
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene and graphene oxide induce ROS production in human HaCaT skin keratinocytes: the role of xanthine oxidase and NADH dehydrogenase†
Marco
Pelin
 ab,
Laura
Fusco
a,
Cristina
Martín
cd,
Silvio
Sosa
a,
Javier
Frontiñán-Rubio
d,
Jose Miguel
González-Domínguez
d,
Mario
Durán-Prado
e,
Ester
Vázquez
cd,
Maurizio
Prato
*bfg and
Aurelia
Tubaro
*a
ab,
Laura
Fusco
a,
Cristina
Martín
cd,
Silvio
Sosa
a,
Javier
Frontiñán-Rubio
d,
Jose Miguel
González-Domínguez
d,
Mario
Durán-Prado
e,
Ester
Vázquez
cd,
Maurizio
Prato
*bfg and
Aurelia
Tubaro
*a
aDepartment of Life Sciences, University of Trieste, 34127 Trieste, Italy. E-mail: mpelin@units.it; lfusco@units.it; ssosa@units.it; tubaro@units.it
bDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, 34127 Trieste, Italy. E-mail: prato@units.it
cDepartamento de Química Orgánica, Facultad de Ciencias y Tecnologías Químicas, Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain. E-mail: Cristina.MJimenez@uclm.es
dInstituto Regional de Investigación Científica Aplicada (IRICA), Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain. E-mail: Javier.Frontinan@uclm.es; JoseMiguel.Gonzalez@uclm.es; Ester.Vazquez@uclm.es
eCell Biology area, Faculty of Medicine, University of Castilla-La Mancha, 13071, Ciudad Real, Spain. E-mail: Mario.Duran@uclm.es
fCIC BiomaGUNE, Parque Tecnológico de San Sebastián, Paseo Miramón, 182, 20009 San Sebastián, Guipúzcoa, Spain
gBasque Foundation for Science, Ikerbasque, Bilbao 48013, Spain
First published on 11th June 2018
Abstract
The extraordinary physicochemical properties of graphene-based nanomaterials (GBNs) make them promising tools in nanotechnology and biomedicine. Considering the skin contact as one of the most feasible exposure routes to GBNs, the mechanism of toxicity of two GBNs (few-layer-graphene, FLG, and graphene oxide, GO) towards human HaCaT skin keratinocytes was investigated. Both materials induced a significant mitochondrial membrane depolarization: 72 h cell exposure to 100 μg mL−1 FLG or GO increased mitochondrial depolarization by 44% and 56%, respectively, while the positive control valinomycin (0.1 μg mL−1) increased mitochondrial depolarization by 48%. Since the effect was not prevented by cyclosporine-A, it appears to be unrelated to mitochondrial transition pore opening. By contrast, it seems to be mediated by reactive oxygen species (ROS) production: FLG and GO induced time- and concentration-dependent cellular ROS production, significant already at the concentration of 0.4 μg mL−1 after 24 h exposure. Among a panel of specific inhibitors of the major ROS-producing enzymes, diphenyliodonium, rotenone and allopurinol significantly reverted or even abolished FLG- or GO-induced ROS production. Intriguingly, the same inhibitors also significantly reduced FLG- or GO-induced mitochondrial depolarization and cytotoxicity. This study shows that FLG and GO induce a cytotoxic effect due to a sustained mitochondrial depolarization. This seems to be mediated by a significant cellular ROS production, caused by the activation of flavoprotein-based oxidative enzymes, such as NADH dehydrogenase and xanthine oxidase.
Introduction
Graphene is an allotrope of carbon consisting of a single atom thick sheet of sp2-bonded carbons organized in a closely packed honeycomb two-dimensional lattice ranging in the nanosize dimension. This promising material, derived from graphite,1–4 has been attracting increasing interest in recent years, due to its unique electronic, mechanical and physicochemical properties.5–7New kinds of two-dimensional graphene-based nanomaterials (GBNs), including the highly oxidized form of graphene (graphene oxide, GO), have been produced to modify and expand graphene properties. The potential applications of GBNs range among various fields, including nanoelectronics, energy technology and nanobiomedicine. However, safety issues for human health need to be addressed, because the GBNs market and the potential human exposure to these materials are expected to significantly increase over the next years. In particular, the skin represents the body's first line of defense and can be considered one of the major exposure routes to these materials during their production, use and discharge, but little is known about their cutaneous toxicity.
Therefore, the potential dermatotoxicity of these novel materials and the relevant mechanism(s) need to be addressed. In this view, we recently demonstrated the ability of a few layer graphene (FLG) and different GOs to induce a sustained cytotoxicity related to a reduced mitochondrial activity in human skin HaCaT keratinocytes, a preliminary in vitro model to investigate skin toxicity.8 It is widely known that one of the main causes of mitochondrial dysfunction is represented by oxidative stress.9 Intriguingly, it has been suggested that oxidative stress plays a major role in the cytotoxicity of other carbon-based nanomaterials.10 The link between reactive oxygen species (ROS) overproduction and mitochondrial damage is well known. Doubtlessly, mitochondria are both the source and target of ROS, the latter being able to induce an oxidative stress-caused mitochondrial damage. Therefore, it is crucial to shed light on the role of ROS in the mechanism of cytotoxicity induced by GBNs.
Herein, the mechanisms underlying GBN-induced mitochondrial damage in human HaCaT skin keratinocytes have been investigated as one of the feasible intracellular pathways of cytotoxicity consequent to nanomaterials exposure. For this purpose, we selected two different materials, representative of GBNs, a research grade FLG and a commercially available graphene oxide (GO), which were shown to induce a significant oxidative damage at the mitochondrial level consequent to NADH dehydrogenase- and xanthine oxidase-dependent ROS production.
Experimental
Chemicals
Few layer graphene (FLG) was obtained by ball-milling, under solvent-free conditions, as a powder easily dispersible in culture media.11–13 The FLG sample was thoroughly characterized (ESI 1†): thermogravimetric analysis (TGA) was performed with a TGA Q50 (TA Instruments, USA) at 10 °C per minute under a nitrogen atmosphere, from 100 °C to 800 °C; Raman spectra were studied using an InVia Raman microscope (Renishaw plc, UK). At least 28 Raman measurements were collected at 532 nm with a 100× objective and an incident power of 1%. FLG was studied by high-resolution transmission electron microscopy (HRTEM) JEM 2100 (JEOL Ltd, JP), and the lateral dimension distribution of the flakes was analyzed using Fiji software. In addition, quantitative elemental analysis and total reflection X-ray fluorescence (TXRF) of FLG were performed using a LECO CHNS-932 analyzer and a Bruker-S2 PicoFox TXRF spectrometer, respectively. Graphene oxide (GO) was received from Graphenea (San Sebastián, Spain), and was characterized by average lateral dimensions of 979 nm, as assessed by HRTEM and the O/C atomic ratio obtained from XPS analysis equal to 0.51, as reported in our previous study.8If not otherwise reported, all cell culture reagents were from Euroclone (Milan, Italy).
Cell cultures
The human skin HaCaT cell line was purchased from Cell Line Service (DKFZ, Eppelheim, Germany) and maintained in DMEM high glucose with the addition of 10% fetal bovine serum, 200 mM 1% L-glutamine, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 IU mL−1 1% penicillin and 10 mg mL−1 streptomycin. Cell cultures were maintained according to standard procedures in a humidified incubator at 37 °C and with 5% CO2. For cytotoxicity experiments, the cells were seeded at a density of 5 × 103 cells per well in 96-well plates.
000 IU mL−1 1% penicillin and 10 mg mL−1 streptomycin. Cell cultures were maintained according to standard procedures in a humidified incubator at 37 °C and with 5% CO2. For cytotoxicity experiments, the cells were seeded at a density of 5 × 103 cells per well in 96-well plates.
Cellular internalization of GBNs
000g, obtaining the mitochondrial (MF) and cytoplasm/membrane (pellet and supernatant, respectively) fractions. MF were washed by adding 500 μL of fractionation buffer, re-suspended, passed 10 times through a 25 gauge needle and then centrifuged again at 10000g for 10 minutes. The buffer was removed and mitochondrial pellets were re-suspended in the mitochondrial buffer (SFB with 10% glycerol and 0.1% SDS added), sonicated at 4 °C and deposited on a fragment of silicon wafer (Si-Mat CZ) previously cleaned with isopropanol for Raman measurements. As a negative control, to exclude the possible contamination of GBNs between the subcellular fractions, FLG or GO were added to untreated cells immediately before the fractionation process.
JC-1 mitochondrial membrane potential assay
Mitochondrial depolarization in HaCaT cells was evaluated using a JC-1 Mitochondrial Staining Kit (Sigma-Aldrich; Milan, Italy) following the manufacturer's instructions. As a preliminary set of experiments, the potential interference of FLG or GO with the assay was evaluated in an acellular system by incubating each GBN (1–100 μg mL−1) with the JC-1 probe for 20 minutes at 37 °C. Subsequently, after cell exposure to GBNs (1–100 μg mL−1) for increasing exposure times (24–72 h), the medium was removed, the cells were washed three times with PBS (200 μL per well) before adding 100 μL per well of 0.5 μM JC-1 working solution for 20 minutes at 37 °C. As a positive control, 0.1 μg mL−1 valinomycin was used. The cells were then washed twice with ice-cold culture medium and the fluorescence was immediately measured by using a Fluorocount Microplate Fluorometer (Packard, Germany). Red fluorescence given by JC-1 aggregates (intact mitochondria) was detected with an excitation wavelength of 530 nm and an emission wavelength of 590 nm whilst the green fluorescence was given by JC-1 monomers (disrupted mitochondria) with a 485 nm and 570 nm filter combination. The results are expressed as a ratio between red and green fluorescence and are reported as % relative to negative control (cells not exposed to GBNs).Nitro blue tetrazolium (NBT) assay
ROS production by HaCaT cells exposed to GBNs was evaluated by the NBT reduction assay, as previously reported.14,15 As a preliminary set of experiments, the potential interference of GBNs with the assay was evaluated in an acellular system by incubating FLG or GO (0.4–100 μg mL−1) with the NBT reagent for 4 h at 37 °C. Subsequently, the cells were exposed to FLG or GO (0.4–100 μg mL−1) for 24 h. Then, the cells were washed three times with PBS (200 μL per well) before adding fresh medium (100 μL per well) containing NBT (final concentration: 0.5 mg mL−1). After 4 h, the crystals were solubilized by 140 μL DMSO and 120 μL KOH (2 M). The absorbance was read by using an Automated Microplate Reader EL 311s (Bio-Tek Instruments, Winooski, VT, USA) at 630 nm. The results are expressed as % of ROS production as compared to negative control (cells not exposed to GBNs).Dichlorofluorescin diacetate (DCFDA) assay
ROS production by HaCaT cells exposed to GBNs was evaluated by the 2′,7′-dichlorofluorescin diacetate (DCFDA) assay, as previously described.14 As a preliminary set of experiments, the potential interference of GBNs with the assay was evaluated in an acellular system by incubating FLG or GO (0.4–100 μg mL−1) with the DCFDA probe for increasing time intervals up to 72 h at 37 °C. Subsequently, the cells were incubated with medium (200 μL per well) containing DCFDA (final concentration: 100 μM) for 30 min at 37 °C in the dark. The cells were then washed twice with PBS containing Ca2+ and Mg2+ (200 μL per well) and exposed to GBNs (0.4–100 μg mL−1) in complete medium without phenol red for increasing exposure times (3–72 h). Fluorescence was read after increasing intervals of time by using a Fluorocount Microplate Fluorometer (Packard, Germany) with an excitation wavelength of 485 nm and an emission wavelength of 570 nm. The results are expressed as % of ROS production as compared to negative control (cells not exposed to GBNs). The cells exposed to each concentration of FLG or GO without any probe were used to assess any absorbance properties of the tested GBNs and the relevant data were considered as blanks.Luminol assay
ROS production by HaCaT cells exposed to GBNs was also evaluated by the luminol assay. Preliminarily, the potential interference of FLG and GO with the assay was evaluated in an acellular system by incubating each GBN (0.4–100 μg mL−1) with luminol for 15 minutes at 37 °C. Subsequently, the cells were seeded overnight at a density of 1 × 104 cells per well in 96-well white plates and exposed to GBNs (0.4–100 μg mL−1) for 24 h before adding luminol (1 μM) to each well. Chemiluminescence was recorded 15 minutes after luminol addition, by using a multiwell luminometer (Wallac 1450 Microbeta counter, PerkinElmer, Milan, Italy). The results are expressed as % of ROS production as compared to negative control (cells not exposed to GBNs).CCK-8 assay
The effect of GBNs on HaCaT cell viability was evaluated by the Cell Counting Kit (CCK)-8 assay (Sigma Aldrich; Milan, Italy) following the manufacturer's instructions. After exposure to FLG or GO (1–100 μg mL−1), the cells were washed three times with PBS (200 μL per well) and incubated for 4 h with fresh medium (100 μL per well) containing 10 μL of the probe. Absorbance was subsequently read at 450 nm by using an Automated Microplate Reader EL 311s (Bio-Tek Instruments, Winooski, VT, USA). Data are reported as % of cell viability as compared to negative control (cells not exposed to GBNs).Statistical analysis
The results are presented as mean ± SE from at least three independent experiments performed in triplicate. Statistical analysis was performed by a two-way ANOVA followed by Bonferroni's post-test (GraphPad Prism version 6.00) and statistical significance was considered for p < 0.05.Results
Characterization of FLG
The FLG sample was thoroughly characterized using different techniques such as Raman spectroscopy, TGA, HRTEM, elemental analysis and TXRF (Fig. 1).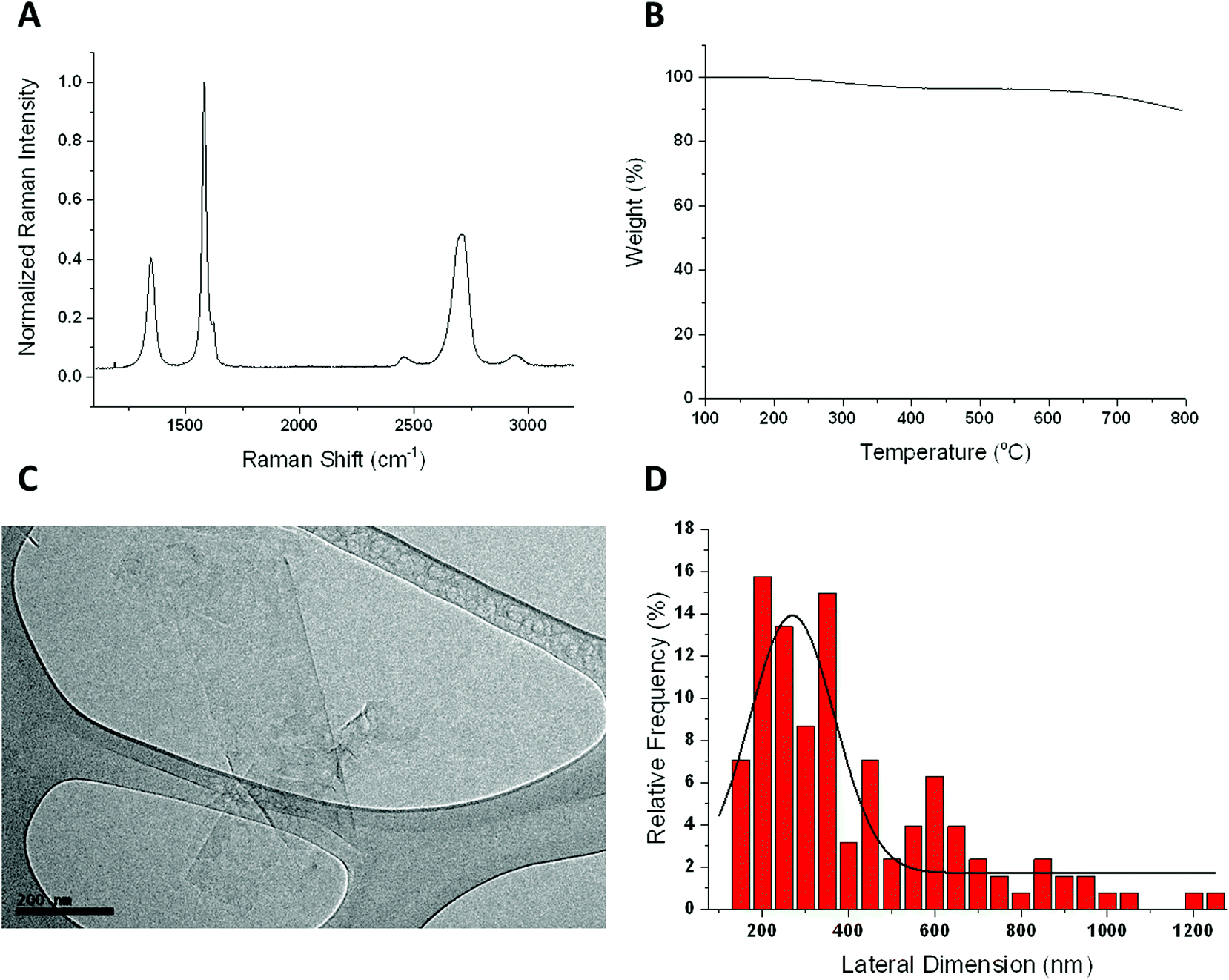 | ||
| Fig. 1 Chemical characterization of FLG by Raman spectroscopy as an average of 28 spectra (A), thermogravimetric analysis (B), representative HRTEM image (C) and lateral size distribution (n = 107) of FLG (D). | ||
The Raman spectrum shows the two most intense bands of graphene, the G peak at around 1580 cm−1 and the 2D band at around 2700 cm−1. The intensity ratio I(2D)/I(G) falls below 1, as described for few layer graphene.16 In fact, only 3 layers were calculated for our FLG.17 The disorder-induced D band, appears at 1345 cm−1, being the intensity ratio I(D)/I(G) of 0.4, which reveals a low density of defects. Elemental analysis shows average values of 95.7 ± 0.51% C, 0.42 ± 0.042% H, 0.43 ± 0.014% N and 0.22 ± 0% S. The oxygen content, less than 3.5%, is compatible with the weight loss at 600 °C observed by TGA. These results corroborate the low number of defects presented in the FLG sheets. HRTEM analysis was used to determine the lateral dimensions of FLG from at least 100 different sheets. A lateral size distribution between 200 and 1200 nm was obtained with an average lateral dimension of 391 ± 238 nm and a major population <1 μm, significantly lower than that of GO (average lateral dimension of 979 nm).8 Finally, since a stainless-steel flask is used during the ball-milling process, TXRF was performed to ensure the absence of metals, especially Fe, in the FLG sample, revealing a Fe concentration of only 0.026 mg L−1 (ESI 1†).
Internalization of GBNs in HaCaT cells
To investigate if GBNs can be internalized into HaCaT keratinocytes, the cells were fractionated into nuclear, mitochondrial, cytoplasm and membrane fractions after 24 h exposure to the materials and their presence in each fraction was assessed by Raman spectroscopy. Both FLG and GO were detected in all the fractions, suggesting that they can be internalized into HaCaT cells (data not shown). As a representative of GBNs, herein only the results related to FLG are presented. Fig. 2 shows the typical morphology of FLG in the mitochondrial fraction (MF), as shown by the black spot represented in the micrograph (panel A). In addition, FLG Raman fingerprints were detected in around 50% of all the aforesaid black spots, contained in MF, demonstrating that FLG is interacting with mitochondria (panel B). This result indicates the presence of FLG in HaCaT MF. It is also interesting to note that the cellular environment also has its own Raman bands, which are visible in the FLG profile, possibly explaining the somewhat higher D-Band as compared to the starting FLG. In contrast, in the negative control, only the profile corresponding to the MF was found, but not the FLG characteristic spectrum. | ||
| Fig. 2 (A) Optical microscopy image of mitochondrial fraction (MF) deposited on silicon oxide, showing typical morphologies (centre of the image) which correspond to FLG, according to their Raman spectrum. Scale bar = 20 μm. (B) Raman spectra of FLG alone, MF alone, FLG found in MF and MF negative control. The FLG spectrum is an average of more than 20 spectra, whereas the other spectra are an average of 5 repetitions. Every spectrum is normalized to its own highest-intensity band. | ||
These data cannot demonstrate if FLG is inside the mitochondria or attached to their outer membrane. However, together with FLG detection in the membrane, cytosol and nuclei, they unequivocally show that FLG is internalized into HaCaT cells.
FLG and GO induce mitochondrial membrane depolarization, not dependent on mitochondrial permeability transition pore opening
The mitochondrial effects of FLG and GO on HaCaT cells were evaluated by means of mitochondrial depolarization (JC-1 fluorescence shift) after different exposure times, up to 72 h. As reported in Fig. 3, both FLG and GO (1–100 μg mL−1) induced a concentration-dependent mitochondrial membrane depolarization already after 24 h exposure (29 and 31% at 100 μg mL−1 for FLG and GO, respectively). The effect was also time-dependent and, after 72 h exposure, the highest concentration (100 μg mL−1) of FLG or GO induced 48% (p < 0.001) or 57% (p < 0.001) mitochondrial depolarization, respectively. As a positive control, valinomycin (0.1 μg mL−1) induced 48% (p < 0.001) mitochondrial depolarization.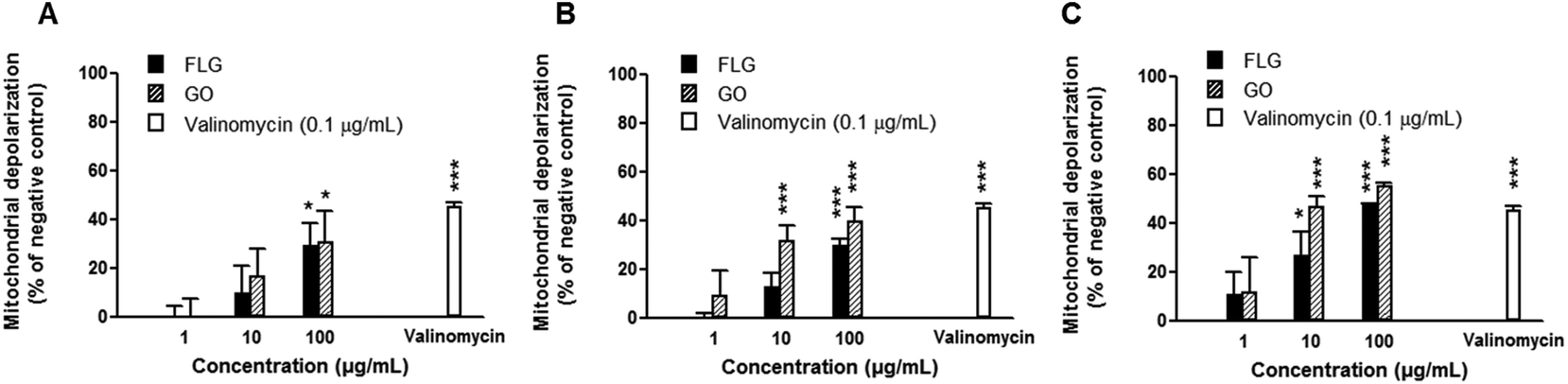 | ||
| Fig. 3 Mitochondrial depolarization in HaCaT cells exposed to FLG or GO (1–100 μg mL−1) for 24 (A), 48 (B) and 72 h (C). As a positive control, the cells were exposed to valinomycin (0.1 μg mL−1). Results are presented as % of JC-1 fluorescence shift with respect to negative control (cells not exposed to GBNs) calculated on the ratio between red (530/590 nm) and green (485/570 nm) fluorescence. Data are the mean ± SE of 3 independent experiments performed in triplicate. Statistical differences vs. untreated control: *p < 0.05; ***p < 0.001 (two-way ANOVA and Bonferroni's post-test). | ||
To investigate the mechanism of mitochondrial depolarization induced by FLG and GO, their effects were evaluated in the presence of cyclosporine A (Cs A), a well-known inhibitor of mitochondrial permeability transition pore (MPTP) formation. HaCaT cells were pre-exposed to Cs A (0.2 μg mL−1) for 1 h and subsequently to FLG or GO (1–100 μg mL−1) and Cs A up to 72 h. As shown in Fig. 4, the effects of FLG and GO on mitochondrial depolarization after 72 h exposure were not reverted by Cs A (p > 0.05), suggesting that FLG- and GO-increased mitochondrial depolarization is not mediated by MPTP formation. Similar results were obtained after 24 and 48 h exposure (ESI 2†).
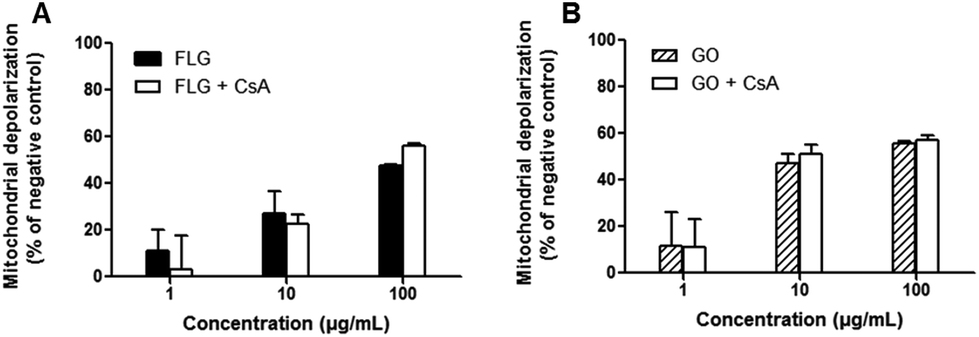 | ||
| Fig. 4 Role of MPTP opening in the mitochondrial depolarization in HaCaT cells exposed to FLG (A) or GO (B). HaCaT cells were pre-exposed to 0.2 μg mL−1 Cs A for 1 h and then exposed to GBNs and Cs A for 72 h. Mitochondrial membrane depolarization induced by FLG or GO in the presence of Cs A is compared to that induced by FLG or GO alone. Results are presented as % of fluorescence shift with respect to the negative control (cells not exposed to GBNs). Results are the mean ± SE of 3 independent experiments performed in triplicate. No statistical differences were observed between the cells exposed to GBNs with or without Cs A (two-way ANOVA and Bonferroni's post-test). | ||
FLG and GO induce a concentration- and time-dependent reactive oxygen species production
To verify whether mitochondrial membrane depolarization is mediated by reactive oxygen species (ROS) overproduction, FLG and GO were evaluated for their ability to increase ROS levels in HaCaT keratinocytes after 24 h exposure. The amount of specific ROS was evaluated by three methods: NBT reduction assay, luminol assay and DCFDA fluorescence assay, able to specifically measure superoxide anion, hydrogen peroxide and peroxynitrite/hydroxyl radicals, respectively. As shown in Fig. 5, FLG induced a significant concentration-dependent increase of ROS production starting from the concentration of 33 μg mL−1 (DCFDA and NBT assay, p < 0.05) or above 1 μg mL−1 (luminol assay, p < 0.05). GO significantly increased ROS production at the concentration of 33 μg mL−1 and above, recorded by all the assays (p < 0.05). At the highest concentration tested (100 μg mL−1), FLG increased ROS production by 9% (NBT assay, p < 0.01), 39% (luminol assay, p < 0.001) and 25% (DCFDA assay, p < 0.01). GO (100 μg mL−1) increased ROS production by 36% (p < 0.01), 21% (p < 0.01) and 39% (p < 0.001), as assessed by the NBT, luminol and DCFDA assays, respectively. Only at this concentration, GO was significantly more active than FLG in increasing ROS production as assessed by NBT (p < 0.01). The positive control 2,2′-azobis(2-amidinopropane)dihydrochloride (AAPH, 0.3 mg mL−1) increased ROS production by 32% (p < 0.001), 91% (p < 0.001) and 148% (p < 0.001), as assessed by the NBT, luminol and DCFDA assays, respectively. Preliminary experiments carried out in acellular systems revealed no ROS production detectable by each assay at all GBNs concentrations. Only a significant reduction of the signal was observed by the DCFDA assay at the highest FLG concentrations (33–100 μg mL−1), as reported below. | ||
| Fig. 5 Reactive oxygen species (ROS) production in HaCaT cells exposed to FLG (A) or GO (B) for 24 h, evaluated by NBT reduction assay, DCFDA fluorescence and luminol assay. As positive control, the cells were exposed to AAPH (0.3 mg mL−1). Data are expressed as % of ROS increase in comparison to negative control (cells not exposed to GBNs) and are the mean ± SE of at least 3 independent experiments performed in triplicate. Statistical differences: *p < 0.05; **p < 0.01; ***p < 0.001 (two-way ANOVA and Bonferroni's post-test). | ||
To deeply investigate the effects of GBNs, the kinetics of ROS production by HaCaT cells exposed to FLG or GO (0.4–100 μg mL−1) was investigated after increasing intervals of time (3 to 72 h exposure), using a time-dependent DCFDA assay. As shown in Fig. 6, FLG- and GO-induced ROS production was time-dependent at all the concentrations tested. In particular, as compared to the negative control cells not exposed to GBNs, the incubation of HaCaT cells with GO significantly increased ROS production already after 12 h (p < 0.05, 100 μg mL−1), while FLG induced a significant ROS production starting from 24 h exposure (p < 0.05, at 0.4 μg mL−1 and above). Cell exposure to the highest concentration (100 μg mL−1) of FLG or GO for 72 h increased ROS production by 85% or 124%, respectively (p < 0.001). These effects were lower than that induced by the positive control AAPH (0.3 mg mL−1), which increased ROS production by 304% after 72 h exposure (p < 0.001). On the whole, ROS production induced by GO was significantly higher than that induced by FLG, starting from the concentration of 33 μg mL−1 after 48 h exposure (p < 0.05).
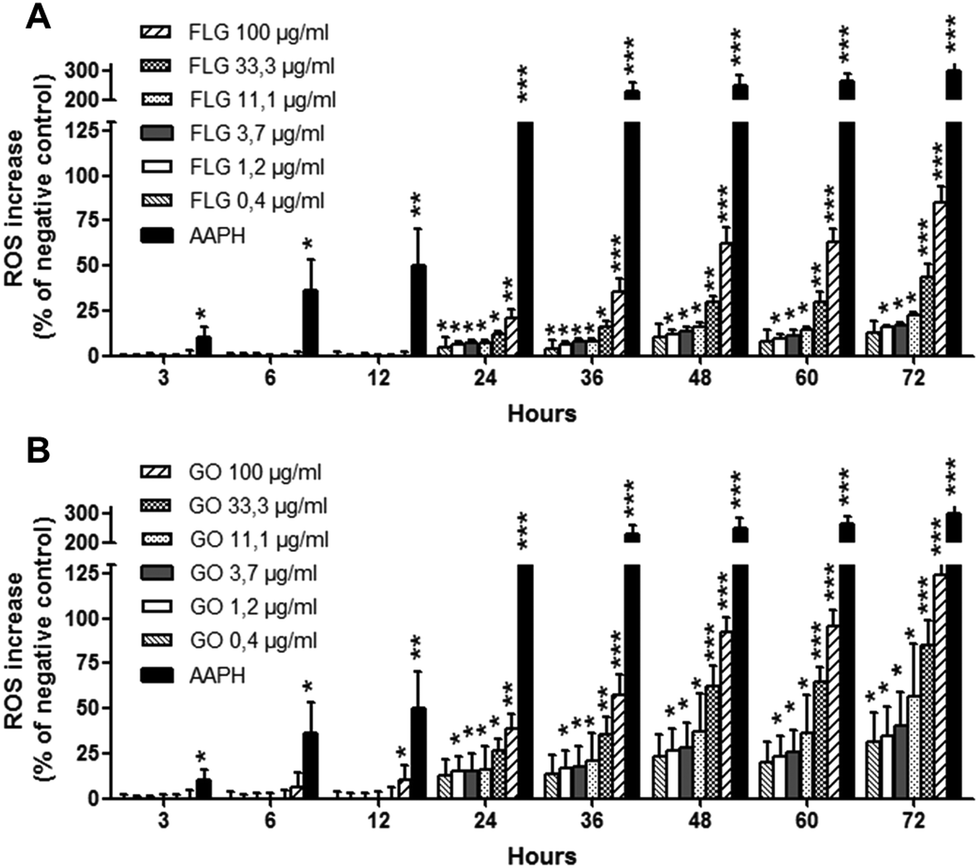 | ||
| Fig. 6 Kinetics of ROS production in HaCaT cells exposed to FLG (A) or GO (B) up to 72 h, evaluated by the DCFDA assay. Results are expressed as % of ROS increase with respect to negative control (cells not exposed to GBNs) and are the mean ± SE of at least 3 independent experiments performed in triplicate. As positive control, the cells were exposed to AAPH (0.3 mg mL−1). Statistical differences: *p < 0.05; **p < 0.01; ***p < 0.001 (two-way ANOVA and Bonferroni's post-test). | ||
ROS production in HaCaT cells exposed to FLG or GO was not dependent on the spontaneous ability of GBNs to generate free radicals, since neither FLG nor GO induced ROS production in cell-free culture medium up to 72 h. In contrast, only a significant signal reduction was observed for FLG at the concentration of 33 μg mL−1 and above (ESI 3†).
FLG- and GO-induced ROS production is mediated by the activation of NADH dehydrogenase and xanthine oxidase
The putative role of specific HaCaT cell ROS-generating enzymes in FLG- and GO-induced ROS production was investigated using selective inhibitors of these enzymes. In particular, HaCaT cells were pre-exposed for 1 h to diphenyliodonium chloride (DPI; 1 μg mL−1), an inhibitor of flavoprotein enzymes, apocynin (1 μg mL−1), a selective inhibitor of NADPH oxidase (NOX), L-NG-monomethyl arginine citrate (L-NMMA; 75 μg mL−1), a selective inhibitor of nitric oxide synthase (NOS), rotenone (2 μg mL−1), a selective inhibitor of mitochondrial complex I (NADH dehydrogenase), indomethacin (35 μg mL−1), an inhibitor of cyclooxygenases (COXs), or allopurinol (10 μg mL−1), an inhibitor of xanthine oxidase (XO). Subsequently, the cells were exposed to FLG or GO (1–100 μg mL−1) and the same specific inhibitors for 72 h, and ROS production was measured by the DCFDA assay. The effects were evaluated as % increase in ROS production with respect to the negative control (cells exposed to each selected inhibitor, without GBNs). As shown in Fig. 7 (panels A and B), the inhibitor of flavoprotein-based enzymes DPI, the inhibitor of NADH dehydrogenase, rotenone, and the inhibitor of xanthine oxidase, allopurinol, significantly reduced or even abolished ROS production induced by FLG or GO at 10 μg mL−1 and above. In particular, DPI, rotenone and allopurinol reduced ROS overproduction induced by the highest FLG concentration (100 μg mL−1) by 53% (p < 0.001), 95% (p < 0.001) and 96% (p < 0.001), respectively. ROS overproduction induced by GO (100 μg mL−1) was reduced by DPI, rotenone and allopurinol by 57% (p < 0.001), 75% (p < 0.001) and 81% (p < 0.001), respectively. The other inhibitors did not significantly affect FLG- or GO-induced ROS production.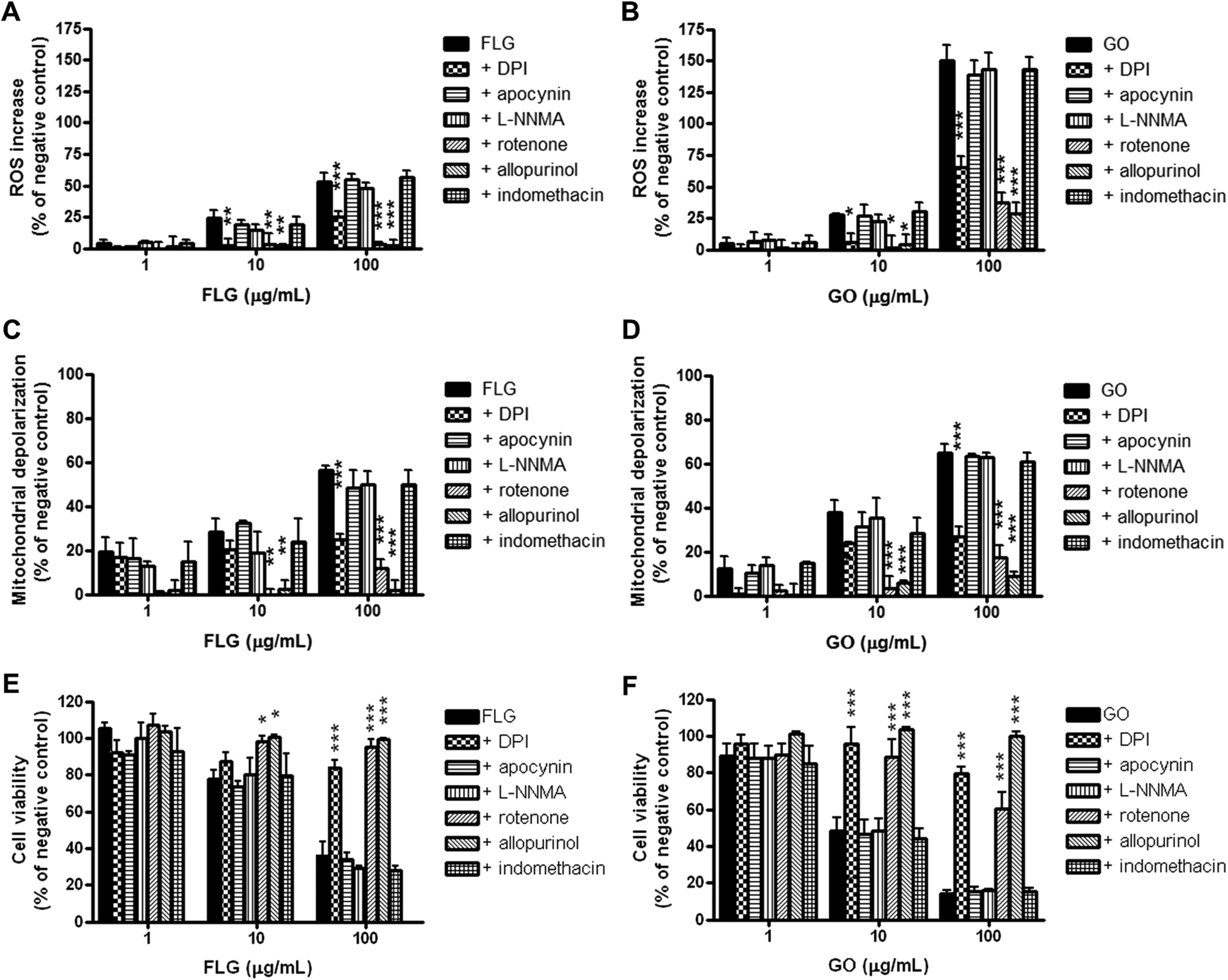 | ||
| Fig. 7 Effect of selected inhibitors of ROS-generating enzymes on FLG- and GO-induced ROS production, mitochondrial depolarization and cytotoxicity in HaCaT cells. The cells were pre-exposed to 5.0 × 10−6 M DPI, 1.0 × 10−5 M apocynin, 1.0 × 10−4 M L-NMMA, 5.0 × 10−6 M rotenone, 1.0 × 10−4 M indomethacin or 1.0 × 10−4 M allopurinol for 1 h and subsequently exposed to FLG or GO and the same selected ROS-generating enzyme inhibitor for 72 h. ROS production was measured by DCFDA assay (panels A and B), mitochondrial depolarization by JC-1 fluorescence shift (panels C and D) and cell viability by CCK-8 assay (panels E and F). Data are expressed as % difference of each measured parameter with respect to relevant negative control (cells exposed to each selected inhibitor, without GBNs) and are the mean ± SE of 4 independent experiments performed in triplicate. Statistical differences: *p < 0.05; **p < 0.01; ***p < 0.001 (two-way ANOVA and Bonferroni's post-test). | ||
FLG- and GO-induced mitochondrial damage is mediated by the activation of NADH dehydrogenase and xanthine oxidase
To investigate the relationship between FLG- or GO-induced oxidative stress and FLG- or GO-induced mitochondrial damage in HaCaT cells, the changes in mitochondrial membrane potential were evaluated by the JC-1 assay, in the presence of the ROS-producing enzyme inhibitors used in the previous set of experiments. The cells were pre-exposed to each inhibitor for 1 h and subsequently exposed to FLG or GO (1–100 μg mL−1) and the same inhibitor for 72 h. The effects were evaluated as % increase in mitochondrial depolarization with respect to the negative control (cells exposed to each selected inhibitor, without GBNs). As observed for the ROS production by the DCFDA assay (Fig. 7, panels A and B), DPI, rotenone and allopurinol also significantly reduced the mitochondrial depolarization induced by FLG or GO at 10 μg mL−1 and above (Fig. 7, panels C and D). In particular, mitochondrial depolarization evoked by the highest FLG concentration (100 μg mL−1) was reduced by DPI, rotenone and allopurinol by 56% (p < 0.001), 79% (p < 0.001) and 96% (p < 0.001), respectively. Similarly, DPI, rotenone and allopurinol reduced mitochondrial depolarization induced by 100 μg mL−1 GO by 59% (p < 0.001), 73% (p < 0.001) and 87% (p < 0.001), respectively. In contrast, the other inhibitors did not affect FLG- or GO-induced mitochondrial depolarization.FLG- and GO cytotoxicity is mediated by the activation of NADH dehydrogenase and xanthine oxidase
To investigate the relationship between FLG- or GO-induced oxidative stress and their cytotoxicity, HaCaT cells were pre-exposed for 1 h to the ROS-producing enzyme inhibitors used in the previous set of experiments, and subsequently exposed to FLG or GO (1–100 μg mL−1) and the same selective inhibitors for 72 h. Cell viability was then evaluated by the CCK-8 assay. The effects were evaluated as % difference of cell viability as compared to the negative control (cells exposed to each selected inhibitor, without GBNs). As shown in Fig. 7 (panels E and F), FLG and GO induced a concentration-dependent decrease of viable cells, with GO being significantly more cytotoxic than FLG at the highest concentrations (10 and 100 μg mL−1; p < 0.01). This cytotoxic effect was significantly counteracted by DPI, rotenone and allopurinol in cells exposed to FLG or GO at 10 μg mL−1 and above. In particular, as compared to the negative control, exposure to the highest FLG concentration (100 μg mL−1) reduced viable cells to 36%, whereas viable cells after FLG and DPI, rotenone or allopurinol co-exposure were significantly increased up to 84% (p < 0.001), 95% (p < 0.001) and 99% (p < 0.001), respectively. Similarly, cell exposure to 100 μg mL−1 GO reduced cell viability to 14% while their co-exposure with GO and DPI, rotenone or allopurinol counteracted the GBN effect, significantly increasing cell viability up to 80% (p < 0.001), 60% (p < 0.001) or 100% (p < 0.001), respectively. In contrast, the other inhibitors did not counteract FLG- or GO-induced cytotoxicity.Discussion
In recent years, GBNs have attracted attention for their promising applications in biomedical and optoelectronic fields. However, despite the occupational cutaneous exposure and their potential skin applications, mainly as wound healing applications and skin sensors as well as artificial and electronic skin,18,19 data on their cutaneous toxicity are still limited. Indeed, skin biocompatibility of GBNs is largely unexplored, although other carbon-based nanomaterials, such as carbon nanotubes, may lead to adverse skin reactions of different severities.20,21 Recently, we provided the first comparative study of the effects of different GBNs on human HaCaT keratinocytes, a widely used in vitro model to predict skin toxicity. On this in vitro skin model, GBNs exerted a significant cytotoxicity, with variable potencies depending on their oxidation state.8 Considering that mitochondrial damage is a common feature of the mechanism of toxicity of carbon-based nanomaterials,22,23 the present study was aimed to investigate the putative mechanism(s) of mitochondrial damage induced by a research grade FLG and a commercially available GO on skin keratinocytes.In HaCaT cells exposed to these materials, both FLG and GO were detected in cell membranes, cytosol, mitochondria and nucleus, as observed by Raman spectroscopy. FLG and GO induced a concentration-dependent mitochondrial membrane depolarization already after 24 h. Prolonging the exposure to 72 h, the effect also appeared to be time-dependent. These data suggest that FLG and GO can induce a significant mitochondrial depolarization, as a possible cause of mitochondrial damage. These results are supported by previous studies reporting the ability of other GBNs to induce mitochondrial depolarization, such as carboxyl graphene,24 pristine graphene25 and few layer graphene (obtained by the arc discharge method).26 To evaluate the involvement of MPTP opening as a causative mechanism of FLG- and GO-induced mitochondrial depolarization, a set of experiments was carried out co-exposing HaCaT cells with Cs A, a well-known inhibitor of MPTP opening.27 However, in our model, MPTP opening does not seem to be involved in the mitochondrial damage, since Cs A was unable to prevent FLG- and GO-induced mitochondrial depolarization at any concentration and exposure time to GBNs.
It is well known that one of the main causes of mitochondrial damage is the increased ROS cellular levels.9 Hence, in the effort to investigate an alternative putative mechanism of mitochondrial depolarization, the role of ROS was evaluated. Both FLG and GO induced a concentration-dependent increase of ROS production in HaCaT skin keratinocytes after 24 h exposure. For the first time, this effect was assessed by three different ROS detection methods: spectrophotometric (NBT), chemiluminescent (luminol) and fluorometric (DCFDA).
To deeply investigate the role of ROS in the mechanism of mitochondrial dysfunction, the kinetics of their production was evaluated using the DCFDA assay, the most suitable method to perform a time course measurement. FLG and GO induced a significant time-dependent ROS production already after 24 and 12 h exposure, respectively, an effect significantly lower than that of the positive control AAPH, a free radical initiator. However, an underestimation of the effect of GBNs due to their ability to quench the dye fluorescence cannot be excluded. In fact, in line with the literature data on the quenching properties of GBNs,28 FLG significantly reduced the DCFDA signal under cell-free conditions, starting from the concentration of 33 μg mL−1 (ESI 3†), which suggests a possible underestimation of cellular ROS production induced by GBNs, at least at the highest concentrations. In contrast, no interference between FLG or GO and NBT or luminol was observed, suggesting the absence of possible false positive or negative results. In addition, possible particle overload, especially at the highest concentration, should be considered as an additional factor that might influence the effects of GBNs. Notwithstanding, a significant ROS production was observed also at concentrations as low as 0.4 μg mL−1, at which particle overload cannot be expected.
Intriguingly, 12–72 h exposure to GO induced a ROS production higher than that induced by FLG, with a significant difference starting from 48 h exposure. This result is in agreement with the finding that the higher density of oxygen-containing functional groups on the GO surface in comparison to FLG can enhance the production of ROS.29 Altogether, these observations are in line with previous results, supporting the oxidation state of GBNs as an important feature for their cytotoxic potency,8,30 even though other physicochemical properties (i.e. dimension, aggregation, shape, etc.) could affect their cytotoxicity.
These results are in agreement with previous studies reporting ROS overproduction induced by FLG (obtained with different methodologies) in PC12 cells31 and Human Umbilical Vein Endothelial Cells (HUVEC) after 24 h exposure,26 and by other GO (Hummers’ method) after 24 h exposure in A549 cells,32 MCF7 cells,33 HeLa cells,34 MDA-MB-231 cells35 and BEAS-2B cells.36 Furthermore, Li and coworkers (2012) have shown that commercial pristine graphene can induce ROS production and depletion of the mitochondrial membrane potential in murine RAW 264.7 macrophages after 24 h exposure, triggering apoptosis by activation of the mitochondrial pathway.25 In immune cells (Jurkat T lymphocytes and THP-1 monocytes), the mitochondrial pathway seemed to be affected by a GO prepared using a modified Hummers’ method, by altering the oxidative phosphorylation.37 Similarly, GO seems to induce mitochondrial alterations by increasing the activity of mitochondrial electron transport complexes in MHS cells.38 It has been reported that oxidative stress may also be indirectly derived by a decreased activity of antioxidant enzymes, such as superoxide dismutase and glutathione peroxidase, as reported for GO.22,36 However, no information on the molecular mechanism of ROS generation induced by GBNs is available, so far. Hence, the putative molecular mechanism(s) of cellular ROS production induced by GBNs was investigated. With this aim, six well-known inhibitors of the major ROS-generating enzymes expressed in HaCaT cells were evaluated for their ability to reduce GBNs-induced oxidative stress. Only DPI, rotenone and allopurinol significantly reduced or even abolished GBNs-increased ROS levels. The effects of rotenone and allopurinol suggest a role for mitochondrial electron transport chain complex I (NADH dehydrogenase) and xanthine oxidase in GBNs-induced ROS production. This observation is further supported by the ability of DPI to inactivate flavoprotein-based enzymes, such as mitochondrial NADH dehydrogenase and xanthine oxidase.39,40 Intriguingly, the same inhibitors also significantly reduced or even abolished GBNs-induced mitochondrial depolarization and cytotoxicity. These data suggest that ROS production, mediated by mitochondrial NADH dehydrogenase and xanthine oxidase, might also be involved in the mechanism of mitochondrial damage induced by GBNs leading to reduced cell viability. Altogether, for the first time, these results identify a possible intracellular pathway of ROS-dependent mitochondrial damage evoked by GBNs, which involves the activation of flavoprotein-based enzymes. However, even though GBNs can interact with the plasma membrane to reach the cytoplasm, mitochondria and nucleus of HaCaT cells, their ability to directly activate these enzymes cannot be excluded. Tentatively, the mitochondrial dysfunction driven by their activation could be hypothesized as a secondary stress response to the insult given by GBNs entrance inside the cells or by their perturbation of cell membrane, as previously observed.8 In fact, xanthine oxidase is reported to be located not only in the cytoplasm, but also on the plasma membrane.41–43 In addition, we cannot exclude the involvement of other mechanism(s) in the mitochondrial damage induced by GBNs, including a disruption of osmotic balance consequent to plasma membrane damage.8 Similarly, other cellular structures and pathways that could be involved in FLG- and GO-induced cytotoxicity cannot be excluded.
On the whole, these results should be taken into account when considering GBNs for possible biomedical applications at the skin level, e.g. for wound healing.44 Indeed, the sustained ROS production and the consequent mitochondrial depolarization might hinder the use of GBNs in such applications. In addition, it is well known that ROS production is a key element in triggering and modulating inflammation. Moreover, activated keratinocytes may interact with inflammatory/immunity cells, such as monocytes, macrophages and/or resident dendritic cells (i.e. Langerhans cells). In this scenario, the effects of GBNs on some of these cells have already been investigated.45 Indeed, GBNs were shown to upregulate critical genes implicated in immune responses and induce the release of pro-inflammatory cytokines from healthy donors’ immune cells.46 FLG was also shown to exert a selective killer action towards monocytes.47,48 Similarly, pro-inflammatory effects of GBNs were also observed on macrophages, with GO being able to activate the inflammasome49 and enhance the production of inflammatory cytokines50 in a size-dependent manner. Altogether, these observations support the hypothesis of pro-inflammatory properties of GBNs at the cutaneous level. Thus, further studies are needed to assess the safety of the biomedical applications of GBNs at the skin level.
It is noteworthy that the oxidative enzymatic machinery generating ROS is required for the effective degradation of carbonaceous nanomaterials. For instance, it is reported that xanthine oxidase can oxidize multi-walled carbon nanotubes (MWCNTs).51 Similarly, the superoxide/nitric oxide/peroxynitrite-driven pathway, mainly catalyzed by inducible nitric oxide synthase (iNOS) and NADPH oxidase, seems to be involved in the oxidative biodegradation of single-walled carbon nanotubes (SWCNTs).52 However, these biodegradation pathways have been characterized only in immune and inflammatory cells, so far. To the best of our knowledge, no data are available on the possible interaction of GBNs with these enzymes in non-immune/inflammatory cells, such as skin keratinocytes, which are the first line of body defense. In this view, the biodegradation pathways of GBNs in skin keratinocytes might involve flavoprotein-based oxidative enzymes, such as xanthine oxidase, triggering oxidative stress conditions and the consequent mitochondrial damage (Fig. 8).
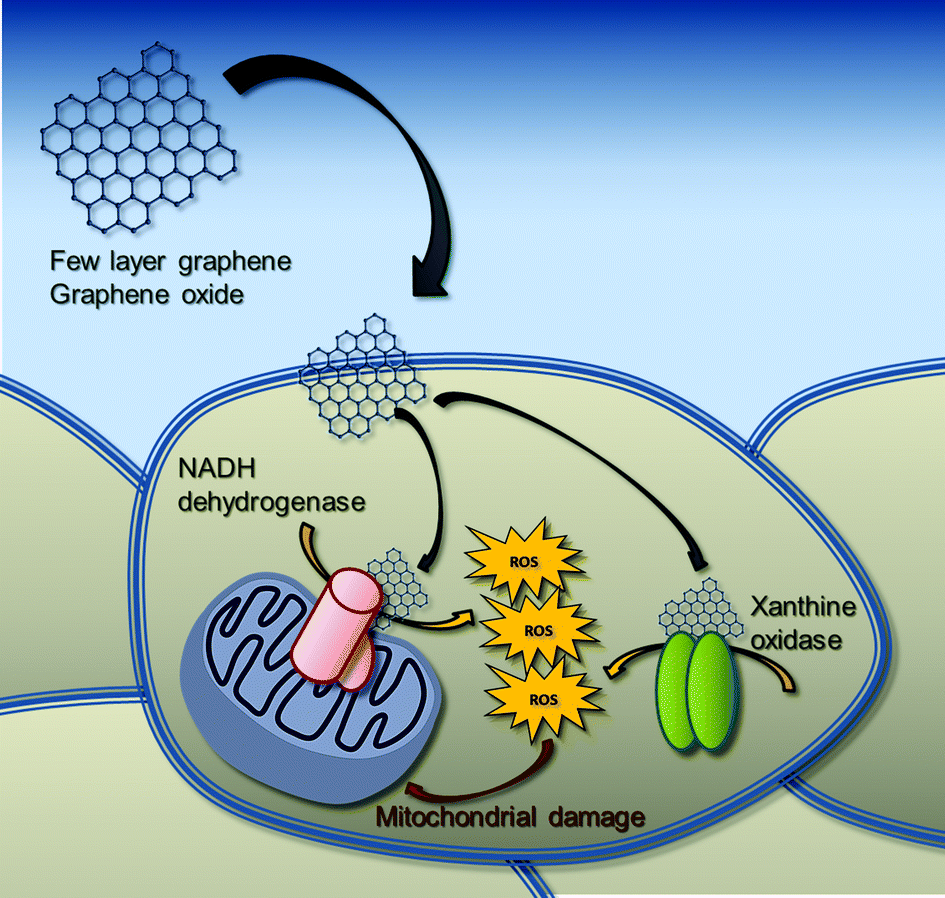 | ||
| Fig. 8 Proposed putative mechanism of mitochondrial damage induced by FLG and GO in HaCaT skin keratinocytes. GBNs induce significant intracellular increase of ROS production, mediated mainly by the activation of flavoprotein-based oxidative enzymes, such as NADH dehydrogenase and xanthine oxidase. The consequent increased ROS levels induce a significant mitochondrial membrane depolarization and mitochondrial damage. | ||
Conclusions
In conclusion, the results of this study may represent a significant step forward in the hazard identification and mechanism of toxicity of GBNs. In particular, FLG and GO are able to internalize inside HaCaT cells, inducing a significant intracellular ROS production, which in turn induces a sustained mitochondrial depolarization not mediated by MPTP opening. ROS production seems to be mediated mainly by the activation of flavoprotein-based oxidative enzymes, such as NADH dehydrogenase and xanthine oxidase, known to be present at the mitochondrial and cytosolic/cell membrane level, respectively. The selective activation of these intracellular enzymes suggests that GBNs-induced mitochondrial dysfunction is not consequent to a direct mitochondrial effect, but a secondary cellular response induced by the interaction of GBNs with HaCaT skin keratinocytes.Author contributions
MP, AT and MP defined the experimental design. LF, MP and SS carried out the biological analysis and interpreted the results. CM and EV carried out the FLG preparation and chemical characterization. JMGD, JFR and MDP carried out the cellular internalization analysis of GBNs. MP, AT, LF and MP wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the European Union H2020 Program under grant agreement no. 696656-Graphene Flagship Core1. The authors thank Graphenea (San Sebastián, Spain) for a generous gift of GO.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef PubMed
.
- G. Brumfiel, Nature, 2009, 458, 390–391 CrossRef PubMed
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef PubMed
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef PubMed
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef PubMed
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef PubMed
- D. D. L. Chung, J. Mater. Sci., 2004, 39, 2645–2661 CrossRef
- M. Pelin, L. Fusco, V. León, C. Martín, A. Criado, S. Sosa, E. Vázquez, A. Tubaro and M. Prato, Sci. Rep., 2017, 7, 40572 CrossRef PubMed
- M. Ott, V. Gogvadze, S. Orrenius and B. Zhivotovsky, Apoptosis, 2007, 12, 913–922 CrossRef PubMed
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef PubMed
- V. León, M. Quintana, M. A. Herrero, J. L. Fierro, A. de la Hoz, M. Prato and E. Vázquez, Chem. Commun., 2011, 47, 10936–10938 RSC
- V. León, A. M. Rodriguez, P. Prieto, M. Prato and E. Vázquez, ACS Nano, 2014, 8, 563–571 CrossRef PubMed
- V. León, J. M. González-Domínguez, J. L. Fierro, M. Prato and E. Vázquez, Nanoscale, 2016, 8, 14548–14555 RSC
- M. Pelin, C. Ponti, S. Sosa, D. Gibellini, C. Florio and A. Tubaro, Toxicol. Appl. Pharmacol., 2013, 266, 1–8 CrossRef PubMed
- M. Pelin, S. De Iudicibus, L. Fusco, E. Taboga, G. Pellizzari, C. Lagatolla, S. Martelossi, A. Ventura, G. Decorti and G. Stocco, Chem. Res. Toxicol., 2015, 28, 1186–1195 Search PubMed
- U. Mogera, R. Dhanya, R. Pujar, C. Narayana and G. U. Kulkarni, J. Phys. Chem. Lett., 2015, 6, 4437–4443 Search PubMed
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef PubMed
- S. R. Shin, Y. C. Li, H. L. Jang, P. Khoshakhlagh, M. Akbari, A. Nasajpour, Y. S. Zhang, A. Tamayol and A. Khademhosseini, Adv. Drug Delivery Rev., 2016, 105, 255–274 CrossRef PubMed
- J. Kim, J. Lee, D. Son, M. K. Choi and D. H. Kim, Nano Convergence, 2016, 3, 4 CrossRef PubMed
- D. J. Eedy, Contact Dermatitis, 1996, 35, 362–363 CrossRef PubMed
- A. A. Kasparov, T. B. Popova, N. V. Lebedeva, E. V. Gladkova and E. B. Gurvich, Vopr. Onkol., 1989, 35, 445–450 Search PubMed
- L. Ou, B. Song, H. Liang, J. Liu, X. Feng, B. Deng, T. Sun and L. Shao, Part. Fibre Toxicol., 2016, 13, 57 CrossRef PubMed
- B. Zhang, P. Wei, Z. Zhou and T. Wei, Adv. Drug Delivery Rev., 2016, 105, 145–162 CrossRef PubMed
- T. Lammel, P. Boisseaux, M. L. Fernández-Cruz and J. M. Navas, Part. Fibre Toxicol., 2013, 10, 27 CrossRef PubMed
- Y. Li, Y. Liu, Y. Fu, T. Wei, L. Le Guyader, G. Gao, R. S. Liu, Y. Z. Chang and C. Chen, Biomaterials, 2012, 33, 402–411 CrossRef PubMed
- A. Sasidharan, S. Swaroop, P. Chandran, S. Nair and M. Koyakutty, Nanomedicine, 2016, 12, 1347–1355 CrossRef PubMed
- K. G. Norman, J. A. Canter, M. Shi, G. L. Milne, J. D. Morrow and J. E. Sligh, Mitochondrion, 2010, 10, 94–101 CrossRef PubMed
- M. A. Creighton, J. R. Rangel-Mendez, J. Huang, A. B. Kane and R. H. Hurt, Small, 2013, 9, 1921–1927 CrossRef PubMed
- X. Zou, L. Zhang, Z. Wang and Y. Luo, J. Am. Chem. Soc., 2016, 138, 2064–2077 CrossRef PubMed
- S. Das, S. Singh, V. Singh, D. Joung, J. M. Dowding, D. Reid, J. Anderson, L. Zhai, S. I. Khondaker, W. T. Self and S. Seal, Part. Part. Syst. Charact., 2013, 30, 148–157 CrossRef
- Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef PubMed
- Y. Chang, S. T. Yang, J. H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef PubMed
- S. Gurunathan, J. W. Han, V. Eppakayala and J. H. Kim, Int. J. Nanomed., 2013, 8, 1015–1027 CrossRef PubMed
- X. Zhang, W. Hu, J. Li, L. Tao and Y. Wei, Toxicol. Res., 2012, 1, 62–68 RSC
- J. Wu, R. Yang, L. Zhang, Z. Fan and S. Liu, Toxicol. Mech. Methods, 2015, 25, 312–319 CrossRef PubMed
- S. Mittal, V. Kumar, N. Dhiman, L. K. Chauhan, R. Pasricha and A. K. Pandey, Sci. Rep., 2016, 6, 39548 CrossRef PubMed
- M. Orecchioni, D. Bedognetti, L. Newman, C. Fuoco, F. Spada, W. Hendrickx, F. M. Marincola, F. Sgarrella, A. F. Rodrigues, C. Ménard-Moyon, G. Cesareni, K. Kostarelos, A. Bianco and L. G. Delogu, Nat. Commun., 2017, 8, 1109 CrossRef PubMed
- M. C. Duch, G. R. Budinger, Y. T. Liang, S. Soberanes, D. Urich, S. E. Chiarella, L. A. Campochiaro, A. Gonzalez, N. S. Chandel, M. C. Hersam and G. M. Mutlu, Nano Lett., 2011, 11, 5201–5207 CrossRef PubMed
- S. Chakraborty and V. Massey, J. Biol. Chem., 2002, 277, 41507–41516 CrossRef PubMed
- V. B. O'Donnell, G. C. Smith and O. T. Jones, Mol. Pharmacol., 1994, 46, 778–785 Search PubMed
- S. Vickers, H. J. Schiller, J. E. Hildreth and G. B. Bulkley, Surgery, 1998, 124, 551–560 CrossRef PubMed
- P. A. Grange, C. Chéreau, J. Raingeaud, C. Nicco, B. Weill, N. Dupin and F. Batteux, PLoS Pathog., 2009, 5, e1000527 CrossRef PubMed
- M. Rinnerthaler, J. Bischof, M. K. Streubel, A. Trost and K. Richter, Biomolecules, 2015, 5, 545–589 CrossRef PubMed
- C. Zhong, D. Shi, Y. Zheng, P. J. Nelson and Q. Bao, Nanoscale Res. Lett., 2017, 12, 533 CrossRef PubMed
- M. Orecchioni, C. Ménard-Moyon, L. G. Delogu and A. Bianco, Adv. Drug Delivery Rev., 2016, 105, 163–175 CrossRef PubMed
- M. Orecchioni, D. A. Jasim, M. Pescatori, R. Manetti, C. Fozza, F. Sgarrella, D. Bedognetti, A. Bianco, K. Kostarelos and L. G. Delogu, Adv. Healthcare Mater., 2016, 5, 276–287 CrossRef PubMed
- J. Russier, V. León, M. Orecchioni, E. Hirata, P. Virdis, C. Fozza, F. Sgarrella, G. Cuniberti, M. Prato, E. Vázquez, A. Bianco and L. G. Delogu, Angew. Chem., Int. Ed., 2017, 56, 3014–3019 CrossRef PubMed
- J. Yan, L. Chen, C. C. Huang, S. C. Lung, L. Yang, W. C. Wang, P. H. Lin, G. Suo and C. H. Lin, Colloids Surf., B, 2017, 153, 300–309 CrossRef PubMed
- S. P. Mukherjee, K. Kostarelos and B. Fadeel, Adv. Healthcare Mater., 2018, 7, 1700815 CrossRef PubMed
- J. Ma, R. Liu, X. Wang, Q. Liu, Y. Chen, R. P. Valle, Y. Y. Zuo, T. Xia and S. Liu, ACS Nano, 2015, 9, 10498–10515 CrossRef PubMed
- A. R. Sureshbabu, R. Kurapati, J. Russier, C. Ménard-Moyon, I. Bartolini, M. Meneghetti, K. Kostarelos and A. Bianco, Biomaterials, 2015, 72, 20–28 CrossRef PubMed
- I. I. Vlasova, A. A. Kapralov, Z. P. Michael, S. C. Burkert, M. R. Shurin, A. Star, A. A. Shvedova and V. E. Kagan, Toxicol. Appl. Pharmacol., 2016, 299, 58–69 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: ESI 1. Total reflection X-ray fluorescence (TXRF) of FLG. ESI 2. ROS production induced by FLG (A) and GO (B) under cell-free conditions. See DOI: 10.1039/c8nr02933d |
| This journal is © The Royal Society of Chemistry 2018 |