
Hierarchically constructed NiO with improved performance for catalytic transfer hydrogenation of biomass-derived aldehydes†
Jian
He
ab,
Monia Runge
Nielsen
c,
Thomas Willum
Hansen
c,
Song
Yang
 *b and
Anders
Riisager
*a
*b and
Anders
Riisager
*a
aCentre for Catalysis and Sustainable Chemistry, Department of Chemistry, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark. E-mail: ar@kemi.dtu.dk
bState Key Laboratory Breeding Base of Green Pesticide & Agricultural Bioengineering, Key Laboratory of Green Pesticide & Agricultural Bioengineering, Ministry of Education, State-Local Joint Laboratory for Comprehensive Utilization of Biomass, Center for Research & Development of Fine Chemicals, Guizhou University, Guiyang 550025, PR China. E-mail: jhzx.msm@gmail.com
cNational Centre for Nano Fabrication and Characterization, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark
First published on 12th February 2019
Abstract
A 3D nano-/micrometer-scaled NiO material with urchin-like structure was prepared via a facile, green synthesis route, and served as a highly efficient and durable catalyst for catalytic transfer hydrogenation (CTH) of bio-based furfural (FF) to furfuryl alcohol (FAOL) using 2-propanol as H-donor and solvent. The as-prepared NiO possessed a good active-site accessibility owing to a high surface area and large amount of acid–base sites, resulting in high FF conversion of 97.3% with 94.2% FAOL yield at 120 °C and 3 h of reaction, which was a superior catalytic performance compared to commercial NiO nanoparticles. Besides, the excellent catalytic performance of the sea urchin-like NiO was validated for gram-scale FAOL synthesis, and recyclability test confirmed the catalyst to be reusable for multiple reaction runs without significant activity loss after intermediary calcination in air. Notably, the introduced catalytic system was also applicable to CTH of alternative bio-derived aldehydes.
1 Introduction
Considerable attention is nowadays directed towards the development of renewable feedstock from biomass resources that can serve as substitutes for limited fossil-based resources for the production of bio-fuels and fine chemicals.1–3 Furfural (FF), the acid-hydrolysis product of biomass-derived C5-based carbohydrates, is recognized as one of the most promising feedstock in the manufacture of a broad spectrum of fuels and valuable chemicals via upgrading strategies such as, e.g., hydrogenation, hydrogenolysis and aldolization.4–6 Furfuryl alcohol (FAOL), as a typical representative reduction derivative of FF, can serve as a versatile precursor in the synthesis of synthetic fibers, resins, adhesives and vitamin C,7–10 and act as an intermediate linking C5-carbohydrates to the down-stream products of bio-based furan derivatives (e.g., levulinic acid and γ-valerolactone) (Scheme 1).11,12 In this regard, hydrogenation of FF into FAOL has received widespread attention. | ||
| Scheme 1 Cascade synthesis of FAOL from bio-based carbohydrates and application of FAOL. | ||
The multi-functionality of the FF molecule (i.e., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and C–O) can result in several hydrogenation products such as FAOL, tetrahydrofurfuryl alcohol (TFAOL), 2-methylfuran (2-MF) and 2-methyltetrahydrofuran (2-MTHF).13–15 This renders the selectivity formation of FAOL from FF a remaining challenging as low product yield is often obtained.16,17 So far, many heterogeneous catalysts have been developed for the transformation of FF into FAOL using gaseous hydrogen as H-donor.18–21 However, low reaction selectivity towards the target product and the potential danger of handling pressurized, external H2 gas owing to its flammable and explosive properties coupled with the high cost of transportation and storage, encourage exploitation of alternative strategies for the production of FAOL from FF.22 Recently, catalytic transfer hydrogenation (CTH) has emerged as a very interesting alternative approach using abundant, renewable and low-cost alcohol as combined H-donor and solvent during the process. This results in more safe and convenient operation in comparison with the direct use of gaseous H2 as H-donor in terms of cost and handling.23–26 Moreover, it is proven that carbonyl groups can be highly selectivity reduced to hydroxyl groups during the CTH process, which is strongly associated to the Meerwein–Ponndorf–Verley (MPV) reaction in organic chemistry.27 Many heterogeneous catalysts have already been exploited for CTH of FF to FAOL with alcohol (Table S1†), but although substantial progression have been made most catalytic systems are generally subjected to undesired harsh reaction conditions (i.e., T ≥ 140 °C and/or t ≥ 6 h) and require large catalyst dosage to work efficiently. In addition, most of the reported catalysts involve tedious preparation procedures limiting their practical application. Thus, there is still great potential to improve the synthesis of FAOL from FF by CTH.
C, CO and C–O) can result in several hydrogenation products such as FAOL, tetrahydrofurfuryl alcohol (TFAOL), 2-methylfuran (2-MF) and 2-methyltetrahydrofuran (2-MTHF).13–15 This renders the selectivity formation of FAOL from FF a remaining challenging as low product yield is often obtained.16,17 So far, many heterogeneous catalysts have been developed for the transformation of FF into FAOL using gaseous hydrogen as H-donor.18–21 However, low reaction selectivity towards the target product and the potential danger of handling pressurized, external H2 gas owing to its flammable and explosive properties coupled with the high cost of transportation and storage, encourage exploitation of alternative strategies for the production of FAOL from FF.22 Recently, catalytic transfer hydrogenation (CTH) has emerged as a very interesting alternative approach using abundant, renewable and low-cost alcohol as combined H-donor and solvent during the process. This results in more safe and convenient operation in comparison with the direct use of gaseous H2 as H-donor in terms of cost and handling.23–26 Moreover, it is proven that carbonyl groups can be highly selectivity reduced to hydroxyl groups during the CTH process, which is strongly associated to the Meerwein–Ponndorf–Verley (MPV) reaction in organic chemistry.27 Many heterogeneous catalysts have already been exploited for CTH of FF to FAOL with alcohol (Table S1†), but although substantial progression have been made most catalytic systems are generally subjected to undesired harsh reaction conditions (i.e., T ≥ 140 °C and/or t ≥ 6 h) and require large catalyst dosage to work efficiently. In addition, most of the reported catalysts involve tedious preparation procedures limiting their practical application. Thus, there is still great potential to improve the synthesis of FAOL from FF by CTH.
The structural features of solids, including morphology, surface area and component dispersion, have a significant influence on their catalytic performance.28,29 In this perspective, 3D micro or nano-materials with hierarchical construction have shown outstanding activity in catalysis thanks to their large surface area, highly available active sites and improved mass transportation.30 For instance, has flower-like Co3O4–CeO2 composite demonstrated to be an efficient catalyst for the degradation of 1,2,4-trichlorobenzene,31 flower-like MgO catalyst was efficient for dimethyl carbonate synthesis,32 and 3D flower-like micro/nano Ce–Mo composite oxide has been developed as an efficient catalyst for one-pot transformation of fructose to 2,5-diformylfuran.33
In previous work,34 we reported that commercially available NiFe2O4 nanoparticles could efficiently catalyze the CTH of FF to FAOL with 2-propanol. Following our continuous interesting in the development of highly efficient and low cost catalysts for the CTH of FF, we prepared in this study 3D sea urchin-like and flower-like NiO catalysts by facile and environmental-friendly methods without the use of strong base or pungent ammonia hydroxide. The 3D sea urchin-like NiO was thoroughly characterized by various techniques and demonstrated to be highly catalytically active for CTH of FF to FAOL with 2-propanol as H-donor, yielding 94.2% FAOL under mild reaction conditions (120 °C, 3 h).
2 Experimental
2.1 Materials
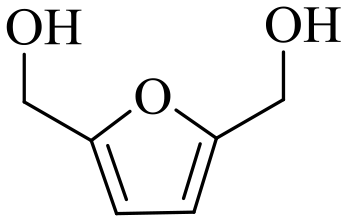
Nickel(II) chloride (98%), PEG–PPG–PEG pluronic P-123 (Maverage = 5800 g mol−1), NiO (99.8%, <50 nm particle size), furfuryl alcohol (FAOL, 98%), 5-methylfurfural (5-MFF, 99%), naphthalene (>99%, internal standard), benzaldehyde (>99%), benzyl alcohol (≥99%), 4-methoxybenzaldehyde (98%), 4-methoxybenzyl alcohol (98%), 3,4-dimethoxybenzaldehyde (99%), 3,4-dimethoxybenzyl alcohol (96%), 2,4,6-trimethylbenzaldehyde (98%), 2,4,6,-trimethylbenzyl alcohol (99%), cinnamic alcohol (98%), heptanal (≥95%), 2-propanol (99.5%), methanol (≥99.8%), ethanol (99.5%) and acetone (≥99%) were purchased from Sigma-Aldrich. Cinnamaldehyde (>98%) was received from Merck. 5-Hydroxymethylfurfural (HMF, ≥95%), 2,5-furandicarboxaldehyde (DFF, ≥95%), 5-methylfuran-2-methanol (≥95%) and 2,5-furandimethanol (≥95%) were obtained from Bepharm Ltd. Urea (≥99%), furfural (FF, ≥99%), 2-butanol (≥99.5%), tert-butyl alcohol (≥99.7%) and n-heptanol (>99%) were procured from Fluka.2.2 Catalyst preparation
In a typical synthesis, 1.296 g (10 mmol) NiCl2, 1.80 g (30 mmol) urea and 2.5 g pluronic P-123 were dissolved in deionized water (80 mL) with magnetic stirring (500 rpm) at room temperature for 2 h. The resulting green transparent solution was then transferred into a 200 mL Teflon lined stainless steel autoclave and heated at 90 °C for 12 h. Subsequently, the autoclave was quickly cooled to room temperature in a water bath, the green product collected by filtration followed by washing with deionized water until the pH of the filtrate was neutral following by drying at 80 °C in air for 5 h (86% yield of solid). Finally, the as-prepared sample was calcined at 300 or 400 °C in air for 4 h and the obtained catalyst were denoted as NiO(P)-300 or NiO(P)-400, respectively. For comparison, NiO-300 was prepared according to the above described procedure but in the absence of pluronic P-123.2.3 Catalyst characterization
Powder X-ray diffraction (XRD) patterns of samples were acquired on a Huber G670 diffractometer with Cu Kα radiation (λ = 0.154184 nm) and recorded in the 2θ range from 5 to 80°. N2 adsorption–desorption measurements were completed on a Micromeritics ASAP 2020 instrument, wherein the samples were firstly degassed at 200 °C in vacuum for 4 h and adsorption–desorption isotherms subsequently acquired at −196 °C. The Brunauer–Emmett–Teller (BET) method was applied to evaluate the specific surface area of the samples. Thermal gravimetric (TG) analysis of samples were performed on a METTLER TOLEDO thermal analyzer in dynamic air atmosphere (30 mL min−1) in the temperature range 25–600 °C with a heating rate of 10 °C min−1. NH3/CO2-Temperature programmed desorption (TPD) measurements of samples were conducted on a Micromeritics AutoChem II 2920 apparatus equipped with a thermal conductivity detector (TCD). Prior to TPD analysis, the samples were heated at 300 °C under a He flow (25 mL min−1) for 1 h, subjected to a mixture of NH3/CO2–He gas flow (15 mL min−1) for 1 h at 50 °C followed by flushing with pure He gas (25 mL min−1) to remove physically adsorbed NH3/CO2. Finally, the desorption of NH3/CO2 were performed by heating the samples to 300 °C at a rate of 10 °C min−1 and maintaining it here for 1 h. SEM (scanning electron microscopy) images of samples were recorded on a FEI Nova NANO SEM 600 with the thru-the-lens camera with an accelerating voltage at 5 kV. TEM (transmission electron microscopy) images were recorded on a FEI Tecnai T20 G2 microscope at 200 kV with a LaB6/CeB6 Thermionic filament. X-ray photoelectron spectroscopy (XPS) spectra of samples were recorded on a ThermoScientific K-Alpha spectrometer with Al Kα radiation (1484.6 eV), in which C 1s at 284.8 eV was applied as calibration peak. Metal leaching from the NiO(P)-300 catalyst in the filtrate after reaction was determined by inductively coupled plasma optical emission mass spectrometry (ICP-MS) on a Thermo Scientific iCAP Q ICP-MS with the aid of acid digests.2.4 Catalytic activity test and product analysis
The CTH of FF was performed in 15 mL ACE pressure tube equipped with magnetically driven stirrer and oil-bath. In brief, 0.096 g (1 mmol) FF, specified amount of catalyst (0.005–0.03 g), 5 mL 2-propanol (as H-donor and solvent), and 0.02 g naphthalene (as internal standard) were mixed in the reactor. After sealing, the reactor was placed in a preheated oil bath (100–140 °C) for a specific reaction time at a stirring rate of 500 rpm (time zero when the reactor was immersed in the oil bath). After the desired time of reaction, the reactor was quickly cooled to room temperature in a water bath. Aliquot of the reaction mixtures was quantitatively analyzed by GC (Agilent 6890N) fitted with FID detector, wherein the FF conversion and FAOL yield were calculated on basis of standard curves of commercially pure compounds. Furthermore, the liquid products in the reaction mixture were identified by GC-MS (Agilent 6850-5975C). Both the GC and the GC-MS was equipped with a HP-5MS capillary column (30.0 m × 250 μm × 0.25 μm).The recyclability of the NiO(P)-300 catalyst was tested over six consecutive reaction runs at 120 °C with a reaction time of 3 h. For the first four runs, the used catalyst was after each run separated by centrifugation, washed with ethanol and acetone (5 mL) in sequence, subsequently dried at 80 °C for 2 h and then used for the next run. For the fifth and sixth recycle runs, catalyst regeneration were achieved by calcination in air at 300 °C for 4 h. Product analysis was performed as described above.
The CTH of other aldehydes was performed in a similar way as described above, but with a 50 mL stainless steel autoclave with mechanical stirring (500 rpm) using 2 mmol substrate, 0.06 g catalyst, 10 mL 2-propanol and reaction temperature of 140 or 150 °C.
3 Results and discussion
3.1 Catalyst characterization
XRD patterns obtained of the as-prepared catalysts and commercial NiO nanoparticles are shown in Fig. 1a. NiO(P)-400 and commercial NiO had five characteristic peaks at 2θ = 37.3, 43.2, 62.7, 75.4 and 79.4°, which are indexed as (111), (200), (220), (311) and (222) planes of the crystalline rhombohedral phase of NiO,35,36 respectively, consistent with JCPDS card 44-1159. In contrast, only three of the characteristic peaks were clearly observed for the samples of NiO(P)-300 and NiO-300, indicating less crystallinity of NiO when calcined at lower temperature.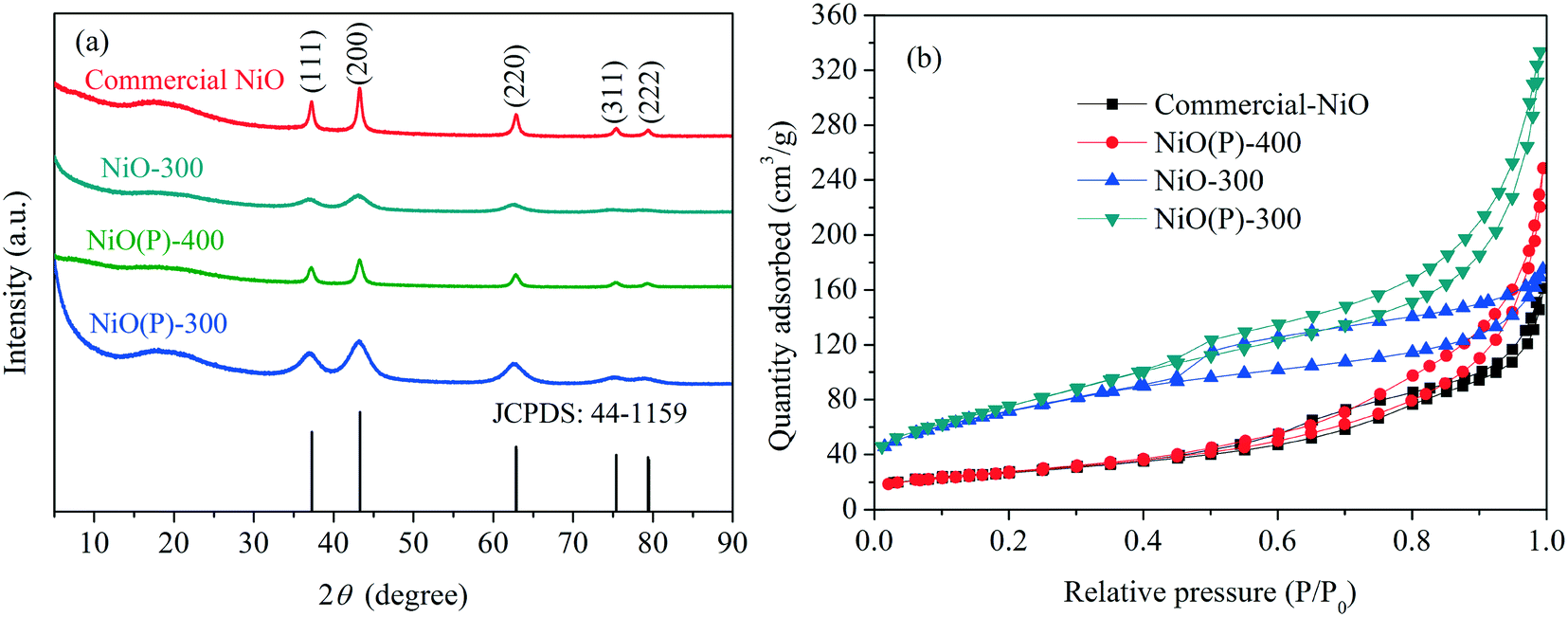 | ||
| Fig. 1 (a) XRD patterns and (b) N2 adsorption–desorption isotherms of the as-prepared catalysts and NiO nanoparticles. | ||
Nitrogen physisorption isotherms of the catalyst samples measured by the BET method (Fig. 1b) showed type IV isotherms with H3-type hysteresis loop, implying that irregular mesoporous structures formed between the particles. The obtained BET surface areas of the samples are compiled in Table 1. As expected, higher calcination temperature greatly diminished the surface area of the as-prepared NiO (entries 2 and 3, Table 1). Besides, the surface area of NiO(P)-300 was larger than that of NiO-300 (entries 2 and 4, Table 1), revealing that the addition of pluronic P-123 during the course of preparation increased the surface area of the formed material.
| Entry | Catalyst | BET surface areaa (m2 g−1) | Acid amountb (mmol g−1) | Base amountb (mmol g−1) |
|---|---|---|---|---|
| a BET surface areas were determined by N2 physisorption. b Acidity and basicity were evaluated by NH3- and CO2-TPD, respectively. | ||||
| 1 | Commercial NiO | 94.8 | 0.192 | 0.059 |
| 2 | NiO(P)-300 | 273.8 | 0.918 | 0.072 |
| 3 | NiO(P)-400 | 95.5 | 0.303 | 0.065 |
| 4 | NiO-300 | 257.8 | 0.923 | 0.080 |
The morphologies of the as-prepared catalysts were visualized by SEM and TEM analysis (Fig. 2). The SEM image of NiO(P)-300 (Fig. 2a) revealed a well-defined 3D nanometer-scale structure, which was assembled from a core with many densely arranged nano-needles as “spikes”. The 3D nanometer-scale structure of NiO(P)-300 was clearly confirmed by its TEM image (Fig. 2b) consisting of a core of about 120 nm in diameter with “spikes” having diameters of about 50 nm. Thus, the construction of the NiO(P)-300 catalyst looked like “sea urchin”. NiO(P)-400 possessed less pronounced sea urchin appearance, which seemed to be destroyed at increased calcination temperature. Instead it presented an agglomerated shape (Fig. 2c and d), which could account for the lower surface area of NiO(P)-400 (entry 3, Table 1) compared to NiO(P)-300. Fig. 2e and f showed that the construction units of NiO-300 were 3D hierarchical flower-like structures with diameters in the range of 1.2–1.4 μm, which were composed of many densely packed nanosheets like “petals”. The distinct differences in catalyst construction between NiO(P)-300 and NiO-300 reflected that pluronic P-123 significantly influenced the catalyst structure during the course of preparation by decreasing the size of particles as well as enlarging the surface area.
 | ||
| Fig. 2 (a) SEM image of NiO(P)-300; (b) TEM image of NiO(P)-300; (c) SEM image of NiO(P)-400; (d) TEM image of NiO(P)-400; (e) SEM image of NiO-300; (f) TEM image of NiO-300. | ||
The acid and base properties of the catalysts were further measured using NH3- and CO2-TPD (Fig. S1 and S2†). As shown in Table 1, the acidity and basicity of the NiO catalysts were strongly associated to the calcination temperature as evident from the NiO(P)-300 and NiO-300 samples, which possessed almost the same amount of acid and base sites (entries 2 and 4, Table 1), while NiO(P)-400 in comparison had less acid and base sites (entry 3, Table 1).
Furthermore, the Ni species in NiO(P-300) and commercial NiO nanoparticles were examined by XPS analysis. The XPS spectra of the Ni 2p3/2 region (Fig. 3) exhibited a main peak at about 854 eV and an associated satellite peak at about 861 eV for both NiO(P-300) and commercial NiO nanoparticles.35,37 Overall, four deconvoluted peaks were needed to reproduce the shape of the Ni 2p3/2 signal, where the peaks at about 854.0 and 855.5 eV in the main peak region were characteristic of Ni2+ species and the other two deconvoluted peaks located at six to eight eV higher binding energy relative to the main peaks (i.e., 854.0 and 855.5 eV) are generally assigned to satellite peaks.37,38 No peak at 852.6 eV assignable to metallic Ni38 was observed in either of the samples.
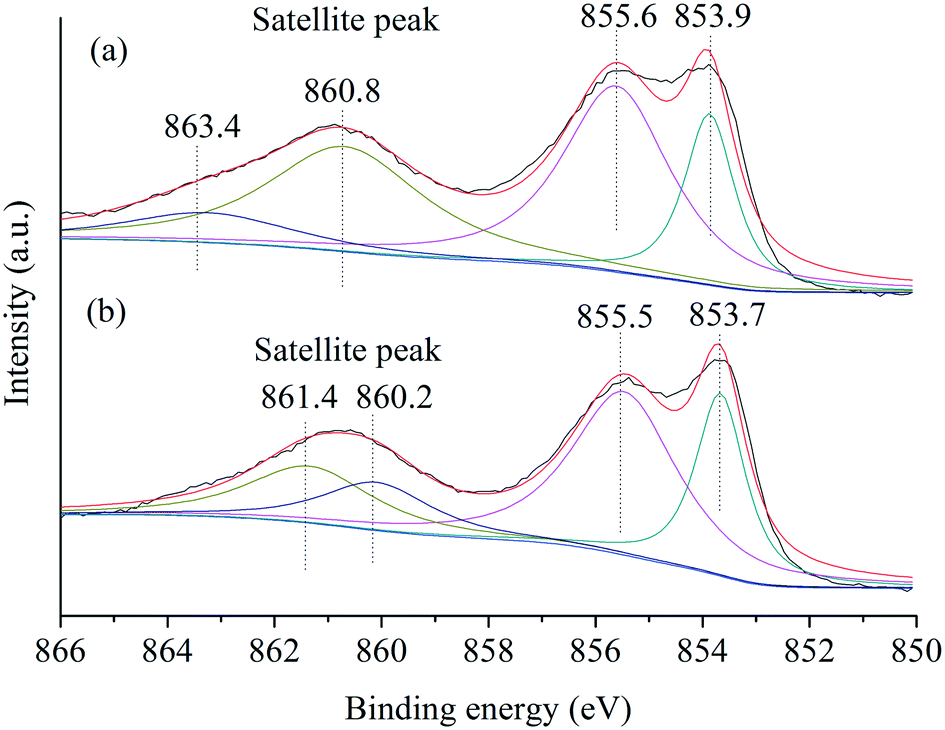 | ||
| Fig. 3 XPS spectra of Ni 2p3/2 of (a) NiO(P)-300 and (b) commercial NiO samples. | ||
3.2 Catalyst evaluation for CTH of FF
Initial investigations were commenced by using the as-prepared NiO catalysts for CTH of FF to FAOL in 2-propanol at 120 °C (Table 2). A reference experiment confirmed that the formation of FAOL did not take place without catalyst present (entry 1, Table 2). In contrast, considerable amount of FAOL formed using the different NiO catalysts (entries 2–5, Table 2), confirming that the acid–base properties of the metal oxides were capable of facilitating the CTH reaction relying on MPV reduction.39–41 Especially, 72.6% FF conversion with 70.2% FAOL yield (96.7% selectivity) was obtained in the presence of NiO(P)-300 under mild reaction conditions (120 °C, 1 h) resulting in a high FAOL formation rate of 585 μmolg−1 min−1 and a relatively high TOF value of 2.6 h−1 (entry 2, Table 2). The excellent catalytic performance of NiO(P)-300 was attributed to a combination of high surface area (273.8 m2 g−1) and high concentration of acid (0.918 mmol g−1) and base sites (0.072 mmol g−1) (entry 2, Table 1). Notably, the catalytic behavior of NiO(P)-300 far outperformed commercial NiO nanoparticles (FAOL formation rate of 142 μmolg−1 min−1 and TOF of 0.6 h−1) (entry 5, Table 2). Similarly, good results were also obtained with NiO-300 which possessed a relative high surface area and equally many acid and base sites as NiO(P)-300, affording 56.9% yield of FAOL (90.7% selectivity) along with a FAOL formation rate of 474 μmolg−1 min−1 and a TOF value of 2.1 h−1 (entry 4, Table 2). Hence, both as-prepared NiO with sea urchin (i.e., NiO(P)-300) or flower (i.e., NiO-300) construction exhibited superior performance to commercial NiO nanoparticles with a mean particle size of about 15 nm (Fig. S3†) in the CTH of FF to FAOL, possibly as a consequence of their special constructions in combination with large density of acid–base sites. Noteworthy, as-prepared NiO with flower construction (i.e., NiO-300) afforded a slightly lower selectivity toward FAOL compared to the other tested NiO catalysts (entries 2–5, Table 2). This was likely associated to the large particle size of NiO-300 as evidenced from Fig. S4,† in well agreement with nano-catalysis offering excellent selectivity.22,42 As the as-prepared nanometer-scaled NiO with sea urchin construction (i.e., NiO(P)-300) showed the best catalytic performance, this catalyst was selected for further study.| Entry | Catalyst | Conv. (%) | Yield (%) | Select. (%) | FAOL formation rateb (μmolg−1 min−1) | TOFc (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 1 mmol FF, 0.02 g catalyst, 5 mL 2-propanol, 120 °C, 1 h. b Calculated from the FAOL yield obtained after 1 h. c Turn-over frequency (TOF) as (mole of FAOL)/(mole of catalyst × reaction time). | ||||||
| 1 | None | 4.5 | 0.0 | 0.0 | — | — |
| 2 | NiO(P)-300 | 72.6 | 70.2 | 96.7 | 585 | 2.6 |
| 3 | NiO(P)-400 | 11.2 | 10.4 | 92.9 | 87 | 0.4 |
| 4 | NiO-300 | 62.7 | 56.9 | 90.7 | 474 | 2.1 |
| 5 | Commercial NiO | 17.7 | 17.0 | 96.0 | 142 | 0.6 |
3.3 CTH of FF with NiO(P)-300
 | ||
| Fig. 4 (a) Effect of reaction temperature and reaction time on CTH of FF to FAOL (1 mmol FF, 0.02 g NiO(P)-300, 5 mL 2-propanol); (b) effect of alcohol as H-donor on CTH of FF to FAOL (1 mmol FF, 0.02 g NiO(P)-300, 5 mL alcohol, 120 °C, 3 h, formation rate calculated from the FAOL yield obtained after 3 h); (c) effect of catalyst dosage on the CTH of FF to FAOL (1 mmol FF, 5 mL 2-propanol, 120 °C, 3 h, mass ratio of FF to catalyst ranging from 19.2 to 3.2). | ||
Variation of the alcohol acting as solvent and H-donor in the tested system had a significant effect on the catalytic behavior (Fig. 4b). No FAOL was formed using tert-butanol as the reaction media, confirming that it could not facilitate CTH. On the other hand, excellent FAOL selectivities were achieved with 2-propanol and 2-butanol, confirming the good H-donating capability of secondary alcohols. Notably, a moderate FF conversion was observed at 120 °C in 2-butanol while ∼90% FF conversion was achieved at 160 °C (Table S3†). This difference was probably a consequence of the relatively high viscosity and/or steric hindrance effect of 2-butanol compared to the other alcohols (e.g., 2-propanol), resulting in relatively large mass dispersion limitation at low reaction temperature (120 °C). At higher reaction temperature (e.g., 160 °C) where the viscosity of 2-butanol is significantly lower43,44 the effect was less important. Ethanol performed also good as solvent/H-donor (75.4% yield of FAOL, 209 μmolg−1 min−1 of FAOL formation rate), however superior result (94.2% yield of FAOL, 262 μmolg−1 min−1) was observed with 2-propanol as preferred H-donor solvent.
The effect of NiO(P)-300 dosage on the CTH of FF at 120 °C was subsequently studied and the results are shown in Fig. 4c. No FAOL product was formed without added catalyst into the reaction system, as expected. In contrast, FF conversion and FAOL yield increased with increasing catalyst dosage and reached 97.3 and 94.2%, respectively, when the mass ratio of FF to catalyst was 4.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (i.e., use of 0.02 g catalyst). Further increase in catalyst dosage to 0.03 g (i.e., mass ratio of FF to catalyst of 3.2:1) only marginally increased the yield to 95.5% after 3 h, but resulted in high FAOL yield (e.g., 85.7%) within a shorter time (e.g., 1 h). Also, the FAOL selectivity increased with increasing catalyst dosage, and the relative low FAOL selectivity at lower catalyst dosage was attributed to competing acetalization of FF with 2-propanol as revealed from GC-MS analysis.
:1 (i.e., use of 0.02 g catalyst). Further increase in catalyst dosage to 0.03 g (i.e., mass ratio of FF to catalyst of 3.2:1) only marginally increased the yield to 95.5% after 3 h, but resulted in high FAOL yield (e.g., 85.7%) within a shorter time (e.g., 1 h). Also, the FAOL selectivity increased with increasing catalyst dosage, and the relative low FAOL selectivity at lower catalyst dosage was attributed to competing acetalization of FF with 2-propanol as revealed from GC-MS analysis.
Upon recycling, a gradual decline in FF conversion and FAOL yield was, however, observed for the first four reaction runs (Fig. 5a) resulting in moderate FF conversion and FAOL yield of 41.5 and 36.9%, respectively, while the FAOL selectivity maintained around 95%. Notably, the catalyst activity could be recovered almost completely after regeneration by calcination at 300 °C in air, which resulted in FAOL yield of 88.3 and 86.8% after the fifth and sixth reaction run, respectively, when applying intermediate calcination. Thus in-depth characterization of used and generated catalysts were conducted with an attempt to shed light on the reason of activity variation during the recycling experiments.
 | ||
| Fig. 5 (a) Catalytic performance of NiO(P)-300 in the CTH of FF to FAOL during recycling. (b) XRD patterns of fresh, used and regenerated NiO(P)-300 catalysts. (c) SEM image of used NiO(P)-300 catalyst. (d) TEM image of used NiO(P)-300 catalyst. (e) TG curves of fresh, used and regenerated NiO(P)-300 catalysts. (f) N2 adsorption–desorption isotherms of fresh, used and regenerated NiO(P)-300 catalysts. Reaction conditions: 1 mmol FF, 0.02 g NiO(P)-300, 5 mL 2-propanol, 120 °C, 3 h. | ||
XRD (Fig. 5b) and XPS analysis (Fig. S8†) of the spent and regenerated catalysts revealed no crystallographic changes compared to the fresh catalyst and no reduced Ni species (no characteristic metallic Ni peak at 852.6 eV), respectively, strongly confirming that reduction of Ni2+ did not take place during the reaction. Besides, SEM and TEM analysis (Fig. 5c and d) confirmed preservation of the sea urchin-like constructions in the used samples, while ICP-MS analysis confirmed negligible nickel leaching (<0.5 ppm) into the filtrate after 3 h at 120 °C. Combined, this firmly demonstrated that the NiO(P)-300 catalyst was stable during the reaction. However, a significant weight loss was indeed found by TG analysis for the used catalyst in comparison to the fresh and regenerated catalysts (Fig. 5e), and an apparent color change was also observed (Fig. S9†). Retrospectively, it is speculated that organic residues adsorbed on the surface of the NiO(P)-300 catalyst resulted in the loss of activity and calcination at 300 °C restored the catalyst activity. This deduction was validated by the variation in the surface areas of the fresh (273.8 m2 g−1), used (228.5 m2 g−1) and regenerated catalyst (246.4 m2 g−1) (Fig. 5f).
In addition to FF, NiO(P)-300 was successfully employed for CTH of various other aldehydes, including α,β-unsaturated carbonyls, as shown from the results listed in Table 3. For instance, were excellent yields (92.8–97.4%) of the corresponding reduction products obtained in CTH of 2,5-furandicarboxaldehyde (entry 3), benzaldehyde (entry 4), 4-methoxybenzaldehyde (entry 5), 3,4-dimethoxybenzaldehyde (entry 6) and heptanal (entry 9) under relative mild reaction conditions (≤150 °C). Although lower conversions were observed for other aldehydes (entries 1, 2, 7 and 8), excellent selectivities of the corresponding reduction products were attained confirming that the NiO(P)-300 catalyst had good versatility in the CTH of aldehydes.
| Entry | Substrate | Product | Temp. (°C) | Time (h) | Conv. (%) | Yield (%) | Sel. (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 2 mmol substrate, 0.06 g NiO(P)-300, 10 mL 2-propanol. b 1 mmol substrate. c HMF yield. | |||||||
| 1b |
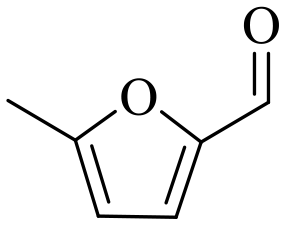
|
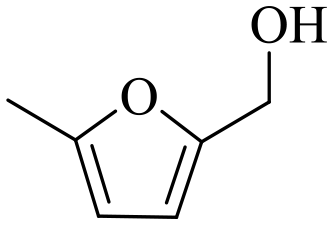
|
150 | 5 | 60.5 | 58.1 | 96.0 |
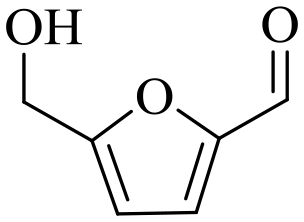
| 2b |
|
|
150 | 4 | 77.9 | 73.5 | 94.4 |
| 3b |
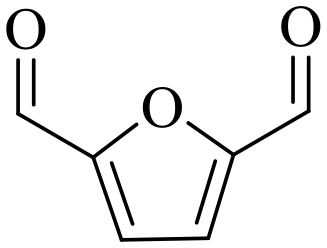
|
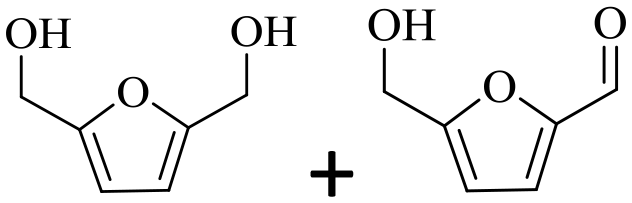
|
150 | 4 | >99 | 79.6 (16.7)c | 96.3 |
| 4 |

|
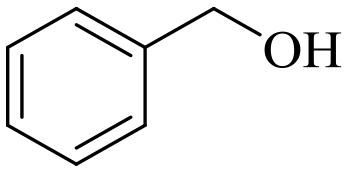
|
140 | 3 | 96.5 | 95.7 | 99.2 |
| 5 |
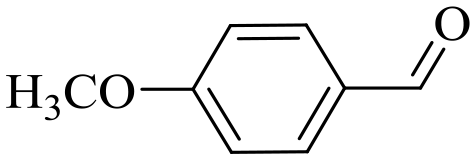
|
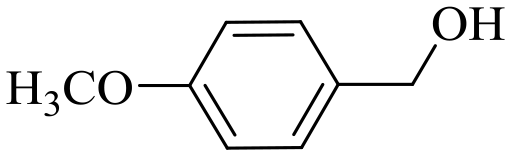
|
150 | 2 | >99 | 97.1 | 98.1 |
| 6 |
|
|
150 | 2 | >99 | 97.4 | 98.4 |
| 7 |
|
|
150 | 8 | 46.1 | 45.2 | 98.0 |
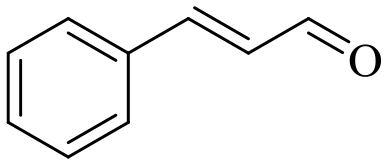
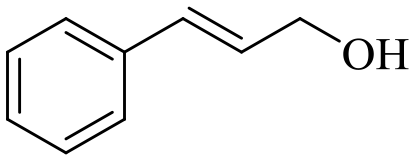
| 8 |
|
|
140 | 8 | 71.6 | 70.5 | 98.5 |
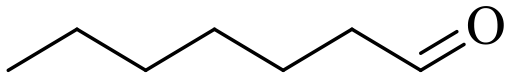
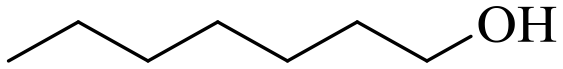
| 9 |
|
|
150 | 3 | 96.2 | 92.8 | 96.5 |
Inspired by the general excellent catalytic performance of NiO(P)-300 in CTH, we further attempted to use NiO(P)-300 combined with other acid catalysts for one-pot transformation of FF to alkyl levulinate, which is hailed as another versatile feedstock for the manufacture of bio-fuels and chemicals.11 Since isopropyl levulinate (IPL) was not commercially available, pure IPL was prepared according to the method reported by Geboers et al.45 and the purity confirmed by NMR analysis (Fig. S11†). As shown in Table S4,† nearly full FF conversion was achieved over NiO(P)-300 at 140 °C after 2 h of reaction, and the conversion of formed FAOL into IPL was subsequently catalyzed by common acid catalysts such as, e.g. Amberlyst 15, H-Beta, H-ZSM-5, H-MOR and H-Y. Eventually, a IPL yield of 61.5% was obtained from FF over NiO(P)-300 and Amberlyst 15 at 140 °C after 6 h in a two-step procedure, which is close to previously reported results.46 The superior activity found for Amberlyst 15 compared to zeolites was mainly attributed to its larger amount of acidic sites and stronger acidity (Table S4†). In line with this, the dominant product from reaction with H-Beta (12.5) was the precursor of IPL, i.e. 2-(isopropoxymethyl)furan, derived from etherification of FAOL with 2-propanol. In contrast, IPL was the main product in the presence of Amberlyst 15 as its large amount of acidic sites and strong acidity further facilitated the transformation into IPL (Fig. S12†). In addition to the acid amount and acid strength, the narrow channels of zeolites constituting relatively large diffusion limitation probably also contributed to their low catalytic performance.46 Anyway, the results clearly demonstrated the potential of NiO(P)-300 to be employed in further upgrading of biomass-derived compounds with the combination of other type of catalysts, further supporting the versatility of NiO(P)-300.
The results obtained from the above characterization and catalytic testing in combination with knowledge from literature22,23,26,27 allowed us to propose a plausible reaction mechanism for the CTH process with NiO(P)-300, assuming that the mechanism was associated to MPV reduction facilitated by acidic (Ni2+) and basic sites (O2−). As delineated in Scheme 2, 2-propanol was adsorbed to the surface of NiO(P)-300 catalyst and then activated by acidic–basic sites (Ni2+–O2−) within the catalyst to give 2-propoxide, while the carbonyl group in FF was concomitantly activated by acidic sites (Ni2+). Next, H-transfer between activated FF and generated 2-propoxide occurred involving a six-member intermediate on the surface of the catalyst forming FAOL and acetone. Normally, minor amount of 2-(diisopropoxymethyl)furan was observed throughout the studies as a by-product (Fig. S5†), generated by acetalization of FF with 2-propanol over acidic sites of the catalyst. Additionally, trace amount of 2-(isopropoxymethyl)furan, originated from etherification of FAOL with 2-propanol over acidic sites of the catalyst, was detected when the reaction took place in presence of a large amount of NiO(P)-300 catalyst (i.e., 0.03 g) with long reaction time (i.e., ≥3 h).
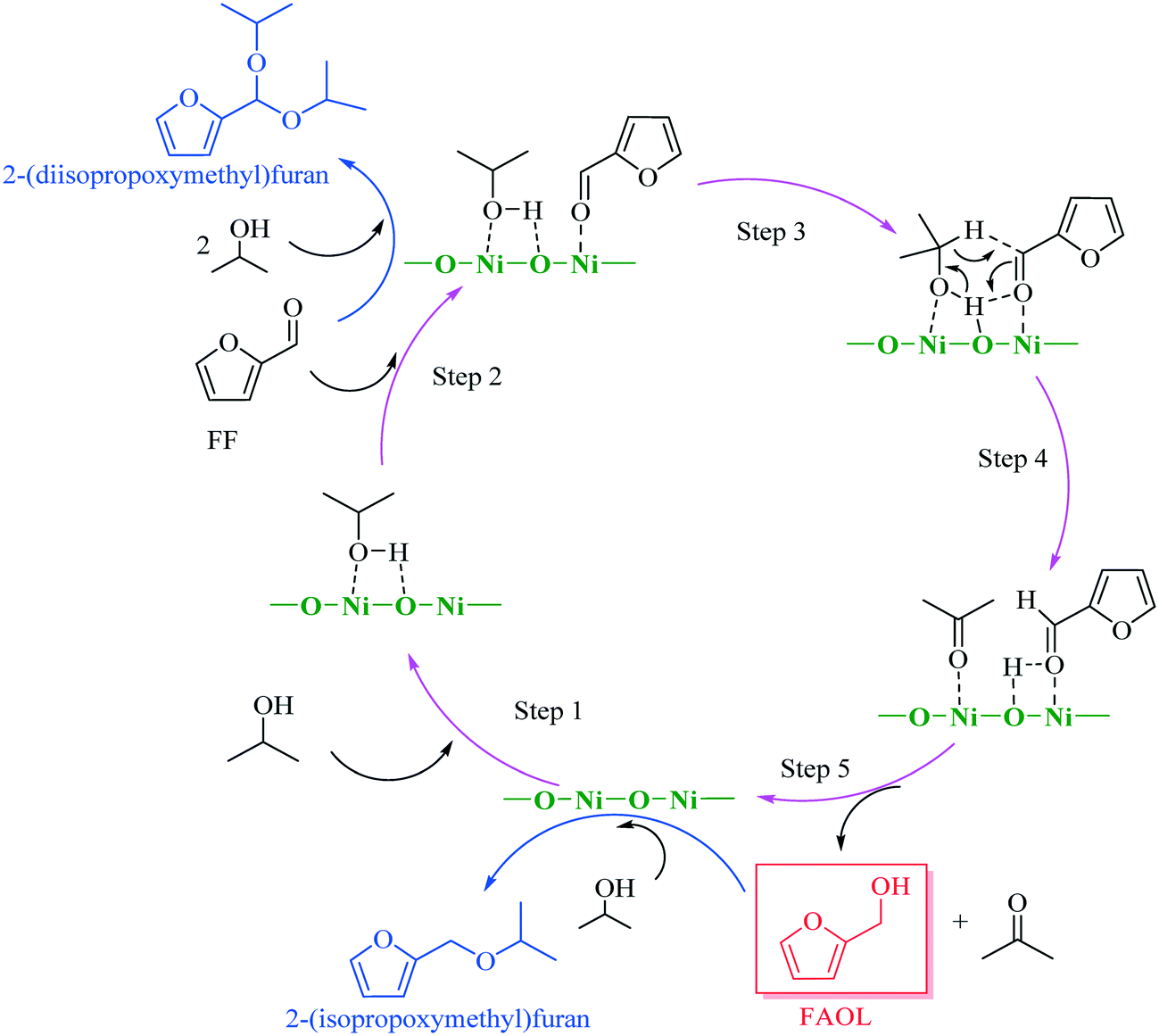 | ||
| Scheme 2 Plausible reaction mechanism for the CTH of FF to FAOL with NiO(P)-300 in 2-propanol. | ||
It is well-known that Ni-based catalysts, preferable RANEY® Ni,49,50 are efficient catalysts for transfer hydrogenation/hydrogenolysis using alcohols as H-donor, and several Ni-based catalysts have already been reported for CTH of FF to FAOL as compiled in Table 4.51–55 Notably, the NiO(P)-300 catalyst developed in this work exhibited however superior catalytic performance in CTH of FF to FAOL compared to previously reported Ni-based catalysts. Specifically, the NiO(P)-300 catalyst did not require pre-reduction and H2 gas (or high temperature carbon reduction under inert gas) could therefore be omitted during catalyst preparation making the process cost-effective. Therefore, the facile, low-cost and green preparation method together with high catalytic performance render the developed NiO(P)-300 catalyst a promising substitute of Ni(0)-based catalysts for CTH.
| Entry | Catalyst | Cat. reduction | Cat. amounta (wt%) | C initial FFb (mol L−1) | H-Donor | Temp. (°C) | Time (h) | Conv. (%) | Yield (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Relative to initial mass of FF. b Initial molar concentration of FF. c This work. | |||||||||||
| 1 | Ni–Ru/C | 600 °C/N2/3 h | 28.9 | 0.36 | Benzyl alcohol | 150 | 12 | — | 81.1 | — | 51 |
| 2 | Ni–Cu/Al2O3 | 320 °C/15 bar H2/3 h | 22.3 | 1.17 | 2-Propanol | 200 | 4 | 95.4 | 95.4 | 100 | 52 |
| 3 | Ni/Fe2O3 | No reduction | 33.3 | 0.40 | 2-Propanol | 180 | 7.5 | 46 | 33 | 71.7 | 53 |
| 4 | Ni–Cu | 250 °C/1 bar H2/1 h | 40.0 | 0.43 | 2-Propanol | 110 | 2.3 | 50 | 50 | 100 | 54 |
| 5 | 5% Ni/AC | 450 °C/1 bar H2/1 h | 12.5 | 0.42 | 2-Propanol | 260 | 5 | 95 | 20 | 21.1 | 55 |
| 6 | 10% Ni–15% W/AC | 450 °C/1 bar H2/3 h | 12.5 | 0.42 | 2-Propanol | 260 | 5 | 83 | 25 | 30.1 | 55 |
| 7 | NiFe2O4 | No reduction | 31.2 | 0.2 | 2-Propanol | 180 | 6 | 99 | 94 | 94.9 | 34 |
| 8 | NiO(P)-300 | No reduction | 20.8 | 0.2 | 2-Propanol | 120 | 3 | 97.3 | 94.2 | 96.8 | |
4 Conclusions
In this work, 3D hierarchical structures of NiO catalysts (sea urchin- and flower-like) have successfully been prepared via a facile yet environmental-friendly hydrothermal method in the absence of strong or stimulating alkali supply, and demonstrated to be highly active for CTH of FF to FAOL using 2-propanol as both H-donor and solvent. The 3D hierarchical construction and low calcination temperature (300 °C) contributed to high surface area and large amount of acid–base sites as well as facile active-site accessibility in the catalysts, which were found to play critical roles in the efficient production of FAOL from CTH of FF. A high FF conversion of 91.8% along with 88.9% FAOL yield was achieved under very mild reaction conditions (100 °C, 5 h) using the sea urchin-like NiO(P)-300 catalyst, and FF conversion and FAOL yield were increased to 97.3 and 94.2%, respectively, at 120 °C after only 3 h. Moreover, the effectiveness and heterogeneous nature of the catalyst were confirmed by a low value of activation energy (41.8 kJ mol−1), successful gram-scale testing, one-pot conversion of FF into IPL in combination with Amberlyst 15 and filtration experiment, respectively. In addition, the catalyst proved reusable several times without significant activity loss after facile regeneration by calcination (300 °C, in air). Importantly, the catalytic system proved also applicable to CTH of various other aldehydes. In perspective, we envisage the low-cost and efficient catalyst prepared by a green strategy in this work to be a potentially attractive catalyst for industrial CTH of FF as well as other aldehydes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JH gratefully acknowledges financial support from The Chinese State Scholarship (No. 201606670008), SY acknowledges the National Natural Science Foundation of China (No. 21576059 and 21666008), and AR acknowledges the Department of Chemistry, Technical University of Denmark for support.References
- T. Renders, S. V. den Bosch, S. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC
.
- H. Li, S. Yang, S. Saravanamurugan and A. Riisager, ACS Catal., 2017, 7, 3010–3029 CrossRef CAS
- H. Li, A. Riisager, S. Saravanamurugan, A. Pandey, R. S. Sangwan, S. Yang and R. Luque, ACS Catal., 2018, 8, 148–187 CrossRef CAS
- L. Zhang, G. Xi, Z. Chen, D. Jiang, H. Yu and X. Wang, Chem. Eng. J., 2017, 307, 868–876 CrossRef CAS
- S. S. Chen, T. Maneerung, D. C. W. Tsang, Y. S. Ok and C. Wang, Chem. Eng. J., 2017, 328, 246–273 CrossRef CAS
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC
- S. T. Thompson and H. H. Lamb, J. Catal., 2017, 350, 111–121 CrossRef CAS
- W. Gong, C. Chen, Y. Zhang, H. Zhou, H. Wang, H. Zhang, Y. Zhang, G. Wang and H. Zhao, ACS Sustainable Chem. Eng., 2017, 2172–2180 CrossRef CAS
- P. N. Romano, J. M. A. R. de Almeida, Y. Carvalho, P. Priecel, E. F. Sousa-Aguiar and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 3387–3392 CrossRef CAS PubMed
- Z. Zhang, J. Song, Z. Jiang, Q. Meng, P. Zhang and B. Han, ChemCatChem, 2017, 9, 2448–2452 CrossRef CAS
- E. Jorge, T. D. M. Lima, C. Lima, L. Marchini, W. N. Castelblanco, D. G. Rivera, E. A. Urquieta-González, R. S. Varma and M. W. Paixao, Green Chem., 2017, 19, 3856–3868 RSC
- S. Song, L. Di, G. Wu, W. Dai, N. Guan and L. Li, Appl. Catal., B, 2017, 205, 393–403 CrossRef CAS
- Z. Jiang, W. Wan, Z. Lin, J. Xie and J. G. Chen, ACS Catal., 2017, 7, 5758–5765 CrossRef CAS
- N. Pino, S. Sitthisa, Q. Tan, T. Souza, D. López and D. E. Resasco, J. Catal., 2017, 350, 30–40 CrossRef CAS
- X. Chang, A. Liu, B. Cai, J. Luo, H. Pan and Y. Huang, ChemSusChem, 2016, 9, 3330–3337 CrossRef CAS PubMed
- P. T. Sulmonetti, S. H. Pang, M. T. Claure, S. Lee, D. A. Cullen, P. K. Agrawal and C. W. Jones, Appl. Catal., A, 2016, 517, 187–195 CrossRef
- L. Grazia, D. Bonincontro, A. Lolli, T. Tabanelli, C. Lucarelli, S. Albonetti and F. Cavani, Green Chem., 2017, 19, 4412–4422 RSC
- J. Wu, G. Gao, J. Li, P. Sun, X. Long and F. Li, Appl. Catal., B, 2017, 203, 227–236 CrossRef CAS
- J. J. Musci, A. B. Merlo and M. L. Casella, Catal. Today, 2017, 296, 43–50 CrossRef CAS
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, A. Chutia, N. Jian, R. E. Palmer, M. Perdjon, A. Thetford, N. Dimitratos, A. Villa and P. P. Wells, ACS Catal., 2017, 7, 2266–2274 CrossRef CAS
- C. P. Jiménez-Gómez, J. A. Cecilia, D. Durán-Martín, R. Moreno-Tost, J. Santamaría-González, J. Mérida-Robles, R. Mariscal and P. Maireles-Torres, J. Catal., 2016, 336, 107–115 CrossRef
- G. Wang, X. Deng, D. Gu, K. Chen, H. Tüysüz, B. Spliethoff, H. Bongard, C. Weidenthaler, W. Schmidt and F. Schüth, Angew. Chem., Int. Ed., 2016, 55, 11101–11105 CrossRef CAS PubMed
- H. Li, J. He, A. Riisager, S. Saravanamurugan, B. Song and S. Yang, ACS Catal., 2016, 6, 7722–7727 CrossRef CAS
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS
- J. He, H. Li, A. Riisager and S. Yang, ChemCatChem, 2018, 10, 430–438 CrossRef CAS
- L. Hu, T. Li, J. Xu, A. He, X. Tang, X. Chu and J. Xu, Chem. Eng. J., 2018, 352, 110–119 CrossRef CAS
- M. Koehle and R. F. Lobo, Catal. Sci. Technol., 2016, 6, 3018–3026 RSC
- B. P. Bastakoti, H. Huang, L. Chen, K. C. Wu and Y. Yamauchi, Chem. Commun., 2012, 48, 9150–9152 RSC
- M. Ko, L. Mendecki and K. A. Mirica, Chem. Commun., 2018, 54, 7873–7891 RSC
- S. Zhang, H. Gao, J. Li, Y. Huang, A. Alsaedi, T. Hayat, X. Xu and X. Wang, J. Hazard. Mater., 2017, 321, 92–102 CrossRef CAS PubMed
- S. Lin, G. Su, M. Zheng, D. Ji, M. Jia and Y. Liu, Appl. Catal., B, 2012, 123–124, 440–447 CrossRef CAS
- Z. Cui, Z. Chen, C. Cao, W. Song and L. Jiang, Chem. Commun., 2013, 49, 6093–6095 RSC
- Z. Yang, W. Qi, R. Su and Z. He, ACS Sustainable Chem. Eng., 2017, 5, 4179–4187 CrossRef CAS
- J. He, S. Yang and A. Riisager, Catal. Sci. Technol., 2018, 8, 790–797 RSC
- M. A. Peck and M. A. Langell, Chem. Mater., 2012, 24, 4483–4490 CrossRef CAS
- C. Yuan, X. Zhang, Q. Wu and B. Gao, Solid State Ionics, 2006, 177, 1237–1242 CrossRef CAS
- S. Song, S. Yao, J. Cao, L. Di, G. Wu, N. Guan and L. Li, Appl. Catal., B, 2017, 217, 115–124 CrossRef CAS
- H. W. Nesbitt, D. Legrand and G. M. Bancroft, Phys. Chem. Miner., 2000, 27, 357–366 CrossRef CAS
- M. J. Gilkey, P. Panagiotopoulou, A. V. Mironenko, G. R. Jenness, D. G. Vlachos and B. Xu, ACS Catal., 2015, 5, 3988–3994 CrossRef CAS
- A. Prasertsab, J. A. Cecilia, C. P. Jiménez-Gómez, C. García-Sancho, R. Moreno-Tost and P. Maireles-Torres, Appl. Catal., A, 2018, 556, 1–9 CrossRef
- N. S. Biradar, A. M. Hengne, S. S. Sakate, R. K. Swami and C. V. Rode, Catal. Lett., 2016, 146, 1611–1619 CrossRef CAS
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC
- H. Habibi, A. Hekmat-Nazemi, A. Kamran-Pirzaman and A. H. Mohammadi, J. Mol. Liq., 2016, 220, 558–565 CrossRef CAS
- C. K. Zéberg-Mikkelsen, S. E. Quiñones-Cisneros and E. H. Stenby, Fluid Phase Equilib., 2002, 194–197, 1191–1203 CrossRef
- J. Geboers, X. Wang, A. B. de Carvalho and R. Rinaldi, J. Mol. Catal. A: Chem., 2014, 388–389, 106–115 CrossRef CAS
- M. Paniagua, J. A. Melero, J. Iglesias, G. Morales, B. Hernández and C. López-Aguado, Appl. Catal., A, 2017, 537, 74–82 CrossRef CAS
- X. Tang, H. Chen, L. Hu, W. Hao, Y. Sun, X. Sun, L. Lin and S. Liu, Appl. Catal., B, 2014, 147, 827–834 CrossRef CAS
- V. A. Ivanov, J. Bachelier, F. Audry and J. C. Lavalley, J. Mol. Catal., 1994, 91, 45–59 CrossRef CAS
- Z. Yang, Y. Huang, Q. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328–5330 RSC
- X. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 RSC
- Z. Gao, L. Yang, G. Fan and F. Li, ChemCatChem, 2016, 8, 3769–3779 CrossRef CAS
- H. P. R. Kannapu, C. A. Mullen, Y. Elkasabi and A. A. Boateng, Fuel Process. Technol., 2015, 137, 220–228 CrossRef
- D. Scholz, C. Aellig and I. Hermans, ChemSusChem, 2014, 7, 268–275 CrossRef CAS PubMed
- S. A. Khromova, M. V. Bykova, O. A. Bulavchenko, D. Y. Ermakov, A. A. Saraev, V. V. Kaichev, R. H. Venderbosch and V. A. Yakovlev, Top. Catal., 2016, 59, 1413–1423 CrossRef CAS
- Y. Wang, P. Prinsen, K. S. Triantafyllidis, S. A. Karakoulia, A. Yepez, C. Len and R. Luque, ChemCatChem, 2018, 10, 3459–3468 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02536c |
| This journal is © The Royal Society of Chemistry 2019 |