
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleobase deaminases: a potential enzyme system for new therapies
Vandana Gaded
and
Ruchi Anand
 *
*
Department of Chemistry, Indian Institute of Technology Bombay, Mumbai, Maharashtra 400076, India. E-mail: ruchi@chem.iitb.ac.in; Fax: +91-22-2576-7152; Tel: +91-22-2576-7165
First published on 28th June 2018
Abstract
Nucleobase deaminases are essential enzymes that are involved in the catabolic pathway and stringently regulate the concentration of the nucleobase derivative pool, which is paramount for nucleotide recycling. This review presents an overview of the structure, function and mechanism of CDA deaminases and their potential as enzyme systems for the development of new antimicrobial therapies. The evolutionary divergence of human nucleobase deaminases with respect to bacterial enzymes has been used as a central theme towards the development of strategies for potential drug targets. Especially, differences in their tertiary fold, active site architectures and mechanisms of regulation have been highlighted in this study. Overall, deaminases present a unique opportunity as drug targets because of their functional plasticity and fidelity.
 Vandana Gaded | Vandana Gaded is currently a PhD student under Prof. Ruchi Anand in the Department of Chemistry, Indian Institute of Technology Bombay (IITB), Mumbai. She was born in Ambernath (Mumbai), Maharashtra. She obtained her Bachelor's degree from Smt. C.H.M. College and her Master's degree from Khalsa College, Mumbai. Her research interests include structural biology, biochemistry and enzymology. Her PhD work emphasizes on understanding the structure–function relationships of nucleobase deaminases, which form an important part of the nucleotide metabolic pathway and are paramount to the survival of organisms. |
 Ruchi Anand | Ruchi Anand is an Associate Professor in the Department of Chemistry at the Indian Institute of Technology Bombay (IITB), India. She obtained her PhD degree from the Department of Chemistry and Chemical Biology, Cornell University. She then pursued postdoctoral research at Memorial Sloan Kettering Cancer Center, New York and UPENN, Philadelphia. She is a recipient of the RSC Madam Curie Award for Women Scientists and has been a member of the Editorial Advisory Board of ACS Sensors since 2016. Her research group at IIT Bombay focuses on gaining structural and functional insights into proteins involved in nucleobase deamination, antimicrobial resistance and biosynthetic pathways by employing a combination of X-ray crystallography, biochemistry and biophysical tools. A part of her research also focuses on bacterial transcription factors that has paved the way to structure-guided development of biosensors for aromatic pollutants. |
1. Introduction
Drug resistance is increasing worldwide at an alarming rate.1 There is a constant surge of pathogenic strains that are becoming resistant to even the newest lines of antibiotics.2 It is projected if no action is taken to circumvent this silent epidemic, more than 300 million deaths will be attributed to resistant bacteria by 2050.1,3 One strategy that can be adopted to combat drug resistance is to develop new therapies that pathogens have never encountered and that are therefore likely to be effective.4 An efficient way to undertake this endeavour is to search for new pathways that are essential for the survival of bacteria but differ evolutionarily between humans and pathogens.5,6 Towards this goal, enzymes involved in regulating the overall nucleobase pool are an enticing prospect.7 The regulation of purine and pyrimidine nucleotides is central to organisms because these nucleotides are prime energy carriers and form the building blocks of nucleic acids.5 In the cell, the concentration of the nucleobase pool is under stringent control because this pool is utilized for synthesis of the genetic materials DNA and RNA.8,9 Living cells have dedicated pathways to synthesize biomolecules via de novo biosynthetic pathways; however, these pathways require enormous amounts of energy input.10 Hence, to meet this increased energy demand, especially for nucleic acid synthesis, most cells have salvage and catabolic pathways to recover nucleobase compounds that are obtained from diet or released during nucleic acid degradation.11–13 These pathways work in parallel to the biosynthetic route and help maintain the total cellular purine and pyrimidine derivative pool.14,15 In most cases, these pathways are essential yet evolutionarily distinct between eukaryotes and prokaryotes.16 The differences in substrate specificity, overall structure and mechanism, etc. across the phyla can therefore be exploited towards development of specific drug targets.12,16Nucleobase deaminases form an important part of the catabolic pathway and aid the salvage of nucleobases.5 Nucleobase deamination is catalysed via metal-assisted hydrolysis of amino groups of heterocyclic bases in nucleosides, nucleotides and nucleic acids.12,17 Nature has evolved a variety of deaminases; based on their structural folds, these enzymes are categorized into two major sub-groups: the amidohydrolase (AHS) and cytidine deaminase-like (CDA) superfamilies.9,12 Although both these families use metal-assisted deamination as their primary mode of catalysis, they show extreme sequence and structural divergence. The AHS deaminases have a distorted triosephosphate isomerase (TIM) fold and in some cases contain multiple metal centers, such as zinc or iron.18–20 Meanwhile, the CDA superfamily members have a relatively compact alpha–beta–alpha layered fold with 3 helices and 4 strands in the order 2134.9 Moreover, they display a ubiquitous metal binding signature comprising residues HXE and PCXXC, which is a hallmark of this family and is responsible for chelating the zinc ion that drives the deamination reaction.21 Because of this evolutionary divergence, these enzymes present unique opportunities to develop drugs at the levels of both sequence and three-dimensional structure. For example, cytosine deaminases are exclusively present in fungi and prokaryotes and are completely absent in higher organisms.22,23 Yeast cytosine deaminase (yCD), which belongs to the CDA family, is an effective prodrug system that is widely used in cancer therapy. The yCD gene therapy approach has been very successful and is currently being employed in the clinic, especially for prostate cancer.24 Overall, due to the stark difference between the primary folds of human and bacterial deaminases, nucleobase deaminases have emerged as attractive drug targets.23,25
Enzymes from the CDA superfamily are widely spread across bacteria and are the focus of this review. In particular, the CDA superfamily is further divided into two major divisions based on the presence of helix 4; it causes parallel stacking of strands 4 and 5 when present, while antiparallel stacking occurs in its absence.9 Interestingly, the CDA superfamily consists of both mononucleotide and nucleic acid deaminases. Mononucleotide deaminases, which are crucial in nucleotide metabolism, include guanine, cytosine, cytidine, dCMP (deoxycytidylate), riboflavin, and blasticidin.9 Meanwhile, nucleic acid deaminases, which perform in situ deamination (editing) of bases in polynucleotides, play a major role in gene diversity and antivirus defence.9 Functional analysis of the CDA superfamily enzymes has revealed that this family of enzymes are highly specific towards their substrates and exhibit very low degrees of promiscuity.12,26 This is not surprising, especially considering that the nucleobase pool maintained by these enzymes is utilized for incorporation of bases during replication; hence, nature has endowed them with fidelity.9,12 Further, structural analysis of the CDA superfamily shows that while maintaining the basic CDA fold, particular deaminases have tailored specificities by addition or removal of critical structural features near their active sites; therefore, substrate specificity is achieved by exhibiting structural plasticity.9,12,26 For example, cytosine deaminases have an extra helix that seals the active site, thus inhibiting entry of bulkier nucleobases.23 tRNA adenosine deaminases have an extended C-terminal helix that interacts with RNA, and guanine deaminases have a lid that opens and closes in each reaction cycle; its active site is tailored to accommodate only guanine.12,27
In recent years, with the dramatic increase in annotated protein sequences, the practice of employing bioinformatics tools has widely emerged to organize information based on function. The application of methods such as sequence similarity networks is being exploited widely to visualize functional trends across diverse protein superfamily members.28 Several groups, such as those of Raushel, Jacobson and Chen, have applied this method to determine the functions of important proteins and elucidate novel pathways.10,29,30 In addition, by employing this evolutionary cataloguing approach in the past few years, Anand and co-workers have constructed a sequence similarity network and successfully annotated the function and established the structure–function relationship of uncharacterised deaminases in the cog0590 network (Fig. 1).5,12 This review summarises the current status of select nucleobase deaminases. Their unique structural features, important catalytic residues and implications in mechanisms, along with a comparative analysis and their potential and current roles in therapeutics, are described.
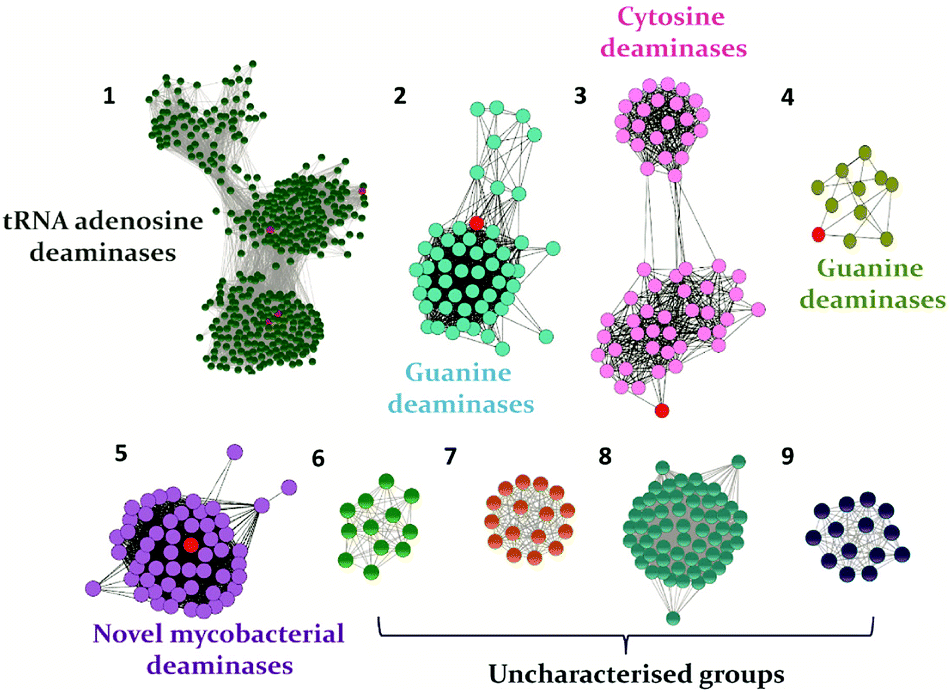 | ||
| Fig. 1 The sequence similarity network of the CDA superfamily cog0590 constructed at an e-value cutoff of 10−40. Groups are named based on the characterized protein (highlighted in red) present in each group. In each group, the nodes represent the proteins, while the edges represent the BLASTP linkages. | ||
2. Cytosine deaminase
Cytosine deaminase (CD) catalyzes the hydrolytic deamination of cytosine to uracil and ammonia (Scheme 1A).22,31,32 Cytosine deaminases in the CDA superfamily are mostly of fungal origin, while those in the AHS superfamily are of bacterial origin.25,33 From the distinct structural folds of the AHS and CDA superfamilies, it appears that bacterial and fungal CDs evolved separately. CDs in both superfamilies are capable of deaminating the prodrug 5-fluorocytosine to 5-fluorouracil (Scheme 1A). In the CDA superfamily, the best-studied system is yeast CD of cog0590 (Fig. 1). It deaminates 5-methylcytosine in addition to cytosine (Scheme 1A).23,31 However, unlike the CDA superfamily, CDs in the AHS superfamily are unable to deaminate 5-methylcytosine because of architectural differences in their active sites.34 Raushel and co-workers have shown that AHS CDs possess an additional aspartate residue, D314, in Escherichia coli CD (eCD) that prevents the deamination of 5-methylcytosine. Recent discoveries by this group have shown that a separate group of enzymes exists in the AHS superfamily which exclusively deaminate this epigenetic base.34 An important distinction among CDs is that they provide an important mechanism for pyrimidine salvage in microbes but are completely absent in mammalian cells.16,22 Because CDs are present only in fungi and prokaryotes, they are being explored for antimicrobial drug design and gene therapy applications.35,36 In particular, as mentioned earlier, they are used in suicide gene therapy (SGT) against cancer due to their ability to convert the antifungal agent 5-fluorocytosine (5FC) to the potent antimetabolite 5-fluorouracil (5FU).35–37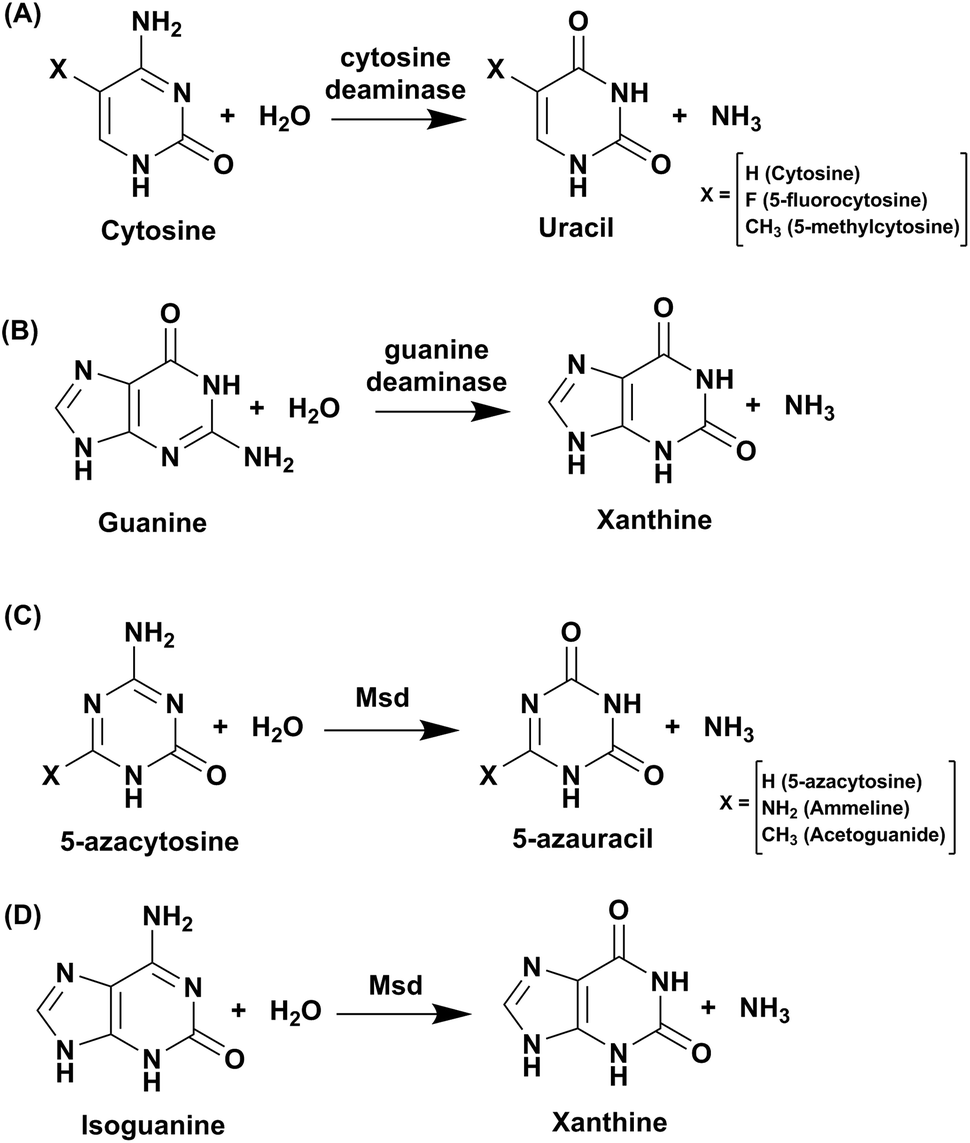 | ||
| Scheme 1 Reactions catalysed by CDA superfamily deaminases. | ||
2.1 Structural analysis of cytosine deaminase
In the CDA superfamily, only the crystal structure of yCD is available; it has been determined in complex with a transition state analogue by S. H. Liaw's group. yCD exists as a dimer that is formed by the head to tail association of two monomers and is stabilized by stacking interactions (Fig. 2A).23 The structure of each monomer consists of a central β-sheet sandwiched by α-helices on both sides. Analysis of the active site architecture reveals that the signature zinc binding motif is conserved and that zinc adopts a square bi-pyramidal geometry that is coordinated to histidine, two cysteines and a water molecule. The water molecule serves as a nucleophile in the deamination reaction.23 In the crystal structure of CD in complex with the analogue 2-hydroxypyrimidine, the ligand has been trapped as a hydrated adduct, 3,4-dihydrouracil (DHU), which acts as a transition state analogue.16,23 It has been shown that the C-terminal helix in each monomer governs the substrate specificity of CD, and residues from the C-terminus (W152, D155, and I156, present on helix 6) provide necessary interactions for the stabilization and binding of cytosine.16,23 Helix 6, on which these residues reside, is responsible for sealing the active site entrance and limits the pocket size so that larger substrates are unable to enter, thereby conferring specificity to cytosine (Fig. 2B).16,23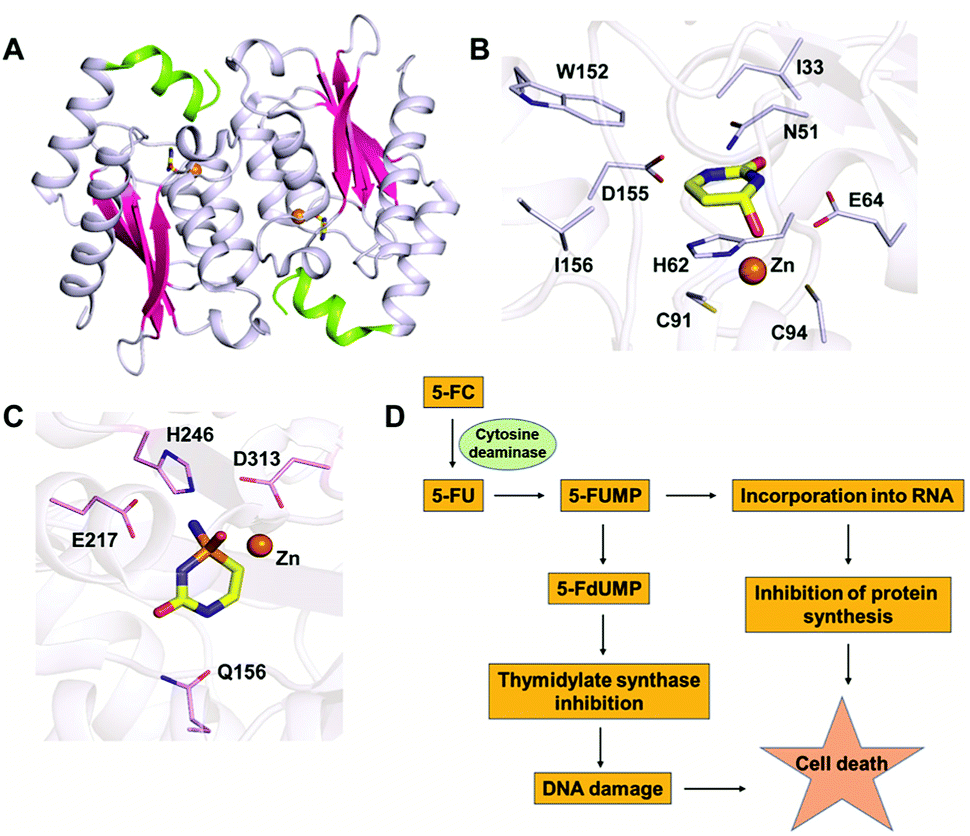 | ||
| Fig. 2 Structural analysis of CD. (A) Cartoon representation of yCD (PDB ID: 1UAQ). α-helices are shown in blue-white, while β-strands are shown in pink. The C-terminal helix 6, which serves as a gate, is highlighted in green. (B) Active site representation of yCD in complex with 3,4-dihydrouracil. (C) Active site of eCD of the AHS superfamily (PDB ID: 3O7U) in complex with the transition state inhibitor phosphonocytosine. Carbon atoms of the interacting residues are shown in blue-white in yeast CD and in light pink in E. coli CD. The carbon atoms of the ligand are shown in yellow in all the complexes, whereas zinc is shown as an orange sphere. (D) Schematic of the mechanism of action of 5-fluorocytosine (5-FC) in suicide gene therapy employing CD. | ||
2.2 Mechanism of cytosine deamination
Based on the structure of yCD with the transition state analog 3,4-dihydrouracil (DHU), Liaw's group proposed a cytosine deamination mechanism where yCD catalyzes the reaction via a tetrahedral intermediate. A conserved glutamate (E64) serves as a proton shuttle and initiates the reaction by abstracting a proton from the zinc-bound water.23 This leads to protonation of the N3 atom, followed by a decrease of the N3–C4 double bond character. In turn, the C4 atom becomes more susceptible to nucleophilic attack for formation of the tetrahedral intermediate. Finally, transfer of the proton to NH2 assisted via E64 leads to cleavage of the carbon–nitrogen bond (Scheme 2A).23 Additionally, using the structural information of yCD, H. Yan's group employed ONIOM calculations and proposed that cytosine deamination proceeds via a sequential mechanism.38 In their detailed analysis, they stated that the protonation of N3, which reduces the N3–C4 double bond order, first generates a positive charge in cytosine, which further decreases the distance between the zinc-bound water and C4 atom and thereby facilitates the reaction. This is followed by the nucleophilic attack of C4 by the zinc-coordinated hydroxide and, finally, the cleavage of the C4–N4 bond, resulting in the release of ammonia. In their proposed mechanism, protonation of the amino group in the tetrahedral intermediate and cleavage of the C4–N4 bond, which occur in a concerted fashion, represent the likely rate determining step for deamination.38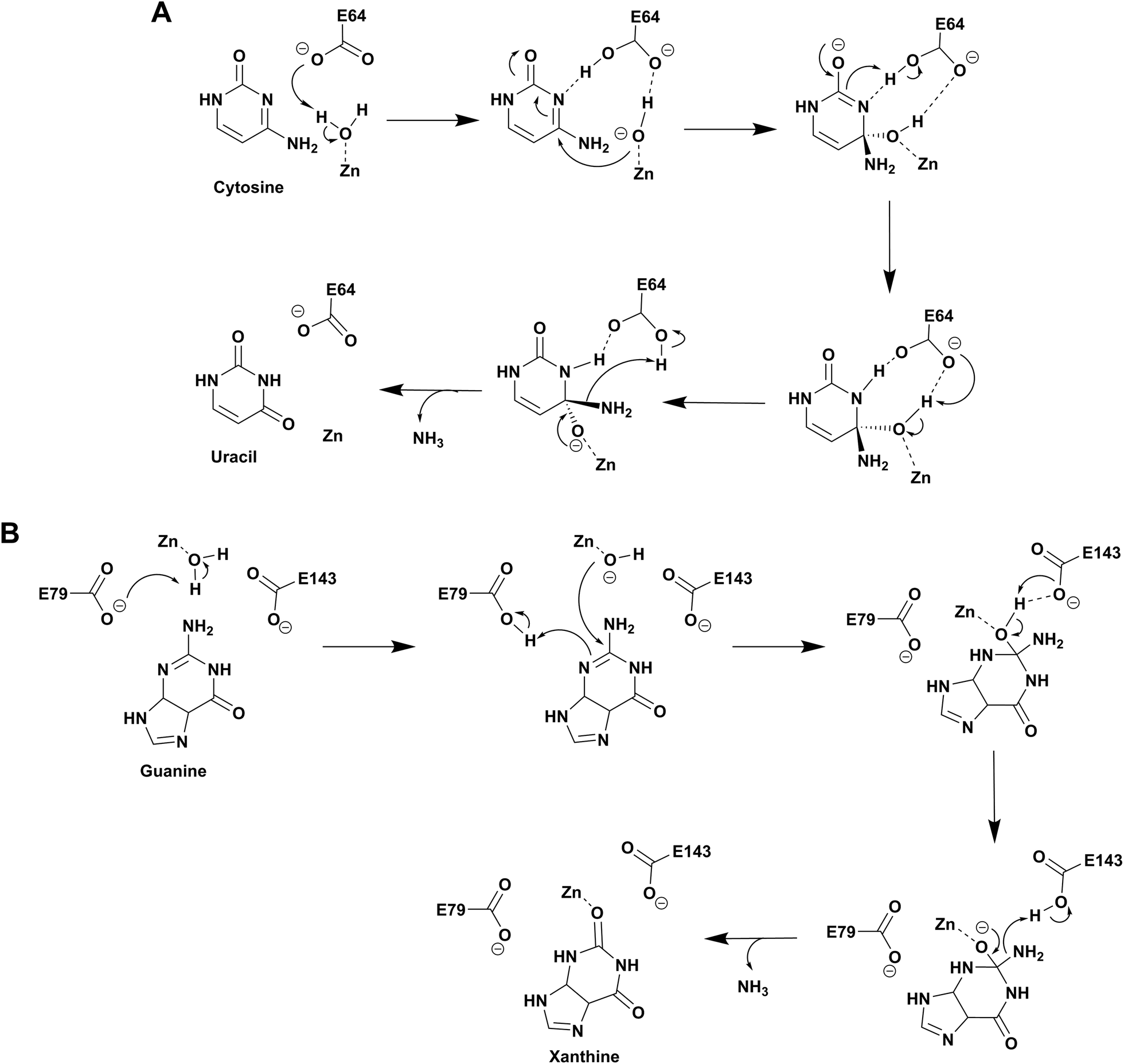 | ||
| Scheme 2 Proposed catalytic mechanism involving one glutamic acid residue in yCD (A) and two glutamic acid residues in NE0047 (B). | ||
As evident from the DHU bound structure and supporting data by Himo's group, in contrast to yCD, which requires only one carboxylate group (E64) as a proton shuttle, the mechanism of action of eCD of the AHS superfamily is seen to utilize two negatively charged amino acids for catalysis.22,39 Moreover, eCD requires either Fe2+ or Zn2+ for catalytic activity, exhibiting maximum efficiency with Fe2+ in the active site. In the case of eCD, the active site is present at the mouth of the enzyme barrel and contains Fe2+, which is coordinated with the hydrophilic water molecule. In the active site, H246 and D313 are poised to serve as general acid/base groups to activate the metal-bound water molecule and the amino leaving group.17,22 In addition, E217 is positioned to deliver the proton to the N3 atom of the pyrimidine ring. In addition, the carbamoyl moiety at N1/C2 is stabilized and hydrogen bonded with the side chain of Q156 (Fig. 2C).17,22 Based on density functional theory calculations, the proton transfer is proposed to be concerted in eCD. Meanwhile, in yCD, this process occurs in a stepwise fashion.39 Overall, the mechanistic and structural differences between the two CDs are the prime reason that they exhibit differential substrate specificity.
2.3 Application in suicide gene therapy
The ability of CDs to convert the prodrug 5-fluorocytosine (5-FC) into the potent antimetabolite 5-fluorouracil (5-FU) has gained wide applications in therapeutics. 5-FC is non-toxic; however, its conversion to 5-FU renders it extremely mutagenic. The 5-FC to 5-FU conversion is known to result in de-regulation of the nucleotide pool, leading to apoptosis.25,35–37 This therapy is being used by introducing CD in tumor cells via viral and non-viral gene delivery systems. Intracellularly, the mechanism by which this drug functions involves halting DNA synthesis. 5-FU is metabolized by endogenous enzymes to 5FdUMP (fluorodeoxyuridine monophosphate), an irreversible inhibitor of thymidylate synthetase, thereby restricting the production of dTMP (deoxythymidine monophosphate).40 This further results in downstream depletion of dTTP pools and stalls DNA synthesis, which ultimately leads to apoptosis (Fig. 2D). Introduction of CD in tumour cells after 5-FC treatment has been used as intra-tumour chemotherapy for several cancer cell types, such as colon, prostate and rectal.35,36,40 To increase the efficiency of CDs, several efforts have been made where random mutagenesis techniques were employed to create alternate versions that can be more effective toward catalysis of 5-FC.24,25 Specifically, in yCD, it has been shown that a yCD-triple mutant (A23L/I140L/V108I) and the yCD–D92E mutant provide enhanced 5-FC sensitivity.25,41 Moreover, studies by Fuchita and co-workers have corroborated these findings and shown that in both tumorigenic cells and mice models, modified versions of CD can be used in combination with 5-FC as an enhanced enzyme–prodrug combination for targeted cancer therapy.24 Both structural and mutagenesis experiments have successfully shown that mutations in CD can result in improved 5-FC activity both in vitro and in vivo, thereby opening doors to the possibility of engineering improved enzyme–prodrug systems.24,25,37 As a means of enhancing anticancer activity, several modifications to the basic 5-FC scaffold have been made towards controlled and site-specific delivery of the drug. Various prodrugs of 5-FU, such as capecitabine, ftorafur, and peptide-conjugated 5-FC, have been designed in order to achieve increased absorption and bioavailability.423. Guanine deaminase
Guanine deaminase (GD), also known as cypin, is another key zinc-dependent enzyme involved in the purine catabolic pathway that is essential for all organisms.12,43 GDs catalyse the conversion of guanine to xanthine and help to control guanine-containing metabolites (Scheme 1B). By producing xanthine and ammonia, this reaction irreversibly eliminates the guanine base from further re-utilization as a guanylate nucleotide in mammals.12,43 GDs adopt both CDA and AHS superfamily folds, with the human GD belonging to the latter family.12,44 GDs are essential enzymes for both bacterial and eukaryotic phyla. In mammals, GDs exhibit tissue-specific and development-dependent expression. Because of this evolutionary divergence and selective expression in mammalian tissues, GDs are used as potent markers in diagnosis.11,44 In higher eukaryotes, GDs play an important role in the development of neuronal morphology by regulating branching of dendrites. There are reports of the death of an infant who was found to be deficient in brain GD activity.11,45,46 In addition to its role in the maturing brain, GD is believed to be involved in proper liver function because increased levels of GD activity have been correlated with liver disease and transplant rejection. Due to its near absence in normal human serum, erythrocytes and lymphoid cells, serum GD activity in humans is among the most sensitive indicators of liver disease.11,45,46 Owing to the important role played by GDs in human biology, if GDs are to be exploited as drug targets, divergence between bacterial and human forms is paramount.11,45–47 Therapies where GD-based prodrug systems are employed include imidazole derivatives, such as 5-amino-4-imidazole carboxamide (AICA), and acyclic inhibitors, such as valaciclovir, aciclovir, and mercaptopurine. These options have been explored with the aim to enhance the bioavailability of purine antagonists such as 8-azaguanine, which in turn aids in chemotherapy of cancer.48,49 Here, we compare and contrast the structural and mechanistic aspects of GDs to explore them as crucial drug targets.3.1 Structural characteristics of guanine deaminase
The GDs of human and bacterial species are evolutionarily divergent; however, they share a common zinc-dependent catalytic deamination route. In humans, GDs possess the AHS fold, and the zinc-coordinating residues are nested within the TIM (β/α)8-barrel structural fold.47,50 Meanwhile, most bacterial GDs belong to the CDA superfamily and coordinate zinc via the catalytic signature, a hallmark of this superfamily.12,51 In CDA GDs, the protein structure resembles the conserved α–β–α layer possessed by the CDA superfamily members. The active site zinc is coordinated via a histidine, 2 cysteines and a catalytic water molecule.48,49 Within the CDA superfamily itself, GDs exhibit evolutionary variation. There are two evolutionarily divergent structures of GDs reported in cog0590 that fall into two separate groups in the sequence similarity network.12,51 The crystal structure of Bacillus subtilis GD (bGD), which belongs to group 2 (Fig. 1), is the first reported GD in this family; it exists as an intertwined dimer that exhibits domain swapping through the C-terminal.51 The active site is composed of residues from both monomers and is inter-subunit in nature. The domain swapping is reported to facilitate the recruitment of guanine-interacting residues in the active site and is responsible for structural stability and substrate specificity (Fig. 3A).51 In the case of the other GD, group 4, the only reported structure is a Nitrosomonas europaea GD (NE0047). In this case, domain swapping does not occur, and all the active site residues are contained in a single subunit. However, unlike bGD, the commonly found dimerization interface in CDA is further augmented in NE0047 by additional stabilization via an exclusive N-terminal β-sandwich that is unique to it.12 It appears that both bGD and NE0047 have a functionally important common feature of an extended dimerization interface that further intertwines the two subunits together (Fig. 3B) and possess total dimeric buried surface areas of 3900 Å2 and 3755 Å2, respectively, which are around 40% greater than those of other CDs.12,23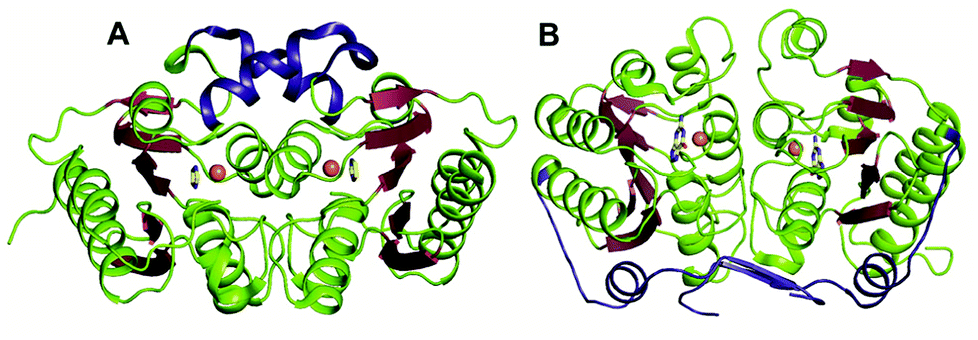 | ||
| Fig. 3 Evolutionary divergence and unique dimerization interfaces in GDs. (A) Cartoon representation of bGD (PDB ID: 1WKQ) and (B) NE0047 (PDB ID: 4HRQ). α-helices are shown in green and β-strands are shown in raspberry in both structures. The additional dimerization features are highlighted in blue. Zinc is shown as orange spheres and the bound ligands are shown in yellow. | ||
Among the two types of GDs, extensive mechanistic and structural work is available on NE0047; this system can therefore be employed as a model system for furthering drug development.12,26 Based on the crystal structure of NE0047 in complex with 8-azaguanine (the guanine analogue) and several mutagenesis and biochemical studies, a mechanism of guanine deamination has been proposed.12 Two glutamic acid residues (E79 and E143) have been demonstrated to be required for catalysis (Scheme 2B). In NE0047, E79 initiates the reaction by abstracting a proton from the zinc-bound water. This results in the formation of a nucleophilic hydroxide ion, which subsequently attacks the C2 atom of guanine. Here, E79 is proposed to stabilize this transition state and shuttle the same proton to the N3 atom of guanine. Further, E143 facilitates collapse of the tetrahedral intermediate via abstraction of the proton from the activated hydroxide that bridges the zinc ion with the C2 atom of guanine and delivers it to the amino group of guanine, resulting in the release of ammonia (Scheme 2B).12 A hallmark of GDs is that unlike yCD, they require two negatively charged residues, which facilitates proton transfer during the course of the reaction.12,23 A conserved asparagine residue (N66) additionally aids in neutralizing the negative charge built during the progress of the reaction. The overall active site architecture is hydrophobic in nature and is padded by the presence of two phenylalanine residues, F48 and F141, in NE0047.12 It appears that this mechanism is also conserved in AHS GDs. Although the placement of the residues in the active site is completely different from that of CDA GDs, the requirements appear to be met.47,50 Both GDs employ two negatively charged proton shuttles (E243 and D330 in human GD; E79 and E143 in NE0047), and the O6 oxygen of guanine is stabilized via amino acids containing the same head group (N66 in NE0047 and Q87 in human GD) (Fig. 4A and B).12,47 Therefore, to develop exclusive therapies that only target bacterial GDs, instead of targeting mechanism, the differences in their folds, substrate entries, and pocket architectures should be exploited. It has been observed that to further fine-tune their substrate specificity, bacterial GDs have developed an exclusive C-terminal loop that opens to allow entry of the substrate and closes during the progress of the reaction to prevent entry of solvent.12 This mode of action appears to be exclusive to bacterial GDs. Mutagenesis experiments where the C-terminal loop has been deleted show that removal of this loop results in complete loss of activity. The presence of the C-terminal lid is essential for deamination.12 This conformational change helps to achieve the metal-dependent high-energy intermediate, which is essential to facilitate the reaction, and the flap protects the active site from the surrounding solvent (Fig. 5A and B). Furthermore, this C-terminal loop is critical in limiting the size of the ligand and aids in ascertaining the fidelity and progress of the reaction.12,51 Inhibitors that target these exclusive features of bGD will be paramount in determining selectivity.
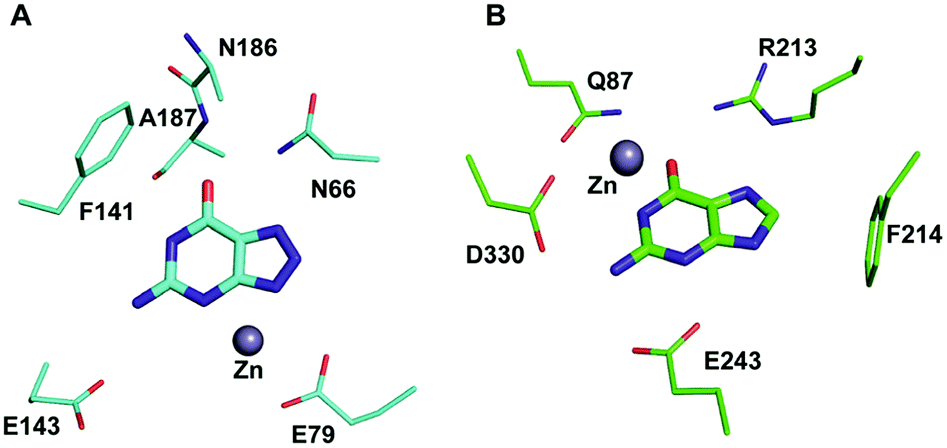 | ||
| Fig. 4 Architectural differences between human and bacterial GDs. (A) The active site of NE0047 in complex with 8-azaguanine (PDB ID: 4HRQ). (B) Active site representation of human GD (AHS superfamily) with guanine (PDB ID: 4AQL). Carbon atoms of the interacting residues and ligands are shown in cyan in NE0047 and in green in human GD. Zinc is represented as a grey sphere in both. | ||
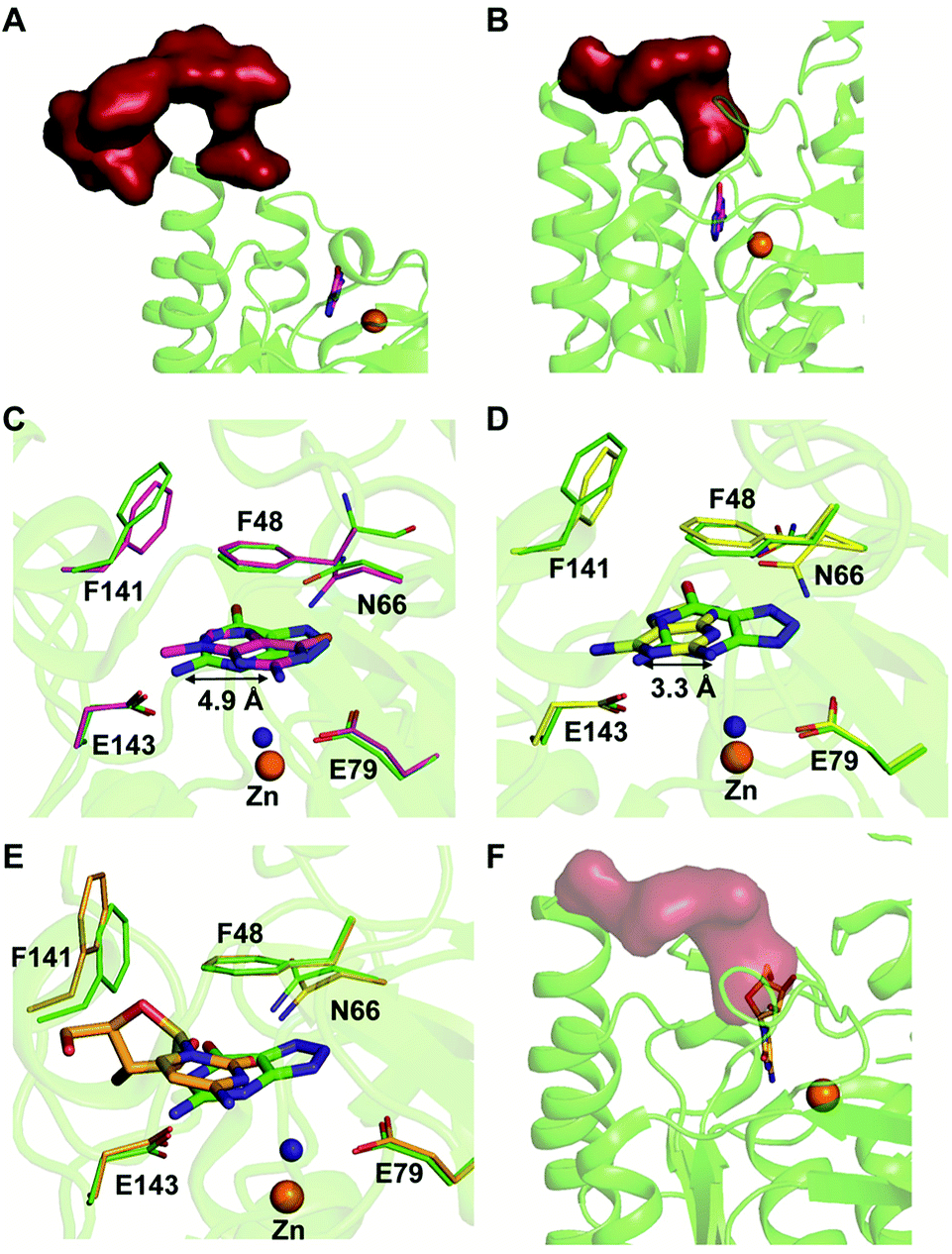 | ||
| Fig. 5 Mechanism of action of NE0047. (A) Open form of NE0047 in subunit A. (B) Closed form of NE0047 in subunit B. (C) Active site superposition of NE0047–8-azaguanine (green) and NE0047–9-methylguanine (magenta) complex. (D) Active site superposition of NE0047–8-azaguanine (green) and NE0047–2,6-diaminopurine (yellow) complex. (E) Active site superposition of NE0047–8-azaguanine (green) and NE0047–cytidine (blue-white) complex. (F) Model of the C-terminal loop showing the clash with the ribose moiety of cytidine in the NE0047–cytidine complex. | ||
3.2 Substrate specificity of guanine deaminase
Extensive work has been performed by Bitra et al. on NE0047 guanine deaminase, where a series of structures and activity data with guanine analogues have demonstrated that NE0047 is very sensitive to perturbation of the guanine scaffold.12,26 It has been shown that GDs can bind to a variety of nucleosides and nucleobase analogues but only catalyze deamination of guanine. Even small variations in the guanine scaffold, such as addition of a methyl group or an amino group, are not tolerated. A series of crystal structures in complex with guanine analogues and with representative nucleosides sheds light on the structural basis of substrate specificity.12,26 The structures of NE0047 in complex with guanine analogues (9-methylguanine and 2,6-diaminopurine) show that the GD active site is tailor-made to only allow guanine to fit in a catalytically competent conformation.26 In the case of the NE0047–9-methylguanine complexed structure (PDB ID: 4LC5), it is apparent that the ligand has been re-oriented by approximately 90°; this drastic change has caused the amino group (to be potentially deaminated) to shift by 4.9 Å in comparison to the original position adopted in the NE0047–8-azaguanine complexed structure (PDB ID: 4HRQ) (Fig. 5C).26 Similarly, in the case of NE0047–2,6-diaminopurine complex (PDB ID: 4LCP), a 3.3 Å displacement of the amino group in comparison to the original position results in complete loss of activity (Fig. 5D).26 Although guanine analogues can fit in the active site of NE0047, as evident from the crystal structures, the failure to adopt a conformation where the proton shuttles can facilitate the reaction renders them inactive. It appears that the active site is tailored in such a compact fashion that any small variation disturbs the arrangement, leading to complete loss of activity, and improper orientation causes the analogues to act as inhibitors.26 Moreover, it was observed that the cross talk between the two dimers in the GD was also destroyed and that the modified ligands bound preferentially to only one of the active sites, thereby locking the enzyme in an unproductive state.12,26 Studies reveal that in addition to the proper positioning of the ligand in the active site, closure of the C-terminal loop is also essential for catalysis.12 This became more apparent when X-ray structures with cytidine (PDB ID: 4LD2) highlighted that the amino group to be deaminated was properly positioned in the active site but still was unable to be turned over by the GD.26 Closer examination revealed that the bulky size of cytidine impeded the closure of the C-terminal loop due to the apparent clash with the ribose moiety (Fig. 5E and F).12,26 Thus, the X-ray structural data along with the enzymatic assay explains the molecular reason behind the fidelity of GDs. These studies not only bring forth a battery of inhibitors that can be further tested on an in vivo scale; they also highlight strategies that can be used to develop potent inhibitors that will strongly bind to the GD but will be unable to be catalytically turned over.26 The unique features of bacterial GDs and their dissimilarity to human GDs have been exploited for the design of potential drug targets.4. Deaminases exclusive to pathogens
Evolutionary analysis is a very powerful method that can help reveal enzymes and pathways that are exclusive to a particular set of organisms.5,30 By creating sequence-based similarity networks, Raushel and co-workers have been very successful in discerning the function of unique groups of deaminases.52,53 Using the above approach, one of their findings was the enzyme Pa0142 from Pseudomonas aeruginosa, which belongs to the AHS superfamily. This enzyme was found to deaminate the mutagenic bases 8-oxoguanine and isocytosine.52 Similarly, this method of classifying enzymes was applied to the CDA superfamily for categorizing deaminating/hydrolytic enzymes according to function.5 The network of cog0590 revealed that apart from the usual guanine, cytosine, cytidine and tRNA adenosine deaminases, the common orthologous group contained several uncharacterized groups that clustered together and were sequence-divergent (Fig. 1). Surprisingly, one of these groups contained a large concentration of several mycobacterial enzymes with unknown functions.5 Recent investigation of this group by Anand and co-workers led to the discovery of a mycobacterial enzyme, Msd, from Mycobacterium smegmatis that specifically deaminates mutagens such as 5-azacytosine and isoguanine-type scaffolds and is non-reactive towards natural bases.5 The unique ability of this enzyme in the Mycobacterium genus to exclusively accept unnatural bases prompted the need to determine the reason for the existence of these deaminases.54.1 Structural and functional analyses of Msd
The structure of Msd was determined at a resolution of 1.9 Å; it was necessary to employ Zn-SAD phasing because several parts of the structure were different from other known cog0590 enzymes.5 Structural analysis reveals that Msd exhibits the core α–β–α CDA fold and has additional parts which exhibit topological differences. It has a unique helix loop insertion (αD) in the structure which has been shown to be flexible and therefore is proposed to act as a flap above the active site (Fig. 6A).5 Another distinctive feature of Msd is that it possesses a relatively large active site in comparison to other known deaminases, the purpose of which remains unclear.5 However, this space may be exploited to design future inhibitors of varying sizes that can be accommodated in Msd. A close examination of the active site of Msd revealed that it has characteristics of both guanine and cytosine deaminases but that it acts on neither.5 The hydrophobic stacking residues in Msd, such as F29 and W95, are similar to those of GDs.5,12 Analogous to GDs, Msd harbours two glutamates, although they are differentially located in the active site (Fig. 6D). The positioning of the metal-coordinated glutamate appeared to be closer to that of yCDs. However, as mentioned earlier, the active site is a hybrid between those of GDs and CDs and resembles no known deaminases.5 Therefore, identification of its function from its structure could not be achieved. Due to this evolutionary diversity in the Mycobacterium genus, Msd is unique and, as mentioned above, conducive to drug development.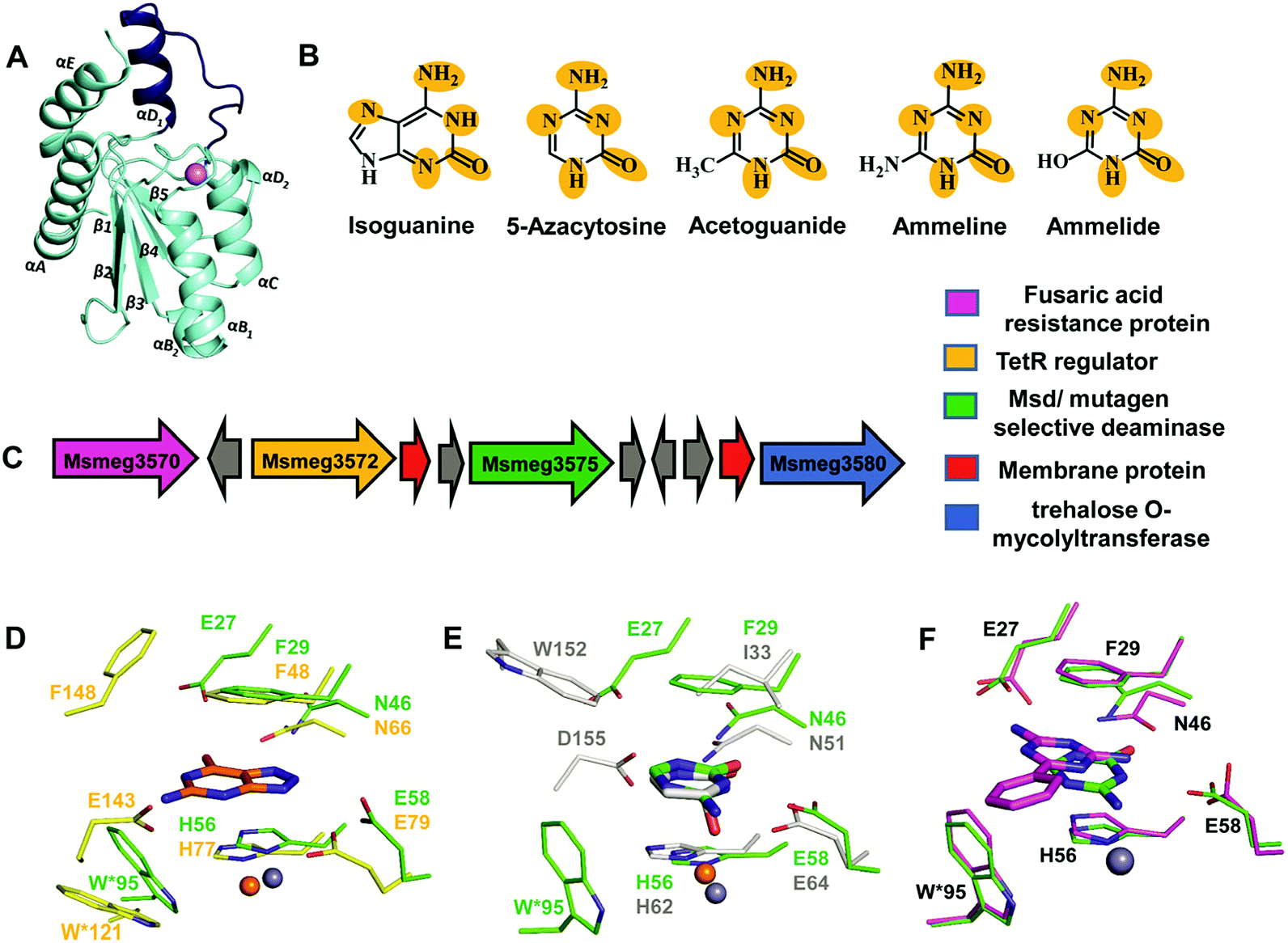 | ||
| Fig. 6 Structure and function of Msd. (A) Cartoon representation of the monomer of Msd (PDB ID: 5XKO). The unique helix-loop insertion (αD1) is highlighted in blue. (B) Selective substrates catalysed by Msd with the requisite chemical scaffolds encircled. (C) Genome context for Msd. (D) Active site superposition of Msd and 8-azaguanine–NE0047 complex (PDB ID: 4HRQ). (E) Active site superposition of yCD (PDB ID: 1UAQ) with 5-azacytosine–Msd complex (PDB ID: 5XKP). Carbon atoms of the interacting residues and ligand are in green in Msd, yellow in NE0047 and blue-white in yCD. Zinc is represented as a grey sphere in Msd and as an orange sphere in NE0047 and yCD. (F) Active site superposition of Msd–benzoguanamine (PDB ID: 5XKR) and the Msd–5-azacytosine complex. Carbon atoms of the interacting residues and ligand are shown in green in the Msd–5-azacytosine complex and in magenta in the Msd–benzoguanamine complex. | ||
To identify its function, this mycobacterial enzyme was subjected to rigorous screening, both virtual and enzymatic. It was found that it could deaminate only the mutagenic purine isoguanine and the pyrimidine 5-azacytosine (Scheme 1C and D).5 Msd was very specific for these bases and was completely non-reactive towards natural bases, nucleosides or nucleotides. Msd could also deaminate 5-azacytosine derivatives such as acetoguanide, ammeline, and ammelide (Scheme 1C and Fig. 6B).5 These compounds belong to the 1,3,5-triazine class of compounds, which are being exploited as anticancer and antimicrobial agents in their derivatized forms.54,55 Upon analysing the chemical nature of the substrates, it was concluded that all the compounds which underwent deamination by Msd exhibit a typical azapyrimidine-like scaffold; isoguanine, a purine substrate of Msd, also displays similar conjugation chemistry to s-triazines (Fig. 6B).5 The function determination of Msd raised several questions; for example, why does it not deaminate natural bases such as cytosine but acts on 5-azacytosine? What is the reason for acquiring this activity, and why do mycobacterium species in particular possess this enzyme? To understand some of these aspects, analyses of the substrate specificity via structural, mutagenesis and biochemical analysis were performed. Additionally, the genome location of the enzyme was analysed.
4.2 Substrate selectivity of Msd
To understand the basis of substrate specificity, which may provide clues to the reason for the existence of Msd, detailed examination of the chemical nature of the substrates catalysed by Msd was undertaken. The results reveal that it possesses strategically placed keto and amino functional groups between the two electron-withdrawing nitrogen atoms.5 Because Msd has a large and a less shielded active site, it catalyzes deamination in a rather solvent-exposed environment. This is in contrast to GDs and CDs, which have evolved to limit the sizes of their active sites to eliminate unwanted solvent molecules.5,12,23 Yet, the stringency of the substrates catalysed by Msd is maintained via controlling the chemistry of the substrates. To avoid deamination of natural bases such as cytosine, Msd has evolved to tune its energetics to only facilitate deamination of bases with increased electrophilic character.5 The azapyrimidine-like scaffold has an additional nitrogen atom adjacent to the carbon atom where the attack occurs, increasing the electrophilicity of the aza-scaffold-containing base.5 This in turn facilitates nucleophilic addition to this electrophilic carbon centre; thus, the deamination reaction is more facile. It was observed that compounds which lacked this chemical property did not undergo deamination.5 For example, pyrimidines such as cytosine, isocytosine and 5-methylcytosine, which harbour similar scaffolds but lack the additional nitrogen atom found in s-triazine compounds, are non-reactive.5 In contrast to the pyrimidines, isoguanine exhibits the presence of the N7 atom in addition to the keto group at position 2 of the purine scaffold. Although N7 is more distant than N5 in the azapyrimidine scaffold, its electrophilic character is still maintained, albeit to a lesser extent; hence, the deamination reaction is driven at a 5-fold decreased catalytic efficiency in comparison to 5-azacytosine.5Further, the crystal structures of Msd in complex with 5-azacytosine and ammeline provide additional structural perspective on the substrate selectivity.5 They explain why Msd is unable to deaminate cytosine. As mentioned earlier, the Msd active site is more open and lacks the C-terminal tail that is critical for activity in yCD.5,23 An active site comparison with yCD shows that the conserved aspartic residue in the C-terminal tail plays a crucial role in stabilizing the cytosine moiety and determines the substrate specificity (Fig. 6E). This whole topological feature, along with its supporting residues, is completely absent from Msd.5,23 Because Msd lacks these specific interacting residues, it is unable to anchor the cytosine moiety and thus cannot turn over the naturally occurring pyrimidine (Fig. 6E).5 Moreover, to gain insights into the stringency of the substrate scaffold, the crystal structure of Msd in complex with benzoguanamine (2,4-diamino-6-phenyl-1,3,5-triazine) was determined by Gaded and co-workers.5 Benzoguanamine possesses an NH2 group in place of the C2 carbonyl group (as in other substrates of Msd); this different functional group leads to differential orientation of the molecule in the active site, rendering it catalytically incompetent (Fig. 6F).5 The structure of the benzoguanamine–Msd complex in conjunction with mutagenesis and biochemical experiments leads to the conclusion that the second glutamic acid in the active site (E27) plays a major role in anchoring the bulkier purine ring rather than participating directly in catalysis.5 Thus, Msd, like yCD, most likely uses only one glutamic acid residue as a proton shuttle.5,23 Another observation was that the distance of the amino group in benzoguanamine from the catalytic water and glutamate, which are essential for catalysis, is sub-optimal; thus, it is an inhibitor.5 Overall, these studies provide clues towards the design of inhibitors that can be used to block Msd function. The above facts suggest that Msd may accept larger substrates with azapyrimidine-like scaffolds; this avenue remains to be explored.
4.3 Msd as a drug target
To unravel the reason why Msd is present in the Mycobacterium genus, its genome location was analysed. It was observed that the enzyme is part of a conserved gene cluster that is involved in the efflux of xenobiotic compounds.5 Investigation of the gene cluster shows that Msd is surrounded by genes such as tetracycline family (TetR) regulators, outer membrane assembly, and ABC transporters, which play a central role in efflux pump formation and regulation.5 The fusaric acid resistance protein, which is known to efflux fusaric acid (a mycotoxin), and trehalose o-mycolyltransferase, which plays an important role in outer membrane assembly, are also part of this broad spectrum gene cluster (Fig. 6C).5,56–58 It appears that this enzyme screens for s-triazine scaffold compounds, deaminates them, and thereby primes them to be exported out of the cell. For certain soil organisms and Mycobacterium species, which are constantly exposed to mutagens, this pathway appears to provide protection by imparting innate resistance against this class of compounds.5 Inhibitors against Msd are likely to sensitize the organism against triazine compounds; this theory requires testing. This opens the whole class of aza-drugs as treatment options against the Mycobacterium genus and may lead to alternative therapies. Another potential use of Msd lies in cancer therapy as a potential replacement for the CD-prodrug system. Currently, CDs compete with the natural pool of cytosine to preferentially deaminate 5-fluorocytosine.24 Replacement by Msd and 5-azacytosine will likely achieve a similar scenario without the shortcomings of the former system. These avenues require further study and may open doors to the large number of possibilities that this system offers.5. Conclusions and outlook
In conclusion, the CDA superfamily comprises essential enzymes that represent potential enzyme systems that can be exploited for drug therapy. Especially, the deaminases (cytosine, guanine) overviewed here are essential regulators of the nucleobase derivative pool, and Msd is a unique enzyme that is present in mycobacteria. The stark differences in fold and active site architecture between humans and bacteria among these deaminases provides new frameworks for the development of drug targets. In this regard, CDs are already being used as efficient enzyme–prodrug systems as therapy for several cancers. Moreover, the recently identified mycobacterial enzyme (Msd), which has selectivity towards mutagens and s-triazine compounds, has opened doors towards understanding innate resistance mechanisms. Inhibitors against this enzyme can provide promising leads to develop therapies to sensitize this bacterium against the s-triazine class of drugs.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.Acknowledgements
We thank IIT Bombay and DBT-NER, Government of India for the funding. We also thank Prof. Pradeep Kumar P. I. for helpful suggestions.References
- E. C. Bragginton and L. J. V. Piddock, Lancet Infect. Dis., 2014, 14, 857–868 CrossRef PubMed
.
- C. C. Boehme, P. Nabeta, D. Hillemann, M. P. Nicol, S. Shenai, F. Krapp, J. Allen, R. Tahirli, R. Blakemore, R. Rustomjee, A. Milovic, M. Jones, S. M. O'Brien, D. H. Persing, S. Ruesch-Gerdes, E. Gotuzzo, C. Rodrigues, D. Alland and M. D. Perkins, N. Engl. J. Med., 2010, 363, 1005–1015 CrossRef PubMed
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef PubMed
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed
- V. Gaded and R. Anand, J. Am. Chem. Soc., 2017, 139, 10762–10768 CrossRef PubMed
- K. Honer zu Bentrup and D. G. Russell, Trends Microbiol., 2001, 9, 597–605 CrossRef PubMed
- B. A. Moffatt and H. Ashihara, Arabidopsis Book, 2002, vol. 1, p. e0018 Search PubMed
- L. Holm and C. Sander, Proteins: Struct., Funct., Bioinf., 1997, 28, 72–82 CrossRef
- L. M. Iyer, D. Zhang, I. B. Rogozin and L. Aravind, Nucleic Acids Res., 2011, 39, 9473–9497 CrossRef PubMed
- D. S. Hitchcock, H. Fan, J. Kim, M. Vetting, B. Hillerich, R. D. Seidel, S. C. Almo, B. K. Shoichet, A. Sali and F. M. Raushel, J. Am. Chem. Soc., 2013, 135, 13927–13933 CrossRef PubMed
- J. Berger Sosamma, G. Carter Joyce and H. Lowry Oliver, J. Neurochem., 2006, 44, 1736–1740 CrossRef
- A. Bitra, B. Hussain, A. S. Tanwar and R. Anand, Biochemistry, 2013, 52, 3512–3522 CrossRef PubMed
- A. Cortes, E. Gracia, E. Moreno, J. Mallol, C. Lluis, E. I. Canela and V. Casado, Med. Res. Rev., 2015, 35, 85–125 CrossRef PubMed
- M. P. Gleeson, N. A. Burton and I. H. Hillier, Phys. Chem. Chem. Phys., 2003, 5, 4272–4278 RSC
- J. Guz, M. Jurgowiak and R. Olinski, Postepy Hig. Med. Dosw., 2012, 66, 275–286 CrossRef
- G. C. Ireton, M. E. Black and B. L. Stoddard, Structure, 2003, 11, 961–972 CrossRef PubMed
- R. S. Hall, A. A. Fedorov, C. Xu, E. V. Fedorov, S. C. Almo and F. M. Raushel, Biochemistry, 2011, 50, 5077–5085 CrossRef PubMed
- S. S. Kamat, A. Bagaria, D. Kumaran, G. P. Holmes-Hampton, H. Fan, A. Sali, J. M. Sauder, S. K. Burley, P. A. Lindahl, S. Swaminathan and F. M. Raushel, Biochemistry, 2011, 50, 1917–1927 CrossRef PubMed
- H. Pospisilova and I. Frebort, Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia, 2007, vol. 151, pp. 3–10 Search PubMed
- C. Ribard, M. Rochet, B. Labedan, B. Daignan-Fornier, P. Alzari, C. Scazzocchio and N. Oestreicher, J. Mol. Biol., 2003, 334, 1117–1131 CrossRef PubMed
- N. Navaratnam and R. Sarwar, Int. J. Hematol., 2006, 83, 195–200 CrossRef PubMed
- G. C. Ireton, G. McDermott, M. E. Black and B. L. Stoddard, J. Mol. Biol., 2002, 315, 687–697 CrossRef PubMed
- T. P. Ko, J. J. Lin, C. Y. Hu, Y. H. Hsu, A. H. Wang and S. H. Liaw, J. Biol. Chem., 2003, 278, 19111–19117 CrossRef PubMed
- M. Fuchita, A. Ardiani, L. Zhao, K. Serve, B. L. Stoddard and M. E. Black, Cancer Res., 2009, 69, 4791–4799 CrossRef PubMed
- T. S. Stolworthy, A. M. Korkegian, C. L. Willmon, A. Ardiani, J. Cundiff, B. L. Stoddard and M. E. Black, J. Mol. Biol., 2008, 377, 854–869 CrossRef PubMed
- A. Bitra, A. Biswas and R. Anand, Biochemistry, 2013, 52, 8106–8114 CrossRef PubMed
- H. C. Losey, A. J. Ruthenburg and G. L. Verdine, Nat. Struct. Mol. Biol., 2006, 13, 153–159 CrossRef PubMed
- H. J. Atkinson, J. H. Morris, T. E. Ferrin and P. C. Babbitt, PLoS One, 2009, 4, e4345 CrossRef PubMed
- T.-L. Mai, G.-M. Hu and C.-M. Chen, J. Proteome Res., 2016, 15, 2123–2131 CrossRef PubMed
- S. Zhao, R. Kumar, A. Sakai, M. W. Vetting, B. M. Wood, S. Brown, J. B. Bonanno, B. S. Hillerich, R. D. Seidel, P. C. Babbitt, S. C. Almo, J. V. Sweedler, J. A. Gerlt, J. E. Cronan and M. P. Jacobson, Nature, 2013, 502, 698 CrossRef PubMed
- J. Kream and E. Chargaff, J. Am. Chem. Soc., 1952, 74, 5157–5160 CrossRef
- T. A. Kunkel and M. Diaz, Mol. Cell, 2002, 10, 962–963 CrossRef PubMed
- D. J. Porter and E. A. Austin, J. Biol. Chem., 1993, 268, 24005–24011 Search PubMed
- D. S. Hitchcock, A. A. Fedorov, E. V. Fedorov, S. C. Almo and F. M. Raushel, Biochemistry, 2014, 53, 7426–7435 CrossRef PubMed
- T. Nishiyama, Y. Kawamura, K. Kawamoto, H. Matsumura, N. Yamamoto, T. Ito, A. Ohyama, T. Katsuragi and T. Sakai, Cancer Res., 1985, 45, 1753–1761 Search PubMed
- Q. T. Trinh, E. A. Austin, D. M. Murray, V. C. Knick and B. E. Huber, Cancer Res., 1995, 55, 4808–4812 Search PubMed
- L. Yao, Y. Li, Y. Wu, A. Liu and H. Yan, Biochemistry, 2005, 44, 5940–5947 CrossRef PubMed
- S. Sklenak, L. Yao, R. I. Cukier and H. Yan, J. Am. Chem. Soc., 2004, 126, 14879–14889 CrossRef PubMed
- B. Manta, F. M. Raushel and F. Himo, J. Phys. Chem. B, 2014, 118, 5644–5652 CrossRef PubMed
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef PubMed
- A. Korkegian, M. E. Black, D. Baker and B. L. Stoddard, Science, 2005, 308, 857–860 CrossRef PubMed
- M. Malet-Martino and R. Martino, Oncologist, 2002, 7, 288–323 CrossRef PubMed
- B. F. Akum, M. Chen, S. I. Gunderson, G. M. Riefler, M. M. Scerri-Hansen and B. L. Firestein, Nat. Neurosci., 2004, 7, 145 CrossRef PubMed
- J. R. Fernandez, B. Byrne and B. L. Firestein, J. Mol. Evol., 2009, 68, 227–235 CrossRef PubMed
- S. Kumar, V. Josan, K. C. Sanger, K. K. Tewari and P. S. Krishnan, Biochem. J., 1967, 102, 691–704 CrossRef PubMed
- G. Yuan, J. C. Bin, D. J. McKay and F. F. Snyder, J. Biol. Chem., 1999, 274, 8175–8180 CrossRef PubMed
- J. R. Fernandez, E. S. Sweet, W. J. Welsh and B. L. Firestein, Bioorg. Med. Chem., 2010, 18, 6748–6755 CrossRef PubMed
- L. Egeblad, M. Welin, S. Flodin, S. Graslund, L. Wang, J. Balzarini, S. Eriksson and P. Nordlund, PLoS One, 2012, 7, e37724 CrossRef PubMed
- A. K. Saxena, S. Ahmad, K. Shanker and K. Kishor, Pharmacol. Res. Commun., 1984, 16, 243–252 CrossRef PubMed
- J. L. Seffernick, A. G. Dodge, M. J. Sadowsky, J. A. Bumpus and L. P. Wackett, J. Bacteriol., 2010, 192, 1106–1112 CrossRef PubMed
- S. H. Liaw, Y. J. Chang, C. T. Lai, H. C. Chang and G. G. Chang, J. Biol. Chem., 2004, 279, 35479–35485 CrossRef PubMed
- R. S. Hall, A. A. Fedorov, R. Marti-Arbona, E. V. Fedorov, P. Kolb, J. M. Sauder, S. K. Burley, B. K. Shoichet, S. C. Almo and F. M. Raushel, J. Am. Chem. Soc., 2010, 132, 1762–1763 CrossRef PubMed
- D. S. Hitchcock, A. A. Fedorov, E. V. Fedorov, L. J. Dangott, S. C. Almo and F. M. Raushel, Biochemistry, 2011, 50, 5555–5557 CrossRef PubMed
- R. Banerjee, N. J. Pace, D. R. Brown and E. Weerapana, J. Am. Chem. Soc., 2013, 135, 2497–2500 CrossRef PubMed
- S. N. Khattab, H. H. Khalil, A. A. Bekhit, M. M. El-Rahman, A. El-Faham and F. Albericio, Molecules, 2015, 20, 15976–15988 CrossRef PubMed
- Y. Yamaryo-Botte, A. K. Rainczuk, D. J. Lea-Smith, R. Brammananth, P. L. van der Peet, P. Meikle, J. E. Ralton, T. W. T. Rupasinghe, S. J. Williams, R. L. Coppel, P. K. Crellin and M. J. McConville, ACS Chem. Biol., 2015, 10, 734–746 CrossRef PubMed
- B. Youenou, S. Favre-Bonte, J. Bodilis, E. Brothier, A. Dubost, D. Muller and S. Nazaret, Genome Biol. Evol., 2015, 7, 2484–2505 CrossRef PubMed
- N. Zeytuni and R. Zarivach, Structure, 2012, 20, 397–405 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2018 |