
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the unusual role of bases in activation/deactivation of catalytic systems: O–NHC coupling in M/NHC catalysis†
Victor M.
Chernyshev
a,
Oleg V.
Khazipov
a,
Maxim A.
Shevchenko
a,
Andrey Yu.
Chernenko
a,
Alexander V.
Astakhov
a,
Dmitry B.
Eremin
 b,
Dmitry V.
Pasyukov
a,
Alexey S.
Kashin
b and
Valentine P.
Ananikov
*ab
b,
Dmitry V.
Pasyukov
a,
Alexey S.
Kashin
b and
Valentine P.
Ananikov
*ab
aPlatov South-Russian State Polytechnic University (NPI), Prosveschenya 132, Novocherkassk, 346428, Russia
bZelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, Moscow, 119991, Russia. E-mail: val@ioc.ac.ru
First published on 6th June 2018
Abstract
Numerous reactions are catalyzed by complexes of metals (M) with N-heterocyclic carbene (NHC) ligands, typically in the presence of oxygen bases, which significantly shape the performance. It is generally accepted that bases are required for either substrate activation (exemplified by transmetallation in the Suzuki cross-coupling), or HX capture (e.g. in a variety of C–C and C-heteroatom couplings, the Heck reaction, C–H functionalization, heterocyclizations, etc.). This study gives insights into the behavior of M(II)/NHC (M = Pd, Pt, Ni) complexes in solution under the action of bases conventionally engaged in catalysis (KOH, NaOH, t-BuOK, Cs2CO3, K2CO3, etc.). A previously unaddressed transformation of M(II)/NHC complexes under conditions of typical base-mediated M/NHC catalyzed reactions is disclosed. Pd(II) and Pt(II) complexes widely used in catalysis react with the bases to give M(0) species and 2(5)-oxo-substituted azoles via an O–NHC coupling mechanism. Ni(NHC)2X2 complexes hydrolyze in the presence of aqueous potassium hydroxide, and undergo the same O–NHC coupling to give azolones and metallic nickel under the action of t-BuOK under anhydrous conditions. The study reveals a new role of NHC ligands as intramolecular reducing agents for the transformation of M(II) into “ligandless” M(0) species. This demonstrates that the disclosed base-mediated O–NHC coupling reaction is integrated into the catalytic M/NHC systems and can define the mechanism of catalysis (molecular M/NHC vs. “NHC-free” cocktail-type catalysis). A proposed mechanism of the revealed transformation includes NHC-OR reductive elimination, as implied by a series of mechanistic studies including 18O labeling experiments.
Introduction
Complexes of transition metals with N-heterocyclic carbene (NHC) ligands are commonly applied as catalysts for a variety of C–C and C-heteroatom bond formations, C–H functionalizations, the Mizoroki–Heck reaction, atom-economic additions, and many other transformations.1 Fine organic synthesis and its industrial applications are indispensable for obtaining complex molecular structures of pharmaceutical substances, natural product substitutes, smart materials, and molecular electronic components, among many other examples of paramount significance.2 The universal application of M/NHC complexes in catalysis is attributed to the great stability of M–NHC bonds and the high variability of the steric and electronic parameters of NHC ligands, which allows effective tuning of the catalyst activity.1,3The vast majority of practically relevant M/NHC catalyzed reactions are conducted in the presence of bases, including strong bases like alkali metal hydroxides or alkoxides.1–5 The bases play important roles of capturing HX from the catalytic cycle, facilitating the transmetallation process, and inducing the formation of nucleophilic reagents.4b,6 The nature of the base can significantly shape the catalytic performance of the M/NHC system in terms of activity and selectivity.1–7
Several published studies of catalytic systems consider the effects of the base on the metal center of the catalyst and reveal few additional roles of bases in metal-catalyzed reactions. Participation of bases in ligand exchange, with its effects on the activity and selectivity, is a well-established phenomenon. For example, coordination of tert-butoxide anions to nickel promotes the oxidative addition of H2 in C–O activation reactions.8 Coordination of tertiary amines apparently blocks the β-hydride elimination in the Mizoroki–Heck reaction, and this explains why the replacement of CsOPiv bases by Bu3N switches the reaction toward Michael addition.7c Yet another important role of bases, the catalyst activation via reduction of M(+)L complexes to active M(0)L molecular complexes, should be considered. In this type of catalyst activation, bases like alcoholates or amines can act as either direct reductants via hydride transfer,4a,9 or promoters of reductive elimination of the “leaving” π-coordinated allyl ligands via nucleophilic addition to the allyl π-system.10
Within the framework of the current concept, the M/NHC core is thought to confer the catalytic activity, whereas the bases are thought to be responsible for the chemical versatility. Boosts of catalytic performance observed with M/NHC complexes are typically attributed to ligand effects on the chemical transformation studied or optimization of reaction conditions. Recently, the ability of aliphatic amine bases to induce cleavage of M–NHC bonds with the fast release of catalytically active NHC-free metal species has been reported.11 Decomposition of M/NHC complexes in the presence of strong bases was reported in a few cases.12 However, activation/deactivation of M/NHC complexes with bases, which would cause transformations of NHC ligands and affect the M–NHC bonds, remain understudied.
In the present work, we reveal a transformation of M/NHC (M = Pd, Pt, Ni) complexes taking place in the presence of most widely applied bases, such as KOH, NaOH, t-BuOK, Cs2CO3, K2CO3, etc. The transformation reported here actually may take place in a number of already known catalytic systems;5 however, it was not reported previously.
A new base-mediated reaction of O–NHC coupling is discovered, leading to “NHC-free” metal species and azolones (Scheme 1) under conditions typical for M/NHC catalyzed reactions. By virtue of the O–NHC coupling, NHC ligands act as intrinsic reducing agents to produce M(0) species. “Ligandless” metal species are renowned for their high chemical activity and outstanding catalytic performance. The revealed O–NHC coupling reaction can occupy a dominant place in the whole process by controlling the generation of active centers or initiating deactivation of a catalytic system, depending on particular catalytic mechanisms.
 | ||
| Scheme 1 Discovered O–NHC coupling reactions in M/NHC systems. | ||
Results and discussion
Transformations of M/NHC complexes under the action of bases
A variety of NHC ligands (16 examples) and M/NHC complexes of Pd, Pt, Ni (25 examples) were tested to estimate the scope of the revealed reaction (Scheme 2). A set of typical bases including KOH, NaOH, K2CO3, Cs2CO3, t-BuOK, and t-BuONa were employed in these experiments (Table S1†). Solvents, temperatures and reaction times (40–140 °C, 3–24 h) used here correspond to typical reaction conditions used for a variety of reported catalytic reactions.1–4,6,7,13 | ||
| Scheme 2 Overview of the ligand precursors (1) and metal complexes (2–6) utilized in the present study. | ||
For all of the bases, decomposition of the metal complexes leading to precipitation of agglomerated M(0) nanoparticles (M = Pd, Pt, Ni) or Ni(OH)2 was observed. Compositions and structures of the metal precipitates were determined by X-ray fluorescence spectroscopy (XRF), field-emission scanning electron microscopy (FE-SEM, Fig. 1 and ESI†), and Raman spectroscopy.
 | ||
| Fig. 1 FE-SEM images of palladium (a) and platinum (b) precipitates isolated from reaction mixtures after heating complex 2a with KOH in pyridine at 100 °C for 24 h and complex 5a with t-BuOK in 1,4-dioxane at 100 °C for 20 h, respectively. | ||
Reactions of complexes 2–6 with bases can be divided into three different types depending on the main reaction product structures (Scheme 3). Plausible stoichiometry for the reactions is presented in Scheme S2.†
 | ||
| Scheme 3 Reactions of M/NHC complexes with KOH (Conditions A) and t-BuOK (Conditions B). HPLC and isolated (blue color) yields are shown for each case. HPLC/isolated yields are calculated assuming that, theoretically, 1 mole of azolone 7a, c, and f is formed from 1 mole of the complex 4a and b, and 6a and b. Conditions A: KOH, H2O, pyridine, 100 °C; Conditions B: t-BuOK, 1,4-dioxane, 100 °C. For additional details and other reaction conditions see the Experimental section, Table S1 and Fig. S1.† | ||
Type 1 reactions involved palladium complexes 2 and 3 and platinum complexes 5a and b to yield free metal particles of Pd(0) and Pt(0) (Fig. 1) and azolones 7a–o with all of the bases (Scheme 3, Table S1†).
Type 2 reactions were observed for bis-NHC complexes 4a and b and 6a and b. Heating complexes 4a and b with KOH and t-BuOK, and heating complexes 6a and b with t-BuOK resulted in formation of Pd(0) and Ni(0) precipitates, correspondingly, and azolones 7a, c and f (quantitatively detected as 1 mol per mole of the initial complex) as the main products. However, diamines 8a–c were also detected by HPLC and GC-MS in the reaction mixtures (Scheme 3, Table S1†).
A Type 3 reaction was observed for nickel complex 6a, which transformed into Ni(OH)2 and diamine 8a when heated with aqueous KOH in dioxane (Scheme 3). Only trace amounts of 1,3-dimethylbenzimidazolone 7a (yields up to 9%) were detected by HPLC and GC-MS in the reaction mixture.
The main product yields strongly depended on the structure of the initial complex, the base, and the solvent. Comparing the reactivities of the complexes toward t-BuOK in Type 1 and Type 2 reactions, it could be concluded that Pd complexes 2 and the halogen-bridged complex 3 were the most reactive, bis-NHC Pd complexes 4 were significantly less reactive, and Pt complexes 5 and Ni complexes 6 were the least reactive (Table S1†). Notably, the reaction proceeded even at 40 °C and was accelerated upon heating (see variable temperature experiments, Fig. S1†). As an illustration, compound 2a was converted almost completely within ∼20 min when heated with t-BuOK in dioxane at 100 °C to give benzimidazolone 7a in ∼80% yield, while it required 3 h for complexes 4a and b to give good yields of azolones 7a and c (86–87%) (Table S1†). Even more, promoting the reaction with Pt complexes 5a and b and Ni complexes 6a and b required heating for ∼20 h and ∼48 h, respectively.
Concerning the possible influence of the structure of NHC ligands, metal complexes with bulky substituents, especially DiPP and Mes (compounds 2j–m), reacted somewhat slower. The yields of azolones 7 dramatically depended on the heterocycle type of the NHC ligands. The highest yields of azolones 7 were obtained for complexes 2a–i, 3, 4a and b, 5a and b, and 6a and b with NHC ligands of benzimidazole type, which formed stable benzimidazol-2-ones 7a–h. For complexes 2j–r comprising NHC ligands of imidazole and 1,2,4-triazole series (except the 2j–m complexes with bulky substituted N,N′-aryl-imidazoles), only trace yields of the corresponding azolones 7k–o were detected by GC-MS (see ESI†). Low yields of imidazol-2-ones and triazol-5-ones were related to their instability and solvolysis (discussed below in more detail).
By comparing the effects of different bases on 2a–i Pd complexes it could be noticed that stronger bases like t-BuOK, t-BuONa, KOH, and NaOH provided higher reaction rates and higher benzimidazol-2-ones yields (43–97%) than mild and less soluble K2CO3 and Cs2CO3 (∼0–57%) (Table S1†). Remarkably, Pt complexes 5a and b were almost unreactive toward KOH in dioxane within 20 h, while they afforded 42% and 65% HPLC yields of azolones 7a and d with t-BuOK under the same conditions (Table S1†). The Type 3 reaction observed for complex 6a, heated with aqueous KOH in dioxane, may be considered as a fresh example of Ni–NHC hydrolysis (see a more detailed discussion below).14
Yields of azolone 7a in the reaction between complex 2a and t-BuOK in 2-propanol (62%) and DMF (58%) were lower than those in dioxane (91%) and pyridine (95%) (Table S1†). The lower yields may be explained by concurrent partial reduction of Pd(II)NHC to Pd(0) species and azolium salts/NHC in these solvents.15 Azolium salts and NHCs subsequently undergo solvolysis to form diaminobenzenes (see discussion below).
Mechanistic study of azolone formation
Consideration of the results raises a question: what pathways lead from the metal complexes to azolones in the presence of strong oxygen bases? It should be noted that azolium salts 1 heated with KOH and t-BuOK under the conditions that favor decomposition of complexes 2–6 produce quite low yields of azolones (≤10%), probably via the disproportionation reaction.16 This result makes it impossible to explain the high yields of azolones 7 by simple solvolysis of complexes 2–6 and subsequent disproportionation of azolium salts.To evaluate the formation of azolones 7 directly in the reaction of M/NHC complexes with bases rather than during the isolation procedures or preparation of analytic samples, we performed ESI-MS online monitoring of the reaction for a selection of complexes, 2a, b, o, and r, and 4a (Fig. S2–S11†). ESI-MS online monitoring is a powerful method for detecting charged or ionizable reactants, intermediates, and products directly in the course of metal-catalyzed reactions.17 A solution of a particular complex in THF was heated at 100 °C with continuous transfusing to a mass spectrometer. After ∼1 min, an aqueous solution of KOH (or a suspension of t-BuOK in THF) was injected into the reaction mixture, initiating almost immediate appearance of the corresponding azolone 7a, b, l, or o (details in the ESI†). To corroborate that the base was the source of oxygen, we also carried out isotope experiments with 18O labeled KOH. Indeed, ESI-MS online monitoring of the reaction between the compound 2a and a solution of KOH in H218O (97% of 18O in the aqueous solution) revealed the formation of 18O azolone 7a (Fig. 2). The reaction was also performed preparatively in pyridine using a solution of potassium in H218O. The azolone 7a-[18O] was obtained in 47% isolated yield (Scheme 4). The possibility of isotope exchange in the product under reaction conditions was rejected by conducting a corresponding control experiment with isolated 7a-[16O] and K18OHaq (see ESI for details†). Eventually, the ESI-MS online monitoring and isotope experiments unambiguously confirmed the direct formation of azolones 7 in the reaction between M/NHC complexes and oxygen bases.
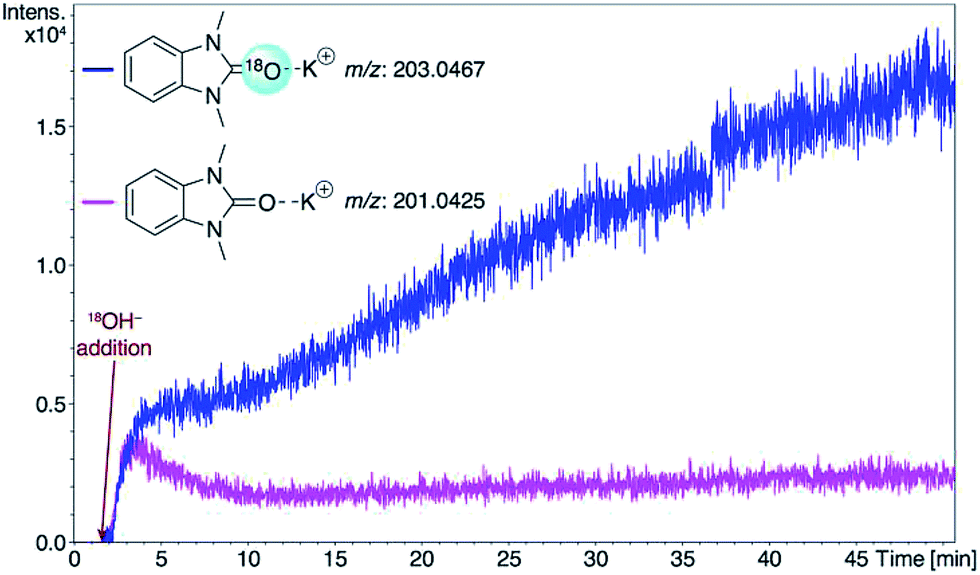 | ||
| Fig. 2 Real-time abundances of 7a-[18O] and 7a-[16O] (in ionic [M + K]+ form) in the reaction between complex 2a and K18OHaq in THF at 100 °C. Solution of KOH in H218O was added after 1.2 min of recording. | ||
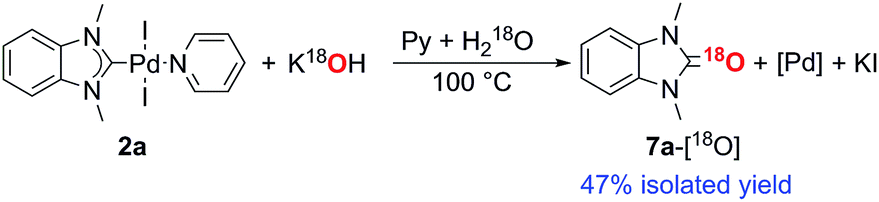 | ||
| Scheme 4 Reaction of complex 2a with K18OH. | ||
Thus, the experiments clearly revealed a previously undescribed O–NHC coupling reaction resulting from the action of strong oxygen bases on the M/NHC complexes (Scheme 5). The assumed stoichiometries for various combinations of M/NHC complexes and bases are presented in Scheme S2.† In this reaction, NHC ligands play a previously undiscussed role of metal reductants, transforming M(II) into M(0) with the formation of azolones 7 as oxidized derivatives of NHC.
 | ||
| Scheme 5 Plausible mechanisms of the oxygen base-mediated O–NHC coupling of M/NHC complexes. | ||
The most probable mechanism of this reaction, in our opinion, includes a reversible exchange of ligand L (or X) by RO− with subsequent C–O cross-coupling of M-coordinated NHC and RO− accompanied by the reductive elimination of M(0) species (“Reductive elimination” mechanism, Scheme 5). Indeed, ESI-MS online monitoring of the reaction between 4a and KOH in THF revealed the formation of intermediate hydroxy-complex 9 (in the [M + K]+ form) (Scheme 6, Fig. S4†). Similar intermediate complexes were observed in reactions of other Pd–NHC complexes with KOH. Collision-induced dissociation tandem mass spectrometry (MS/MS) applied to the ion 9 suggests the direct formation of azolone 7a from 9 as a major fragmentation pathway (Scheme 6, Fig. S5†). Similar mechanisms of C–C and C–H cross-couplings of NHC have been previously presented as the most probable pathways of deterioration of M/NHC complexes in the course of catalytic reactions.18
 | ||
| Scheme 6 Formation of hydroxy-complex 9 and its transformation into azolone 7a during the reaction of complex 4a with KOH (ESI-MS online and MS/MS experiments). | ||
Yet, an alternative mechanism of azolone formation may include direct nucleophilic attack of RO− on the C(carbene) atom of M-coordinated NHC with subsequent dissociation of M–C bonds accompanied by elimination of M(0) species (“Nucleophilic” mechanism, Scheme 5). However, direct nucleophilic attack on the carbene atom of NHC ligands coordinated with metal M should be sterically hindered by bulky N-substituents of the NHC-ligand and M-coordinated co-ligands X and L. Therefore, we think that the “nucleophilic” mechanism is less probable. We also cannot exclude a completely different mechanism of the oxidation of electron rich [NHC-OR]− anions by M(II) species. The anions may be formed from dissociation of M–NHC bonds and reversible addition of RO− to the released NHC. Despite the strongly nucleophilic character of NHCs, some examples of their electrophilic behavior can be found in the literature.19 The structures of azolones 7 are confirmed by 1H, 13C NMR, and ESI-MS spectra.
Mechanistic study of diamines and other cleavage product formation
Formation of diaminobenzenes 8 from bis-NHC complexes 4a and b, and 6a and b in the course of Type 2 reactions may be explained by a transformation shown in Scheme 7. At the first stage, O–NHC coupling with a base gives an azolone (1 mol per mole of M(NHC)2X2) and a NHC (in equilibrium with its protonated form, i.e. azolium cation). At the second stage, the released NHC undergoes solvolysis to give diamine 8 under strongly basic conditions via the known reaction mechanism (Schemes 7 and S1, and details in the ESI†).16 | ||
| Scheme 7 Formation of diamines 8 from complexes 4 and 6. | ||
Reaction of nickel complex 6a with aqueous KOH giving Ni(OH)2 and diamine 8a (Scheme 8) could be predicted from previously published data on facile hydrolysis of Ni/NHC complexes producing azolium salts and Ni(OH)2 in the presence of water.14 Benzimidazolium salt 1a, the intermediate product of the reaction, undergoes solvolysis to diamine 8a.16 The solvolysis is accompanied by the formation of small amounts (∼9% HPLC yield) of azolone 7avia the parallel disproportionation mechanism (Scheme S1 and discussion in the ESI†).16
 | ||
| Scheme 8 Reaction of complex 6a with KOH. | ||
To check the proposed schemes of the formation of diamines 8 from benzimidazolium salts, a model reaction of azolium salts 1a, c, and f with t-BuOK in 1,4-dioxane was examined. Diamines 8a–c (82–85% HPLC yields) and azolones 7a, c, and f (8–10% HPLC yields) were the major products, in full compliance with the proposed schemes.
Possible impacts of the revealed base effect on catalytic reactions
The three different reactions chosen to investigate the impact of the revealed O–NHC coupling on catalytic performance were: the Mizoroki–Heck reaction (1), the Suzuki–Miyaura cross-coupling (2), and C–H functionalization (3) as shown in Scheme 9. Pd/NHC complex 2j which is frequently used in catalysis was selected as a precatalyst for the reactions (1)–(3). | ||
| Scheme 9 Possible impacts of O–NHC coupling on Pd-catalyzed C–C coupling reactions. (a) Pretreatment of the catalyst with t-BuOK for 10 min at 100 °C. (b) Pretreatment of the catalyst with t-BuOK for 3 h at 100 °C. | ||
The reactions (1)–(3) were performed in two distinct manners, methods A and B (Scheme 9). In the method A, freshly prepared solution of the complex 2j in pyridine was used as the catalyst. In the method B, the precatalyst 2j was heated with t-BuOK in pyridine, neutralized by pyridine hydrochloride, and the solution obtained was introduced to the reaction mixtures (see the Experimental part for details). It is important to note that decomposition of the complex 2j to give azolone 7i in the course of the pretreatment (method B) was confirmed by GC-MS. The obtained experimental data are shown in Scheme 9.
In the Mizoroki–Heck reaction (1) performed without initial pre-activation (method A), complex 2j was almost inactive, only a minor yield of the product 12 was observed. However, in the experiment with the t-BuOK pretreatment (method B), the catalytic activity of the precatalyst solution was significantly higher. Thus, in the Mizoroki–Heck reaction (1), O–NHC coupling leads to activation of the Pd/NHC catalyst, apparently by promoting the formation of “NHC-free” Pd active species. High performances in cocktail-type systems is a distinctive feature of the Mizoroki–Heck reaction.18c,20 Removal of the NHC ligands favours generation of a cocktail-type system and facilitates the product formation.
In the Suzuki–Miyaura reaction (2), formation of product 14 was observed in both cases. Since transmetallation usually represents a rate-limiting step of this cross-coupling reaction, M/NHC catalytic species and ligand-free cocktail-type systems may show similar performances. Although it affects the formation of “NHC-free” metal species in solution, O–NHC coupling has no determining effect on the outcomes of the Suzuki–Miyaura reaction.
In the C–H arylation reaction (3) carried out in the presence of t-BuOK, only a fresh solution of the complex 2j was fairly active (Scheme 9). The catalytic solution obtained after the t-BuOK pretreatment (method B) was less active, and its activity significantly decreased with prolongation of the pretreatment time from 10 min to 3 h. Formation of azolone 7i due to slow decomposition of 2j in the course of the reaction (method A) was also detected by GC-MS. Thus, in the C–H functionalization reaction (3) O–NHC coupling causes deactivation of Pd/NHC catalytic systems. The results suggest that Pd/NHC are catalytically active species, while ligand-free or cocktail-type systems are less efficient or inactive.
The results outlined in Scheme 9 clearly corroborate the importance of the base-mediated O–NHC coupling for the catalytic activity of M/NHC complexes. Base-mediated M–NHC bond cleavage revealed in the current study can largely determine the behavior of catalytically active metal species.
On the one hand, the O–NHC coupling reaction can provoke decomposition of M/NHC complexes and thus accelerate deactivation of catalytic systems where only molecular M/NHC complexes are capable of driving the catalytic cycle (classical homogeneous molecular M/NHC catalysis). In such systems, when strong bases like alkali metal alkoxides or hydroxides are applied, O–NHC coupling can represent a serious problem that needs to be solved for providing stability and high performance of the catalytic systems.
On the other hand, the opposite effect of the O–NHC coupling may be anticipated in reactions where M/NHC complexes serve as a source of “NHC-free” active metal species without typical metal–NHC bonds. As an example, the activity of Pd/NHC systems in the Mizoroki–Heck reaction is determined predominantly by the cleavage of the metal–NHC bond and generation of cocktail-type catalytic systems, in which the “NHC-free” palladium species act as truly active catalytic centers.18c The proposed pathways leading from M/NHC complexes to the cocktails of active metal species include transformation of the NHC ligands via C–H or C–C coupling and formation of corresponding azolium salts. The plausible role of aliphatic amine bases as hydride sources and reductants of metal cations in M/NHC complexes to produce azolium salts and “NHC-free” active metal species via H–NHC (C–H) coupling was reported recently.11 As described here, a principally new mechanism for generation of the “NHC-free” active species can be implemented in the presence of oxygen bases like KOH, t-BuOK, or alkali metal carbonates. In such systems, NHC ligands play a role in the reduction of M(II) to M(0) by means of the O–NHC coupling reaction affording “NHC-free” M(0) active species and azolones.
Conclusions
Complexes of metals with N-heterocyclic carbenes, M(NHC)X2L [M = Pd(II), Pt(II), Ni(II)], react with oxygen bases typically applied in various catalytic processes to give M(0) nanoparticles and azol-2(5)-ones. In this previously undescribed O–NHC coupling reaction, NHC ligands play the role of two-electron intramolecular reductants of the coordinated metal dications. The base anions serve as a source of oxygen. The azol-2(5)-ones forming from the NHC of imidazole and 1,2,4-triazole series are labile compounds that decompose with the destruction of heterocycles.Among Pd(II), Pt(II), and Ni(II) complexes examined in this work, Pd complexes are found to be the most reactive and thus more prone to the O–NHC coupling. A combination of Pd with NHC ligands gives a flexible catalytic system, whose nature can be tuned from the Pd/NHC to “NHC-free” molecular or cluster/nanoparticle catalysis.
The base-mediated O–NHC coupling has a significant impact on the performance of the M/NHC catalytic systems. On one hand, the reaction provokes deactivation of these catalytic systems, where participation of molecular M/NHC complexes in the transition state of a catalytic reaction is essential, like in the chiral NHC–metal-based asymmetric catalysis. Certainly, O–NHC coupling should be a problem for homogeneous M/NHC catalysis when highly active palladium complexes and strong bases like alkali metal alcoholates or hydroxides are applied. The present study clearly demonstrates that the suppression of O–NHC coupling is an important, overlooked priority in maintaining the high efficiency of well-defined M/NHC systems in the catalysis of numerous reactions mediated by strong oxygen bases.
On the other hand, the base-mediated O–NHC coupling can serve as a driving force in the activation of M/NHC systems for the reactions catalyzed by “NHC-free” metal species. The latter scheme is valid, for example, for the high-temperature Mizoroki–Heck reaction.
Indeed, the correct choice of base is essential for maximizing the performance of the M/NHC catalytic systems and we anticipate many studies in this regard in the nearest future.
Experimental section
General information
1H and 13C{1H} NMR spectra were acquired on a Bruker DRX 500 instrument at 500 MHz and 125 MHz, respectively, in DMSO-d6 or CDCl3. 1H and 13C{1H} chemical shifts are given in ppm relative to the residual peak of the solvent (δ 2.5 for DMSO and 7.26 for CHCl3) for the proton spectra and the 13C{1H} DMSO-d6 signal (δ 39.5) or the CDCl3 signal (δ 77.2) for the carbon spectra.High-resolution mass spectra were obtained on a Bruker maXis Q-TOF or microTOF instruments (Bruker Daltonik GmbH, Bremen, Germany) equipped with an electrospray ionization (ESI) ion source. The experiments were performed in positive (+) MS ion mode (HV Capillary: 4500 V; Spray Shield Offset: −500 V) with a scan range of m/z 50–1500. External calibration of the mass spectrometer was achieved using a low-concentration tuning mix solution (Agilent Technologies). Unless otherwise stated, direct syringe injection was applied for the analyzed solutions in MeCN (flow rate: 3 μL × min−1). Nitrogen was applied as the nebulizer gas (0.4 bar) and dry gas (4.0 L × min−1, 180 °C). All the MS spectra were recorded with 1 Hz frequency and processed using Bruker Data Analysis 4.0 software.
HPLC analyses were accomplished using an Agilent 1260 Infinity LC system equipped with a reversed-phase Zorbax SB-C18 column (50 × 4.6 mm) thermostated at 35 °C and a detection wavelength of 280 nm. Gradient elution with a 1 mL min−1 flow rate was applied. The mobile phase A consisted of 10% MeCN aqueous solution, the phase B was neat MeCN, and the phase C was 0.02 M NaClO4 aqueous solution. The following gradient conditions were used: ramp over 3 min from 80% A and 20% C to 80% B and 20% C; then hold 5 min.
GC-MS experiments were carried out with an Agilent 7890A GC system, equipped with an Agilent 5975C mass-selective detector (electron impact, 70 eV) and a HP-5MS column (30 m × 0.25 mm × 0.25 μm film) using He as carrier gas at a flow of 1.0 mL min−1.
For electron microscopy measurements, the samples were mounted on a 25 mm aluminum specimen stub, fixed by conductive carbon tape and coated with a thin film (15 nm) of carbon. The observations were carried out using a Hitachi SU8000 field-emission scanning electron microscope (FE-SEM). Images were acquired in secondary electron mode at 10 kV accelerating voltage and at a working distance of 4–5 mm. The morphology of the samples was studied taking into account the possible influence of the carbon coating on the surface. EDS-SEM studies were carried out using an Oxford Instruments X-max 80 EDS system at 10 kV accelerating voltage and at a working distance of 15 mm.
Raman spectra were recorded using an InVia Raman microscope (Renishaw, UK) equipped with a 20 mW 514.5 nm argon laser. All the spectra were collected using a 50× objective lens, 20 s of acquisition time, and 3 accumulations. A silicon wafer was used for calibration.21
C, H, and N elemental analyses were performed using a PerkinElmer 2400 elemental analyzer. Melting points were determined in open capillary tubes using a Thiele apparatus and are uncorrected. The Ni, Pd, and Pt contents in metal precipitates were determined by means of energy dispersive X-ray fluorescence spectroscopy (ARL Quant′X EDXRF Analyzer, Thermo Scientific). For the analyses, the weighed samples of metal precipitates were dissolved in aqueous aqua regia.
Experiments concerning reactions of M/NHC complexes with bases were performed under an argon atmosphere using standard Schlenk techniques. Solvents were dried over activated 3 Å molecular sieves and degassed by argon bubbling within 20 min. Preparative column chromatography was performed using silica gel 60 (230–400 mesh, Merck).
1-Methylbenzimidazole,22 1,3-dimethyl-1H-benzimidazol-3-ium iodide (1a),23 1,3-diethyl-1H-benzimidazol-3-ium bromide (1b),24 1,3-di-n-propyl-1H-benzimidazol-3-ium iodide (1c),11 1,3-di(propan-2-yl)-1H-benzimidazol-3-ium bromide (1d),25 1,3-dibutyl-1H-benzimidazol-3-ium bromide (1f),14 1-butyl-3-methyl-1H-benzimidazol-3-ium iodide (1g),14 1,3-dibenzyl-1H-benzimidazol-3-ium chloride (1h),18c 1,3-dibenzyl-1H-benzimidazol-3-ium bromide (1i),18c 1,3-bis[2,6-di(propan-2-yl)phenyl]-1H-imidazol-3-ium chloride (1j),14 1,3-bis(2,4,6-trimethylphenyl)-1H-imidazol-3-ium chloride (1k),14 1,3-dimethyl-1H-imidazol-3-ium iodide (1l),14 1,4-dimethyl-4H-1,2,4-triazol-1-ium iodide (1n),14 1,4-dibenzyl-4H-1,2,4-triazol-1-ium chloride (1o),18c 4-benzyl-1-tert-butyl-4H-1,2,4-triazol-1-ium bromide (1p),14 (1,3-dimethyl-1,3-dihydro-2H-benzimidazol-2-ylidene)diiodo(pyridine)palladium (2a),18c (1,3-dipropyl-1,3-dihydro-2H-benzimidazol-2-ylidene)(diiodo)(pyridine)palladium (2c),11 dibromo[1,3-di(propan-2-yl)-1,3-dihydro-2H-benzimidazol-2-ylidene](pyridine)palladium (2d),26 dibromo(1,3-dibutyl-1,3-dihydro-2H-benzimidazol-2-ylidene)(pyridine)palladium (2f),18c dichloro(1,3-dibenzyl-1,3-dihydro-2H-benzimidazol-2-ylidene)(pyridine)palladium (2h),18c dibromo(1,3-dibenzyl-1,3-dihydro-2H-benzimidazol-2-ylidene)(pyridine)palladium (2i),18c {1,3-bis[2,6-di(propan-2-yl)phenyl]-1,3-dihydro-2H-imidazol-2-ylidene}dichloro(pyridine)palladium (2j),27 {1,3-bis[2,6-di(propan-2-yl)phenyl]-1,3-dihydro-2H-imidazol-2-ylidene}(dichloro)(3-chloropyridine)palladium (2k),28 [1,3-bis(2,4,6-trimethylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene](dichloro)(pyridine)palladium (2l),28 [1,3-bis(2,4,6-trimethylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene](dichloro)(3-chloropyridine)palladium (2m),28 (1,3-dimethyl-1,3-dihydro-2H-imidazol-2-ylidene)diiodo(pyridine)palladium (2n),18c dichloro(1-butyl-3-methyl-1,3-dihydro-2H-imidazol-2-ylidene)(pyridine)palladium (2o),18c (2,4-dimethyl-2,4-dihydro-3H-1,2,4-triazol-3-ylidene)diiodo(pyridine)palladium (2p),18c dichloro(2,4-dibenzyl-2,4-dihydro-3H-1,2,4-triazol-3-ylidene)(pyridine)palladium (2q),18c (4-benzyl-2-tert-butyl-2,4-dihydro-3H-1,2,4-triazol-3-ylidene)dibromo(pyridine)palladium (2r),18c di-μ-bromo(dibromo)bis[1,3-di(propan-2-yl)-1,3-dihydro-2H-benzimidazol-2-ylidene]dipalladium (3),25 bis(1,3-dimethyl-1,3-dihydro-2H-benzimidazol-2-ylidene)diiodopalladium (4a),29 bis(1,3-dipropyl-1,3-dihydro-2H-benzimidazol-2-ylidene)(di-iodo)palladium (4b),11 (1,3-dimethyl-1,3-dihydro-2H-benzimidazol-2-ylidene)diiodo(pyridine)platinum (5a),30 dibromo[1,3-di(propan-2-yl)-1,3-dihydro-2H-benzimidazol-2-ylidene](pyridine)platinum (5b),31 bis(1,3-dimethyl-1,3-dihydro-2H-benzimidazol-2-ylidene)diiodonickel (6a),32 dibromo[bis(1,3-dibutyl-1,3-dihydro-2H-benzimidazol-2-ylidene)]nickel (6b),14 1-butyl-3-methyl-1,3-dihydro-2H-imidazol-2-one (7l),33 and 2,4-dibenzyl-2,4-dihydro-3H-1,2,4-triazol-3-one (7n)34 were synthesized as described in the literature. All other chemicals were commercially available.
1-Methyl-3-(propan-2-yl)-1H-benzimidazol-3-ium iodide (1e)
A mixture of 1-methylbenzimidazole (1.3 g, 0.01 mol), isopropyl iodide (3.4 g, 0.02 mol) and acetonitrile (15 mL) was heated under reflux for 24 h. Then the reaction mixture was evaporated to dryness in vacuo. The residue obtained was recrystallized from the MeCN–acetone 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture, washed with acetone and dried in vacuo. Yield 2.69 g (89%) of colorless crystals, mp 195–198 °C. 1H NMR (DMSO-d6): δ 1.62–1.63 (m, 6H, 2CH3), 4.09 (s, 3H, CH3), 5.04–5.12 (m, 1H, CH), 7.67–7.71 (m, 2H, Ar), 8.02–8.05 (m, 1H, Ar), 8.13–8.15 (m, 1H, Ar), 9.89 (s, 1H, H-2). 13C{1H} NMR (DMSO-d6): δ 21.6, 33.4, 50.3, 113.5, 113.7, 126.26, 126.34, 130.2, 131.9, 141.1. ESI-MS(TOF) calcd for C11H15N2 [M − I]+m/z 175.1230, found m/z 175.1227.
:2 mixture, washed with acetone and dried in vacuo. Yield 2.69 g (89%) of colorless crystals, mp 195–198 °C. 1H NMR (DMSO-d6): δ 1.62–1.63 (m, 6H, 2CH3), 4.09 (s, 3H, CH3), 5.04–5.12 (m, 1H, CH), 7.67–7.71 (m, 2H, Ar), 8.02–8.05 (m, 1H, Ar), 8.13–8.15 (m, 1H, Ar), 9.89 (s, 1H, H-2). 13C{1H} NMR (DMSO-d6): δ 21.6, 33.4, 50.3, 113.5, 113.7, 126.26, 126.34, 130.2, 131.9, 141.1. ESI-MS(TOF) calcd for C11H15N2 [M − I]+m/z 175.1230, found m/z 175.1227.
General procedure for the preparation of compounds 2b, e, and g
A stirred mixture of corresponding azolium salts 1b, e, and g (1.05 mmol), KBr or KI (5 mmol), anhydrous K2CO3 (690 mg, 5 mmol), PdCl2 (177 mg, 1 mmol) and dry pyridine (8 mL) was heated for 16 h at 80 °C in a sealed glass vial. After cooling to 20 °C, the reaction mixture was diluted with CH2Cl2 (20 mL) and passed through a short pad of silica gel eluting with CH2Cl2 until the yellow product was completely recovered. The obtained solution was evaporated to dryness under vacuum (rotary evaporator) at 40 °C. The residue obtained was treated with hexane (∼10 mL). The precipitate formed was collected by filtration and recrystallized from the CH2Cl2–hexan 1:3 mixture.
General procedure for the reaction of complexes 2–6 with KOH or t-BuOK
For the reactions of complexes 2–6 with bases under other conditions, see Table S1 in the ESI.†
Online ESI-MS monitoring of the O–NHC coupling reaction
Online monitoring of ions in the reaction progress was performed as follows.A 10 mL Schlenk tube equipped with a magnetic stir bar was thoroughly flushed with Ar. Then degassed THF (4 mL) and a 3.07 × 10−3 M solution (freeze–pump–thaw degassed) of the corresponding Pd/NHC complexes 2a, b, o, and r, and 4a (100 μL) in THF were charged into the tube under an argon backflush, and the tube neck was closed with a septum. A red PEEK capillary (72 cm) was pulled into the tube through the septum and immersed into the reaction mixture. The tube was placed in a hot glycerol bath. Then, the tube was heated at 100 °C under continuous magnetic stirring. After heating for ∼1 min, a degassed 3.57 × 10−1 M solution of the corresponding base (100 μL) was injected into the flask via a syringe through the septum. For KOH, solutions in H2O or H218O were used. For t-BuOK, a THF suspension was applied. The reaction was monitored until the entire solution volume was transfused into the mass spectrometer. Azolones were monitored as signals of [M + K]+ ions.
A solution of 40 mg of 8a × 2HCl in 10% aqueous KOH (0.5 mL) was extracted by C6D6 (1 mL). The extract of N,N′-dimethylbenzene-1,2-diamine (8a, free base) was dried by Na2SO4 and then analyzed by NMR. 1H NMR (C6D6): δ 2.41 (s, 6H, 2CH3), 6.62–6.65 (m, 2H, Ar), 6.98–7.01 (m, 2H, Ar). 13C{1H} NMR (C6D6): δ 30.7, 110.9, 119.6, 138.8. ESI-MS(TOF) calcd for C8H13N2 [M + H]+m/z 137.1073, found m/z 137.1074.
A solution of 40 mg of the dihydrochloride (8b × 2HCl) in 0.5 mL of 10% aqueous KOH was extracted by C6D6 (1 mL). The extract of N,N′-dipropylbenzene-1,2-diamine (8b, free base) was dried by Na2SO4 and then analyzed by NMR. 1H NMR (C6D6): δ 0.80 (t, J = 7.4 Hz, 6H, 2CH3), 1.33–1.41 (m, 4H, 2CH2), 2.78–2.80 (m, 4H, 2CH2), 6.70–6.73 (m, 2H, Ar), 6.97–7.01 (m, 2H, Ar). 13C{1H} NMR (C6D6): δ 11.9, 23.1, 46.4, 112.4, 119.7, 138.1.
Influence of the O–NHC coupling reaction on the catalytic activity of Pd–NHC complexes in the Mizoroki–Heck reaction
A 7 mL screw-capped glass tube equipped with a magnetic stirring bar was charged with K2CO3 (138 mg, 1 mmol), PhI (102 mg, 0.5 mmol), n-butyl acrylate (96 mg, 0.75 mmol) and naphthalene (12.8 mg, 0.1 mmol) in DMF (0.9 mL). A solution of the corresponding Pd catalyst (2.5 μmol, 0.5 mol%) in pyridine (0.1 mL) prepared according to the method A or B was added to the reaction tube. The tube was sealed with a screw cap, fitted with a septum and placed immediately in a thermostated oil bath with vigorous stirring at 60 °C, and that instant was taken as the starting time of the reaction. After 20 hours, samples of the reaction mixtures (1 μL) were taken up from the tubes, diluted with MeCN (1 mL) and then subjected to analysis. The yields (%) of coupling product 12 were determined by GC-MS (naphthalene as internal standard).
Influence of the O–NHC coupling reaction on the catalytic activity of Pd–NHC complexes in the Suzuki–Miyaura reaction
A 7 mL screw-capped glass tube equipped with a magnetic stirring bar was charged with a solution of 4-bromo-N,N-dimethylaniline (50 mg, 0.25 mmol), phenylboronic acid (43 mg, 0.35 mmol) and naphthalene (16 mg, 0.125 mmol), i-PrOH (0.5 mL), and a solution of KOH (28 mg, 0.5 mmol) in H2O (0.4 mL). A solution of the Pd catalyst (0.625 μmol, 0.25 mol%) prepared according to the method A or B was added to the reaction tube. The tube was purged with argon and sealed with a screw cap, fitted with a septum and placed immediately in a thermostated oil bath with vigorous stirring at 80 °C, and that instant was taken as the starting time of the reaction. After 2 hours, samples of the reaction mixtures (1 μL) were taken up from the tubes, diluted with MeCN (1 mL) and then subjected to analysis. The yields (%) of coupling product 14 were determined by GC-MS (naphthalene as internal standard).
Influence of the O–NHC coupling reaction on the catalytic activity of Pd–NHC complexes in the arylation of ketones
A 7 mL screw-capped glass tube equipped with a magnetic stirring bar was charged with a solution of t-BuONa (53 mg, 0.55 mmol), PhCl (56 mg, 0.5 mmol), acetophenone (66 mg, 0.55 mmol) and naphthalene (12.8 mg, 0.1 mmol) in 1,4-dioxane (1.9 mL). A solution of the Pd catalyst (5 μmol, 1 mol%) prepared according to the method A or B was added to the reaction tube. The tube was purged with argon and sealed with a screw cap, fitted with a septum and placed immediately in a thermostated oil bath with vigorous stirring at 100 °C, and that instant was taken as the starting time of the reaction. After 3 hours, samples of the reaction mixtures (1 μL) were taken up from the tubes, diluted with MeCN (1 mL) and then subjected to analysis. The yields (%) of coupling product 16 were determined by GC-MS (naphthalene as internal standard).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was supported by the Russian Science Foundation RSF grant no. 14-23-00078 (mechanistic study and investigation of the catalytic reactions) and by the Russian Foundation for Basic Research, RFBR grants 16-29-10786 (synthesis and transformations of metal complexes) and 17-33-50047 (ESI-MS studies of reaction intermediates). The authors also thank the Shared Research Center “Nanotechnologies” of the Platov South-Russian State Polytechnic University and the Department of Structural Studies of Zelinsky Institute of Organic Chemistry for analytical services. The authors are grateful to Olga E. Eremina (Chemistry Department, Lomonosov Moscow State University) for the Raman spectroscopy analysis.References
- (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef; (b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC; (c) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef PubMed; (d) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef PubMed; (e) N-Heterocyclic Carbenes: From Laboratory to Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, Royal Society of Chemistry, London, UK, 2017 Search PubMed; (f) A. Nasr, A. Winkler and M. Tamm, Coord. Chem. Rev., 2016, 316, 68 CrossRef; (g) E. Peris, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.6b00695; (h) V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890 CrossRef; (i) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 0025 CrossRef PubMed; (j) S. Hameury, P. de Fremont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632 RSC; (k) Hydrofunctionalization, ed. V. P. Ananikov and M. Tanaka, Springer, Berlin, Heidelberg, 2013 Search PubMed; (l) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef PubMed.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef PubMed; (b) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493 CrossRef PubMed; (c) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef PubMed; (d) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027 CrossRef.
- (a) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef; (b) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687 CrossRef; (c) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef PubMed.
- (a) C. Valente, M. Pompeo, M. Sayah and M. G. Organ, Org. Process Res. Dev., 2014, 18, 180 CrossRef; (b) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef PubMed.
- Literature search via SciFinder and Scopus revealed hundreds of studies in which M/NHC catalyzed reactions were performed in the presence of excess of a strong base like tert-BuOK, tert-BuONa, KOH, NaOH (sometimes, up to 3 mol of the base per mol of coupling reagent was used).
- (a) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633 CrossRef; (b) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef PubMed; (c) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef PubMed; (d) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef PubMed.
- (a) A. Tudose, L. Delaude, B. André and A. Demonceau, Tetrahedron Lett., 2006, 47, 8529 CrossRef; (b) Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 12215 CrossRef PubMed; (c) A. L. Gottumukkala, J. G. de Vries and A. J. Minnaard, Chem.–Eur. J., 2011, 17, 3091 CrossRef PubMed; (d) C. Jahier, O. V. Zatolochnaya, N. V. Zvyagintsev, V. P. Ananikov and V. Gevorgyan, Org. Lett., 2012, 14, 2846 CrossRef PubMed; (e) O. V. Zatolochnaya, E. G. Gordeev, C. Jahier, V. P. Ananikov and V. Gevorgyan, Chem.–Eur. J., 2014, 20, 9578 CrossRef PubMed.
- L. Xu, L. W. Chung and Y.-D. Wu, ACS Catal., 2016, 6, 483 CrossRef.
- (a) J. Pytkowicz, S. Roland, P. Mangeney, G. Meyer and A. Jutand, J. Organomet. Chem., 2003, 678, 166 CrossRef; (b) S. Raoufmoghaddam, S. Mannathan, A. J. Minnaard, J. G. de Vries and J. N. H. Reek, Chem.–Eur. J., 2015, 21, 18811 CrossRef PubMed.
- (a) M. S. Viciu, R. F. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Organometallics, 2002, 21, 5470 CrossRef; (b) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef PubMed; (c) A. J. DeAngelis, P. G. Gildner, R. Chow and T. J. Colacot, J. Org. Chem., 2015, 80, 6794 CrossRef PubMed; (d) P. R. Melvin, D. Balcells, N. Hazari and A. Nova, ACS Catal., 2015, 5, 5596 CrossRef.
- O. V. Khazipov, M. A. Shevchenko, A. Y. Chernenko, A. V. Astakhov, D. V. Pasyukov, D. B. Eremin, Y. V. Zubavichus, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2018, 37, 1483 CrossRef.
- (a) R. Garrido, P. S. Hernández-Montes, Á. Gordillo, P. Gómez-Sal, C. López-Mardomingo and E. de Jesús, Organometallics, 2015, 34, 1855 CrossRef; (b) E. Steeples, A. Kelling, U. Schilde and D. Esposito, New J. Chem., 2016, 40, 4922 RSC.
- (a) J. C. Lewis, S. H. Wiedemann, R. G. Bergman and J. A. Ellman, Org. Lett., 2004, 6, 35 CrossRef PubMed; (b) F. Zeng and Z. Yu, J. Org. Chem., 2006, 71, 5274 CrossRef PubMed; (c) Y.-M. Liu, Y.-C. Lin, W.-C. Chen, J.-H. Cheng, Y.-L. Chen, G. P. A. Yap, S.-S. Sun and T.-G. Ong, Dalton Trans., 2012, 41, 7382 RSC.
- A. V. Astakhov, O. V. Khazipov, E. S. Degtyareva, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2015, 34, 5759 CrossRef.
- (a) A. M. Zawisza and J. Muzart, Tetrahedron Lett., 2007, 48, 6738 CrossRef; (b) M. Górna, M. S. Szulmanowicz, A. Gniewek, W. Tylus and A. M. Trzeciak, J. Organomet. Chem., 2015, 785, 92 CrossRef.
- A. A. Konstantinchenko, A. S. Morkovnik, A. F. Pozharskii and B. A. Tertov, Chem. Heterocycl. Compd., 1985, 21, 1398 CrossRef.
- (a) L. S. Santos, Eur. J. Org. Chem., 2008, 2008, 235 CrossRef; (b) V. V. Kachala, L. L. Khemchyan, A. S. Kashin, N. V. Orlov, A. A. Grachev, S. S. Zalesskiy and V. P. Ananikov, Russ. Chem. Rev., 2013, 82, 648 CrossRef; (c) K. L. Vikse, Z. Ahmadi and J. S. McIndoe, Coord. Chem. Rev., 2014, 279, 96 CrossRef; (d) C. Iacobucci, S. Reale and F. De Angelis, Angew. Chem., Int. Ed., 2016, 55, 2980 CrossRef PubMed.
- (a) B. R. M. Lake, M. R. Chapman and C. E. Willans, in Organomet. Chem., The Royal Society of Chemistry, 2016, vol. 40, p. 107 Search PubMed; (b) D. J. Nelson, J. M. Praetorius and C. M. Crudden, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, The Royal Society of Chemistry, 2017, vol. 27, ch. 2, p. 46 Search PubMed; (c) A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2017, 36, 1981 CrossRef; (d) E. G. Gordeev, D. B. Eremin, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2017, 37, 787 CrossRef.
- (a) U. Siemeling, C. Farber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC; (b) D. Martin, N. Lassauque, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2012, 51, 6172 CrossRef PubMed.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2 CrossRef.
- O. E. Eremina, A. V. Sidorov, T. N. Shekhovtsova, E. A. Goodilin and I. A. Veselova, ACS Appl. Mater. Interfaces, 2017, 9, 15058 Search PubMed.
- M. J. Saif and K. R. Flower, Transition Met. Chem., 2013, 38, 113 CrossRef.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608 CrossRef PubMed.
- J. A. V. Er, A. G. Tennyson, J. W. Kamplain, V. M. Lynch and C. W. Bielawski, Eur. J. Inorg. Chem., 2009, 2009, 1729 CrossRef.
- H. V. Huynh, Y. Han, J. H. H. Ho and G. K. Tan, Organometallics, 2006, 25, 3267 CrossRef.
- Y. Han, H. V. Huynh and G. K. Tan, Organometallics, 2007, 26, 6447 CrossRef.
- Y. Shi, Z. Cai, Y. Peng, Z. Shi and G. Pang, J. Chem. Res., 2011, 35, 161 CrossRef.
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2006, 12, 4743 CrossRef PubMed.
- H. V. Huynh, J. H. H. Ho, T. C. Neo and L. L. Koh, J. Organomet. Chem., 2005, 690, 3854 CrossRef.
- M. Bouché, G. Dahm, A. Maisse-François, T. Achard and S. Bellemin-Laponnaz, Eur. J. Inorg. Chem., 2016, 2016, 2828 CrossRef.
- Y. Han, H. V. Huynh and G. K. Tan, Organometallics, 2007, 26, 4612 CrossRef.
- H. V. Huynh, C. Holtgrewe, T. Pape, L. L. Koh and E. Hahn, Organometallics, 2006, 25, 245 CrossRef.
- I. M. AlNashef, M. A. Hashim, F. S. Mjalli, M. Q. A.-h. Ali and M. Hayyan, Tetrahedron Lett., 2010, 51, 1976 CrossRef.
- J. B. H. Hřebabecký, Collect. Czech. Chem. Commun., 1985, 50, 779 CrossRef.
- M. Guiliano, G. Mille, J. Chouteau, J. Kister and J. Metzger, Appl. Spectrosc., 1981, 35, 486 CrossRef.
- Z. Wu, Z. An, X. Chen and P. Chen, Org. Lett., 2013, 15, 1456 CrossRef PubMed.
- S. T. Manjare, H. B. Singh and R. J. Butcher, Tetrahedron, 2012, 68, 10561 CrossRef.
- J. Duchek and A. Vasella, Helv. Chim. Acta, 2011, 94, 977 CrossRef.
- G. Vernin, H. Domlog, C. Siv, J. Metzger and A. K. El-Shafei, J. Heterocycl. Chem., 1981, 18, 85 CrossRef.
- (a) J.-P. Li, Y. Huang, M.-S. Xie, G.-R. Qu, H.-Y. Niu, H.-X. Wang, B.-W. Qin and H.-M. Guo, J. Org. Chem., 2013, 78, 12629 CrossRef PubMed; (b) Y. Li, W. Du and W.-P. Deng, Tetrahedron, 2012, 68, 3611 CrossRef; (c) H. M. Lima and C. J. Lovely, Org. Lett., 2011, 13, 5736 CrossRef PubMed; (d) Y. Kandri Rodi, F. Ouazzani Chahdi, E. M. Essassi, S. V. Luis, M. Bolte and L. El Ammari, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o3234 Search PubMed.
- W. Zeng, E. Wang, R. Qiu, M. Sohail, S. Wu and F.-X. Chen, J. Organomet. Chem., 2013, 743, 44 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data for reaction mechanisms and stoichiometry, copies of NMR spectra and Raman spectra, GC-MS and ESI-MS data, and FE-SEM/EDS data. See DOI: 10.1039/c8sc01353e |
| This journal is © The Royal Society of Chemistry 2018 |