
Fe-Catalyzed insertion of fluoromethylcarbenes generated from sulfonium salts into X–H bonds (X = Si, C, P)†
Yaya
Duan
 a,
Jin-Hong
Lin
*a,
Ji-Chang
Xiao
*a and
Yu-Cheng
Gu
b
a,
Jin-Hong
Lin
*a,
Ji-Chang
Xiao
*a and
Yu-Cheng
Gu
b
aKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: jchxiao@sioc.ac.cn; jlin@sioc.ac.cn; Tel: +86-21-54925340 Tel: (+86)21-5492-5541
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 20th June 2017
Abstract
The Fe-catalyzed insertion of fluoromethylcarbenes including trifluoromethylcarbene and difluoromethylcarbene generated in situ from sulfonium salts (Ph2S+CH2CF3−OTf and Ph2S+CH2CF2H −OTf) into X–H (X = Si, C and P) bonds is described. The insertion of both carbenes into the Si–H bond occurred smoothly, and trifluoromethylcarbene could also insert into C–H and P–H bonds.
Fluoromethylcarbenes, including trifluoromethylcarbene (CF3CH:)1 and difluoromethylcarbene (HCF2CH:),2 have proven to be attractive synthetic tools for the incorporation of trifluoromethyl (CF3) and difluoromethyl (HCF2) fragments, both of which are valuable functionalities in medicinal chemistry,3 materials chemistry,4 and so on. Although X–H bond (X = C, Si, etc.) functionalization has received a great deal of attention due to its high efficiency and atom economy, and significant efforts have been directed toward the development of efficient methods for the insertion of carbenes into the X–H bond,5 the insertion of fluoromethylcarbenes into the X–H bond remains challenging.
Trifluoromethyldiazomethane (CF3CHN2)1d,g,6 and difluoromethyldiazomethane (HCF2CHN2)2,7 have served as versatile intermediates in a variety of transformations. It was recently found that they can act as a trifluoromethylcarbene precursor1 and a difluoromethylcarbene precursor,2 respectively. But the insertion of fluoromethylcarbenes into the X–H bond has been limited to CF3CHN2.1h–j In 2012, the group of Ma reported that the Cu-catalyzed insertion of trifluoromethylcarbene produced from CF3CHN2 into the Csp–H bond occurred smoothly to afford the desired product in high yields.1h In 2015, Wang and co-workers described the insertion into N–H and O–H bonds catalyzed by a silver complex.1i Shortly afterwards, Gouverneur et al. found that the insertion strategy could be successfully applied to Si–H, B–H, P–H, S–H, and N–H bonds.1j Apparently, CF3CHN2 is efficient for the insertion into X–H bonds (X = C, Si, P, etc.). However, it is a potentially explosive and toxic gas, limiting its synthetic utility. Therefore, the development of mild protocols for the insertion of fluoromethylcarbenes into X–H bonds is highly desirable.
We have shown that fluorinated carbenes can be produced from fluorinated ylides including phosphonium ylides8 and sulfonium ylides9 under mild conditions. On the basis that trifluoromethyl sulfonium ylide (Ph2S+CH−CF3) and difluoromethyl sulfonium ylide (Ph2S+CH−CF2H) could be converted by FeCl(TPP) into trifluoromethylcarbene (Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCF3)9a and difluoromethylcarbene (FeCHCF2H),9c respectively, we have now investigated the use of both sulfonium ylides as fluoromethylcarbene precursors in the insertion into X–H bonds (X = Si, C, and P). Ylides Ph2S+CH−CF3 and Ph2S+CH−CF2H were in situ generated from sulfonium salts Ph2S+CH2CF3−OTf (I) and Ph2S+CH2CF2H −OTf (II), respectively, via deprotonation by CsF.
CHCF3)9a and difluoromethylcarbene (FeCHCF2H),9c respectively, we have now investigated the use of both sulfonium ylides as fluoromethylcarbene precursors in the insertion into X–H bonds (X = Si, C, and P). Ylides Ph2S+CH−CF3 and Ph2S+CH−CF2H were in situ generated from sulfonium salts Ph2S+CH2CF3−OTf (I) and Ph2S+CH2CF2H −OTf (II), respectively, via deprotonation by CsF.
We previously found that a reductant was not required in the Fe-catalyzed transformation of trifluoromethylcarbene,9a but it was necessary in the reaction of difluoromethylcarbene.9c Interestingly, in the Fe-catalyzed insertion of trifluoromethylcarbene into the Si–H bond in DMA, the presence of the reductant Na2S2O4 could slightly increase the yield (Table 1, entry 3 vs. 1). A comparable yield was obtained in DMF (entry 4). Increasing the loading of salt I and CsF could lead to the increase in the yields (entries 6–8 vs. 3). The yield was further increased slightly by increasing the amount of the catalyst FeCl(TPP) (TPP = 5,10,15,20-tetraphenyl-21H,23H-porphine) from 1 mol% to 2 mol% (entry 9 vs. 8). However, 3 mol% of catalyst loading did not increase the yield (entry 10 vs. 9). The absence of the reductant Na2S2O4 resulted in a lower yield (entry 11 vs. 9). Room temperature was found to be the appropriate reaction temperature. Irrespective of whether the temperature was elevated or lowered, the yields were decreased (entries 12–14 vs. 9).
|
|
||||
|---|---|---|---|---|
| Entry | Reductant | x | Ratiob | Yieldc (%) |
a Reaction conditions: 1a (0.2 mmol), sulfonium salt I, FeCl(TPP), reductant, and CsF in DMA (1.5 mL) at rt for 2 h.
b Molar ratio of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :salt I:CsF.
c The yields were determined by 19FNMR.
d DMF was used as the reaction solvent instead of DMA.
e The reaction temperature was 40 °C.
f The reaction temperature was 50 °C.
g The reaction temperature was 0 °C. :salt I:CsF.
c The yields were determined by 19FNMR.
d DMF was used as the reaction solvent instead of DMA.
e The reaction temperature was 40 °C.
f The reaction temperature was 50 °C.
g The reaction temperature was 0 °C.
|
||||
| 1 | — | 1 | 1:1:1.1 |
41 |
| 2 | Zn | 1 | 1:1.1:1.2 |
43 |
| 3 | Na2S2O4 | 1 | 1:1.1:1.2 |
50 |
| 4d | Na2S2O4 | 1 | 1:1.1:1.2 |
45 |
| 5 | Na2S2O4 | 1 | 1.3:1:1.2 |
56 |
| 6 | Na2S2O4 | 1 | 1:1.3:1.5 |
66 |
| 7 | Na2S2O4 | 1 | 1:1.5:1.8 |
70 |
| 8 | Na2S2O4 | 1 | 1:2:2.5 |
79 |
| 9 | Na2S2O4 | 2 | 1:2:2.5 |
83 |
| 10 | Na2S2O4 | 3 | 1:2:2.5 |
81 |
| 11 | — | 2 | 1:2:2.5 |
70 |
| 12e | Na2S2O4 | 2 | 1:2:2.5 |
53 |
| 13f | Na2S2O4 | 2 | 1:2:2.5 |
45 |
| 14g | Na2S2O4 | 2 | 1:2:2.5 |
64 |
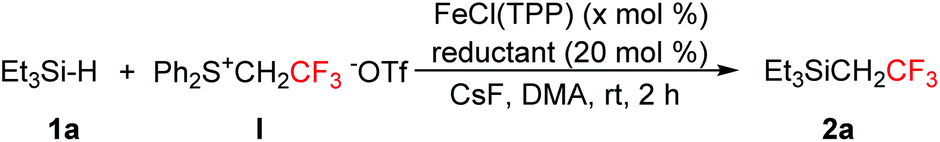
With the optimal reaction conditions in hand (Table 1, entry 9), we then investigated the substrate scope for the insertion of fluoromethylcarbenes into the Si–H bond. As shown in Scheme 1, trialkylsilanes could be smoothly converted into the desired products in moderate to good yields (2a–2g). Severe steric effects would result in the complete suppression of Si–H bond insertion (2h). For a slightly hindered substrate, a moderate yield was obtained under slightly modified reaction conditions (2i). Phenylsilane showed a much lower reactivity and the modified conditions gave the expected product in only 50% yield (2j). The product was so volatile that we failed to isolate it from the reaction solvent toluene. Besides trifluoromethylcarbene, difluoromethylcarbene could also be inserted well into the Si–H bond (2k–2m).
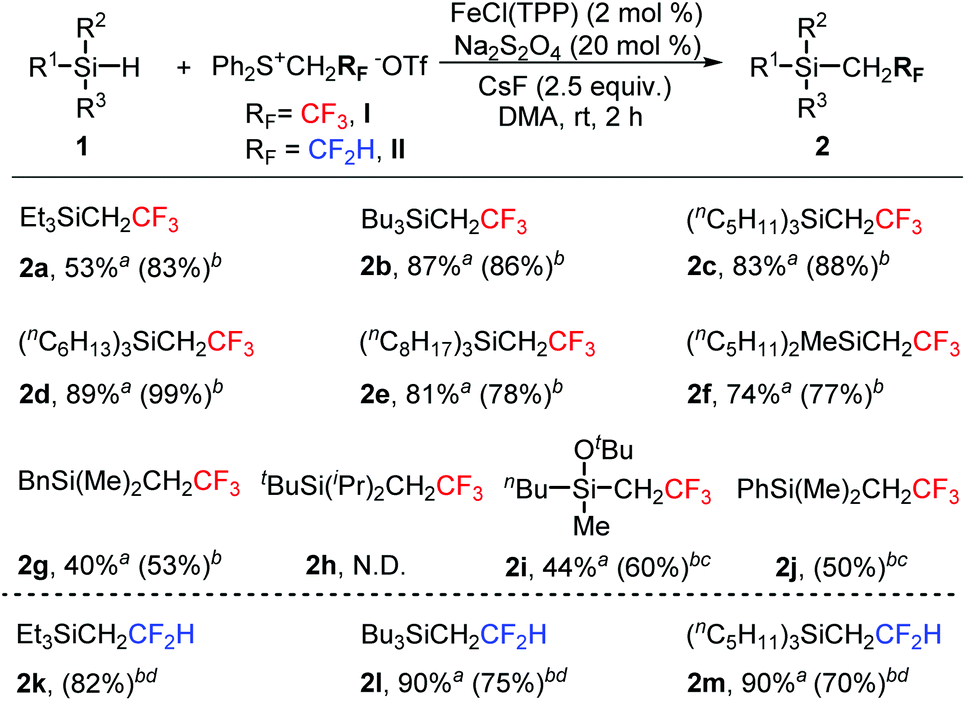 | ||
| Scheme 1 Substrate scope for the insertion of methylcarbenes into the Si–H bond. aIsolated yields; bthe yields in parentheses were determined by 19F NMR spectroscopy; cCs2CO3 was used instead of CsF, toluene was used instead of DMA, and the reaction temperature was 80 °C; dZn was used instead of Na2S2O4 and DMF was used instead of DMA. | ||
Organosilicon compounds are highly attractive scaffolds and have found widespread applications in organic synthesis,10 materials chemistry,11 and pharmaceuticals.12 Si–H bond functionalization is one of the most straightforward protocols to synthesize organosilicon derivatives. This carbene insertion strategy is worth paying attention since it allows for the convenient formation of the Si–C bond and the incorporation of fluoromethyl groups.
Inert C–H bond functionalization is a challenging research area and a powerful tool for organic synthesis.13 The insertion of fluoromethylcarbenes into the inert Csp3–H bond was also investigated. Although a large number of reaction conditions were screened, we could not identify optimal conditions to obtain a high yield (see the ESI†). Fortunately, we found that moderate yields could be obtained for the insertion of trifluoromethylcarbene into the benzyl C–H bond (Scheme 2, 4a–4b). For the alkyl C–H bond, the yield was quite low (4c). In these reactions, substrates 3 have to be used as the reaction solvent. Therefore, it was quite difficult to isolate the products from substrates 3 due to their similar polarity and the high volatility of products 4.
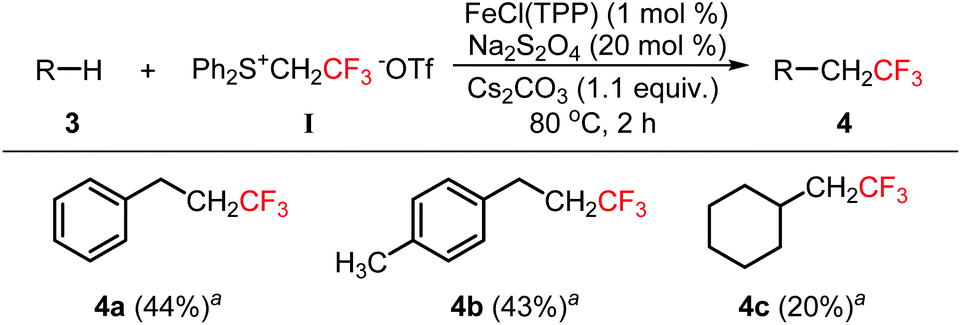 | ||
| Scheme 2 Insertion of trifluoromethylcarbene into the inert C–H bond. aThe yields were determined by 19F NMR spectroscopy. | ||
Phosphines are widely used in synthetic chemistry. For example, they can act as ligands in organometallic chemistry,14 and as catalysts in the Morita–Baylis–Hillman reaction.15 P–H functionalization is apparently an efficient strategy to prepare organophosphines. Trifluoromethylcarbene was found to be able to insert into the P–H bond to furnish the desired product in a moderate yield (Scheme 3).
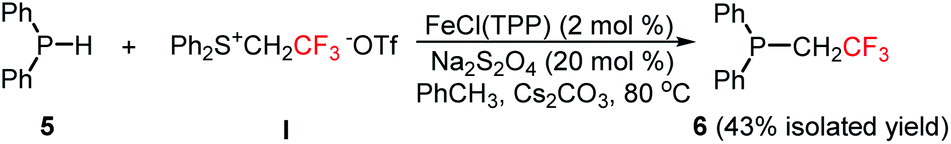 | ||
| Scheme 3 Insertion of trifluoromethylcarbene into the P–H bond. | ||
Other X–H (X = N or S) bond insertions were also investigated. For the insertion into the N–H bond in arylamines such as 4-chlorophenylamine (4-ClC6H4NH2), no desired product was produced. The S–H bond in thiophenol (PhSH) could react with sulfonium salt I to give sulfur ether (PhSCH2CF3). But the reaction may not proceed via trifluoromethylcarbene insertion into the S–H bond since thiophenol could act as a nucleophile to directly attack salt I under basic conditions.
Based on the above results and our previous observations,9a,c we propose that the reaction may proceed through a concerted X–H insertion. The Fe-carbene (FeCHRF, RF = CF3 or CF2H) generated in situ is highly reactive, and thus would be readily trapped by the X–H bond (Scheme 4). The cleavage of the X–H bond and the formation of C–H and C–X bonds would occur simultaneously to give the final products.
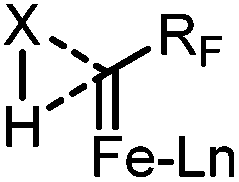 | ||
| Scheme 4 The proposed transition state. | ||
Conclusions
In conclusion, we have described the Fe-catalyzed insertion of fluoromethylcarbenes including trifluoromethylcarbene and difluoromethylcarbene into Si–H, C–H and P–H bonds. This work represents the first protocol for the insertion of difluoromethylcarbene into the Si–H bond, and the mild strategy for the insertion of fluoromethylcarbenes into X–H (X = Si, C and P) bonds. The fluoromethylcarbene insertion strategy may find synthetic utility in other research areas.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the National Basic Research Program of China (2015CB931900, 2012CBA01200), the National Natural Science Foundation (21421002, 21472222, 21502214, 21672242), the Chinese Academy of Sciences (XDA02020105, XDA02020106), the Science and Technology Commission of Shanghai Municipality (15DZ1200102), the Key Research Program of Frontier Sciences (CAS) (QYZDJ-SSW-SLH049), and the Syngenta Ph.D. Fellowship awarded to Y. Duan for financial support.Notes and references
- (a) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938–941 CrossRef CAS PubMed; (b) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 4294–4296 CrossRef CAS PubMed; (c) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 9085–9088 CrossRef CAS PubMed; (d) B. Morandi, J. Cheang and E. M. Carreira, Org. Lett., 2011, 13, 3080–3081 CrossRef CAS PubMed; (e) B. Morandi, B. Mariampillai and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 1101–1104 CrossRef CAS PubMed; (f) Z. Chai, J.-P. Bouillon and D. Cahard, Chem. Commun., 2012, 48, 9471–9473 RSC; (g) S. A. Kunzi, B. Morandi and E. M. Carreira, Org. Lett., 2012, 14, 1900–1901 CrossRef CAS PubMed; (h) C. B. Liu, W. Meng, F. Li, S. Wang, J. Nie and J.-A. Ma, Angew. Chem., Int. Ed., 2012, 51, 6227–6230 CrossRef CAS PubMed; (i) H. Luo, G. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 14503–14507 CrossRef CAS PubMed; (j) S. Hyde, J. Veliks, B. Liegault, D. Grassi, M. Taillefer and V. Gouverneur, Angew. Chem., Int. Ed., 2016, 55, 3785–3789 CrossRef CAS PubMed.
- (a) K. J. Hock, L. Mertens and R. M. Koenigs, Chem. Commun., 2016, 52, 13783–13786 RSC; (b) L. Mertens, K. J. Hock and R. M. Koenigs, Chem. – Eur. J., 2016, 22, 9542–9545 CrossRef CAS PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons Ltd, United Kingdom, 2009 Search PubMed; (c) X.-L. Qiu, X. Yue and F.-L. Qing, in Chiral Drugs: Chemistry and Biological Action, ed. G.-Q. Lin, Q.-D. You and J.-F. Cheng, John Wiley & Sons, Inc., Hoboken, New Jersey, 2011, pp. 195–252 Search PubMed; (d) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2013, 114, 2432–2506 CrossRef PubMed; (e) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- (a) K. Herd, in Organofluorine Chemistry, Springer, 1994, pp. 287–314 Search PubMed; (b) M. Matsui, J. Fluorine Chem., 1999, 96, 65–69 CrossRef CAS; (c) A. Sikder and N. Sikder, J. Hazard. Mater., 2004, 112, 1–15 CrossRef CAS PubMed.
- (a) Y. Landais and D. Planchenault, Tetrahedron Lett., 1994, 35, 4565–4568 CrossRef CAS; (b) F. Z. Dorwald, Metal Carbenes in Organic Synthesis, WILEY-VCH Verlag GmbH, Weinheim (Federal Republic of Germany), 1999 Search PubMed; (c) G. Bertrand, Carbene Chemistry-From fleeting intermediate to powerful reagents, Marcel Dekker, Inc., 2002 Search PubMed; (d) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2903 CrossRef CAS PubMed; (e) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (f) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed; (g) R. A. Moss and M. P. Doyle, Contemporary carbene chemistry, John Wiley & Sons, Inc., Hoboken, New Jersey, 2014 Search PubMed.
- (a) B. Morandi and E. M. Carreira, Org. Lett., 2011, 13, 5984–5985 CrossRef CAS PubMed; (b) G. A. Molander and L. N. Cavalcanti, Org. Lett., 2013, 15, 3166–3169 CrossRef CAS PubMed; (c) G. Wu, Y. Deng, C. Wu, X. Wang, Y. Zhang and J. Wang, Eur. J. Org. Chem., 2014, 4477–4481 CrossRef CAS; (d) Z. Chen, Y. Zheng and J.-A. Ma, Angew. Chem., Int. Ed., 2017, 56, 4569–4574 CrossRef CAS PubMed.
- (a) P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2015, 54, 6558–6561 CrossRef CAS PubMed; (b) J. Li, X.-L. Yu, J. Cossy, S.-Y. Lv, H.-L. Zhang, F. Su, P. K. Mykhailiuk and Y. Wu, Eur. J. Org. Chem., 2017, 266–270 CrossRef CAS.
- (a) J. Zheng, J. Cai, J. H. Lin, Y. Guo and J. C. Xiao, Chem. Commun., 2013, 49, 7513–7515 RSC; (b) J. Zheng, J. H. Lin, J. Cai and J. C. Xiao, Chem. – Eur. J., 2013, 19, 15261–15266 CrossRef CAS PubMed; (c) X.-Y. Deng, J.-H. Lin, J. Zheng and J.-C. Xiao, Chem. Commun., 2015, 51, 8805–8808 RSC; (d) J. Zheng, J.-H. Lin, L.-Y. Yu, Y. Wei, X. Zheng and J.-C. Xiao, Org. Lett., 2015, 17, 6150–6153 CrossRef CAS PubMed; (e) J. Zheng, L. Wang, J.-H. Lin, J.-C. Xiao and S. H. Liang, Angew. Chem., Int. Ed., 2015, 54, 13236–13240 CrossRef CAS PubMed; (f) X. Y. Deng, J. H. Lin and J. C. Xiao, Org. Lett., 2016, 18, 4384–4387 CrossRef CAS PubMed; (g) J. Zheng, R. Cheng, J.-H. Lin, D.-H. Yu, L. Ma, L. Jia, L. Zhang, L. Wang, J.-C. Xiao and S. H. Liang, Angew. Chem., Int. Ed., 2017, 56, 3196–3200 CrossRef CAS PubMed.
- (a) Y. Duan, J. H. Lin, J. C. Xiao and Y. C. Gu, Org. Lett., 2016, 18, 2471–2474 CrossRef CAS PubMed; (b) C.-B. Yue, J.-H. Lin, J. Cai, C.-P. Zhang, G. Zhao, J.-C. Xiao and H. Li, RSC Adv., 2016, 6, 35705–35708 RSC; (c) Y. Duan, J. H. Lin, J. C. Xiao and Y. C. Gu, Chem. Commun., 2017, 53, 3870–3873 RSC.
- (a) M. Bois and T. Skrydstrup, Chem. Rev., 1995, 95, 1253–1277 CrossRef; (b) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835–846 CrossRef CAS PubMed; (c) J. W. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732–4739 CrossRef CAS PubMed.
- (a) M. A. Brook, Silicon in organic, organometallic, and polymer chemistry, J. Wiley, 2000 Search PubMed; (b) F. Hoffmann, M. Cornelius, J. Morell and M. Froba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- S. Fujii and Y. Hashimoto, Future Med. Chem., 2017, 9, 485–505 CrossRef CAS PubMed.
- (a) J.-Q. Yu and Z. Shi, CH activation, Springer, 2010 Search PubMed; (b) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC; (c) B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885–898 CrossRef CAS PubMed; (d) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed; (e) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed; (f) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468–3517 CrossRef CAS PubMed; (g) T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141–4144 CrossRef CAS PubMed; (h) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789–1792 CrossRef CAS PubMed; (i) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230–234 CrossRef CAS PubMed; (j) S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200–16203 CrossRef CAS PubMed.
- (a) C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS; (b) T. Hayashi, Acc. Chem. Res., 2000, 33, 354–362 CrossRef CAS PubMed.
- (a) Y. L. Shi and M. Shi, Adv. Synth. Catal., 2007, 349, 2129–2135 CrossRef CAS; (b) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005–1018 CrossRef CAS PubMed; (c) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qo00430c |
| This journal is © the Partner Organisations 2017 |