
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Retracted Article: Eosin Y catalysed photoredox synthesis: a review
Vishal Srivastava
a and
Praveen P. Singh
 *b
*b
aDepartment of Chemistry, United College of Engineering & Management, Naini, Allahabad-211010, U.P., India
bDepartment of Chemistry, United College of Engineering & Research, Naini, Allahabad-211010, U.P., India. E-mail: ppsingh23@gmail.com
First published on 19th June 2017
Abstract
In recent years, photoredox catalysis using eosin Y has come to the fore front in organic chemistry as a powerful strategy for the activation of small molecules. In a general sense, these approaches rely on the ability of organic dyes to convert visible light into chemical energy by engaging in single-electron transfer with organic substrates, thereby generating reactive intermediates. In this perspective, we highlight the unique ability of photoredox catalysis to expedite the development of completely new reaction mechanisms, with particular emphasis placed on multicatalytic strategies that enable the construction of challenging carbon–carbon and carbon–heteroatom bonds.
 Vishal Srivastava | Vishal Srivastava is working as Assistant Professor, Department of Chemistry, United College of Engineering and Management (AKT University). He has completed Graduation (B.Sc.), Post-Graduation (M.Sc.) in Organic Chemistry and Doctoral Degree (D.Phil.) from Department of Chemistry, University of Allahabad, India. His current research work involves the designing of novel photoredox catalysed synthetic organic compounds. Photoredox catalysis and organocatalysis represent two powerful fields of molecular activation that have found widespread application in the areas of inorganic and organic chemistry. |
 Praveen P. Singh | Praveen P. Singh is an Assistant Professor in the Department of Chemistry at the United College of Engineering and Research (A.K.T. University), Allahabad, India. He obtained his B.Sc., M.Sc. in Organic Chemistry from T. D. College (V. B. S Purvanchal University) Jaunpur and D.Phil. from Department of Chemistry, University of Allahabad, India in 2009. His current research interests include the development of synthetic receptors for the recognition of biological target structures and the application of visible light chemical photocatalysis towards organic synthesis. |
1. Introduction
Visible light photoredox catalysis has recently received much attention in organic synthesis owing to ready availability, sustainability, non-toxicity and ease of handling of visible light1–13 but the general interest in the field started much earlier.14 Unlike thermal reactions, photoredox processes occur under mild conditions and do not require radical initiators or stoichiometric chemical oxidants or reductants.Ruthenium and iridium polypyridyl complexes are commonly employed visible light photocatalysts and their chemistry and application in organic synthesis has recently been summarized.15–17
However, the transition metal based photocatalysts disadvantageously exhibit high cost, low sustainability and potential toxicity. Recently, a superior alternative to transition metal photoredox catalysts, especially metal-free organic dyes particularly eosin Y has been used as economically and ecologically superior surrogates for Ru(II) and Ir(II) complexes in visible-light promoted organic transformations involving SET18–21 (single electron transfer). These organic dyes have got much more attention with the last few years also due to easy handling, eco-friendly and have great potential for applications in visible-light-mediated organic synthesis22–24 which fulfils the basic principle of green chemistry. In this article, we discuss recent applications of eosin Y as a visible light photocatalyst in organic synthesis. The general scheme outlining the important photocatalytic reactions with eosin Y as photocatalyst is given in Fig. 1.
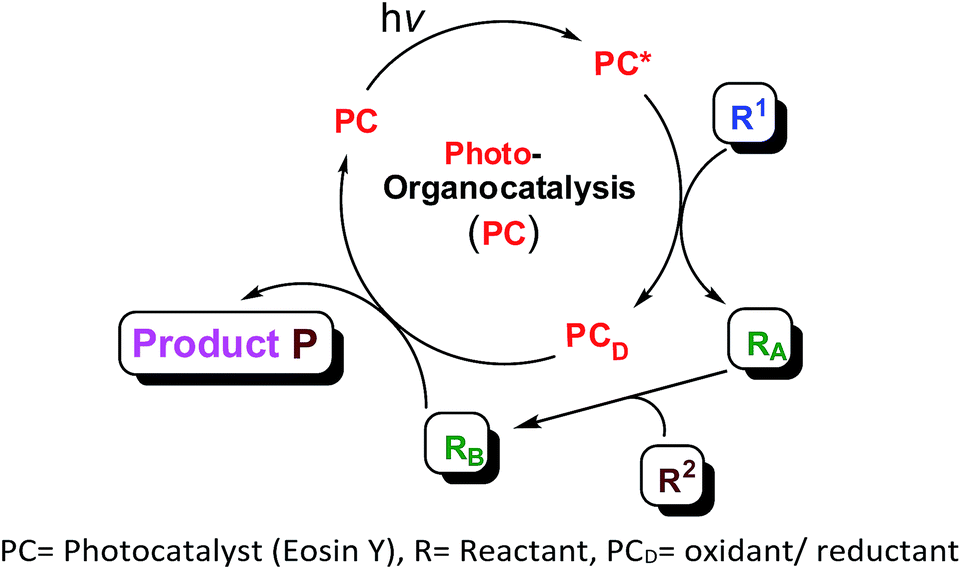 | ||
| Fig. 1 General scheme involving application of eosin Y in photo-organocatalysis. | ||
The possibilities of generation of following intermediates in a photocatalytic reaction may occurs by oxidative and reductive quenching (Fig. 2).
 | ||
| Fig. 2 Intermediates involved in photo-organocatalysis. | ||
The organic dye eosin Y has been found more synthetic utility in photocatalysed organic reactions due to its better yield capacity in comparison to other organic dye of fluorescein family.50
The photochemistry of eosin Y is well investigated: upon excitation by visible light, eosin Y undergoes rapid intersystem crossing to the lowest energy triplet state, which has a life time of 24 μs.25–27 Eosin Y absorbs green light; the UV-Vis spectrum shows a characteristic peak at 539 nm with a molar extinction coefficient ε = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 803 M−1 cm−1. Upon excitation eosin Y becomes more reducing and more oxidizing compared to in its ground state. The redox potentials of the excited state can be estimated from the standard redox potentials of the ground state, determined by cyclic voltammetry, and the triplet excited state energy. The measured ground state and the estimated excited state oxidation and reduction potentials are given in Scheme 1.28,29 In addition, the photoexcited state of eosin Y may also undergo energy transfer.30
803 M−1 cm−1. Upon excitation eosin Y becomes more reducing and more oxidizing compared to in its ground state. The redox potentials of the excited state can be estimated from the standard redox potentials of the ground state, determined by cyclic voltammetry, and the triplet excited state energy. The measured ground state and the estimated excited state oxidation and reduction potentials are given in Scheme 1.28,29 In addition, the photoexcited state of eosin Y may also undergo energy transfer.30
 | ||
| Scheme 1 Different forms of eosin Y and the redox potentials of eosin Y in CH3CN–H2O (1:1) in ground and corresponding excited states. | ||
2. Acid–base chemistry
A major difference between organic and metallic photocatalysts is the pronounced acid–base chemistry of the former due to the availability of electron lone pairs at heteroatoms. For example, eosin Y and other fluorescein dyes exist as an equilibrating mixture of four components:31 two neutral forms Y (e.g., spirocyclic eosinYH2spiro and ring-opened eosinYH2) and upon sequential deprotonations the monoanionic eosinYHNa and dianionic eosinYNa2. The negative charge at the long-wavelength absorbing xanthene core exerts a significant effect on the photophysical properties. pKa values of 2.0 and 3.8 were derived.32 The neutral forms of fluoresceins adopt spirocyclic structures31 in which the xanthenoid π-system is disrupted and visible absorption and photocatalytic activity are extinguished (Scheme 2).33 Unfortunately, the recent literature has not entirely appreciated the relevance of acid–base behavior in photocatalysis. The first reports of eosin Y photocatalysis involved α-amine oxidations, which proceeded in the presence of stoichiometric amines to ensure sufficient formation of the dibasic eosin Y.34,35 In some occasions, weakly basic reactants such as sulfinates can enable conversion to the photoactive forms of eosin Y.36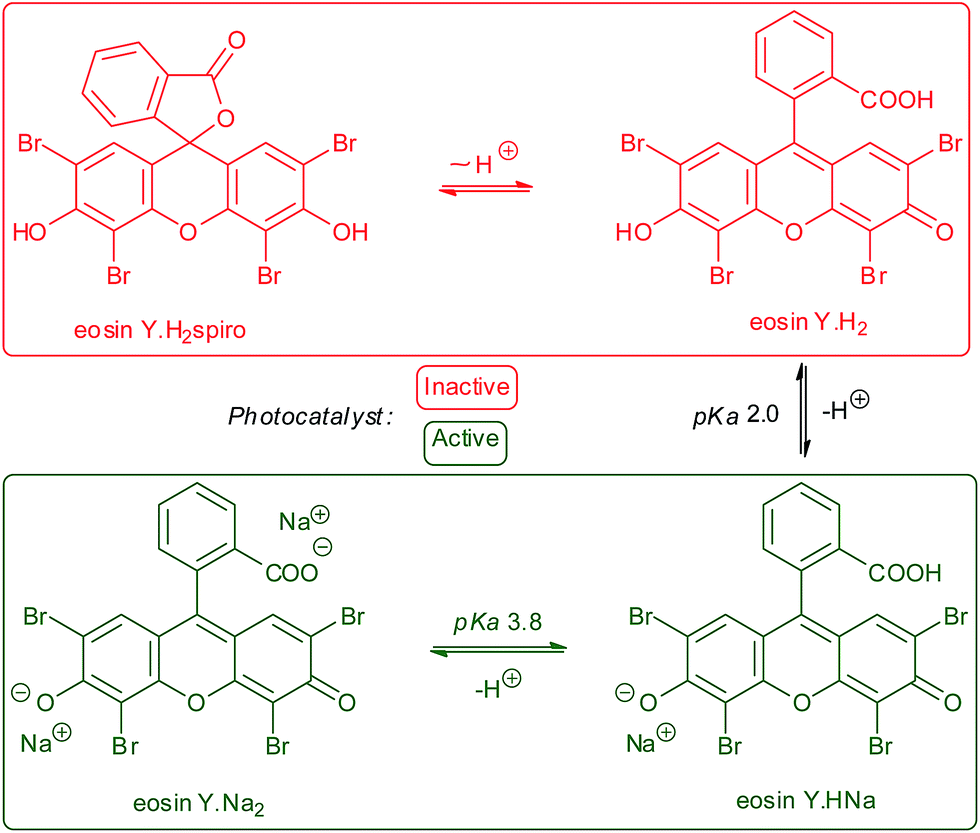 | ||
| Scheme 2 Acid–base behaviour of eosin Y. | ||
3. Early work
The first applications of photoredox catalysis to organic synthesis were reported almost 40 years ago, and these seminal publications laid the foundations for the recent developments in the field of modern photoredox catalysis. In 1978, Kellogg demonstrated that the visible light induced reduction of phenacyl sulfonium salts by 1,4-dihydropyridines (Scheme 3).37 Irradiation of a mixture of 3.1 and 3.2 in CD3CN or CD3COCD3 without any photosensitizer provided the reduced product 3.3 in quantitative yield after 48 h using normal room light (neon fluorescent lamp at ca. 2 m distance) at 25 °C. Addition of 1 mol% of Na2–eosin Y accelerated the reaction resulting in complete conversion within 1 h of irradiation. The authors speculated that light induced single electron transfer (SET) steps are responsible for the formation of the reduced product and suggested an acceleration effect upon addition of the photocatalyst. However, the exact role of the photocatalyst in the reaction mechanism remains undisclosed.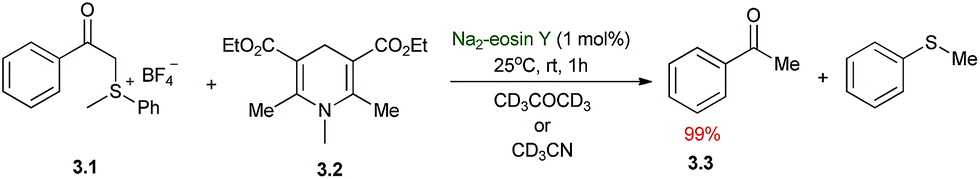 | ||
| Scheme 3 Kellogg 1978 – reductive desulfuration. | ||
4. Reductive deoxygenation
In 2011, Ananthakrishnan and co-workers reported a photoredox catalytic reduction of 4-nitrophenol to 4-aminophenol, which was catalyzed by EY2-bound to a cationic resin (Scheme 4, method B).38 A large stoichiometric excess of NaBH4 (250 equiv) was used as the sacrificial reductant, but this process was greatly improved upon in 2014 with the conditions shown in Scheme 4, method A, which uses triethanolamine (TEOA) as the stoichiometric reductant.39 The reaction is chemoselective and tolerates the presence of other functional groups, such as carbonyls, halogen atoms, and nitriles. The nitro group is a better electron acceptor. Important factors to achieve the optimal reaction yield are the pH value of the reaction mixture in the deoxygenated ethanol–water (3:2, v/v) mixture and the amount of added TEOA. Nitro groups of substrates bearing either electron donating or electron withdrawing substituents are smoothly reduced.
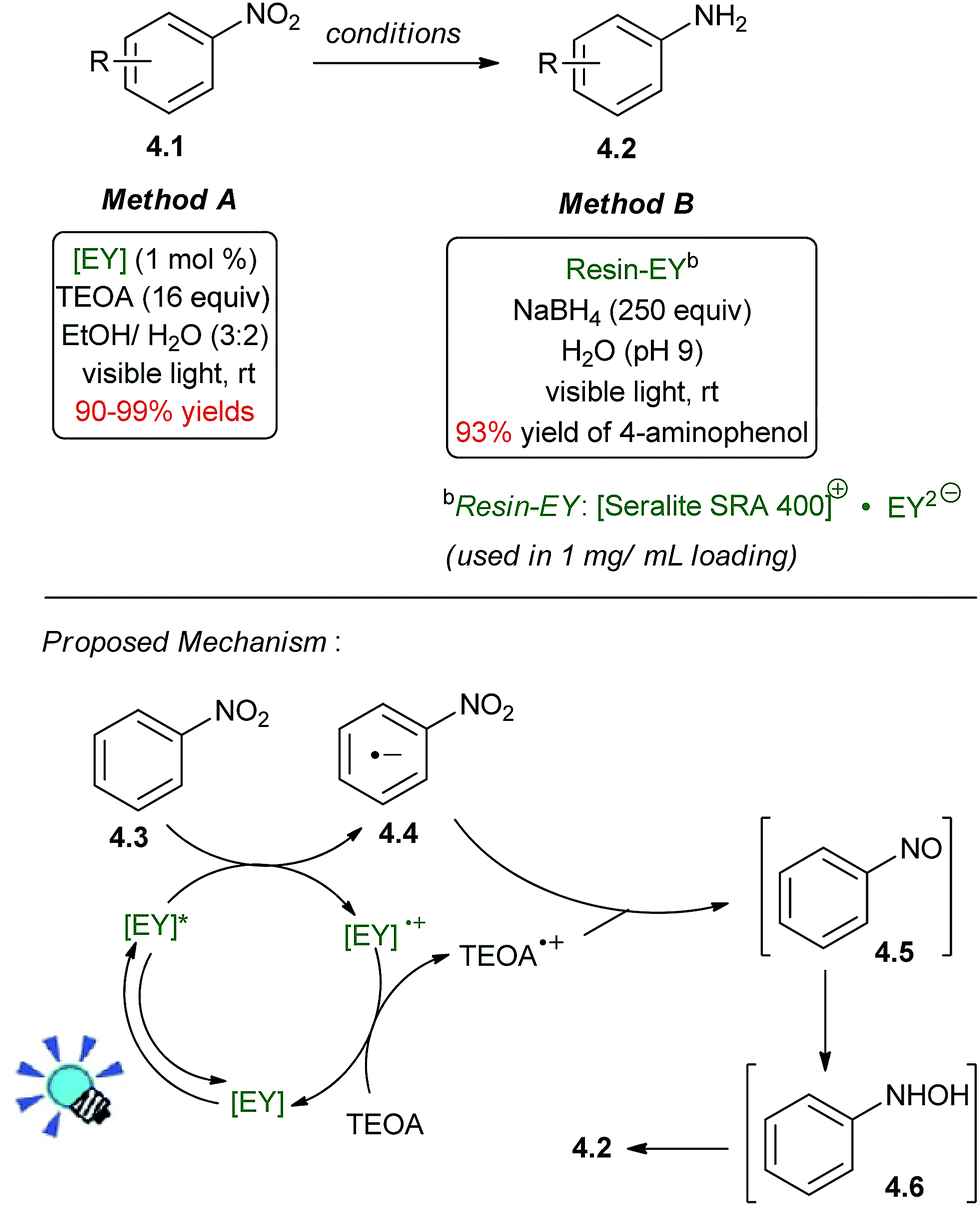 | ||
| Scheme 4 Hydrogenation of nitroarenes to anilines. | ||
Accompanying photophysical studies revealed that electron transfer from TEOA to 3[EY]* was second orders of magnitude slower than electron transfer from 3[EY]* to nitrobenzene. Accordingly, the mechanism is likely to involve an oxidative PETs (photoinduced electron transfer) cycle, in which electrons are repeatedly donated from 3[EY]* to the arene intermediates. Ultimately, the production of the aniline from a single equivalent of nitroarene is a 6-electron reduction which proceeds through nitrosobenzene 4.5 and N-phenylhydroxylamine 4.6 as intermediates, and these species were detected by 1H NMR at partial conversion.
5. Reductive desulfonylation
The sulfone group is removed using metal containing reducing agents, such as Bu3SnH, Al (Hg), or Sm/HgCl2. Preceded by the work of Ohno using Ru(bpy)32+ in the 1980s, a recent report details that arylketosulfones 5.1 are desulfonylated to arylketones 5.2 with TBA–eosin Y as a photoredox catalyst and di-isopropylethylamine (i-Pr2NEt) as a sacrificial reductant (Scheme 5).40 Irradiation of a mixture of 5.1, TBA–eosin Y and diisopropylethylamine under an inert atmosphere using a 3 W blue LED in CH3CN furnishes the desired product 5.2 in good yields. Sulfonylated aliphatic ketones give no reaction due to their very negative reduction potential of −1.94 V vs. SCE not accessible by the excited state of TBA–eosin Y. | ||
| Scheme 5 Reductive desulfonylation of ketosulfones. | ||
The mechanism for the desulfonylation reaction is proposed in Scheme 5. Irradiation of TBA–eosin Y generates its excited state, which is oxidatively quenched by β-arylketosulfones resulting in the formation of the cation radical of TBA–eosin Y and the radical anion of 5.7. A SET from diisopropylethylamine to the radical cation of TBA–eosin Y regenerates the photocatalyst and closes the cycle. Finally, the radical anion 5.8 undergoes desulfonylation to produce a ketone radical which abstracts a hydrogen atom from the radical cation of diisopropylethylamine affording the desired ketone 5.10. The radical cation of the TBA–eosin Y was identified in the presence of β-arylketosulfones by laser-flash photolysis. The observed absorption at 460 nm corresponds to the reported value for the eosin Y radical cation.
6. Reductive dehalogenation
Neumann and co-workers reported the catalytic dehalogenation of α-halo carbonyl compounds 6.1 using HEH and i-Pr2NEt as sacrificial reductants (Scheme 6).41 A number of different organic dyes gave quantitative yields of acetophenone 6.3 from phenacyl bromide, including a perylene diimide, fluorescein (FLH2), and eosin Y (EYH2). EYH2 was the most robust under the photolytic conditions, and a short survey of phenacyl bromides and α-chlorophenylacetates gave high yields of the dehalogenated products 6.3–6.6, leaving other reducible groups intact. With consideration of the reduction potential of phenacyl bromide,42 it was assumed that a carbon centered radical could be generated by reduction of the α-halo compound from EYH2˙− produced upon PET to EYH2* from i-Pr2NEt. Both HEH and i-Pr2NEt˙+ are likely to play a role in HAT (hydrogen atom transfer) to furnish the product.43,44 This system was implemented in a flow reactor45 to the effect of greatly reduced reaction times (40 s, compared to 18 h in batch for 6.3) and improved yields for one substrate 6.5.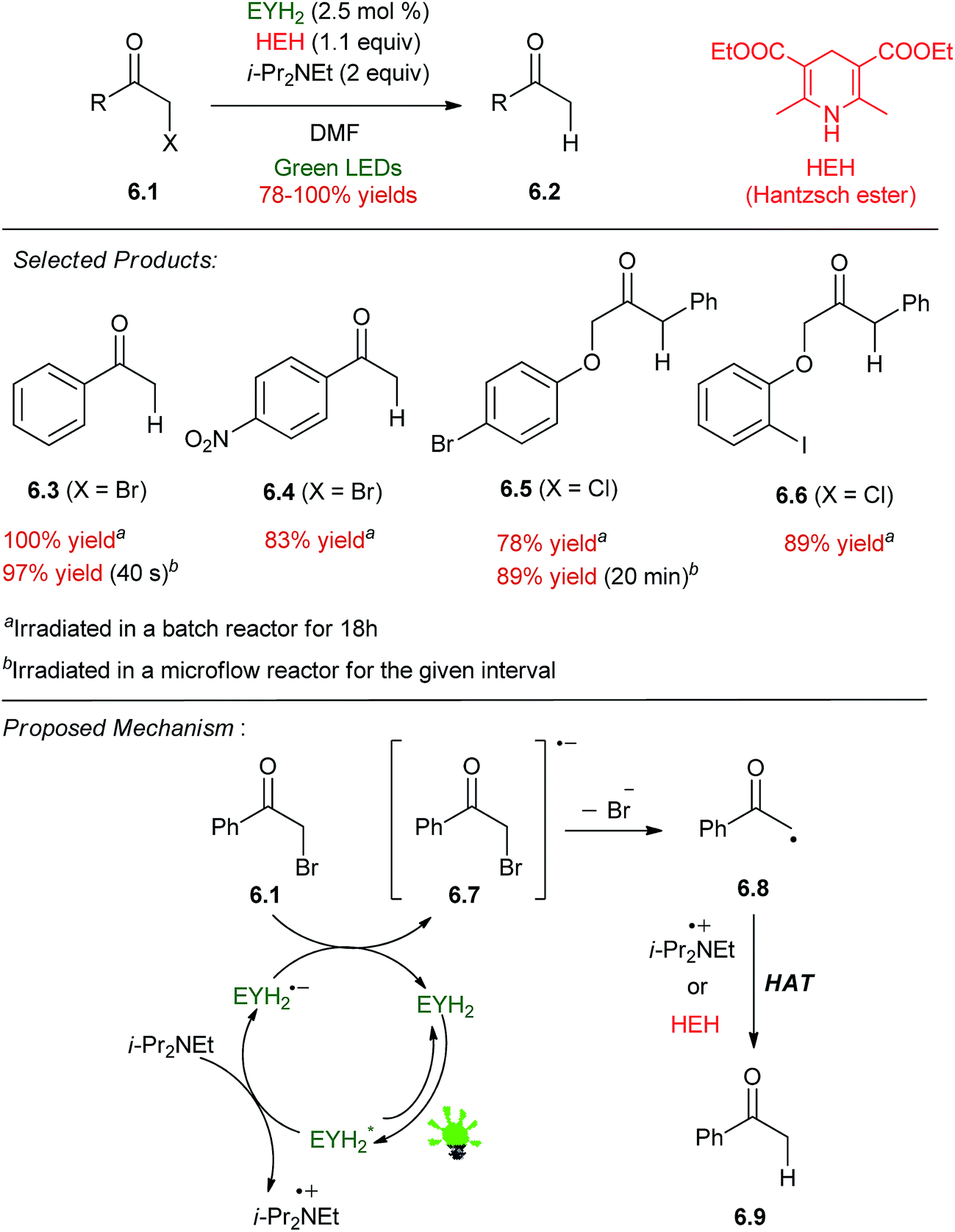 | ||
| Scheme 6 Reductive dehalogenation of α-halo carbonyl compounds. | ||
7. Oxidative transformation of phenylboronic acid to phenol
Paula and coworkers reported the visible light and eosin Y based photoredox activation of PhI(OAc)2 for the conversion of arylboronic acids 7.1 to phenols 7.2. A mixture of arylboronic acid (1 mmol), diacetoxyiodobenzene (PhI(OAc)2, 0.5 mmol), K2CO3 (1 mmol) and eosin-Y (0.01 mmol) in CH3CN (5 mL) was degassed by bubbling Ar gas for 15 minutes after which the mixture was irradiated with a 14 W CFL (compact fluorescent lamp) from a distance of 5 cm for 6 h. The reaction mixture was then removed from the light source and water was added to it. The aqueous mixture was then extracted with ethyl acetate following which the ethyl acetate layer was dried with anhydrous Na2SO4 and concentrated. This was followed by column chromatography over silica gel to afford the product. The purified products were characterized by 1H and 13C NMR spectra which corresponded well with the reported values.The mechanism of the reaction appears to be complex and could proceed through the pathway outlined in Scheme 7.46 Initially, the boronic acid reacts to form the boronate salt, CO2 and H2O which coordinates to the former. Electron transfer from the photoexcited eosin Y to PhI(OAc)2 leads to the formation of an CH3COO− and a CH3 radical. H-Abstraction from the boron complexed H2O followed by migration of the phenyl group and back electron transfer to eosin Y generates the phenyl ether which is then able to complex with the CH3COO−. Further electron transfer to the boron complexed CH3COO− from photoexcited eosin Y generates a CH3 radical which again abstracts an H-atom from the boron complexed water and forms the product via a pathway similar to the previous.
 | ||
| Scheme 7 Photoredox activation of PhI(OAc)2 for the conversion of arylboronic acids to phenols. | ||
8. Carbonylation of arene diazonium salts
Aromatic esters are key building blocks in the synthesis of fine chemicals, agrochemicals, pharmaceuticals, and materials. The method involves a redox reaction driven by visible light and catalyzed by eosin Y which affords alkyl benzoates from arene diazonium salts, carbon monoxide, and alcohols under mild conditions.Under irradiation with green light (LED, λmax = 525 nm, 3.8 W), solutions of 8.1 in methanol were treated under an atmosphere of CO at room temperature to give methyl 4-methoxybenzoate 8.2. Commercial eosin Y (4 mol%, employed as the disodium salt) was used as the metal-free photoredox catalyst. Unwanted dimerization (Ar2) and reduction (Ar–H) was suppressed at higher dilutions by the higher relative concentrations of CO and the alcohol. Lower pressures of CO resulted in low conversion and competing hydrodediazotation (10–25%) and biaryl coupling (ca. 5%). The optimized conditions were applied to the synthesis of various alkyl benzoates (Scheme 8).47 Several functional groups and electron-poor and -rich substituents (nitro, chloro, bromo, esters, benzylic protons) were tolerated in the substrates. Tertiary esters can also be prepared in high yields 8.3. Recently, the Wu et al.48 reported their studies on visible-light induced photoredox carbonylation reactions.
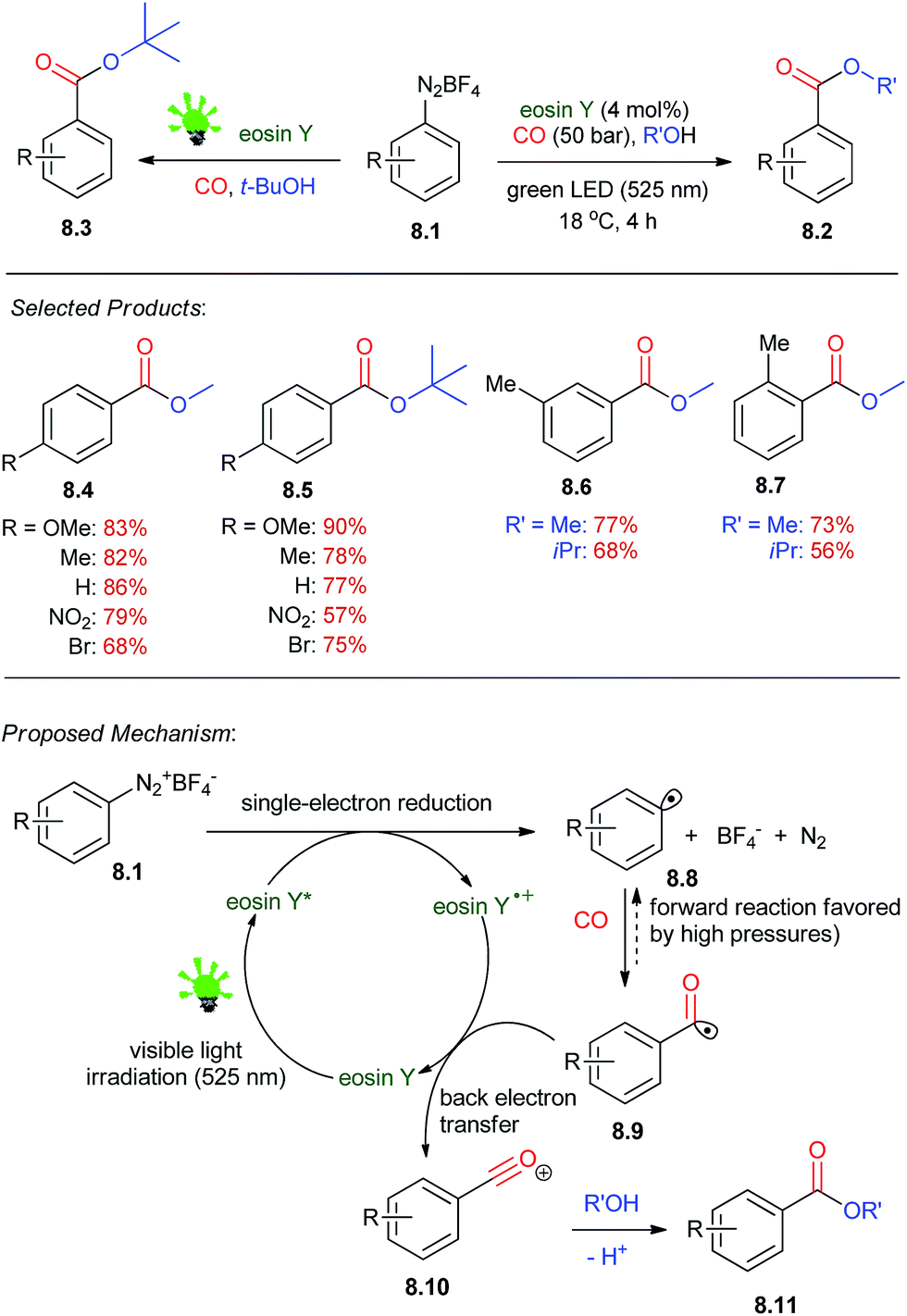 | ||
| Scheme 8 Visible-light-driven eosin-catalyzed carbonylation of arene diazonium salts. | ||
9. Oxidative synthesis of α-chloro and α-alkoxy aryl ketones
For visible-light-induced radical-triggered chemoselective domino process to access α,α-di-functionalized ketones, Wang and co-workers reported a direct approach to synthesize α-chloro or α-alkoxy aryl ketones based on the electronic properties of the substrates. A series of substrates withstood the reaction conditions to give the corresponding products in moderate to good yields. The subsequent control experiments were carried out to gain some insight into the mechanism (Scheme 9).49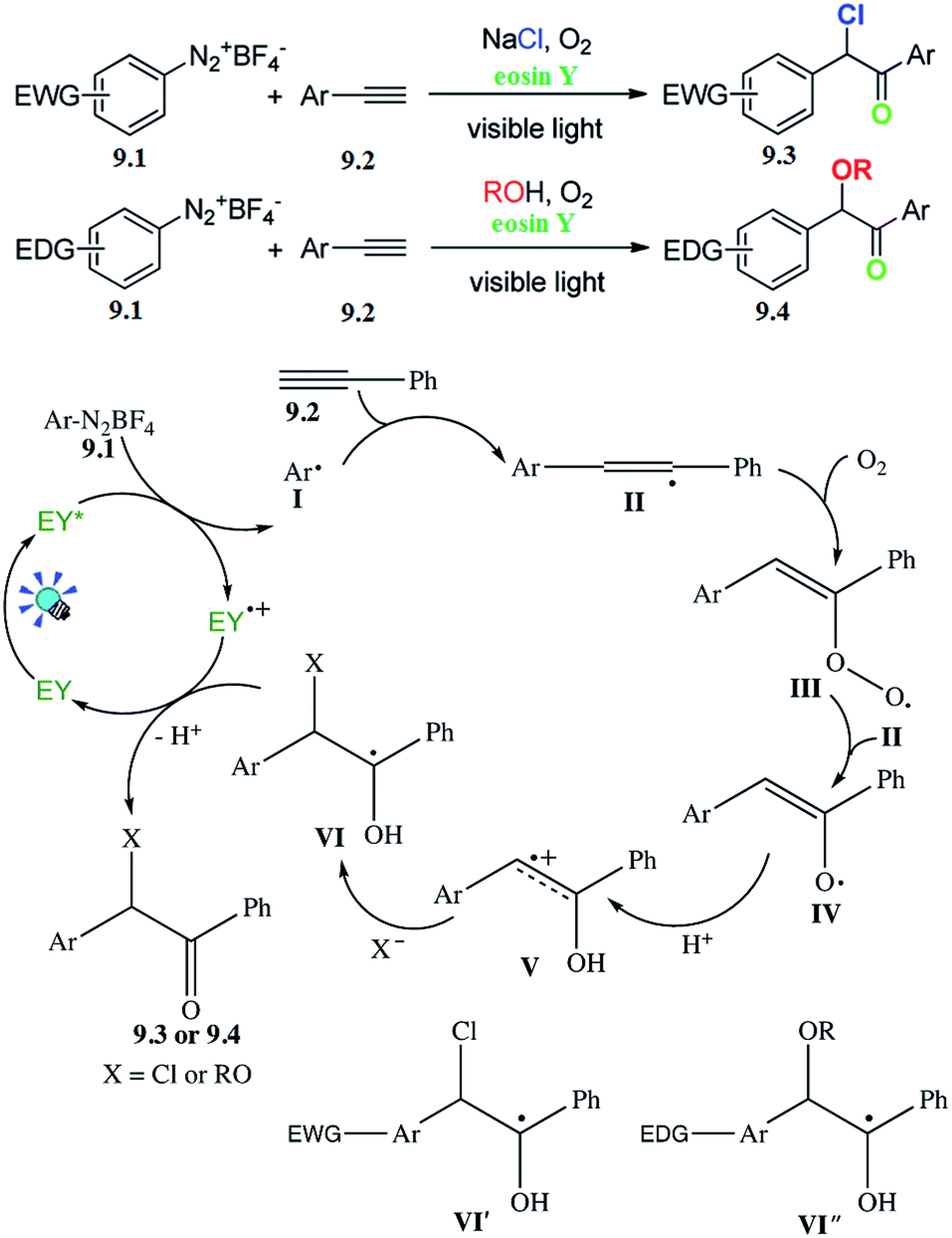 | ||
| Scheme 9 Synthesis of α-chloro and α-alkoxy aryl ketones. | ||
10. Oxidative synthesis of aryl ketones
Aryl ketones are common structural motifs in natural products and are versatile building blocks in the synthesis of more complex natural products, pharmaceuticals, agricultural chemicals, dyes, and other commercially important materials. Liua and co-workers reported the photocatalysed synthesis of aryl ketones. The effort was initiated by using phenyl diazonium tetrafluoroborate 10.1 with benzene 10.2 as a model reaction in the presence of eosin Y (1 mol%) under irradiation with 5 W white LED light and a CO pressure of 70 atm in MeCN at room temperature. The desired benzophenone 10.3 was formed in 74% yield after 16 h.On the basis of these preliminary results, and those of previous studies, they proposed the mechanism shown in (Scheme 10).50 Initially, photoexcitation of eosin Y by visible light generates excited [eosin Y*]. Then the electron-deficient phenyl diazonium tetrafluoroborate 10.1 accepts one electron from the excited [eosin Y*]. This single-electron transfer (SET) results in the generation of a phenyl radical (Ph˙) and the oxidized dye radical cation [eosin Y+˙]. The resulting phenyl radical (Ph˙) is rapidly trapped by CO to give a benzoyl radical A. Further oxidation of A by [eosin Y+˙] results in the benzylidyneoxonium B. Finally, electronic trapping of B by benzene gives the desired aryl ketone 10.3.
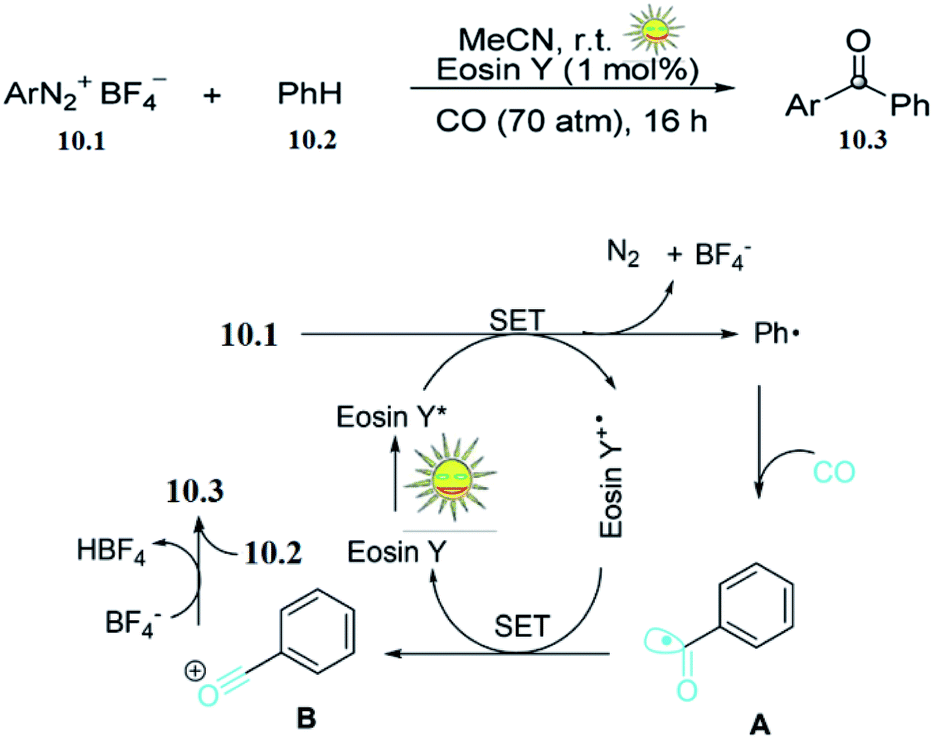 | ||
| Scheme 10 Oxidative synthesis of aryl ketones. | ||
11. Oxidation of alkynes
1,2-Diketones play an important role in pharmaceutical chemistry. Sun and co-workers reported photocatalysed synthesis of diketones. These compounds were synthesized by the oxidation of corresponding 1,2-diphenylethyne 11.1 (0.2 mmol, 35.6 mg), 4-chlorobenzenethiol (0.4 mmol, 57.4 mg), and eosin Y (2 mol%, 2.8 mg) were added in MeCN (2 mL). The mixture was stirred under blue LED irradiation for 8 h under ambient air. After completion of the reaction, the reaction mixture was extracted with (dichlorometane) DCM (15 mL × 3) to afford the pure product 11.2.The plausible reaction mechanism is described in Scheme 11.51 Initially, eosin Y was converted to excited state eosin Y* upon irradiation of visible light, and this eosin Y* underwent reductive quenching by thiophenol to afford the radical cation A and form an eosin Y radical anion. The eosin Y radical anion was oxidized to the ground state by aerobic oxygen to complete the photoredox cycle and a superoxide radical anion (O2−˙) was generated simultaneously. The resulting radical cation A was deprotonated by O2−˙ to give the thiophenyl radical B. Subsequently, a thiylperoxyl radical C was formed through the reversible trapping of B with O2. The addition of radical C to 1,2-diphenylethyne 11.1 generated a vinyl radical D. The rearrangement of D via the homolytic O–O bond cleavage and the radical transfer formed radical intermediate E. The desired product 1,2-diphenylethane-1,2-dione 11.2 was finally produced by the elimination of thiophenyl radical B from E. In addition, a byproduct di(4-chlorophenyl)disulfide F which came from the homocoupling of radical B was also separated from the reaction mixture.
 | ||
| Scheme 11 Oxidation of alkynes. | ||
12. Oxidation of benzyl alcohols
The synthesis of carbonyl compounds from alcohols represents one of the most addressed problems in organic synthesis as well as in process and medicinal chemistry. Shah and co-workers started with the use of 1-phenyl ethanol 12.1 as model substrate, 27 W household CFL as light source and eosin Y (5 mol%) as photocatalyst in presence of 1 equiv. of TBHP (tert-butyl hydroperoxide) as an oxidant using ACN as solvent. The reaction resulted in the synthesis of acetophenone 12.2 in 20–97% yield after 72 h. A possible explanation of the reaction is cleavage of TBHP by photo-excited eosin Y*, leading to the generation of tert-butoxy radical (t-BuO˙) and hydroxyl radical (OH˙). The t-BuO˙ radical then abstracts hydrogen from benzylic position to give benzylic radical, which on subsequent loss of water molecule aided by the OH˙ results in the generation of corresponding carbonyl compounds (Scheme 12).52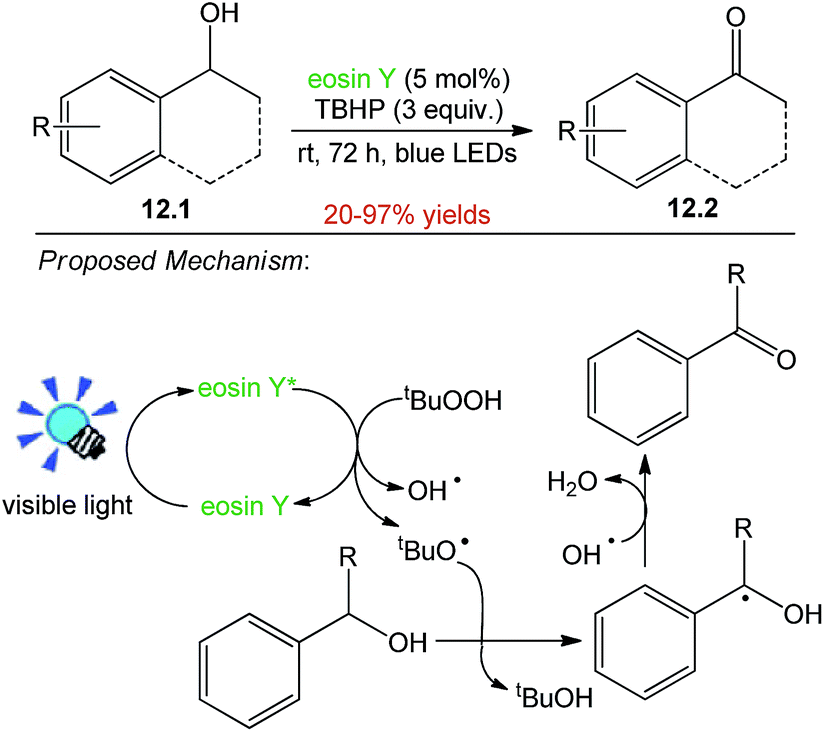 | ||
| Scheme 12 Oxidation of benzyl alcohols. | ||
13. Oxidative synthesis of α-alkoxybenzamides
Saturated nitrogen heterocycles are ubiquitous in natural products, bioactive molecules, and drugs. Zhang and co-workers utilized the eosin Y as the organophotoredox catalyst because it has been already demonstrated that the aryl radicals can be generated by the reaction of aryl diazonium salts with eosin Y. tBuONO was chosen as the commercially available nitrosating agent. Radical translocation of readily prepared o-aminobenzamide 13.1 (0.4 mmol) with methanol 13.2 (2.0 mmol) in the presence of blue LED lights (450 nm) at room temperature under N2 was investigated first. Gratifyingly, the desired product 13.3 was obtained in 20–78% yields. Solvent effects were then investigated, and the results indicate that CH3NO2 is the best solvent for this transformation.The plausible reaction mechanism is proposed in Scheme 13.53 First, the o-aminobenzamide 13.1 reacts with tBuONO and sequentially anion exchange takes place with the anion of TsOH to the diazonium salt I and tBuOH. Subsequently, the diazonium salt I is reduced through the single electron transfer (SET) process by the excited state of eosin Y to the aryl radical II, which abstracts a hydrogen atom from a remote aliphatic C–H bond to the alkyl radical III (racial translocation event). The alkyl radical III was further oxidized through SET by eosin Y radical cation to the iminium intermediate IV. However, an alternative pathway involving the transformation of III into IV by radical chain propagation cannot be excluded at the current stage. Finally, IV is trapped by alcohol 13.2 to form the product 13.3.
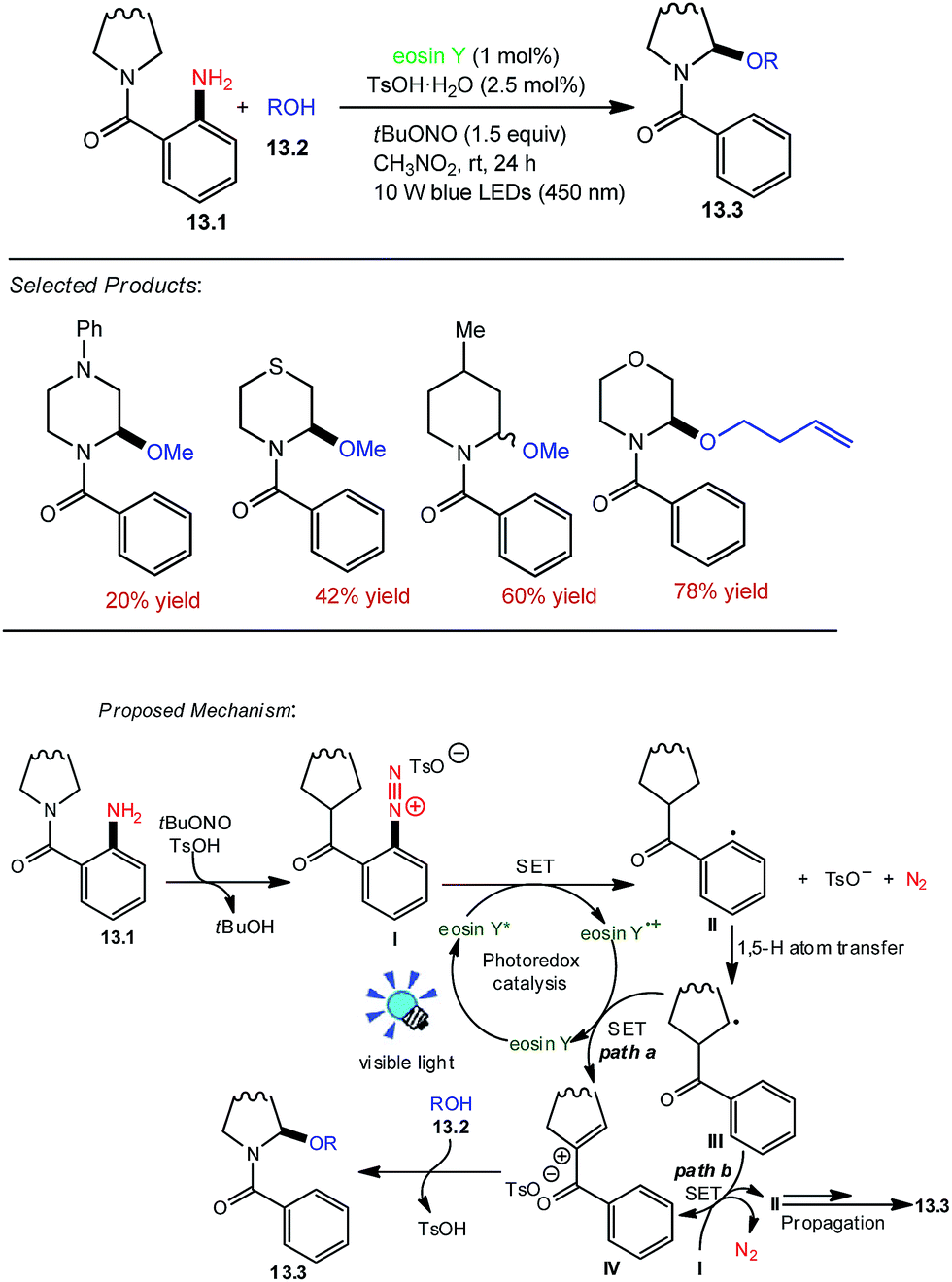 | ||
| Scheme 13 Oxidative synthesis of α-alkoxybenzamides. | ||
14. Oxidative synthesis of β-ketosulfones
The β-ketosulfone, one of the most valuable sulfur-containing compounds, has been widely used in the fields of synthetic and pharmaceutical chemistry.Wang and co-workers focused the photocatalysed oxidative synthesis of β-ketosulfones under nitrogen atmosphere, styrene 14.1 (0.2 mmol), benzenesulfinic acid 14.2 (0.3 mmol), eosin Y (1.0 mol%), TBHP (3.0 equiv.), solvent (2.0 mL), EtOH/H2O (v1/v2 = 4:1), at room temperature for 24 h. The desired product 14.3 was obtained in 42–84% yields.
Under the visible-light irradiation, eosin Y was converted to the excited eosin Y*. A single electron transfer between eosin Y* and TBHP afforded a tert-butoxyl radical and hydroxyl anion. Then sulfinic acids 14.2 reacted with a tert-butoxyl radical to furnish the corresponding sulfonyl radical A. Subsequent radical addition of A to alkenes 14.1 produced carbon-centered radical B, which could be further transformed into carbocation intermediate C through single electron transfer (SET) with eosin Y˙+. Subsequently, nucleophilic attack of hydroxyl anion and H2O on the carbocation intermediate produced the intermediate E, which was transformed into the desired product 14.3 under the oxidative conditions in Scheme 14.54
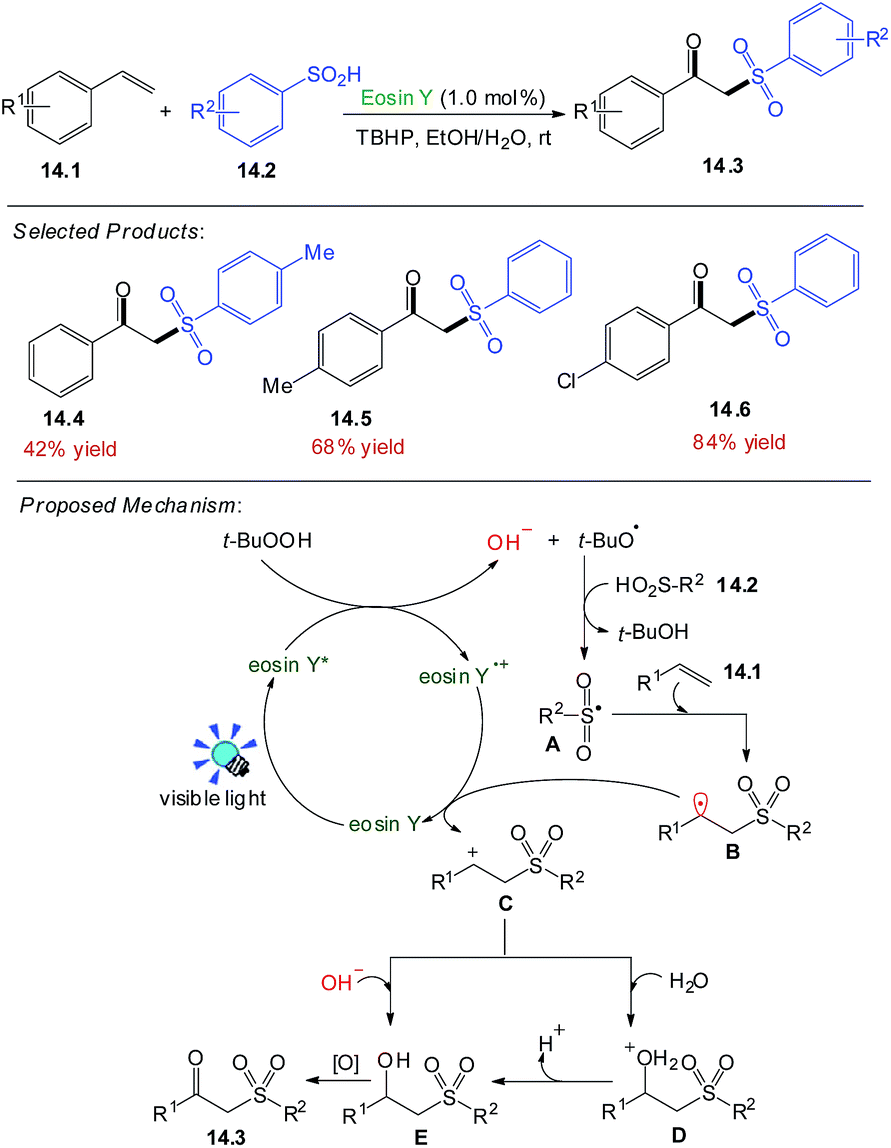 | ||
| Scheme 14 Oxidative synthesis of β-ketosulfones. | ||
15. Difunctionalization of alkynes with alkyl bromides
Wang, Meng and co-workers reported the visible light promoted difunctionalization of alkynes with alkyl bromides. Amino-brominated aromatic β,β-dicyanoalkene is a new reactant and is attractive for potential utilization in organic transformations. The reaction was carried out with solution of amino-brominated aromatic β,β-dicyanoalkene 15.1 (0.40 mmol) with alkyne 15.2 (0.80 mmol) in 2.0 mL of DCE (1,2-dichloroethane) was added Na2–eosin Y (0.02 mmol), which after completion of reaction and purification give pure product 15.3.The proposed mechanism is depicted in Scheme 15.55 Photoexcitation of Na2–eosin Y by visible light provides [Na2–eosin Y]*, which is then oxidatively quenched by 15.1 to produce [Na2–eosin Y]+ and a radical intermediate A. This radical A then undergoes a regioselective addition to alkyne to form B. It should be noted that the final product 15.3 can be obtained from B via two possible pathways. First, intermediate B reacts with 15.1 by a radical chain transfer to give product 15.3 along with the regeneration of radical A.
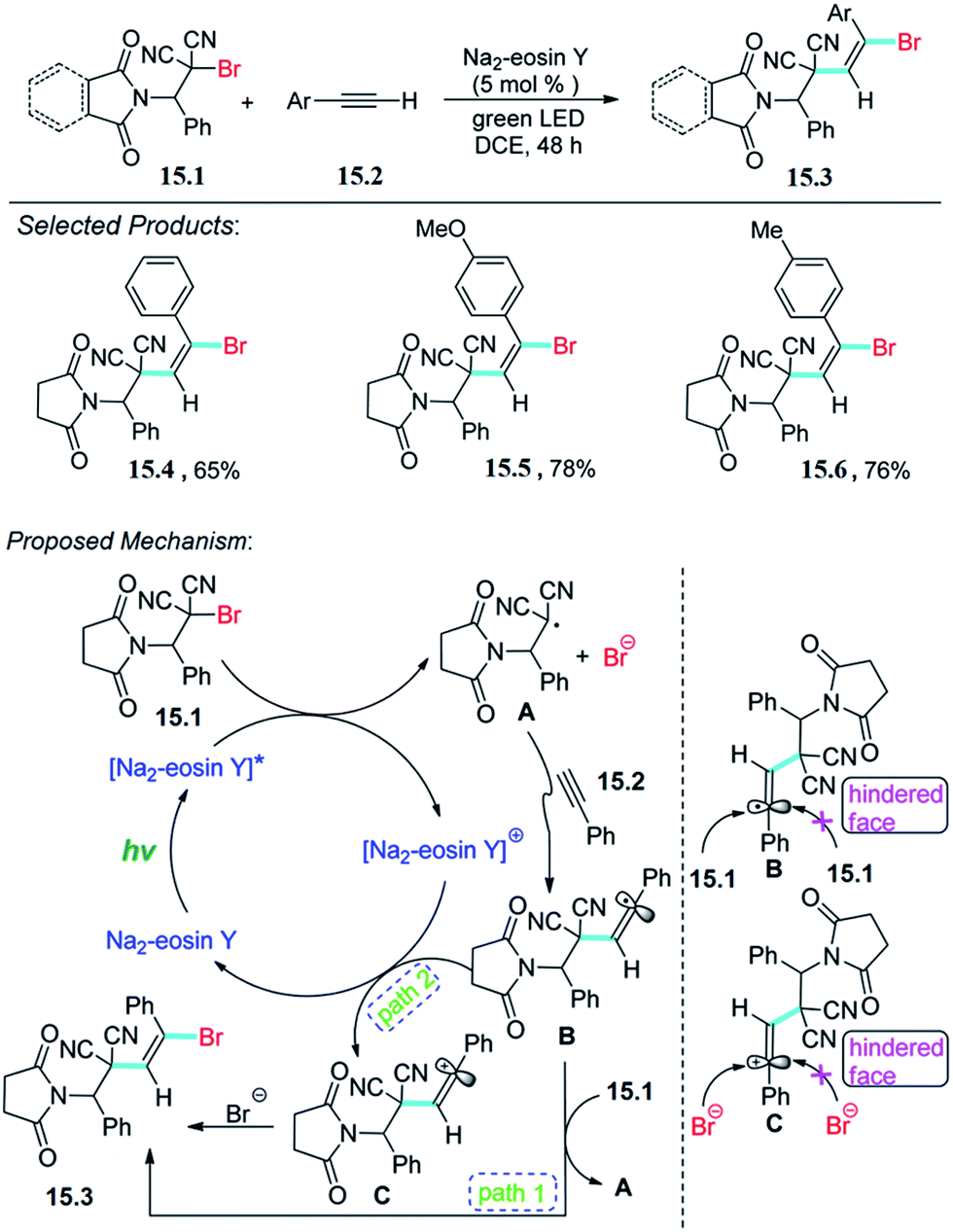 | ||
| Scheme 15 Difunctionalization of alkynes with alkyl bromides. | ||
Alternatively, oxidation of the intermediate B produces vinyl carbocation C, which reacts with the produced bromine anion to provide the final product 15.3. Either with path 1 or path 2, the double bond in the final product is in the E-configuration, which may be ascribed to steric hindrance.
16. Thiocyanation of imidazoheterocycles
Thiocyanation reaction is one of the most useful carbon-sulfur bond-forming reactions. Thiocyanates are the building blocks of many heterocyclic compounds that show a wide range of biological activities and also consist in many natural products. Hajra and co-workers focused on photocatalysed thiocyanation of heterocyclic compounds. An oven-dried 5 mL round-bottom flask was charged with 2-phenylimidazo[1,2-a]pyridine 16.1 (0.5 mmol, 97 mg), ammonium thiocyanate (1.5 mmol, 114 mg), and eosin Y (5 mol%, 17 mg) in CH3CN (2 mL), and the reaction mixture was stirred under blue LED irradiation for 3 h under ambient air, which after completion of reaction to afford the pure thiocyanated product 16.2 (117 mg, 93%) as a red solid.A plausible mechanistic path has been outlined in Scheme 16.56 Initially, eosin Y is being photoexcited in the presence of blue LED light. The thiocyanate anion is being oxidized to thiocyanate radical by the SET mechanism from anion to photoactivated eosin Y via a reductive quenching cycle. The resulting thiocyanate radical interacts with 16.1 to produce the radical intermediate B. Subsequently, B is oxidized to the intermediate C, which affords the product 16.2 via deprotonation. Aerobic oxygen probably plays a crucial role to complete the photoredox cycle by oxidation of the eosin Y radical anion to the ground state.
 | ||
| Scheme 16 Thiocyanation of imidazoheterocycles. | ||
17. Carboxylation of styrenes
The direct carboxylation of alkenes is a formidable challenge despite its high potential as a practical method for the preparation of unsaturated carboxylic acid derivatives. At first Tang and co-workers reported the visible light induced carboxylation of alkene using styrene 17.1 and CBr4 with eosin Y disodium (10 mol%) as a photocatalyst under irradiation from a fluorescent bulb (18 W) and under a nitrogen atmosphere for 15 h in DMSO at 50 °C as the model the highest yield of the target product 17.2.A plausible mechanism for the carboxylation is shown (Scheme 17).57 The excited [eosin Y disodium]* or Co(II) transfers an electron to the halide, generating a radical that adds to the alkene under electronic and steric control. The resulting radical combines with the halide with concurrent electron transfer back to [eosin Y disodium]+ or Co(III), thus, regenerating the catalyst or Co(II). Then, the intermediate 17.3 is eliminated, giving compound C, which undergoes SET (single electron transfer) reduction to generate the alkyl radical. Oxidation of the alkyl radical by [eosin Y disodium]+ generates carbocation E, accompanied by formation of the catalyst. Nucleophilic attack of DMSO to carbocation E affords alkoxysulfonium F, which also may be obtained from 17.3 by CoI2-catalyzed hydrolysis reaction. Then, a reaction similar to Kornblum (DMSO) oxidation proceeds to give compound G. Finally, nucleophilic attack by DMSO leads to the desired product 17.2.
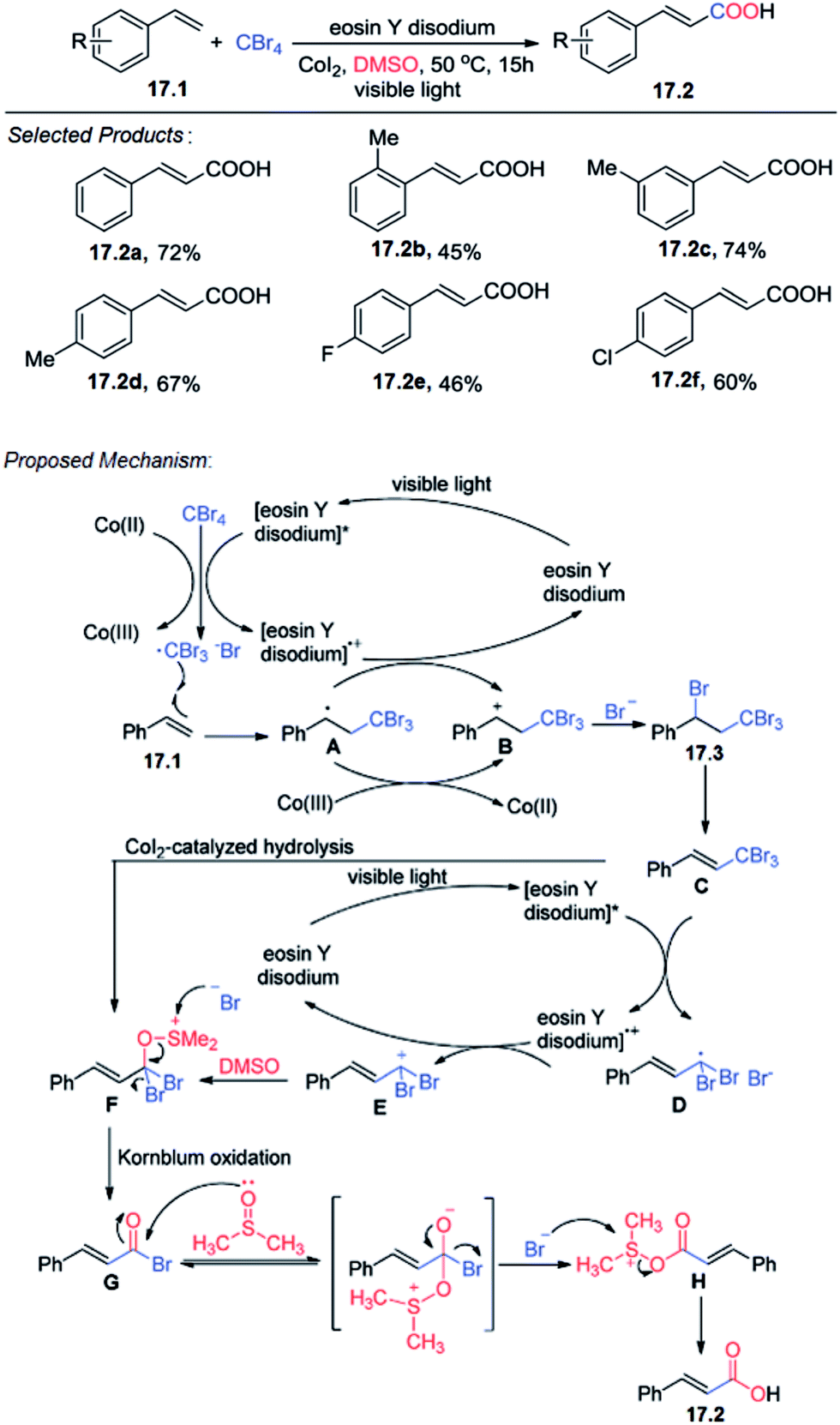 | ||
| Scheme 17 Carboxylation of styrenes. | ||
18. Decarboxylative alkylation
Carboxylic acids are among the most abundant, renewable feedstocks on our planet. They are non-toxic, stable and inexpensive, therefore it is a valuable starting materials for “green chemistry”. König and co-workers reported for the first time a metal-free, photo-catalytic, decarboxylative alkylation which is applicable for a broad variety of natural carboxylic acids involving amino acids. The reaction conditions of the decarboxylative alkylation, the N-(acyloxy)phthalimide of N-Boc-protected proline 18.1 and n-butyl acrylate 18.2 served as test substrates. A mixture of both the compounds, the base DIPEA (N,N-diisopropylethylamine) and a homogeneous photocatalyst was irradiated with LEDs under a nitrogen atmosphere. For this reaction, no heterogeneous catalysts have been investigated, although an application would be conceivable. The best product yield of 96% 18.3 was obtained with photocatalyst eosin Y (EY) and CH2Cl2 as the solvent.They proposed the following mechanism for the decarboxylative alkylation of N-(acyloxy)phthalimides with electron-deficient alkenes (Scheme 18).58 Irradiation of a photocatalyst (PC) with visible light generates the excited catalyst PC*, which is reductively quenched by the sacrificial electron donor DIPEA to give DIPEA+˙. Regeneration of the PC is presumably achieved by reduction of N-(acyloxy)phthalimide 18.1, which gives the corresponding radical anion. Splitting of the N–O bond and subsequent elimination of CO2 generates alkyl radical 18.1˙. This radical can attack the double bond of Michael acceptor 18.2 to form the intermediate 18.3˙. It is assumed that the hydrogen atom abstraction from DIPEA+˙ or the solvent finally enables the formation of product 18.3. This mechanistic proposal was confirmed by several spectroscopic studies. Single electron transfer (SET) from DIPEA to the excited PC should be exergonic according to the redox potentials of DIPEA (+0.72 V vs. SCE in CH3CN) and the excited PC EY (eosin Y*/eosin Y˙−: +0.83 V vs. SCE in CH3CN–H2O, 1:1).
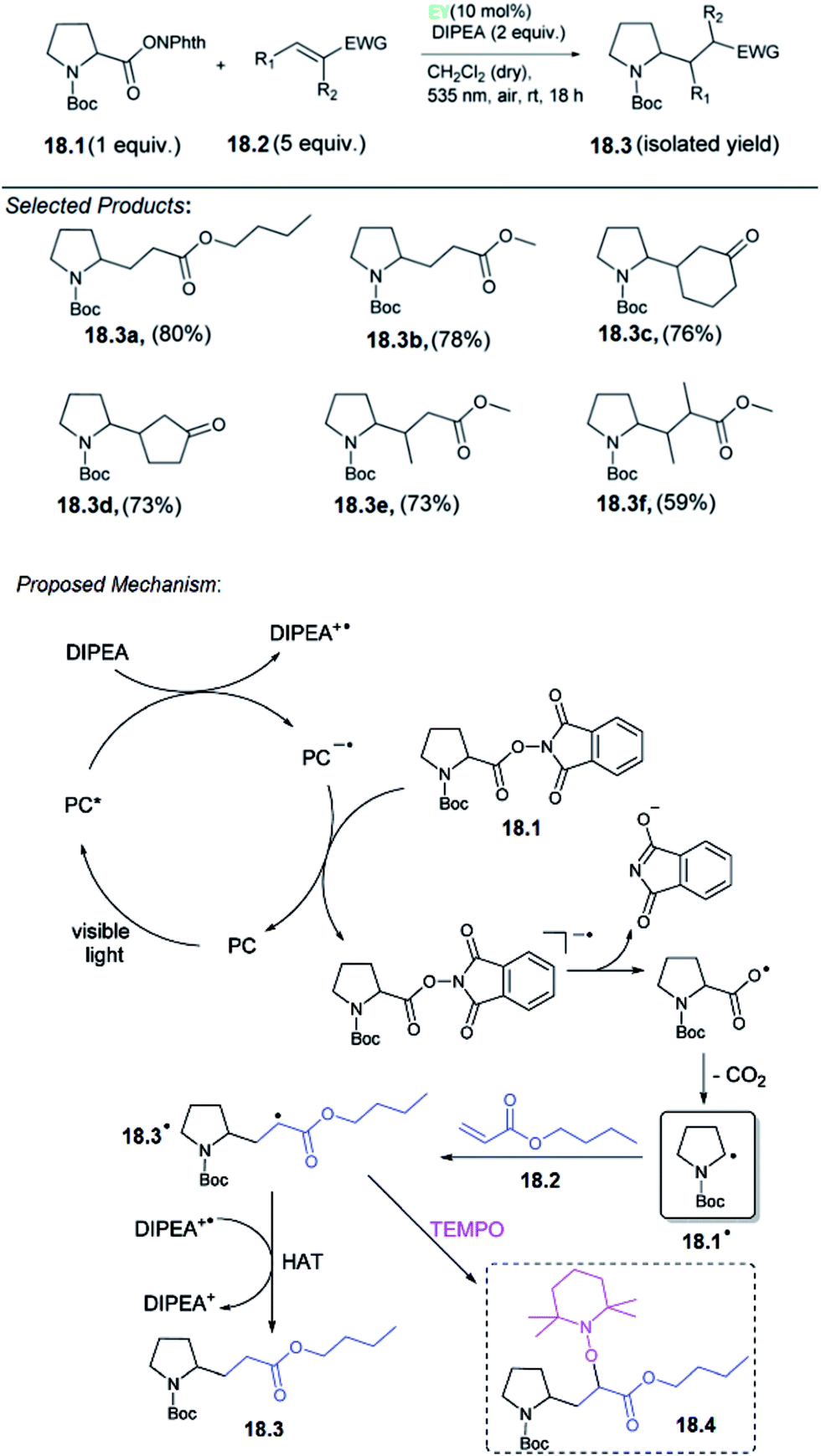 | ||
| Scheme 18 Decarboxylative alkylation. | ||
19. Difunctionalization of styrenes using O2 and CS2
The 1,3-oxathiolane moiety is present in a variety of biologically active compounds and natural products. They show antibacterial, tuberculostatic, antifungal and neuroprotective activities. Moreover, 1,3-oxathiolane-2-thiones (cyclic dithiocarbonates) are useful intermediates for the synthesis of alkanes, alkenes, and thiols and have found application in materials science. Styrenes are readily available substrates, which allow the introduction of two functional groups in a single step. Consequently, several communications have appeared exploring the difunctionalization of styrenes. Recently, anionic species like ArSO2–, NCS– and RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CRS– have been converted to the corresponding radicals under photoredox catalysis. Inspired by the fact that sulphur compounds can be easily converted into radicals employing eosin Y (EY) as a visible light photoredox catalyst, L. D. S. Yadav and co-workers hypothesized that the xanthate anion formed in situ from CS2 and alcoholates could be converted into its radicals, which could be trapped with styrenes and finally cyclized under aerobic conditions to give 1,3-oxathiolane-2-thiones.
CRS– have been converted to the corresponding radicals under photoredox catalysis. Inspired by the fact that sulphur compounds can be easily converted into radicals employing eosin Y (EY) as a visible light photoredox catalyst, L. D. S. Yadav and co-workers hypothesized that the xanthate anion formed in situ from CS2 and alcoholates could be converted into its radicals, which could be trapped with styrenes and finally cyclized under aerobic conditions to give 1,3-oxathiolane-2-thiones.
To test this idea and optimize the reaction conditions, styrene 19.1 was selected as the model substrate. The key reaction was performed with a mixture of 19.1 (1.0 mmol), an excess amount of CS2 (1.0 mL), Cs2CO3 (1.0 equiv.) and a catalytic amount of eosin Y (2 mol%) in MeOH (3 mL). The reaction mixture was irradiated with green LEDs [2.50 W, λ = 535 nm] for 20 h in open air under stirring at room temperature. The desired product 1,3-oxathiolane-2-thione 19.2 in good to excellent yields (68–95%). Styrenes 19.1 with an electron-donating group (EDG) on the aromatic ring appear to react faster and afford marginally higher yields in comparison with those bearing an electron-withdrawing group (EWG). However, the reaction does not work with aliphatic alkenes, this is probably because of the less stability of the radical intermediate formed with aliphatic alkenes than that in the case of styrenes. Moreover, the greater stability of the radical formed at the α-position of an aromatic or heteroaromatic ring plausibly governs the high regioselectivity in the formation of product 19.2 (Scheme 19).59
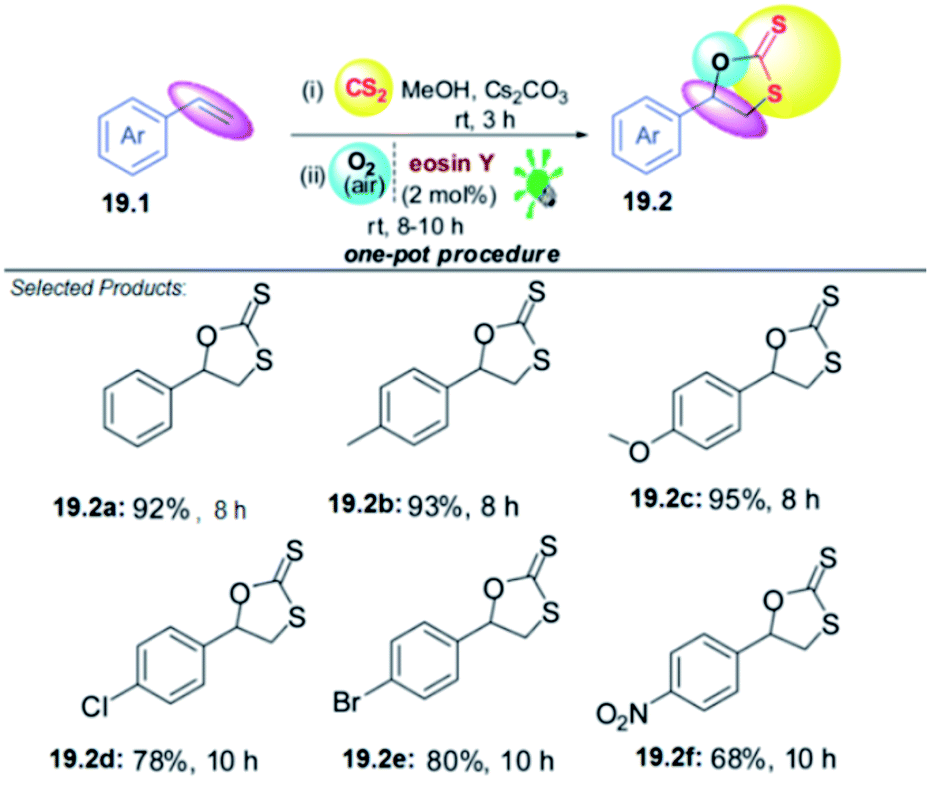 | ||
| Scheme 19 Difunctionalization of styrenes using O2 and CS2. | ||
20. Cyclization of arylsulfonyl chlorides with o-azidoarylalkynes
Polysubstituted indoles are not only common motifs in natural products and pharmaceuticals, but also useful building blocks for the construction of highly complex target structures. The importance of indole motifs has initiated substantial research efforts directed towards the development of efficient synthetic strategies to prepare this heteroarene.G. Li and co-workers reported photocatalysed cyclization of arylsulfonyl chlorides. As an initial test reaction, the simple benzenesulfonyl chloride 20.1 (0.35 mmol) was treated with the o-azidophenylalkyne 20.2 (0.3 mmol), 1,4-cyclohexadiene (1,4-CHD) (0.45 mmol), MeCN (2.0 mL) in the presence of eosin Y (3 mol%) and the inorganic base Na2HPO4 (0.3 mmol) at room temperature, Ar atmosphere was irradiated with 5 W blue LED (λmax = 455 nm) light for 14 h. The desired product 2,3-diphenyl-1H-indole 20.3 was obtained in good yields.
The proposed mechanism for this visible-light initiated cyclization reaction is given in Scheme 20.60 In this scheme SET reduction of benzenesulfonyl chloride 20.1 will occur upon photocatalyst excitation using visible light irradiation, delivering the phenyl radical (Ph˙) and [eosin Y]˙+. Subsequently, the addition of Ph˙ to the alkynyl moiety of 20.2 forms intermediate A, which immediately undergoes the intramolecular cyclization of the alkenyl radical with the azido moiety to produce the N-radical intermediate B with extrusion of N2 gas. H-Atom abstraction from 1,4-CHD will form the desired product 20.3 and generate the radical C, which closes the photoredox cycle by SET with PC˙+.
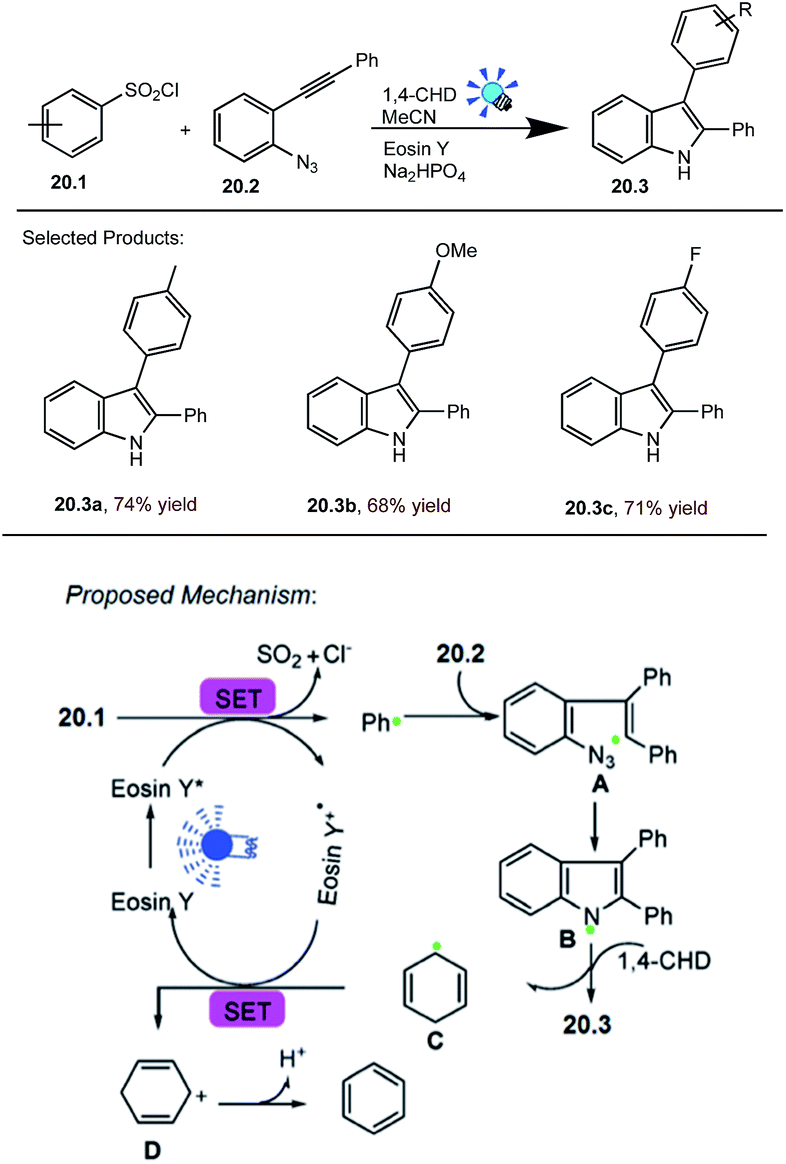 | ||
| Scheme 20 Cyclization of arylsulfonyl chlorides with o-azidoarylalkynes. | ||
21. Dual C–C bond formation via selective C(sp3)–H bond cleavage
Wang, Li and co-workers reported photocatalysed C–C bond formation through H bond cleavage. Direct cleavage and functionalization of C–H bond has evolved to be one of the most efficient and straightforward synthetic approach to carbon–carbon and carbon–heteroatom bond formations, and it does not require substrate prefunctionalization, and minimize the number of synthetic steps.A reaction tube was charged with N-methyl-N-phenylmethacryl-amide 21.1 (0.25 mmol), THF 21.2 (3.0 mL), TBHP (3.0 equiv.), 4 Å molecular sieve (60 mg), eosin-Y (3.0 mol%). The mixture was stirred under the irradiation of 25 W blue LED for 12 h. Then it was concentrated in vacuo to yield the crude product, which was further purified by column chromatography on silica gel (petroleum ether/EtOAc = 2:1) to give the desired product 21.3.
A plausible mechanism for this reaction is proposed in (Scheme 21).61 Initially, the excited-state eosin Y* formed under blue LED irradiation donates an electron to TBHP, giving tert-butyloxyradical. The formed tert-butyloxy radical then abstracts a hydrogen from α-C(sp3)–H of THF 21.1 to generate alkoxyalkyl radical intermediate A, which was trapped by TEMPO to form a radical adduct 21.4, determined by HPLC-HRMS analysis. Subsequently, an addition of alkoxyalkyl radical A to the CC bond of amide 21.2 produces an alkyl radical B, followed by intramolecular cyclization with an aryl ring to afford a radical intermediate C. The generated C is oxidized by a cation radical of eosin Y˙+ to deliver a cationic intermediate D through a SET process. Finally, D is deprotonated to afford alkylated oxindole 21.3 via an aromatization process along with the formation of water.
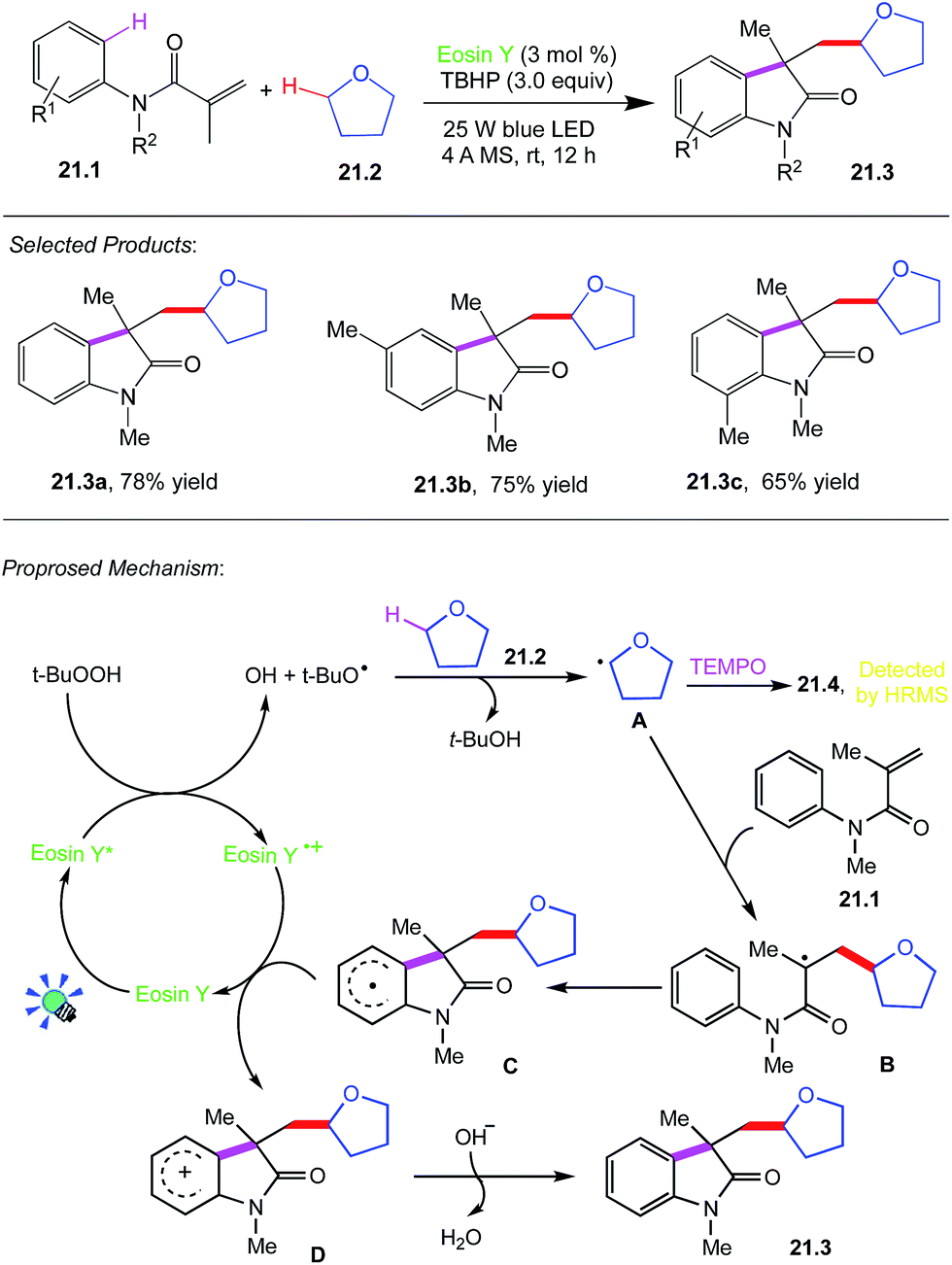 | ||
| Scheme 21 Dual C–C bond formation via selective C(sp3)–H bond cleavage. | ||
22. Aerobic oxidative cyclization of 2-aminobenzothiazole
Heterocycles bearing thiazole, sulphur and nitrogen moieties constitute the core structure of a number of pharmacologically and biologically active interesting compounds. Benzothiazole derivatives possess a wide spectrum of biological applications such as antitumor, schictosomicidal, anti-inflammatory, anticonvulsant, antidiabetic, antipsychotic, diuretic, and antimicrobial activities. Srivastava and co-workers reported photocatalysed synthesis of 2-aminobenzothiazole, which was not been reported by photooxidation reaction so far. In general, organosulfur/nitrogen compounds have been frequently used as precursors in radical reactions because they form radicals very readily.A solution of an arylisothiocyanate 22.1 (1.0 mmol) and a secondary amine 22.2 (1.0 mmol) in DMF (3 mL) was heated at 65 °C for 2–5 h to form the corresponding N-arylthioureas (as monitored by TLC). Then, eosin Y (2.0 mol%) and iPr2Net (2.0 equiv.) were added and the mixture was irradiated with green LEDs (2.4 W, 120 lm) with stirring under an air atmosphere at rt for 10–18 h. After completion of the reaction (monitored by TLC), water (5 mL) was added and the mixture was extracted with EtOAc (3 × 5 mL). The combined organic phase was dried over MgSO4, filtered and evaporated under reduced pressure. The resulting product was purified by silica gel column chromatography using a gradient mixture of hexane/ethyl acetate as eluent to afford an analytically pure 22.4.
A plausible mechanism involving photoredox catalysis for the oxidative cyclization of N-arylthioureas is depicted in Scheme 22.24 On absorption of visible light, the organophotoredox catalyst eosin Y (EY) is excited to its singlet state 1EY* which through inter system crossing (ISC) comes to its more stable triplet state 3EY* and undergoes a single electron transfer (SET). 3EY* may undergo both reductive and oxidative quenching. A SET from A to 3EY* generates thioacyl radical B, which undergoes intramolecular cyclization (5-endo-trig) to form C followed by attack of (O2˙−) to give the product 22.4, successively. The formation of superoxide radical anion (O2˙−) during the reaction was confirmed by the detection of the resulting H2O2 using KI/starch indicator. The reaction is very mild and applicable to aryl and alkyl, tolerates considerable functional group variations like, MeO, Me, Cl and NO2 in the substrate 22.1, which results the desired product 22.4 in good to excellent yields (78–97%). However, arylisothiocyanate 22.1 and a secondary amine 22.2 with an electron-donating group on the aromatic ring appear to react faster and afford marginally higher yields in comparison to those bearing an electron withdrawing group.
 | ||
| Scheme 22 Aerobic oxidative cyclization of 2-aminobenzothiazole. | ||
23. Trifluoromethylation of disubstituted morpholines
Organic compounds with CF3 groups are very important in the production of agrochemicals and pharmaceuticals. Fluorine is the most abundant halogen in the earth's crust and is widely used during lead optimization in drug discovery. The proposed trifluoromethylation strategy, reported by Srivastava and co-workers was first evaluated with substituted-4-tosylmorpholine-3-thiol 23.1, eosin Y, and a green LEDs (2.4 W, 120 lm) with CF3I was used as a CF3 source. The use of an organic base effectively suppressed by-product formation. Optimal conditions were obtained when 2.0 equivalents of iPr2NEt (DIEA) was introduced to the reaction, resulting in the selective tri-fluoromethylation of substituted-4-tosylmorpholine-3-thiol 23.2 within 30–180 min. DMF was the best solvent in terms of the reaction time and yield, hence it was used throughout the synthesis.This clearly shows that the reaction is very mild and applicable to aryl and alkyl, tolerates considerable functional group variations like, MeO, Br, Me, Cl and NO2 in the substrate 23.1, which results the desired product 23.2 in good to excellent yields (72–97%). On the basis of the above observations and the literature precedents, a plausible mechanism involving photocatalytic tri-fluoromethylation of substituted-4-tosylmorpholine-3-thiol is depicted in Scheme 23.62
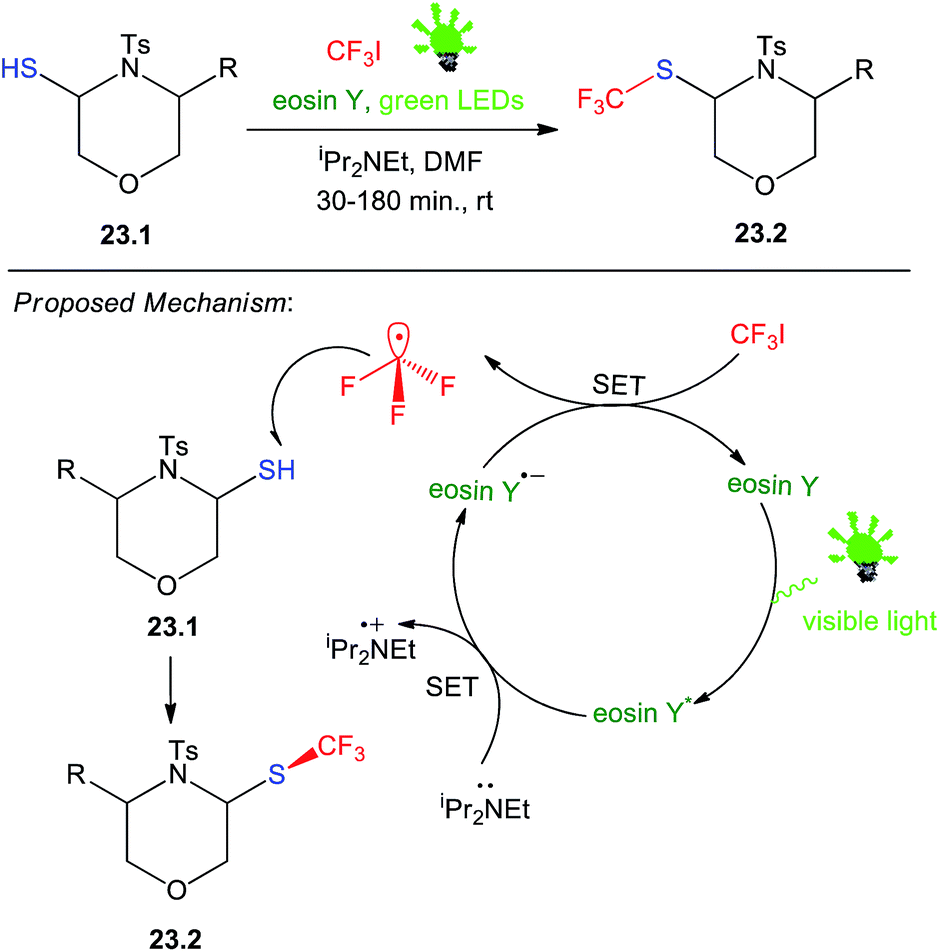 | ||
| Scheme 23 Trifluoromethylation of disubstituted morpholines. | ||
On absorption of visible light, the organophotoredox catalyst eosin Y (EY) is excited to its singlet state 1EY* which through inter system crossing (ISC) comes to its more stable triplet state 3EY* and undergoes a single electron transfer (SET). Reductive quenching of the exited state of eosin Y occurs via a nitrogen base. Next, oxidizing the eosin Y species to its ground state generates an electrophilic CF3 radical.63–65 This CF3 radical can subsequently react with the substituted-4-tosylmorpholine-3-thiol substrate to yield the desired product 23.2.
24. B–C coupling via a boron-centered carboranyl radical
Xie and co-workers reported that boron-centered carboranyl radical can be generated in situ by photoredox catalysis and it can undergo electrophilic substitution with a broad range of (hetero)arenes to efficiently produce 3-(hetero)aryl-o-carboranes. More importantly, this general and convenient procedure provides a metal-free approach to 3-heteroaryl-o-carboranes, and may find applications in medicine and materials science. Inspired by the photocatalytic transformations of aryldiazonium salts into the corresponding aryl radicals,66 researchers wondered whether a boron-centered carboranyl radical (o-C2B10H11˙) could be generated by visible-light-induced photoredox catalysis from 3-diazonium-o-carborane tetrafluoroborate ([3-N2-o-C2B10H11][BF4], 24.1).67 Herein, they described the generation of such a carboranyl radical by photoredox catalysis and its reactions with (hetero)arenes for high-yielding syntheses of 3-(hetero)aryl-o-carboranes.Reaction conditions are diazonium salt 24.1 (0.1 mmol), heteroarene 24.2 (0.5 mmol for thiophene derivatives, 0.2 mmol for other heteroarenes), eosin Y (2 mol%), CH3CN (2 mL), 12 W LED (530 nm), room temperature for 2 h. Excellent yield of the isolated product 24.3 was obtained.
The C–H carboranylation of heteroarenes with 24.1 using eosin Y is expected to proceed through a radical mechanism and preliminary mechanistic investigations support this assumption. Addition of 1.2 equivalents of 2,2,6,6-tetramethylpiperdinoxyl (TEMPO) to the reaction mixture completely suppressed this carboranylation process and the TEMPO-trapped adduct 24.4 was isolated in 89% yield. A plausible reaction mechanism is proposed in Scheme 24.68 Irradiation of eosin Y by visible light generates a photoexcited species, eosin Y*, which can undergo single-electron transfer (SET) with 24.1 (Ered1/2 = −0.23 V vs. SCE in CH3CN) to afford the boron-centered carboranyl radical A. Addition of A to the arene 24.2 gives the intermediate B. Oxidation of B by either the eosin Y radical cation (path a) or the diazonium salt 1 through a radical-chain transfer (path b) generates the carbocation intermediate C. Deprotonation of C leads to the final rearomatized product 24.3. The possibility that 24.3 is produced from the Friedel–Crafts reaction via the carboranyl boronium cation68 D can be ruled out. If D was formed in the reaction, it would be trapped by the solvent CH3CN to generate the corresponding solvated adduct. However, no such species was detected.
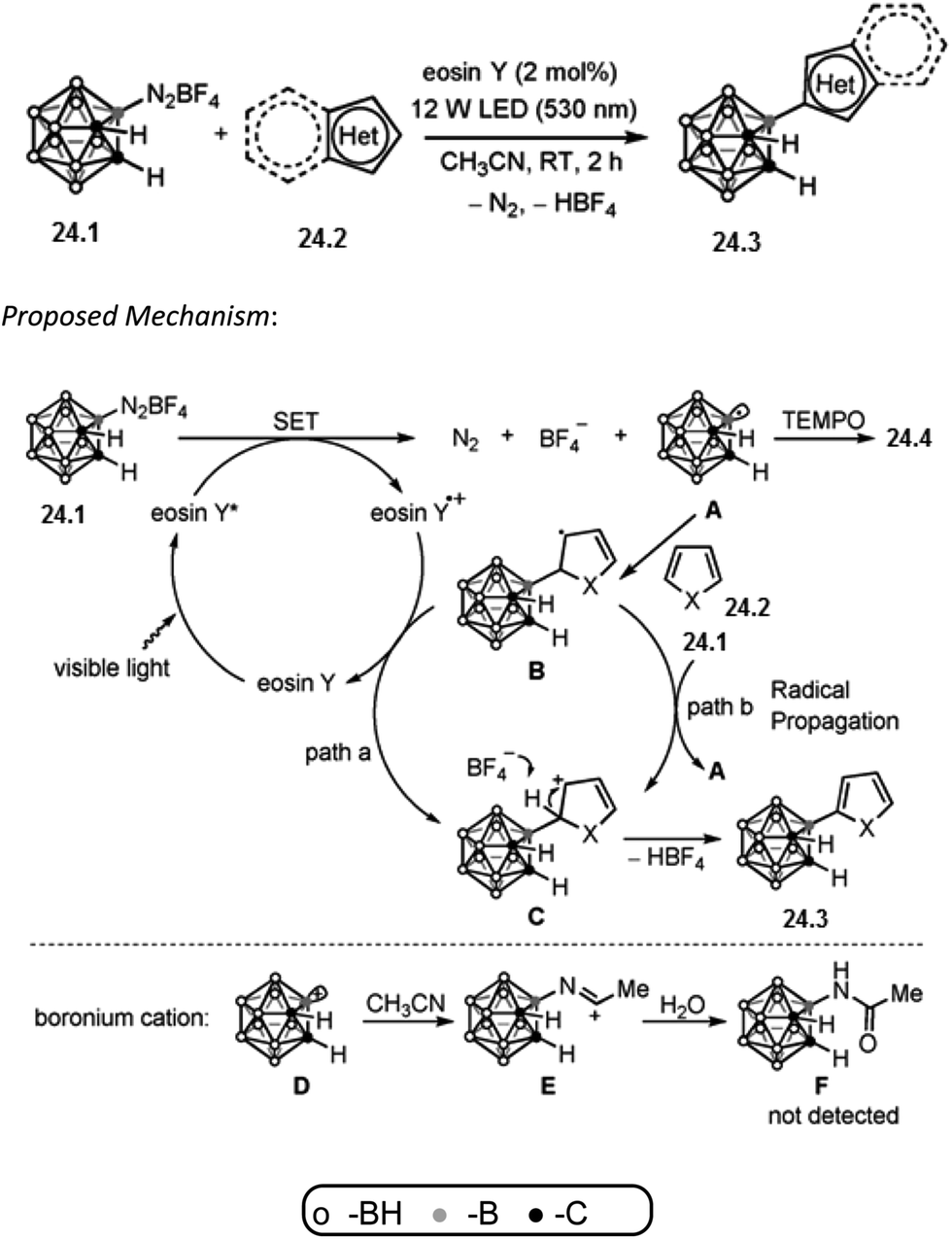 | ||
| Scheme 24 B–C coupling via a boron-centered carboranyl radical. | ||
25. Conclusions
Visible light photoredox catalysis with metal complexes has already received a lot of attention as a tool for organic synthetic transformations. For several applications eosin Y serves as an attractive alternative to redox active metal complexes and even outperforms them in some cases.69–72 Eosin Y photocatalysis has been applied to generate reactive intermediates including aryl radicals, iminium ions, trifluoromethyl radicals, and enone radical anions, which are utilized in arene C–H functionalization, [2 + 2] cyclo addition, amine functionalization, hydroxylation, reduction, and oxidation reactions.The photocatalyst eosin Y has got much more attention due to easy handling, less expansive, eco-friendly, non-toxicity, as well as it can be reusable by covalent immobilization of it on reduced graphene oxide and demonstrate the obtained hybrid can be used as a recyclable photocatalyst with excellent catalytic performances.73
In addition, eosin Y catalysis has been merged with other modes of catalysis, such as enamine catalysis and hydrogen bond promoted catalysis, to achieve enantioselective reactions. The use of eosin Y photocatalysis in continuous flow technology has been described. Overall, the good availability, strong absorption in the visible part of the spectrum and suitable redox potential values for a variety of organic transformations make eosin Y an appealing and green photocatalyst for organic synthesis.
Acknowledgements
We sincerely thank Emerging Science Society for financial support.References
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS.
- J. Xie, H. Jina, P. Xu and C. Zhu, Tetrahedron Lett., 2014, 55, 36 CrossRef CAS.
- M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef.
- D. P. Hari and B. Konig, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed.
- D. P. Hari, T. Hering and B. König, Angew. Chem., Int. Ed., 2014, 53, 725–728 CrossRef PubMed.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 CAS.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 CAS.
- T. Hering, D. P. Hari and B. König, J. Org. Chem., 2012, 77, 10347–10352 CrossRef CAS PubMed.
- M. Rueping, C. Vila and T. Bootwicha, ACS Catal., 2013, 3, 1676–1680 CrossRef CAS.
- F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859–917 CrossRef.
- F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859–917 CrossRef.
- C. K. Prier, D. A. Rankic and D. W. Macmillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81(16), 6898–6926 CrossRef CAS PubMed.
- X.-J. Yang, B. Chen, L.-Q. Zheng, L.-Z. Wu and C.-H. Tung, Green Chem., 2014, 16, 1082 RSC.
- Y. C. Teo, Y. Pan and C. H. Tan, ChemCatChem, 2013, 5, 235 CrossRef CAS.
- K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293 RSC.
- D.-T. Yang, Q.-Y. Meng, J.-J. Zhong, M. Xiang, Q. Liu and L.-Z. Wu, Eur. J. Org. Chem., 2013, 7528 CrossRef CAS.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgense and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS PubMed.
- V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88(1), 59–65 CrossRef CAS.
- V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88(3), 227–233 CrossRef CAS.
- A. Penzkofer, A. Beidoun and M. Daiber, J. Lumin., 1992, 51, 297–314 CrossRef CAS.
- A. Penzkofer, A. Beidoun and S. Speiser, Chem. Phys., 1993, 170, 139–148 CrossRef CAS.
- A. Penzkofer and A. Beidoun, Chem. Phys., 1993, 177, 203–216 CrossRef CAS.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- J. Zhang, L. Wang, Q. Liu, Z. Yang and Y. Huang, Chem. Commun., 2013, 49, 11662–11664 RSC.
- N. Klonis and W. H. Sawyer, J. Fluoresc., 1996, 6, 147–157 CrossRef CAS PubMed.
- V. R. Batistela, D. S. Pellosi, E. D. de Souza, W. F. da Costa, S. M. de Oliveira Santin, V. R. de Souza, W. Caetano, H. P. M. de Oliveira, I. S. Scarminio and N. Hioka, Spectrochim. Acta, Part A, 2011, 79, 889–897 CrossRef CAS PubMed.
- M. Májek, F. Filace and A. Jacobi von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef PubMed.
- D. P. Hari and B. König, Org. Lett., 2011, 13, 3852–3855 CrossRef CAS PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- A. U. Meyer, K. Straková, T. Slanina and B. König, Chem.–Eur. J., 2016, 22, 8694–8699 CrossRef CAS PubMed.
- D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef.
- S. Gazi and R. Ananthakrishnan, Appl. Catal., B, 2011, 105, 317–325 CrossRef CAS.
- X.-J. Yang, B. Chen, L.-Q. Zheng, L.-Z. Wu and C. H. Tung, Green Chem., 2014, 16, 1082–1086 RSC.
- D.-T. Yang, Q.-Y. Meng, J.-J. Zhong, M. Xiang, Q. Liu and L.-Z. Wu, Eur. J. Org. Chem., 2013, 7528–7532 CrossRef CAS.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- D. D. Tanner and H. K. Singh, J. Org. Chem., 1986, 51, 5182–5186 CrossRef CAS.
- S. Fukuzumi, S. Mochizuki and T. Tanaka, J. Phys. Chem., 1990, 94, 722–726 CrossRef CAS.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- M. Neumann and K. Zeitler, Org. Lett., 2012, 14, 2658–2661 CrossRef CAS PubMed.
- A. Paula, D. Chatterjeea, Rajkamal, T. Haldera, S. Banerjeeb and S. Yadava, Tetrahedron Lett., 2015, 56, 2496–2499 CrossRef.
- M. Majek and A. J. Wangelin, Angew. Chem., Int. Ed., 2014, 53, 1–6 CrossRef.
- J.-B. Peng, X. Qi and X.-F. Wu, ChemSusChem, 2016, 9, 1–6 CrossRef PubMed.
- T.-F. Niu, D.-Y. Jiang, S.-Y. Li, B.-Q. Ni and L. Wang, Chem. Commun., 2016, 52, 13105 RSC.
- L. Gu, C. Jinb and J. Liua, Green Chem., 2015, 17, 3733–3736 RSC.
- X. Liu, T. Cong, P. Liu and P. Sun, J. Org. Chem., 2016, 81(16), 7256–7261 CrossRef CAS PubMed.
- S. Devari, M. A. Rizvi and B. A. Shah, Tetrahedron Lett., 2016, 57, 3294–3297 CrossRef CAS.
- F. Q. Huang, X. Dong, L. W. Qi and B. Zhang, Tetrahedron Lett., 2016, 57, 1600–1604 CrossRef CAS.
- D. Yang, B. Huang, W. Wei, J. Li, G. Lin, Y. Liu, J. Ding, P. Sun and H. Wang, Green Chem., 2016, 18, 5630–5634 RSC.
- K. Wang, L. G. Meng and L. Wang, J. Org. Chem., 2016, 81(16), 7080–7087 CrossRef CAS PubMed.
- S. Mitra, M. Ghosh, S. Mishra and A. Hajra, J. Org. Chem., 2015, 80, 8275–8281 CrossRef CAS PubMed.
- C. Song, P. Chen and Y. Tang, RSC Adv., 2017, 7, 11233–11243 RSC.
- J. Schwarz and B. König, Green Chem., 2016, 18, 4743–4749 RSC.
- A. K. Yadav and L. D. S. Yadav, Green Chem., 2016, 18, 4240–4244 RSC.
- L. Gu, C. Jin, W. Wang, Y. He, G. Yangc and G. Li, Chem. Commun., 2017, 53, 4203–4206 RSC.
- D. Xia, Y. Li, T. Miao, P. Li and L. Wang, Green Chem., 2017, 19, 1732–1739 RSC.
- V. Srivastava, P. K. Singh and P. P. Singh, Asian J. Chem., 2016, 28(10), 2159–2163 CrossRef CAS.
- N. J. W. Straathof, H. P. L. Gemoets, X. Wang, J. C. Schouten, V. Hessel and T. Noel, ChemSusChem, 2014, 7, 1612 CrossRef CAS PubMed.
- N. J. W. Straathof, D. J. G. P. van Osch, A. Schouten, X. Wang, J. C. Schouten, V. Hessel and T. Noel, J. Flow Chem., 2014, 4, 12 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- D. Zhao, J. Zhang and Z. Xie, Angew. Chem., Int. Ed., 2014, 53, 8488 CrossRef CAS PubMed.
- D. Zhao and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 3166–3170 CrossRef CAS PubMed.
- B. Ringstrand and P. Kaszyński, Acc. Chem. Res., 2013, 46, 214 CrossRef CAS PubMed.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169–171 CrossRef CAS.
- Y. C. Teo, Y. Pan and C. H. Tan, ChemCatChem, 2013, 5, 235–240 CrossRef CAS.
- D. Cantillo, O. de Frutos, J. A. Rincon, C. Mateos and C. O. Kappe, Org. Lett., 2014, 16, 896–899 CrossRef CAS PubMed.
- Z. Li, W. Zhang, Q. Zhao, H. Gu, Y. Li, G. Zhang, F. Zhang and X. Fan, ACS Sustainable Chem. Eng., 2015, 3(3), 468–474 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |