
Light-weight 3D Co–N-doped hollow carbon spheres as efficient electrocatalysts for rechargeable zinc–air batteries†
Shengmei
Chen
 abc,
Junye
Cheng
abcd,
Longtao
Ma
c,
Shanke
Zhou
e,
Xiuwen
Xu
c,
Chunyi
Zhi
c,
Wenjun
Zhang
abc,
Linjie
Zhi
*e and
J. Antonio
Zapien
*abc
abc,
Junye
Cheng
abcd,
Longtao
Ma
c,
Shanke
Zhou
e,
Xiuwen
Xu
c,
Chunyi
Zhi
c,
Wenjun
Zhang
abc,
Linjie
Zhi
*e and
J. Antonio
Zapien
*abc
aCenter of Super-Diamond and Advanced Films (COSDAF), City University of Hong Kong, Kowloon, Hong Kong 999077. E-mail: apjazs@cityu.edu.hk
bCity University of Hong Kong, Shenzhen Research Institute, Shenzhen, Guangdong, P. R. China
cDepartment of Materials and Science Engineering, City University of Hong Kong, Kowloon, Hong Kong 999077
dGuangdong Provincial Key Laboratory of Micro/Nano Optomechatronics Engineering, College of Mechatronics and Control Engineering, Shenzhen University, Shenzhen 518060, China
eCAS Center of Excellence for Nanoscience, Key Laboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: zhilj@nanoctr.cn
First published on 20th March 2018
Abstract
Rational design of cost-effective, nonprecious metal-based catalysts with a desirable oxygen reduction reaction (ORR) performance by a simple and economical synthesis route is a great challenge for the commercialization of future fuel cell and metal–air batteries. Herein, light-weight 3D Co–N-doped hollow carbon spheres (Co–NHCs) have been fabricated via a facile emulsion approach followed by carbonization. The prepared 0.1-Co–NHCs catalyst with suitable Co doping content exhibits favorable ORR catalytic activity (onset potential of 0.99 V and half-wave potential of 0.81 V vs. RHE), comparable to that of commercial Pt–C (onset potential of 1.02 V and half-wave potential of 0.83 V vs. RHE) and rivals that of Pt–C with better cycling stability. The excellent performance of the catalyst is attributed to the synergetic effect of Co and N doping with a high total ratio of active sites, high surface area and good conductivity of the material. More impressively, the assembled rechargeable zinc–air batteries based on the 0.1-Co–NHCs catalyst outperform those afforded by commercial Pt–C. The progress presented in this reported work is of great importance in the development of outstanding non-noble metal based electrocatalysts for the fuel cell and metal–air battery industry.
1 Introduction
The increasing energy crisis has stimulated intense research for the development of sustainable and economical energy conversion and storage systems such as fuel cells, metal–air batteries and supercapacitors.1–6 Among these systems, metal (M)–air batteries (M = Li, Zn, Mg, Al, etc.) hold great potential and economic value since they possess high theoretical energy densities and strong long-term stability.7–10 Moreover, the low cost and environmental benignity of metal anodes make metal–air batteries suitable for practical domestic and industrial applications.11 However, commonly utilized noble metal based cathode electrocatalysts with high cost and poor durability largely suppress the economic benefit of metal–air batteries.12,13To date, a wide range of nonprecious metal based catalysts have been explored, including transition-metal-coordinating macrocyclic compounds,14–16 metal–nitrogen-doped carbons (M–N/C),17–20 transition-metal oxide-, carbide-, chalcogenide-, or phosphide-carbon hybrids,21–26 and metal-free heteroatom doped carbon materials.27–30 Amongst them, M–N-doped hollow carbon spheres (M–NHCs) have attracted significant interest due to the synergetic M and N co-doping effect, high surface area, large internal void and homogeneous distribution of active sites.31–33 Synthesis of such hollow structured catalysts frequently involves the use of hard templates. However, removal of the hard template is time consuming and harmful to the environment due to the use of strong acids.34–36 For example, Zhu et al. fabricated hollow carbon nanospheres as ORR electrocatalysts using polystyrene as a hard template.37 Unfortunately, this method was quite complicated involving synthesizing the polystyrene template first and removing it finally, which caused the Co atoms to distribute poorly and damage the hollow carbon structures, resulting in lower than the expected performance. Therefore, efforts to design a facile route to prepare M–NHCs are highly desirable.
Herein, we report a facile two step method to synthesize light-weight 3D Co–N-doped hollow carbon spheres as electrocatalysts for the ORR. First, we use the emulsion method to synthesize polyaniline-co-polypyrrole (PACP) hollow spheres. This is followed by calcination of the above hollow copolymer after soaking with different concentrations of a cobalt acetate solution. We found that the 0.1-Co–NHCs electrocatalyst exhibited remarkably enhanced ORR activity in terms of the onset potential, half-wave potential and limiting current density compared with others. The improved ORR performance of the 0.1-Co–NHCs electrocatalyst is ascribed to the synergistic effect of the N and Co co-doping, a high total ratio of active sites, high surface area and good conductivity of the material. Finally, 0.1-Co–NHCs are applied as the cathode catalyst in a rechargeable zinc–air battery and its performance outperforms those afforded by commercial Pt–C catalysts. This indicates that the material prepared by us has the potential to replace current Pt based electrocatalysts and promote the commercial application of metal–air batteries.
2 Experimental
2.1 Preparation of materials
Aniline, pyrrole, Triton X-100, ammonium persulphate (APS) and cobalt acetate tetrahydrate were purchased from Sigma-Aldrich. All chemicals were analytical grade and used directly without further purification.Polyaniline-co-polypyrrole (PACP) hollow spheres were synthesized by using APS as an oxidizing agent to polymerize a mixture of aniline and pyrrole in the presence of TX-100.38 Typically, 0.38 ml of aniline and 0.29 ml of pyrrole were added into a 100 ml beaker with 60 ml of deionized water containing 0.06 g of TX-100. The resulting solution was stirred in an ice bath for 30 min. Then, the precooled APS solution (1.9 g of APS dissolved in 15 ml of deionized water) was added to the above solution and was maintained at 0 °C for 12 h without stirring. Finally, the product was washed with deionized water via vacuum filtering several times and dried at 60 °C for 24 h. 0.1-Co–NHCs were obtained by calcination of PACP (soaked with 0.1 g ml−1 of cobalt acetate tetrahydrate solution for 24 h) at 900 °C for 2 h. The method to prepare other Co-NHCs was the same as that of 0.1-Co-NHCs only with different concentrations of cobalt acetate tetrahydrate and calcination temperatures. It is expected that different concentrations of cobalt acetate tetrahydrate will lead to different Co doping contents.
2.2 Physical characterization of the materials
The methodology and structure of the obtained materials were characterized by scanning electron microscopy (SEM) (Philips XL30 FEG) and transmission electron microscopy (TEM, JEOL TEM 2100F FEG operated with an accelerating voltage of 200 kV) whereas electron energy loss spectroscopy (EELS) was used to characterize the composite elements. The crystal structure of the prepared materials was analyzed using X-ray diffraction (Bruker, D2 Phaser) with Cu Kα (λ = 1.5418 Å) radiation. Additional information on the carbon structure was obtained from Raman spectra (Renishaw inVia™ confocal Raman microscope) using a 514 nm wavelength excitation laser. The chemical compositions of the obtained materials were analyzed by X-ray photoelectron spectroscopy (XPS) (VG ESCALAB 220i-XL). The N2 adsorption–desorption isotherms were obtained by using a Micrometric ASAP 2020 instrument. The specific surface area was obtained via the Brunauer–Emmett–Teller (BET) method using adsorption data and the pore size distribution was derived from the adsorption data by using the Barrett–Joyner–Halenda (BJH) method.2.3 Electrochemical measurements
For ORR measurements, a CHI 760D electrochemical workstation was employed to conduct all the electrochemical measurements including cyclic voltammetry (CV), linear sweep voltammetry (LSV), and rotating ring-disk electrode (RRDE). A typical 3-electrode system was used with a glassy carbon electrode (GCE) of 3 mm in diameter loaded with different catalysts as the working electrode whereas Ag/AgCl (3 M KCl) and Pt sheet were used as the reference electrode and counter electrode, respectively. All measurements were performed using 0.1 M KOH as the electrolyte and the recorded potentials were converted to reversible hydrogen electrode (RHE). The loading masses of all the obtained materials were 217 μg cm−2 for CV and LSV measurements while were 304 μg cm−2 for RRDE measurement. For comparison, the loading masses used for commercial Pt–C were 140 μg cm−2 for all measurements.The CV measurements were carried out at a scan rate of 100 mV s−1 in a saturated N2 or O2 electrolyte. The LSV measurements were conducted at a scan rate of 5 mV s−1 at various rotating speeds (400, 900, 1600, 2500, and 3600 rpm) in an O2 saturated electrolyte.
The transferred electron numbers (n) per O2 molecule involved in the oxygen reduction at each electrode were determined from the Koutecky–Levich (K–L) equation given in eqn (1):39,40
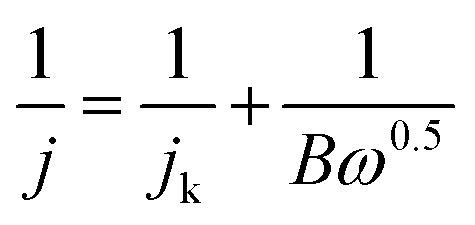 | (1) |
| B = 0.2nF(DO2)2/3ν−1/6CO2 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), DO2 represents the diffusion coefficient of O2 in 0.1 M KOH (DO2 = 1.9 × 10−5 cm2 s−1), ν represents the kinetic viscosity (0.01 cm2 s−1), and CO2 represents the bulk concentration of O2 (1.2 × 10−6 mol cm−3). The constant 0.2 is adopted when the rotating speed is expressed in rpm.
485 C mol−1), DO2 represents the diffusion coefficient of O2 in 0.1 M KOH (DO2 = 1.9 × 10−5 cm2 s−1), ν represents the kinetic viscosity (0.01 cm2 s−1), and CO2 represents the bulk concentration of O2 (1.2 × 10−6 mol cm−3). The constant 0.2 is adopted when the rotating speed is expressed in rpm.
The RRDE measurements were carried out with a Pt ring surrounded by a 4 mm diameter GCE. The transferred electron numbers (n) were determined using eqn (3):
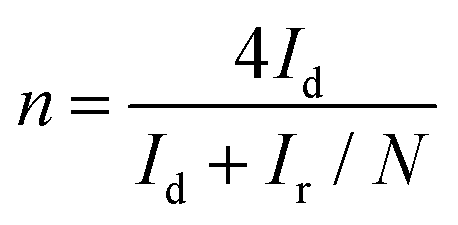 | (3) |
The HO2− yield was determined using eqn (4):
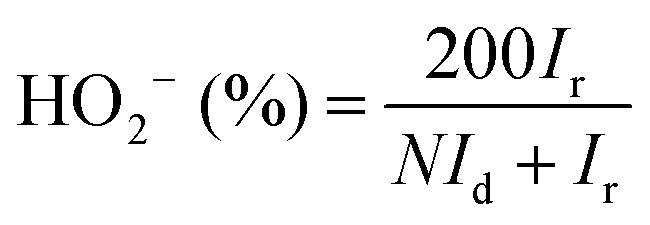 | (4) |
2.4 Zn–air battery fabrication
The air electrode was fabricated by spraying a 0.1-Co–NHCs slurry on carbon cloth and drying at room temperature for 24 hours. The 0.1-Co–NHCs slurry was prepared by dispersing 8 mg 0.1-Co–NHCs catalyst into a 1 ml mixture solution of isopropanol, distilled water and Nafion solution (5 wt%) (10:40:3). The air electrode as the cathode, the Zn plate electrode as the anode and 6.0 M KOH with a 0.2 M Zn(Ac)2 additive as the electrolyte were used for rechargeable Zn–air batteries.
The charge–discharge polarization was determined utilizing an electrochemical workstation (CHI 760e, Chenhua). The galvanostatic test was performed using a Land 2001A battery test system at room temperature.
The power density (P) of a zinc–air battery was calculated using eqn (5):
| P = I·V | (5) |
3 Results and discussion
3.1 Materials preparation
A light-weight 3D Co–N-doped hollow carbon sphere electrocatalyst was synthesized via a facile two step method and the schematic illustration of the preparation is shown in Scheme 1. First, we used the emulsion method to synthesize polyaniline-co-polypyrrole (PACP) hollow spheres. The PACP hollow spheres are formed due to different hydrophobic properties of aniline and pyrrole in the presence of hydrophobic Triton X-100 micelles. At first, the hydrophobic pyrrole was located in the inner side of micelles whereas the hydrophilic aniline was located in the outer side of the micelles. After addition of the hydrophilic oxidant APS, the copolymerization of aniline and pyrrole took place at the micelle–water interface and the hollow structure was formed as the pyrrole quickly diffused from the inside to the surface of the micelles.41 Then, the Co–N-doped hollow carbon spheres were obtained by calcination of the above hollow copolymer after soaking with a cobalt acetate solution at 900 °C for 2 h. For comparison, we have synthesized different Co–N-doped hollow carbon spheres with different concentrations of cobalt acetate (0.00 g ml−1, 0.05 g ml−1, 0.10 g ml−1, and 0.15 g ml−1) for the soaking process, which are denoted as NHCs, 0.05-Co–NHCs, 0.1-Co–NHCs and 0.15-Co–NHCs, respectively. It is proved that different concentrations of cobalt acetate tetrahydrate lead to different Co doping contents as confirmed by the XPS result as expected.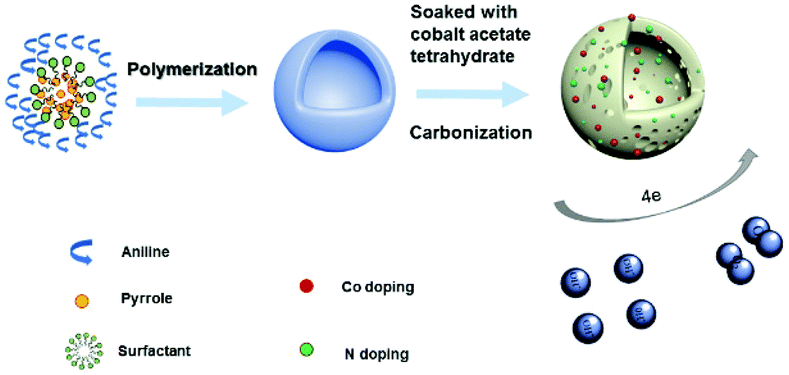 | ||
| Scheme 1 Schematic illustration of the preparation of Co–NHCs. | ||
3.2 Materials characterization
The morphology and structure from SEM and TEM analysis of the prepared materials are presented in Fig. 1, for NHCs, Fig. 1(a and b), and 0.1-Co–NHCs, Fig. 1(c and d), that show their well-defined hollow nanosphere structure. Clearly, the total and inner diameters of the sphere and the hollow structure are maintained after Co doping and similar to those of their counterparts without Co doping. Besides, the SEM images of 0.05-Co–NHCs (Fig. S1(a)†), 0.1-Co–NHCs (Fig. 1(c)) and 0.15-Co–NHCs (Fig. S1(b)†) indicate that the diameters of the sphere are independent of Co doping. It is noteworthy that this soft template method is quite facile to synthesize the uniform hollow nanosphere structure as it does not require a tedious template removal process. Fig. 1(e) shows the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and the corresponding energy dispersive spectrometry (EDS) maps of 0.1-Co–NHCs, which confirms the presence of C, N and Co. Interestingly, the C, N and Co elements are distributed relatively homogeneously in the 0.1-Co–NHCs catalyst. It is speculated that the homogeneously dispersed N and Co moieties are the active sites for improving ORR performance.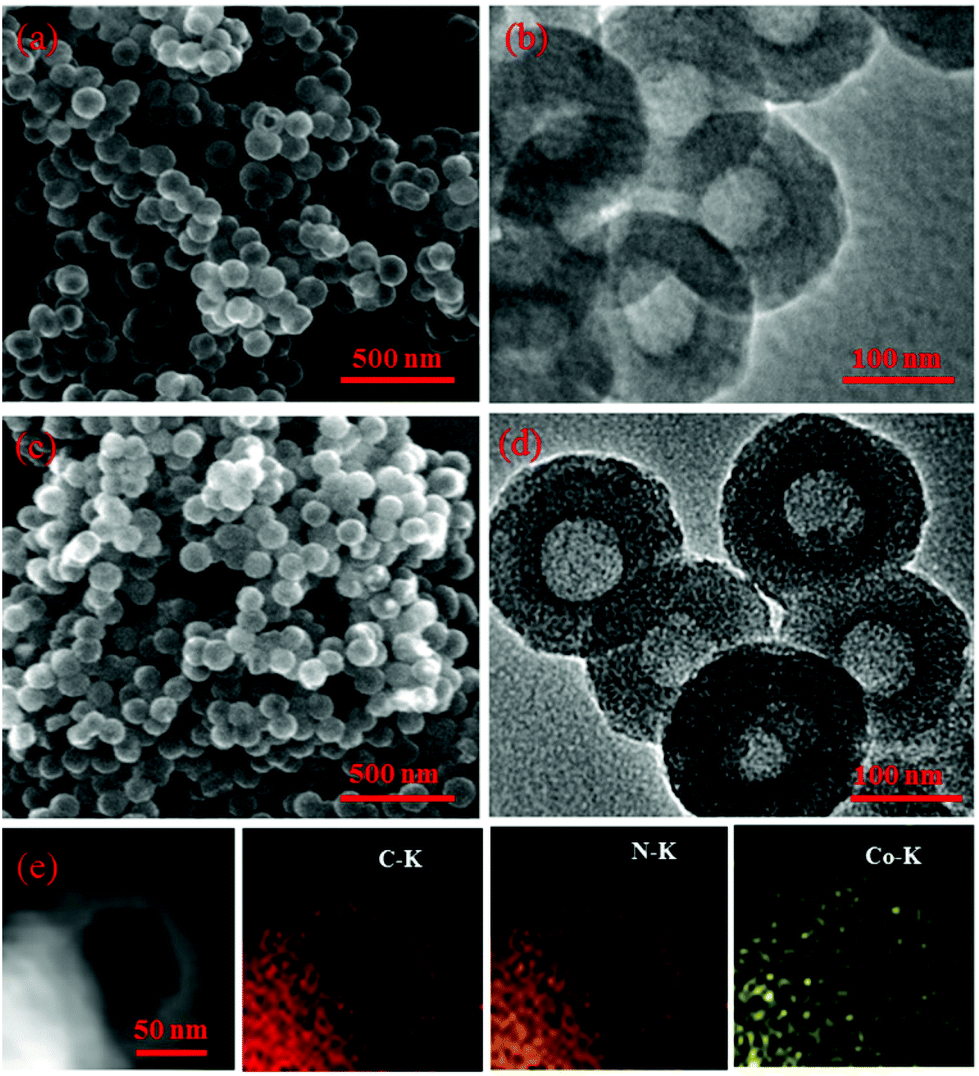 | ||
| Fig. 1 SEM and TEM images of NHCs (a, b) and 0.1-Co–NHCs (c, d). HAADF-STEM image and the corresponding EDS mapping of C, N and Co of 0.1-Co–NHCs are shown in (e). | ||
Additional structural characterization was conducted by XRD and Raman spectroscopy. Fig. 2(a) depicts the XRD patterns of the NHCs and the Co–NHCs doped with different concentrations of Co. The major diffraction peak at around 25 °C in all the samples is assigned to the (002) diffraction planes of graphitic carbon, which suggests that the co-polymers are fully carbonized. Fig. 2(b) shows the D band (1340 cm−1) and G band (1595 cm−1) of the carbon skeleton, corresponding to the disordered carbon and the crystallized graphitic sp2 carbon, respectively.42 The intensity ratio of the D band to G band decreases from 1.00 to 0.96 upon increasing the concentration of cobalt acetate from 0.00 to 0.15 g ml−1, suggesting an enhanced graphitization degree by introducing Co doping, which is good for material conductivity. Fig. 2(c) and (d) show the N2 adsorption and desorption isotherms and pore diameter distributions of the resulting samples NHCs, 0.05-Co–NHCs, 0.1-Co–NHCs and 0.15-Co–NHCs. Fig. 2(c) shows that all the samples display a type IV isotherm with hysteresis, which demonstrates the existence of porous structures. The total BET surface areas are 819.5, 782.2, 744.9 and 677.2 m2 g−1 for NHCs, 0.05-Co–NHCs, 0.1-Co–NHCs and 0.15-Co–NHCs, respectively. Fig. 2(d) shows the pore diameter distribution of these samples and it indicates that the diameters of the pore decrease with increasing concentration of cobalt acetate. The gradual decreasing surface area and the pore diameter with increasing concentration of cobalt acetate indicate the filling effect of the Co doping. The more the Co doping content, the smaller the surface area and pore diameter.
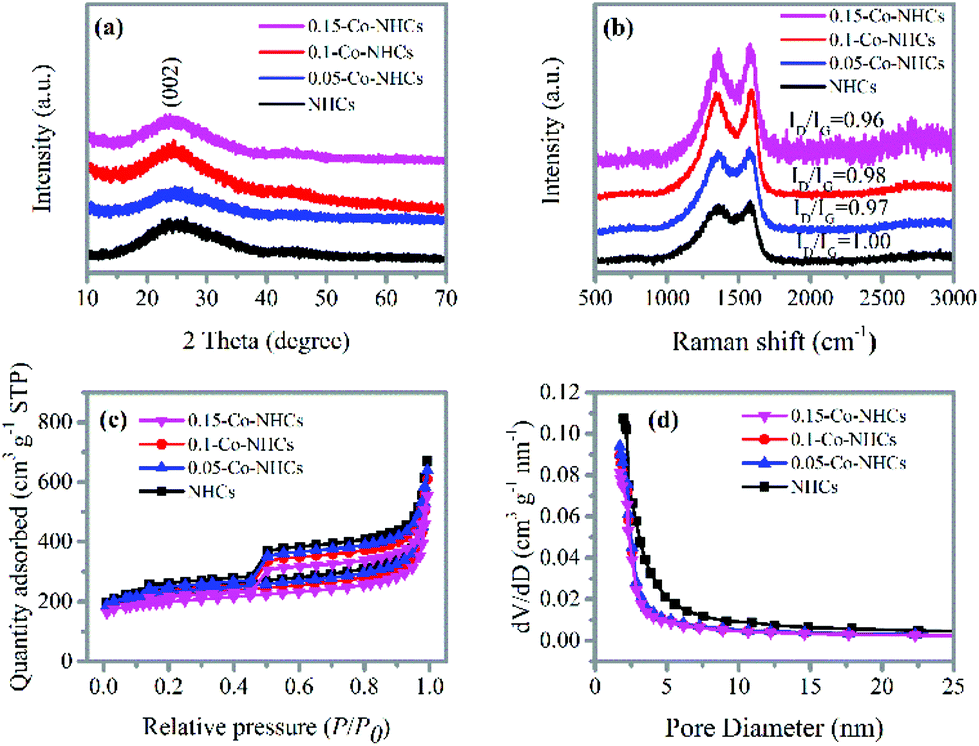 | ||
| Fig. 2 (a) XRD patterns and (b) Raman spectra of different samples; (c) N2 adsorption and desorption isotherms, and (d) pore diameter distributions of different samples. | ||
The survey XPS spectra in Fig. 3(a) show the peaks corresponding to C 1s, N 1s, and O 1s in all the prepared materials. In contrast, the Co 2p peak is absent only in the NHCs sample as expected. The high-resolution N 1s spectrum of 0.1-Co–NHCs shown in Fig. 3(b) can be divided into four peaks with energies of 398.2 eV, 399.3 eV, 401 eV and 403.4 eV, which correspond to pyridinic N (9.7%), Co–N (32.7%), quaternary N (39.9%) and oxidized pyridinic N (17.8%), respectively.43 The Co–N peak indicates that some N atoms bond to Co atoms to form Co–N moieties. The Co–N moieties are considered to be the active sites for the ORR.44 The high-resolution N 1s spectra of the 0.05-Co–NHCs and 0.15-Co–NHCs samples (see Fig. S2(a and c)†) are similar to that of 0.1-Co–NHCs only with different moiety ratios whereas no trace of the Co–N peak can be found in the high-resolution N 1s spectrum of the NHCs sample as shown in Fig. 3(c). The peaks at about 780.9 eV and 786.7 eV in the high-resolution Co 2p of 0.1-Co–NHCs correspond to Co 2p3/2 and Co 2p1/2. It indicates that the existence of Co(III) and Co(II) species mainly originate from CoOx or CoCxNy (780.5 eV) and CoNx (782.5 eV) (Fig. 3(d)). It is further proved that the presence of the Co–N species has been considered as the active sites for the ORR.44 The appearance of CoOx is assigned to the superficial cobalt oxide in the surface oxidation layer of the Co–NHCs, because Co is susceptible to air.45 The high-resolution Co 2p spectra of 0.05-Co–NHCs and 0.15-Co–NHCs are similar to that of 0.1-Co–NHCs as shown in Fig. S2(b and d).† The calculated composition analysis and N 1s peak deconvolution are given in Table S1.† It is noteworthy that the Co doping content increases with the increasing soaking concentration of the cobalt acetate solution as expected. Besides, the total ratio of the ORR active sites (Co–N moieties and pyridinic N) increases first and finally decreases with further increase in the Co doping content, which is ascribed to the pyridinic-N groups doped at the edges of the carbon materials having the potential to form Co–N groups embedded between the graphitic pores.46 It is proved that the Co–N moieties and pyridinic N were the active sites for the ORR.47,48
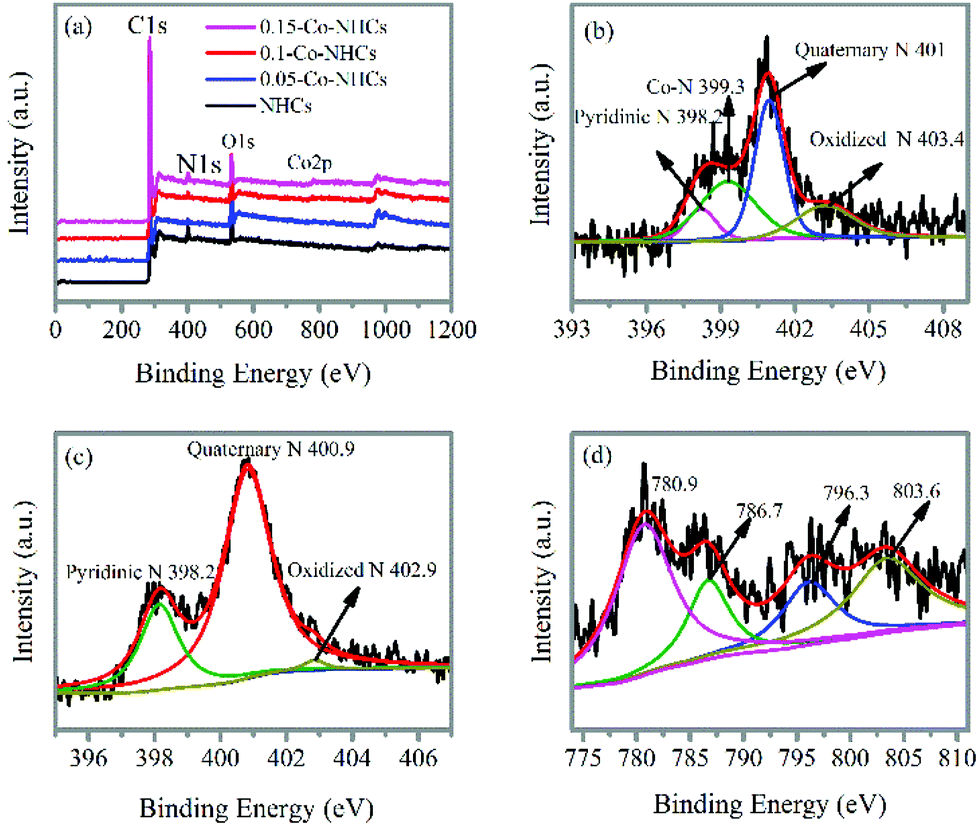 | ||
| Fig. 3 (a) XPS survey spectra of NHCs, 0.05-Co–NHCs, 0.1-Co–NHCs and 0.15-Co–NHCs. High-resolution XPS N 1s of 0.1-Co–NHCs (b) and NHCs (c). High-resolution XPS Co 2p of 0.1-Co–NHCs (d). | ||
3.3 Comparison of materials’ ORR performance
To evaluate the ORR performance of the prepared materials, we have carried out CV and LSV measurements. The CV curves were first obtained in the 0.1 M KOH solution as shown in Fig. 4(a). All the catalysts display CV curves with obvious reduction peaks in the O2 saturated electrolyte, suggesting the efficient reduction of O2. We can observe that the reduction peak potentials of the catalysts first positively increase with the increasing Co doping content and finally decrease with a further increase in Co doping content. The 0.1-Co–NHCs sample possesses the best ORR performance with its most positive reduction peak potential and the highest reduction peak current density. This phenomenon is also evidenced in the LSV curves recorded at a rotation rate of 1600 rpm as shown in Fig. 4(b). The onset and half-wave potential of catalysts first positively increase with the increasing Co doping content and finally decrease with further increasing Co doping content. Furthermore, the 0.1-Co–NHCs sample shows the best catalytic activity with the most positive onset potential and half-wave potential compared to NHCs, 0.05-Co–NHCs and 0.15-Co–NHCs. Both CV and LSV measurements prove that the Co doping content affects the ORR performance. A suitable Co doping content results in good ORR performance. This is because increasing the Co content will increase the material graphitization degree as indicated by the Raman results, which will increase the material conductivity. Furthermore, increasing the Co content will also increase the synergetic effects of N and Co co-doping. However, increasing the Co content will decrease the material surface area as indicated by the BET results. Although increasing Co content will increase the synergetic effects of N and Co co-doping, the total ratio of the ORR active sites (Co–N moieties and pyridinic N) increases first and finally decreases with further increasing Co doping content as proved by the XPS results. Also, the semicircular diameters of EIS measurement of the prepared materials decrease first with increasing Co content and increase with further increasing Co doping content (Fig. S3†), indicating that the interface charge transfer resistances of the materials decrease first and then increase with further increasing Co content. Furthermore, the electrochemical active surface areas of the prepared materials are increased first and decrease finally with increasing Co content as indicated by the double layer capacitance Cdl: 7.01 mF cm−2 for NHCs, 12.00 mF cm−2 for 0.05-Co–NHCs, 22.28 mF cm−2 for 0.1-Co–NHCs and 14.22 mF cm−2 for 0.15-Co–NHCs (Fig. S4†). Therefore, the Co doping content affects the ORR performance by changing the conductivity, surface area and the total ratio of active sites of the materials. To make sure of high surface area and material conductivity we need to control the doping content of the Co. The 0.1-Co–NHCs sample exhibits the best catalytic performance among the synthesized catalysts because it possesses suitable Co doping content which results in appreciable material conductivity, surface area and the total ratio of ORR active sites. To further study the ORR performance of the prepared materials, RRDE measurements were also carried out at different rotation speeds, as shown in Fig. 4(c) and Fig. S5.† The corresponding K–L plots obtained from the RRDE measurements are shown in Fig. 4(d) and Fig. S5.†Fig. 4(d) shows the transferred electron number of the 0.1-Co–NHCs sample is about 3.75 close to 4, suggesting a four-electron transfer pathway. To determine the good carbonization temperature for preparing our materials, we also investigated the ORR performance of materials obtained at 700, 800 and 900 and 1000 °C. The detailed ORR performance is presented in Fig. S6† and it indicates that the sample obtained at 900 °C exhibits better catalytic performance compared to those prepared at 700 °C, 800 °C and 1000 °C. Therefore, all our investigated samples are obtained at 900 °C.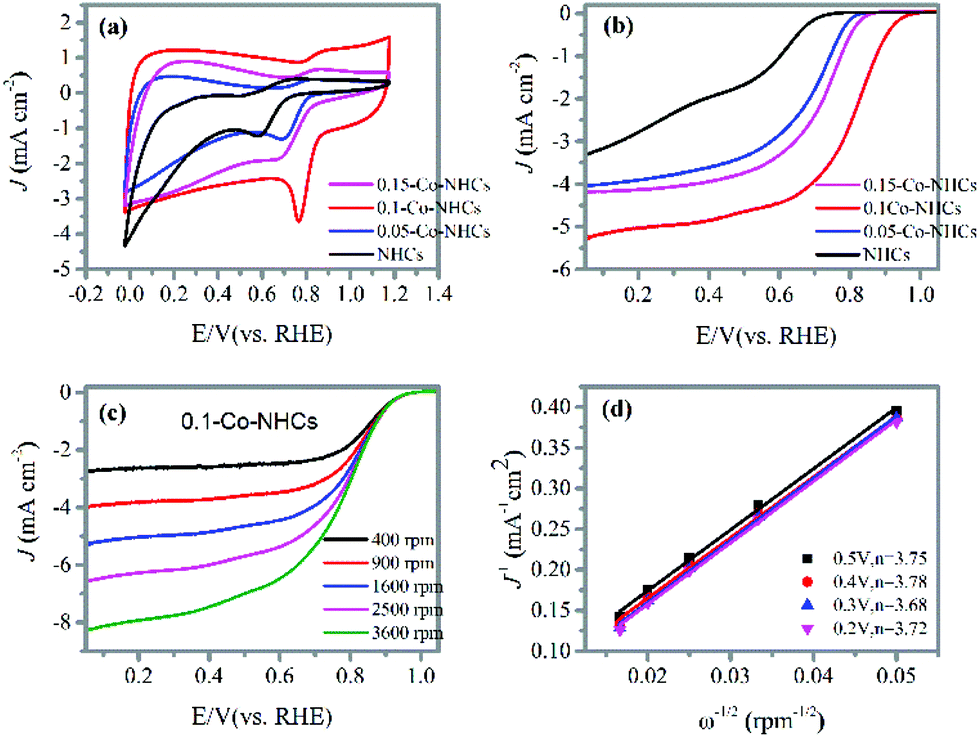 | ||
| Fig. 4 (a) CV curves of the NHCs, 0.05-Co–NHCs, 0.1-Co–NHCs and 0.15-Co–NHCs catalysts recorded at 100 mV s−1 in O2 saturated 0.1 M KOH solution; (b) LSV curves of the NHCs, 0.05-Co–NHCs, 0.1-Co–NHCs and 0.15-Co–NHCs catalysts at a speed of 1600 rpm. (c) LSV curves of the 0.1-Co–NHCs catalyst at different rotation speeds; (d) K–L plots of the 0.1-Co–NHCs catalyst at different potentials with the corresponding transferred electron number. | ||
3.4 ORR and OER performances of the target material
Remarkably, the 0.1-Co–NHCs sample exhibits an ORR performance comparable to that of Pt–C. The CV curves presented in Fig. 5(a) show that the reduction peak potential of the 0.1-Co–NHCs sample is 0.79 V vs. RHE comparable to that of Pt–C (0.81 V vs. RHE). Furthermore, the LSV curves indicate the onset and half-wave potential of 0.99 V and 0.81 V vs. RHE, respectively, for the 0.1-Co–NHCs catalyst, which is also comparable to the corresponding values of the Pt–C catalyst (onset potential of 1.02 V and half-wave potential of 0.83 V vs. RHE), as shown in Fig. 5(b). Additional information on Pt–C is presented in Fig. S7† including the CV curves, LSV curves and K–L plot. The performance of 0.1-Co–NHCs is comparable to that of most of the other reported catalysts (Table S2†). The RRDE tests (Fig. 5(c) and its inset) uncover that the transferred electron number (n) and yield of HO2− of 0.1-Co–NHCs are 3.8–3.9 per oxygen molecule and 12.5%–5.9%, respectively, while those of Pt–C are 3.7–4.0 and 13.7%–5%, respectively, in a wide potential range (0.1 V–0.8 V). This is consistent with the results from the K–L plots. It suggests that the ORR catalyzed by 0.1-Co–NHCs is mainly a 4 e− process, which is similar to that of the commercial Pt–C catalyst. The long-term stability of 0.1-Co–NHCs and Pt–C catalysts is measured by chronoamperometric tests, as shown in Fig. 5(d). In a continuous 24 h test, the 0.1-Co–NHCs sample retains 76% of its initial current and surpasses that of commercial Pt–C (65% of initial current retention), further reinforcing the excellent performance of 0.1-Co–NHCs. Interestingly, the 0.1-Co–NHCs sample also exhibits considerable OER performance with a low overpotential of 420 mV to drive a catalytic current density of 10 mA cm−2, which is only a little bit higher than that of the benchmark IrO2 (360 mV of overpotential), as indicated in Fig. 5(e). The corresponding Tafel slope is 96 mV dec−1 for the 0.1-Co–NHCs sample whereas it is 67 mV dec−1 for IrO2, as shown in Fig. 5(f). It indicates that the 0.1-Co–NHCs sample shows considerable performance for the OER.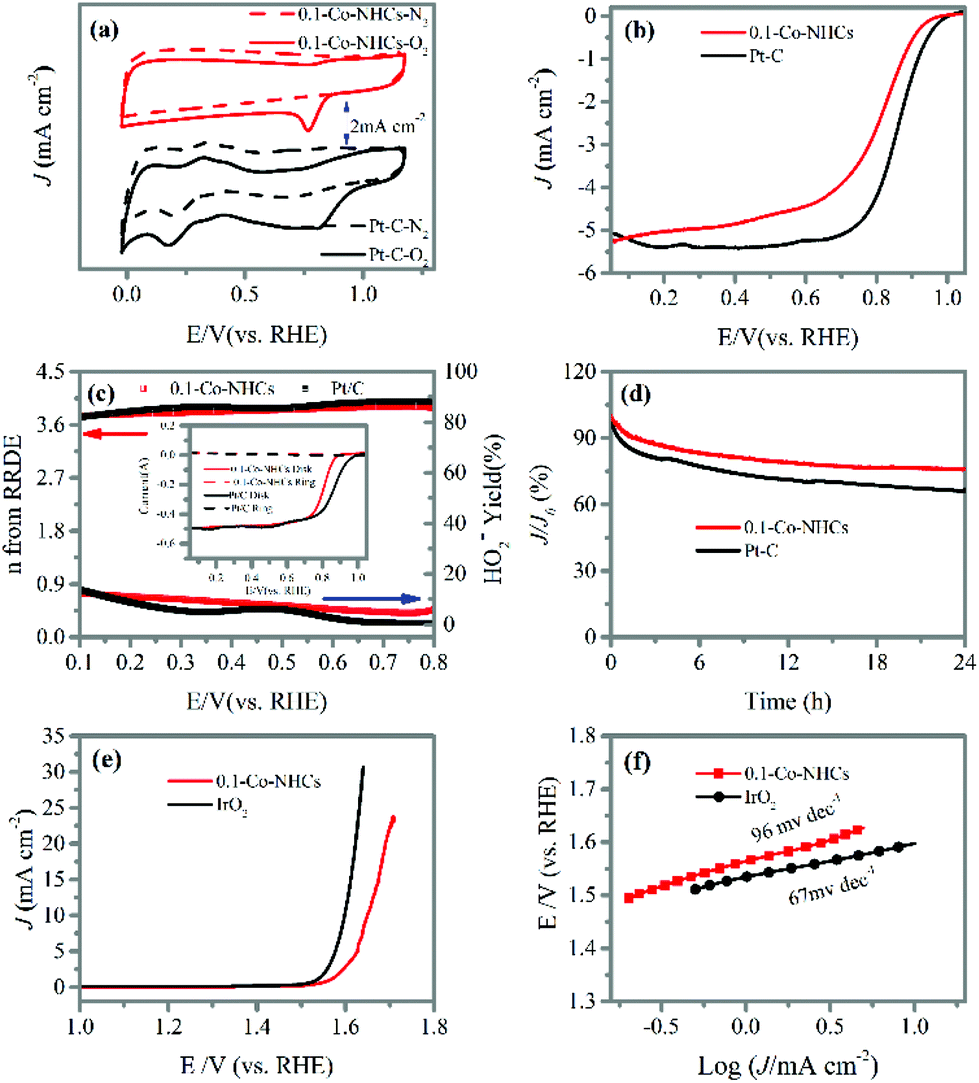 | ||
| Fig. 5 (a) CV curves of the 0.1-Co–NHCs and Pt–C catalysts recorded at 100 mV s−1 in N2 and O2 saturated 0.1 M KOH solution; (b) LSV curves of the 0.1-Co–NHCs and Pt–C catalysts at a speed of 1600 rpm; (c) transferred electron number (n) and HO2− yield derived from its inset RRDE tests of 0.1-Co–NHCs and Pt–C catalysts at 1600 rpm; (d) chronoamperometric stability tests of the 0.1-Co–NHCs and Pt–C catalysts at 1600 rpm at 0.7 V (vs. RHE) in O2 saturated 0.1 M KOH solution; (e) OER polarization curves of 0.1-Co–NHCs and IrO2 for the OER at 1600 rpm in O2 saturated 0.1 M KOH solution after IR-correction; and (f) the corresponding Tafel plots. | ||
3.5 Rechargeable zinc–air battery performance of the target material
The high ORR and considerable OER performance of the 0.1-Co–NHCs catalyst suggest it can be used in a rechargeable zinc–air battery. Accordingly, we prepared zinc–air batteries with the 0.1-Co–NHCs catalyst and commercial Pt–C catalysts. Fig. 6(a) shows three main components of the batteries: air electrode comprising an oxygen catalyst as the cathode, aqueous 6 M KOH solution as the electrolyte, and a zinc plate as the anode. The resulting discharge polarization and power density are shown in Fig. 6(b). It is observed that the 0.1-Co–NHCs catalyst battery has a higher power density than that assembled with the commercial Pt–C with the values of 239.8 mW cm−2 and 156 mW cm−2, respectively. The corresponding galvanodynamic charge–discharge properties of the assembled zinc–air batteries are presented in Fig. 6(c), revealing that the 0.1-Co–NHCs catalyst battery conducts the charge–discharge process with a smaller voltage gap than that of commercial Pt–C, indicating higher efficiency of the charge–discharge process. The stability of the battery was evaluated by using the charge–discharge cycles at a current density of 10 mA cm−2 with 11 min per cycle, as shown in Fig. 6(d). The voltage gap of the 0.1-Co–NHCs catalyst battery is ∼0.84 V. The voltage is stable for 50 hours, ∼300 cycles, which is comparable with the performance of a commercial Pt–C + IrO2 battery, indicating a stable charge–discharge process for the 0.1-Co–NHCs catalyst battery. The performance of a zinc–air battery assembled with the 0.1-Co–NHCs catalyst is comparable to that of most of the zinc–air batteries assembled with the other reported catalysts (Table S3†). In Fig. 6(e), we can see that the assembled battery has an open-circuit of 1.556 V and it can power different colored LEDs using two of such zinc–air batteries in series (the larger view of Fig. 6e also given in ESI Fig. S8†).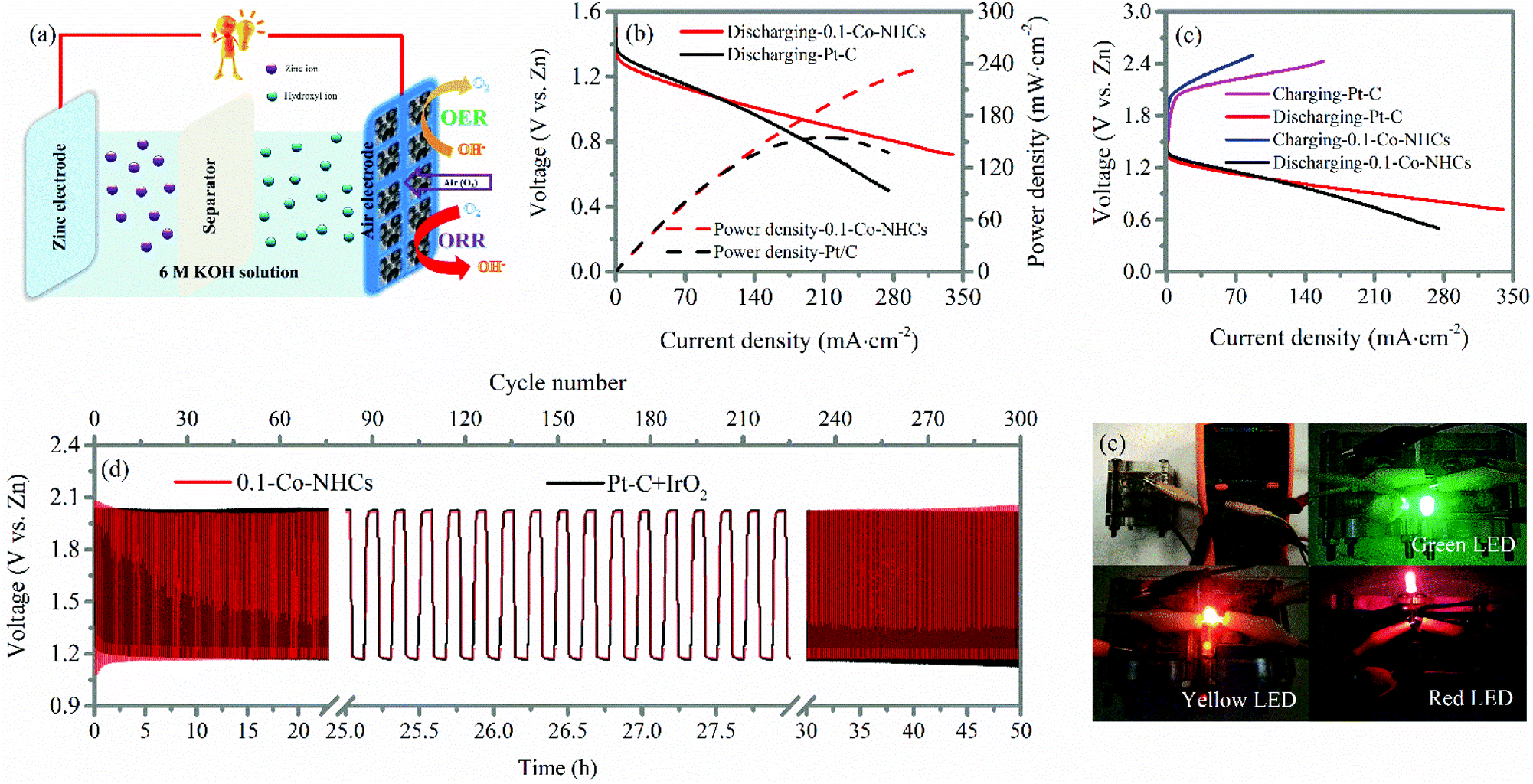 | ||
| Fig. 6 (a) Schematic diagram of the structure of the assembled zinc–air battery; (b) battery discharge polarization and the corresponding power density curves of the zinc–air battery with 0.1-Co–NHCs and Pt–C versus the Zn electrode; (c) galvanodynamic charge–discharge profile of the 0.1-Co–NHCs and Pt–C catalysts; (d) performance cycling tests of the Zn–air battery at a current density of 10 mA cm−2; and (e) digital image of the assembled zinc–air battery using 0.1-Co–NHCs as the catalyst to display open-circuit voltage and different colored LEDs powered by two of the assembled zinc–air batteries in series. | ||
4 Conclusion
We demonstrated a facile and effective method to synthesize Co and N co-doped hollow carbon spheres. We also found that the Co doping content of the prepared materials affects the ORR performance by changing the conductivity, surface area and the total ratio of active sites of the materials. It indicates that the 0.1-Co–NHCs sample with suitable Co doping content shows the best ORR performance. The excellent performance of the 0.1-Co–NHCs catalyst is ascribed to the high graphitization degree, high BET surface area with high internal void and synergetic effects of Co and N co-doping with the highest total ratios of pyridinic N and Co–N active sites. Moreover, the assembled rechargeable zinc–air batteries using 0.1-Co–NHCs as the cathode catalyst exhibit a high power density of 239.8 mW cm−2, small charge–discharge voltage gap (0.84 V@10 mA cm−2) and excellent stability, which outperforms those of the commercial Pt–C catalyst. The progress represented by this work is of great importance in the development of outstanding non-noble metal based electrocatalysts for the fuel cell and metal–air battery industry.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This work was supported by the Basic Research Project of Knowledge Innovation Program of Shenzhen City (JCYJ20160229165250876).Notes and references
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- S. H. Noh, C. Kwon, J. Hwang, T. Ohsaka, B.-J. Kim, T.-Y. Kim, Y.-G. Yoon, Z. Chen, M. H. Seo and B. Han, Nanoscale, 2017, 9, 7373–7379 RSC.
- S. Repp, E. Harputlu, S. Gurgen, M. Castellano, N. Kremer, N. Pompe, J. Worner, A. Hoffmann, R. Thomann, F. M. Emen, S. Weber, K. Ocakoglu and E. Erdem, Nanoscale, 2018, 10, 1877–1884 RSC.
- R. Genc, M. O. Alas, E. Harputlu, S. Repp, N. Kremer, M. Castellano, S. G. Colak, K. Ocakoglu and E. Erdem, Sci. Rep., 2017, 7, 11222 CrossRef PubMed.
- Z.-Q. Liu, H. Cheng, N. Li, T. Y. Ma and Y.-Z. Su, Adv. Mater., 2016, 28, 3777–3784 CrossRef CAS PubMed.
- B. Liu, P. Yan, W. Xu, J. Zheng, Y. He, L. Luo, M. E. Bowden, C.-M. Wang and J.-G. Zhang, Nano Lett., 2016, 16, 4932–4939 CrossRef CAS PubMed.
- Y. Xu, Y. Zhao, J. Ren, Y. Zhang and H. Peng, Angew. Chem., Int. Ed., 2016, 55, 7979–7982 CrossRef CAS PubMed.
- Y. Li, X. Zhang, H.-B. Li, H. D. Yoo, X. Chi, Q. An, J. Liu, M. Yu, W. Wang and Y. Yao, Nano Energy, 2016, 27, 8–16 CrossRef CAS.
- J. Fu, D. U. Lee, F. M. Hassan, L. Yang, Z. Bai, M. G. Park and Z. Chen, Adv. Mater., 2015, 27, 5617–5622 CrossRef CAS PubMed.
- J. Park, M. Park, G. Nam, J.-s. Lee and J. Cho, Adv. Mater., 2015, 27, 1396–1401 CrossRef CAS PubMed.
- X. Liu, M. Park, M. G. Kim, S. Gupta, G. Wu and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 9654–9658 CrossRef CAS PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. Gyu Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 Search PubMed.
- P.-J. Wei, G.-Q. Yu, Y. Naruta and J.-G. Liu, Angew. Chem., Int. Ed., 2014, 53, 6659–6663 CrossRef CAS PubMed.
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- C. Cao, L. Wei, G. Wang and J. Shen, Nanoscale, 2017, 9, 3537–3546 RSC.
- M. Wang, Y. Yang, X. Liu, Z. Pu, Z. Kou, P. Zhu and S. Mu, Nanoscale, 2017, 9, 7641–7649 RSC.
- H. Wang, Y. Liang, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2011, 50, 10969–10972 CrossRef CAS PubMed.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., 2012, 124, 11940–11943 CrossRef.
- Z. Pu, Q. Liu, C. Tang, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, J. Power Sources, 2014, 257, 170–173 CrossRef CAS.
- R. Ning, C. Ge, Q. Liu, J. Tian, A. M. Asiri, K. A. Alamry, C. M. Li and X. Sun, Carbon, 2014, 78, 60–69 CrossRef CAS.
- R. Ning, J. Tian, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, Langmuir, 2013, 29, 13146–13151 CrossRef CAS PubMed.
- L. Ma, H. Fan, J. Wang, Y. Zhao, H. Tian and G. Dong, Appl. Catal., B, 2016, 190, 93–102 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- L. Ma, H. Fan, K. Fu, S. Lei, Q. Hu, H. Huang and G. He, ACS Sustainable Chem. Eng., 2017, 5, 7093–7103 CrossRef CAS.
- J. Sanetuntikul, T. Hang and S. Shanmugam, Chem. Commun., 2014, 50, 9473–9476 RSC.
- Y. Wang, A. Kong, X. Chen, Q. Lin and P. Feng, ACS Catal., 2015, 5, 3887–3893 CrossRef CAS.
- G. A. Ferrero, K. Preuss, A. Marinovic, A. B. Jorge, N. Mansor, D. J. L. Brett, A. B. Fuertes, M. Sevilla and M.-M. Titirici, ACS Nano, 2016, 10, 5922–5932 CrossRef CAS PubMed.
- J. Zhu, H. Zhou, C. Zhang, J. Zhang and S. Mu, Nanoscale, 2017, 9, 13257–13263 RSC.
- C. Zhou, S. Geng, X. Xu, T. Wang, L. Zhang, X. Tian, F. Yang, H. Yang and Y. Li, Carbon, 2016, 108, 234–241 CrossRef CAS.
- S. Wang, D. Yu and L. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., 2010, 122, 2619–2623 CrossRef.
- C. Zhou, J. Han, G. Song and R. Guo, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3563–3572 CrossRef CAS.
- Y. Hou, Z. Wen, S. Cui, S. Ci, S. Mao and J. Chen, Adv. Funct. Mater., 2015, 25, 872–882 CrossRef CAS.
- X. X. Liu, J. B. Zang, L. Chen, L. B. Chen, X. Chen, P. Wu, S. Y. Zhou and Y. H. Wang, J. Mater. Chem. A, 2017, 5, 5865–5872 CAS.
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- X. Li, Q. Jiang, S. Dou, L. Deng, J. Huo and S. Wang, J. Mater. Chem. A, 2016, 4, 15836–15840 CAS.
- M. Li, X. Bo, Y. Zhang, C. Han, A. Nsabimana and L. Guo, J. Mater. Chem. A, 2014, 2, 11672–11682 CAS.
- J. Wang, L. Li, X. Chen, Y. Lu, W. Yang and X. Duan, Nano Res., 2017, 10, 2508–2518 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr01140k |
| This journal is © The Royal Society of Chemistry 2018 |