
Triphenylamine based conjugated microporous polymers for selective photoreduction of CO2 to CO under visible light†
Chunhui
Dai‡
 a,
Lixiang
Zhong‡
b,
Xuezhong
Gong
b,
Lei
Zeng
b,
Can
Xue
b,
Shuzhou
Li
*b and
Bin
Liu
*a
a,
Lixiang
Zhong‡
b,
Xuezhong
Gong
b,
Lei
Zeng
b,
Can
Xue
b,
Shuzhou
Li
*b and
Bin
Liu
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: cheliub@nus.edu.sg
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: lisz@ntu.edu.sg
First published on 18th November 2019
Abstract
Organic π-conjugated polymers (CPs) have been intensively explored for a variety of critical photocatalytic applications in the past few years. Nevertheless, CPs for efficient CO2 photoreduction have been rarely reported, which is mainly due to the lack of suitable polymers with sufficient solar light harvesting ability, appropriate energy level alignment and good activity and selectivity in multi-electron-transfer photoreduction of CO2 reaction. We report here the rational design and synthesis of two novel triphenylamine (TPA) based conjugated microporous polymers (CMPs), which can efficiently catalyze the reduction of CO2 to CO using water vapor as an electron donor under ambient conditions without adding any co-catalyst. Nearly 100% selectivity and a high CO production rate of 37.15 μmol h−1 g−1 are obtained for OXD-TPA, which is significantly better than that for BP-TPA (0.9 μmol h−1 g−1) as a result of co-monomer change from biphenyl to 2,5-diphenyl-1,3,4-oxadiazole. This difference could be mainly ascribed to the synergistic effect of a decreased optical band gap, improved interface charge transfer and increased CO2 uptake for OXD-TPA. This contribution is expected to spur further interest in the rational design of porous conjugated polymers for CO2 photoreduction.
For the past few decades, artificial photocatalysis has been well established as a sustainable technology for CO2 conversion into carbonaceous chemical fuels such as CO, CH3OH, CH4, etc.1–6 The technology is particularly appealing as it not only holds great promise to handle environmental issue caused by excess CO2 release, but also provides fuels for energy supply. However, high energy input is required for CO2 transformation owing to its remarkable stability with the dissociation energy of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond up to 750 kJ mol−1,6 which makes it difficult to develop efficient photocatalysts. Since the pioneering report of heterogeneous CO2 photoreduction using TiO2 as the catalyst,7 numerous inorganic semiconductors have been under active exploration. However, most inorganic photocatalysts suffer from limited tunability, potential heavy metal toxicity and wide band gap,7–10 which hinder their practical applications and thus require the exploration of alternatives.
O bond up to 750 kJ mol−1,6 which makes it difficult to develop efficient photocatalysts. Since the pioneering report of heterogeneous CO2 photoreduction using TiO2 as the catalyst,7 numerous inorganic semiconductors have been under active exploration. However, most inorganic photocatalysts suffer from limited tunability, potential heavy metal toxicity and wide band gap,7–10 which hinder their practical applications and thus require the exploration of alternatives.
Conjugated microporous polymers (CMPs), a subclass of structurally diverse polymeric materials, have many advantages such as permanent porosity, large surface area, strong visible light activity, facile synthesis, tunable optoelectronic properties, and high chemical and thermal stability, making them attractive candidates for a range of applications, such as gas sequestration and separation,11–15 heterogeneous catalytic reaction,16–20 sensing,21,22 and energy storage and conversion.23–27 Recent studies have revealed that porous conjugated polymers (CPs) are promising photocatalysts for CO2 reduction and their photocatalytic activities could be effectively enhanced by rational molecular design. For instance, Liu et al. reported photocatalytic CO2 reduction using a series of pyrene-based porous CPs in a CO2-saturated ionic liquid, which yielded CO with a rate of 47.37 μmol h−1 g−1 and reaction selectivity reached 98.3% (>420 nm).28 By copolymerization between 2,4,6-triphenyl-1,3,5-triazine and different co-monomers, the charge separation of the resulting polymers could be improved to give a maximum CO production rate of 18.2 μmol h−1 g−1 with 81.6% selectivity in the presence of CoCl2 and dipyridyl as the co-catalysts.29 Crystalline covalent organic framework (COF) based CO2-reduction photocatalysts were reported by Huang et al., in which Re(bpy)(CO)3Cl was incorporated into the 2D COF to reduce CO2 using triethanolamine as a sacrificial agent and 98% selectivity was obtained.30 Despite this exciting progress, these CO2 reduction reactions were carried out in CO2-saturated liquid media, which required the use of organic electron donors, co-catalysts or ionic liquids, adding cost and complexity. Moreover, the limited solubility of CO2 in liquid media is unfavorable for achieving high reaction efficiency. By contrast, the reduction reaction performed on a gas–solid interface relies on CO2 chemisorption and activation by the photocatalysts, which eliminates the need for liquid media. This simple and environmentally-friendly approach uses only a trace amount of water vapor as the electron donor, which significantly inhibits the competing process of water reduction and favors the selectivity of CO2 reduction.1,3 However, to the best of our knowledge, the exploration of efficient polymers for CO2 reduction on a gas–solid system remains a great challenge.31 Very recently, Eosin Y-based conjugated porous polymers were developed for CO2 photoreduction, which resulted in a CO production rate of 33 μmol h−1 g−1 with 92% selectivity.31 To achieve better photocatalytic performance, new polymers and useful molecular design strategies to reveal the critical parameters that control their photoactivities are highly desirable.
In this study, we present two specially designed CMPs as visible-light-active photocatalysts for the reduction of CO2 to CO only in the presence of water vapor under ambient conditions. The polymer structures are shown in Scheme 1. Triphenylamine (TPA) based CMPs were developed owing to their inherent advantages of high surface area for effective CO2 absorption, facile preparation, and fine optimization of energy levels.32,33 The optical band gaps of the as-prepared CMPs could be tuned from 2.38 to 2.22 eV by replacing biphenyl (BP) with 2,5-diphenyl-1,3,4-oxadiazole (OXD). More importantly, the CO production rate could be enhanced to 37.15 μmol h−1 g−1 with ∼100% selectivity under visible light irradiation (>420 nm). In addition, we provide a comprehensive understanding of the photocatalytic activity of polymers by complementary experimental and computational studies, which is critical for future development of conjugated polymers for CO2 photoreduction.
 | ||
| Scheme 1 Chemical structures of the conjugated microporous polymers for CO2 photoreduction in this study. | ||
The synthetic details and characterization of the monomers and conjugated microporous polymers are given in the ESI.† Tris(4-ethynylphenyl)amine was prepared by the Pd(II)-catalyzed coupling of tris(4-bromophenyl)amine and ethynyltrimethylsilane and subsequent deprotection promoted by K2CO3.34 Starting from 4-bromobenzoyl chloride, 2,5-bis(4-bromophenyl)-1,3,4-oxadiazole was obtained in three high yielding steps as depicted in Scheme S1.† A condensation reaction between 4-bromobenzoyl chloride and hydrazine hydrate afforded 1,2-bis(4-bromobenzoyl)hydrazide, followed by cyclodehydration to give 2,5-bis(4-bromophenyl)-1,3,4-oxadiazole in 71% yield.35 BP-TPA and OXD-TPA were synthesized from tris(4-ethynylphenyl)amine with 4,4′-dibromodiphenyl and 2,5-diphenyl-1,3,4-oxadiazole, respectively, by the Sonogashira coupling reaction in an Et3N/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixture with Pd(PPh3)4 and CuI as catalysts. The resulting product was successively washed with water, acetone and methanol followed by Soxhlet extraction (THF and then CH2Cl2). BP-TPA and OXD-TPA are insoluble in all common solvents such as DMF, THF and CH2Cl2. The structural characterization of the two polymers was studied by solid-state 13C NMR spectroscopy, FT-IR spectroscopy, and elemental analysis. Both polymers show aromatic peaks at 120–150 ppm and peaks at ∼91 ppm for quaternary alkynes (Fig. S1 and S2†), consistent with the CMPs synthesized by the Sonogashira polymerization.34 In addition, the peak at 163 ppm in the NMR spectrum of OXD-TPA is ascribed to the carbon atom of oxadiazole. Fourier-transform infrared (FT-IR) spectra analysis confirmed the alkynyl linker of the two polymers by the evidence of stretching vibrations of triple bonds appearing around 2200 cm−1 (Fig. S3†). BP-TPA and OXD-TPA show similarly spherical morphologies revealed by scanning electron microscopy (SEM) measurement (Fig. S4†). Negligible Pd residual was detected in both polymer networks and the content is 0.34% for BP-TPA and 0.54% for OXD-TPA, respectively. The powder X-ray diffraction (PXRD) patterns demonstrated that both polymers exhibited broad diffraction peaks with low intensities in the wide region, indicative of their amorphous nature (Fig. S6†). Thermal gravimetric analysis (TGA) demonstrated their excellent thermostability with 5% weight loss for BP-TPA and OXD-TPA observed at 463 °C and 412 °C, respectively (Fig. S7†).
:1, v/v) mixture with Pd(PPh3)4 and CuI as catalysts. The resulting product was successively washed with water, acetone and methanol followed by Soxhlet extraction (THF and then CH2Cl2). BP-TPA and OXD-TPA are insoluble in all common solvents such as DMF, THF and CH2Cl2. The structural characterization of the two polymers was studied by solid-state 13C NMR spectroscopy, FT-IR spectroscopy, and elemental analysis. Both polymers show aromatic peaks at 120–150 ppm and peaks at ∼91 ppm for quaternary alkynes (Fig. S1 and S2†), consistent with the CMPs synthesized by the Sonogashira polymerization.34 In addition, the peak at 163 ppm in the NMR spectrum of OXD-TPA is ascribed to the carbon atom of oxadiazole. Fourier-transform infrared (FT-IR) spectra analysis confirmed the alkynyl linker of the two polymers by the evidence of stretching vibrations of triple bonds appearing around 2200 cm−1 (Fig. S3†). BP-TPA and OXD-TPA show similarly spherical morphologies revealed by scanning electron microscopy (SEM) measurement (Fig. S4†). Negligible Pd residual was detected in both polymer networks and the content is 0.34% for BP-TPA and 0.54% for OXD-TPA, respectively. The powder X-ray diffraction (PXRD) patterns demonstrated that both polymers exhibited broad diffraction peaks with low intensities in the wide region, indicative of their amorphous nature (Fig. S6†). Thermal gravimetric analysis (TGA) demonstrated their excellent thermostability with 5% weight loss for BP-TPA and OXD-TPA observed at 463 °C and 412 °C, respectively (Fig. S7†).
The porosity of BP-TPA and OXD-TPA was evaluated by N2 sorption isotherm measurements at 77 K. As shown in Fig. 1a, both polymer networks give rise to type I isotherms and exhibit a steep increase in the low relative pressure regions (<0.01) due to their microporosity. The apparent BET surface area of BP-TPA is 512 m2 g−1, which is lower than that of OXD-TPA (686 m2 g−1).
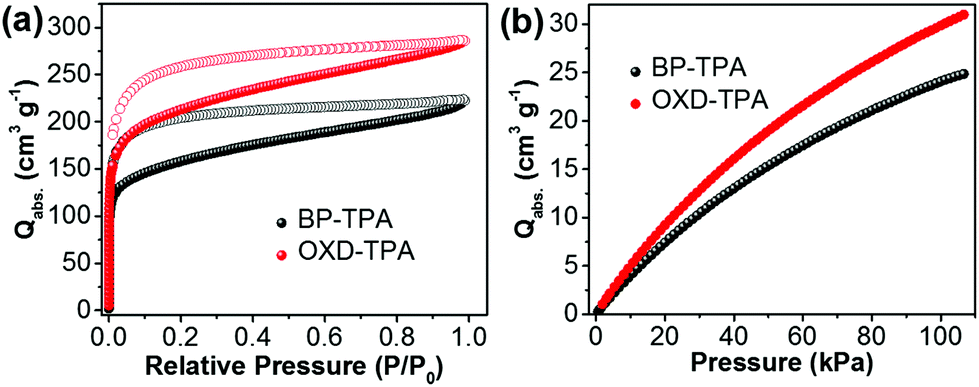 | ||
| Fig. 1 (a) N2 sorption isotherm curves of the polymers obtained at 77 K. (b) CO2 adsorption isotherm curves of the polymers at 298 K. | ||
Both polymers have similar pore size distributions with pore widths at about 1.1, 1.5 and 2.7 nm, respectively (Fig. S5†). The CO2 uptake capacities of the two polymers at 298 K were evaluated by CO2 adsorption isotherms. OXD-TPA exhibited a higher volumetric CO2 uptake than BP-TPA (Fig. 1b). In particular, the difference of CO2 uptake became more distinct with increasing pressure. At P = 100 kPa, the CO2 uptake capacities of BP-TPA and OXD-TPA were 24.1 and 30.3 cm3 g−1, respectively. The enhanced CO2 absorbing ability of OXD-TPA compared to BP-TPA is attributed to its higher BET surface area and the dipole-induced electrostatic interactions between the N or O atoms of the oxadiazole unit and the C atoms of CO2.36–38
The UV/vis diffuse reflectance spectra (DRS) and photoluminescence spectra of BP-TPA and OXD-TPA are displayed in Fig. 2a. OXD-TPA with extended π-conjugation shows broader absorption than BP-TPA. Accordingly, the optical band gaps estimated from the absorption onset of the DRS spectra are 2.38 eV for BP-TPA and 2.22 eV for OXD-TPA. Upon excitation at 380 nm, BP-TPA exhibits emission centered at 562 nm, which is slightly blue-shifted to 557 nm for OXD-TPA. Notably, the emission of OXD-TPA is significantly stronger than that of BP-TPA. This is largely due to the twisted conformation between phenyl and oxadiazole in 2,5-diphenyl-1,3,4-oxadiazole, which interrupts the π-conjugation and molecular stacking, thus blue-shifts the emission and enhances the fluorescence intensity.39,40 In addition, time-resolved photoluminescence decay spectra demonstrated a slightly longer fluorescence lifetime of 0.82 ns for OXD-TPA than that of BP-TPA (0.61 ns) (Fig. S8†). The LUMO levels of polymers were evaluated using cyclic voltammetry (Fig. S9†). Both exhibited well-defined reversible reduction waves with the LUMO levels of −0.41 V (vs. NHE) for BP-TPA and −0.44 V (vs. NHE) for OXD-TPA (Table 1). Accordingly, the HOMO levels of BP-TPA and OXD-TPA were calculated to be 1.97 V and 1.78 V, respectively. The energy band structures of both polymers are well positioned for the reduction of CO2 to CO, indicating that they are thermodynamically capable of generating CO from CO2 photoreduction under light irradiation (Fig. 2b).41 Photoelectrochemical measurements were conducted to compare the photogenerated charge carrier transport of the two polymers. As shown in Fig. 2c, the Nyquist plot of OXD-TPA exhibits a smaller diameter of the semicircular than that of BP-TPA, revealing a decrease of the charge-transfer resistance by replacing biphenyl with 2,5-diphenyl-1,3,4-oxadiazole. The improved photoinduced charge transport can be further supported by an increased transit photocurrent response of OXD-TPA as shown in Fig. 2d, which reveals a visible-light photocurrent of ∼2 μA cm−2 for OXD-TPA and ∼0.5 μA cm−2 for BP-TPA.
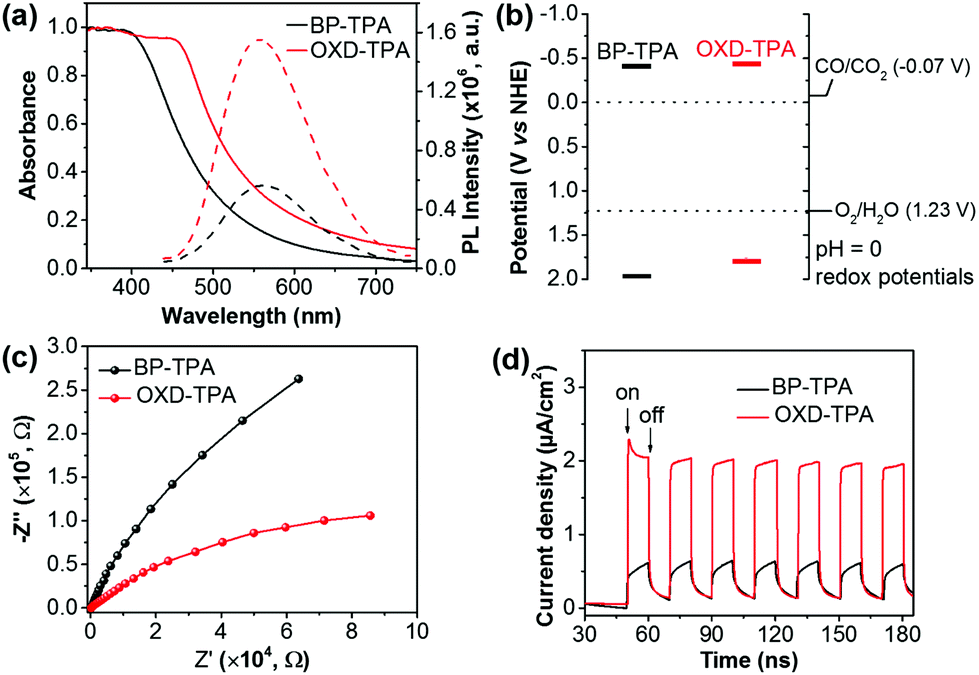 | ||
| Fig. 2 (a) UV/vis diffuse reflectance spectra (solid line) and photoluminescence (PL) spectra (dashed lines; λex = 380 nm) of BP-TPA and OXD-TPA in the solid state. (b) Energy levels of BP-TPA and OXD-TPA relative to the potentials of CO2 reduction to CO and water oxidation, respectively, vs. the NHE at pH = 0. (c) EIS Nyquist plots and (d) periodic transient photocurrent response of polymer electrodes in 0.5 M Na2SO4 aqueous solution. | ||
| Polymers |
E
ga (eV) |
E
red, onsetb (V) |
LUMO/HOMOc (V, vs. NHE) | S BET (m2 g−1) | CO2 uptaked (cm3 g−1) |
|---|---|---|---|---|---|
| a Estimated from the UV/vis diffuse reflectance spectra onset (Eg = 1240/λonset). b Versus Fc/Fc+ in acetonitrile solution, 50 mV s−1 scan rate. c E NHE = EFc/Fc+ + 0.63 V, EHOMO = ELUMO − Eg, opt. d At P = 100 kPa. | |||||
| BP-TPA | 2.38 | −1.04 | −0.41/1.97 | 2.38 | 24.1 |
| OXD-TPA | 2.22 | −1.07 | −0.44/1.78 | 2.22 | 30.3 |
The CO2 photoreduction reactions over the as-synthesized polymers were carried out in the presence of CO2 and water vapor under ambient conditions. Interestingly, CO was detected as the only reductive product in the photocatalytic system (Fig. S10b†). As shown in Fig. 3a, after the four-hour irradiation of visible light (>420 nm), OXD-TPA produced 148.6 μmol g−1 CO, which is much more than that by BP-TPA (3.6 μmol g−1). At 420 nm, the quantum efficiencies are about 0% and 0.19% for BP-TPA and OXD-TPA (Fig. S19†), respectively. To the best of our knowledge, the photocatalytic activity of OXD-TPA (37.15 μmol h−1 g−1) is much better than that of commonly used g-C3N4 and many newly reported CPs under similar conditions (Table S1†).
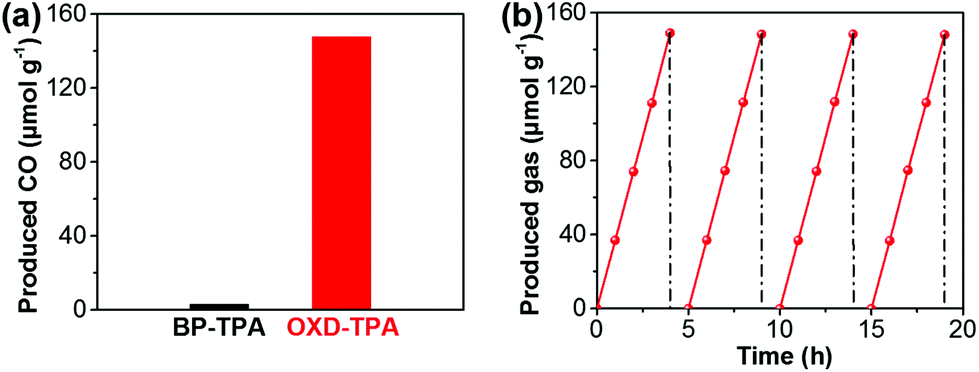 | ||
| Fig. 3 (a) CO production from the photocatalytic CO2 reduction over BP-TPA and OXD-TPA after 4 h irradiation (>420 nm). Reaction conditions: 15 mg polymer, CO2 (1.0 atm), water vapor, room temperature. (b) Time course of photocatalytic CO2 reduction over OXD-TPA. | ||
To confirm the photocatalytic process, we have performed a series of control experiments. No reductive products were detected under dark conditions. Upon replacing CO2 with N2, only a trace amount of CO could be detected, indicating that CO2 is the reductive agent in the system. When no H2O is added to the reaction system, negligible CO could be measured, which suggests that H2O plays an important role as an electron donor. Notably, H2 and other carbonaceous products, such as CH4, HCHO, HCOOH, and CH3OH were not found in our tests, demonstrating that the separated electrons on the surface of the polymer network are almost exclusively used for CO2 reduction to CO and ∼100% selectivity was obtained for OXD-TPA in the photocatalytic reaction.
To study the source of generated CO during the photocatalytic process, an isotopic experiment with 13CO2 (Fig. S11†) was conducted under the same reaction conditions (>420 nm). When 12CO2 was replaced by 13CO2 in the reactor, a gas product with a retention time of 4.026 min was detected, which was different from that of 12CO at 3.958 min (Fig. S10b and c†). In addition, after adding concentrated NaOH aqueous solution into the reactor to remove unreacted 13CO2, the gas sample was subsequently analyzed by MS spectrometry and a peak at m/z = 29 was found (Fig. S12†), which was assigned to 13CO. This peak was not due to the fragmentation of 13CO2 during the MS analysis as the unreacted 13CO2 was completely removed prior to the analysis. Based on all these results, it could be concluded that CO originated from the reduction of CO2 on OXD-TPA.
The photocatalytic CO2 reduction stability and recyclability of OXD-TPA was evaluated by four cyclic testing (4 h-irradiation in each run). The photocatalytic reactor was treated under vacuum and then refilled with the mixture of CO2 and water vapor after each run. Steady CO production is observed over four runs (Fig. 3b). After the photocatalysis experiments, the recycled polymer did not show any significant structural changes (Fig. S13–S15†), indicating that OXD-TPA is highly stable during the photocatalytic process.
DFT calculations were performed to understand the electronic properties of both polymers. The optimized structures are shown in Fig. S16.† The excited electrons for both BP-TPA and OXD-TPA are distributed all over the backbone, whereas the excited holes are mainly localized in TPA due to its strong electron-donating feature (Fig. S17a and S17b†). Moreover, compared to the BP moiety, the more electronegative OXD moiety impairs the density of excited electrons within the TPA part, which decreases the overlap between the excited electrons and holes in OXD-TPA as compared to that of BP-TPA, indicating less recombination of photogenerated charges in OXD-TPA.
We further studied the adsorption energy of COOH*, which is proposed to be the intermediate for CO2 reduction to CO, on different positions of BP-TPA and OXD-TPA (Fig. 4a and b), and the corresponding adsorption energies are shown in Fig. 4c. The most stable configurations of COOH* are shown in the insert of Fig. 4c and Fig. S17c.† The most stable adsorbed COOH* has similar configurations but the adsorption is ∼0.2 eV more stable on OXD-TPA, which facilitates the reduction of CO2.
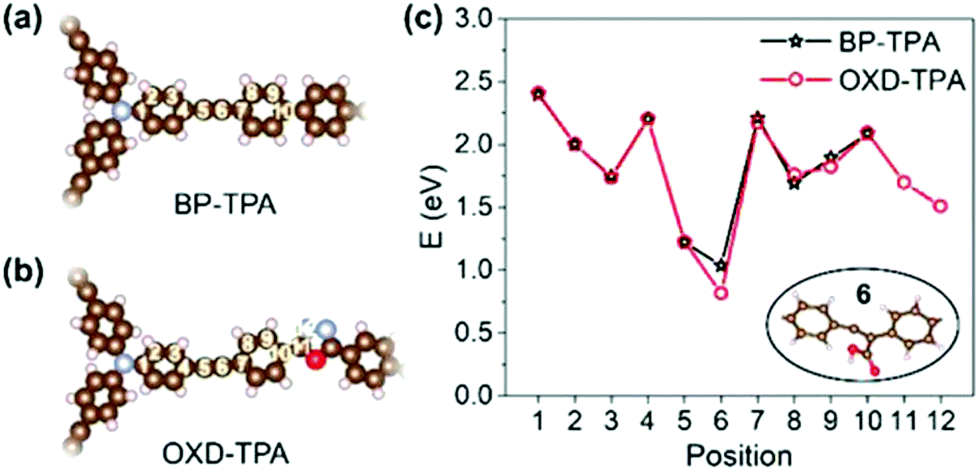 | ||
| Fig. 4 Different adsorption sites on (a) BP-TPA and (b) OXD-TPA, and (c) the corresponding adsorption energies for COOH*. | ||
DFT calculations revealed that the band gaps for BP-TPA and OXD-TPA are 2.47 and 2.38 eV, respectively, which are in agreement with the optical bandgaps obtained in our experiments (2.38 eV for BP-TPA and 2.22 eV for OXD-TPA as shown in Table 1). Notably, the theoretically predicted HOMOs and LUMOs for BP-TPA and OXD-TPA are depicted in Fig. S18a.† The HOMO potential of BP-TPA (0.79 V) is approximate to the one required for water oxidation (0.82 V, neutral conditions), while it is 0.27 V more positive for OXD-TPA. The LUMO potentials of both BP-TPA (−1.68 V) and OXD-TPA (−1.31 V) are much more negative than the reduction potential of CO2 to CO (−0.53 V). The results reinforced the sufficient redox ability of the two polymers to trigger the photocatalytic reactions in our system. The possible CO2 photoreduction process for the polymers is described as follows: the CO2 gas can be absorbed and activated by the porous polymers. Upon the illumination of visible light, the photogenerated electrons (e−) could migrate from the LUMO levels to the HOMO levels of the polymer. Subsequently, the absorbed CO2 captures electrons on the polymer surface, forming CO and H2O (CO2 + 2H+ + 2e− → CO + H2O), while the photoinduced holes were utilized for oxidizing water to generate oxygen and hydrogen ions via the half-reaction (2H2O + 4h+ → O2 + 4H+) (Fig. S18b†). The low water concentration in the reaction system could efficiently inhibit the competitive reaction to give H2, CH3OH, CH4, etc. This is beneficial for the high selectivity of polymers. The much better photocatalytic performance of OXD-TPA could be attributed to its smaller band gap, increased dissociation of photogenerated excitons, and more stable COOH*.
Conclusions
In conclusion, we present the rational design of two novel triphenylamine based CMPs for CO2 photoreduction to CO under visible light irradiation. The CO production rate could be significantly enhanced from 0.9 to 37.15 μmol h−1 g−1 (>420 nm) with ∼100% selectivity, which is much better than the newly reported CPs in similar photocatalytic systems (Table S1†). The enhancement in the photocatalytic activity of polymers resulting from molecular tuning was fully investigated by experiments and theoretical calculations. Accordingly, we expect that this study will stimulate research interest in designing stable and highly efficient conjugated polymers for CO2 photoreduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Singapore National Research Foundation (R279-000-444-281 and R279-000-483-281), the National University of Singapore (R279-000-482-133), the Singapore MOE AcRF-Tier1 (2018-T1-001-072, RG 9/18) and MOE AcRF-Tier2 (MOE2016-T2-2-056).Notes and references
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888 CrossRef CAS PubMed.
- M. Marszewski, S. Cao, J. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485 CrossRef CAS.
- R. Shi, G. I. N. Waterhouse and T. Zhang, Sol. RRL, 2017, 1, 1700126 CrossRef.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610 CrossRef CAS PubMed.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485 CrossRef CAS.
- A. Li, R. F. Lu, Y. Wang, X. Wang, K. L. Han and W. Q. Deng, Angew. Chem., Int. Ed., 2010, 49, 3330 CrossRef CAS PubMed.
- R. Dawson, E. Stockel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239 RSC.
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084 CrossRef CAS PubMed.
- Y. H. Xu, S. B. Jin, H. Xu, A. Nagai and D. L. Jiang, Chem. Soc. Rev., 2013, 42, 8012 RSC.
- Y. Yuan, H. Huang, L. Chen and Y. Chen, Macromolecules, 2017, 50, 4993 CrossRef CAS.
- L. Chen, Y. Yang, Z. Guo and D. Jiang, Adv. Mater., 2011, 23, 3149 CrossRef CAS PubMed.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Mater., 2015, 27, 6265 CrossRef CAS PubMed.
- Y.-B. Zhou, Y.-Q. Wang, L.-C. Ning, Z.-C. Ding, W.-L. Wang, C.-K. Ding, R.-H. Li, J.-J. Chen, X. Lu, Y.-J. Ding and Z.-P. Zhan, J. Am. Chem. Soc., 2017, 139, 3966 CrossRef CAS PubMed.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735 CrossRef CAS.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738 CrossRef CAS PubMed.
- C. Gu, N. Huang, J. Gao, F. Xu, Y. Xu and D. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4850 CrossRef CAS PubMed.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753 CrossRef CAS PubMed.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819 RSC.
- C. Gu, N. Huang, Y. Chen, L. Qin, H. Xu, S. Zhang, F. Li, Y. Ma and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 13594 CrossRef CAS PubMed.
- C. Zhang, Y. He, P. Mu, X. Wang, Q. He, Y. Chen, J. Zeng, F. Wang, Y. Xu and J.-X. Jiang, Adv. Funct. Mater., 2018, 28, 1705432 CrossRef.
- J. Zhu, C. Yang, C. Lu, F. Zhang and Z. Yuan, Acc. Chem. Res., 2018, 51, 3191 CrossRef CAS PubMed.
- Y. Chen, G. Ji, S. Guo, B. Yu, Y. Zhao, Y. Wu, H. Zhang, Z. Liu, B. Han and Z. Liu, Green Chem., 2017, 19, 5777 RSC.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem. – Eur. J., 2018, 24, 17454 CrossRef CAS PubMed.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614 CrossRef CAS PubMed.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632 CrossRef CAS PubMed.
- T. Geng, Z. Zhu, W. Zhang and Y. Wang, J. Mater. Chem. A, 2017, 5, 7612 RSC.
- Z. Chen, W. Li, Y. Dai, N. Xu, C. Su, J. Liu and C. Zhang, Electrochim. Acta, 2018, 286, 187 CrossRef CAS.
- Z. Xie, Y. Wei, X. Zhao, Y. Li, S. Ding and L. Chen, Mater. Chem. Front., 2017, 1, 867 RSC.
- W.-Y. Wong and Y.-H. Guo, J. Mol. Struct., 2008, 890, 150 CrossRef CAS.
- M. Saleh, H. M. Lee, K. C. Kemp and K. S. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 7325 CrossRef CAS PubMed.
- S. Hug, L. Stegbauer, H. Oh, M. Hirscher and B. V. Lotsch, Chem. Mater., 2015, 27, 8001 CrossRef CAS.
- K. Yuan, C. Liu, L. Zong, G. Yu, S. Cheng, J. Wang, Z. Weng and X. Jian, ACS Appl. Mater. Interfaces, 2017, 9, 13201 CrossRef CAS PubMed.
- C. Wang, L.-O. Pålsson, A. S. Batsanov and M. R. Bryce, J. Am. Chem. Soc., 2006, 128, 3789 CrossRef CAS PubMed.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304 CrossRef CAS.
- W. Fan, Q. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and supporting figures. See DOI: 10.1039/c9gc03131f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |