
A helically-twisted ladder based on 9,9′-bifluorenylidene: synthesis, characterization, and carrier-transport properties†
Jinjia
Xu
a,
Atsuro
Takai
 *a,
Alisa
Bannaron
ab,
Takafumi
Nakagawa
c,
Yutaka
Matsuo
c,
Manabu
Sugimoto
d,
Yoshitaka
Matsushita
a and
Masayuki
Takeuchi
*a
*a,
Alisa
Bannaron
ab,
Takafumi
Nakagawa
c,
Yutaka
Matsuo
c,
Manabu
Sugimoto
d,
Yoshitaka
Matsushita
a and
Masayuki
Takeuchi
*a
aMolecular Design and Function Group, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, 305-0047, Japan. E-mail: TAKAI.Atsuro@nims.go.jp; TAKEUCHI.Masayuki@nims.go.jp
bInstitute for Materials Chemistry and Engineering, Kyushu University, 6-1 Kasuga-koen, Kasuga-city, Fukuoka 816-8580, Japan
cDepartment of Mechanical Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
dFaculty of Advanced Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
First published on 26th January 2018
Abstract
We synthesized a ladder-shaped 9,9′-bifluorenylidene cyclic dimer (CBF), in which the two 9,9′-bifluorenylidene units are connected directly with two covalent bonds. CBF has a pair of enantiomers which are alternately packed in a perpendicular fashion in the crystal, and these enantiomers exhibit rapid interconversion in solution. Owing to the ladder-type connection, the two 9,9′-bifluorenylidene units in CBF electronically interact upon redox, and thus the structure of CBF is changed stepwise during the oxidation and reduction processes, which was confirmed by electrochemistry and quantum chemical calculations. The π-network of CBF in the solid state leads to its hole transport properties.
The construction of molecular-based materials that exhibit dynamic behavior on a meso- to macroscopic scale, like natural molecular machines, is the ultimate challenge in the field of organic materials chemistry. One of the most promising approaches toward this aim is to develop macromolecular systems in which artificial molecular machines are well organized.1 Molecular machines have been embedded in liquid crystalline matrices,2 incorporated in polymer chains,3 and highly organized in crystals4 or supramolecular assemblies;5 such systems have been reported to exhibit meso- to macroscopic dynamic behavior upon external stimuli. Among them, molecular rotors based on overcrowded alkenes are representative molecular machines that have been used in various systems.1–6 An overcrowded alkene stores strain energy because of the steric crowding around a sp2 carbon–carbon double bond, which leads to rotation or twisting of the movable units around the double bond upon photoirradiation or heating.7 However, it still remains difficult to propagate the strain energy through macromolecular materials in a cooperative manner. In this context, we surmised that if overcrowded alkenes can be connected directly with two covalent bonds, they would form a helically-twisted ladder macromolecule8 so that the twisting motion of the movable units could induce the expansion and contraction of the macromolecules (Fig. 1a and Fig. S1, ESI†).
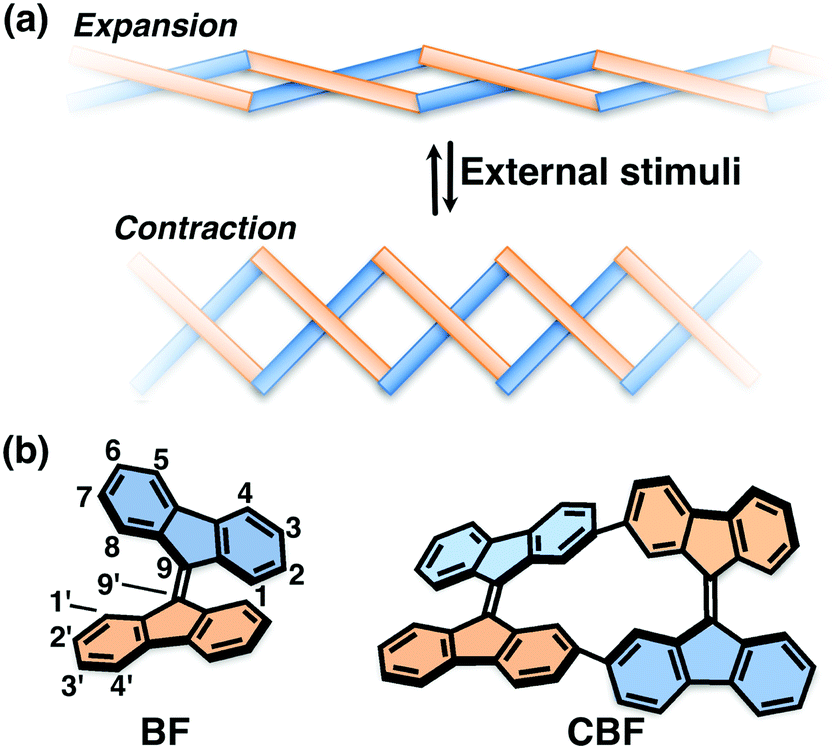 | ||
| Fig. 1 (a) Schematic illustration of a stimuli responsive ladder-shaped macromolecule. (b) Structures of BF and CBF. | ||
As a proof of concept, we designed a ladder-shaped 9,9′-bifluorenylidene cyclic dimer (CBF) as shown in Fig. 1b. 9,9′-Bifluorenylidene (BF) is one of the simplest overcrowded alkenes composed of hydrocarbons, and exhibits dynamic twisting motion around a C9–C9′ double bond upon heating and redox.9 Associated with the torsional angle between the two fluorene π-planes in BF, its optoelectronic properties and spin states change remarkably.10 Therefore, BF recently has attracted attention as a building block for organic photovoltaics or singlet fission materials.11BF-based molecules have good solubility in common organic solvents even in the absence of substituents, which is also a great advantage from the viewpoints of synthetic accessibility and solution processability. We established the synthetic method of newly designed CBF and studied its structure by single-crystal X-ray analysis. The structural change upon external stimuli such as redox is discussed based on quantum chemical calculations, and electrochemistry. We also studied the carrier-transport properties of the thin films of CBF.
The synthetic method of CBF is shown in Scheme 1. Precursor 1 was obtained by the homo-dimerization of 2-bromo-9-fluorenone through the formation of the corresponding thioketone by Lawesson's reagent.11b The NMR spectrum of 1 (Fig. S2, ESI†) indicates the existence of two conformers: 1cis in which two bromine groups are on the same side and 1trans in which they are on the opposite side. The ratio between 1cis and 1trans is calculated to be 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in chloroform-d (CDCl3) at 298 K. The major conformer is 1trans probably because it is less sterically hindered. The thermal cis–trans isomerization of 1 was not observed in the range from 298 to 363 K (Fig. S3, ESI†). The Yamamoto coupling reaction of 1 (a mixture of 1cis and 1trans) in the presence of bis(1,5-cyclooctadiene)nickel(0): Ni(COD)2 and ligands in dimethylformamide (DMF) and toluene afforded a macrocyclic 9,9′-bifluorenylidene dimer (CBF) and the BF-based oligomers (Scheme 1).12,13CBF was purified by silica-gel column chromatography and recycling gel permeation chromatography (GPC) using CHCl3 as an eluent with an isolated yield of 19%. CBF is presumably a coupling product of 1cis themselves. Considering that the reactant was a mixture of 1cis and 1trans and the cis–trans isomerization might not occur under the reaction conditions (363 K), the reaction yield of CBF seems to be reasonable. The experimental details and characterization data are provided in the ESI.†CBF is soluble in common organic solvents such as dichloromethane (CH2Cl2) and toluene and fully characterized (Fig. 2 and Fig. S4, S5 for NMR and Fig. S6 for MALDI/TOF–MS, ESI†).
:4 in chloroform-d (CDCl3) at 298 K. The major conformer is 1trans probably because it is less sterically hindered. The thermal cis–trans isomerization of 1 was not observed in the range from 298 to 363 K (Fig. S3, ESI†). The Yamamoto coupling reaction of 1 (a mixture of 1cis and 1trans) in the presence of bis(1,5-cyclooctadiene)nickel(0): Ni(COD)2 and ligands in dimethylformamide (DMF) and toluene afforded a macrocyclic 9,9′-bifluorenylidene dimer (CBF) and the BF-based oligomers (Scheme 1).12,13CBF was purified by silica-gel column chromatography and recycling gel permeation chromatography (GPC) using CHCl3 as an eluent with an isolated yield of 19%. CBF is presumably a coupling product of 1cis themselves. Considering that the reactant was a mixture of 1cis and 1trans and the cis–trans isomerization might not occur under the reaction conditions (363 K), the reaction yield of CBF seems to be reasonable. The experimental details and characterization data are provided in the ESI.†CBF is soluble in common organic solvents such as dichloromethane (CH2Cl2) and toluene and fully characterized (Fig. 2 and Fig. S4, S5 for NMR and Fig. S6 for MALDI/TOF–MS, ESI†).
 | ||
| Scheme 1 Synthesis of CBF and the corresponding oligomers. Reagents and conditions: (a) Lawesson's reagent, toluene, reflux, overnight, and then copper powder, toluene, reflux, 3 h; (b) Ni(COD)2, 2,2′-bipyridyl, COD, DMF, and toluene, 48 h, 363 K. | ||
 | ||
| Fig. 2 1H NMR spectra of (a) CBF and (b) BF in CD2Cl2 at 298 K. | ||
The 1H NMR spectra of CBF and BF are shown in Fig. 2. The seven sets of sharp peaks of CBF indicate the symmetric structure of CBF. No significant changes were observed in the variable-temperature 1H NMR spectra of CBF (Fig. S7, ESI†). Each proton signal of CBF was assigned together with the two-dimensional correlated NMR spectrum (1H, 1H COSY) of CBF (Fig. S4, ESI†). The proton signals of Ha, Hb, Hc, and Hd of CBF appeared at almost identical positions to those of BF because of similar chemical environments. New aromatic proton peaks of He, Hf, and Hg were observed for CBF. The proton peak of He (8.78 ppm) is shifted downfield compared with that of Ha (8.36 ppm) due to the neighboring aromatic rings.
The structure of CBF was determined by single-crystal X-ray structural analysis (Fig. 3 and Table S1, ESI†). The single crystal of CBF was obtained by slow vapor diffusion of acetonitrile into a toluene solution of CBF at room temperature. The two central carbon–carbon bonds (C9–C9′) of CBF are 1.336(8) Å, indicating their double bond character. The both torsional angles around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of each 9,9′-bifluorenylidene unit (C8–C9–C9′–C8′) in CBF are 33° (Fig. 3a). These values are comparable with that of BF (32°),14 and also with that (34°) obtained from the density functional theory (DFT) calculation of CBF (Fig. S8, ESI†). Four independent CBF molecules exist in one unit cell, and a pair of enantiomers CBF-(P) and CBF-(M)15 are alternately packed in a perpendicular fashion (Fig. 3b and c). The adjacent fluorene π-planes of CBF stack with typical π–π stacking distances (3.328 Å as a minimum value).10 We could obtain only tiny bar-shaped crystals, probably owing to the rapid interconversion between CBF-(P) and CBF-(M) in solution. Indeed, we were unable to separate these enantiomers by using chiral high-performance liquid chromatography (HPLC) under more than 50 different conditions.
C bond of each 9,9′-bifluorenylidene unit (C8–C9–C9′–C8′) in CBF are 33° (Fig. 3a). These values are comparable with that of BF (32°),14 and also with that (34°) obtained from the density functional theory (DFT) calculation of CBF (Fig. S8, ESI†). Four independent CBF molecules exist in one unit cell, and a pair of enantiomers CBF-(P) and CBF-(M)15 are alternately packed in a perpendicular fashion (Fig. 3b and c). The adjacent fluorene π-planes of CBF stack with typical π–π stacking distances (3.328 Å as a minimum value).10 We could obtain only tiny bar-shaped crystals, probably owing to the rapid interconversion between CBF-(P) and CBF-(M) in solution. Indeed, we were unable to separate these enantiomers by using chiral high-performance liquid chromatography (HPLC) under more than 50 different conditions.
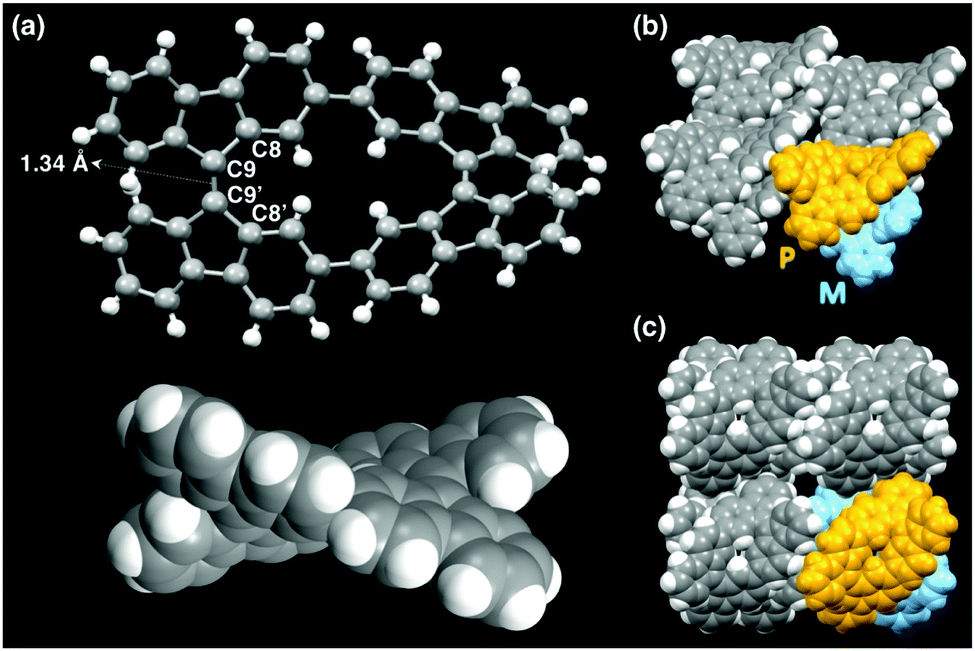 | ||
| Fig. 3 (a) Crystal structure of CBF-(M). (b) and (c) Molecular packing views, in which CBF-(P) and CBF-(M) are colored in yellow and blue, respectively. | ||
Next we studied the optical properties of CBF and BF. Fig. 4a shows the UV-vis absorption spectra of CBF and BF in CH2Cl2 at 298 K. Both CBF and BF exhibited intense absorption in the UV-vis region. The broad absorption of CBF over 400 nm can be fitted with two Gaussian-shaped lines at 438 nm and 483 nm (shoulder: sh), while the absorption band of BF can be fitted with a symmetric Gaussian-shaped line at 458 nm. These spectral features are well reproduced by time-dependent density functional theory (TDDFT) calculations (Fig. S9, ESI†), although the theoretical value overestimates the experimental one by ca. 0.3 eV. The molecular orbitals of CBF (HOMO−1, HOMO, LUMO, and LUMO+1) indicate that there are nodes between the two 9,9′-bifluorenylidene units, and thus there is little interaction between them in the neutral state (Fig. 4b). The HOMO−1 and HOMO, as well as the LUMO and LUMO+1, are almost degenerate, which results in several transitions over 400 nm. Emissions of CBF and BF were very weak in solution and in the film states even at low temperature.16
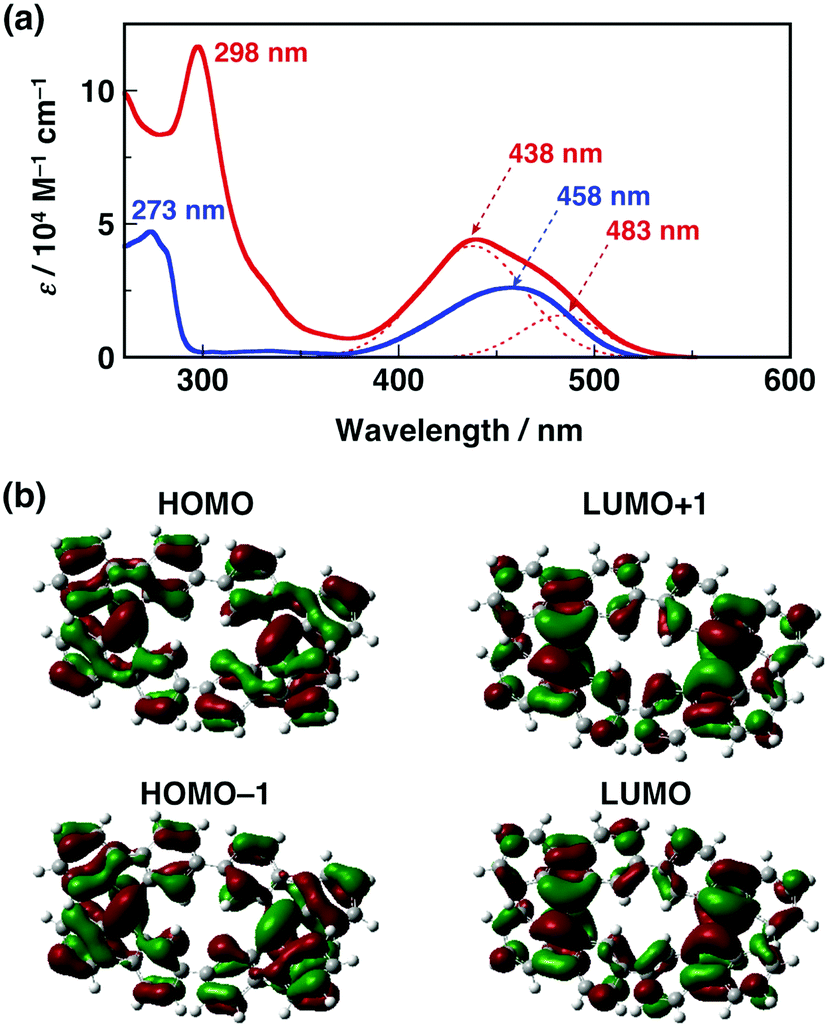 | ||
| Fig. 4 (a) UV-Vis spectra of CBF (red line) and BF (blue line) with simulated Gaussian curves (red dashed lines) in CH2Cl2 (10 μM) at 298 K. (b) Molecular orbitals of CBF calculated by DFT at the M06-2×/6-31G** level. | ||
We next studied the electronic properties of CBF and BF by means of electrochemistry. The cyclic voltammogram (CV) and the differential pulse voltammogram (DPV) of CBF and BF were measured under deaerated conditions in CH2Cl2 at 298 K. As shown in Fig. 5, both CBF and BF showed reversible first reduction processes. The potential for the first reduction process of BF was observed at −1.35 V, which is attributable to the formation of the radical anion (BF˙−). The initial reduction process of CBF is split into two discrete one-electron steps. The DPV of CBF can be simulated by two Gaussian-shaped reduction waves: two reduction potentials at −1.49 V and −1.62 V were obtained with the same current intensity (Fig. S10, ESI†). The first reduction potential of CBF is positively shifted compared to that of BF by 0.07 V, indicating that CBF˙− is more stabilized as a result of the electronic interaction between the two 9,9′-bifluorenylidene units. In contrast, the second reduction potential of CBF (−1.62 V) is negatively shifted than the first reduction potentials of CBF and BF, owing to the electrostatic repulsion between the two 9,9′-bifluorenylidene reduced species. Thus, the two 9,9′-bifluorenylidene units connected in a ladder fashion interact with each other upon reduction. It is known that the one electron reduction leads to twisting of BF because of the strain release and steric hindrance (Fig. 1).10,11a The DFT calculations strongly indicate the structural changes in CBF upon reduction; the torsional angles around the CC bond of each 9,9′-bifluorenylidene unit in CBF are 34°, while those in CBF˙− and CBF2− are 36° and 39°, respectively, as shown in Fig. S11 (ESI†). The initial oxidation process of CBF exhibited broad redox waves at around +0.94 V, while that of BF exhibited a reversible one-electron step at +0.90 V. The broad redox waves of CBF might result from the electronic interaction between the two 9,9′-bifluorenylidene units. Indeed, the DPV of CBF measured at a slower scan rate of 50 mV s−1 exhibited the splitting of oxidation waves as well as reduction waves (Fig. S12, ESI†). All optical and electronic data of CBF and BF are summarized in Table 1. The HOMO and LUMO of CBF were calculated to be −5.74 eV and −3.31 eV, which are reasonable energy levels for electronic device fabrication.18
 | ||
| Fig. 5 CV (top) and DPV (bottom) of (a) CBF and (b) BF in deaerated CH2Cl2 (0.5 mM) containing 0.1 M TBAPF6 with a scan rate of 100 mV s−1. | ||
Finally, we evaluated carrier mobilities of thin films of CBF and BF by means of space charge limited current (SCLC) measurements.18,19 Thin films of CBF and BF were fabricated using spin-coating techniques. The smoothness of the thin films was highly dependent on the solvents used to cast. A thin film of CBF (50 nm thick) cast from chlorobenzene was smooth and exhibited a hole mobility of 1.3 × 10−5 cm2 V−1 s−1 (Fig. S13, ESI†). In contrast, the thin films of CBF cast from o-dichlorobenzene were nonuniform crystalline films which precluded their SCLC measurements. Such a difference in the film morphologies and carrier mobilities of CBF may correlate with the variation in the crystal packing structures. Indeed, a single crystal of CBF grown in an o-dichlorobenzene solution includes the solvent molecule that inhibits π-stacking between the CBF molecules (Fig. S14 and Table S2, ESI†),20 which is significantly different from that grown in toluene or CHCl3 solutions. On the other hand, the smooth thin films of BF were not obtained by spin-coating chlorobenzene solutions. The hole mobility of a thin film of BF (100 nm thick) cast from CHCl3 was 7.1 × 10−5 cm2 V−1 s−1 (Fig. S15, ESI†), which is in the same order as that of CBF.
Conclusions
We successfully synthesized a novel ladder-shaped bis(9,9′-bifluorenylidene) dimer (CBF) by the Yamamoto coupling reaction. Single-crystal X-ray analysis revealed the twisted and cyclic structure of CBF. A rapid isomerization between the enantiomeric pairs of CBF-(P) and CBF-(M) occurred in solution, while they were packed alternatively in a perpendicular fashion in the crystal. The double helical structure of CBF changed upon redox, as a result of the electronic interactions between the neighboring 9,9′-bifluorenylidene units. The CBF exhibited hole transporting properties with a value of 1.3 × 10−5 cm2 V−1 s−1. Thus, our concept presented here would provide a versatile strategy toward solution processable, synthetically feasible, and optoelectronically functionable three-dimensional materials. Preparation of the corresponding ladder-shaped cyclic oligomers and polymers based on twisted overcrowded alkenes is currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr Takeshi Yasuda (NIMS) for fruitful discussion, DAICEL Corporation for the trial on chiral HPLC experiments. Mr Toshikane Suzuki (NIMS) is greatly acknowledged for his contribution in the initial stages of this work. This research was supported by Grants-in-Aid for Scientific Research on Innovative Areas “π-System Figuration” from JSPS (26102009 for M. T. and 26102015 for M. S.) and on Challenging Research (Exploratory, JP17K19116 for Y. M.), the Green Asia program (Kyushu University) for A. B., the MEXT “NIMS Molecule and Material Synthesis Platform” program, and the Materials Analysis Station of NIMS.Notes and references
- (a) A. Cnossen, W. R. Browne and B. Feringa, in Molecular Machines and Motors: Recent Advances and Perspectives, ed. A. Credi, S. Silvi and M. Venturi, Springer International Publishing, Cham, Switzerland, 2014, vol. 354, pp. 139 Search PubMed; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed.
- (a) R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS PubMed; (b) H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149 CrossRef CAS PubMed , and references therein.
- (a) T. J. White and D. J. Broer, Nat. Mater., 2015, 14, 1087 CrossRef CAS PubMed , and references therein; ; (b) K. Iwaso, Y. Takashima and A. Harada, Nat. Chem., 2016, 8, 625 CrossRef CAS PubMed; (c) J. T. Foy, Q. Li, A. Goujon, J. R. Colard-Itte, G. Fuks, E. Moulin, O. Schiffmann, D. Dattler, D. P. Funeriu and N. Giuseppone, Nat. Nanotechnol., 2017, 12, 540 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174 CrossRef CAS PubMed , and references therein.
- (a) A. Goujon, G. Du, E. Moulin, G. Fuks, M. Maaloum, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2016, 55, 703 CrossRef CAS PubMed; (b) A. Takai, T. Kajitani, T. Fukushima, K. Kishikawa, T. Yasuda and M. Takeuchi, J. Am. Chem. Soc., 2016, 138, 11245 CrossRef CAS PubMed; (c) J. Chen, F. K.-C. Leung, M. C. A. Stuart, T. Kajitani, T. Fukushima, E. van der Giessen and B. L. Feringa, Nat. Chem., 2018, 10, 132 CrossRef CAS PubMed.
- (a) T. van Leeuwen, A. S. Lubbe, P. Štacko, S. J. Wezenberg and B. L. Feringa, Nat. Rev. Chem., 2017, 1, 0096 CrossRef; (b) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060 CrossRef CAS PubMed , and references therein.
- (a) P. U. Biedermann, J. J. Stezowski and I. Agranat, Chem. – Eur. J., 2006, 12, 3345 CrossRef CAS PubMed; (b) D. Lenoir, P. J. Smith, J. F. Liebman, A. Nicolaides, R. P. Johnson and K. M. Konrad, in Strained Hydrocarbons, Wiley-VCH, Weinheim, Germany, 2009, pp. 103 Search PubMed; (c) C. Wentrup, M. J. Regimbald-Krnel, D. Müller and P. Comba, Angew. Chem., Int. Ed., 2016, 55, 14600 CrossRef CAS PubMed.
- (a) A. D. Schlüter, M. Löffler and V. Enkelmann, Nature, 1994, 368, 831 CrossRef; (b) M. J. Marsella, Acc. Chem. Res., 2002, 35, 944 CrossRef CAS PubMed; (c) T. Ikeda, S. Shinkai, K. Sada and M. Takeuchi, Tetrahedron Lett., 2009, 50, 2006 CrossRef CAS; (d) S. Ito, S. Hiroto, S. Lee, M. Son, I. Hisaki, T. Yoshida, D. Kim, N. Kobayashi and H. Shinokubo, J. Am. Chem. Soc., 2015, 137, 142 CrossRef CAS PubMed; (e) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed; (f) M. Rickhaus, M. Mayor and M. Juricek, Chem. Soc. Rev., 2016, 45, 1542 RSC; (g) F. Ishiwari, N. Takeuchi, T. Sato, H. Yamazaki, R. Osuga, J. N. Kondo and T. Fukushima, ACS Macro Lett., 2017, 6, 775 CrossRef CAS.
- (a) I. Agranat and Y. Tapuhi, J. Org. Chem., 1979, 44, 1941 CrossRef CAS; (b) R. Korenstein, K. A. Muszkat and S. Sharafy-Ozeri, J. Am. Chem. Soc., 1973, 95, 6177 CrossRef CAS; (c) P. U. Biedermann, J. J. Stezowski and I. Agranat, Eur. J. Org. Chem., 2001, 15 CrossRef CAS; (d) G. L. Eakins, M. W. Cooper, N. N. Gerasimchuk, T. J. Phillips, B. E. Breyfogle and C. J. Stearman, Can. J. Chem., 2013, 91, 1059 CrossRef CAS.
- A. Takai, D. J. Freas, T. Suzuki, M. Sugimoto, J. Labuta, R. Haruki, R. Kumai, S. Adachi, H. Sakai, T. Hasobe, Y. Matsushita and M. Takeuchi, Org. Chem. Front., 2017, 4, 650 RSC.
- (a) F. G. Brunetti, X. Gong, M. Tong, A. J. Heeger and F. Wudl, Angew. Chem., Int. Ed., 2010, 49, 532 CrossRef CAS PubMed; (b) H. U. Kim, J.-H. Kim, H. Suh, J. Kwak, D. Kim, A. C. Grimsdale, S. C. Yoon and D.-H. Hwang, Chem. Commun., 2013, 49, 10950 RSC; (c) C.-Y. Chiu, H. Wang, F. G. Brunetti, F. Wudl and C. J. Hawker, Angew. Chem., Int. Ed., 2014, 53, 3996 CrossRef CAS PubMed; (d) C. Li, Z. P. Mao, H. J. Chen, L. P. Zheng, J. Y. Huang, B. Zhao, S. T. Tan and G. Yu, Macromolecules, 2015, 48, 2444 CrossRef CAS; (e) K. Rakstys, M. Saliba, P. Gao, P. Gratia, E. Kamarauskas, S. Paek, V. Jankauskas and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2016, 55, 7464 CrossRef CAS PubMed; (f) S. Kawata, J. Furudate, T. Kimura, H. Minaki, A. Saito, H. Katagiri and Y.-J. Pu, J. Mater. Chem. C, 2017, 5, 4909 RSC; (g) H. Minaki, S. Kawata, J. Furudate, A. Saito, H. Katagiri and Y.-J. Pu, Chem. Lett., 2017, 46, 1126 CrossRef CAS.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed.
- MALDI/TOF–MS and GPC showed signals corresponding to BF-based oligomers such as a trimer and a tetramer, although they could not be isolated.
- J.-S. Lee and S. C. Nyburg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 560 CrossRef.
- CBF-(P) indicates an enantiomer in which two 9,9′-bifluorenylidene units are arranged in a right-handed helical manner, while CBF-(M) in a left-handed helical manner.
- J. Conyard, I. A. Heisler, W. R. Browne, B. L. Feringa, S. Amirjalayer, W. J. Buma, S. Woutersen and S. R. Meech, J. Phys. Chem. A, 2014, 118, 5961 CrossRef CAS PubMed.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551 CrossRef CAS.
- T. Suzuki, H. Okada, T. Nakagawa, K. Komatsu, C. Fujimoto, H. Kagi and Y. Matsuo, Chem. Sci., 2018, 9, 475 RSC.
- P. E. Burrows, Z. Shen, V. Bulovic, D. M. McCarty, S. R. Forrest, J. A. Cronin and M. E. Thompson, J. Appl. Phys., 1996, 79, 7991 CrossRef CAS.
- The supplementary crystallographic data of CBF that includes o-dichlorobenzene have been deposited with the Cambridge Crystallographic Data Centre, accession number CCDC 1817815†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis, characterization, NMR chart, additional physicochemical data and pertinent calculation results. CCDC 1590344 and 1817815. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00583k |
| This journal is © the Partner Organisations 2018 |