2 | R3 | t (h) | Yield of 2b (%) |
|
a Reaction conditions: Unless otherwise specified, the reaction was carried out using 1 (1 mmol), Togni's reagent II (2.0 mmol), TBAI (1 mmol), K3PO4 (2 mmol), Cu(OAc)2 (0.10 mmol) and L2 (0.20 mmol) in 7 mL of CH2Cl2 under an argon atmosphere.
b Isolated yield.
c The reaction was carried out using 1b (5 mmol, 1.0604 g), Togni's reagent II (10.0 mmol), TBAI (5 mmol), and Cu(OAc)2 (0.5 mmol) and L2 (1.0 mmol) in 35 mL of CH2Cl2 under an argon atmosphere.
d The reaction was carried out using 3 equiv. of K3PO4 and 3 equiv. of Togni's reagent II.
e The reaction gave a pair of Z/E stereoisomers with a ratio of 1:3.
|
||||
| 1 | Me, Me | Me (1b) | 12 | 86 (79c)(2b) |
| 2 | –(CH2)5– | Me (1c) | 24 | 84 (2c) |
| 3 | Me, Me | Et (1d) | 13.5 | 82 (2d) |
| 4 | Me, Me | Bn (1e) | 12 | 74 (2e) |
| 5 | Et, Et | Et (1f) | 24 | 70 (2f) |
| 6 | n-Bu, n-Bu | Et (1g) | 24 | 79 (2g) |
| 7 | –(CH2)3– | Et (1h) | 17 | 64 (2h) |
| 8 | –(CH2)4– | Et (1i) | 15 | 60 (2i) |
| 9 | –(CH2)5– | Et (1j) | 13 | 74 (2j) |
| 10d | i-Pr, i-Pr | Et (1k) | 24 | 51 (2k) |
| 11 | Ph, Me | Me (1l) | 12 | 66e (2l) |
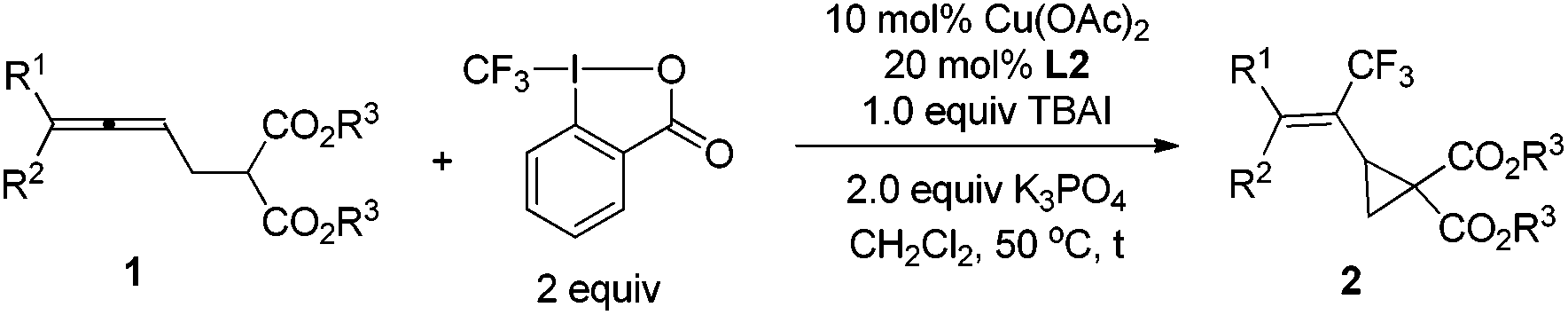
As stated in the introduction, one unique character of the strained three-membered ring is the selective cleavage of C–C bonds in cyclopropanes with an easy incorporation of other molecules to afford a series of complex molecules bearing the trifluoromethyl group. After some screening of the reported Lewis acid catalysts for such transformations,5,11 we observed that reactions catalyzed by 10 mol% of Sc(OTf)3 in DCE afforded the ring-opening products under very milder conditions: when trifluoromethylated vinylic cyclopropane 2b was exposed to benzaldehyde, a highly substituted tetrahydrofuran product cis-5a12 was formed highly diastereoselectively in 85% yield; with nitrone, cis-tetrahydro-1,2-oxazines cis-6a12 and cis-6b were formed in 81% and 84% yields from 2b and 2c, respectively; the reaction of 2b with N-methylindole afforded the ring-opened functionalized indole product 7a12 in 90% yield (Scheme 2 and Fig. 1).
 | ||
| Scheme 2 Sc(OTf)3-catalyzed reactions of 2. | ||
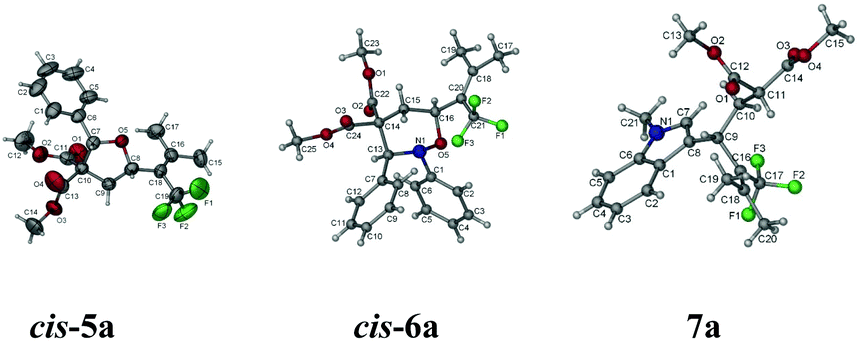 | ||
| Fig. 1 ORTEP representation of cis-5a, cis-6a, and 7a. | ||
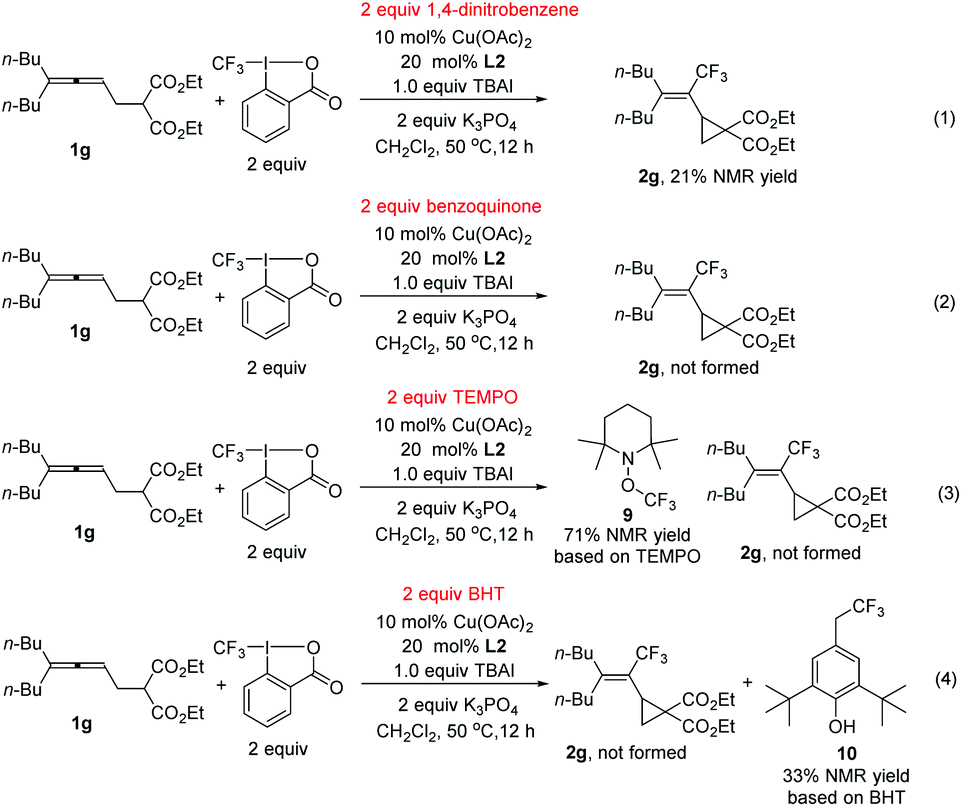
During the study, we also identified the Togni's reagent II-based by-product by the X-ray diffraction study unambiguously as methylene bis(2-iodobenzoate) 8 (Scheme 3A).12 Control experiments showed that in the absence of the copper complex, potassium 2-iodobenzoate didn't react with CH2Cl2 (Scheme 3B, eqn (1)). When potassium 2-iodobenzoate was exposed under the standard conditions without the allene and K3PO4, compound 8 was formed in 31% yield. In order to further study the mechanism, radical scavengers were added under the standard reaction conditions (Scheme 4). With 1,4-dinitrobenzene, the reaction was somewhat suppressed to yield 21% of 2g (Scheme 4, eqn (1)). With benzoquinone, the trifluoromethylative cyclization reaction was completely shut down (Scheme 4, eqn (2)). With TEMPO, the reaction didn't occur and the radical trapping TEMPO-CF3 adduct 9 was formed in 71% yield (Scheme 4, eqn (3)). With BHT, the trifluoromethylative product 2g was not formed while a BHT-CF3 adduct 10 was observed in 33% yield as judged by the analysis of the crude product comparing the signals with those reported in the literature13 (Scheme 4, eqn (4)). These results indicated that the reaction may proceed via a radical pathway in the beginning.
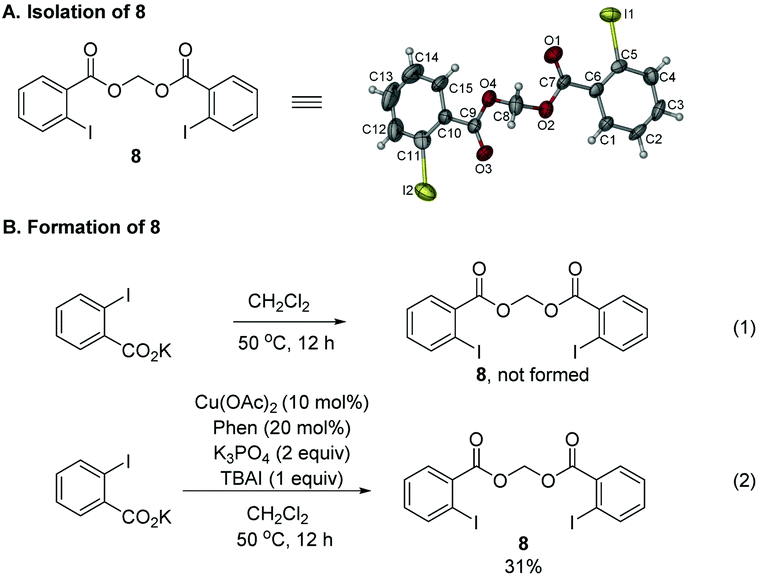 | ||
| Scheme 3 Formation of by-product 8: (A) ORTEP representation. (B) Control experiments. | ||
 | ||
| Scheme 4 Radical trapping experiments. | ||
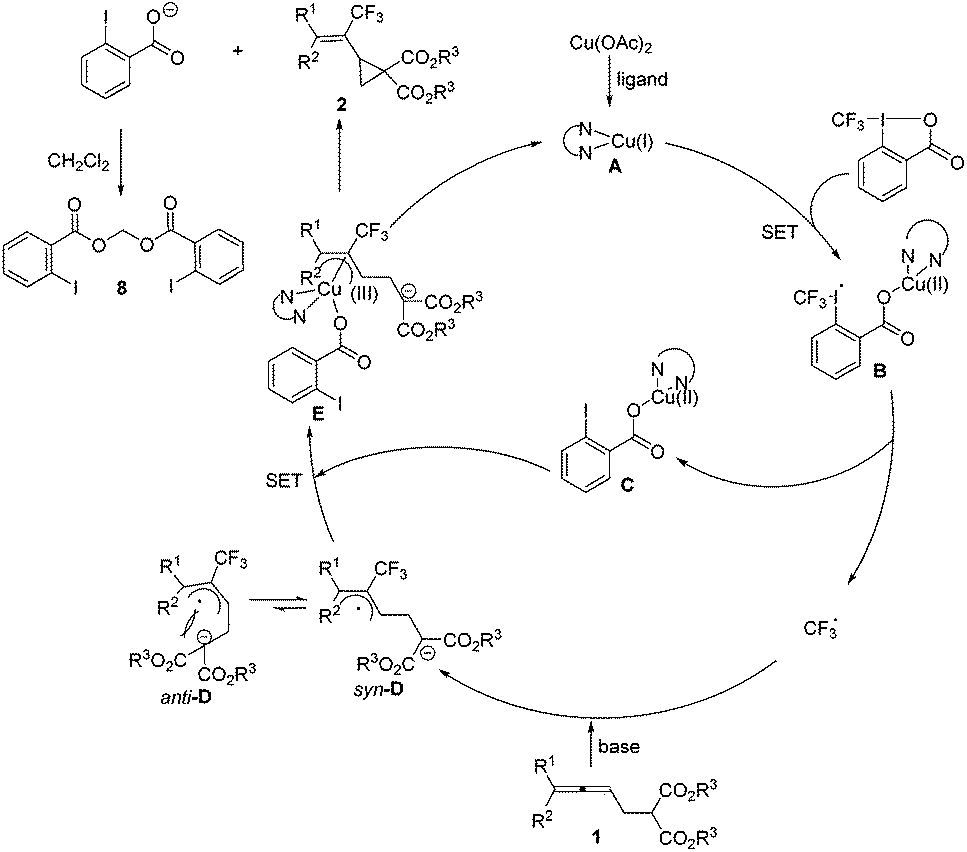
A mechanism was then proposed on the basis of the above results (Scheme 5). Initially, the in situ reduction14 or disproportionation15 of Cu(OAc)2 forms the highly reactive Cu(I), which would coordinate with the ligand forming a catalytically active copper(I) species A. Then a radical intermediate B could be generated by the reaction of A with Togni's reagent II, which would further release the CF3 radical and (2-iodobenzoyloxy)copper(II) C. Allene 1 would be attacked by the trifluoromethyl radical and its nucleophilic unit would be deprotonated with the base to form the thermodynamically more stable π-allylic radical syn-D. The intermediate syn-D would further undergo oxidation with Cu(II) species C yielding the π-allylic copper(III) intermediate E, which would undergo an intramolecular nucleophilic attack to release the cyclopropane products 2 and the o-iodobenzoic acid anion associated with the regeneration of the catalytically active Cu(I) species A. The reaction of two molecules of the o-iodobenzoic acid anion with CH2Cl2 would generate the isolated by-product 8.16 The unfavored formation of anti-D excludes the formation of 3-type of a 5-membered ring. However, it should be noted that the mechanism requires more studies and there may be other possibilities.
 | ||
| Scheme 5 Proposed mechanism. | ||
In conclusion, we have demonstrated an efficient copper-catalyzed introduction of a trifluoromethyl group into organic skeletons through the cyclization of allenes and C–C bond cleavage-based transformations via the formation of the strained trifluoromethylated vinyl cyclopropanes with an excellent regioselectivity under ambient conditions. Further studies in this area are ongoing in our laboratory.
Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 21690063) and the National Natural Basic Research Program of China (2015CB856600) is greatly appreciated.Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (c) K. Uneyama, Organofluorine Chemistry, Blackwell, Oxford, U. K., 2006 Search PubMed; (d) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, U. K., 2009 Search PubMed; (e) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (f) M. Hird, Chem. Soc. Rev., 2007, 36, 2070 RSC; (g) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS; (h) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (i) R. Filler and R. Saha, Future Med. Chem., 2009, 1, 777 CrossRef CAS PubMed; (j) J. L. Aceña, S.-A. Fuentes and S. Fustero, Curr. Org. Chem., 2010, 14, 928 CrossRef; (k) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC.
- For selected reviews, see: (a) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) D. Cahard and V. Bizet, Chem. Soc. Rev., 2014, 43, 135 RSC; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed.
- For selected recent reviews on trifluoromethylation, see: (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (e) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (f) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (g) C. Alonso, E. Marigorta de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (h) X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163 RSC. For selected recent reports on trifluoromethylation of alkenes with CF3+, see: (i) A. T. Parsons, T. D. Senecal and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 2947 CrossRef CAS PubMed; (j) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2013, 52, 12414 CrossRef CAS PubMed; (k) Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270 CrossRef CAS PubMed; (l) W. Kong, N. Fuentes, A. García-Domínguez, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 2487 CrossRef CAS PubMed; (m) B. Sahoo, J.-L. Li and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 11577 CrossRef CAS PubMed; (n) M. Asano, R. Tomita, T. Koike and M. Akita, J. Fluorine Chem., 2015, 179, 83 CrossRef CAS; (o) Q. Lefebvre, N. Hoffmann and M. Rueping, Chem. Commun., 2016, 52, 2493 RSC; (p) L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed; (q) T. Kawamoto, R. Sasaki and A. Kamimura, Angew. Chem., Int. Ed., 2017, 56, 1342 CrossRef CAS PubMed. For selected recent reports on trifluoromethylation of arenes with CF3+, see: (r) K. Zhang, X.-H. Xu and F.-L. Qing, J. Org. Chem., 2015, 80, 7658 CrossRef CAS PubMed; (s) L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809 CrossRef CAS PubMed; (t) J. Lin, Z. Li, J. Kan, S. Huang, W. Su and Y. Li, Nat. Commun., 2017, 8, 14353, DOI:10.1038/ncomms14353. For selected recent reports on trifluoromethylation of other compounds with CF3+, see: (u) Vinyl azides: Y.-F. Wang, G. H. Lonca and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 1067 CrossRef CAS PubMed; (v) 3-Alkenoic acids: Z. He, P. Tan and J. Hu, Org. Lett., 2016, 18, 72 CrossRef CAS PubMed; (w) Enol triflates: X. Su, H. Huang, Y. Yuan and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 1338 CrossRef CAS PubMed.
- (a) A. L. Handlon, L. T. Schaller, L. M. Leesnitzer, R. V. Merrihew, C. Poole, J. C. Ulrich, J. W. Wilson, R. Cadilla and P. Turnbull, ACS Med. Chem. Lett., 2016, 7, 83 CrossRef CAS PubMed; (b) B. J. M. G. Cals and S. B. Nabuurs, PCT Int. Appl, WO2015082533A120150611, 2015 Search PubMed; (c) R. Kaul, E. J. McEachern, J. Sun, D. J. Vocadlo, Y. Zhou and Y. Zhu, PCT Int. Appl, WO2015095963A120150702, 2015 Search PubMed; (d) F. B. Laforge and W. F. Barthel, J. Org. Chem., 1947, 12, 199 CrossRef CAS PubMed; (e) L. Crombie, M. Elliott, S. H. Harper and H. W. B. Reed, Nature, 1948, 162, 222 CrossRef CAS PubMed; (f) A. H. Glickman, T. Shono, J. E. Casida and J. J. Lech, J. Agric. Food Chem., 1979, 27, 1038 CrossRef CAS PubMed; (g) G. W. Ivie and L. M. Hunt, J. Agric. Food Chem., 1980, 28, 1131 CrossRef CAS PubMed; (h) P. D. Bentley, R. Cheetham, R. K. Huff, R. Pascoe and J. D. Sayle, Pestic. Sci., 1980, 11, 156 CrossRef CAS; (i) E. L. Plummer and R. R. Stewart, J. Agric. Food Chem., 1984, 32, 1116 CrossRef CAS.
- For selected reviews on the chemistry of cyclopropanes, see: (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (c) F. Brackmann and A. de Meijere, Chem. Rev., 2007, 107, 4538 CrossRef CAS PubMed; (d) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (e) B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318 RSC.
- For reviews, see: (a) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 CrossRef CAS PubMed; (b) J. A. Marshall, Chem. Rev., 2000, 100, 3163 CrossRef CAS PubMed; (c) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590 CrossRef CAS; (d) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (e) R. W. Bates and V. Satchareon, Chem. Soc. Rev., 2002, 31, 12 RSC; (f) H. U. Reissig, W. Schade, G. M. O. Amombo, R. Pulz and A. Hausherr, Pure Appl. Chem., 2002, 74, 175 CrossRef CAS; (g) L. K. Sydnes, Chem. Rev., 2003, 103, 1133 CrossRef CAS PubMed; (h) S. Ma, Acc. Chem. Res., 2003, 36, 701 CrossRef CAS PubMed; (i) L. Brandsma and N. A. Nedolya, Synthesis, 2004, 735 CrossRef CAS; (j) L. Wei, H. Xiong and R. Hsung, Acc. Chem. Res., 2003, 36, 773 CrossRef CAS PubMed; (k) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 CrossRef PubMed; (l) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed; (m) S. Ma, Aldrichimica Acta, 2007, 40, 91 CAS; (n) S. Ma, Acc. Chem. Res., 2009, 42, 1679 CrossRef CAS PubMed; (o) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (p) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed; (q) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef CAS PubMed.
- (a) K. Tsuchii, M. Imura, N. Kamada, T. Hirao and A. Ogawa, J. Org. Chem., 2004, 69, 6658 CrossRef CAS PubMed; (b) N. Zhu, F. Wang, P. Chen, J. Ye and G. Liu, Org. Lett., 2015, 17, 3580 CrossRef CAS PubMed; (c) Y. Wang, M. Jiang and J. Liu, Adv. Synth. Catal., 2014, 356, 2907 CrossRef CAS; (d) Z. He, P. Tan and J. Hu, Org. Lett., 2016, 18, 72 CrossRef CAS PubMed; (e) J. Jacquet, S. Blanchard, E. Derat, M. D.-E. Murr and L. Fensterbank, Chem. Sci., 2016, 7, 2030 RSC; (f) R. Tomita, T. Koike and M. Akita, Chem. Commun., 2017, 53, 4681 RSC.
- For formation of γ-lactones: Q. Yu and S. Ma, Chem. – Eur. J., 2013, 19, 13304 CrossRef CAS PubMed.
- (a) S. Ma and Z. Zhao, Org. Lett., 2000, 2, 2495 CrossRef CAS PubMed; (b) S. Ma, N. Jiao, S. Zhao and H. Hou, J. Org. Chem., 2002, 67, 2837 CrossRef CAS PubMed; (c) S. Ma, N. Jiao, Q. Yang and Z. Zheng, J. Org. Chem., 2004, 69, 6463 CrossRef CAS PubMed.
- For selected examples on iodo-trifluoromethylation using Togni's reagent, see: (a) P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef CAS PubMed; (b) N. O. Ilchenko, P. G. Janson and K. J. Szabó, J. Org. Chem., 2013, 78, 11087 CrossRef CAS PubMed; (c) H. Egami, Y. Usui, S. Kawamura, S. Nagashima and M. Sodeoka, Chem. – Asian J., 2015, 10, 2190 CrossRef CAS PubMed.
- (a) P. D. Phhlhaus and J. S. Johnson, J. Org. Chem., 2005, 70, 1057 CrossRef PubMed; (b) I. S. Young and M. A. Kerr, Angew. Chem., Int. Ed., 2003, 42, 3023 CrossRef CAS PubMed; (c) M. A. Kerr and R. G. Keddy, Tetrahedron Lett., 1999, 40, 5671 CrossRef CAS.
- For X-ray single crystal data for cis-5a, cis-6a, 7a, and 8, see the ESI.†.
- H. Egami, T. Ide, Y. Kawatoa and Y. Hamashima, Chem. Commun., 2015, 51, 16675 RSC.
- For reduction, see: (a) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2860 CrossRef CAS PubMed; (b) G. Li, C. Jia, K. Sun, Y. Lv, F. Zhao, K. Zhou and H. Wu, Org. Biomol. Chem., 2015, 13, 3207 RSC; (c) X.-F. Xia, S.-L. Zhu, J.-B. Liu, D. Wang and Y.-M. Liang, J. Org. Chem., 2016, 81, 12482 CrossRef CAS PubMed.
- For disproportionation, see: (a) X. Ribas, D. A. Jackson, B. Donnadieu, J. Mahía, T. Parella, R. Xifra, B. Hedman, K. O. Hodgson, A. Llobet and T. D. P. Stack, Angew. Chem., Int. Ed., 2002, 41, 2991 CrossRef CAS; (b) L. M. Huffman and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 9196 CrossRef CAS PubMed; (c) A. Casitas, A. E. King, T. Parella, M. Costas, S. S. Stahl and X. Ribas, Chem. Sci., 2010, 1, 326 RSC.
- Y. Zhang, J. Han, Y. Xu and Y. Wei, J. Chem. Res., 2012, 36, 303 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation and characterisation data as well as 1H and 13C NMR spectra of all compounds. CCDC 1012501, 1012706, 1012812 and 1012813. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00419b |
| ‡ These two authors contributed equally. |
| This journal is © the Partner Organisations 2017 |