
Enzyme-inspired flavin–polydopamine as a biocompatible nanoparticle photocatalyst†
Leander
Crocker
a,
Philipp
Koehler
b,
Patrick
Bernhard
 a,
Antonina
Kerbs
a,
Tijmen
Euser
b and
Ljiljana
Fruk
*a
a,
Antonina
Kerbs
a,
Tijmen
Euser
b and
Ljiljana
Fruk
*a
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK. E-mail: lf389@cam.ac.uk
bNanophotonics Centre, Cavendish Laboratory, University of Cambridge, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK
First published on 17th June 2019
Abstract
A new approach aimed at designing an enzyme-inspired photocatalyst is presented that exploits the inherent photocatalytic activity of flavin and the facile polymerization of dopamine to afford hybrid cofactor-containing nanoparticles. The flavin–polydopamine system benefits from ease of synthesis, tunability in terms of size and activity, and excellent temporal control over the catalyzed reactions. This novel, versatile photocatalyst exhibits both photooxidation and photoreduction of chromogenic enzymatic substrates. In addition, the prepared hybrid nanoparticles are shown to be non-toxic, paving the way to their use in a wider range of applications beyond green catalysis, such as antioxidant adjuvants to various therapeutic approaches.
New conceptsThe use of nanomaterials as enzyme-like catalysts has risen in popularity over the last decade, with particular focus on inorganic nanomaterials (nanozymes). However, the versatility and efficiency of enzymes are difficult to match and there are continuous efforts to propose new design strategies. By taking inspiration from natural flavoenzymes, we designed a novel biocompatible nanoparticle system that is light-responsive and shows tuneable activity. Our approach employs polydopamine (PDA) nanoparticles as a flavin carrier, which mimics the tryptophan environment of natural flavin enzymes. Nanoparticles of tuneable size and flavin content are prepared via a facile co-polymerisation of dopamine and flavin–dopamine conjugates. Temporal control over both the oxidation and reduction is demonstrated using visible light irradiation and two different enzyme substrates. In addition, a preliminary in vitro assay confirmed a high level of biocompatibility, as well as potential antioxidant effect. Our findings open a new route to the design of biocompatible and tuneable nano-sized, enzyme-inspired photocatalysts, which exhibit a versatile reactivity profile, are tuneable both in terms of structure and reactivity, and are characterized by ease of fabrication and low toxicity. This profile makes them suitable for use in green catalysis, but also in nanomedicine and design of organic energy harvesting systems. |
Enzymes are not only protein catalysts that drive biochemical processes responsible for the sustenance of life, but due to their specificity, biodegradability, and efficiency both in terms of substrate conversion and energy usage, they are highly desirable for green chemistry applications. However, the key disadvantages of wider enzyme use out of the lab-setting are difficulties associated with their isolation in large quantities, post-reaction separation, low stability under non-physiological conditions such as high temperature or pH, and sensitivity to organic solvents.1,2 Several approaches have been developed to address these issues, each with their own set of advantages and drawbacks. For example, more robust enzymes and enzyme-inspired systems have been designed either by immobilization or encasing natural enzymes,3 by use of directed evolution to make efficient non-natural enzymes,4 or by utilization of various materials to prepare enzyme mimics.5,6 The latter has particularly benefited from developments in nanotechnology and the emergence of inorganic materials with enzyme-like activities known as nanozymes.7,8 The most prominent examples of nanozymes to date are iron oxide (Fe3O4) nanoparticles, with high catalytic activity similar to those of peroxidases and catalase, ceria nanoparticles (CeO2), and metallic nanoparticles such as gold (Au NP) and silver (Ag NPs). Over the past decade, these materials have been explored as low-cost and robust alternatives to natural enzymes, with the focus on industrial applications for chemical synthesis,9,10 water remediation,11 or as fuel additives to reduce pollution.12 Lately, the attention of researchers working on nanozymes has shifted more towards nanomedicine and therapeutic applications in biofilm disruption, antioxidation, tumor removal and tissue engineering.8,13 Despite their attractiveness due to ease of synthesis and storage, one of the drawbacks of nanozyme use is their poorly understood toxicological profile including their long-term toxicity and environmental impact.14,15 This calls for new strategies and rational design approaches to afford a new generation of biocompatible nanozymes tailored to a particular application.16 To get closer to this goal, we have set out to rationally design a new generation of hybrid nanozymes combining an organic enzyme component with a nanostructured material, which is not a passive carrier of an enzymatic centre, but actively engages in the catalytic process.
We took inspiration from flavoenzymes, an important group of oxidoreductases that catalyse a large variety of different types of redox reactions, thanks to their isoalloxazine-based flavin cofactors, flavin adenine mononucleotide (FMN) and flavin dinucleotide (FAD). As a result of their wide-ranging activity profile, flavoenzymes are implemented within a range of biological processes such as metabolic pathways, respiratory chain reactions and photochemical processes.17–20 The latter has contributed to the development of flavin photocatalysts as an efficient, green and mild strategy to afford light-driven transformations such as E/Z isomerisation, [2+2] cycloaddition, electrophilic chlorination and sulfide oxidation.21 However, although efficient homogenous systems have been explored, there are issues related to such systems such as limited separation of the products from catalysts hindering both the product purity and the reuse of the catalysts. There are only a few examples of heterogeneous flavin photocatalysts and they are yet to show efficiency and selectivity similar to their natural counterparts.22–24 There are examples, however, of very efficient ‘dark’ catalytic flavin systems utilising artificial and biomimetic polymeric structures.25–27 With this in mind, and to further advance the promising field of flavin photocatalysis, we aimed to develop an enzyme-inspired photocatalytic system by embedding flavin within an active nanostructured polymer matrix employing an easy-to-use copolymerization strategy.
We have chosen polydopamine (PDA) as the carrier polymer due to its reported biocompatibility,28,29 and structural and electronic properties.30 Although PDA has extensively been used as a molecular adhesive because of its ability to form coatings on virtually any surface,31 a more interesting property from a synthetic chemistry point of view is its intrinsic catalytic activity.32–34 Composed of a sequence of extended π-systems, PDA can be considered an amorphous organic semiconductor and has already been employed as an electron gate for artificial photosynthesis,35 and as a mediator in various catalytic systems based on metallic nanoparticles,36 and even whole cells.37 Although the exact mechanism of PDA's action in these applications has not yet been deciphered, it has been shown that PDA accelerates the rate of electron transfer by acting as an electron acceptor.35
Based on these studies, we hypothesized that the combination of flavin and PDA would result in an interesting system that displays a wider activity profile significantly enhancing the current repertoire of catalytic flavin systems. Within natural flavoenzymes, function and catalytic activity are defined by specific interaction between a flavin cofactor and surrounding amino acids. For example, a key reaction enabling switching from a dark to light-adapted state of BLUF (sensors of blue light using FAD) photoreceptors, which guides biological signaling pathways, is the electron transfer between tyrosine and a flavin cofactor.38 The structure of PDA (Fig. 1A) mimics the tyrosine moieties suggesting that in a flavin–PDA conjugate, the polymer can actively engage in the catalytic activity and electron transfer. We therefore set out to prepare a flavin–dopamine conjugate, and formulate our catalysts in the form of flavin–PDA nanoparticles (FLPDA NPs). Such nanoparticles are characterized by an increased surface area aiding the charge transfer (CT) and substrate interaction with the photo-excited flavin species whilst offering more flexibility in terms of future applications. For example, the surface of the nanoparticles could be additionally modified to contain particular cell targeting moieties or stabilizing shells to tailor the stability and physiochemical properties of the system to a desired application.
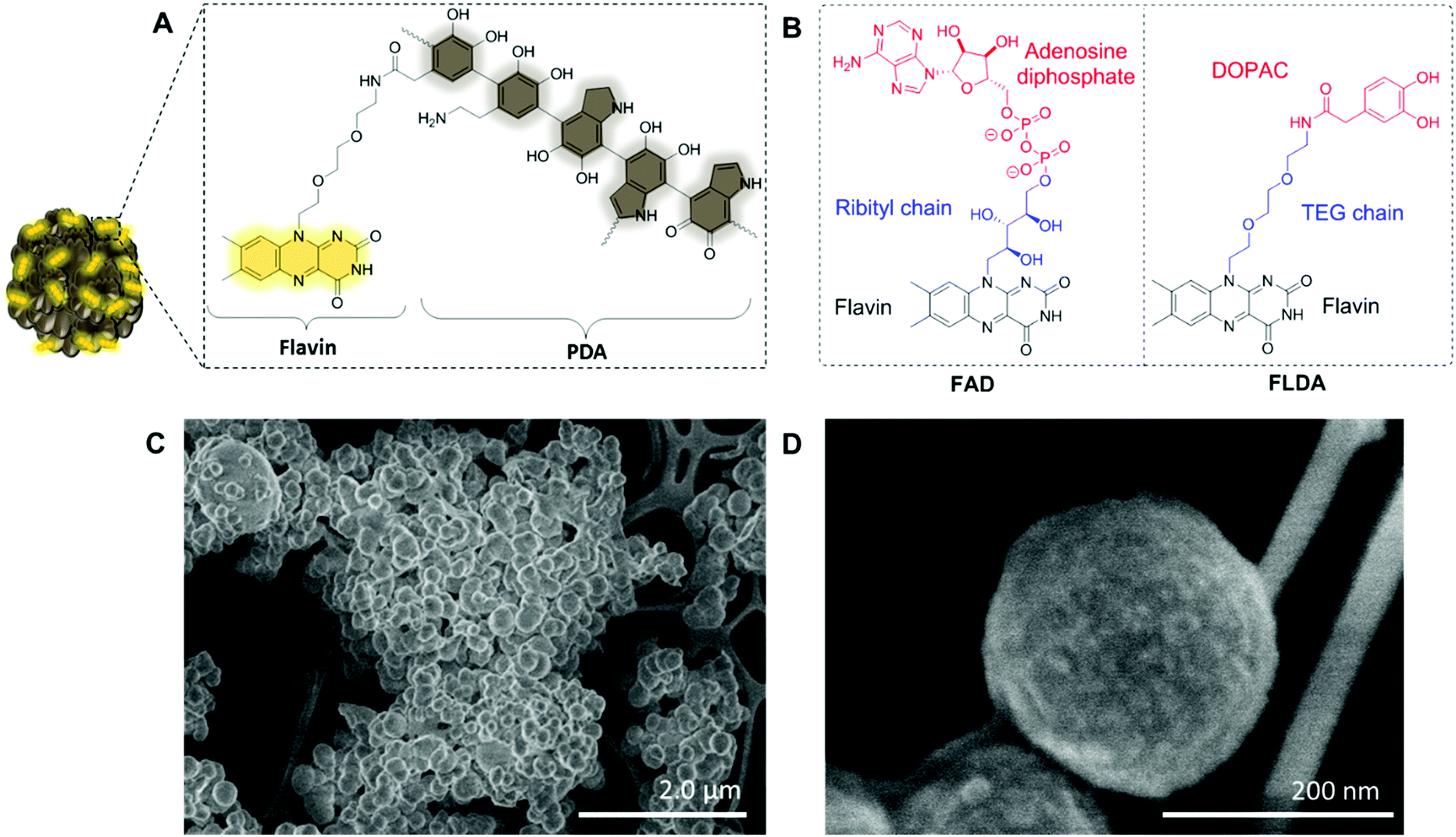 | ||
| Fig. 1 (A) Illustration of flavin–polydopamine (FLPDA) NPs highlighting the polydopamine structure. (B) Comparison of naturally occurring flavin cofactor FAD and FLDA synthesized in this paper. The STEM images of the NPs prepared using different ratios of FLDA and dopamine: (C) FLPDA-5, scale bar = 2 μm, (D) FLPDA-10, scale bar = 200 nm. | ||
To prepare the FLPDA NPs, a flavin–dopamine monomer (FLDA, Fig. 1B, right) was first prepared that mimics the structure of the naturally occurring FAD (Fig. 1B, left). The ribityl chain was replaced with a triethylene glycol (TEG) moiety, and adenosine diphosphate with the dopamine analogue, 3,4-dihydroxyphenylacetic acid (DOPAC) (see ESI,† Section 1.3 for synthetic details). Following the synthesis of a monomer, a copolymerization of FLDA with dopamine (DA) was performed at room temperature using a water/ethanol solvent system in the presence of ammonia as previously described for bare polydopamine (PDA) NP formation.39 As shown in Fig. 1C and D, spherical FLPDA NPs ranging from 100 to 200 nm were obtained depending on the ratio of used monomers.
In order to explore the effect of the amount of FLDA on the co-polymerisation and formation of NPs, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10:1 and 20:1 molar ratios of DA to FLDA were used, resulting in NPs referred to as FLPDA-5, FLPDA-10, and FLPDA-20 respectively. The prepared samples were washed extensively and lyophilized to avoid any thermal decomposition of the polymer that could occur through vacuum heating. They were noticeably browner in color than black unmodified PDA and their zeta potential values ranged from −35.7 to −41.5 mV for FLPDA-5 and FLPDA-20 respectively indicating high stability and dispersibility (see Table S1, ESI†). The size and shape of the FLPDA particles were further investigated by STEM (Fig. 1C and D and ESI†) showing that all the samples contained spherical NPs of similar size (approx. 200 nm) with relatively large size distribution (±∼50 nm). Some conjoined particles and the largest size distribution was observed in FLPDA-5 (see Table S1, ESI†). In contrast, PDA synthesized under the same conditions displays a narrow size distribution with an average size of 110 ± 18 nm (see Fig. S4, ESI†), which indicates that the presence of flavin moieties affects the polymerization mechanism and oligomer aggregation to form particles with less defined shape and size. This is most likely due to H-bonding and electrostatic interactions between the flavin group and oligomeric units, and our ongoing work is focused on optimizing the polymerization procedure further.
:1, 10:1 and 20:1 molar ratios of DA to FLDA were used, resulting in NPs referred to as FLPDA-5, FLPDA-10, and FLPDA-20 respectively. The prepared samples were washed extensively and lyophilized to avoid any thermal decomposition of the polymer that could occur through vacuum heating. They were noticeably browner in color than black unmodified PDA and their zeta potential values ranged from −35.7 to −41.5 mV for FLPDA-5 and FLPDA-20 respectively indicating high stability and dispersibility (see Table S1, ESI†). The size and shape of the FLPDA particles were further investigated by STEM (Fig. 1C and D and ESI†) showing that all the samples contained spherical NPs of similar size (approx. 200 nm) with relatively large size distribution (±∼50 nm). Some conjoined particles and the largest size distribution was observed in FLPDA-5 (see Table S1, ESI†). In contrast, PDA synthesized under the same conditions displays a narrow size distribution with an average size of 110 ± 18 nm (see Fig. S4, ESI†), which indicates that the presence of flavin moieties affects the polymerization mechanism and oligomer aggregation to form particles with less defined shape and size. This is most likely due to H-bonding and electrostatic interactions between the flavin group and oligomeric units, and our ongoing work is focused on optimizing the polymerization procedure further.
To validate the presence of flavin moieties, particles were analyzed by UV-Vis absorption and fluorescence spectroscopy. The UV-Vis spectrum of our FLDA monomer shows absorption bands at λmax = 445 nm and 373 nm corresponding to the transitions from the ground state (S0) to the S1 (λmax ∼ 442–450 nm) and S2 (λmax ∼ 360–375 nm) excited states (see Fig. S6A, ESI†).18 These bands are redshifted to ∼456 nm for the S0 → S1 transition and ∼376 nm for the S0 → S2 transition in FLPDA (Fig. S6B, ESI†). This can be explained by an increase in proton donation from PDA,40 and by the electron-withdrawing inductive effects on the flavin moieties due to incorporation into the highly conjugated PDA system.41
The fluorescence emission spectrum of FLDA is characterized by emission maxima at ∼527 nm (λex = 450 nm) which correlates well to other known flavin compounds (see Fig. S7A, ESI†).42 The FLPDA samples also exhibit this signal, confirming the presence of flavin moieties in the particles (see Fig. S7B, ESI†). An earlier study in which flavin compounds were incubated with melanin (structurally analogous to PDA) showed that the fluorescent properties of flavins did not change upon binding and there was no significant fluorescence quenching by the polymer.43 That study, however, focused on the non-covalent binding of the flavin derivatives to melanin, and not on the covalent attachment used in our work. In order to validate the assumption that flavin content can be indeed quantified via fluorescence measurements of the particles, a DOPAC-free flavin analogue, FLOH was prepared and compared to the DOPAC conjugated monomer FLDA (see Fig. S8A, ESI†). We observed that the fluorescence intensity was much lower for FLDA when compared to the same concentration of FLOH, despite the absorbance of the compounds being very similar. This observation indicates some intramolecular isoalloxazine-catechol fluorescence quenching similar to that found for FAD and adenosine due to a possible formation of a ‘closed’ structure (see Fig. S8B and C, ESI†).44 Kozik et al. suggested that the key phenolic and indolic groups in the melanin polymer do not incur any fluorescence quenching due to their inability to form CT complexes with the isoalloxazine ring.43 Although we initially found out that there was a fluorescence decrease when FLOH was incubated with the PDA NPs, this was no longer valid after we corrected for the absorbance of PDA at the excitation and emission wavelengths. The fluorescence spectrum shows a slight redshift to 527 nm similar to that of FLDA and FLPDA, but at the same intensity as FLOH alone (Fig. S9, ESI†). This demonstrates that the flavin moiety bound covalently to the polymer experiences the same quenching as the monomer flavin bound to the dopamine since the formation of the CT complex between isoalloxazine and other units appears to be negligible.
Taking this into account, a fluorescence calibration curve was obtained using known concentrations of FLDA to approximate the flavin concentration within the nanoparticles. As expected, FLPDA-5 contains a higher proportion of flavin than FLPDA-10 and FLPDA-20; approximately 1.11, 0.84, and 0.71 μmol mg−1FLPDA, respectively (see Fig. S10 and Table S2, ESI†). Clearly, the relative amounts of flavin do not exactly match the initial monomer ratios used for the synthesis. This can be explained by base-catalyzed cleavage and/or hydrolysis of some flavin moieties during the polymerization, which was previously reported for other flavins.45,46 The presence of flavin within the nanoparticles was additionally confirmed by FTIR measurements showing characteristic C–H stretching vibration bands at 2924 and 2855 cm−1, the sharp bands at 1545 cm−1 and 1580 cm−1 relating to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) modes in the isoalloxazine ring and flavin carbonyl ν(CO) and ν(CC) at 1711 and 1680 cm−1, respectively (see Fig. S11, ESI† for more details).
N) modes in the isoalloxazine ring and flavin carbonyl ν(CO) and ν(CC) at 1711 and 1680 cm−1, respectively (see Fig. S11, ESI† for more details).
Following the preparation and characterization of the FLPDA NPs, we set out to explore their photocatalytic activity. This was done by exploiting the fluorescent compound resorufin (RF, absorption λmax = 572 nm) and using the precursor compound Amplex Red (AR) to monitor oxidation, and resazurin (RZ) to monitor the reduction reactions (Scheme 1). In a typical experiment, 1–10 μg mL−1FLPDA NPs were incubated with AR (100 μM) in 10 mM KPi buffer at pH 7.4 (2 mL) and then irradiated with a blue laser diode (458 nm), monitoring the absorption spectrum in real time (see Fig. 2A and ESI† for set up details). Our hypothesis that FLPDA would be able to carry out photooxidation was confirmed by conversion of AR to RF upon irradiation with FLPDA-5 showing the highest conversion rate of 3.69 μM h−1 using an excitation intensity of 1.19 mW cm−2, due to the higher concentration of flavin moieties (see Fig. 2B). Control experiments were also performed at the same excitation intensity using the FLDA monomer, unmodified PDA and a blank sample containing AR without any catalyst. The latter was carried out as it has been previously reported that trace amounts of excited state RF can catalyse the oxidation of AR.47 In our case, however, the blank measurement showed a much slower rate of conversion (0.24 μM h−1) and no reaction was observed for PDA (10 μg mL−1) (Fig. 2B). The latter was expected due to high photon absorption capability of PDA, which has already been successfully utilized for the design of light-harvesting devices.48–50
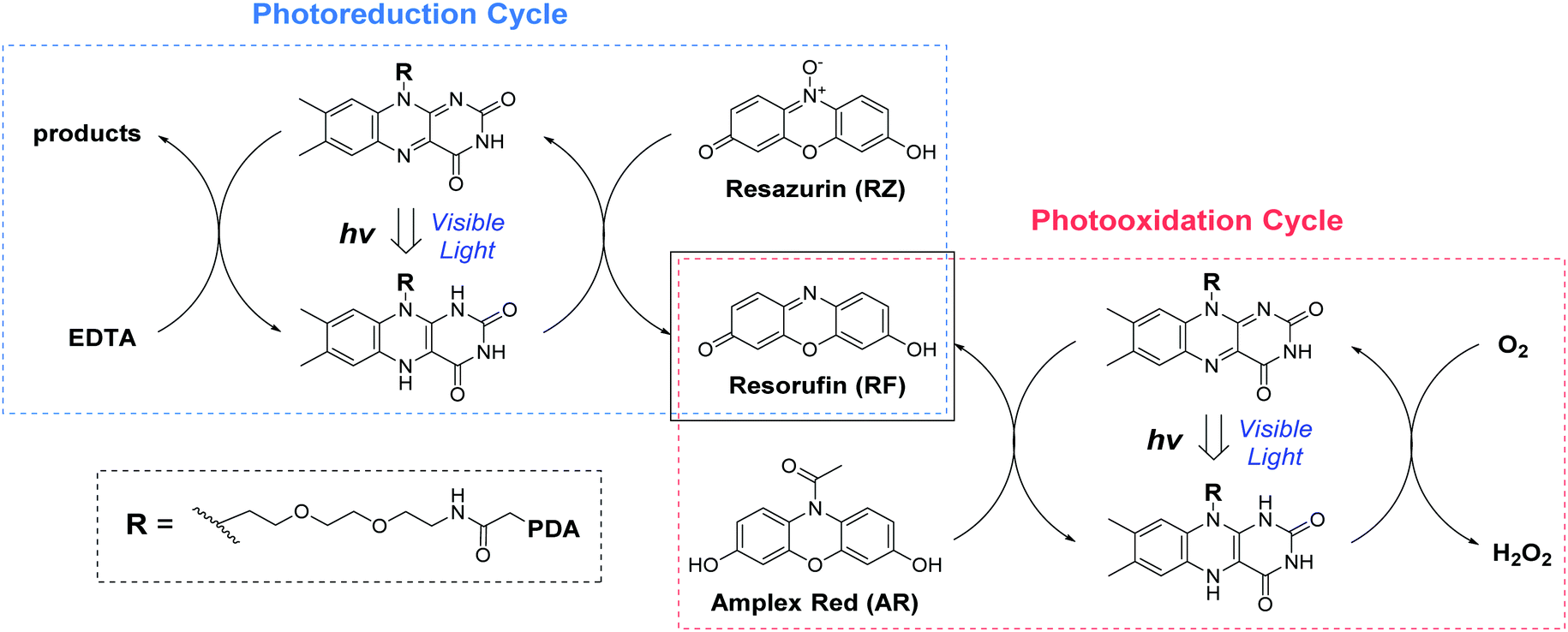 | ||
| Scheme 1 The photo-redox catalytic cycles of resorufin (RF) production starting from Amplex Red (AR photooxidation) and resazurin (RZ photoreduction) in the presence of FLPDA. | ||
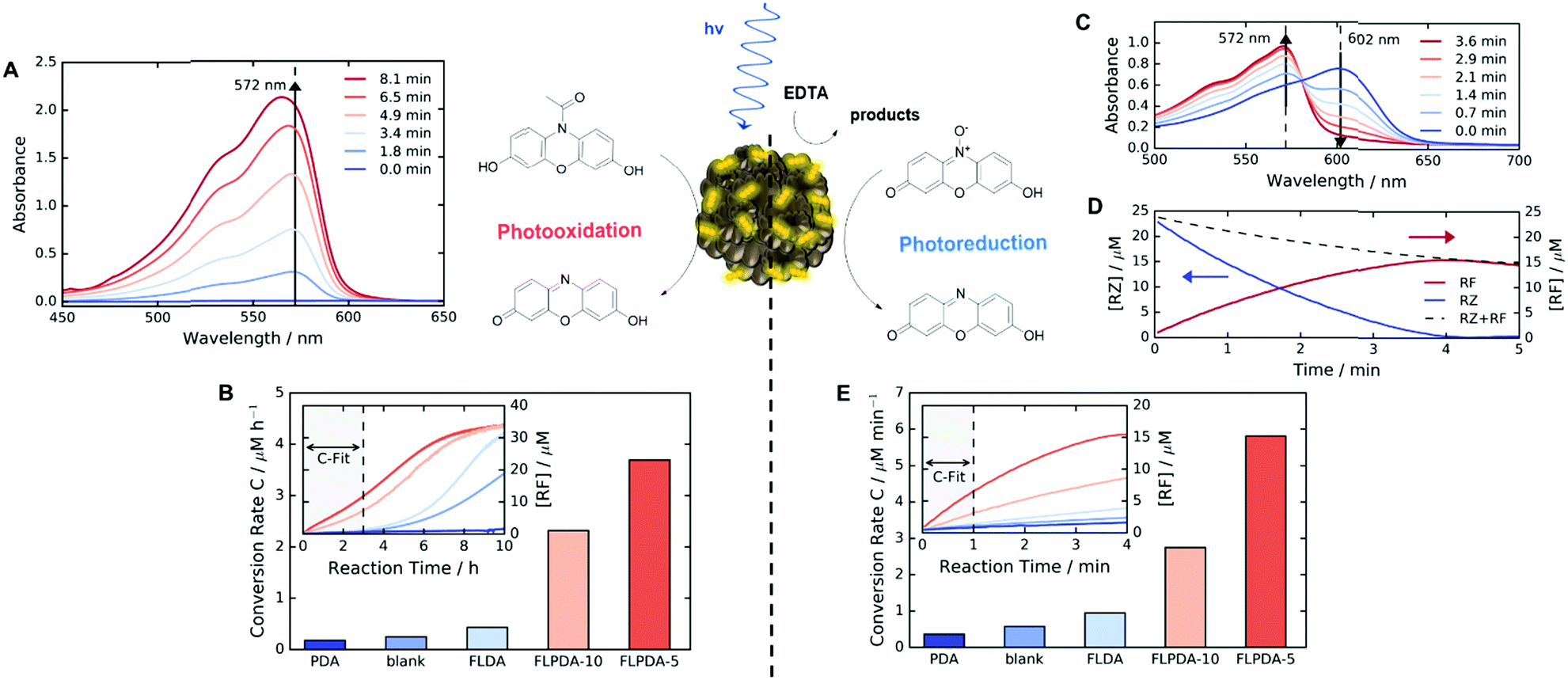 | ||
| Fig. 2 (A) Plot of the absorbance changes observed for the photooxidation of AR to RF by FLPDA-5 (10 μg mL−1) in KPi buffer (10 mM, pH 7.4). (B) Comparative plot of AR photooxidation in the presence of FLPDA photocatalysts and controls (10 μg mL−1, [FLDA] – 10 mol%). (C) Plot of the absorbance changes observed for the photoreduction of RZ to RF by FLPDA-5 (10 μg mL−1) in the presence of EDTA (100 μM) in KPi buffer (10 mM, pH 7.4). (D) Plot showing the relative consumption of RZ and production of RF by FLPDA-5 (10 μg mL−1) in the presence of EDTA (100 μM) in KPi buffer (10 mM, pH 7.4). (E) The comparative plot of RZ photoreduction in the presence of the FLPDA photocatalysts and controls (10 μg mL−1, [FLDA] – 10 mol%). All experiments were done using a 458 nm light source with 6.40 mW cm−2 excitation intensity, apart from (B) which was obtained using 2.95 mW cm−2 excitation intensity. | ||
When compared to FLPDA, a much slower reaction, with only 0.43 μM RF being produced per hour, was observed with the FLDA monomer (10 mol%). This large difference indicates a potential synergistic relationship between PDA and flavin within the hybrid material, which results in stabilization of reactive intermediates and/or enhancement of CT. To gain a better understanding of this effect, we further investigated the kinetic profile of AR photooxidation by FLPDA-5 using the Michaelis–Menten model. This showed that our system does display enzyme-like kinetic behaviour indicating substrate binding, leading to reaction and subsequent product dissociation. The Km value obtained from this fit was 166.7 μM with Vmax of 1.66 μM min−1 (see Fig. S12A and B, ESI†). Compared to enzymes with high specificity to Amplex Red, such as horseradish peroxidase (HRP), these values indicate less specificity and efficiency (6.4 μM and 2.2 μM min−1 respectively) but still in a range to be considered as an effective nanozyme.51 Overall, we envisage that aromatic stacking between AR, PDA and flavin allows favourable orientation for catalysis, therefore, enhancing the activity of FLPDA over homogenous FLDA – similar to previously reported flavin–PPh3-functionalised gold nanoclusters.52 It can also be assumed that non-specific binding of AR to the PDA polymer alone is responsible for a decrease in the activity for FLPDA-10 (less flavin, more PDA) when compared with FLPDA-5 (less PDA, more flavin).
We also observed that higher laser power accelerates the conversion process, which clearly demonstrates the photocatalytic character of FLPDA as one would expect more absorbed photons to lead to higher activity (see Fig. S13, ESI†). Also, no reaction occurred between the catalyst and substrate in the dark over a period of >18 hours (see Fig. S14, ESI†). It should also be noted that the STEM images of the recovered NPs do not show significant changes in the shape or morphology after irradiation (see Fig. S5C and D, ESI†).
Photoreductive capabilities of our catalytic system were also investigated due to the importance of catalytic reduction reactions in organic chemistry. Although these reactions are often used to yield a range of synthons and feedstocks, they traditionally employ costly and toxic rare earth metals or hydride complexes as catalysts.53 Only recently, photo-biocatalytic approaches employing enzymes emerged as safer and milder alternatives. In one such example, reduction of the CC bond was achieved using old yellow enzyme homologue from B. subtilis and ethylenediaminetetraacetic acid (EDTA) as an electron donor.54 To explore the potential of our hybrid nanoparticles for photoreductions, we employed a blue dye resazurin (RZ, absorption λmax = 602 nm) as a substrate and EDTA as the electron donor to afford RF (Scheme 1).
Similarly to previous experiments, 1–10 μg mL−1FLPDA NPs were irradiated with a blue laser diode (458 nm) at different excitation intensities but now in the presence of RZ (25 μM) and EDTA (100 μM) in degassed 10 mM KPi buffer at pH 7.4 (2 mL) under N2 atmosphere. The spectral changes were then monitored in real time, following the decrease of RZ absorption at 602 nm and the increase of RF absorption at 572 nm (see Fig. 2C). Fast reduction of the RZ substrate and production of RF was observed showing complete conversion after 4 min of irradiation (Fig. 2D). The decrease in RF concentration after 4 min can be attributed to bleaching. Similar to the AR photooxidation reactions, the STEM images of the recovered NPs do not show significant changes in the shape or morphology (see Fig. S5E and F, ESI†).
Control runs were also performed (Fig. 2E) showing that PDA (10 μg mL−1) displayed almost no activity whereas, some conversion of RZ to RF (0.58 μM min−1 compared to 5.7 μM min−1 for FLPDA-5) was observed in the blank measurement containing EDTA, but no catalyst. As previously reported, this is due to the interaction of triplet state RZ and electron dense tertiary aliphatic amines.55 Irradiation of RZ without EDTA led to RZ bleaching and no conversion to RF (data not shown). As with our photooxidation assay, the conversion rate was doubled for FLPDA-5 compared to FLPDA-10 due to a larger number of flavin active sites. The same amount of FLDA monomer showed 6 times slower conversion rate (0.95 μM min−1) providing further evidence of the enhanced activity of FLPDA through the substrate–PDA–flavin interactions.
Furthermore, we investigated the switchability of our catalytic system using FLPDA in the presence of the AR substrate. A clear increase in absorption of the product RF at 572 nm is observed when the system is irradiated (ON state) and no further reaction takes place when the laser is switched off for 30 min (OFF state) as evidenced by the absorbance plateau (see Fig. 3). This demonstrates that the catalytic activity of FLPDA towards oxidation of the substrate is light-dependent and has an excellent capacity for temporal control, a feature that is widely sought after in green chemistry to minimize the cost and maximize the efficiency of a reaction.56 Another factor that is valuable in green catalysis is the recyclability of a catalyst. Accordingly, we tested the recyclability of FLPDA-5 in the RZ photoreduction reaction by removing the catalyst post-reaction via centrifugation. The catalyst was then washed and reused in subsequent reactions, maintaining good activity for up to 4 runs (see Fig. S15, ESI†). Due to the inherent losses experienced during catalyst recovery by washing/centrifugation our future work is focussed on developing a continuous flow system using immobilised FLPDA.
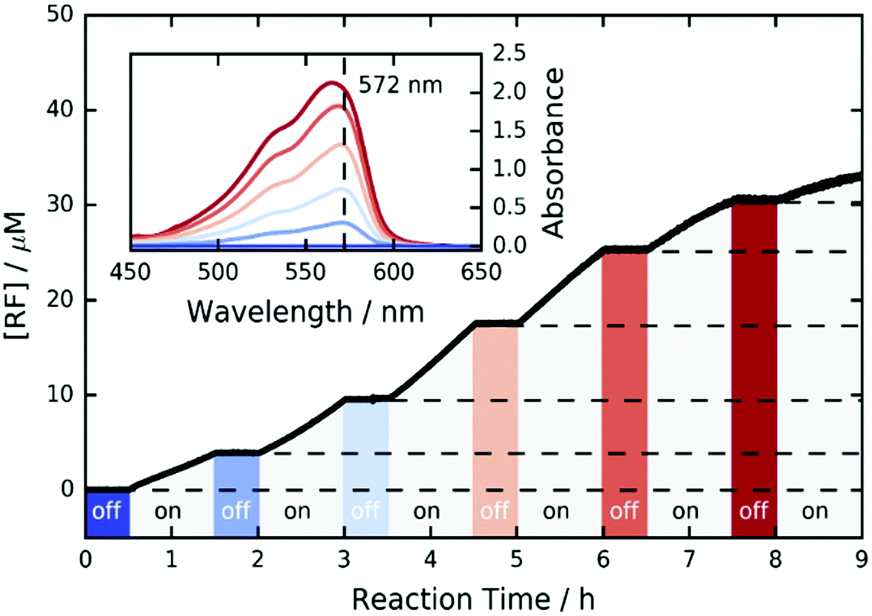 | ||
| Fig. 3 ON/OFF cycle data for the photooxidation of AR by FLPDA-5 (10 μg mL−1) in KPi Buffer (10 mM, pH 7.4) using 2.95 mW cm−2 excitation intensity (458 nm). | ||
The promising results regarding FLPDA's activity prompted us to explore the photodecomposition of the system. The photostability of flavin compounds is often an issue when these compounds are utilised in photocatalysis. Namely, upon irradiation the absorbance of flavin at ∼445 nm decreases due to dealkylation to either lumichrome or lumiflavin as well as scission of the alloxazine ring.57 To see if the same happens with dopamine-conjugates, we irradiated the monomer FLDA and the analogue FLOH alone in a solution using 458 nm light (50.3 mW) under N2 atmosphere and in the air over 30 min at pH 7.4. Remarkably, in both air and under N2 atmosphere, FLDA shows almost no photodecomposition when compared to FLOH (see Fig. S16, ESI†). This could be explained by possible CT between alloxazine and catechol in a ‘closed’ conformation akin to FAD as rationalised based on our fluorescence data. The decrease in absorbance was far greater for FLOH in the air when compared to N2, most likely due to the photoinduced ROS formation increasing the rate of decomposition.
The same study was also performed for FLPDA-5 by recording the UV-Vis absorbance and fluorescence spectra of the particles before and after 30 minutes irradiation, as well as the supernatant after centrifugation of the particles (see Fig. S17, ESI†). The absorbance spectra of the particles in both air and nitrogen still showed the presence of characteristic flavin bands (Fig. S17A, ESI†). However, the fluorescence of the particles after the reaction had decreased (Fig. S17B, ESI†). Analysis of the supernatants showed emission at ∼524 nm most likely indicating the presence of the lumiflavin indicating that some flavins were cleaved (Fig. S12B, ESI†).57 This could be remedied by the inclusion of a sacrificial electron donor such as EDTA which effectively limited the extent of this degradation (see Fig. S17A and B, ESI†).58 Nevertheless, it is clear that the incorporation of PDA does enhance the photostability of the flavin moieties through the formation of CT complexes increasing the recyclability. Our current work is looking at different dopamine–flavin conjugation sites and the possible effects this could have on both the catalytic activity and the photodecomposition.
Finally, considering the potential uses of such a system in nanomedicine and water remediation, we performed a preliminary investigation of the inherent toxicity of the FLPDA nanoparticles using three in vitro assays. First, FLPDA was PEGylated to increase the stability in cell culture media (DMEM) and reduce immunogenicity when applied in vivo applications.59 This was achieved through the covalent attachment of amino-terminated mPEG5000 to FLPDA by incubation in 10 mM TRIS buffer at pH 8.5 for 18 h. Cell viability was investigated via an MTS assay after 24 h of incubation with the particles. As shown in Fig. 4, (black diamonds) PEG-FLPDA has a negligible effect on the cell proliferation over a wide dosage range until the concentration of particles is significantly increased (to 1.0 mg mL−1) at which point the viability is reduced to 54.6%. We also tested the cytotoxicity of PEG-FLPDA using a lactate dehydrogenase (LDH) assay, which measures the release of LDH and signals significant cell damage. This was only observed for very high concentration dosages of 1.0 mg mL−1 after incubation for 24 h (Fig. 4, blue dots), correlating well to the MTS assay.
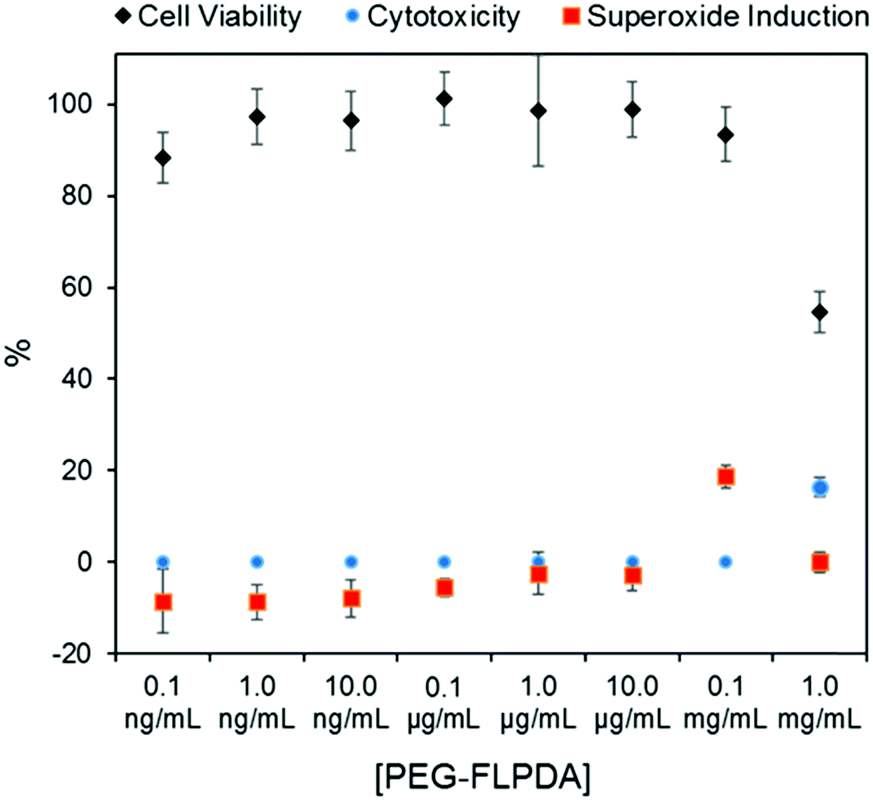 | ||
| Fig. 4 Summary of in vitro assay data for A549 cells showing cell viability (via MTS assay), cytotoxicity (via LDH assay) and superoxide generation in the presence of PEG-FLPDA after 24 h. | ||
To complement these assays, we additionally explored the effect of PEG-FLPDA on the cell oxidative stress using a superoxide detection assay to monitor the induction or scavenging of superoxide within the cells due to the presence of the particles. The data in Fig. 4 (red squares) show that the amount of superoxide decreases (relative to untreated controls) upon incubation with up to 10.0 μg mL−1PEG-FLPDA. This indicates that FLPDA has antioxidant properties similar to PDA and melanin.60–62 However, at a concentration of 0.1 mg mL−1, incubated cells showed an increased accumulation of superoxide, although this seems not to impact the viability of the cells. Decrease of the superoxide production again at 1.0 mg mL−1 is correlated to the widespread cell death at this high concentration. However, these three assays clearly indicate that the FLPDA NPs are non-toxic below concentrations of 1.0 mg mL−1, and even show some antioxidative effect below 0.1 mg mL−1. These first studies indicate the high potential of these flavin-containing hybrid systems not only for photocatalysis but also for potential use in water remediation and biomedical applications.
Conclusions
In conclusion, we have designed a novel, effective and versatile organic photocatalyst based on the combination of two natural species, flavin and polydopamine. Such an enzyme-inspired catalyst can be prepared using a straightforward, green chemistry protocol, is easy to purify and handle, can be used both for photooxidation and photoreduction, and can be activated on demand. To our knowledge, this is the first example of an organic polymer nanoparticle with embedded flavin being used as a photocatalyst for oxidation and reduction of the organic species. In vitro assays performed to demonstrate the non-toxicity of the system have also indicated some antioxidative properties, prompting further studies exploring the use of this material for biomedical applications. We believe that this enzyme-inspired material could find application beyond organic catalysis, in particular within biomedical research as an effective antioxidant adjuvant or drug nanocarrier, and in combination with suitable substrates in the design of organic photovoltaic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC DTC Studentship (LC) and Newton Trust Postdoctoral Funding (AK). TE acknowledges the support from the Winton Programme for the Physics of Sustainability and the Isaac Newton Trust. PK acknowledges the Cambridge NanoDTC (EPSRC Grant EP/L015978/1). We wish to thank the staff of NMR and MS facilities in the Department of Chemistry, the University of Cambridge for their assistance, and the EM suite staff in the Department of Physics, University of Cambridge for their help with STEM imaging.References
- M. T. Reetz, Chem. Rec., 2016, 16, 2449–2459 CrossRef CAS PubMed
.
- S. Li, X. Yang, S. Yang, M. Zhu and X. Wang, Comput. Struct. Biotechnol. J., 2012, 3, e201209017 CrossRef PubMed
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS
-
F. H. Arnold, Angew. Chem., Int. Ed., 2017, 57, 4143–4148 Search PubMed
-
R. Breslow, Artificial Enzymes, Wiley-VCH, Weinheim, 2006 Search PubMed
- E. Kuah, S. Toh, J. Yee, Q. Ma and Z. Gao, Chem. – Eur. J., 2016, 22, 8404–8430 CrossRef CAS PubMed
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC
- X. Wang, Y. Hu and H. Wei, Inorg. Chem. Front., 2016, 3, 41–60 RSC
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed
- L. Pasquato, P. Pengo and P. Scrimin, Supramol. Chem., 2005, 17, 163–171 CrossRef CAS
- W. Zhang, J. Nanopart. Res., 2003, 5, 323–332 CrossRef CAS
- V. Sajith, C. B. Sobhan and G. P. Peterson, Adv. Mech. Eng., 2010, 2, 581407 CrossRef
- J. Golchin, K. Golchin, N. Alidadian, S. Ghaderi, S. Eslamkhah, M. Eslamkhah and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2017, 45, 1069–1076 CrossRef CAS
- M. A. Maurer-Jones, I. L. Gunsolus, C. J. Murphy and C. L. Haynes, Anal. Chem., 2013, 85, 3036–3049 CrossRef CAS PubMed
- A. Sukhanova, S. Bozrova, P. Sokolov, M. Berestovoy, A. Karaulov and I. Nabiev, Nanoscale Res. Lett., 2018, 13, 1–21 CrossRef CAS PubMed
- B. Liu and J. Liu, Nano Res., 2017, 10, 1125–1148 CrossRef CAS
- C. Walsh, Acc. Chem. Res., 1980, 13, 148–155 CrossRef CAS
- P. F. Heelis, Chem. Soc. Rev., 1982, 11, 15–39 RSC
- V. Massey, Biochem. Soc. Trans., 2000, 28, 283–296 CrossRef CAS PubMed
-
E. Silva and A. M. Edwards, Flavins Photochemistry and Photobiology, Royal Society of Chemistry, Cambridge, 2006 Search PubMed
- B. König, S. Kümmel, E. Svobodová and R. Cibulka, Phys. Sci. Rev., 2018, 3, 20170168 Search PubMed
- H. Schmaderer, P. Hilgers, R. Lechner and B. König, Adv. Synth. Catal., 2009, 351, 163–174 CrossRef CAS
- J. Špačková, E. Svobodová, T. Hartman, I. Stibor, J. Kopecká, J. Cibulková, J. Chudoba and R. Cibulka, ChemCatChem, 2017, 9, 1177–1181 CrossRef
- M. Kurfiřt, J. Špačková, E. Svobodová and R. Cibulka, Monatsh. Chem., 2018, 149, 863–869 CrossRef
- B. J. Jordan, G. Cooke, J. F. Garety, M. A. Pollier, N. Kryvokhyzha, A. Bayir, G. Rabani and V. M. Rotello, Chem. Commun., 2007, 1248–1250 RSC
- Y. Arakawa, K. Minagawa and Y. Imada, Polym. J., 2018, 50, 941–949 CrossRef CAS
- Y. Chevalier, Y. L. T. Ki, D. L. Nouen, J. P. Mahy, J. P. Goddard and F. Avenier, Angew. Chem., Int. Ed., 2018, 57, 16412–16415 CrossRef CAS PubMed
- S. Hong, K. Y. Kim, H. J. Wook, S. Y. Park, K. D. Lee, D. Y. Lee and H. Lee, Nanomedicine, 2011, 6, 793–801 CrossRef CAS PubMed
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384–9395 CrossRef CAS PubMed
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed
- Z. Yang, J. Sun, X. Liu, Q. Su, Y. Liu, Q. Li and S. Zhang, Tetrahedron Lett., 2014, 55, 3239–3243 CrossRef CAS
- R. Mrówczyński, A. Bunge and J. Liebscher, Chem. – Eur. J., 2014, 20, 8647–8653 CrossRef PubMed
- Y. Du, H. C. Yang, X. L. Xu, J. Wu and Z. K. Xu, ChemCatChem, 2015, 7, 3822–3825 CrossRef CAS
- J. H. Kim, M. Lee and C. B. Park, Angew. Chem., Int. Ed., 2014, 53, 6364–6368 CrossRef CAS PubMed
- A. Kunfi, V. Szabó, Á. Mastalir, I. Bucsi, M. Mohai, P. Németh, I. Bertóti and G. London, ChemCatChem, 2017, 9, 3236–3244 CrossRef CAS
- L. Wang, Z.-Y. Hu, X.-Y. Yang, B.-B. Zhang, W. Geng, G. Van Tendeloo and B.-L. Su, Chem. Commun., 2017, 53, 6617–6620 RSC
- T. Mathes, I. H. M. Van Stokkum, M. Stierl and J. T. M. Kennis, J. Biol. Chem., 2012, 287, 31725–31738 CrossRef CAS PubMed
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. Lu, Adv. Mater., 2013, 25, 998–1003 CrossRef CAS PubMed
- A. Kotaki, M. Naoi and K. Yagi, J. Biochem., 1970, 68, 287–292 CrossRef CAS PubMed
- L. N. Mataranga-Popa, I. Torje, T. Ghosh, M. J. Leitl, A. Späth, M. L. Novianti, R. D. Webster and B. König, Org. Biomol. Chem., 2015, 13, 10198–10204 RSC
- A. Kotaki and K. Yagi, J. Biochem., 1970, 68, 509–516 CrossRef CAS PubMed
- A. Kozik, W. Korytowski, T. Sarna and A. S. Bloom, Biophys. Chem., 1990, 38, 39–48 CrossRef CAS PubMed
- G. Weber, Biochem. J., 1950, 47, 114–121 CrossRef CAS PubMed
- S. B. Smith and T. C. Bruice, J. Am. Chem. Soc., 1975, 14, 2875–2881 CrossRef
- T. Harayama, Y. Tezuka, T. Taga and F. Yoneda, J. Chem. Soc., Perkin Trans. 1, 1987, 75–83 RSC
- F. A. Summers, B. Zhao, D. Ganini and R. P. Mason, Methods Enzymol., 2013, 526, 1–17 CAS
- M. Lee, J. U. Kim, J. S. Lee, B. Il Lee, J. Shin and C. B. Park, Adv. Mater., 2014, 26, 4463–4468 CrossRef CAS PubMed
- A. Xie, K. Zhang, F. Wu, N. Wang, Y. Wang and M. Wang, Catal. Sci. Technol., 2016, 6, 1764–1771 RSC
- M. Lee, J. U. Kim, K. J. Lee, S. Ahn, Y. B. Shin, J. Shin and C. B. Park, ACS Nano, 2015, 9, 6206–6213 CrossRef CAS PubMed
- H. S. Rho, A. T. Hanke, M. Ottens and H. Gardeniers, PLoS One, 2016, 11, e0153437 CrossRef PubMed
- Y. Imada, M. Osaki, M. Noguchi, T. Maeda, M. Fujiki, S. Kawamorita, N. Komiya and T. Naota, ChemCatChem, 2015, 7, 99–106 CrossRef CAS
-
R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed
- M. M. Grau, J. C. Van Der Toorn, L. G. Otten, P. Macheroux, A. Taglieber, F. E. Zilly, I. W. C. E. Arends and F. Hollmann, Adv. Synth. Catal., 2009, 351, 3279–3286 CrossRef CAS
- C. Bueno, M. L. Villegas, S. G. Bertolotti, C. M. Previtali, M. G. Neumann and M. V. Encinas, Photochem. Photobiol., 2002, 76, 385–390 CrossRef CAS PubMed
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC
- M. A. Sheraz, S. H. Kazi, S. Ahmed, Z. Anwar and I. Ahmad, Beilstein J. Org. Chem., 2014, 10, 1999–2012 CrossRef PubMed
- S. Alonso-De Castro, E. Ruggiero, A. Ruiz-De-Angulo, E. Rezabal, J. C. Mareque-Rivas, X. Lopez, F. López-Gallego and L. Salassa, Chem. Sci., 2017, 8, 4619–4625 RSC
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed
- Y. Liu, K. Ai, X. Ji, D. Askhatova, R. Du, L. Lu and J. Shi, J. Am. Chem. Soc., 2017, 139, 856–862 CrossRef CAS PubMed
- M. D’Ischia, A. Napolitano, V. Ball, C. T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed
- K. Y. Ju, Y. Lee, S. Lee, S. B. Park and J. K. Lee, Biomacromolecules, 2011, 12, 625–632 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00199a |
| This journal is © The Royal Society of Chemistry 2019 |