
Enantioselective reduction of N-alkyl ketimines with frustrated Lewis pair catalysis using chiral borenium ions†
Dan M.
Mercea
 a,
Michael G.
Howlett
a,
Adam D.
Piascik
a,
Daniel J.
Scott
a,
Alan
Steven
b,
Andrew E.
Ashley
a and
Matthew J.
Fuchter
*a
a,
Michael G.
Howlett
a,
Adam D.
Piascik
a,
Daniel J.
Scott
a,
Alan
Steven
b,
Andrew E.
Ashley
a and
Matthew J.
Fuchter
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London, W12 0BZ, UK. E-mail: m.fuchter@imperial.ac.uk
bAstraZeneca PT&D, Charter Way, Macclesfield, SK10 2NA, UK
First published on 22nd May 2019
Abstract
Enantioselective reduction of ketimines was demonstrated using chiral N-heterocyclic carbene (NHC)-stabilised borenium ions in frustrated Lewis pair catalysis. High levels of enantioselectivity were achieved for substrates featuring secondary N-alkyl substituents. Comparative reactivity and mechanistic studies identify key determinants required to achieve useful enantioselectivity and represent a step forward in the further development of enantioselective FLP methodologies.
The concept of ‘frustration’ continues to be useful in the development of transition metal free Lewis acid/base pairs with fascinating reactivity.1 Pioneered by Stephan and co-workers,2 ‘frustration’ is often seen as a result of steric demands and this view currently serves as the dominant design principle.3 The frustrated Lewis pair (FLP) formalism not only applies to the discovery of new reactions, but also to rationalise chemistry that pre-dates the coining of the term, a notable example being Piers-type hydrosilylation.4 FLPs have now been exploited in both stoichiometric and catalytic manifolds, in applications as diverse as polar hydrogenation (of imines,5a carbonyls,5b alkenes and alkynes5c), C–H bond activation,5d and polymer chemistry.5e Key to this work is the reduction of substrates using H2 and hydrosilanes, a topic that has been extensively reviewed by Oestreich et al.6
Despite many impressive developments in FLP chemistry and catalysis, enantioselective FLP-catalysed reduction is still significantly limited (Fig. 1). Chiral borane FLPs were pioneered by Klankermayer and co-workers,7a followed by ferrocene-derived boranes7b and ansa-ammonium borates7c from other groups. However, many of these catalysts showed limited enantioselectivity. The most successful scaffold for chiral borane FLP catalysts to date is the binaphthyl core. Binaphthyl-derived, C6F5-substituted boranes were first investigated by Piers and co-workers as catalysts for the asymmetric allylstannation of aromatic aldehydes7d followed by reports of chiral borepine catalysts (Oestreich, 1a)7e and acyclic designs (Repo and Du; 1b7f and 1c,7g respectively). Beside this scaffold, a novel bicyclic bisborane has also been reported recently by Peng, Wang et al., for N-aryl ketimine reduction.7h It is notable amongst these examples that, with the exception of FLP 1b,7f asymmetric ketimine reduction is limited to substrates with an N-aryl group. Herein we report a method to use a chiral borenium catalyst in the FLP reduction of N-alkyl ketimines, which gives good to high enantioselectivity for secondary alkyl ketimine substrates. We compare hydrosilylation and hydrogenation pathways, which give important insight into the factors that underpin good enantioselectivity. We believe these results will further contribute to the development of FLP catalysts for highly enantioselective reduction methodologies.
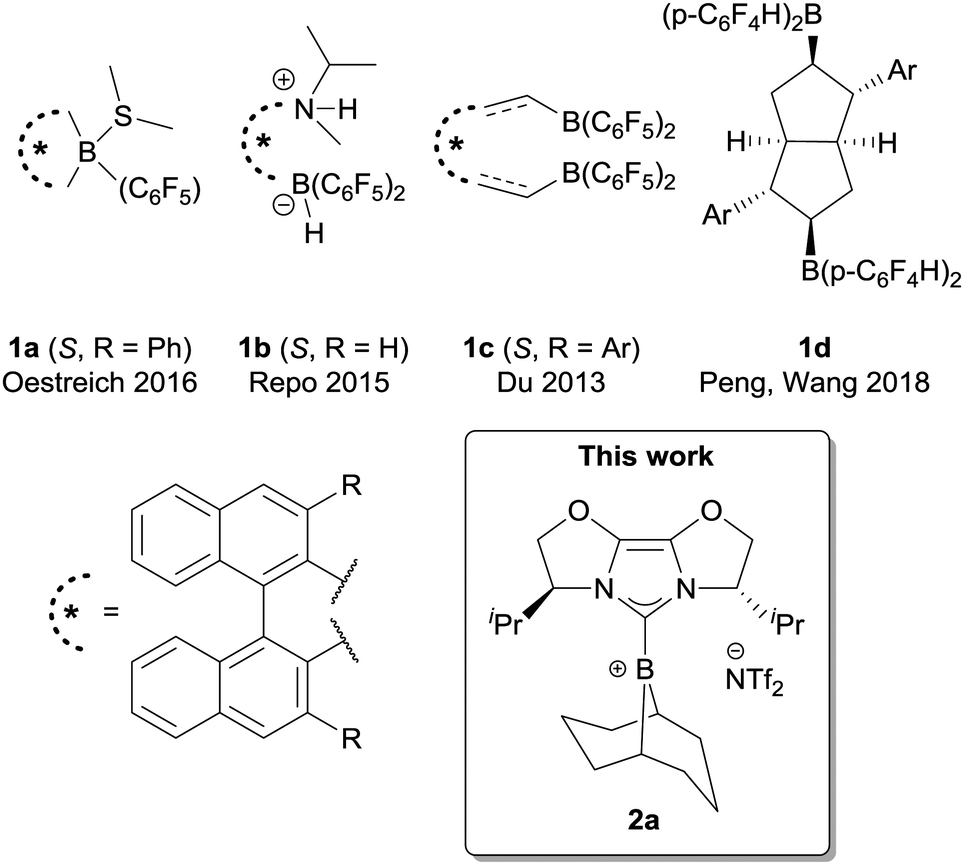 | ||
| Fig. 1 Examples of chiral FLP Lewis acid catalyst used in FLP reductions. | ||
One of the key issues with the prior designs for chiral borane FLP catalysts is the challenging synthesis required to assemble the Lewis acid. We considered chiral NHC-stabilised borenium ions to hold promise as alternative asymmetric FLP catalysts, given their effectiveness in FLP-catalysed reduction chemistry,8a and potential modularity: a large variety of chiral NHCs are available.9a We chose to initially survey chiral NHCs of the IBiox class9b for generating our borenium catalysts (2a–c, Scheme 1) given our previous work on such NHCs,9c and the fact that the corresponding borohydride 3a has been shown to act as a stoichiometric enantioselective reducing agent towards ketones.10a During the early phases of our project, Stephan, Crudden, Melen and co-workers reported the attempted asymmetric reduction of ketimines using borenium 2d as part of a large screening effort of chiral borenium FLP catalysts.8b Catalyst 2d hydrogenated N-phenyl ketimine 4a with poor selectivity (56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44 e.r.), and showed no activity towards N-benzyl ketimine 4b, even under 102 atm H2 pressure (298 K, CH2Cl2).8b Given this outcome, we sought to further understand how the reducing agent, catalyst components (NHC, borenium counterion, etc.) and ketimine substrate could be developed to enable good reactivity and enantioselectivity.
:44 e.r.), and showed no activity towards N-benzyl ketimine 4b, even under 102 atm H2 pressure (298 K, CH2Cl2).8b Given this outcome, we sought to further understand how the reducing agent, catalyst components (NHC, borenium counterion, etc.) and ketimine substrate could be developed to enable good reactivity and enantioselectivity.
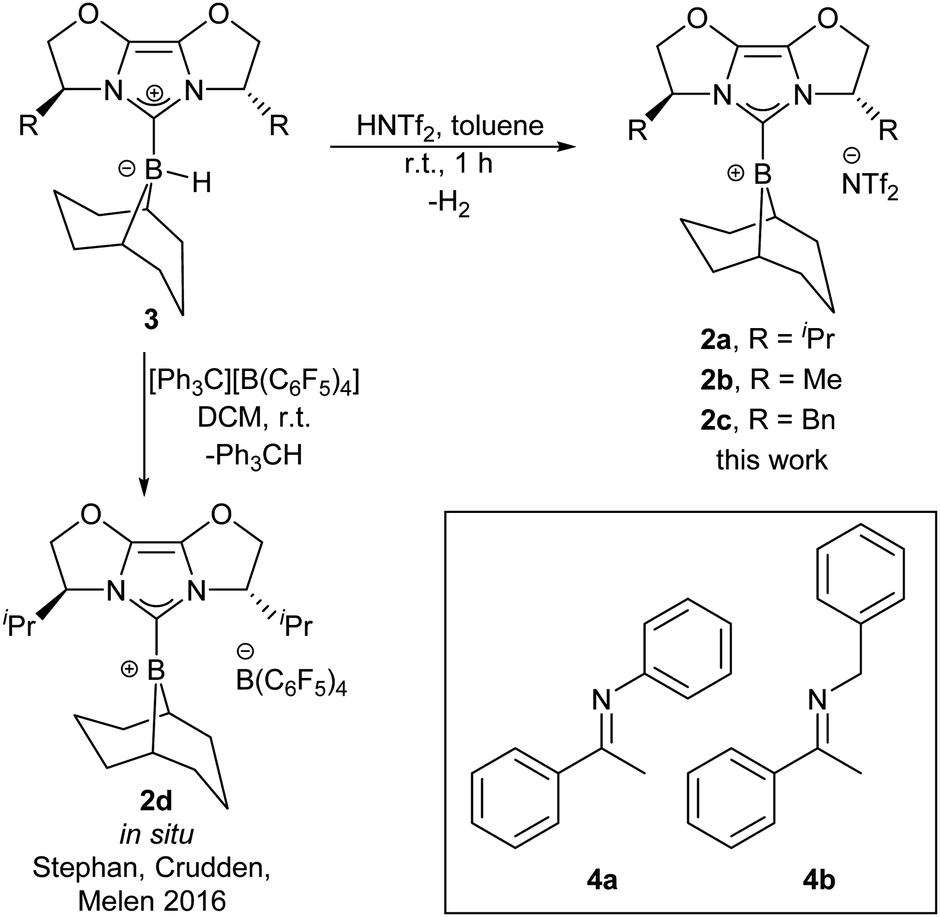 | ||
| Scheme 1 Catalyst synthesis and literature model substrates (Tf = SO2CF3). | ||
Borenium catalyst 2a was prepared from hydride 3a by treatment with HNTf2 in 73% yield on gram scale.10b‡ The borenium ion nature of 2a was confirmed by 11B NMR spectroscopy (δ = 73 ppm (br), CD2Cl2), DOSY NMR studies (Dc/Da = 1.06),10b,11 and X-ray crystallography on the isolated species (see ESI†). Compound 2a displayed good solubility in halogenated solvents: chloroform, dichloromethane, and 1,2-difluorobenzene (1,2-DFB). A Gutmann–Beckett measurement,12 which provides a measure of Lewis acidity in terms of an acceptor number (AN), revealed 2a to be a weaker Lewis acid (AN = 70.7) than the common FLP borane B(C6F5)3 (AN = 78.113). We additionally synthesised borenium catalysts 2b and 2c which vary in steric bulk of the chiral NHC (Scheme 1) and investigated counterions more coordinating than NTf2. However, a preliminary assessment of NHC–borenium ions with OTf, OMs, or OTs counterions revealed that such complexes are unable to cleave H2 in combination with ketimine substrates (Fig. S13, ESI†), due to coordination of the counterion of the borenium (see ESI†).
With appropriate chiral NHC–borenium catalysts 2 in hand, we surveyed the enantioselective hydrosilylation of N-aryl ketimine 4a and N-alkyl ketimines 4b and 4c (Table 1). Good to excellent reactivity was observed using PhMe2SiH and a catalyst loading of 4 mol%, which is comparable to other studies (Fig. 1). Substrate 4a was reduced with low enantioselectivity and a slight preference for the R product enantiomer (Table 1, entries 1 and 4). The use of a bulkier hydrosilane (entries 5 and 10) led to a reduced reaction rate with similar enantioselectivity. More promising results were obtained however when the substrate was changed to an N-alkyl derivative. Although N-benzyl substrate 4b exhibited excellent reactivity in toluene (entry 6), only low enantioselectivity was observed. Interestingly, the major enantiomer obtained (S) was of opposite configuration to that obtained for substrate 4a. While toluene versus 1,2-DFB gave comparable results for 4a, 1,2-DFB gave an increased enantioselectivity (e.r. 81:19, S:R, entry 7) in the reduction of 4b. Increasing the bulk of the N-alkyl substituent further improved enantioselectivity. Notably, N-cyclohexyl substrate 4c gave a high enantioselectivity (e.r. 90:10, S:R, entry 9) comparable with other leading chiral borane FLP catalysts (Fig. 1). Similar enantioselectivity was observed for catalysts 2a–c.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Subst. | Solvent | Conv. (time) | e.r. (R:S)a |
| Reactions were carried out on 0.375 mmol scale (0.63 M), conversion to amine product(s) assessed by NMR; unless otherwise stated the hydrosilane used was PhMe2SiH; products 5 were obtained following workup with MeOH and chromatography.a Determined by chiral HPLC, the N-acetylated derivative of 5c was used; enantiomers assigned by comparison of optical rotation values with the literature.b Reaction carried out with stirring on double the scale.c Catalyst formed in situ from the corresponding borohydride and HNTf2.d Hydrosilane used was Ph2MeSiH. | |||||
| 1b | 2a | 4a | Toluene | 98% (44 h) | 65:35 |
| 2 | 2b | 4a | Toluene | 71% (24 h) | 57:43 |
| 3 | 2c | 4a | Toluene | 100% (24 h) | 64.5:35.5 |
| 4 | 2a | 4a | 1,2-DFB | 100% (19 h) | 57:43 |
| 5d | 2a | 4a | 1,2-DFB | 89% (286 h) | 59:41 |
| 6 | 2a | 4b | Toluene | 100% (6 h) | 38.5:61.5 |
| 7 | 2a | 4b | 1,2-DFB | 100% (34 h) | 19:81 |
| 8 | 2a | 4c | Toluene | 53% (74 h) | 21:79 |
| 9 | 2a | 4c | 1,2-DFB | 100% (145 h) | 10:90 |
| 10d | 2a | 4c | 1,2-DFB | 68% (316 h) | 10:90 |
Given these promising results, substrate scope was further explored in the enantioselective hydrosilylation of N-alkyl ketimines using 2a (Table 2). Catalyst 2a showed comparably high enantioselectivity (e.r. 90:10–93:7) in the reduction of a range of bulky secondary N-alkyl ketimines (Table 2, entries 1–4). Although primary N-alkyl substituents gave slightly lower selectivity (Table 2, entries 5 and 6), we highlight that the enantioselectivity observed (e.r. 79:21–82:18, entries 5 and 6) is comparable to the only other example of FLP asymmetric reduction of N-alkyl ketimines.7f While a bulky substituent on the nitrogen atom improves enantioselectivity, excessive bulk prevents reaction (Table 2, entry 7). Extending the aliphatic chain that derives from the ketone component of the ketimine (substrate 4i) also leads to a reduction in enantioselectivity.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Subst. | Ar | R′ | R | Conv. (%) | Yield (%) | e.r. (S:R)a |
| Reactions were carried out on 0.375 mmol scale (0.63 M), conversion to amine product(s) assessed by NMR; unless otherwise stated solvent was 1,2-DFB, and the reaction time was 34 h; products 5 were obtained following workup with MeOH and chromatography.a Determined using chiral HPLC following acetylation; enantiomers assigned by comparison of optical rotation values with the literature.b 145 h.c Reaction solvent was DCM.d 31 h.e 2 h.f ∼11% (PhMe2Si)2O impurity.g Enantiomers not assigned. | |||||||
| 1b | 4c | Ph | H | c-Hex | 100 | 91f | 90:10 |
| 2 | 4d | Ph | H | i-Pr | 91 | 47 | 90:10 |
| 3 | 4e | Ph | H | c-Pent | 100 | 71 | 90:10 |
| 4c,d | 4f | 2-Np | H | c-Hex | 77 | 75 | 93:7 |
| 5c,e | 4b | Ph | H | Bn | 97 | ∼100 | 82:18 |
| 6 | 4g | Ph | H | Bu | 100 | 79 | 79:21 |
| 7 | 4h | Ph | H | CHPh2 | 0 | — | — |
| 8 | 4i | Ph | Me | c-Hex | 61 | 62 | 64:36g |

Given the competency of catalyst 2a for the asymmetric hydrosilylation of N-alkyl ketimines, we additionally assessed the potential to use H2 as the reductant. Increasing the catalyst loading to 10 mol% allowed for a range of substrates to be hydrogenated (Table 3). Similarly to hydrosilylation, the reduction of secondary N-alkyl ketimines derived from acetophenone (4c,e) gave good reactivity and enantioselectivity (e.r. 90:10, Table 3 entries 1 and 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Subs. | R′ | R | Conv. (%) | Yield (%) | e.r. (S:R)a |
| Reactions carried out on a 0.375 mmol scale (0.63 M) using SPR (screening pressure reactor) equipment, conversion to amine product assessed by NMR.a Determined using chiral HPLC following acetylation; enantiomers assigned by comparison of optical rotation values with the literature.b At 10 bar H2, NMR tube (re-pressurised after 192 h): 97%, 212 h, e.r. 90:10.c For the reaction carried out in DCM an e.r. value of 91:9 was obtained.d 4 mol% 2a, 90 bar H2, 24 h.e Enantiomers not assigned. |
||||||
| 1 | 4e | H | c-Pent | 100 | 67 | 90:10 |
| 2b,c | 4c | H | c-Hex | 99 | 66 | 89:11 |
| 3d | 4a | H | Ph | 100 | 91 | 41:59 |
| 4 | 4i | Me | c-Hex | 67 | 36 | 65:35e |
| 5 | 4h | H | CHPh2 | 0 | — | — |
| 6 | 4b | H | Bn | 8 | — | — |
| 7 | 4g | H | Bu | 11 | — | — |
Lowering the pressure to 10 bar H2 dramatically reduced the reaction rate. In agreement with the results of Stephan, Crudden, Melen and co-workers,8b hydrogenation of substrate 4a occurred with much lower enantioselectivity (e.r. 59:41, entry 3). Consistent with our results for hydrosilylation, substrate 4i only gave moderate enantioselectivity (e.r. 65:35, entry 4). Changing the steric bulk of the nitrogen substituent to be either larger (entry 5) or smaller (entries 6 and 7) resulted in a substantial drop in product yield.
It is notable that the enantioselectivity observed for substrates 4a and 4c is analogous, regardless of whether a hydrosilane or H2 was used. Oestreich and co-workers have previously shown that borane-promoted imine reduction using hydrosilanes can occur via hydride addition to silyl-iminium 6 and proto-iminium 7 intermediates; the latter also involves the formation of silyl-enamine intermediate 8 (Fig. 2).14 Monitoring the hydrosilylation of substrate 4c using 2a by NMR indicated a comparable mechanism to be operative here (see ESI,† Fig. S6, S7 and S11). Given the operation of both reduction pathways, and the comparison with hydrogenation – where polar reduction can only occur via a proto-iminium 7 intermediate – it would appear that the nature of the imine activating group (+SiR3 or +H) has little or no effect on enantioselectivity. Furthermore, it suggests that improvements in enantioselectivity are due to better substrate/catalyst matching and not due to a potential change in mechanism between hydrogenation and hydrosilylation. The successful enantioselective reduction using catalyst 2a appears to be mostly a result of the presence of a bulky alkyl substituent on the nitrogen atom of the ketimine.
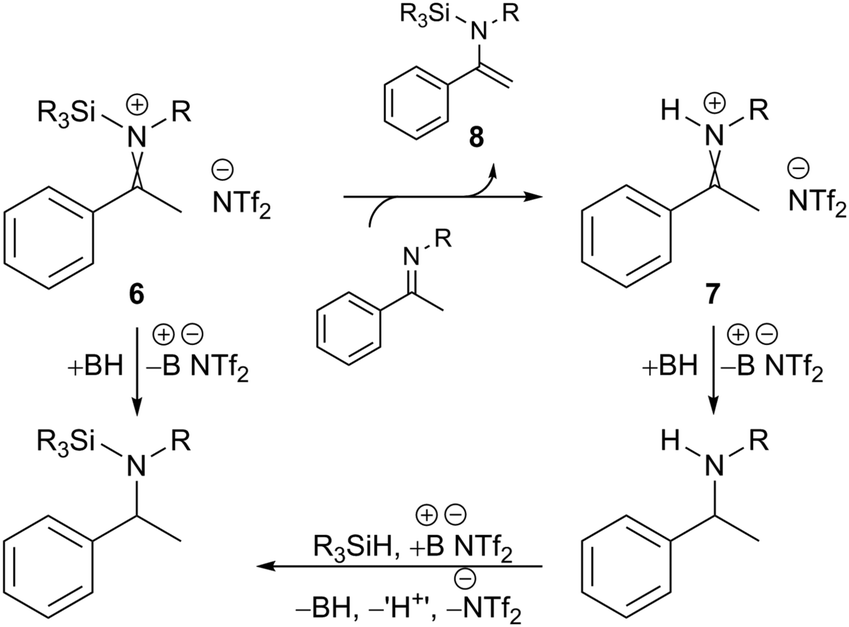 | ||
| Fig. 2 Lewis acid catalysed hydrosilylation using a borenium cation. For a full mechanism see ref. 14 and Fig. S8 (ESI†). | ||
In conclusion, we have explored the use of chiral IBiox NHC-stabilised borenium scaffolds for the enantioselective hydrosilylation and hydrogenation of ketimine substrates. In contrast with prior results of borenium-mediated FLP reductions,8b we have discovered that optimisation of the reaction conditions allowed synthetically useful levels of enantioselectivity to be achieved for a range of substrates. The best results were obtained for substrates featuring secondary N-alkyl substituents; substrates that are poorly explored in prior enantioselective FLP reductions. For such substrates, the level of enantioselectivity obtained is competitive with other chiral borane catalysts (Fig. 1) and comparable for both hydrosilylation and hydrogenation, with higher reactivity observed for the former. In future work, we believe that further optimization of chiral NHC–borenium catalysts may be possible to build upon the results described herein. For example, in preliminary work we have investigated the potential to use a borenium catalyst with the very weakly coordinating counterion {Al[O(CF3)3]4},15 given the need for a non-coordinating counterion for effective catalysis (vide supra). We observed slightly improved enantioselectivity (e.g. product 5c, e.r. = 90:10 NTf2 counterion, 95:5 {Al[O(CF3)3]4} counterion see ESI,† Table S12). We believe our results pave the way for future developments in enantioselective asymmetric reductions using FLP catalysts.
We wish to thank the EPSRC and AstraZeneca for PhD funding (CASE studentship to DMM). MJF would like to thank the EPSRC for an Established Career Fellowship (EP/R00188X/1). AEA would like to thank the Royal Society for a University Research Fellowship (UF/160395). We would also like to thank Kevin Leslie and Fiona Bell (AstraZeneca) for technical support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689–5700 RSC.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS; J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef PubMed; D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef PubMed.
- (a) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338–12348 CrossRef CAS PubMed; (b) D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS; T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 Search PubMed; T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 Search PubMed; (c) J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552–3557 CrossRef CAS PubMed; (d) M.-A. Legare, M.-A. Courtemanche, E. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef CAS PubMed; J. Légaré Lavergne, A. Jayaraman, L. C. Misal Castro, É. Rochette and F.-G. Fontaine, J. Am. Chem. Soc., 2017, 139, 14714–14723 Search PubMed; K. Chernichenko, M. Lindqvist, B. Kótai, M. Nieger, K. Sorochkina, I. Pápai and T. Repo, J. Am. Chem. Soc., 2016, 138, 4860–4868 Search PubMed; (e) Y. Zhang, G. M. Miyake and E. Y. X. Chen, Angew. Chem., Int. Ed., 2010, 49, 10158–10162 CrossRef CAS PubMed; T. Xu and E. Y.-X. Chen, J. Am. Chem. Soc., 2014, 136, 1774–1777 Search PubMed; Q. Wang, W. Zhao, S. Zhang, J. He, Y. Zhang and E. Y.-X. Chen, ACS Catal., 2018, 8, 3571–3578 Search PubMed.
- M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- (a) D. Chen and J. Klankermayer, Chem. Commun., 2008, 2130 RSC; D. Chen, Y. Wang and J. Klankermayer, Angew. Chem., Int. Ed., 2010, 49, 9475–9478 Search PubMed; D. Chen, V. Leich, F. Pan and J. Klankermayer, Chem. – Eur. J., 2012, 18, 5184–5187 Search PubMed; G. Ghattas, D. Chen, F. Pan and J. Klankermayer, Dalton Trans., 2012, 41, 9026 Search PubMed; (b) X. Wang, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2014, 136, 3293–3303 CrossRef CAS PubMed; K.-Y. Ye, X. Wang, C. G. Daniliuc, G. Kehr and G. Erker, Eur. J. Inorg. Chem., 2017, 368–371 Search PubMed; (c) V. Sumerin, K. Chernichenko, M. Nieger, M. Leskelä, B. Rieger and T. Repo, Adv. Synth. Catal., 2011, 353, 2093–2110 CrossRef CAS; (d) D. J. Morrison, W. E. Piers and M. Parvez, Synlett, 2004, 2429–2433 CAS; (e) M. Mewald, R. Fröhlich and M. Oestreich, Chem. – Eur. J., 2011, 17, 9406–9414 CrossRef CAS PubMed; M. Mewald and M. Oestreich, Chem. – Eur. J., 2012, 18, 14079–14084 Search PubMed; L. Süsse, J. Hermeke and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 6940–6943 Search PubMed; (f) M. Lindqvist, K. Borre, K. Axenov, B. Kótai, M. Nieger, M. Leskelä, I. Pápai and T. Repo, J. Am. Chem. Soc., 2015, 137, 4038–4041 CrossRef CAS PubMed; (g) Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed; S. Wei and H. Du, J. Am. Chem. Soc., 2014, 136, 12261–12264 Search PubMed; X. Zhu and H. Du, Org. Biomol. Chem., 2015, 13, 1013–1016 Search PubMed; X. Ren, G. Li, S. Wei and H. Du, Org. Lett., 2015, 17, 990–993 Search PubMed; X. Ren and H. Du, J. Am. Chem. Soc., 2016, 138, 810–813 Search PubMed; X. Liu, T. Liu, W. Meng and H. Du, Org. Biomol. Chem., 2018, 16, 8686–8689 Search PubMed; (h) X. Tu, N. Zeng, R. Li, Y. Zhao, D. Xie, Q. Peng and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 15096–15100 CrossRef CAS PubMed.
- (a) J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed; J. M. Farrell, R. T. Posaratnanathan and D. W. Stephan, Chem. Sci., 2015, 6, 2010–2015 Search PubMed; (b) J. Lam, B. A. R. Günther, J. M. Farrell, P. Eisenberger, B. P. Bestvater, P. D. Newman, R. L. Melen, C. M. Crudden and D. W. Stephan, Dalton Trans., 2016, 45, 15303–15316 RSC.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS; (b) F. Glorius, G. Altenhoff, R. Goddard and C. Lehmann, Chem. Commun., 2002, 2704–2705 RSC; G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed., 2003, 42, 3690–3693 Search PubMed; G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 Search PubMed; (c) J.-N. Levy, C. M. Latham, L. Roisin, N. Kandziora, P. Di Fruscia, A. J. P. White, S. Woodward and M. J. Fuchter, Org. Biomol. Chem., 2012, 10, 512–515 RSC.
- (a) D. M. Lindsay and D. McArthur, Chem. Commun., 2010, 46, 2474 RSC; (b) D. McArthur, C. P. Butts and D. M. Lindsay, Chem. Commun., 2011, 47, 6650 RSC.
- M. Valentini, H. Rüegger and P. S. Pregosin, Helv. Chim. Acta, 2001, 84, 2833–2853 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS; M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef.
- A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O’Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- J. Hermeke, M. Mewald and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 17537–17546 CrossRef CAS PubMed.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS; S. M. Ivanova, B. G. Nolan, Y. Kobayashi, S. M. Miller, O. P. Anderson and S. H. Strauss, Chem. – Eur. J., 2001, 7, 503–510 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details. Data files are openly available (DOI: 10.14469/hpc/5645). CCDC 1902956. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc02900a |
| ‡ Previously borenium 2d was generated in situ.8b Lindsay et al. treated IMes·9-BBN-H with HOTf and characterised the resulting product without isolation.10b |
| This journal is © The Royal Society of Chemistry 2019 |