
Total synthesis of (−)-agelastatin A: an SH2′ radical azidation strategy†
Izuru
Tsuchimochi
ab,
Yuta
Kitamura
b,
Hiroshi
Aoyama
b,
Shuji
Akai
b,
Keiyo
Nakai
a and
Takehiko
Yoshimitsu
*a
aDivision of Pharmaceutical Sciences, Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, Okayama University, 1-1-1 Tsushima-naka, Kita-ku, Okayama 700-8530, Japan. E-mail: yoshimit@okayama-u.ac.jp; Tel: +81-86-251-7930
bGraduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 7th August 2018
Abstract
A reagent generated from TMSN3/KMnO4/BnEt3NCl was found to promote an SH2′ radical azidation of a bromo silyl enol ether to furnish an azido silyl enol ether via olefin transposition. With the present azidation protocol, a new synthetic approach to agelastatin A, a potent antitumor marine alkaloid, has been established.
(−)-Agelastatin A (1), along with its congener agelastatin B (2), was first isolated as a cytotoxic constituent from the Coral Sea sponge Agelas dendromorpha by Pietra and co-workers in 1993 (Fig. 1).1 Thereafter, Molinski and co-workers identified the Indian Ocean sponge Cymbastela sp. as another source that produces 1 along with agelastatins C (3) and D (4), two additional agelastatin members.2 In 2010, Al-Mourabit and co-workers reported the isolation of agelastatins E (5) and F (6) from the New Caledonian sponge A. dendromorpha.3 Early biological assessments of agelastatins conducted by the aforementioned laboratories have revealed that compound 1 exhibits remarkable properties, including antitumor activity,1,3 brine shrimp toxicity,2 and insecticidal activity.2 In addition, Meijer and Pettit have found that agelastatin A (1) is a potent inhibitor of GSK-3β, a pivotal serine/threonine kinase.4 Hale and El-Tanani have reported that agelastatin A (1) dramatically decreases β-catenin levels in cancer cells and inhibits cancer cell proliferation by arresting cell cycle at G2 phase.5
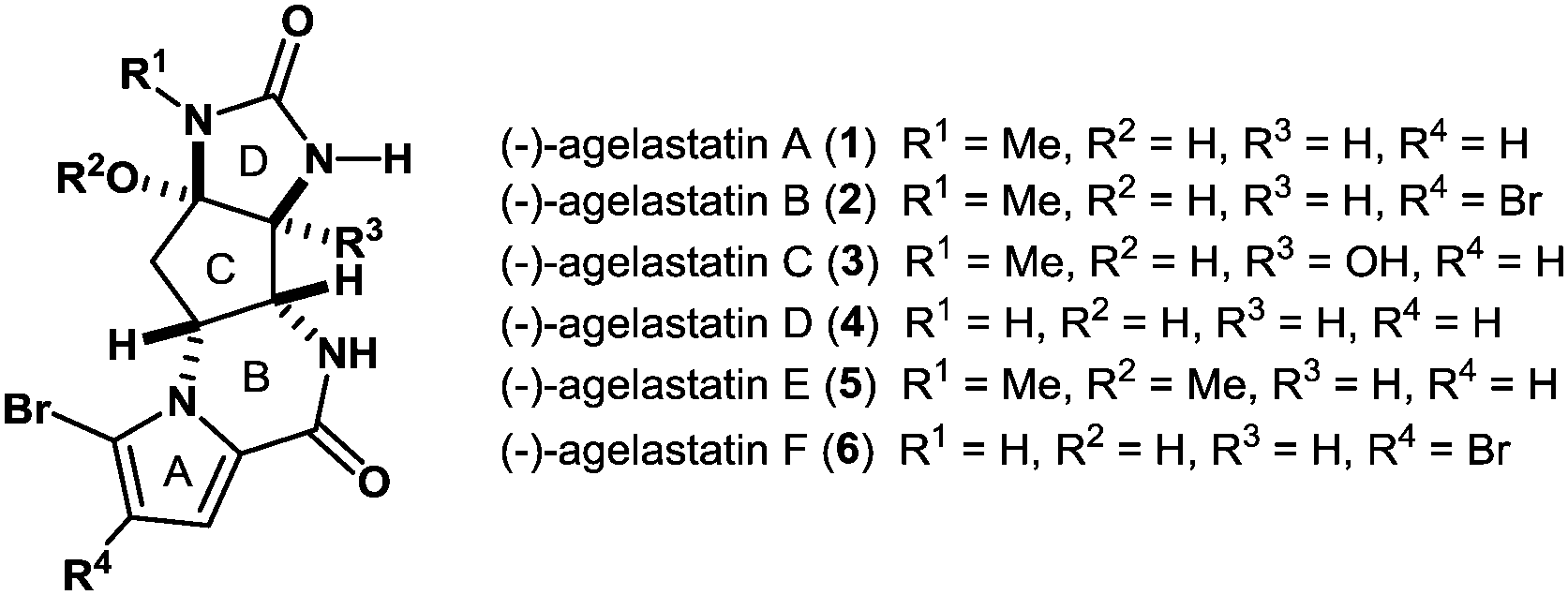 | ||
| Fig. 1 Agelastatin alkaloids. | ||
The biological significance of agelastatin A (1) has made it an attractive target for medicinal studies.6,7 For instance, Movassaghi's comparative cytotoxicity assay of all agelastatin members, i.e., A (1) to F (6), has successfully validated the relevance of agelastatin A (1) as a promising anticancer agent.7s In addition, structure–activity relationship (SAR) studies on agelastatin analogues have recently been disclosed by the groups of Molinski,8 Romo/Liu,9 and Movassaghi,10 boosting the applications of agelastatin particularly to blood cancer chemotherapy.
Our group has also been engaged in synthetic and medicinal studies on 1 and has demonstrated that agelastatin analogues potentially attenuate brain cancer.11 Furthermore, our SAR study has revealed that structural modifications of the N1-substituent of the D-ring of 1 could retain the in vitro and in vivo therapeutic efficacies of agelastatin analogues.12,13 Movassaghi's group has further clarified that D-ring modifications expand the scope of derivatization of agelastatins to access potent analogues.10
In the present study, we have established a new route to agelastatin A (1) through an SH2′ radical azidation protocol using TMSN3/KMnO4/BnEt3NCl that enables the allylic transposition of a bromo silyl enol ether into an azido silyl enol ether, which serves as a useful D-ring precursor of the target natural product (Scheme 1).
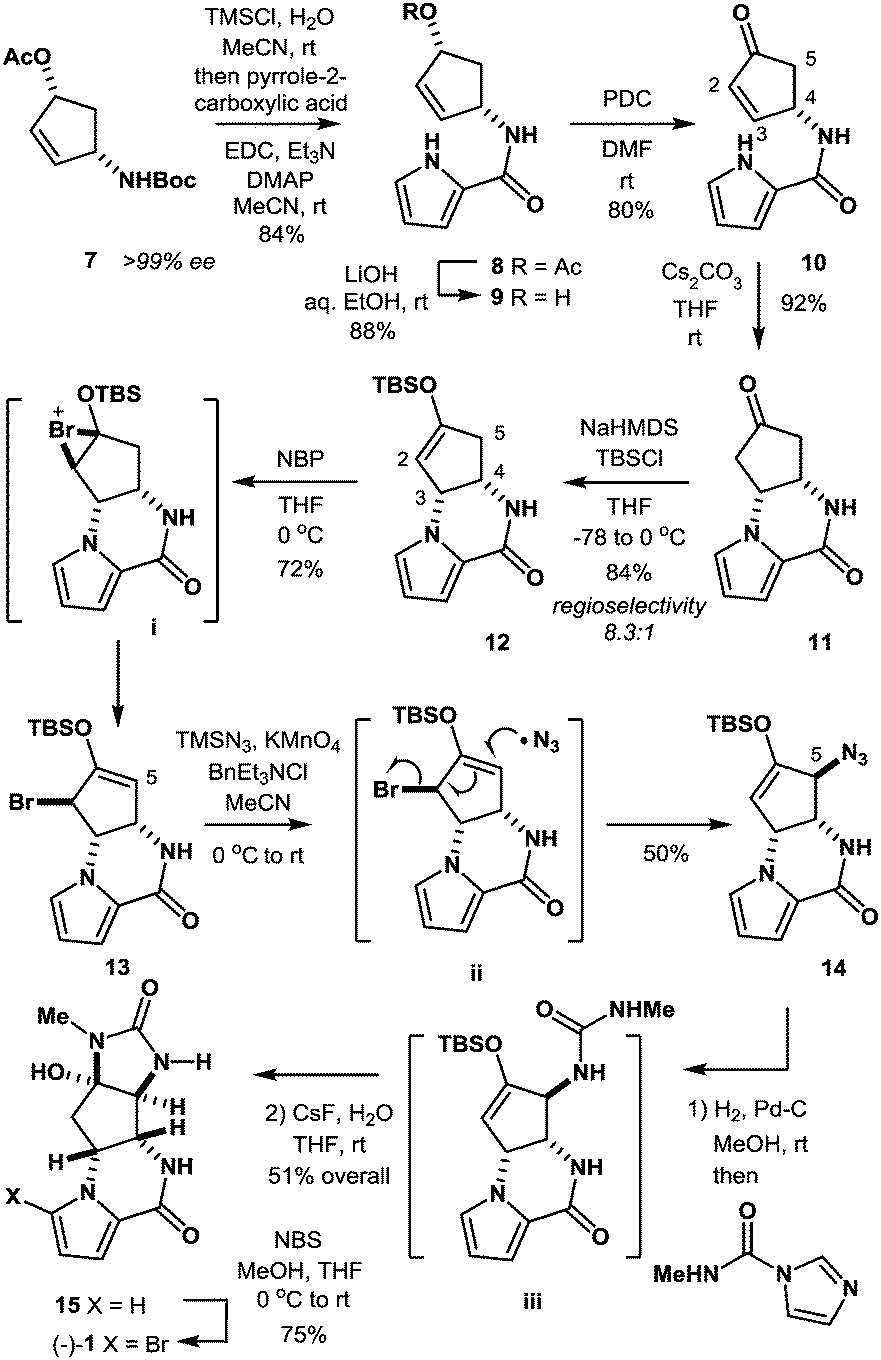 | ||
| Scheme 1 Total synthesis of (−)-agelastatin A (1). | ||
The synthesis was commenced with Boc-protected aminoalcohol derivative 7 (>99% ee).14 The Boc group of 7 was removed with hydrochloric acid (HCl) generated in situ from TMSCl in aq. MeCN to provide an ammonium salt (structure not shown). After evaporation of the solvents under reduced pressure, the resultant crude product was coupled with pyrrole-2-carboxylic acid using EDC, Et3N, and DMAP in MeCN to furnish compound 8 in 84% yield. Then, compound 8 was hydrolyzed with LiOH in aq. EtOH to provide alcohol 9 in 88% yield. PDC oxidation of alcohol 9 in DMF delivered enone 10 in 80% yield, which, upon treatment with Cs2CO3 in THF, gave tricyclic ketone 11via a conjugate addition of the pyrrole nitrogen to the enone double bond. No racemization at C4 position took place in this transformation (9 → 10 → 11), retaining the optical purity of 11 (>99% ee).15 Then, ketone 11 was subjected to enolization with NaHMDS followed by O-silylation with tert-butyldimethylsilyl chloride to produce silyl enol ether 12 along with its minor regioisomer 16 (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 = 8.3:1) (Fig. 2). Obviously, major product 12 was not ideal for further functionalization as it lacked a reactive alkene functionality at C5 position. However, we found that 12 and 16 underwent olefin isomerization with a trace acid probably due to their strained nature.16 Therefore, we envisioned that the brominative olefin transposition of 12 would take place via a bromonium formation followed by deprotonation to allow net olefin transposition that affords an enol ether suitable for C5 functionalization. To our delight, the treatment of silyl enol ether 12 with N-bromophthalimide (NBP) was found to deliver allylic bromide 13 in stereoselective and regiospecific manners as we had expected.
:16 = 8.3:1) (Fig. 2). Obviously, major product 12 was not ideal for further functionalization as it lacked a reactive alkene functionality at C5 position. However, we found that 12 and 16 underwent olefin isomerization with a trace acid probably due to their strained nature.16 Therefore, we envisioned that the brominative olefin transposition of 12 would take place via a bromonium formation followed by deprotonation to allow net olefin transposition that affords an enol ether suitable for C5 functionalization. To our delight, the treatment of silyl enol ether 12 with N-bromophthalimide (NBP) was found to deliver allylic bromide 13 in stereoselective and regiospecific manners as we had expected.
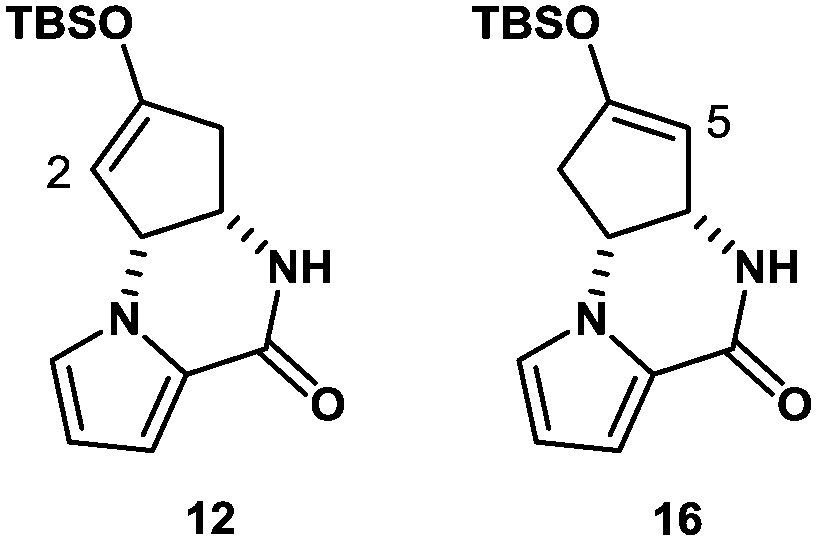 | ||
| Fig. 2 TBS enol ethers 12 and 16 generated from 11. | ||
With compound 13 in possession, the nitrogen functionalization at C5 position was examined to access key intermediate 14 (Table 1). An attempted ionic SN2′ azidation of 13 with NaN3 in DMF was unsuccessful (entry 5), giving rise to a desilylated product. To this end, we expected that the electrophilic nitrogen radical species would preferentially undergo an addition reaction with the electron-rich enol double bond to facilitate SH2′ radical azidation to deliver compound 14.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reagents (equiv.) | Time | Yielda (%) | |||
| 14 | 17 | 18 | 13 | |||
| a Isolated yields after purification by column chromatography. b Recovered unreacted starting material. c The reaction was conducted at r.t. d Bromoketone (60%) was produced. e The reaction was conducted at −78 °C. | ||||||
| 1 | KMnO4 (0.3), BnEt3NCl (0.3), TMSN3 (10), MeCN | 40 min | 50 | 21 | 3 | Trace |
| 2 | KMnO4 (0.1), BnEt3NCl (0.1), TMSN3 (10), MeCN | 40 min | 30 | 4 | 12 | 30 |
| 3 | KMnO4 (0.1), BnEt3NCl (0.1), TMSN3 (10), MeCN, O2 | 40 min | 31 | 5 | 11 | 21 |
| 4 | KMnO4 (0.6), BnEt3NCl (0.6), TMSN3 (10), MeCN | 40 min | 43 | 11 | 4 | 9 |
| 5 | NaN3 (1.1), DMFc | 15 min | — | — | — | —d |
| 6 | BnEt3NCl (0.3), TMSN3 (10), MeCN | 75 min | — | — | — | 90 |
| 7 | TMSN3 (10), MeCN | 70 min | — | — | — | 89 |
| 8 | PhlO (1.2), TMSN3 (2.4), CH2Cl2e | 40 min | 24 | 17 | 14 | 6 |
| 9 | Mn(OAc)3·2H2O (3), TMSN3 (6), MeCNc | 11 h | 42 | 8 | — | — |
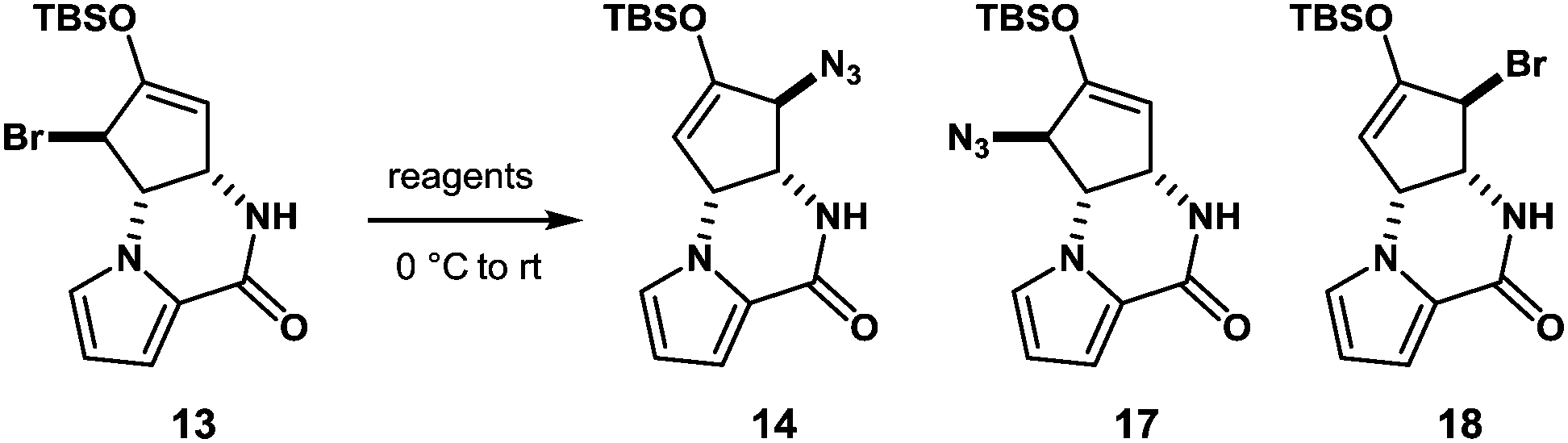
An azide radical is known to be generated from an anionic azide by oxidation processes. The Magnus protocol represents such an example, which utilizes trimethylsilylazide (TMSN3) in combination with iodosylbenzene (PhIO) in CH2Cl2 at low temperature (−78 °C). The Magnus method was proved to afford desired product 14 albeit in moderate yield (entry 8).17 Therefore, we sought a new reagent system to deliver an azido radical and found that the treatment of 13 with TMSN3 (10 equiv.)/KMnO4 (0.3 equiv.)/BnEt3NCl (0.3 equiv.) successfully produced azide 14 in 50% yield along with regioisomeric azide 17 (21%) and bromide 18 (3%)18 (entry 1). In the absence of KMnO4, no reaction took place and unreacted 13 was recovered (entries 6 and 7). When catalytic KMnO4 (0.1 equiv.) was used in combination with TMSN3 (10 equiv.) and BnEt3NCl (0.1 equiv.) in either the presence or absence of molecular oxygen (O2), the chemical yield was low, suggesting that catalysis by O2 in the present radical azidation was not operative (entries 2 and 3). Increasing the amount of Mn(VII) reagent was found to have no impact on the improvement of the chemical yields (entry 4).
It should be mentioned that the addition of TMSN3 to the mixture of KMnO4 and BnEt3NCl at 0 °C caused the evolution of molecular nitrogen (N2) accompanied by a color change of the solution from purple to dark brown, suggesting the production of low-valent manganese species from the Mn(VII) reagent. Although the reactive species responsible for the present radical azidation remains unclear, we assume that permanganate(VII) (MnO4−) reacts with TMSN3 to generate a low-valent mangan azide complex that serves as a metastable azide radical source. To clarify this hypothesis, we measured the amount of nitrogen gas (N2) that was generated from the reagent system. When KMnO4 (0.33 mmol) was treated with BnEt3NCl (0.33 mmol) and a large excess of TMSN3 (11.1 mmol), 20–24 mL (ca. 0.9–1.1 mmol) of molecular nitrogen, which corresponds to ca. 3.0 equiv. relative to 1.0 equiv. of permanganate ion (MnO4−), was generated. Assuming that 1.0 equiv. of permanganate reacts with 5.0 equiv. of TMSN3 to produce 3.0 equiv. of molecular nitrogen, we propose that a pentavalent Mn(V) species is produced from the Mn(VII) species (Scheme 2).
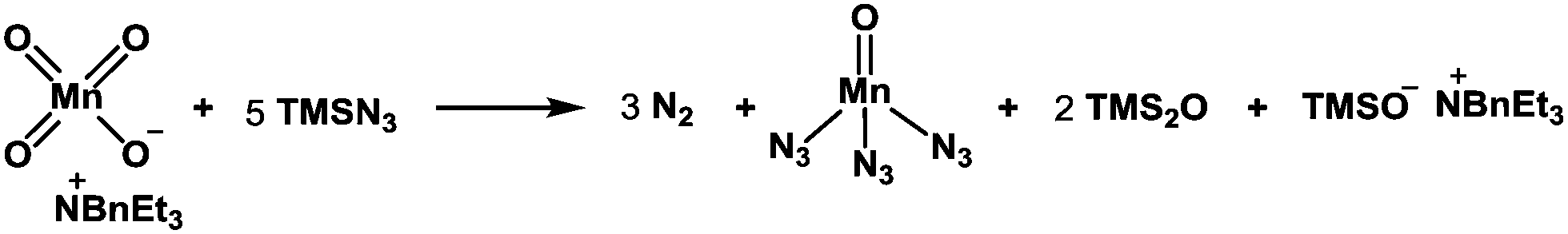 | ||
| Scheme 2 Plausible generation of Mn(V) azide species. | ||
Jiao and co-workers have reported that Mn(III) generated from MnBr2 in the presence of molecular oxygen serves as an effective catalyst to generate an azide radical from TMSN3.19a We have examined Mn(OAc)3·2H2O in combination with TMSN3 (6 equiv.) as a possible source of azido radical and found that desired material 14 could be similarly produced in 42% yield along with 17 (8%) (entry 9).19b This result suggests that Mn(III) azide complex is likely responsible for the present radical azidation. Based on these observations, we currently assume that metastable Mn(V) species is generated from Mn(VII) with excess TMSN3 and that Mn(V) provides 3 equiv. of azido radical to finally become Mn(II), which no longer serves as a radical source. To elucidate the formation of the meta-stable Mn species, we carried out a comparison experiment: after stirring the reagents for 60 min, excess remaining TMSN3 was completely removed under reduced pressure. Then, the residual solid that likely contains the Mn species was diluted with MeCN and mixed with substrate 13. As a result, almost identical yields of products 14 (48%), 17 (22%), and 18 (6%) were obtained as in the case of entry 1, indicating that the Mn(V) azide complex is generated as a reactive meta-stable reagent.
The formation of compounds 17 and 18, which provides an insight into the mechanism of the present azidation, also requires elaboration (Scheme 3). When azide 14 and isomeric azide 17 were separately subjected to the same reaction conditions for 1 h, only a trace amount of corresponding azide 17 and 14 was produced along with the unreacted starting azides, respectively. This indicates that both azides 14 and 17, once produced, were hardly susceptible to the SH2′ azidation. In contrast, when isomeric bromide 18 was treated with the reagent, compounds 14 (34%), 17 (27%), and 18 (12%) were obtained similar to the case of 13. Based on these results, we propose that the addition of an azide radical to bromide 13 generates a Br radical that undergoes rapid addition to substrate 13 to generate regioisomeric bromide 18. Then, 18 is further converted into compound 17via a radical azidation.
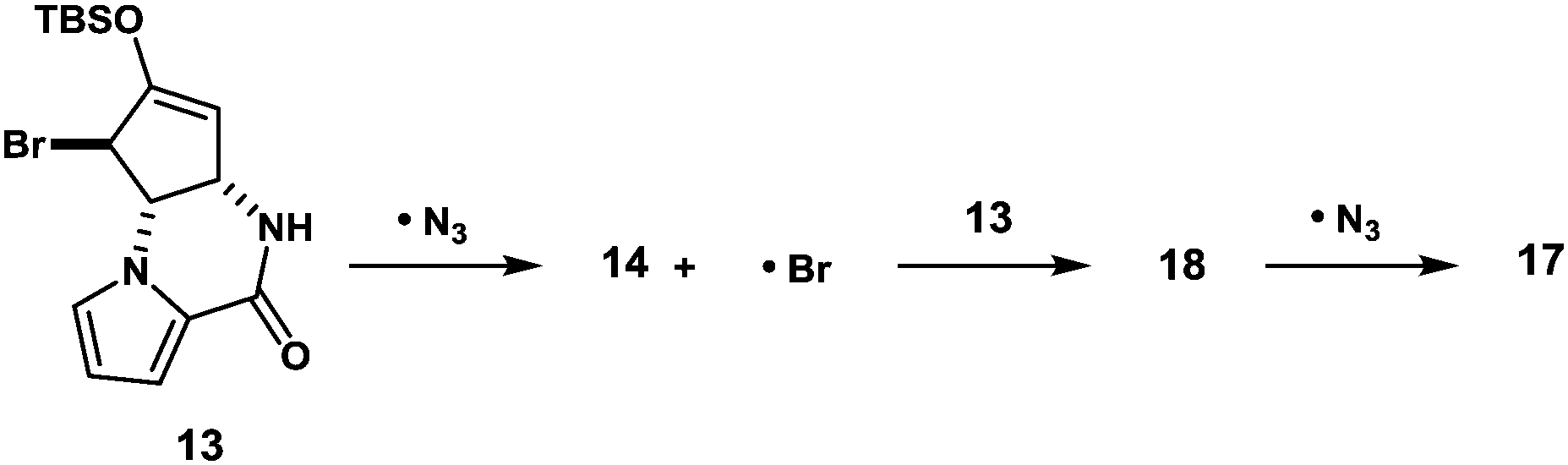 | ||
| Scheme 3 Plausible mechanisms of the production of regioisomeric byproducts 17 and 18. | ||
With azide 14 in possession, we further endeavored to accomplish the total synthesis. Thus, azide 14 was subjected to catalytic hydrogenation followed by one-pot urea formation with Batey's reagent20 and subsequent desilylative cyclization with CsF to afford tetracyclic compound 15 in 51% yield over three steps. It should be mentioned that no purification was required in the three-step sequence, allowing ease of experimental operations. Finally, the known bromination protocol was applied to compound 15 to furnish (−)-agelastatin A (1).
In conclusion, we have established a new approach to (−)-agelastatin A (1) by the strategic implementation of brominative olefin transposition and subsequent SH2′ radical azidation. The present approach features a late-stage construction of D-ring that would allow facile production of D-ring analogues. We believe that the present synthesis would facilitate further development of new agelastatin analogues.
This work was supported by a grant [KAKENHI #15K14977] generously provided by the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), The NOVARTIS Foundation (Japan) for the Promotion of Science, and the Hoansha Foundation.
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
-
(a) M. D’Ambrosio, A. Guerriero, C. Debitus, O. Ribes, J. Pusset, S. Leroy and F. J. Pietra, J. Chem. Soc., Chem. Commun., 1993, 1305 RSC
; (b) M. D’Ambrosio, A. Guerriero, G. Chiasera and F. Pietra, Helv. Chim. Acta, 1994, 77, 1895 CrossRef
- T. W. Hong, D. R. Jímenez and T. F. Molinski, J. Nat. Prod., 1998, 61, 158 CrossRef PubMed
- S. Tilvi, C. Moriou, M. Martin, J. Gallard, J. Sorres, K. Patel, S. Petek, C. Debitus, L. Ermolenko and A. Al-Mourabit, J. Nat. Prod., 2010, 73, 720 CrossRef PubMed
-
(a) L. Meijer, A. M. Thunnissen, A. W. White, M. Garnier, M. Nikolic, L. H. Tsai, J. Walter, K. E. Cleverley, P. C. Salinas, Y. Z. Wu, J. Biernat, E. M. Mandelkov, S. H. Kim and G. R. Pettit, Chem. Biol., 2000, 7, 51 CrossRef PubMed
-
(a) C. K. Mason, S. McFarlane, P. G. Johnston, P. Crowe, P. J. Erwin, M. M. Domostoj, F. C. Campbell, S. Manaviazar, K. J. Hale and M. El-Tanani, Mol. Cancer Ther., 2008, 7, 548 CrossRef PubMed
- For selected reviews on total synthesis of agelastatins, see:
(a) G. Dong, Pure Appl. Chem., 2010, 82, 2231 Search PubMed
-
(a) D. Stien, G. T. Anderson, C. E. Chase, Y. Koh and S. M. Weinreb, J. Am. Chem. Soc., 1999, 121, 9574 CrossRef
- E. P. Stout, M. Y. Choi, J. E. Castro and T. F. J. Molinski, J. Med. Chem., 2014, 57, 5085 CrossRef PubMed
-
(a) M. Jouanneau, B. McClary, J. C. P. Reyes, R. Chen, Y. Chen, W. Plunkett, X. Cheng, A. Z. Milinichik, E. F. Albone, J. O. Liu and D. Romo, Bioorg. Med. Chem. Lett., 2016, 26, 20927 CrossRef PubMed
- A. H. Antropow, K. Xu, R. J. Buchsbaum and M. Movassaghi, J. Org. Chem., 2017, 82, 7720 CrossRef PubMed
-
(a) T. Yoshimitsu, T. Ino and T. Tanaka, Org. Lett., 2008, 10, 5457 CrossRef PubMed
-
(a) Z. Li, T. Kamon, D. A. Personett, T. Caulfield, J. A. Copland, T. Yoshimitsu and H. W. Tun, Med. Chem. Commun., 2012, 3, 233 RSC
-
H. W. Tun, T. Yoshimitsu, D. Shigeoka, T. Kamon, Z. Li, Y. Qiu and T. R. Caulfield, US Pat., US9464093B2, 2016 Search PubMed
-
(a) M. J. Mulvihill, J. I. Gage and M. J. J. Miller, J. Org. Chem., 1998, 63, 3357 CrossRef
- The optical purity was unambiguously confirmed by 1H NMR analysis of Mosher esters derived from an alcohol that was prepared by reduction of ketone 11 with NaBH4 (for the details, see the ESI†).
-
(a) A. Deyine, G. Dujardin, M. Mammeri and J.-M. Poirier, Synth. Commun., 1998, 28, 1817 CrossRef
- P. Magnus, M. B. Roe and C. J. Hulme, J. Chem. Soc., Chem. Commun., 1995, 263 RSC
- The stucture of regioisomeric bromide 18 was unambiguously confirmed by X-ray crystallographic analysis (CCDC 1852628)†.
-
(a) X. Sun, X. Li, S. Song, Y. Zhu, Y.-F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059 CrossRef PubMed
- P. A. Duspara, Md. S. Islam, A. J. Lough and R. A. Batey, J. Org. Chem., 2012, 77, 10362 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of new compounds including NMR spectra. CCDC 1852628. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc05697h |
| This journal is © The Royal Society of Chemistry 2018 |