
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe chemical functionalization of graphene nanoplatelets through solvent-free reaction†
Eunice
Cunha‡
 *a,
He
Ren‡
b,
Fei
Lin
a,
Ian A.
Kinloch
a,
Quanji
Sun
b,
Zhaodong
Fan
*b and
Robert J.
Young
a
*a,
He
Ren‡
b,
Fei
Lin
a,
Ian A.
Kinloch
a,
Quanji
Sun
b,
Zhaodong
Fan
*b and
Robert J.
Young
a
aNational Graphene Institute and School of Materials, University of Manchester, Manchester M13 9PL, UK. E-mail: eunice.cunha-2@manchester.ac.uk
bBeijing Institute of Aeronautical Materials (BIAM), Beijing, China. E-mail: fanzhaodong99@sohu.com
First published on 28th September 2018
Abstract
Graphene nanoplatelets (GNPs) were functionalized through 1,3-dipolar cycloaddition of azomethine ylide using a solvent-free approach and under different reaction conditions. The yield and the functionality of the carboxyl-terminated pyrrolidine ring attached on the surface of GNPs could be affected by varying the reaction temperature as well as the reactant to GNP weight ratio. The functionalized GNPs were characterized extensively using a range of spectroscopic and microscopy techniques.
Introduction
Graphene-based materials have attracted great interest due to their excellent physical, mechanical and electrical properties.1–3 Such properties make these materials suitable for a wide range of applications such as in electronic devices, nanocomposites, supercapacitors, hydrogen storage, among others.4–7Graphite/graphene nanoplatelets (GNP),8 a thin form of graphite flakes with thickness ranging from that of graphene to approximately 100 nm, have recently attracted attention as an economically viable alternative to produce graphene in large quantities.9 Normally these materials are obtained by an expansion process, using heat or microwave irradiation, resulting in GNP or expanded graphite (EG) with an interlayer spacing higher than that of graphite.8–10 Although the low surface energy of these materials may be a limitation for some applications.11 Covalent functionalization of graphene materials provides an effective means of adjusting its surface energy as well as introducing specific functionalities. These functionalities may be an advantage for some applications, such as to enhance the compatibility with solvents and polymer matrices along with the stable attachment of the functional groups that can provide specific functional properties (e.g., fluorescent molecules, dopants, etc.).11–13
Cycloaddition reactions are one of the most important classes of reactions in organic chemistry. Within this class, the 1,3-dipolar cycloaddition (1,3-DCA) reaction of azomethine ylides, first introduced by Huisgen in 1963,14 has been applied for the efficient and high yielding synthesis of different heterocyclic compounds.15 In this reaction, the azomethine ylide is generated in situ through the decarboxylation of immonium salts derived from the condensation of α-amino acids with aldehydes. The wide range of easily accessible starting materials allows the formation of a large variety of different functional groups.15,16 For this reason, the 1,3-DCA reaction has been accepted as an interesting and advantageous approach to functionalize carbon based materials such fullerenes,17,18 carbon nanotubes19–21 as well as graphene related materials.12,22,23 In fact, the 1,3-DCA reaction in exfoliated graphene was first demonstrated by Trapalis et al.22 who estimated, by thermogravimetric analysis, an extent of functionalization of 1 functional group per 40 carbon atoms. Prato and co-workers23 demonstrated that the 1,3-DCA reaction not only occurred at the edges of graphene but also on the basal planes and highlighted an extent of 1 functional group per 128 carbon atoms. Theoretical studies of the 1,3-DCA reaction on the graphene surface have been reported.24–26 While Denis et al.25 reported that the most reactive sites on graphene for the addition of azomethine ylides are defect sites, Houk and co-workers24 found that the edges of graphene flakes are more favourable reaction sites and that azomethine ylides cannot react in the perfect central area of graphene. Later, Denis et al.26 reported that the 1,3-DCA reaction of azomethine ylides on perfect graphene surface can occur due to a cooperative behaviour responsible for the dramatic increase of the reaction energies.
Usually 1,3-DCA reactions on carbon based materials are carried on a small scale owing to the long-time reaction and the large amount of solvent required to disperse the starting carbon based materials.11 Solvent-free approaches can be an alternative way to easily scale up the reaction and decrease the reaction time. Microwave irradiation has been considered as a fast and scalable methodology to functionalize carbon nanotubes27–29 and carbon nanohorns30 through the 1,3-DCA reaction under solvent-free conditions. The effect of microwaves on those materials is, however, complex and it is difficult to control the temperature as well as the homogeneity of the reaction conditions using this source of energy.31 An interesting solid-state 1,3-DCA reaction approach was reported by Paiva et al.32 The solvent-free one-pot reaction was carried out on the surface of carbon nanotubes through the thermal condensation of N-benzyloxycarbonyl glycine and formaldehyde. The authors gave evidence for the formation of two types of functional groups on the surface of the carbon nanotubes, namely a pyrrolidine and a benzyl carbamate, and showed their relative concentrations could be controlled by adjusting the reaction temperature and time. The same group have also been reported the same functionalization procedure on GNPs, taking advantage of the pyrrolidine functionality to react and improve its compatibility with polymer matrices during melt mixing processes.33–37 The functionalized GNPs were reported to lead to a better dispersion into the polypropylene (PP)33,35–37 and polycarbonate (PC)/poly(styrene-acrylonitrile) (SAN)34 matrices when compared to the as received GNPs, although the electrical properties was found to be slightly inferior.34,35
In this work we aimed to study the functionalization of GNP through solvent-free 1,3-DCA reactions using a different α-amino acid – iminodiacetic acid (IDA). The reaction of the IDA with paraformaldehyde (PFA) resulted in the formation of a 1,3-dipolar specie (azomethine ylide) that reacted with the surface of the GNP through cycloaddition to originate the carboxyl-terminated pyrrolidine functionality. The carboxyl functionalities are particularly attractive because they can undergo diverse conjugation reactions. Furthermore, the direct attachment of carboxyl groups to the graphene related materials is challenging. It should be noted that even for graphene oxide (GO) the content of carboxyl groups is generally very low (or even absent).38 In our solvent-free 1,3-DCA reactions the yield and the functionality of the carboxyl-terminated pyrrolidine ring attached on the surface of GNP could be controlled by varying the reaction temperature as well as the reactant to GNPs weight ratio. The structural and morphological changes of the functionalized materials were characterized using a range of spectroscopic and microscopy techniques.
Experimental
Materials
Micronized graphite (grade Micrograf HC11) with an equivalent diameter of about 10 μm and a surface area of 27 m2 g−1 was provided by Nacional de Grafite, Brazil. Since these micronized graphite particles probably have more than 10 layers, they should be formally termed “graphite” nanoplatelets. However, such materials are often termed “graphene” nanoplatelets in the literature39 and so this terminology will also be adopted in this present study. Iminodiacetic acid (IDA) purum ≥98% and paraformaldehyde (PFA) reagent grade, crystalline, were purchased from Sigma Aldrich, ethanol absolute (EtOH) was purchased from VWR Chemicals and acetone analytical reagent grade was purchased from Fisher Scientific.Functionalization of the GNPs
The functionalization of the GNPs was performed based on a procedure reported previously.32 Briefly, GNP HC11 was added to an ethanolic solution/suspension of IDA and PFA (1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.0 molar ratio), under magnetic stirring, and heated gently until the solvent was completely removed. The solid mixture was heated at different temperatures (180 °C, 200 °C, 220 °C and 250 °C) over 5 hours, in a round-bottom flask. The GNP:IDA weight ratio used was 1.0:1.0. Different GNP:IDA weight ratios (1.0:1.5 and 1.0:2.0 weight ratio) were also tested, using the reaction conditions of 200 °C and 5 hours and keeping the same molar ratio of IDA and PFA (1.0:5.0). The resulting functionalized GNP (f-HC11) was washed several times with ethanol, acetone and distilled water, filtered, and dried under vacuum, at 40 °C, overnight.
:5.0 molar ratio), under magnetic stirring, and heated gently until the solvent was completely removed. The solid mixture was heated at different temperatures (180 °C, 200 °C, 220 °C and 250 °C) over 5 hours, in a round-bottom flask. The GNP:IDA weight ratio used was 1.0:1.0. Different GNP:IDA weight ratios (1.0:1.5 and 1.0:2.0 weight ratio) were also tested, using the reaction conditions of 200 °C and 5 hours and keeping the same molar ratio of IDA and PFA (1.0:5.0). The resulting functionalized GNP (f-HC11) was washed several times with ethanol, acetone and distilled water, filtered, and dried under vacuum, at 40 °C, overnight.
Characterization
Thermogravimetric analysis (TGA) was performed on a Modulated TGA Q500 from TA Instruments. The samples were heated at 10 °C min−1 under a constant flow of N2. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Axis Ultra X-ray photoelectron spectrometer; curve fitting was accomplished by CasaXPS software. The samples for Raman characterization were prepared by drop coating of diluted ethanolic suspensions of the pristine HC11 as well as the f-HC11 on a glass slide and dried using a hot-plate. The Raman spectra were obtained on a Renishaw 1000 Raman microprobe system (Renishaw, UK) using an Argon ion laser of 514 nm and a 100× objective lens. For each sample 120 spectra were collected from different HC11 and f-HC11 flakes, randomly, over a sample area of 50 × 20 mm2. The Raman mapping of the pristine and functionalized HC11 flakes was undertaken on a Horiba Labram HR Evolution confocal microscope, equipped with a motorized x-y table, using an excitation laser of 633 nm and a 100× objective lens. The maps were obtained from a 25 × 25 point array with a step size between each point of 0.8 μm. All data were analyzed using the LabSpec 6 software and Lorentzian functions were used to fit the characteristic peaks of the spectra. Scanning electron microscopy (SEM) analysis was performed on the pristine and functionalized HC11, in powder form, using a Philips XL30 FEGSEM microscope equipped with energy dispersive X-ray spectroscopy (EDS) system. Transmission electron microscopy (TEM) samples were prepared by dropping the uniformly-dispersed ethanolic suspensions of pristine and functionalized HC11 onto lacey carbon films (Agar scientific) and dried overnight at room temperature. The samples were analyzed using a FEI Tecnai TF 30 FEG-TEM with acceleration voltage 300 kV. The crystal structure of the samples was investigated by X-ray diffraction (XRD) with a PANalytical X'Pert Pro X'Celerator diffractometer. Atomic Force Microscopy (AFM) images of the GNP HC11 and f-HC11 200 water suspensions deposited and dried onto a silicon wafer substrate were acquired using a NanoWizard® 4 NanoScience AFM (JPK instruments, Berlin) equipped with a digital Vortis™ controller and a piezo-based sample scanner and motorized XY stage. The images were acquired using the tapping and the QI™ imaging mode. The cantilevers were silicon probes (Z566624 BudgetSensors AFM probes, Aldrich, UK) with a 281.4 kHz resonance frequency.Results and discussion
The surface modification of GNP HC11 was carried out through 1,3-DCA reaction under solvent free conditions. The thermal degradation of PFA generates formaldehyde which diffuses through the molten α-amino acid (IDA) forming the 1,3-dipolar species (azomethine ylide) presented in Scheme 1, compound 1. The 1,3 dipole reacts with the surface of the GNP through cycloaddition to originate the carboxyl-terminated pyrrolidine ring attached on the surface of GNP HC11 (compound 2). In this work different reaction conditions were tested to study the effect of temperature as well as the IDA weight ratio in the functionalization reaction. | ||
| Scheme 1 Schematic representation of the production of functionalized GNPs through 1,3-dipolar cycloaddition reaction. | ||
Effect of the temperature
The surface compositions of the pristine and functionalized GNP HC11 were analysed by XPS. Fig. 1 shows the wide scan spectra (Fig. 1a) as well as the high-resolution C 1s spectra (Fig. 1b) of the pristine and functionalized GNP HC11 at different temperatures. The XPS spectrum of the pristine GNP HC11 show peaks corresponding to the binding energies of about 284 eV and 531 eV which are attributed to C 1s and O 1s energy levels, respectively. The oxygen content in the pristine GNP HC11 was found to be very low (less than 2%) respect to carbon. The C 1s core-level spectrum of GNP HC11 (Fig. 1b) is characterized by contributions at 284.5 eV (major contribution, red), and small contributions at 285.2 eV (blue), 286.1 eV (magenta) and 287.2 eV (olive) arising from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (sp2 bonded carbons), C–C (sp3 bonded carbons), C–OH (alkoxy) and CO (carbonyl) moieties, respectively. The weakly pronounced peak at 291.1 eV (violet curve) arises from the π–π* shake-up transition.
C (sp2 bonded carbons), C–C (sp3 bonded carbons), C–OH (alkoxy) and CO (carbonyl) moieties, respectively. The weakly pronounced peak at 291.1 eV (violet curve) arises from the π–π* shake-up transition.
 | ||
| Fig. 1 XPS analysis of pristine and functionalized material: (a) XPS spectra; (b) high resolution C 1s spectra. | ||
The XPS spectra of the functionalized GNP HC11 (f-HC11) at different temperatures show peaks corresponding to the binding energies of about 284 eV, 399 eV and 531 eV which are attributed to C 1s, N 1s and O 1s energy levels, respectively. The functionalized samples also show a significant increase of the oxygen content (up to 9%) and a considerable amount of nitrogen (up to 6%) when compared to the pristine material. All the percentages of the atomic concentration of the chemical elements are presented in Table S1 in ESI.† The C 1s spectra of the f-HC11 become more asymmetric when compared to the pristine material which is related to the increase of the contributions at 284.5 eV (red), 285.2 eV (blue), 286.1 eV (magenta) and 287.2 eV (olive), explained above. A new contribution is clearly identified at 288.9 eV (navy curve) arising from the C(O)–O (carboxyl) moieties derived from the carboxylic acid functionalities.
The reaction yield can be measured from the atomic ratio between the [N]:[C]. As depicted in Table S1,† the [N]:[C] ratio increase with the increase of the temperature reaction, which indicates that increasing the temperature favours the reaction yield. However, an increase the [N]:[O] ratio with the increasing temperature is also observed, particularly at the reaction temperatures 220 °C and 250 °C. The expected [N]:[O] ratio, according to the compound 2 in Scheme 1, is one nitrogen atom to two oxygen atoms. The increase of this ratio with the increase of temperature may indicate some degradation of the functional groups at higher temperatures resulted from the decarboxylation of the functional group. In fact, the IDA itself presents a first thermal degradation step that starts around 200 °C and is related to the loss of carbon dioxide resulted from a partial decarboxylation process.40 Also, Paiva et al.32 reported that increasing the temperature in the solid state 1,3-DCA reaction, namely at temperatures above 210 °C, led to the thermal cleavage of the protecting group of the original amine favouring the formation of pyrrolidine groups at the surface of carbon nanotubes. These results are in agreement with the greater increase of carboxylic acid functionalities found in the f-HC11 200. Increasing the temperature seems to favour the reaction yield however, the decarboxylation of the functional group also increases, losing some of the functionality of these materials.
The thermogravimetric analyses of the pristine and functionalized GNP HC11 are presented in Fig. 2 and the weight losses at 600 °C as well as the extrapolated onset degradation temperatures (Tonset) for the pristine and functionalized materials are shown in Table 1. The TGA curves of the pristine GNP HC11 showed a thermally-stable material with a weight loss at 600 °C of 1.2 ± 0.1%. Conversely, the functionalized GNP HC11 show higher values of weight loss at 600 °C (up to 18%) which is related to the thermal decomposition of the functional groups. A decrease of the weight loss with the increasing of the reaction temperature is also observed and the Tonset values were found to increase with the increasing of the reaction temperature. These results are in agreement with the XPS results. As the decarboxylation process of the functional groups increase with the increase of the reaction temperature, a decrease of the weight loss in the thermal analysis is expected, along with the formation of more thermally-stable functionalized GNP HC11.
 | ||
| Fig. 2 Thermogravimetric analysis of the pristine and functionalized HC11. | ||
| Weight loss at 600 °C (%) | T onset (°C) | |
|---|---|---|
| HC11 pristine | 1.2 ± 0.1 | — |
| f-HC11 180 | 18.1 ± 0.6 | 250 ± 2 |
| f-HC11 200 | 17.4 ± 0.9 | 263 ± 2 |
| f-HC11 220 | 13.1 ± 0.5 | 316 ± 3 |
| f-HC11 250 | 12.0 ± 0.9 | 345 ± 3 |
Raman spectroscopy is an important tool for the characterization of graphene and carbon-based materials. Fig. 3 presents representative Raman spectra of the pristine and functionalized GNP HC11. The three main characteristics bands are identified in both spectra: the D band at around 1350 cm−1, the G band at around 1580 cm−1 and the 2D band at round 2720 cm−1, typical for graphic materials.41,42 The G and 2D band provide information about the level of strain, doping, crystallinity and number of layers,41–43 while the D band is related to lattice defects,44,45 including the formation of sp3 hybridized carbon by covalent functionalization.46–48 The increase of the D band intensity is accompanied by the detection of another band, the D′ band, that appears at around 1620 cm−1. Both D and D′ bands are activated by defects and they are resulted from intervalley (D) and intravalley (D′) double resonance scattering processes.41 As depicted in Fig. 3, the functionalized materials present a more prominent D band and a shoulder in the G band at around 1620 cm−1 is also identified (D′ peak) indicating the formation of sp3 bonds.46 The intensity and the position of the 2D band are not greatly affected when compared to the pristine GNP HC11 indicating that functionalization process does not significantly alter the structural quality of the pristine material.41,42
 | ||
| Fig. 3 Representative Raman spectra of the pristine and functionalized HC11. | ||
The quantification of the density of defects can be achieved by calculating the ratio between the D band and the G band, using either the peak intensity ratio (ID/IG) or the area ratio (AD/AG).44,49 For small levels of disorder or defects the AD/AG can be used as the area under each peak represents all possible Raman scattering occurring for a given process.49,50 For large disorder or defect density, however, ID/IG is normally used since the intensity represents the phonon modes/molecular vibrations involved in most resonant Raman process.49 The full width at half-maximum, denoted in the literature as FWHM or Γ, can also be evaluated as measure of structural disorder,41 since it is reported to increase with the increase of the density of defects.41,44,47,49
In this study we considered the areas of the D band and D′ band normalized by the area of the G band (area ratio) as well as the FWHM of the G band (ΓG) to analyze the density of defects in the functionalized material. Fig. 4 shows the statistical analysis of 120 Raman spectra acquired from pristine and functionalized GNP HC11 samples in terms of AD/AG, AD′/AG and ΓG.
 | ||
| Fig. 4 Statistical analysis of the Raman spectra acquired from the HC11 and f-HC11 samples in terms of AD/AG, AD′/AG and ΓG. | ||
As depicted in Fig. 4, the AD/AG, AD′/AG and the ΓG histograms of the pristine GNP HC11 exhibit an almost symmetric distribution with mean values of 0.24 ± 0.08, 0.03 ± 0.01 and 20 ± 1 cm−1, respectively, a consequence of the homogeneity of the pristine material as well as its low initial defect concentration. The histograms of all functionalized GNP HC11 presented, in general, a shift of the maximum of the distribution to slightly higher mean values of AD/AG, AD′/AG and ΓG and a more asymmetric and broader distribution. This indicates the formation of an arbitrary amount of sp3 hybridized carbon resulting from the covalent functionalization. Interestingly, the f-HC11 250 showed higher mean values of the AD/AG and AD′/AG histograms when compared with the functionalized GNP HC11 at 180 °C, 200 °C and 220 °C, indicating a higher degree of defects. This may be related with the increasing of the reaction yield with increasing temperature, in agreement with the XPS analysis. Although, as mentioned before, this increase of the reaction yield at 250 °C may also indicate some degradation of the carboxyl groups at this temperature resulting from the decarboxylation of the functional group. This may also be related to the decrease of the weight loss of the f-HC11 250 in the TGA analysis.
In order to evaluate the solubility of the functionalized HC11 in water, the carboxyl groups were deprotonated in the presence of sodium hydroxide (NaOH) to yield the corresponding sodium salts. As depicted in Fig. S1 (ESI†) the wettability of the functionalized samples is clearly improved when compared to the pristine HC11.
A more detailed characterization was performed for the functionalized GNP HC11 reacted at 200 °C (f-HC11 200) since this material showed the highest amount of carboxylic acid functionalities, as demonstrated by the XPS analysis (Fig. 1b).
The spatial Raman mapping of a pristine GNP HC11 as well as f-HC11 200 flakes are presented in Fig. 5a. The integrated area of D band and G band of all spectra acquired (around 300 spectra) were fitted with Lorentzian functions using LabSpec 6 software. The AD/AG histograms of the GNP HC11 and f-HC11 200 are presented in Fig. 5b. As demonstrated in the colour-coded Raman mapping shown in Fig. 5a an increase of the overall intensity of the D band along with the decrease of the G band intensities are observed in the functionalized HC11 flake. Moreover, the AD/AG histogram of the f-HC11 200 (Fig. 4b) clearly shows a shift to a higher AD/AG mean value when compared to the pristine HC11 histogram, resulted from the disruption of the sp2 hybridized carbons of the HC11 after the functionalization process.
 | ||
| Fig. 5 (a) Optical micrographs and Raman mapping of HC11 and f-HC11 200 flakes in terms of D band and G band intensities; (b) AD/AG histograms of the same flakes. | ||
The XRD patterns of the pristine GNP HC11 and f-HC11 200 are presented in Fig. 6. The presence of a sharp peak at around 2θ = 26.6° (2θ = 26.7° for the GNP HC11 and 2θ = 26.6° for the f-HC11 200) and a weak peak at 2θ = 55.0° in both samples can be assigned to the (002) and (004) planes of graphitic carbon.51 The calculated d spacing was 0.344 nm and 0.347 nm for the GNP HC11 and f-HC11-200, respectively. These XRD results demonstrate that the crystalline structure of the HC11 was not greatly affected by the functionalization process in agreement with the Raman statistical analysis. Furthermore, the AFM analysis of the GNP HC11 and f-HC11 200 (Fig. S2, ESI†) showed that the thickness of the flakes is very similar for both samples. This may indicate that the functionalization might occur not only at the top surface of the flakes but in the edges as well.
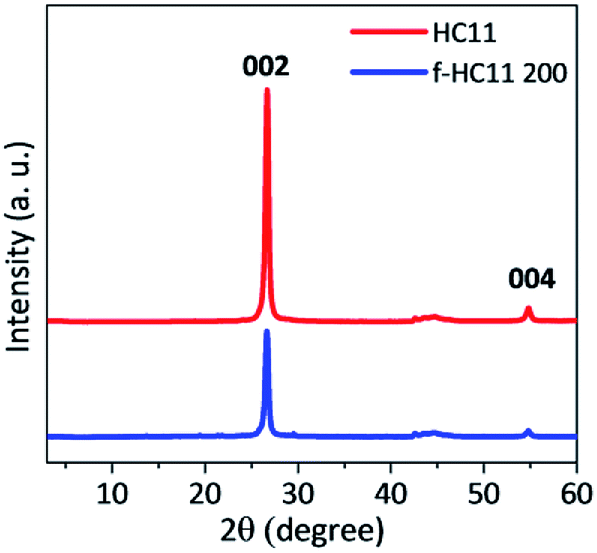 | ||
| Fig. 6 XRD pattern of the pristine HC11 and f-HC11 200. | ||
The SEM images of the pristine and functionalized HC11 are presented in Fig. 7a–d, respectively. The images show that the morphology of the functionalized HC11 is similar to the pristine material demonstrating that the functionalization process does not significantly affect the structural and morphological quality of the pristine HC11. This morphological similarity between both pristine and functionalized HC11 are also demonstrated by the TEM images (Fig. 7e and f, respectively).
 | ||
| Fig. 7 SEM images of (a) and (b) pristine HC11 and (c) and (d) f-HC11 200; TEM images of (e) pristine HC11 and (f) f-HC11 200. | ||
Effect of the reactants weight ratio
The effect of the IDA weight ratio in the functionalization reaction was also analysed. Different GNP HC11:IDA weight ratios (1.0:1.5 and 1.0:2.0) were tested, using the reaction temperature of 200 °C, and compared with the results obtained for the f-HC11 200 presented in the previous section of this paper. The functionalized HC11 samples were denominated as f-HC11 200–1.5, f-HC11 200–2.0 and f-HC11 200–1.0, respectively. The TGA curves shown in Fig. 8a demonstrated an increase of the weight loss with the increase of the IDA weight ratio. Moreover, the XPS results (Fig. 8b) clearly show an increase of the nitrogen atomic concentration from 6.24% to 8.05% and 8.71% for the f-HC11 200–1.0, f-HC11 200–1.5 and f-HC11 200–2.0 samples, respectively, showing that using higher amount of IDA can lead to an increase of functional groups attached to the surface of the GNP HC11. The detailed XPS atomic concentration and atomic ratio of the chemical elements on the pristine and functionalized HC11 are shown in Table S2 in ESI.†Fig. 8c compares the C 1s core-level spectra of the pristine and the functionalized HC11 with different IDA weight ratios. It is clear that the spectra become more asymmetric after the functionalization process and this asymmetry become more pronounced with the increase of IDA ratio, indicating that more functional groups were introduced on the surface of the GNP HC11.
 | ||
| Fig. 8 (a) Thermogravimetric analysis, (b) XPS wide scan spectra, (c) high resolution C 1s spectra for the functionalized HC11 with different IDA weight ratios. | ||
As a higher nitrogen atomic concentration was identified by XPS analysis in the sample f-HC11 200–2.0 we imaged this sample using SEM (Fig. 9a) and performed EDS analysis (Fig. 9b–f). It was demonstrated that the increase of the IDA ratio in the functionalization process resulted in a break of the smoothness of the surface layer of the HC11 flakes (Fig. 9a). The EDS mapping showed the distribution of the main elements identified in the functionalized sample: carbon (C, Fig. 9c), oxygen (O, Fig. 9d) and nitrogen (N, Fig. 9e). As shown in Fig. 9e the nitrogen element seems to be distributed homogenously over the sample appearing to be slightly more concentrated in the edges of the flakes. However, it was also possible to identify nitrogen in the flakes basal plane as well. The overall distribution of the elements identified is presented in the overlapped image (Fig. 9b) of the SEM and EDS analysis. Fig. 9f shows the EDS spectra resulted from the analysis performed in two different single points in the sample (red and blue spots, Fig. 9a). Both spectra clearly show peaks related to the carbon, nitrogen and oxygen elements in agreement with the XPS analysis.
 | ||
| Fig. 9 (a) SEM image; (b) overlap of the EDS mapping of the chemical elements distribution: (c) carbon, (d) oxygen and (e) nitrogen; (f) EDS single spectra of the red and blue single points marked in (a). | ||
Conclusions
In summary we have successfully demonstrated the functionalization of graphene nanoplatelets through solvent free 1,3-DCA reaction using the iminodiacetic acid (IDA) as α-amino acid and paraformaldehyde (PFA). The yield and the functionality of the carboxyl-terminated pyrrolidine ring attached on the surface of GNP could be affected by varying the temperature reaction. Increasing of the temperature from 180 °C to 250 °C was shown to favour the reaction yield, however, the decarboxylation of the functional groups increase for higher temperature reactions (220 °C and 250 °C), losing some of the functionality of these materials. The increase of the IDA weight ratio in the functionalization reaction showed to increase the functional groups attached to the surface of the GNPs and the SEM EDS analysis demonstrated that the functional groups are distributed homogenously over the sample. This fast and scalable approach for graphene functionalization offers considerable scope for the production of graphene-based nanocomposites with enhanced properties.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the Henry Royce Institute (EP/P025021/1) for the AFM facility and the support and funding from the Beijing Institute of Aeronautical Materials (BIAM). The research was performed at the BIAM-Manchester Technical Centre for Graphene Aerospace Materials at the University of Manchester. He Ren acknowledges the funding from China Scholarship Council (CSC).References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- C. N. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2017, 90, 75–127 CrossRef CAS.
- A. Bianco, H. M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- B. Z. Jang and A. Zhamu, J. Mater. Sci., 2008, 43, 5092–5101 CrossRef CAS.
- D. D. L. Chung, J. Mater. Sci., 1987, 22, 4190–4198 CrossRef CAS.
- T. Gatti, N. Vicentini, M. Mba and E. Menna, Eur. J. Org. Chem., 2016, 1071–1090 CrossRef CAS.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2013, 42, 3222–3233 RSC.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed., 1963, 75, 604–637 CAS.
- I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765–2810 CrossRef CAS PubMed.
- K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863–910 CrossRef CAS.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519–526 CrossRef CAS.
- V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760–761 CrossRef CAS.
- I. Kumar, S. Rana and J. W. Cho, Chem.–Eur. J., 2011, 17, 11092–11101 CrossRef CAS.
- N. Tagmatarchis and M. Prato, J. Mater. Chem., 2004, 14, 437–439 RSC.
- V. Georgakilas, A. B. Bourlinos, R. Zboril, T. A. Steriotis, P. Dallas, A. K. Stubos and C. Trapalis, Chem. Commun., 2010, 46, 1766–1768 RSC.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS.
- Y. Cao and K. N. Houk, J. Mater. Chem., 2011, 21, 1503–1508 RSC.
- P. A. Denis and F. Iribarne, Int. J. Quantum Chem., 2010, 110, 1764–1771 CAS.
- P. A. Denis and F. Iribarne, Chem. Phys. Lett., 2012, 550, 111–117 CrossRef CAS.
- F. G. Brunetti, M. A. Herrero, M. Munoz Jde, A. Diaz-Ortiz, J. Alfonsi, M. Meneghetti, M. Prato and E. Vazquez, J. Am. Chem. Soc., 2008, 130, 8094–8100 CrossRef CAS.
- F. G. Brunetti, M. A. Herrero, M. Munoz Jde, S. Giordani, A. Diaz-Ortiz, S. Filippone, G. Ruaro, M. Meneghetti, M. Prato and E. Vazquez, J. Am. Chem. Soc., 2007, 129, 14580–14581 CrossRef CAS PubMed.
- N. Rubio, M. A. Herrero, A. de la Hoz, M. Meneghetti, M. Prato and E. Vazquez, Org. Biomol. Chem., 2010, 8, 1936–1942 RSC.
- N. Rubio, M. A. Herrero, M. Meneghetti, A. Diaz-Ortiz, M. Schiavon, M. Prato and E. Vazquez, J. Mater. Chem., 2009, 19, 4407–4413 RSC.
- E. Vazquez and M. Prato, ACS Nano, 2009, 3, 3819–3824 CrossRef CAS.
- M. C. Paiva, F. Simon, R. M. Novais, T. Ferreira, M. F. Proenca, W. Xu and F. Besenbacher, ACS Nano, 2010, 4, 7379–7386 CrossRef CAS.
- E. Cunha, M. C. Paiva, L. Hilliou and J. A. Covas, Polym. Compos., 2017, 38, 947–954 CrossRef CAS.
- M. C. Paiva, E. P. Cunha, O. Voigt, M. Liebscher, F. Simon, J. Pionteck and P. Potschke, in Proceedings of Pps-32: The 32nd International Conference of the Polymer Processing Society, ed. A.Maazouz, Amer Inst Physics, Melville, 2017, vol. 1914 Search PubMed.
- P. Rodrigues, R. M. Santos, M. C. Paiva and J. A. Covas, Int. Polym. Process., 2017, 32, 614–622 CrossRef CAS.
- R. M. Santos, S. T. Mould, P. Formanek, M. C. Paiva and J. A. Covas, Polymers, 2018, 10, 222 CrossRef.
- R. M. Santos, C. Vilaverde, E. Cunha, M. C. Paiva and J. A. Covas, Soft Matter, 2016, 12, 77–86 RSC.
- A. Y. S. Eng, C. K. Chua and M. Pumera, Nanoscale, 2015, 7, 20256–20266 RSC.
- R. J. Young, M. F. Liu, I. A. Kinloch, S. H. Li, X. Zhao, C. Valles and D. G. Papageorgiou, Compos. Sci. Technol., 2018, 154, 110–116 CrossRef CAS.
- V. Vinciguerra, R. Bucci, F. Marini and A. Napoli, J. Therm. Anal. Calorim., 2006, 83, 475–478 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- L. G. Cancado, A. Jorio, E. H. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H. P. Steinruck, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS.
- J. M. Englert, P. Vecera, K. C. Knirsch, R. A. Schafer, F. Hauke and A. Hirsch, ACS Nano, 2013, 7, 5472–5482 CrossRef CAS.
- S. Niyogi, E. Bekyarova, M. E. Itkis, H. Zhang, K. Shepperd, J. Hicks, M. Sprinkle, C. Berger, C. N. Lau, W. A. deHeer, E. H. Conrad and R. C. Haddon, Nano Lett., 2010, 10, 4061–4066 CrossRef CAS.
- E. H. M. Ferreira, M. V. O. Moutinho, F. Stavale, M. M. Lucchese, R. B. Capaz, C. A. Achete and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 9 Search PubMed.
- D. M. Basko, S. Piscanec and A. C. Ferrari, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 10 CrossRef.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04817g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |