
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic layers stabilize earth-abundant metal–terpyridine diradical complexes for catalytic C–H activation†
Zekai
Lin‡
a,
Nathan C.
Thacker‡
a,
Takahiro
Sawano‡
a,
Tasha
Drake
a,
Pengfei
Ji
a,
Guangxu
Lan
a,
Lingyun
Cao
b,
Shubin
Liu
c,
Cheng
Wang
 b and
Wenbin
Lin
*ab
b and
Wenbin
Lin
*ab
aDepartment of Chemistry, University of Chicago, 929 E. 57th St., Chicago, Illinois 60637, USA. E-mail: wenbinlin@uchicago.edu
bCollaborative Innovation Center of Chemistry for Energy Materials, State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, PR China
cResearch Computing Center, University of North Carolina, Chapel Hill, North Carolina 27599-3420, USA
First published on 30th October 2017
Abstract
We report the synthesis of a terpyridine-based metal–organic layer (TPY-MOL) and its metalation with CoCl2 and FeBr2 to afford CoCl2·TPY-MOL and FeBr2·TPY-MOL, respectively. Upon activation with NaEt3BH, CoCl2·TPY-MOL catalyzed benzylic C–H borylation of methylarenes whereas FeBr2·TPY-MOL catalyzed intramolecular Csp3–H amination of alkyl azides to afford pyrrolidines and piperidines. X-ray absorption near edge structure (XANES), extended X-ray absorption fine structure (EXAFS), X-ray photoelectron spectroscopy, UV-Vis-NIR spectroscopy, and electron paramagnetic spectroscopy (EPR) measurements as well as density functional theory (DFT) calculations identified M(THF)2·TPY-MOL (M = Co or Fe) as the active catalyst with a MII-(TPY˙˙)2− electronic structure featuring divalent metals and TPY diradical dianions. We believe that site isolation stabilizes novel MII-(TPY˙˙)2− (M = Co or Fe) species in the MOLs to endow them with unique and enhanced catalytic activities for Csp3–H borylation and intramolecular amination over their homogeneous counterparts. The MOL catalysts are also superior to their metal–organic framework analogs owing to the removal of diffusion barriers. Our work highlights the potential of MOLs as a novel 2D molecular material platform for designing single-site solid catalysts without diffusional constraints.
Introduction
Over the past two decades, metal–organic frameworks (MOFs) have attracted great interest among scientists and engineers owing to their potential in various applications including gas storage and separation,1–6 heterogeneous catalysis,7–16 nonlinear optics,17,18 chemical sensing,19–21 biomedical imaging,22,23 and drug delivery.24,25 In particular, MOFs have provided an excellent platform for designing single-site solid catalysts for many important organic transformations.26–32 By shutting down intermolecular deactivation pathways via spatial isolation of active sites, MOFs have afforded turnover numbers (TONs) several orders of magnitude higher than their homogeneous analogs.26,29 The catalytic performance of MOFs is, however, still limited by the diffusion rates of large substrates and products within the 3D frameworks.33 Although many strategies have been devised to overcome this diffusion limitation of MOFs, for example, by elongating functional ligands26 or diluting them with catalytically inactive spectator ligands to construct MOFs with larger channels and pores,34 only moderate success has been achieved to date. MOFs constructed from elongated ligands tend to suffer from interpenetration as well as framework distortion, whereas MOFs built from mixed functional and spectator ligands have diminished atom efficiency.We recently showed that diffusional constraint of MOFs could be lifted by reducing one dimension of the MOF crystals to only a few nanometers in thickness to afford a new category of 2D materials, metal–organic layers (MOLs).35 Unlike 3D MOFs, the active sites in ultrathin 2D MOLs are readily accessible to substrates during catalytic reactions. On the other hand, MOLs still inherit the heterogeneous nature, ordered structure, and molecular tunability of MOF catalysts,36–38 and have the potential to provide a rare 2D molecular material platform for designing a new class of single-site solid catalysts without diffusional constraints. We report here the synthesis of a new metal–organic layer, TPY-MOL, based on Hf6(μ3-O)4(μ3-OH)4(HCO2)6 secondary building units (SBUs) and 4′-(4-carboxyphenyl)-[2,2′:6′,2′′-terpyridine]-5,5′′-dicarboxylate (TPY) bridging ligands and the metalation of TPY ligands in TPY-MOL with CoCl2 and FeBr2 to afford highly effective recyclable and reusable MOL catalysts for challenging benzylic C–H borylation and intramolecular sp3 C–H amination reactions (Fig. 1). Spectroscopic and computational studies identified unprecedented CoII/FeII-terpyridine diradical complexes as catalytic active sites for the borylation and amination reactions.
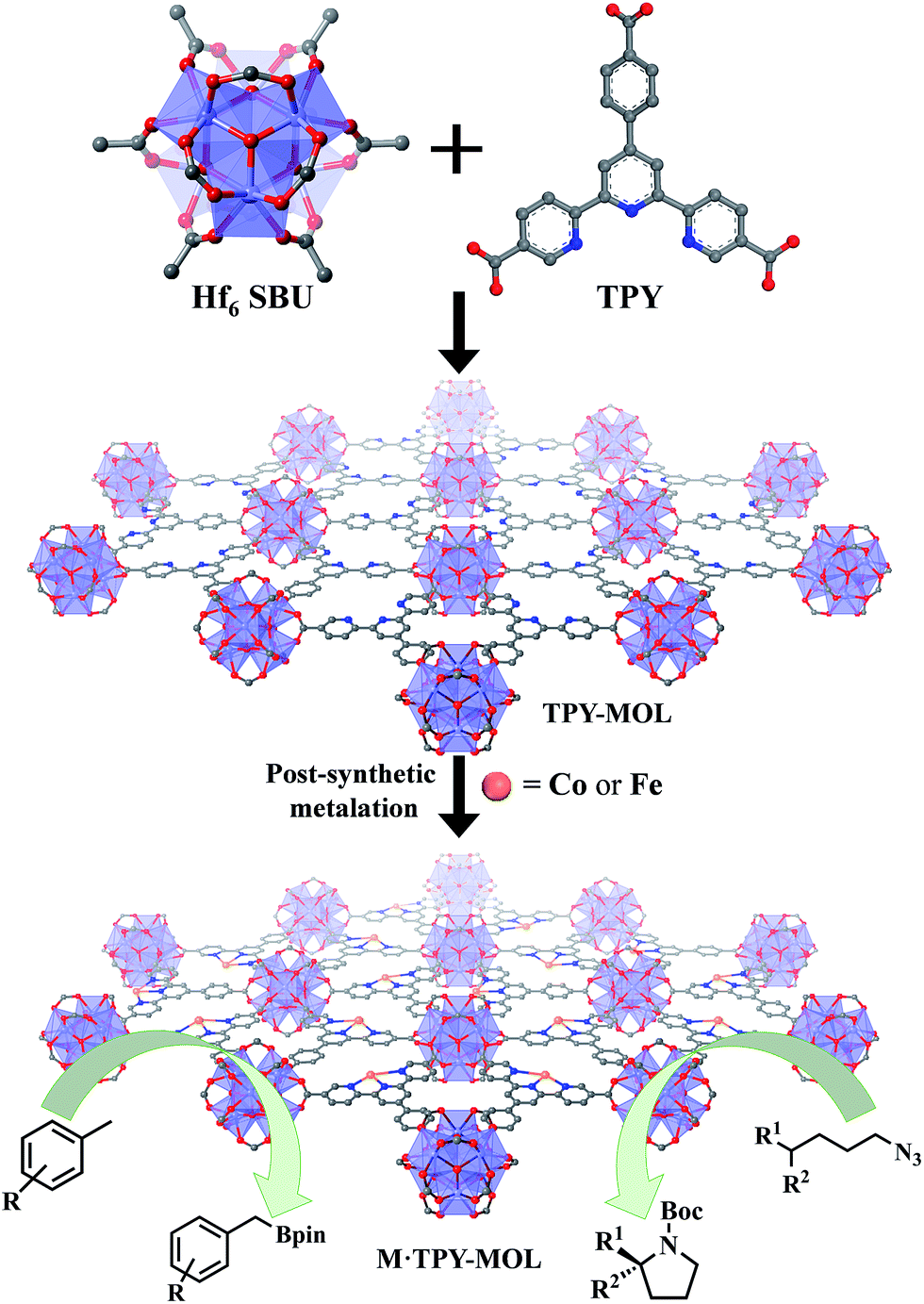 | ||
| Fig. 1 M·TPY-MOLs, constructed from Hf6 SBUs and TPY and then metalated with Co and Fe, were used for benzylic C–H borylation and intramolecular sp3 C–H Amination reactions, respectively. | ||
Owing to their distinct coordination, redox, and photophysical properties, terpyridines (tpy) and their metal complexes have been explored for potential applications in many fields, including polymer science,39,40 optoelectronics,41,42 medicinal chemistry,43,44 nanotechnology,45 and molecular catalysis.41,46,47 Although tpy derivatives provide a potentially interesting ligand platform for designing earth-abundant metal catalysts, few examples have been reported in the literature,47–50 in part due to their strong propensity to undergo disproportionation reactions to form catalytically inactive M(tpy)2 complexes.48,49 Installation of bulky groups on the 6,6′′-positions of tpy could prevent such bimolecular deactivation processes in M-tpy catalysts but often at the expense of their catalytic activities.48 By incorporating a tpy derivative into the TPY-MOL, we effectively shut down the disproportionation decomposition pathway without relying on steric protection at the 6,6′′ positions and obtained highly effective MOL catalysts based on M-tpy complexes (M = Co or Fe) for benzylic C–H borylation and intramolecular sp3 C–H amination reactions. The MOL-based M-tpy catalysts displayed at least 20 times higher catalytic activity and distinct chemoselectivity in benzylic C–H borylation reactions and 50 times higher TONs in intramolecular sp3 C–H amination reactions over their homogeneous analogs.
Results and discussion
Synthesis and postsynthetic metalation of TPY-MOL
TPY-MOL was synthesized in 76% yield by heating a mixture of HfCl4, H3TPY, and formic acid in DMF and water at 120 °C for 24 h. The PXRD pattern of TPY-MOL matched the simulated pattern based on the (hk0) reflections only that are characteristic of 2D MOL structures and aligned well with that of isostructural BTB-MOL (BTB is 1,3,5-benzenetribenzoate, Fig. 2a).35 Transmission Electron Microscopy (TEM) images showed ultra-thin films of TPY-MOL whereas the high resolution TEM (HRTEM) images of TPY-MOL showed a clear lattice with the dark spots corresponding to Hf6 clusters (Fig. 2b and c). The distances between adjacent spots on the HRTEM image (20.1 Å) matched well with that between two adjacent Hf6 SBUs (20.0 Å) in the MOL structural model. Atomic Force Microscopy (AFM) images of TPY-MOL indicated monolayer thickness for many nano-sheets with an average measured thickness of 1.2 nm, corresponding to the van der Waals size of Hf6 SBUs (Fig. 2d and e).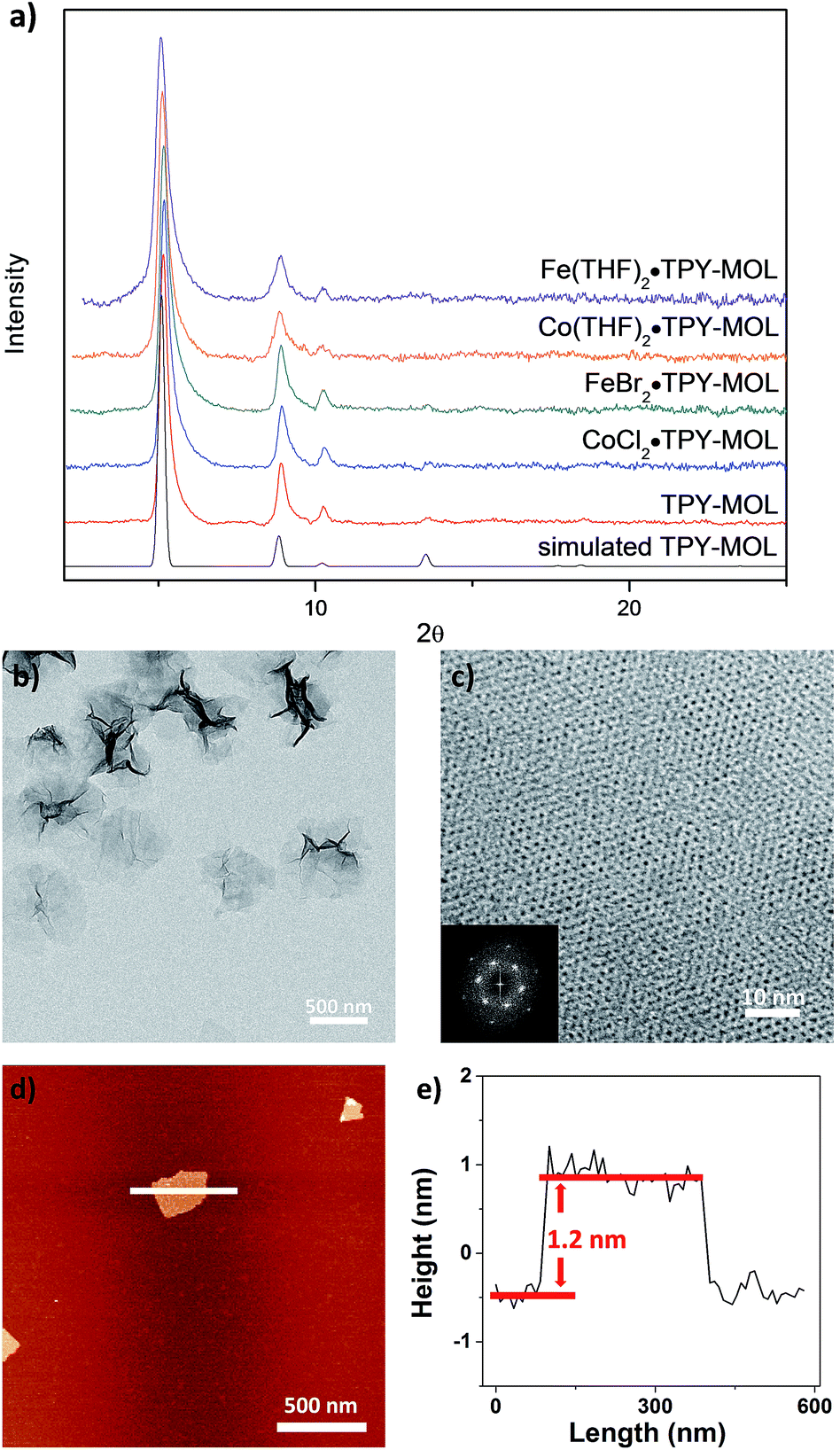 | ||
| Fig. 2 (a) PXRD patterns of TPY-MOL, CoCl2·TPY-MOL, FeBr2·TPY-MOL, Co(THF)2·TPY-MOL, and Fe(THF)2·TPY-MOL in comparison to simulated PXRD pattern for TPY-MOL; (b) TEM image of TPY-MOL; (c) HRTEM image and fast Fourier transform (FFT) pattern of TPY-MOL; (d) tapping-mode atomic-force microscope (AFM) topographic image of TPY-MOL; (e) height profile along the white line of TPY-MOL. | ||
TPY-MOL was readily metalated with CoCl2 or FeBr2(THF)2 (1.05 eqv. w.r.t TPY) to afford CoCl2·TPY-MOL or FeBr2·TPY-MOL with 100% metal loading, as determined by inductively coupled plasma-mass spectrometry (ICP-MS). X-ray absorption near edge structure (XANES) analysis revealed +2 oxidation state for CoCl2·TPY-MOL and FeBr2·TPY-MOL (Fig. 3a and b). The oxidation state assignments were further confirmed by X-ray photoelectron spectroscopy (XPS, Fig. S15, ESI†). Extended X-ray absorption fine structure (EXAFS) fitting indicated the coordination of Co(II) to three N atoms of TPY and two chlorides in CoCl2·TPY-MOL and the coordination of Fe(II) to three N atoms of TPY and two bromides in FeBr2·TPY-MOL (Fig. 3c and d). The similarity of EXAFS-derived bond distances in CoCl2·TPY-MOL (Co–Nc = 2.09 ± 0.01 Å, Co–Nt = 2.16 ± 0.01 Å and Co–Cl = 2.28 ± 0.01 Å) and crystallographically determined CoCl2·tpy distances (Co–Nc = 2.071 Å, Co–Nt = 2.139 Å and Co–Cl = 2.298 Å) validates the EXAFS fitting results.
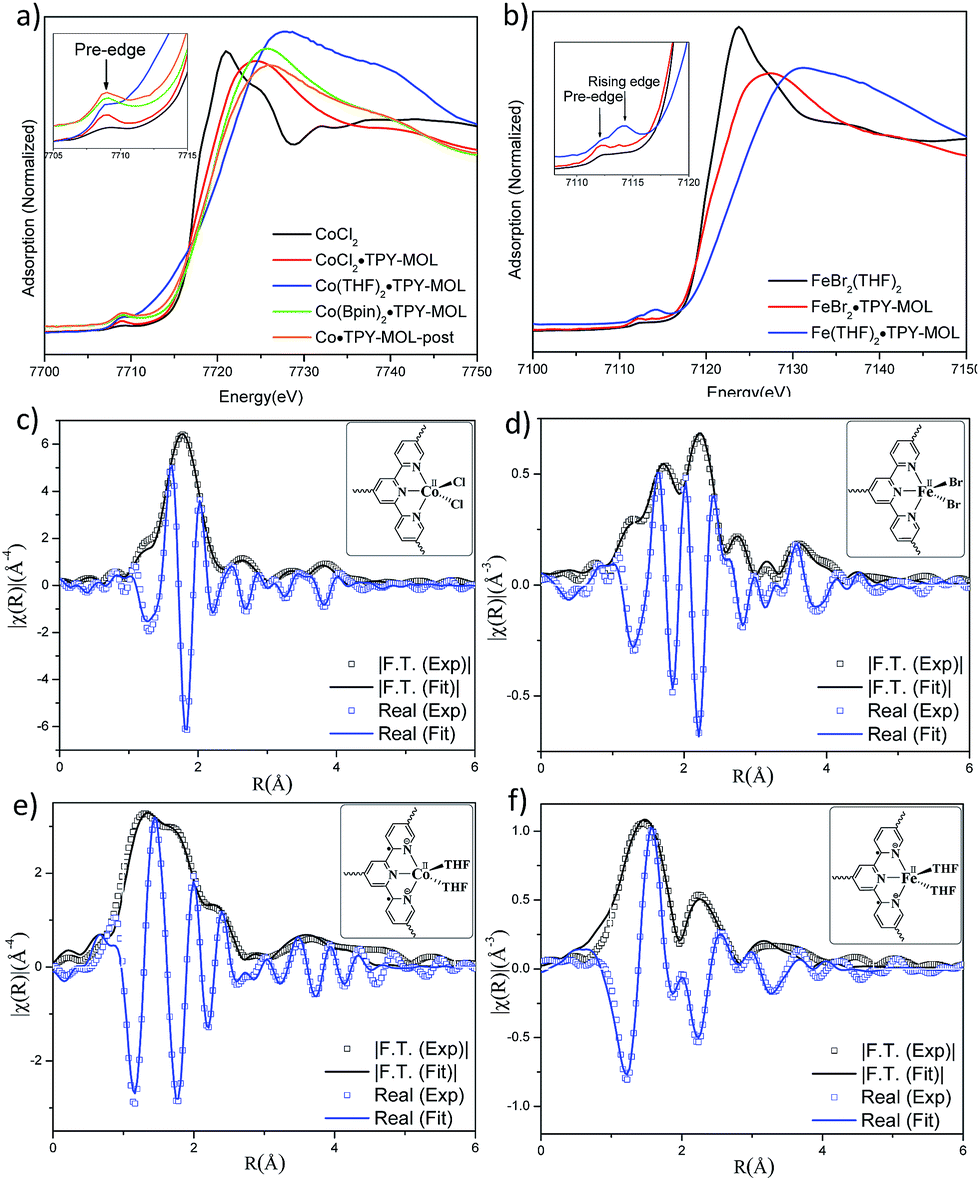 | ||
| Fig. 3 (a) XANES spectra of CoCl2, CoCl2·TPY-MOL, Co(THF)2·TPY-MOL, Co(Bpin)2·TPY-MOL, and Co·TPY-MOL-post; (b) XANES spectra of FeBr2(THF)2, FeBr2·TPY-MOL, and Fe(THF)2·TPY-MOL; (c–f) experimental EXAFS spectra and fits of CoCl2·TPY-MOL, R factor = 0.006 (c), FeBr2·TPY-MOL, R factor = 0.011 (d), Co(THF)2·TPY-MOL, R factor = 0.013 (e) and Fe(THF)2·TPY-MOL, R factor = 0.015 (f) in R space showing the magnitude of Fourier transform (black hollow squares, black solid line) and real components (blue hollow squares, blue solid line). | ||
Co-TPY-MOL catalyzed benzylic C–H borylation
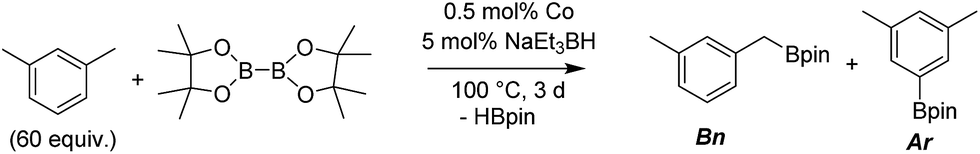
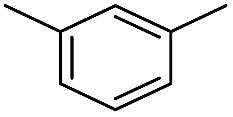
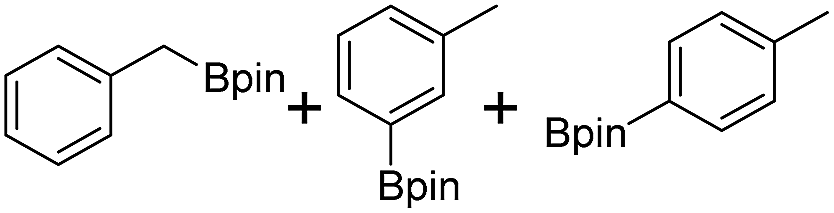
We first investigated C–H borylation of m-xylene by Co·TPY-MOL. Organoboronic compounds are a useful class of intermediates for forming carbon–carbon and carbon–heteroatom bonds through coupling reactions. C–H borylation with boron reagents such as B2pin2 is one of the most direct and convenient methods for the synthesis of organoboronic compounds. Although C–H borylation with arenes has been developed in the past two decades, benzylic C–H borylation is still rare (Table S7, ESI†).27,51–56 Upon activation with NaEt3BH, CoCl2·TPY-MOL (0.5 mol%) catalyzed m-xylene borylation with B2pin2 at 100 °C over 3 days to afford 42% yield of borylated products, with a 4.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity favoring the benzylic position (Table 1, entry 1). The borylated products were obtained in 95% yield with a slightly higher selectivity for benzylic borylation (4.6:1) when the catalyst loading increased to 1.0 mol% (Table 1, entry 2). The activation of CoCl2·TPY-MOL with NaEt3BH is necessary for the borylation reaction (Table 1, entry 3). Under identical conditions, a TPY-MOF control, which is isostructural to the previously reported BTB-MOF in which 2D layers stack in a staggered arrangement to result in a 3D MOF,35 gave no conversion, likely due to slow diffusion of the substrates and products (Table 1, entry 4). The homogeneous analog gave 2% borylated products with a 5.7:1 selectivity favoring the arene C–H bond (Table 1, entry 5). Such moderate arene borylation activity was recently reported for homogenous tpy-Co derivatives.49 Active site isolation in MOLs thus not only increases the TON by more than 20 times (over the homogeneous analog) but also afforded unusual selectivity of borylation for the benzylic C–H bond.
:1 selectivity favoring the benzylic position (Table 1, entry 1). The borylated products were obtained in 95% yield with a slightly higher selectivity for benzylic borylation (4.6:1) when the catalyst loading increased to 1.0 mol% (Table 1, entry 2). The activation of CoCl2·TPY-MOL with NaEt3BH is necessary for the borylation reaction (Table 1, entry 3). Under identical conditions, a TPY-MOF control, which is isostructural to the previously reported BTB-MOF in which 2D layers stack in a staggered arrangement to result in a 3D MOF,35 gave no conversion, likely due to slow diffusion of the substrates and products (Table 1, entry 4). The homogeneous analog gave 2% borylated products with a 5.7:1 selectivity favoring the arene C–H bond (Table 1, entry 5). Such moderate arene borylation activity was recently reported for homogenous tpy-Co derivatives.49 Active site isolation in MOLs thus not only increases the TON by more than 20 times (over the homogeneous analog) but also afforded unusual selectivity of borylation for the benzylic C–H bond.
We further investigated the substrate scope for Co(THF)2·TPY-MOL catalyzed C–H borylation reactions. Benzylic borylated products were produced exclusively for p-xylene, 1-t-butyl-4-methylbenzene, and mesitylene in >90% yields (Table 2, entries 2–4). For p-methoxytoluene, a high selectivity of 59: 6: 1 was obtained for the benzylic borylated product (Table 2, entry 5). For toluene, borylated products were obtained in 92% yield, but the selectivity for the benzylic borylation product was moderate (Table 2, entry 6). These results indicate the influence of steric hindrance on the selectivity of benzylic vs. aromatic borylation by Co(THF)2·TPY-MOL.
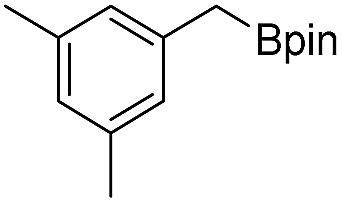
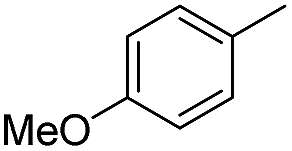
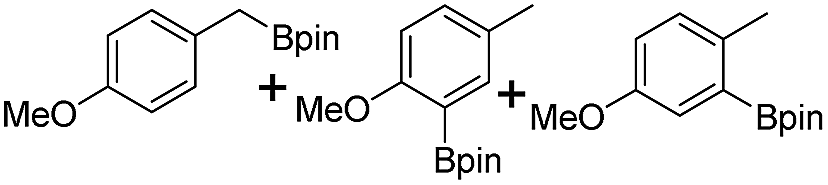
Co·TPY-MOL was recovered and used for at least 10 times without any loss of activity in C–H borylation of p-xylene (Fig. S32, ESI†). We conducted several tests to demonstrate the heterogeneity of Co·TPY-MOL. First, we showed that the PXRD of Co·TPY-MOL recovered from C–H borylation of p-xylene remained the same as that of freshly prepared Co·TPY-MOL (Fig. S33, ESI†). Second, we used ICP-MS to show that the amounts of Co and Hf leaching into the supernatant during the C–H borylation of p-xylene were only 0.092% and 0.037% respectively. Finally, we observed that the removal of Co·TPY-MOL from the reaction mixture after several hours stopped the C–H borylation of p-xylene (Scheme S2, ESI†).
Identification of the Co(THF)2·TPY-MOL catalyst
We studied the catalytically active species by hydrogen quantification, infrared (IR), UV-Vis-NIR, XPS, and electron paramagnetic resonance (EPR) spectroscopy, XANES, EXAFS, and density functional theory (DFT) calculations. One equiv. of H2 was generated upon treatment of CoCl2·TPY-MOL with NaEt3BH, suggesting the formation of Co(THF)x·TPY-MOL via reductive elimination of H2 from the putative CoH2·TPY-MOL intermediate. This 2-electron reduction process was also confirmed by titration of Co(THF)x·TPY-MOL with ferrocenium hexafluorophosphate which resulted in the generation of two equiv. of ferrocene w.r.t to CoTPY-MOL (Fig. S6, ESI†). IR spectra showed no characteristic band of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, ruling out the coordination of dinitrogen to Co. XANES analysis indicated +2 oxidation state for the Co center (Fig. 3a). This oxidation state assignment was further supported by XPS spectroscopy which gave a Co 2p3/2 binding energy of 781.2 eV with the expected shake-up peak for the CoII centers (Fig. 4).
N, ruling out the coordination of dinitrogen to Co. XANES analysis indicated +2 oxidation state for the Co center (Fig. 3a). This oxidation state assignment was further supported by XPS spectroscopy which gave a Co 2p3/2 binding energy of 781.2 eV with the expected shake-up peak for the CoII centers (Fig. 4).
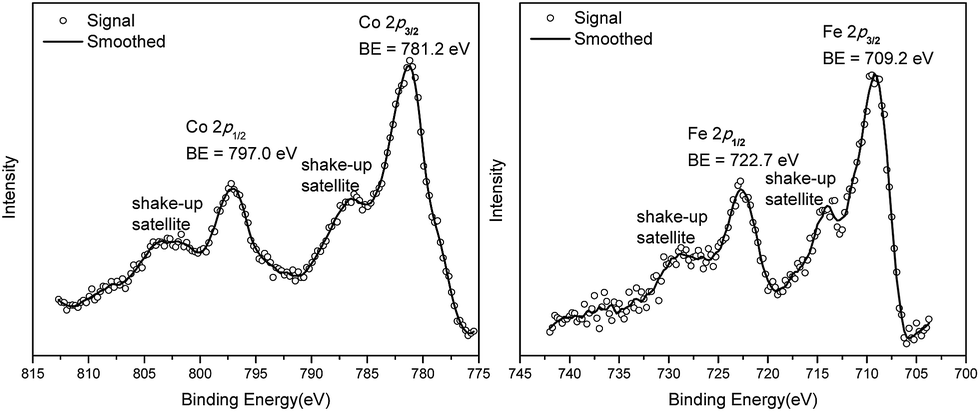 | ||
| Fig. 4 Co 2p and Fe 2p XPS spectra of Co(THF)2·TPY-MOL (left) and Fe(THF)2·TPY-MOL (right). | ||
The EXAFS spectra at the Co K-edge were well fitted with a structural model in which Co coordinates with three N atoms of TPY and two THF molecules (Fig. 3e). Co–N bond distances (Co–Nc = 1.81 ± 0.02 Å, Co–Nt = 1.92 ± 0.02 Å) are shorter than those of the reported [CoI(tpy)2]57 (Co–Nc = 2.003 Å, Co–Nt = 2.130 Å), arguing against the +1 oxidation state for Co(THF)2·TPY-MOL. Furthermore, Co(THF)2·TPY-MOL has shorter Co–N bond distances than those for CoIICl2·TPY-MOL (Co–Nc = 1.90 ± 0.01 Å, Co–Nt = 2.09 ± 0.01 Å), but similar Co–N bond distances to a reported low-spin CoII(tpy)(BH4) complex with the (tpy˙)− ligand (Co–Nc = 1.810 Å, Co–Nt = 1.925 Å).58 The Co–N bond distance analysis thus supports the formulation of the CoII-(tpy˙˙)2− electronic structure for Co(THF)2·TPY-MOL.
We used UV-Vis-NIR spectroscopy to discern the diradical nature of TPY ligands in CoTPY-MOLs (Fig. 5). Co(THF)2·TPY-MOL exhibited two intense, broad bands centered at 552 and 759 nm and a weak but broad band at 1105 nm, indicative of π to π* and π* to π* transitions for the reduced tpy ligand.59–63 In contrast, these bands are absent in CoCl2·TPY-MOL with the neutral TPY ligand (Fig. 5). The proposed (tpy˙˙)2− species was previously observed in reduced M(tpy)2 complexes, such as CrIII(tpy)2, VIV(tpy)2, and TiIV(tpy)2, by Wieghardt and coworkers.62,63 However, we are not aware of any example of M-tpy complexes featuring the (tpy˙˙)2− species.
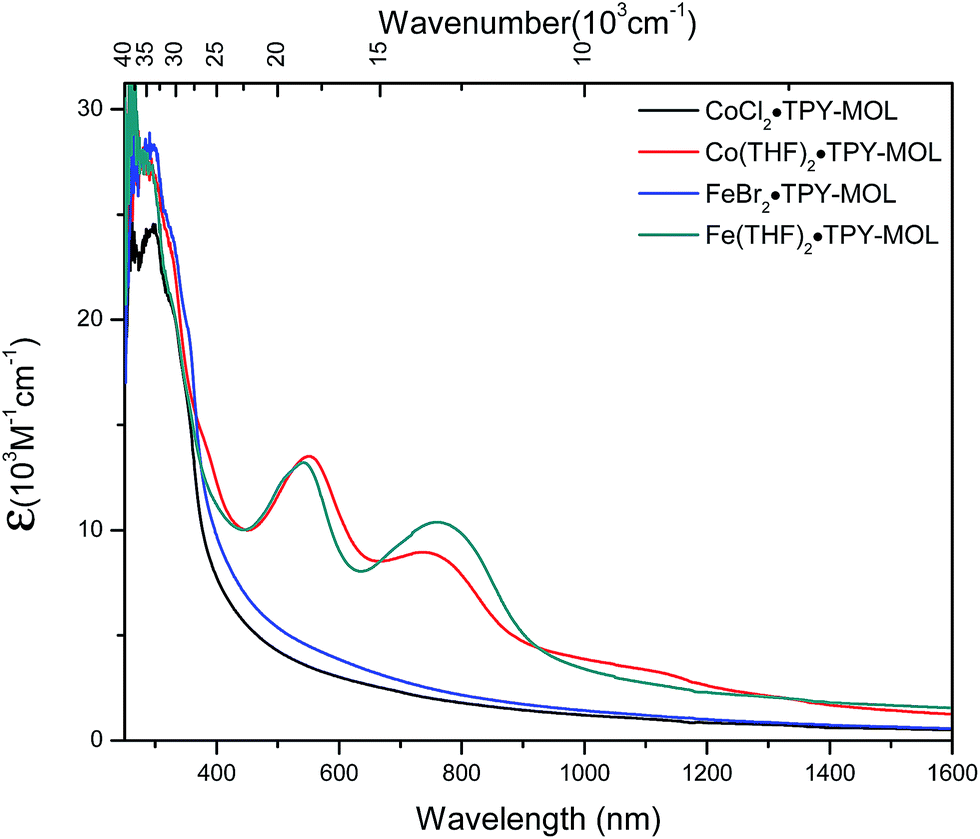 | ||
| Fig. 5 UV-Vis-NIR absorption spectra of CoCl2·TPY-MOL, Co(THF)2·TPY-MOL, FeBr2·TPY-MOL, and Fe(THF)2·TPY-MOL in THF at 25 °C. | ||
Our XANES, EXAFS, and XPS results clearly indicate the CoII oxidation state for Co(THF)2·TPY-MOL whose electronic structure is best described as CoII(THF)2·(TPY˙˙)2−-MOL. The (tpy˙˙)2− diradical dianion can have either a singlet (S = 0) or a triplet (S = 1) ground state, which can potentially be experimentally differentiated by EPR spectroscopy. EPR spectroscopy of Co(THF)2·TPY-MOL gave an isotropic signal with giso = 2.003 at r.t. in toluene suspension. The same MOL sample frozen at 20 K exhibits a stronger isotropic signal with giso = 2.003, confirming that the same species was detected at r.t. and 20 K (Fig. 6). More interestingly, the g value falls in the range of 2.003–2.005,59,64,65 where radicals in extended organic π systems were often observed. The EPR signal intensity was temperature-dependent, which can be fitted with the Bleaney and Bowers equation66 typically used for organic diradicals (Fig. 6). The fitting of temperature-dependent EPR signals indicates that the (TPY˙˙)2− diradical has a singlet ground state with singlet-to-triplet energy gap of 0.04 kcal mol−1. The observed EPR signal is thus attributed to the thermally populated TPY triplet excited state.67 Moreover, a weak signal giso ≈ 2.04 was observed at 20 K, consistent with low-spin CoII centers. Therefore, our EPR data provide strong support to our proposed electronic structure CoII(THF)2·(TPY˙˙)2−-MOL. We have ruled out the possibility of SBU-based free radicals because TPY-MOL treated with NaEt3BH exhibited no signal at r.t. or 20 K (Fig. S16, ESI†).
 | ||
| Fig. 6 X-band EPR spectra of Co(THF)2·TPY-MOL (left) and Fe(THF)2·TPY-MOL (right) suspended in toluene at r.t. and 20 K. Microwave frequency: 9.629 GHz for Co(THF)2·TPY-MOL at r.t.; 9.629 GHz for Co(THF)2·TPY-MOL at 20 K; 9.634 GHz for Fe(THF)2·TPY-MOL at r.t.; 9.630 GHz for Fe(THF)2·TPY-MOL at 20 K. Insets are temperature-dependent EPR intensity plots and their fits to the Bleaney and Bowers equation. The fitting results gave a singlet to triplet (TPY˙˙)2− energy gap of 0.04 and 0.10 kcal mol−1 for Co(THF)2·TPY-MOL (left) and Fe(THF)2·TPY-MOL, respectively. | ||
Density functional theory (DFT) calculations and natural population analyses with the B3LYP/6-311G(d) basis set on Co(THF)2·tpy gave a doublet ground state (GS) with high positive charge distribution (1.24) on the Co center and negative charge distribution (−1.34) on tpy (Table S9, ESI†). A comparison charge distribution on CoCl2·tpy revealed that the Co center in Co(THF)2·tpy maintains +2 oxidation state. A Mulliken spin population analysis and spin density plot revealed that 0.996 unpaired electron resides on the Co center, affording a ground state with a low-spin CoII, d7 doublet (SCo = 1/2) and a tpy diradical dianion singlet (Stpy = 0) (Fig. S47, ESI†). The singlet tpy diradical dianion is not expected to give any EPR signal. Interestingly, the energy of quartet state of Co(THF)2·tpy is calculated to be only 0.40 kcal mol−1 higher than that of the doublet GS. This small energy gap is consistent to that deduced from temperature-dependent EPR signals of Co(THF)2·tpy. The charge distribution of the quartet state is similar to that of the doublet GS with positive charge (1.29) on the Co center and negative charge (−1.40) on tpy (Table S9, ESI†). The calculated bond distances are similar between the quartet state and the doublet GS (Table S11, ESI†). A Mulliken spin density population and spin density plot of the quartet state revealed the residence of the 1.091 unpaired spin on Co center and 1.887 unpaired spins on tpy, affording a low-spin CoII, d7 doublet (SCo = 1/2) and a tpy triplet diradical dianion (Stpy = 1) (Fig. 7). The energetically accessible low-lying triplet excited state of (tpy˙˙)2− was previously proposed for the hypothetical [ZnII(tpy2−)(NH3)2]0.62 DFT calculations thus support the origin of the experimental tpy diradical dianion EPR signal as thermally populated quartet state of CoII(THF)2·tpy˙˙. Moreover, we believe that conjugation of Hf6 SBU to TPY can further stabilize TPY diradical dianion and lower the energy difference between doublet and quartet states of CoII(THF)2·TPY˙˙-MOL.
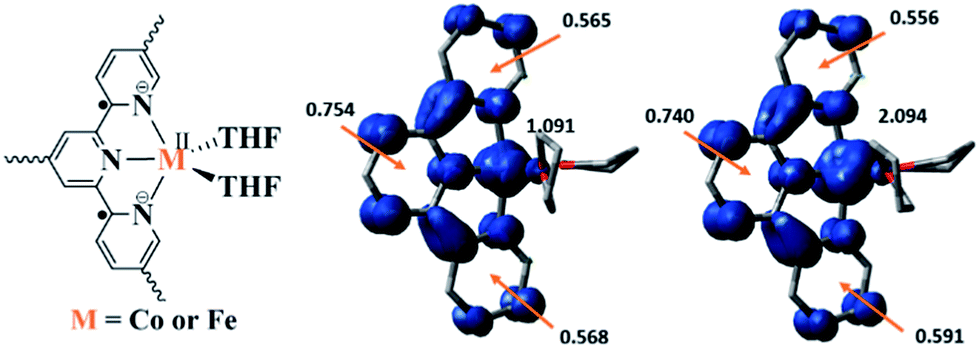 | ||
| Fig. 7 Proposed electronic structure of MII(THF)2·(TPY˙˙)2−-MOL, M = Co or Fe (left); calculated Mulliken spin density distribution and spin density plots (blue: positive; green: negative) of Co(tpy)(THF)2 quartet state (middle) and Fe(tpy)(THF)2 quintet state (right). | ||
We also investigated the activation of CoCl2·tpy molecular complex with NaEt3BH. Upon treating CoCl2·tpy in THF with 10 equiv. of NaEt3BH, the mixture turned dark green immediately with concomitant formation of Co nanoparticles as black precipitate (Fig. S7 and S9, ESI†). The solution was filtered through Celite and evaporated to afford Co(tpy)2 (HR-MS calculated for C30H22N6Co [M+]: 525.1238, found: 525.1257).
Mechanistic studies of Co(THF)2·TPY-MOL catalyzed C–H borylation
To gain insight into the mechanism of the C–H borylation reaction, we carried out several experiments. First, we performed kinetic isotope effect (KIE) studies in order to afford information on the rate-determining step of the C–H borylation reactions. The initial rates of C–H borylations with p-xylene and p-xylene-d8 were determined by running parallel reactions in separate vessels, and the comparison of the initial rates gave a KIE value of 2.7 (Scheme S3, ESI†). Such a primary KIE indicates the involvement of the C–H bond breaking in the rate-determining step.Second, we detected the presence of HBpin by gas chromatography-mass spectrometry (GC-MS) at the end of the C–H borylation reactions. Third, we determined the resting state of the catalyst by EXAFS studies. By treating Co(THF)2·TPY-MOL with 20 equiv. of B2pin2, we obtained the Co(Bpin)2·TPY-MOL product in which Co coordinates to three N atoms of TPY and two Bpin groups according to EXAFS fitting (Fig. S13, ESI†). To determine the resting state of the catalyst, the C–H borylation reaction was stopped at 70% conversion and the organic volatiles were evaporated. EXAFS studies indicated that the remaining residue had the same structure as Co(Bpin)2·TPY-MOL (Fig. S14, ESI†). Finally, EPR spectra of Co(Bpin)2·TPY-MOL did not show any signals corresponding to a TPY-based radical EPR signal (Fig. S16, ESI†), suggesting a typical CoII·TPY complex with negative charge localized on the Bpin ligands.
On the basis of these experimental and calculation results, we propose a catalytic cycle for the C–H active borylation of methylarenes as shown in Scheme 1. The CoCl2·TPY-MOL(I) is activated by NaEt3BH in THF to give the CoH2·TPY-MOL(II) intermediate, which quickly undergoes reductive elimination of H2 to produce the CoII(THF)2·(TPY˙˙)2−-MOL(III) catalyst. Oxidative addition of B2(pin)2 to III results in Co(Bpin)2·TPY-MOL(IV), which is the catalyst resting state for the C–H borylation reactions. σ-Bond metathesis between IV and methylarene proceeds as a rate-determining step to form Co(H)(Bpin)·TPY(V) and the benzylic borylated product. The reaction of V with B2pin2 regenerates the intermediate IV and forms HBpin as a byproduct via σ-Bond metathesis. The transformation of V to IV could alternatively involve a two-step process of reductive elimination of HBpin from V followed by oxidative addition of B2Pin2 to the intermediate to form IV. We are not able to differentiate between the concerted one-step σ-bond metathesis and the two-step reductive elimination/oxidative addition process.
 | ||
| Scheme 1 Proposed mechanism for the Co(THF)2·TPY-MOL catalyzed C–H borylation of arenes with B2pin2. | ||
Fe·TPY-MOL catalyzed intramolecular sp3 C–H amination
TPY-MOL was also metalated with FeBr2(THF)2 to generate FeBr2·TPY-MOL. Similar to the Co(THF)2·TPY-MOL case, when FeBr2·TPY-MOL was treated with 10 equiv. of NaEt3BH, Fe(THF)2·TPY-MOL was generated along with 1 equiv. of H2. This 2-electron reduction process was also confirmed by titration of Fe(THF)2·TPY-MOL with ferrcenium hexafluorophosphate which resulted in the generation of two equiv. of ferrocene. EXAFS fitting indicates Fe coordinates to three N from TPY and two THF molecules for Fe(THF)2·TPY-MOL (Fig. 3f) while infrared spectroscopy indicates no coordination of dinitrogen to Fe centers. The oxidation state of Fe(THF)2·TPY MOL was determined to be +2 by XANES analysis since the pre-edge position for Fe(THF)2·TPY-MOL (7111.6 eV) aligned well with FeBr2(THF)2 (7111.5 eV), FeBr2·TPY-MOL (7111.5 eV) and two reported five-coordinate species (iPrPDI)FeCl2 (7111.8 eV) and (iPrPDI)Fe(N2)2 (7111.9 eV).68 Interestingly, a second feature at 7113.2 eV was observed for Fe(THF)2·TPY-MOL, assignable to the 1s to ligand π* transitions. This feature was also seen in a reported (iPrPDI2−)FeII(N2)2 species (7114.0 eV). It is worth mentioning that [Fe(tpy)2]n+ (n = 0, 1, 2) were all determined to have FeII centers.69 Furthermore, XPS spectroscopy clearly shows FeII oxidation state for Fe(THF)2·TPY-MOL based on characteristic Fe 2P3/2 binding energy of 709.2 eV and shake-up peaks (Fig. 4). The electronic spectrum of Fe(THF)2·TPY-MOL is very similar to that of CoII(THF)2·(TPY˙˙)2−-MOL, indicating the presence of (TPY˙˙)2− diradical dianion on Fe(THF)2·TPY-MOL (Fig. 5). Fe(THF)2·TPY-MOL gave an EPR signal with giso = 2.003 at r. t. in a toluene suspension. The same MOL sample frozen at 20 K exhibited a stronger signal with giso = 2.003 (Fig. 4). The fitting of temperature-dependent EPR signals indicates that the (TPY˙˙)2− diradical has a singlet ground state with singlet-to-triplet energy gap of 0.10 kcal mol−1. The observed EPR signal is thus attributed to the thermally populated TPY triplet excited state (Fig. 6).67 Therefore, the EPR data provide strong evidence of our proposed electronic structure of the FeII(THF)2·(TPY˙˙)2−-MOL catalyst.DFT calculations and natural population analyses with the B3LYP/6-311G(d) basis set on Fe(THF)2·tpy gave a triplet GS with high positive charge distribution (1.29) on the Fe center and negative charge distribution (−1.39) on tpy (Table S10, ESI†). Spin density plot of the GS revealed that 2.013 unpaired electrons reside on the Fe center, affording an intermediate-spin FeII, d6 center (SFe = 1), and a tpy singlet diradical dianion antiferromagnetically coupled to each other (Stpy = 0) (Fig. S51, ESI†). The GS of Fe(THF)2·tpy again is not expected to give any organic radical EPR signal, which contradicts our experimental results. We believe that the experimental tpy EPR signal comes from thermal population of the quintet state of Fe(THF)2·tpy which is only 5.26 kcal mol−1 higher in energy than that of triplet GS, consistent to our EPR analysis. The charge distribution of the quintet state is similar to that of triplet GS with positive charge (1.34) on the Fe center and negative charge (−1.44) on tpy (Table S10, ESI†). A Mulliken spin population analysis and spin density plot revealed that 2.094 unpaired spins reside on the Fe center and 1.887 unpaired spins on tpy, affording an intermediate-spin FeII, d6 compound (SFe = 1), and a tpy triplet diradical dianion (STPY = 1) (Fig. 7), which is consistent with our experimental EPR results. The coordination of Hf6 SBUs to TPY is expected to further stabilize TPY diradical dianion and lower the energy difference between triplet and quintet states of FeII(THF)2·(TPY˙˙)2−-MOL.
Upon activation with NaEt3BH, 2 mol% of FeBr2·TPY-MOL catalyzed intramolecular Csp3–H amination of 1-azido-4-phenylbutane (1a) in the presence of two equivalents of di-tert-butyl dicarbonate (Boc2O) at 90 °C to form Boc-protected α-phenyl pyrrolidine (2a) in 89% yield. This level of activity is 9 times as high as that of the MOF control (Table 3, entry 4). Under identical conditions, the homogeneous tpy-Fe catalyst only afforded the product in 3% yield, probably due to the deactivation of tpy-Fe catalyst via bimolecular pathways (Table 3, entry 5). Indeed, treatment of FeBr2·tpy with 10 equiv. of NaEt3BH produced a mixture Fe(tpy)2 and Fe nanoparticles; such a disproportionation reaction was previously observed for a series of (PDI)FeBr2 complexes.69,70
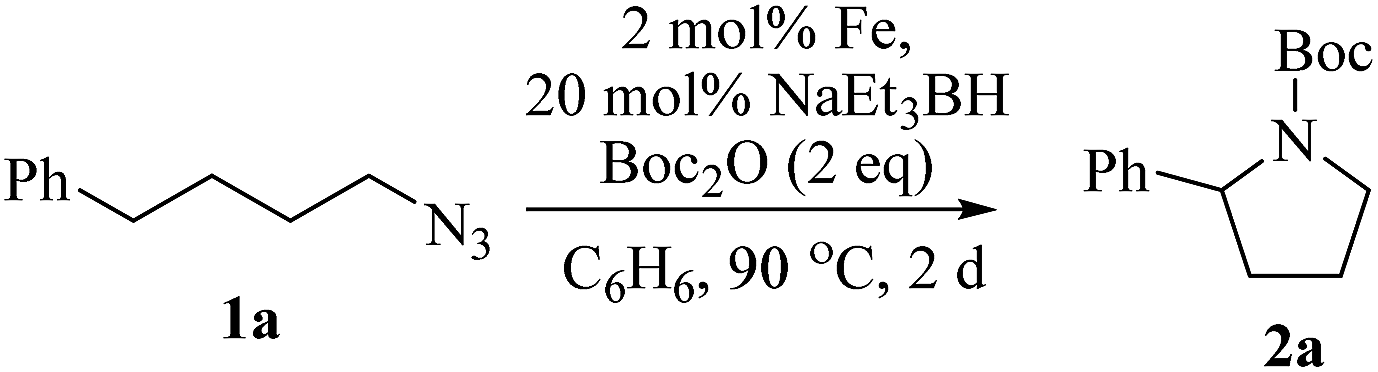
A higher TON of 76 was achieved when the Fe loading was decreased to 1 mol% (Table 3, entry 2). With a much simpler ligand, Fe·TPY-MOL outperformed Betley's Fe-dipyrrinato homogenous catalyst by 13 times71 and our recently reported NacNac-MOF catalysts by 4 times28 in TONs. It is worth noting that FeBr2·TPY-MOL, without activation with NaEt3BH, showed low activity (Table 3, entry 3), suggesting that the formation of Fe-nitrene compound might be a key elementary step of the intramolecular Csp3–H amination reaction.71–77
We further explored the substrate scope of intramolecular Csp3–H amination reactions (Fig. 8). At 2 mol% catalyst loading and in the presence of 2 equiv. of Boc2O, the 2,2-dimethylpyrrolidine (2b) was formed in 57% yield. Due to reactivity of the vinyl substituent in 2c, 5 eq. of Boc2O was required to give modest yield at 2 mol% Fe. Since the MOL catalysts are free from diffusion constraints, substrates with a bulky substituent such as 3,5-diphenylphenyl was also tolerated and gave 75% yield at 5 mol% Fe and 2 eq. of Boc2O.
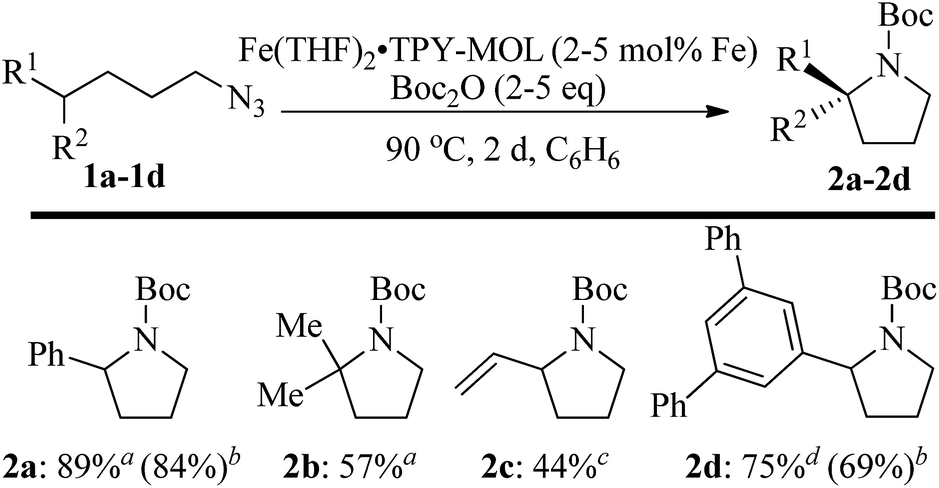 | ||
| Fig. 8 Substrate scope for α-substituted pyrrolidine synthesis. Reaction conditions: aFe (2 mol%), Boc2O (2 equiv.); bisolated yields. cFe (2 mol%), Boc2O (5 equiv.); dFe (5 mol%), Boc2O (2 equiv.). | ||
Piperidines can also be formed via C–H amination with the Fe·TPY-MOL catalyst (Fig. 9). For example, 7-azidohept-1-ene was converted to the exclusively six-member ring product 1-Boc-2-vinylpiperidine in 34% yield. By comparison, Betley's Fe-dipyrrinato homogenous catalyst required a stoichiometric equivalent of catalyst to obtain 45% yield. Furthermore, the 1-Boc-2,2-dimethylpiperidine and 1-Boc-2-phenylpiperidine could also be formed from alkyl azides. In these examples, the pyrrolidine products were also observed.
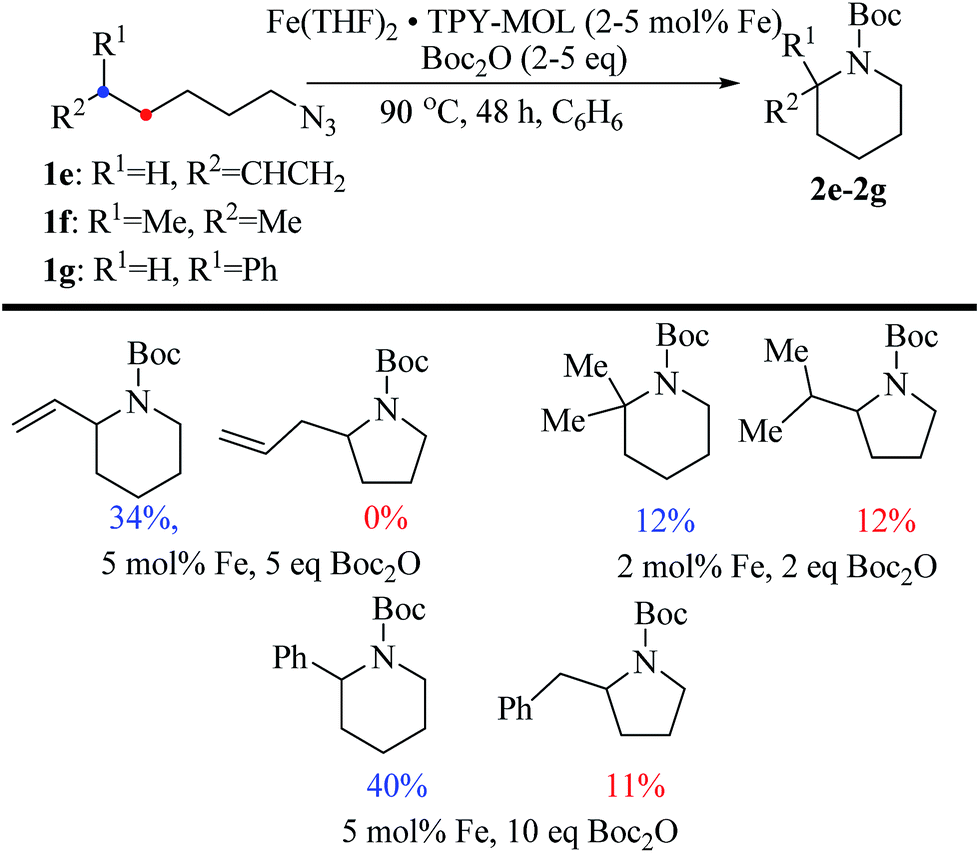 | ||
| Fig. 9 Substrate scope for α-substituted piperidine synthesis. | ||
PXRD pattern of Fe·TPY-MOL catalysts recovered from Csp3–H amination reactions suggested that the integrity of the MOL was maintained under reaction conditions. ICP-MS of the supernatant showed <0.1% of Fe and <0.1% of Hf had leached into the supernatant. Furthermore, The Fe·TPY-MOL catalyst could be recovered and reused four times (Scheme S4, ESI†).
Conclusions
We have synthesized a terpyridine-based TPY-MOL and metalated TPY-MOL with CoCl2 and FeBr2 to generate M·TPY-MOL catalysts for benzylic C–H borylation and Csp3–H amination reactions. Interestingly, M·TPY-MOL catalysts showed significantly higher activity and different chemo-selectivity than homogeneous and MOF controls. Spectroscopic studies and DFT calculations indicated the formation of unprecedented MOL-stabilized MII-(TPY˙˙)2− species featuring divalent metals and TPY diradical dianions. We believe that the formation of novel MII-(TPY˙˙)2− (M = Co or Fe) species endows them with unique and enhanced catalytic activities in Csp3–H borylation and intramolecular amination reactions. Our work demonstrates the ability to engineer MOLs as single-site solid catalysts without diffusional constraints and to elucidate intricate electronic structures of MOL-stabilized metal complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NSF (DMR-1308229), the University of Chicago Materials Research Science and Engineering Center (DMR-1420709) and the National Natural Science Foundation of the P.R. China (21671162, 21471126) for financial support and K. Ni, W. Han, M. Piechowicz, Dr A. S. Filatov and Prof. J. Anderson for experimental help. XAFS data were collected at the APS at ANL on Beamline 10BM-A, B, supported by the Materials Research Collaborative Access Team (MRCAT). MRCAT operations are supported by the Department of Energy and the MRCAT member institutions. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.Notes and references
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- J. L. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- L. Ma, A. Jin, Z. Xie and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 9905 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804 RSC.
- Z. Lin, Z.-M. Zhang, Y.-S. Chen and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 13739 CrossRef CAS PubMed.
- P. Ji, T. Sawano, Z. Lin, A. Urban, D. Boures and W. Lin, J. Am. Chem. Soc., 2016, 138, 14860 CrossRef CAS PubMed.
- M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199 CrossRef CAS PubMed.
- J. A. Johnson, B. M. Petersen, A. Kormos, E. Echeverria, Y. S. Chen and J. Zhang, J. Am. Chem. Soc., 2016, 138, 10293 CrossRef CAS PubMed.
- Q. Han, C. He, M. Zhao, B. Qi, J. Niu and C. Duan, J. Am. Chem. Soc., 2013, 135, 10186 CrossRef CAS PubMed.
- Q. H. Yang, Q. Xu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685 CrossRef CAS PubMed.
- Y. Z. Chen, Z. U. Wang, H. W. Wang, J. L. Lu, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035 CrossRef CAS PubMed.
- C. Wang, T. Zhang and W. Lin, Chem. Rev., 2011, 112, 1084 CrossRef PubMed.
- O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- M. M. Wanderley, C. Wang, C.-D. Wu and W. Lin, J. Am. Chem. Soc., 2012, 134, 9050 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957 CrossRef CAS PubMed.
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024 CrossRef CAS PubMed.
- W. J. Rieter, K. M. Pott, K. M. Taylor and W. Lin, J. Am. Chem. Soc., 2008, 130, 11584 CrossRef CAS PubMed.
- C. B. He, K. D. Lu, D. M. Liu and W. B. Lin, J. Am. Chem. Soc., 2014, 136, 5181 CrossRef CAS PubMed.
- T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241 CrossRef CAS PubMed.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 12610 CrossRef CAS PubMed.
- N. C. Thacker, Z. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2016, 138, 3501 CrossRef CAS PubMed.
- T. Sawano, Z. Lin, D. Boures, B. An, C. Wang and W. Lin, J. Am. Chem. Soc., 2016, 138, 9783 CrossRef CAS PubMed.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocella, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley Iii, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512 CrossRef CAS PubMed.
- R. J. Comito, K. J. Fritzsching, B. J. Sundell, K. Schmidt-Rohr and M. Dincă, J. Am. Chem. Soc., 2016, 138, 10232 CrossRef CAS PubMed.
- C. Wang, M. Zheng and W. Lin, J. Phys. Chem. Lett., 2011, 2, 1701 CrossRef CAS.
- T. Sawano, N. C. Thacker, Z. Lin, A. R. McIsaac and W. Lin, J. Am. Chem. Soc., 2015, 137, 12241 CrossRef CAS PubMed.
- L. Cao, Z. Lin, F. Peng, W. Wang, R. Huang, C. Wang, J. Yan, J. Liang, Z. Zhang, T. Zhang, L. Long, J. Sun and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 4962 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356 CrossRef CAS PubMed.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058 CrossRef CAS PubMed.
- P.-Z. Li, Y. Maeda and Q. Xu, Chem. Commun., 2011, 47, 8436 RSC.
- A. Wild, A. Winter, F. Schlutter and U. S. Schubert, Chem. Soc. Rev., 2011, 40, 1459 RSC.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef CAS PubMed.
- U. S. Schubert, A. Winter and G. R. Newkome, Terpyridine-based Materials: For Catalytic, Optoelectronic and Life Science Applications, Wiley-VCH, 2011 Search PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed.
- I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834 CrossRef CAS PubMed.
- A. Winter, M. Gottschaldt, G. R. Newkome and U. S. Schubert, Curr. Top. Med. Chem., 2012, 12, 158 CrossRef CAS PubMed.
- A. Winter, M. D. Hager, G. R. Newkome and U. S. Schubert, Adv. Mater., 2011, 23, 5728 CrossRef CAS PubMed.
- A. Winter, G. R. Newkome and U. S. Schubert, ChemCatChem, 2011, 3, 1384 CrossRef CAS.
- P. Liu, C.-Y. Zhou, S. Xiang and C.-M. Che, Chem. Commun., 2010, 46, 2739 RSC.
- K. Kamata, A. Suzuki, Y. Nakai and H. Nakazawa, Organometallics, 2012, 31, 3825 CrossRef CAS.
- N. G. Léonard, M. J. Bezdek and P. J. Chirik, Organometallics, 2017, 36, 142 CrossRef.
- C. Zhang, P. Srivastava, K. Ellis-Guardiola and J. C. Lewis, Tetrahedron, 2014, 70, 4245 CrossRef CAS PubMed.
- T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508 RSC.
- W. N. Palmer, J. V. Obligacion, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 766 CrossRef CAS PubMed.
- M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633 CrossRef CAS PubMed.
- T. Ishiyama, K. Ishida, J. Takagi and N. Miyaura, Chem. Lett., 2002, 1082 Search PubMed.
- S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168 CrossRef CAS PubMed.
- W. N. Palmer, C. Zarate and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 2589 CrossRef CAS PubMed.
- J. England, E. Bill, T. Weyhermuller, F. Neese, M. Atanasov and K. Wieghardt, Inorg. Chem., 2015, 54, 12002 CrossRef CAS PubMed.
- E. J. Corey, N. J. Cooper, W. M. Canning, W. N. Lipscomb and T. F. Koetzle, Inorg. Chem., 1982, 21, 192 CrossRef CAS.
- C. Hamacher, N. Hurkes, A. Kaiser, A. Klein and A. Schuren, Inorg. Chem., 2009, 48, 9947 CrossRef CAS PubMed.
- P. S. Braterman, J. I. Song and R. D. Peacock, Inorg. Chem., 1992, 31, 555 CrossRef CAS.
- R. M. Berger and D. R. Mcmillin, Inorg. Chem., 1988, 27, 4245 CrossRef CAS.
- C. C. Scarborough, K. M. Lancaster, S. DeBeer, T. Weyhermueller, S. Sproules and K. Wieghardt, Inorg. Chem., 2012, 51, 3718 CrossRef CAS PubMed.
- M. Wang, T. Weyhermuller, J. England and K. Wieghardt, Inorg. Chem., 2013, 52, 12763 CrossRef CAS PubMed.
- J. E. Leffler, An introduction to free radicals, Wiley-Interscience, New York, 1993 Search PubMed.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175 CrossRef CAS PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
- M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed.
- S. C. E. Stieber, C. Milsmann, J. M. Hoyt, Z. R. Turner, K. D. Finkelstein, K. Wieghardt, S. DeBeer and P. J. Chirik, Inorg. Chem., 2012, 51, 3770 CrossRef CAS PubMed.
- B. M. Wile, R. J. Trovitch, S. C. Bart, A. M. Tondreau, E. Lobkovsky, C. Milsmann, E. Bill, K. Wieghardt and P. J. Chirik, Inorg. Chem., 2009, 48, 4190 CrossRef CAS PubMed.
- J. England, C. C. Scarborough, T. Weyhermüller, S. Sproules and K. Wieghardt, Eur. J. Inorg. Chem., 2012, 4605 CrossRef CAS.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS PubMed.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917 CrossRef CAS PubMed.
- R. E. Cowley, N. A. Eckert, S. Vaddadi, T. M. Figg, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 9796 CrossRef CAS PubMed.
- R. E. Cowley and P. L. Holland, Inorg. Chem., 2012, 51, 8352 CrossRef CAS PubMed.
- S. Wiese, J. L. McAfee, D. R. Pahls, C. L. McMullin, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 10114 CrossRef CAS PubMed.
- Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961 CrossRef CAS PubMed.
- B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. van Leest, B. de Bruin, S. Demeshko, M. A. Siegler and J. I. D. Vlugt, J. Am. Chem. Soc., 2017, 139, 5117 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03537c |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2018 |