
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenome mining, isolation, chemical synthesis and biological evaluation of a novel lanthipeptide, tikitericin, from the extremophilic microorganism Thermogemmatispora strain T81†
Buzhe
Xu‡
ab,
Emma J.
Aitken‡
c,
Benjamin P.
Baker
c,
Claire A.
Turner
c,
Joanne E.
Harvey
 bc,
Matthew B.
Stott
de,
Jean F.
Power
e,
Paul W. R.
Harris
abf,
Robert A.
Keyzers
*bc and
Margaret A.
Brimble
*abf
bc,
Matthew B.
Stott
de,
Jean F.
Power
e,
Paul W. R.
Harris
abf,
Robert A.
Keyzers
*bc and
Margaret A.
Brimble
*abf
aSchool of Chemical Sciences, 23 Symonds Street, Auckland 1010, New Zealand. E-mail: m.brimble@auckland.ac.nz; Tel: +64 9 9238259
bMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: rob.keyzers@vuw.ac.nz; Tel: +64 4 4635117
cSchool of Chemical & Physical Sciences, The Centre for Biodiscovery, Victoria University of Wellington, PO Box 600, Wellington 6140, New Zealand
dSchool of Biological Sciences, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand
eGNS Science, Private Bag 2000, Taupō 3352, New Zealand
fSchool of Biological Sciences, 23 Symonds Street, Auckland 1010, New Zealand
First published on 20th August 2018
Abstract
Genome mining of the New Zealand extremophilic microorganism Thermogemmatispora strain T81 indicated the presence of biosynthetic machinery to produce several different peptidic natural products. Solid-phase culture of T81 led to the isolation of tikitericin 1, a new lanthipeptide characterised by four (methyl)lanthionine bridges. The mass-guided isolation and structural elucidation of tikitericin 1 is described together with its total synthesis via Fmoc-solid-phase peptide synthesis (SPPS). The key non-canonical (methyl)lanthionine residues were synthesised in solution phase via an improved synthetic route and subsequently assembled to construct the peptide backbone using Fmoc-SPPS. N-Terminal truncated analogues of tikitericin (2–5) were also prepared in order to evaluate the contribution of each sequential ring of the polycyclic lanthipeptide to the antibacterial activity.
Introduction
Although several key members have been known for decades, Ribosomally synthesised and Post-translationally modified Peptides (RiPPs) are a recent addition to Nature's natural product biosynthetic repertoire. RiPPs are produced from a genetically encoded linear precursor amino acid sequence which comprise two units, an N-terminal leader peptide and a core peptide unit that goes on to form the primary amino acid sequence of the mature RiPP.1–3 The leader peptide is used for recognition by subsequent post-translational tailoring enzymes. Following biosynthesis via the ribosome, the leader peptide is cleaved off to release the core peptide at a designated leader cleavage sequence, often two contiguous glycine resides (called the “double gly” motif).2,3 Currently, lanthipeptides are classified into one of four classes, depending on the suite of enzymes required to form and install the (Me)Lan units.2 These four classes are all unified, however, by their shared approach to forming (Me)Lan dimers. The biosynthesis of the (Me)Lan structural units is performed post-translationally via enzyme-catalysed dehydration of serine and threonine residues followed by stereospecific intramolecular Michael-type addition of adjacent cysteine thiols to form thioether linkages (Fig. 1).2–5 Over 100 compounds sharing the (Me)Lan chemical moiety have been reported, many of which exhibit antibacterial activity and are therefore referred to as lantibiotics.6,7 Most lantibiotics are believed to exert their activity on pathogenic bacteria via pore formation in the cell membrane and/or inhibition of peptidoglycan biosynthesis.8–11 Several lantibiotics have gathered considerable attention in clinical development for the treatment of infectious diseases due to their potent antimicrobial activity against a wide range of human pathogens8,12–14 and the archetypical member nisin (Fig. 1) has been approved as a food preservation agent.15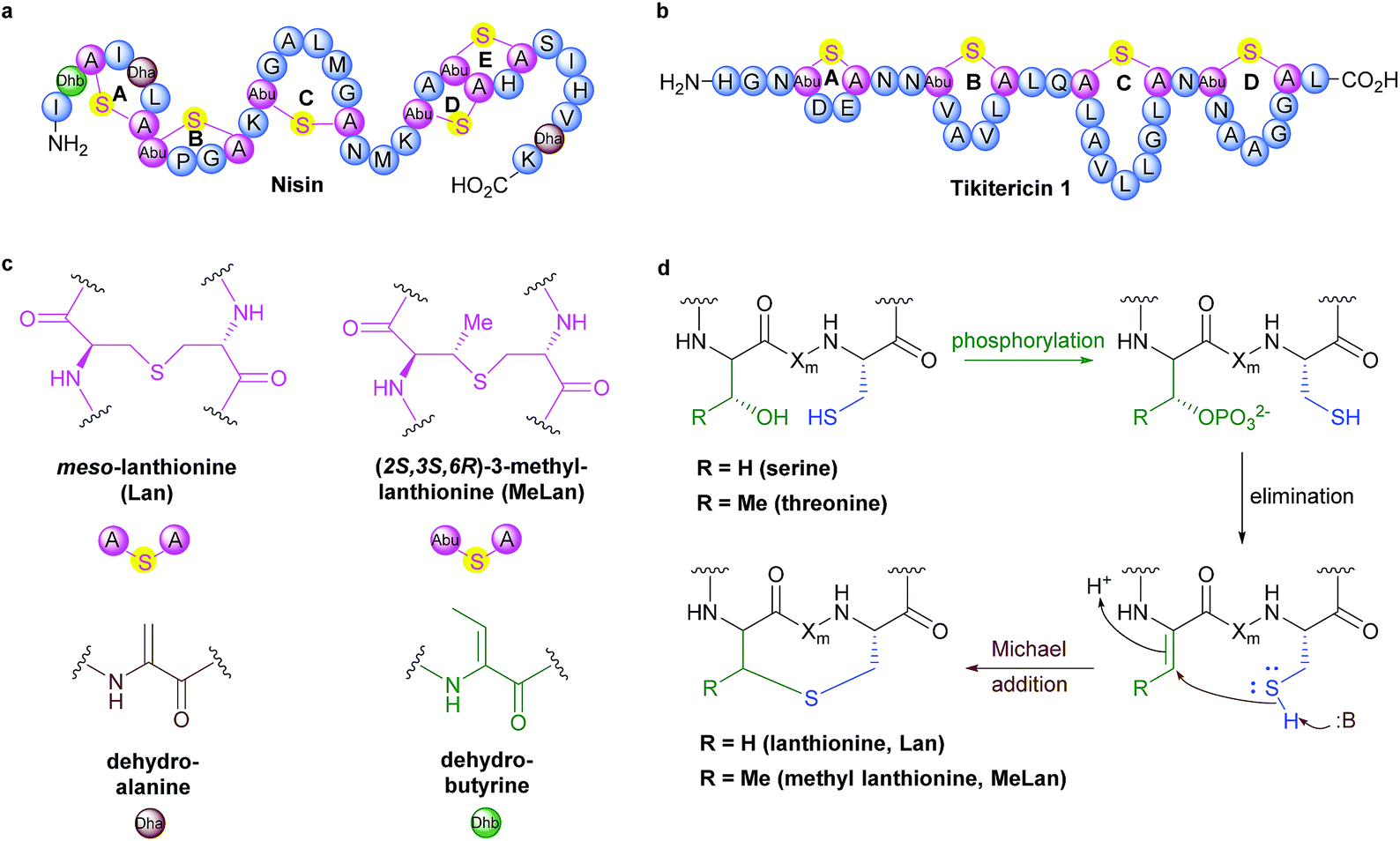 | ||
| Fig. 1 (a) Sequence and ring topologies of nisin. (b) Sequence and ring topologies of tikitericin 1. (c) Chemical structures of unnatural amino acid residues. (d) Biosynthetic pathway to lanthipeptide (Me)Lan residues, Xm indicates a modified residue, adapted from Knerr and van der Donk.2 | ||
Encouraged by their therapeutic activity, a variety of molecular engineering techniques have been developed in vitro and in vivo to make analogues of lantibiotics to construct structure–activity relationships and probe their pharmacological properties.16–22 As a complementary strategy to recombinant methods, chemical synthesis is a powerful platform for the construction of lantibiotics and other cyclic peptides with greater structural variation, including incorporation of non-thioether based bridges.23–28 The lanthipeptides lactocin S,29 lacticin 3147 A1 and A2,30 lacticin 481,31 analogues of epilancin 15X32 and cytolysin33 have all been successfully synthesised via Fmoc-solid-phase peptide synthesis (Fmoc-SPPS), thus providing a general chemical method for the synthesis of complex lantibiotics and analogues thereof.
Thermogemmatispora strain T81 is a thermophilic Gram-positive bacterium isolated from geothermally-heated soils of Tikitere (Hell's Gate), Rotorua, New Zealand34 and exhibits antimicrobial activity against a relatively wide range of extremophilic bacteria such as strains TKA 04.11 and WKT 22.10.35 Genome mining of Thermogemmatispora strain T81 revealed the potential to produce novel RiPP natural products, including the presence of a lanthionine synthetase gene showing 99% homology to a class II lanthipeptide biosynthetic cluster (Fig. S1 and S2, see ESI† for details). In the current study, a new lanthipeptide tikitericin 1 (Fig. 1), featuring four linear (Me)Lan rings, was isolated as a major product from solid-phase culturing of strain T81 and in co-culture with five competitive strains of extremophilic bacteria (see ESI† for details) to investigate whether this RiPP natural product was responsible for the observed antimicrobial activity. We herein report the isolation, structure elucidation, total synthesis and biological evaluation of 1, as well as its N-terminal truncated analogues 2–5 (Fig. 2) to probe their biological activities and determine the key structural components of tikitericin 1 required to confer bioactivity.
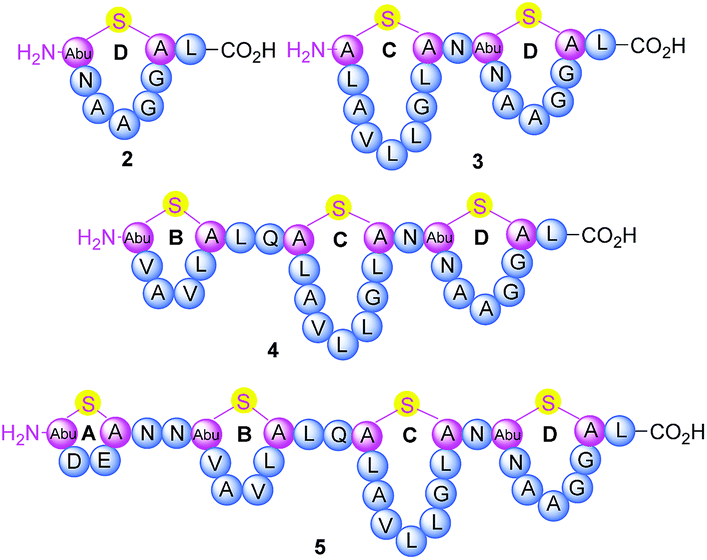 | ||
| Fig. 2 Sequence and ring topologies of N-terminal truncated analogues 2–5 of tikitericin 1. | ||
Results and discussion
Isolation and structure determination of tikitericin 1
The presence of 1 was ascertained from whole cell MALDI-TOF MS analysis of the solid-phase culture of strain T81. Observation of [M + H]+, [M + Na]+ and [M + K]+ adducts at m/z 3429.6, 3451.6 and 3467.6 were consistent with four-fold dehydration and thioether formation of the genome mining predicted core peptide sequence (see ESI† for details). Unfortunately, screening of various different liquid culture media, temperatures and other culture conditions did not result in any production of 1. Subsequently, 1200 Petri dishes of strain T81 were cultured, with the resulting lawns of biomass scraped by hand and extracted into MeOH. Mass spectrometry guided isolation of 1 using a combination of reversed-phase and size exclusion chromatography, culminating in RP-HPLC separation, led to isolation of 400 μg of tikitericin 1 (see ESI† for details).HR-ESI-MS analysis provided a series of multiply protonated and sodiated adduct ions consistent with the formula C144H233N43O46S4 (see ESI† for details). Fortuitously, both MALDI-TOF and collision induced dissociation ESI-TOF tandem mass spectrometry clearly indicated the presence of several key b and y ions, indicative of a “linear” topology36,37 whereby each (Me)Lan link is cyclised locally within a restricted portion of the peptide backbone, without introducing knots into the peptide structure. Such a linear ring topology is rare amongst lanthipeptides.2
Internal thioether rings prevent efficient fragmentation of peptide backbones; therefore, to confirm the primary amino acid sequence of 1, the compound required linearisation. Thermal oxidative ring opening was entirely unsuccessful. Fortunately, Raney Nickel reduction as reported by Fuchs provided a truncated linearised peptide (Val13-Leu35).38 However, cleavage of four key peptide bonds within the backbone chain was not observed in the HR-ESI-MSMS spectrum (see ESI† for details). Finally, base-induced elimination and subsequent trapping of the resultant alkene with thioethanol afforded a fully linearised peptide,39 with cleavage of all peptide linkages observed in the tandem HR-ESI mass spectrum. The b and y ions observed were entirely consistent with the amino acid sequence predicted from bioinformatic analysis of strain T81 (see ESI† for details).
With the predicted linear amino acid sequence of tikitericin 1 confirmed, the last remaining question was the relative configurations of the (Me)Lan amino acid dimers. Assuming a fixed L-configuration for the cysteine nucleophile, there are two possible diastereomers of Lan and four of MeLan possible, moreover some of these different possible diastereomers have been observed in various lanthipeptides40 with the MeLan configuration thought to play an important role in lantibiotic bioactivity,31 although one form of each (viz. 2S,6R-Lan, 2S,3S,6R-3-MeLan) is most common.41 Acid hydrolysis of tikitericin 1 in 6 M HCl was followed by derivatising the C- and N-termini of the resultant amino acids as methyl esters and pentafluoropropionic amides, respectively.40,41 Successful derivatisation was confirmed by HR-ESI-MS. Reversed-phase LC-MS analysis of the derivatised MeLan dimers compared with the retention times of authentic standards indicated that all three residues have the 2S,3S,6R configuration. This was confirmed by observation of a single peak when the tikitericin-derived MeLan residues were co-injected with the corresponding authentic standard (see ESI† for details).
Determination of the configuration of the sole Lan dimer proved more troublesome. Various LC-MS conditions were trialled but unfortunately complete separation of the two possible diastereomers was impossible when using either reversed- or chiral stationary phases, although co-injection of the tikitericin-derived Lan (with (2S,6R)-Lan standard) indicated that it was likely to have the 2S,6R-configuration. The Lan derivative could also not be separated using chiral GC columns (Chirasil-Val or cyclodextrin) due to the upper temperature limits of these column stationary phases. Accordingly, standards of the two possible diastereomers were separated using an HP-VOC column, although without complete baseline separation. Similar to the LC results, the GC analysis suggested the 2S,6R configuration and therefore, although neither LC nor GC analysis was conclusive, we tentatively proposed that this is the configuration of the Lan residue in tikitericin 1. Using the standardised nomenclature proposed by Arnison et al., tikitericin 1 can therefore be described as [SS4-7, SS10-15, S18-26,SS2834C_S]HGNAbuDECNNAbuVAVLCLQALAVLLGLCNAbuNAAGGCL.1
Unfortunately, with such a low production of 1 by strain T81 on solid phase culture, and the fact that a significant number of degradative studies had been carried out to determine the structure of the molecule, we were unable to profile its bioactivity from the natural stock. Our only recourse to assess the biological potential of 1 as a lantibiotic rested on its chemical synthesis.
Chemical synthesis of tikitericin 1 and truncated analogues 2–5
The synthesis of tikitericin 1 involves creating the orthogonally protected MeLan and Lan building blocks in solution, and their subsequent incorporation into Fmoc-SPPS to assemble the polycyclic peptide backbone (Scheme 1). This approach relies heavily on the orthogonal protecting groups of MeLan and Lan being compatible with standard Fmoc peptide synthesis and allowing for installation of the macrocyclic thioether rings.26,29,33,42–46 MeLan 6 and Lan 7 bearing allyl, Alloc and Fmoc as the orthogonal protecting groups, could be incorporated into each ring of tikitericin 1 to form the desired thioether macrocyclic units via selective deprotection of the allyl and Alloc on the corresponding (Me)Lan residues, followed by intramolecular cyclisation with the N-terminus of the elongated peptide chain.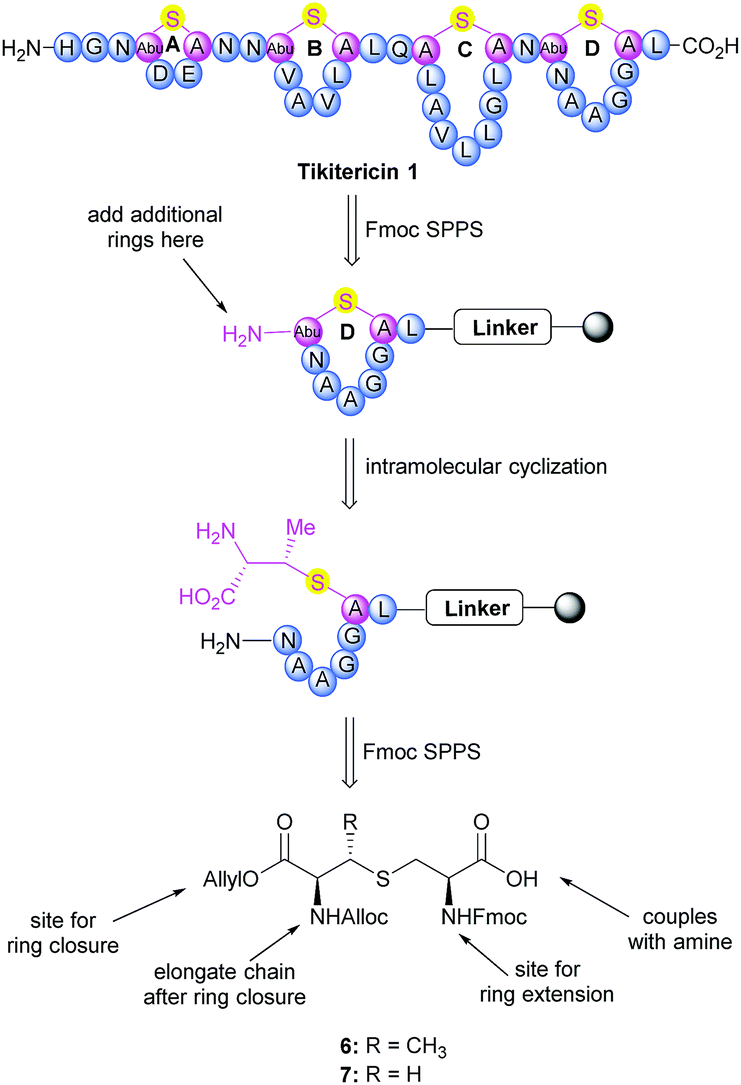 | ||
| Scheme 1 Retrosynthetic analysis of tikitericin 1. | ||
However, efficient chemical synthesis of such orthogonally protected (Me)Lan building blocks with the correct stereochemistry is difficult. We initially followed Vederas's strategy for the preparation of (Me)Lan building blocks, which has already been applied successfully to the total synthesis of lacticin 3147 A1 and A2.42 As tikitericin 1 contains four successive MeLan or Lan rings, one MeLan building block for the D, B and A rings and one Lan building block for the C ring were necessary. Starting from N-Trt-D-Thr or N-Trt-D-Ser , we found that the final step of the reported synthesis, which replaced a 2,4-dinitrobenzenesulfonyl (DNS) protecting group with an Alloc group to obtain compounds 6 and 7, failed in our hands. Additionally, synthesis of the starting materials N-Trt-D-Thr and N-Trt-D-Ser from D-Thr/D-Ser following Mustapa and co-workers' strategy47 was low yielding and difficult to reproduce on a larger scale. An optimised synthetic route to access the MeLan and Lan building blocks 6 and 7 was therefore developed herein and is summarised in Scheme 2.
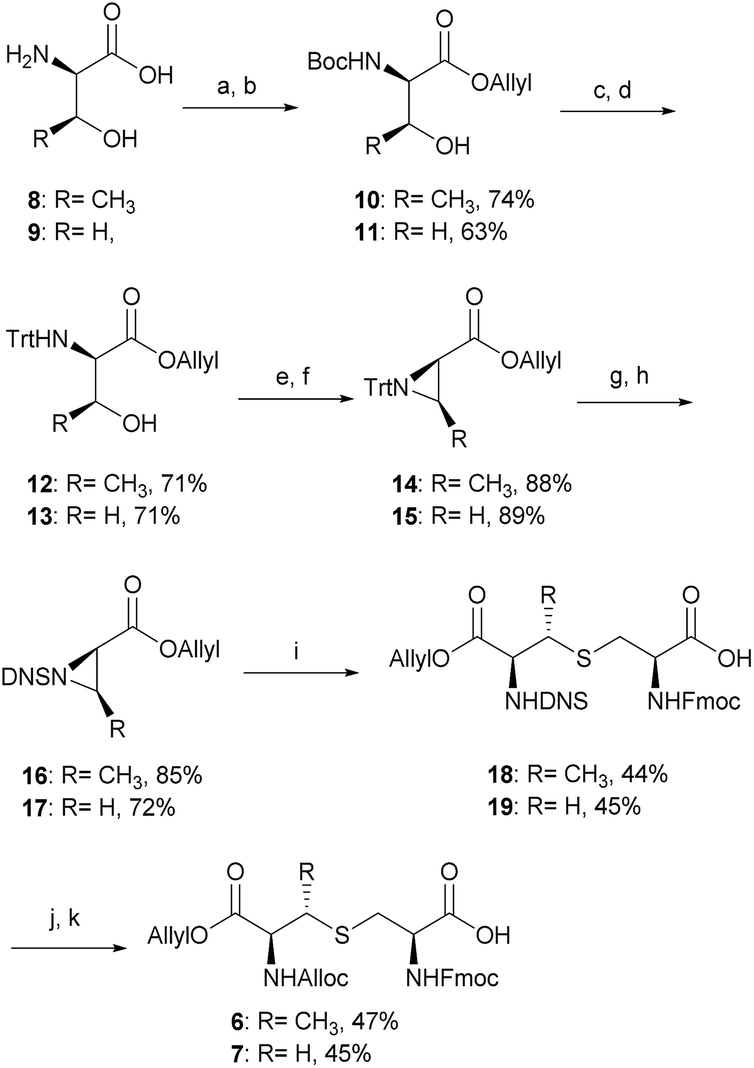 | ||
| Scheme 2 Optimised synthetic route for the orthogonally protected (Me)Lan building blocks 6 and 7. Reagents and conditions: (a) Boc2O, NaOH, RT, 12 h; (b) Allyl-Br, Cs2CO3, DMF, RT, 12 h; (c) TFA, CH2Cl2, RT, 2 h; (d) Trt-Cl, Et3N, EtOAc, 0–25 °C, 12 h; (e) MsCl, CH2Cl2, 0–25 °C, 12 h; (f) Et3N, DMF, 70 °C, 12 h; (g) TFA, CH2Cl2, MeOH, 0 °C, 2 h; (h) DNS-Cl, Na2CO3, EtOAc, RT, 12 h; (i) Fmoc-Cys-OH, BF3·OEt2, CH2Cl2, 0–25 °C, 72 h; (j) thioglycolic acid, Et3N, CH2Cl2, 0 °C, 2 h; (k) Alloc-Cl, NaHCO3, H2O, dioxane, 0–25 °C, 12 h. Alloc, allyloxycarbonyl; Trt, triphenylmethyl; DNS, 2,4-dinitrobenzenesulfonyl. | ||
The α-amino groups on D-Thr 8 or D-Ser 9 were protected as a Boc group with di-tert-butyl dicarbonate (Boc2O), followed by allylation of the carboxylic acid to form the N-Boc-D-Thr-OAllyl 10 or N-Boc-D-Ser-OAllyl 11 in 74% and 63% yield, respectively over two steps (Scheme 2). The Boc group on 10 or 11 was then removed using TFA in CH2Cl2 at room temperature for 2 h, followed by tritylation of the amino group to form the N-Trt-D-Thr-OAllyl 12 or N-Trt-D-Ser-OAllyl 13 intermediates (71% yield over two steps). Subsequent aziridine ring formation using methanesulfonyl chloride to afford Trt-aziridines 14 or 15, Trt to DNS protecting group replacement to afford DNS-aziridines 16 or 17, and aziridine ring-opening by Fmoc-Cys-OH to afford DNS-protected (Me)Lan 18 or 19, were carried out following literature methods with yields similar to those reported.42 The final step to install an Alloc protecting group in place of the DNS on intermediate 18 or 19, however, was carried out using an improved protocol. Initial deprotection of the DNS group on 18 or 19 was achieved by treatment with thioglycolic acid and Et3N in CH2Cl2 at 0 °C for 2 h, followed by washing with Na2CO3 (aq., sat.) to remove the formed 2-((2,4-dinitrophenyl)thio)acetic acid. Subsequent Alloc protecting group installation on the free amino group was carried out by treating the resulting crude material with allyl chloroformate (Alloc-Cl), in H2O and dioxane (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and NaHCO3. In this way orthogonally protected (Me)Lan building blocks 6 and 7 could be produced in 47% and 45% yields respectively, over two steps (Scheme 2, see ESI† for details).
:1) and NaHCO3. In this way orthogonally protected (Me)Lan building blocks 6 and 7 could be produced in 47% and 45% yields respectively, over two steps (Scheme 2, see ESI† for details).
With the (Me)Lan building blocks 6 and 7 in hand, Fmoc-SPPS of tikitericin 1 commenced with attachment of MeLan building block 6 onto TentaGel-PHB resin (Wang linker, 0.25 mmol g−1) preloaded with Fmoc-Leu-OH. The Fmoc-SPPS synthesis proceeded smoothly until the third ring was introduced (ring B, intermediate 27) in which case an additional inseparable by-product was also produced together with the desired analogue 4. Our hypothesis was that a loading of 0.25 mmol g−1 was too high for the on-resin synthesis of this polycyclic peptide. Thus, a low-loaded polystyrene resin (PS) (0.1 mmol g−1) functionalised with Fmoc–Leu–Wang linker, was chosen to circumvent any undesired intermolecular dimerisation taking place during the on-resin intramolecular macrocyclisation step. However, in this case, the formation of the first ring (ring D) did not proceed well and an unidentified impurity was also generated (see ESI† for details). PS resin performs poorly in the synthesis of complex peptides due to its high hydrophobicity and low swelling ability in DMF which is the most common solvent used for SPPS. ChemMatrix is an all PEG-based resin that shows good chemical and mechanical stability and exhibits excellent swelling ability in most solvents due to its amphiphilic nature.48 It therefore performs extremely well for the solid-supported synthesis of highly structured, hydrophobic peptides compared to PS resin.48,49 We therefore selected ChemMatrix for our subsequent synthetic work.
The successful Fmoc-SPPS of tikitericin 1 commenced with attachment of commercially available 4-(hydroxymethyl)phenoxypropanoic acid (HMPP) linker preloaded with Fmoc-Leu onto poly(ethyleneglycol) (PEG)-based ChemMatrix resin (0.1 mmol g−1, Scheme 3). After removal of the Fmoc group on 20 using 20% piperidine in DMF (v/v), the MeLan building block 6 was coupled successfully to the peptidyl resin using (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), 6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt), and 4-methylmorpholine (NMM), followed by standard Fmoc-SPPS to assemble the required amino acids to obtain the linear precursor 21. The allyl and Alloc groups on the MeLan residue of 21 were simultaneously removed using tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) and phenylsilane, followed by removal of the Fmoc group to achieve the resin-bound intermediate 22. Compound 23, containing the first ring D of tikitericin 1, was then obtained via on-resin intramolecular lactamisation using PyBOP/6-Cl-HOBt and NMM. Cleavage of ring D from the resin with TFA, H2O and TIPS, with concomitant removal of all acid labile side chain protecting groups, afforded analogue 2, thereby establishing that the methyllanthionine bridge had been successfully installed. Subsequently, truncated analogues 3, 4, 5 and full length tikitericin 1 were constructed in a similar fashion.
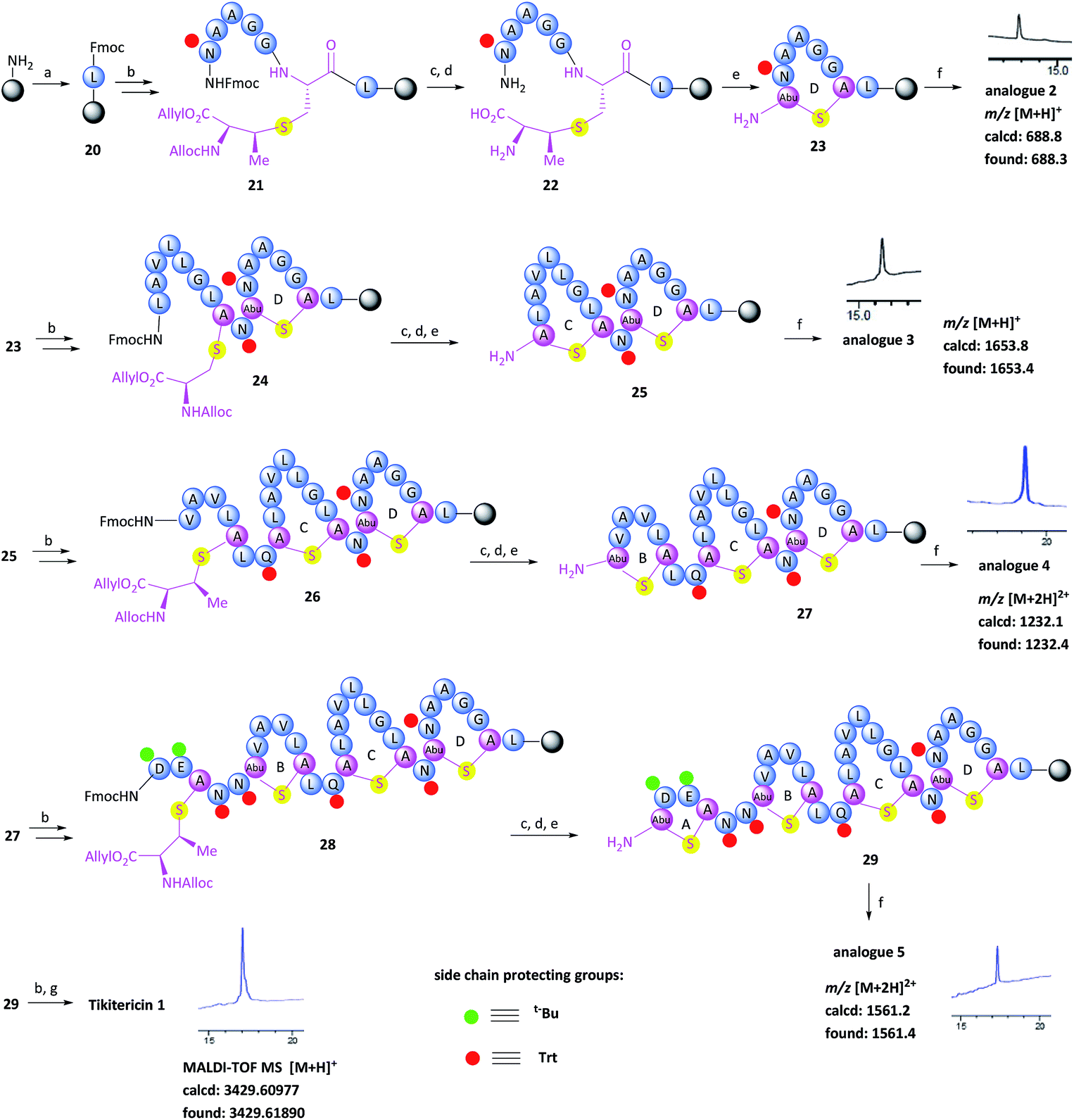 | ||
| Scheme 3 Fmoc-solid-phase peptide synthesis of tikitericin 1. Reagents and conditions: (a) DIC, Fmoc-Leu-HMPP, CH2Cl2, 24 h; (b) Fmoc-SPPS: (i) PyBOP, 6-Cl-HOBt, NMM, DMF, 40 min; (ii) 20% piperidine in DMF (v/v), 2 × 5 min; (c) Pd(PPh3)4, PhSiH3, DMF/CH2Cl2 (v/v, 1/1), 2 h; (d) 20% piperidine in DMF (v/v), 2 × 5 min; (e) PyBOP, 6-Cl-HOBt, NMM, DMF, 2 h; (f) TFA/H2O/TIPS (v/v/v, 95/2.5/2.5), 1 h; (g) TFA/H2O/TIPS/EDT (v/v/v/v, 94/2.5/2.5/1), 1 h. TIPS, triisopropylsilane; EDT, 1,2-ethanedithiol. | ||
Following reversed-phase HPLC purification, MALDI-TOF MS analysis of synthetic tikitericin 1 ([M + H]+ found: 3429.61890 Da; calcd: 3429.60977) compared favourably with the natural product ([M + H]+ 3429.6 Da).35 Subsequent HR-ESI-MS and MS/MS of the intact synthetic 1 returned identical mass spectra to those recorded for the natural product. Furthermore, MS/MS fragmentation studies of the base-eliminated/thioethanol trapped linearised product were also performed on our synthetic material, which confirmed an identical sequence to the natural product (see ESI† for details).
The overall yield of tikitericin 1 was 0.86% based on a resin loading of 0.1 mmol g−1, which is comparable to the reported recoveries for other complex lanthipeptides.30–32,45,46 N-Terminal truncated forms of tikitericin 1 are potentially bioactive; therefore, truncated analogues 2, 3, 4, 5 were all obtained by cleavage of intermediates en route to the final polycyclic compound in yields of 36.1%, 8.9%, 2.8% and 1.4%, respectively.
Synthetic tikitericin 1 and the N-terminal truncated compounds 2, 3, 4, 5 were evaluated via liquid culture antimicrobial assays against Staphylococcus aureus ATCC 6538 using a standard two-fold dilution protocol (see ESI† for details). Unfortunately, no inhibitory activity was observed up to a concentration of 128 μM. While this result suggests that tikitericin 1 is inactive against this S. aureus strain, it may be possible that this peptide is active against other Gram-positive strains of bacteria. A comprehensive study by Mota-Meira et al.50 showed that the minimum inhibitory concentration of the lantibiotics mutacin B-Ny266, nisin A, and antibiotic standards vancomycin and oxacillin against Gram positive bacteria varies significantly between species and strains. Further biological evaluation of tikitericin 1 and its truncated analogues 2, 3, 4, 5 will be reported in due course.
Conclusions
We herein report the isolation of tikitericin 1 from solid-phase cultures of Thermogemmatispora strain T81, and confirmation of its molecular structure by total chemical synthesis using solution phase synthesis of an introduction motif for the thioether ring combined with Fmoc-SPPS. Isolation of 1 (400 μg from 1200 Petri dish cultures) was achieved using a series of mass-guided reversed-phase chromatographic steps, while confirmation of its absolute configuration was achieved using LC-MS and GC-MS analyses of acid hydrosylates in comparison to authentic standards of the (Me)Lan dimers. Orthogonally-protected MeLan and Lan building blocks were prepared in 7.4% and 5.8% yield respectively via an improved 11 step solution phase synthesis starting from commercially available D-Thr or D-Ser. Importantly, these key building blocks could be accessed in large quantities. Incorporation of these building blocks using Fmoc-SPPS on an optimised all PEG-based resin (ChemMatrix) was used to assemble the (Me)Lan polycyclic peptide backbone via on-resin intramolecular cyclisation using standard SPPS conditions and synthetic tikitericin 1 was obtained in 95% purity. The mass spectrometric properties of intact and linearised synthetic tikitericin 1 were concordant with those obtained from natural sources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the late Jim Gray and the Tikitere Trust, the kaitiaki (guardians) of Tikitere Hell's Gate thermal area, for their permission to sample and enthusiasm for our research. We gratefully acknowledge the RSNZ Marsden Fund for a doctoral scholarship support (to B. Xu), VUW and GNS Science for MSc scholarship support (E. Aitken) and the Maurice Wilkins Centre for Molecular Biodiscovery and GNS Science's Geothermal Programme (GRN) for financial support of this project. J. C. Vederas is thanked for provision of authentic standards of the (Me)Lan diastereomers for stereochemical assignment. N. Demarais is thanked for the MALDI-TOF MS and HRESI-MS/MS analysis of the synthetic intact tikitericin 1. S. A. Ferguson is also thanked for the antimicrobial assays of compounds 1–5.Notes and references
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108 RSC.
- P. J. Knerr and W. A. van der Donk, Annu. Rev. Biochem., 2012, 81, 479 CrossRef PubMed.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Chem. Rev., 2017, 117, 5457 CrossRef PubMed.
- A. B. Tabor, Bioorg. Chem., 2014, 55, 39 CrossRef PubMed.
- E. L. Ongey and P. Neubauer, Microb. Cell Fact., 2016, 15, 97 CrossRef PubMed.
- D. Field, P. D. Cotter, C. Hill and R. P. Ross, Front. Microbiol., 2015, 6, 1363 Search PubMed.
- J. M. Willey and W. A. van der Donk, Annu. Rev. Microbiol., 2007, 61, 477 CrossRef PubMed.
- G. Bierbaum and H. G. Sahl, Curr. Pharm. Biotechnol., 2009, 10, 2 Search PubMed.
- E. Breukink, I. Wiedemann, C. van Kraaij, O. P. Kuipers, H. G. Sahl and B. de Kruijff, Science, 1999, 286, 2361 CrossRef PubMed.
- E. Breukink and B. de Kruijff, Nat. Rev. Drug Discovery, 2006, 5, 321 CrossRef PubMed.
- N. I. Martin and E. Breukink, Future Microbiol., 2007, 2, 513 CrossRef PubMed.
- M. M. Al-Mahrous and M. Upton, Expert Opin. Drug Discovery, 2011, 6, 155 CrossRef PubMed.
- M. J. Dawson and R. W. Scott, Curr. Opin. Pharmacol., 2012, 12, 545 CrossRef PubMed.
- P. D. Cotter, C. Hill and R. P. Ross, Curr. Protein Pept. Sci., 2005, 6, 61 CrossRef PubMed.
- J. M. Shin, J. W. Gwak, P. Kamarajan, J. C. Fenno, A. H. Rickard and Y. L. Kapila, J. Appl. Microbiol., 2016, 120, 1449 CrossRef PubMed.
- A. C. Ross and J. C. Vederas, J. Antibiot., 2011, 64, 27 CrossRef PubMed.
- J. Nagao, M. Nishie and K. Sonomoto, Curr. Pharm. Biotechnol., 2011, 12, 1221 Search PubMed.
- D. Field, C. Hill, P. D. Cotter and R. P. Ross, Mol. Microbiol., 2010, 78, 1077 CrossRef PubMed.
- R. Rink, J. Wierenga, A. Kuipers, L. D. Kluskens, A. J. M. Driessen, O. P. Kuipers and G. N. Moll, Appl. Environ. Microbiol., 2007, 73, 5809 CrossRef PubMed.
- F. Oldach, R. Al Toma, A. Kuthning, T. Caetano, S. Mendo, N. Budisa and R. D. Sussmuth, Angew. Chem., Int. Ed., 2012, 51, 415 CrossRef PubMed.
- Y. X. Shi, X. A. Yang, N. Garg and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 2338 CrossRef PubMed.
- M. R. Levengood, P. J. Knerr, T. J. Oman and W. A. van der Donk, J. Am. Chem. Soc., 2009, 131, 12024 CrossRef PubMed.
- A. B. Tabor, Org. Biomol. Chem., 2011, 9, 7606 RSC.
- Z. Dekan, I. Vetter, N. L. Daly, D. J. Craik, R. J. Lewis and P. F. Alewood, J. Am. Chem. Soc., 2011, 133, 15866 CrossRef PubMed.
- P. J. Knerr, A. Tzekou, D. Ricklin, H. C. Qu, H. Chen, W. A. van der Donk and J. D. Lambris, ACS Chem. Biol., 2011, 6, 753 CrossRef PubMed.
- H. Liu, V. R. Pattabiraman and J. C. Vederas, Org. Lett., 2009, 11, 5574 CrossRef PubMed.
- V. R. Pattabiraman, J. L. Stymiest, D. J. Derksen, N. I. Martin and J. C. Vederas, Org. Lett., 2007, 9, 699 CrossRef PubMed.
- N. Ghalit, J. F. Reichwein, H. W. Hilbers, E. Breukink, D. T. S. Rijkers and R. M. J. Liskamp, ChemBioChem, 2007, 8, 1540 CrossRef PubMed.
- A. C. Ross, H. Q. Liu, V. R. Pattabiraman and J. C. Vederas, J. Am. Chem. Soc., 2010, 132, 462 CrossRef PubMed.
- W. Liu, A. S. Chan, H. Liu, S. A. Cochrane and J. C. Vederas, J. Am. Chem. Soc., 2011, 133, 14216 CrossRef PubMed.
- P. J. Knerr and W. A. van der Donk, J. Am. Chem. Soc., 2013, 135, 7094 CrossRef PubMed.
- P. J. Knerr and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 7648 CrossRef PubMed.
- S. Mukherjee, L. Huo, G. N. Thibodeaux and W. A. van der Donk, Org. Lett., 2016, 18, 6188 CrossRef PubMed.
- M. B. Stott, M. A. Crowe, B. W. Mountain, A. V. Smirnova, S. Hou, M. Alam and P. F. Dunfield, Environ. Microbiol., 2008, 10, 2030 CrossRef PubMed.
- E. J. Aitken, MSc thesis, Novel Peptide Natural Products from the NZ Thermophilic Microorganism Thermogemmatispora Strain T81, Victoria University of Wellington, 2013.
- A. R. Johnson and E. E. Carlson, Anal. Chem., 2015, 87, 10668 CrossRef PubMed.
- K. F. Chong and H. W. Leong, J. Bioinf. Comput. Biol., 2012, 10, 1231002 CrossRef PubMed.
- S. W. Fuchs, T. W. Jaskolla, S. Bochmann, P. Kotter, T. Wichelhaus, M. Karas, T. Stein and K. D. Entian, Appl. Environ. Microbiol., 2011, 77, 1698 CrossRef PubMed.
- N. Zimmermann, J. W. Metzger and G. Jung, Eur. J. Biochem., 1994, 228, 786 CrossRef.
- W. Tang and W. A. van der Donk, Nat. Chem. Biol., 2013, 9, 157 CrossRef PubMed.
- N. Garg, W. Tang, Y. Goto, S. K. Nair and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5241 CrossRef PubMed.
- W. Liu, A. S. H. Chan, H. Q. Liu, S. A. Cochrane and J. C. Vederas, J. Am. Chem. Soc., 2011, 133, 14216 CrossRef PubMed.
- P. J. Knerr and W. A. van der Donk, J. Am. Chem. Soc., 2013, 135, 7094 CrossRef PubMed.
- P. J. Knerr and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 7648 CrossRef PubMed.
- A. C. Ross, S. M. McKinnie and J. C. Vederas, J. Am. Chem. Soc., 2012, 134, 2008 CrossRef PubMed.
- V. R. Pattabiraman, S. M. McKinnie and J. C. Vederas, Angew. Chem., Int. Ed. Engl., 2008, 47, 9472 CrossRef PubMed.
- M. F. M. Mustapa, R. Harris, N. Bulic-Subanovic, S. L. Elliott, S. Bregant, M. F. A. Groussier, J. Mould, D. Schultz, N. A. L. Chubb, P. R. J. Gaffney, P. C. Driscoll and A. B. Tabor, J. Org. Chem., 2003, 68, 8185 CrossRef PubMed.
- F. Garcia-Martin, M. Quintanar-Audelo, Y. Garcia-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Cote, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213 CrossRef PubMed.
- F. Garcia-Martin, P. White, R. Steinauer, S. Cote, J. Tulla-Puche and F. Albericio, Biopolymers, 2006, 84, 566 CrossRef PubMed.
- M. Mota-Meira, G. La Pointe, C. Lacroix and M. C. Lavoie, Antimicrob. Agents Chemother., 2000, 44, 24 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02170h |
| ‡ Contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2018 |