
Recent advances in two-dimensional materials and their nanocomposites in sustainable energy conversion applications
Karim
Khan†
 *abcde,
Ayesha Khan
Tareen†
e,
Muhammad
Aslam†
e,
Yupeng
Zhang
cd,
Renheng
Wang
cd,
Zhengbiao
Ouyang
*bc,
Zhongyi
Gou
*a and
Han
Zhang
*cd
*abcde,
Ayesha Khan
Tareen†
e,
Muhammad
Aslam†
e,
Yupeng
Zhang
cd,
Renheng
Wang
cd,
Zhengbiao
Ouyang
*bc,
Zhongyi
Gou
*a and
Han
Zhang
*cd
aAdvanced electromagnetic function laboratory, Dongguan University of Technology (DGUT), Dongguan, Guangdong Province, P.R. China. E-mail: karim_khan_niazi@yahoo.com; guozhongyi@hfut.edu.cn
bCollege of Electronic Science and Technology, and THz Technical Research Center of Shenzhen University, Shenzhen, 518060, P.R. China
cKey Laboratory of Optoelectronics Devices and Systems of Ministry of Education and Guangdong Province Shenzhen University, Shenzhen, 518060, P.R. China. E-mail: wangrh@szu.edu.cn; zbouyang@szu.edu.cn; hzhang@szu.edu.cn
dShenzhen Engineering Laboratory of Phosphorene and Optoelectronics, and SZU-NUS Collaborative Innovation Center for Optoelectronic Science and Technology, Shenzhen University, Shenzhen, 518060, P.R. China
eGovernment Degree college PaharPur, Gomel University, Dera Ismail Khan, K.P.K., Islamic Republic of Pakistan. E-mail: chemistayesha@yahoo.com
First published on 8th November 2019
Abstract
Two-dimensional (2D) materials have a wide platform in research and expanding nano- and atomic-level applications. This study is motivated by the well-established 2D catalysts, which demonstrate high efficiency, selectivity and sustainability exceeding that of classical noble metal catalysts for the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and/or hydrogen evolution reaction (HER). Nowadays, the hydrogen evolution reaction (HER) in water electrolysis is crucial for the cost-efficient production of a pure hydrogen fuel. We will also discuss another important point related to electrochemical carbon dioxide and nitrogen reduction (ECR and N2RR) in detail. In this review, we mainly focused on the recent progress in the fuel cell technology based on 2D materials, including graphene, transition metal dichalcogenides, black phosphorus, MXenes, metal–organic frameworks, and metal oxide nanosheets. First, the basic attributes of the 2D materials were described, and their fuel cell mechanisms were also summarized. Finally, some effective methods for enhancing the performance of the fuel cells based on 2D materials were also discussed, and the opportunities and challenges of 2D material-based fuel cells at the commercial level were also provided. This review can provide new avenues for 2D materials with properties suitable for fuel cell technology development and related fields.
 Karim Khan | Dr Karim Khan (https://orcid.org/0000-0001-8689-9245) was born in Paharpur, Dera-Ismail-Khan, K. P. K., Pakistan. He received his B.Sc. degree (2005) from Gomal University, M.Sc (2009) in Physics from the University of Peshawar, M. Phil (2012) in Solid State Physics from the University of Punjab, Pakistan, and was a researcher at the “National Centre for Physics”, Quaid-e-Azim University, Pakistan. He obtained his Ph.D. (2016) from the Chinese Academy of Sciences and also served as a Postdoctoral Fellow (2019) in Shenzhen University, China. To date, he has published over 45 scientific publications and 2 patents. His current research focuses on 2D electrocatalyst materials and their applications in energy producing/storage/saving devices as well as the development of sensors for biomedical applications. |
 Ayesha Khan Tareen | Dr Ayesha Khan Tareen (https://orcid.org/0000-0002-7238-6366) is a Post-Doctoral fellow in Shenzhen University and completed her Ph.D. (Physical chemistry/Materials) in 2015–2018 from the Ningbo Institute Of Material Technology and Engineering (NIMTE), Chinese Academy Of Sciences (CAS), M. Phil (Analytical Chemistry) in 2013–2015 from “The Islamia University of Bahawalpur” and B.Sc. (Honors) in Chemistry in 2008–2012 from the Government College University Lahore, Pakistan. Her research interests are magnetic/superconductivity and transition metal nitride material synthesis and applications in different directions, especially for magnetic materials, photocatalysts, fuel cells, and dye-sensitized solar cells (DSSC). |
 Muhammad Aslam | Muhammad Aslam received his B.Sc. in Physics and Mathematics (Physical Sciences, 2018) from the Government Degree College Paharpur, Gomal University, Dera-Ismail-Khan, K. P. K., Pakistan and is currently pursuing Master of Science (M.Sc.) in Physics. His current research focuses on 2D materials and their applications in energy producing/storage devices as well as the development of sensors for biomedical application. |
 Yupeng Zhang | Dr Yupeng Zhang (https://orcid.org/0000-0003-2351-5579) received his PhD from Wuhan University. From 2014 to 2017, he was a Postdoctoral Research Fellow at Monash University. Currently, he is a Research Fellow at the College of Physics and Optoelectonic Engineering, Shenzhen University. His research focuses on the synthesis and application of nanomaterials and composites for energy production, saving, and storage, such as OLEDs and high-power/high-energy lithium-ion batteries. |
 Renheng Wang | Dr Renheng Wang received his Ph.D. degree in Metallurgical Engineering from Central South University (CSU) in 2015. From January 2016 to October 2018, he worked as a postdoctoral fellow at Shenzhen University and Nanyang Technological University. He is now a researcher in the College of Physics and Optoelectonic Engineering, Shenzhen University. His research focuses on the synthesis and application of nanomaterials and composites for clean energy storage, such as high-power/high-energy lithium-ion batteries. |
 Zhengbiao Ouyang | Prof. Dr Zhengbiao Ouyang (https://orcid.org/0000-0002-1466-7813) received his B.E. from Harbin Institute of Technology, China, in 1983 and Ph.D. from the University of Electronic Science and Technology of China in 1988. He is currently a Professor at Shenzhen University, Vice Director of the THz Technical Research Center of Shenzhen University, and Vice Director of the Shenzhen Key Laboratory of Micro-Nano Photonic Information Technology. He is a member of the Council of the Chinese Society for Optical Engineering, a Committeeman of the Opt-Electronic Committee of the Chinese Space Aeronautics Association and was a Committeeman of the Adviser Committee for Higher Education in Electronic Science and Technology in the Chinese Ministry of Education (2005–2010). He was awarded 2nd prize of Progress in Science and Technology from the Chinese Ministry of Machine and Electronics in 1990, Alexander-von-Humboldt Fellowship in 1991, and Guangdong Province-Level Scholar in 2007. His research interests focus on photonic crystal materials and their applications in optical integrated devices. |
 Han Zhang | Prof. Dr Han Zhang (https://orcid.org/0000-0002-0166-1973) received his B.S. degree from the Wuhan University in 2006 and Ph.D. from the Nanyang Technological University (NTU) in 2010. He is currently a Director of the Shenzhen Key Laboratory of 2D-Materials and Devices and the Shenzhen Engineering Laboratory of Phosphorene and Optoelectronics, Shenzhen University. His current research focus is the ultrafast and nonlinear photonics of 2D materials. |
1. Introduction
The incidence of technological evolution has augmented from preceding centuries and will perhaps increase to a larger extent in the 21st century and beyond. Currently, in the age of nanotechnology, the search for economical and sustainable energy alternatives for the dwindling fossil fuel reserves has significantly increased.1 The growing global environmental concerns caused by burning fossil fuels demand clean and green energy such as fuel cells that can generate energy efficiently from clean fuels (H2 and methanol). The safe and sustainable future of humanity can be ensured by making new innovations and modifications to the existing energy generation and storage technologies. Worldwide, the energy systems for domestic and industrial applications are going through rapid transitions, which will result in important changes in the traditional energy-producing fuels. The preservation of our future environment and generating large-scale sustainable energy systems are the critical challenges faced today by the 21st century humanity. In the case of energy producing methods, the term “sustainable energy” can be interchanged with the term “renewable energy”. Since the successful application of fuel cells in the Apollo lunar mission, great progress has been made in their commercialization. The increasing world population and industrialization have enhanced the worldwide power demand, which will reach 24 or 26 TW underneath “new and old policies” scenarios by 2040, and the corresponding CO2 emissions will increase to 37 or 44 Gt per year till 2040.2 Therefore, the primary concerns are energy production systems, specifically the climate change due to the use of traditional fossil fuels in energy production. In nature and technology, electrochemical creation and the use of fuels are the most competent ways for energy conversion and environmental protection from pollution. Therefore, the most admirable method by which sustainable energy can be produced and stored is the use of renewable energy sources to produce liquid fuels, “clean hydrogen”, which can be used in portable infrastructures. The chemical bond construction/pattern is essentially the mode of energy storage in materials. Thus, serious momentum exists to expand traditional power supplies, minimizing the dependence on fossil fuels by converting to sustainable energy, especially to fuel cells; however, other renewable energy sources such as solar, wind, and hydroelectric power have some limitations. The superior access to reproducible electricity is very significant, where generally the electricity division accounts for around 12% of the global energy requirement. Additional key energy divisions involving the growth of sustainable energy consist of the transportation (light-duty vehicles, commercial transportation, including rail, heavy-duty road vehicles, marine, and aviation) and chemical industries. The energy requirement for light-duty transportation will probably remain relatively unchanged in the upcoming years; however, that for commercial shipping is predicted to increase by about two-thirds, i.e., 3.2 TW by 2040. Because chemical fuels are considered to be more suitable in this division, the development of sustainable ways to produce clean energy fuels has attracted significant attention. To meet the worldwide requirement for products such as fertilizers, plastics, and other chemicals, the industrial energy requirement is also predicted to increase by two-thirds by 2040 to 2.5 TW.3 Thus, renewable energy for consumption and the production of industrial chemicals is of worldwide significance, such as H2 (50 MT per year), H2O2 (2.2 MT per year), ethylene (C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H4) (115 MT per year), C3H6 (73 MT per year), CH3OH (40 MT per year), and NH3 (175 MT per year), which have a significant role in CO2 emission.4,5 Therefore, the most important direction for the production of renewable energy that can fulfill the future energy requirements is fuel cells.
H4) (115 MT per year), C3H6 (73 MT per year), CH3OH (40 MT per year), and NH3 (175 MT per year), which have a significant role in CO2 emission.4,5 Therefore, the most important direction for the production of renewable energy that can fulfill the future energy requirements is fuel cells.
In fuel cells, fuels are oxidized on the anode, and released electrons transfer through an external circuit to the cathode where O2 is reduced. There are two major energy producing cycles, which rely on carbon and water (H2O) and have been extensively researched. The hydrolysis of H2O is caused hydrogen and oxygen evolution at the cathode (HER) and anode (OER), respectively, in the electrolytic cell. In the opposite process, the H2 (acts as fuel) produced at the anode is oxidized (HOR), while oxygen at the cathode is reduced (ORR) in H2/air fuel cell to produce electricity, with H2O as the major product. The HER and HOR proceed at kinetically high rates; whereas the ORR is slow because it entails multiple electron–proton coupled transfer steps. The combustion of fossil fuels releases CO2, which when emitted by carbonaceous materials can be recycled through the carbon cycle for utilization in the production of fuels (e.g., methanol, methane, and ethanol). Although well-known catalysts such as nanoscale Pt and costly metal oxides (e.g., RuO2 and IrO2) show high catalytic ability, their scarcity and high cost restrict their industrial applications. Thus, new NPMCs with confined dimensions (2D) have been introduced, but their atomic level understanding is still an ongoing debate.1,6–9
To fulfill future electrochemical energy demands, it is crucial to explore advanced electrocatalysts with well-controlled nanostructures for dramatic enhancements in energy and power density, catalytic activity, efficiency, and durability.1,6–10 One renewable energy generation/distribution route called the hydrogen economy concept is receiving significant attention from researchers. The hydrogen economy concept utilizes H2 gas as the main energy source for the efficient running of buildings, homes and vehicles. Unfortunately, due to the many inefficiencies associated with the energy conversion device (electrolyzer) needed to produce pure H2, compatible fuel cell devices to convert H2 into electricity and store the generated electricity are necessary (batteries and supercapacitors (SC)). Furthermore, the development of cheap and active catalysts that rival the state-of-the-art materials for these devices will make the hydrogen economy concept closer to reality. Therefore, the need for alternative, abundant, and financially feasible electrode materials is urgent. 2D materials compared to their bulk counterparts show improved activity as electrocatalysts.11 The energy crisis and global challenges provide excellent opportunities for designing and exploring new electrocatalysts.1,6–10 Electrocatalysis is influenced by the electrode materials, kinetics, and adsorption/desorption at the electrode/electrolyte interface, and it assists in the conversion of chemical and electrical energy in energy conversion and storage devices. Three seemingly simple reactions, the ORR, OER and HER, are significant for green and sustainable energy-producing devices, e.g., fuel cells, water-electrolysis devices, and batteries.12 Accordingly, cost-efficient, alternative, 2D nanosheets materials with large active sites are beneficial for prompt interfacial charge transfer with feasible catalysis. Among the 2D materials, graphene and its derivatives, carbides, nitrides, chalcogenides, and non-noble metal oxides introduce a new generation of electrocatalysts. These 2D materials can act as cathode materials in electrochemical reactions, and reduce the largest portion of electrochemical kinetic loss and system costs. Hence, this has promoted studies on 2D material-based electrocatalysts, and, to some extent, given rise to the recent research boom on the OER and HER for the CO2/N2 electrochemical reduction reactions (ECR/ENR). Most publications focused on discussing different approaches and strategies for the preparation of a specific class of ORR catalysts (e.g., Pt-based core–shell catalysts, nano-structured Pt-alloy electrocatalysts and doped-carbon materials), but few reviews are available regarding the electrocatalytic applications of 2D materials, only one or a few 2D families. In view of the rapid development of 2D electrocatalysts, there is a great need to provide timely updates in this field with emphasis on new design concepts and significant breakthroughs in electrocatalysts. Strategic applications of different families of 2D nanomaterials in electrocatalysis should be analyzed, focusing on the role of edges and surface characteristics. Since the last decade, attention on nanostructured materials (NSMs) has demonstrated that new characteristics are gained at the nanoscale, which change with a change in size or morphology because the size of NSMs determines novel and exciting aspects.13,14 In the middle of the 20th century, for the first time, Gleiter15 gave the classification of NSMs, which was promoted by Skorokhod.16 At that time, this classification was not complete since low-dimensional structures, i.e. fullerenes, nanoflakes and nanotubes, were not included because their effects determined by size are part of the evolution of the physicochemical features of these restricted systems with a change in size.17–20 With the development of new technologies, a new class of low-dimensional materials has resulted in the growth of nanoscale sciences, which are known as 2D materials. The fundamental material parameter is dimensionality, which decides their properties to a significant extent. 2D materials or compounds with the same chemical composition can exhibit different properties.21
Different dimension nanomaterials have different density of states (DOS) due to the number of different electrons present. For example, zero-dimensional (0D) quantum dots,23,24 one-dimensional (1D) nanoribbons, nanotubes, and nanowires, two-dimensional (2D) single-atom-thick materials, and three-dimensional (3D) nano-balls and nanocones (Fig. 1).21,25–42 For 0D NSMs, excited electrons are completely confined in all directions and they are equivalent to atomic/molecular clusters with discreet states, which are well-separated in dissimilar energy states. Michael Faraday in the middle of 19th century presented the first experiments with the nanomaterial colloidal gold, and determined its size-dependent optical properties.43 The 0D NSMs are fabricated via physical and chemical methods due to their well-ordered dimensions. Recently, 0D NSM heterogeneous particle arrays including uniform quantum dots, nanoplatelets, nano-onions, hollow spheres, and core–shell quantum dots have been developed with some potential application from the laboratory to the industrial scale.1,6–10,44–46 Furthermore, regarding the application of 0D materials, i.e. 0D quantum dots, they have been widely applied in solar cells,47 light emitting diodes (LEDs),48 lasers,49,50 and single-electron transistors.51 They are also used in routine applications such as pregnancy test-kits (gold NSMs), antibacterial bandages (silver NSMs), sun-screen lotions formulations (SiO2 NSMs or other oxide ceramic nanoparticles) as few examples of consumer products that contain 0D NSMs.
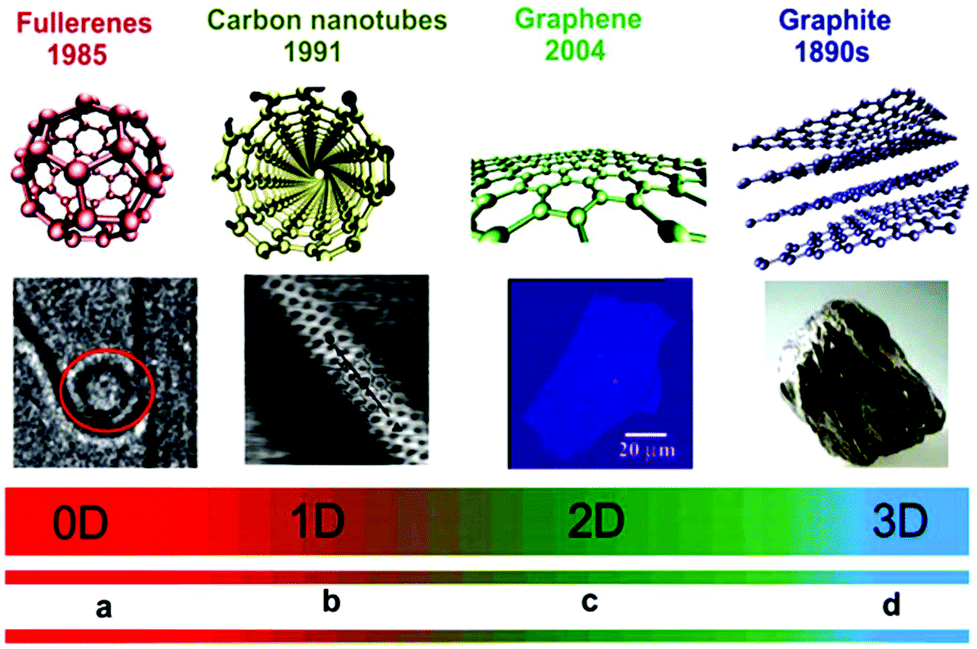 | ||
| Fig. 1 Dimensionality spectrum of sp2 carbon materials. (a) Structure model of a fullerene, where the red circle shows it trapped inside the cavity of an MWCNT. (b) Structure model of CNT and STM image of an SWNT exposed at the surface of a rope. (c) Structure model of graphene and a snapshot (ordinary white light) of a comparatively large graphene multilayer flake of ∼3 nm thickness on an Si wafer substrate. (d) Structure model of graphite and a piece of natural graphite. This figure has been adapted/reproduced from ref. 22 with permission from the Royal Society of Chemistry. | ||
Similarly, the second category of NSMs is 1D nanomaterials, which have nanoscale sizes along two dimensions with a rod- or wire-like appearance. In these nanomaterials, quantum confinement and SSA-related nanoscale effects, which are absent in 0D nanomaterials, are pronounced, allowing them to be integrated into device architectures due to their single bulk-like dimensions.21 1D nanostructured materials, such as nanowires and nanotubes, have attracted substantial attention after groundbreaking studies, especially the discovery of the good transparency with prominent conductivity for transparent and flexible devices in 2D sheet materials. In this review, we mostly focus on the applications of 2D materials; therefore, from here we will discuss in detail the different kinds of 2D materials and their potential industrial applications, especially the establishment of metrics that can facilitate definite targets to be to achieved for technology challenges in energy production and storage devices.
2. Two-dimensional materials and their structures
The search for basic knowledge constantly drives discoveries; thus, here we present a comprehensive introduction to future research guidelines on 2D materials and their potential industrial-scale applications. These discoveries will also serve to categorize key scientific information and landmarks wherever 2D materials will have a considerable effect in the near future. 2D-layered materials are considered a class of materials that are synthetically fabricated14,17,29,52 by scaling down bulk solids into sheets held together by van der Waals forces (vdW).53–58 In bulk materials, are atoms connected with each other through ionic or covalent bonds along the 2D in-plane direction to construct atomic-level structures, while these atomic structures are held together via vdW forces along a third-dimension (out-of-plane). These weak vdW forces within neighbouring layers cleave layers into discrete freestanding few or even single-atom-thick layers through mechanical techniques. More specifically, 2D NSMs are materials with a quasi 2D nanostructure consisting of a small number (<10) of stacked sheets.59 The potential for the new field of 2D, one-atomic-layer-thick materials is not only restricted to graphene. 2D nanomaterials were introduced with the discovery of the atomically thin carbon material called graphene, which possesses remarkable anisotropic physical and electronic properties.60 Interestingly, the library of 2D layered materials grows annually, and now it is consists of more than 150 interesting families. The research regarding 2D material is still in its budding stage with new materials being developed and added to the list each year.The potential of future advanced technologies ensures that materials can be manipulated at the nanometer level and outcomes from new tools and technologies not visualized previously, ranging from nanoparticle-based cancer therapy61,62 to efficient composites, and thus, nanotechnology launches new boundaries for ideas in medicine, electronic devices, etc.23,33,38,62–78 Advanced research and development activity on 2D materials have already shown that these materials are appropriate for large-scale manufacturing. The fabrication of 2D materials is one conspicuous example of the fast progress toward their commercialization, with the random discovery of micro- to large-scale flakes in the laboratory and roll-to-roll manufacturing of nanosheets with sizes approaching the meter-scale. Additionally, the mechanical strength of 2D materials is very high, 200 times higher than that of steel, which gives these materials extraordinary breaking strengths and unpredictable flexible. Their mechanical properties can be further tuned with defects or functional groups, which makes 2D materials strong as lightweight polymer. Furthermore, as ultrathin stretchable membranes, single layer materials are suitable for use in tunable electrochemical systems. They can withstand more than 20% elastic distortion and also present extraordinarily high flexibility.79 At larger strain, these materials are considered to be brittle since they will break like glass because they are very thin. Chemical modification via chemical doping with various metal ions may impart more elastic distortion strength. Besides elasticity in atomic-level-thin layers, they also have confined quantum levels, and hence some remarkable electronic and optical properties.80
Graphene (2004) is a single-atom layer derived from graphite, where carbon atom are connected in a hexagonal honeycomb lattice, and it is a very prominent and mature 2D-layered material, which has opened a new world of 2D materials and owing to its exceptional properties not necessarily found in the familiar 3D world.25,28,30,75,81,82 Besides graphene, its other carbon-based 2D material siblings/family include, graphene oxide (GO), reduced graphene oxide (rGO), graphane, fluorographene, graphyne, and graphdiyne. Their thicknesses range from one to several atoms and their hallmark feature is their unique bonding interactions. These ultrathin sheets typically demonstrate properties that are dissimilar to the parent bulk material due to anisotropy. Notably, the novel structure of graphene endows it with various superior characteristics for example elevated electrical and thermal conductivities, good transparency, high mechanical strength, intrinsic flexibility and large specific surface area (SSA). The rapid development of graphene covers chemistry, physics, biology, materials science and related interdisciplinary fields; however, the large-scale synthesis of big-sized single-layered or few-layered 2D carbon nanosheets is requires for industrial revolution. Graphene is the first and best known 2D material1,8 with a zero band gap semimetal nature, where electrons are highly mobile and exhibit exceptional conductivity.74,83 Inspired by the attractive properties of graphene due to its 2D structure, researchers have been stimulated to hunt for new 2D material families. The 2D class of materials consists of a wide variety of materials with varying chemical compositions and tunable properties.
The unique characteristics of 2D structures such as high surface-volume ratio, shape, surface charge, anisotropic nature and tunable functionalities open up their potential application scope further. Generally 2D materials are categorized by their structure, including graphene, transition metal dichalcogenides (TMDCs), layered double hydroxides (LDHs), gC3N4, LAPONITE® clay, hexagonal boron nitride (h-BN), and black phosphorous (BP). 2D materials consistently have exposed crystal lattices compared to other materials, for example, nanowires, nanoporous materials, and nanotubes, and thus have superior exposed electrocatalytically active sites at a specific loaded catalyst concentration. The ordered and simple molecular framework of 2D materials also endow them with an active SSA according to both theoretical and experimental approaches. An additional reason to investigate and expand 2D electrocatalysts is they are usually cost effective, which make them novel substitutes for precious metal-based catalysts (PMCs). Fig. 2 shows the classification and structures of 2D nanomaterials. Therefore, the rapidly increasing attention on graphene is the last few years has led to the investigation of other 2D atomic crystals, including single- or few-layer TMDs (e.g. molybdenum disulfide (MoS2), tungsten disulfide (WS2), molybdenum diselenide (MoSe2), and tungsten diselenide (WSe2)),85 h-BN, borophene (2D boron), silicene (2D silicon), germanene (2D germanium), metal oxides, LDHs, graphitic carbon nitride (g-C3N4), a family of mono-elemental compounds, BP (or phosphorene), arsenene, antimonene and bismuthene (Xenes), and metal nitrides and carbides (collectively known as MXenes).6,27,64,86 2D nanomaterials have expanded to semiconductor III–VI compounds (InSe, GaSe, etc.),87 transition metal halides (TMHs) (e.g., PbI2 and MgBr2),88 metal oxides (e.g., MnO2 and MoO3),89 perovskite-type oxides (e.g., K2Ln2Ti3O10 and RbLnTa2O7 (Ln: lanthanide ion)),90 hexagonal boron nitride (h-BN or white graphene),91 graphitic carbon nitride (g-C3N4),92 2D polymers and frameworks,93 and MXenes, which present versatile opportunities.94 A TMDC single layer with MX2 stoichiometry consists of a hexagonally stacked transition metal (TM) (e.g. M = Ti, W, Nb, Mo, and Ta) inserted among two planes of chalcogen atoms (X = S, Se and Te).95 In contrast to the semimetal graphene with a zero band gap, 2D TMDs may be semiconductors (e.g., MoS2, MoSe, WS2, and WSe2), semimetals (e.g., TiSe2 and WTe2), or metallic conductors (e.g., VSe2 and NbS2), relying on the coordination setting of the individual metal atom, the particular metal d-electron configuration, or doping the relevant crystal structure.96 In this review, we will discuss in detail the recent progress made to date in the application of 2D materials as electrocatalysts for the ORR, OER, HER, and electrochemical CO2/N2 reduction (ECR/ENR), and also the limitations of traditional catalysts. In this section, we present each family in detail and their corresponding electrocatalytic applications based on important properties.
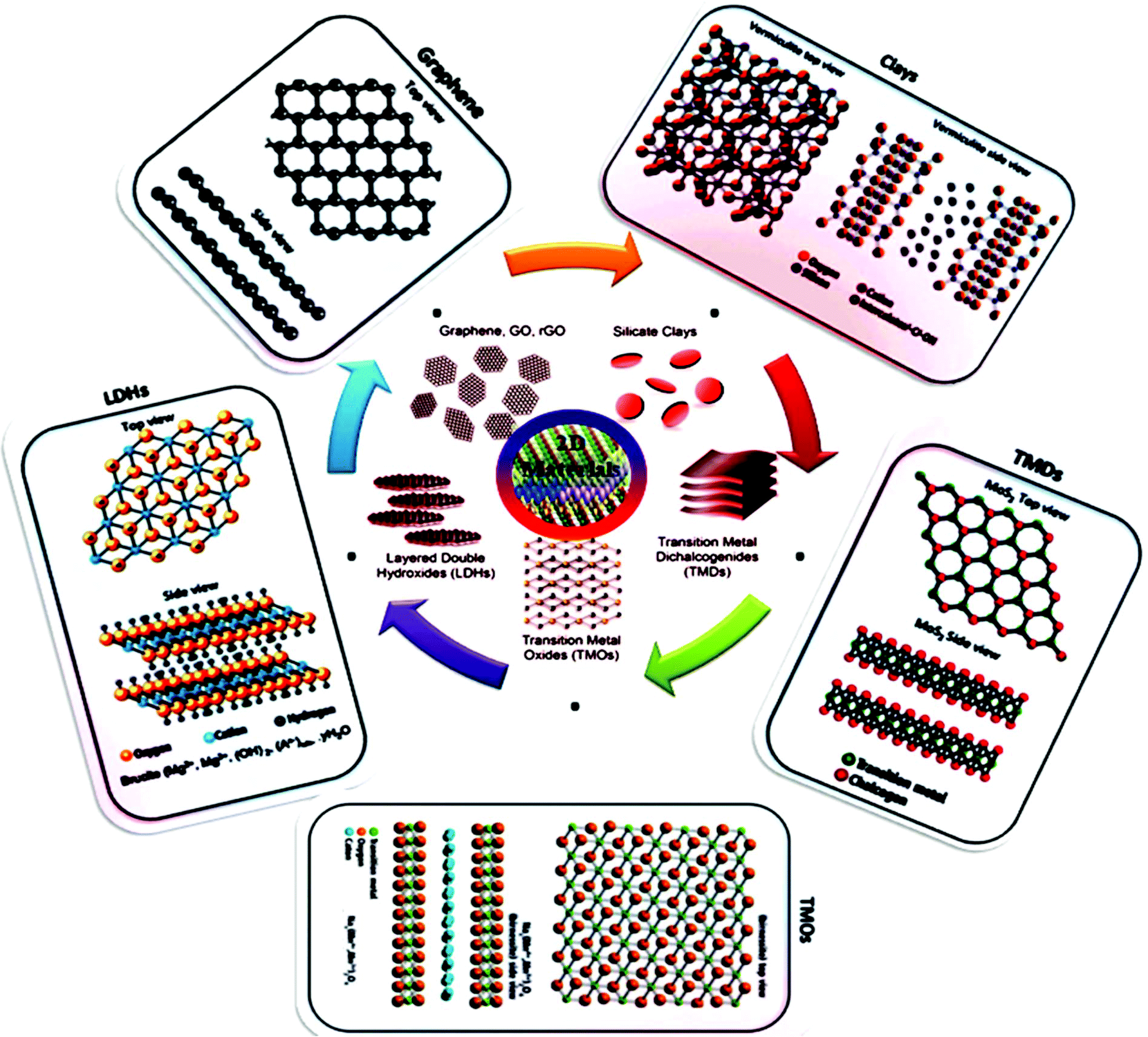 | ||
| Fig. 2 Structures of 2D nanomaterials, including carbon-based nanomaterials (graphene, graphene oxide (GO and rGO), silicate clays, LDHs, TMDCs and TMOs. This figure has been adapted/reproduced from ref. 84 with permission from Wiley. | ||
2.1. Classification of 2D materials based on their electrical properties
Different chemical modification methods give many new opportunities for further basic studies and scientific applications of 2D materials.97,98 Similarly to graphene, the group IV elements, e.g., silicene, germanene, and stanene, called monoelement Xenes, have also been studied in detail. They have to some extent a buckled (β-type) honeycomb structure, which results in their stability under normal circumstances. Other significant 2D materials include binary compounds with a honeycomb structure and IV and III–V group elements.99,100 For example, BP, which belongs to group VA,86 arsenene and antimonene,101 borophene,102 layered silicates and alumino-silicates (for example clays and mica), and LDHs.103 By tweaking the TM or chalcogen atoms, TMDs can achieve a spectrum of properties encompassing insulators, semiconductors, semimetals and true metals (Fig. 3). 2D materials possess insulating (h-BN,104 Ti0.87O20.52–39, calcium niobate (Ca2Nb3O10),105 tantalum oxide (TaO3),106 Ti/Nb mixed oxides,107 HfS2, etc.), semiconductor (g-C3N4,108 TMDCs like MoS2, WSe2, PtSe2, etc.,56,109–113) b-P,86 black arsenic–phosphorene (b-AsxP1−x),114,115 InSe,116,117 MoO3,60 Bi2O2Se,118 Te,119etc., semimetals (PtSe2), true metallic (graphene,80 titanium carbides (Ti3C2),120 RuO2,121 MoO2,122 NbS2, etc.), and superconducting (NbSe2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 116) properties. To date, semiconductor 2D materials and their band gap include long wavelength IR (LWIR, b-As0.87P0.13) to middle wavelength IR (MWIR, b-P), NIR (b-P, few layer MoS2), visible light (TMDCs, g-C3N4), UV (Ti0.87O20.52, Ca2Nb3O10, etc.), and further to deep UV (TaO3, h-BN) materials. This constructs more choices to select 2D materials with diverse characteristics in preferred electronic and optoelectronic devices.
116) properties. To date, semiconductor 2D materials and their band gap include long wavelength IR (LWIR, b-As0.87P0.13) to middle wavelength IR (MWIR, b-P), NIR (b-P, few layer MoS2), visible light (TMDCs, g-C3N4), UV (Ti0.87O20.52, Ca2Nb3O10, etc.), and further to deep UV (TaO3, h-BN) materials. This constructs more choices to select 2D materials with diverse characteristics in preferred electronic and optoelectronic devices.
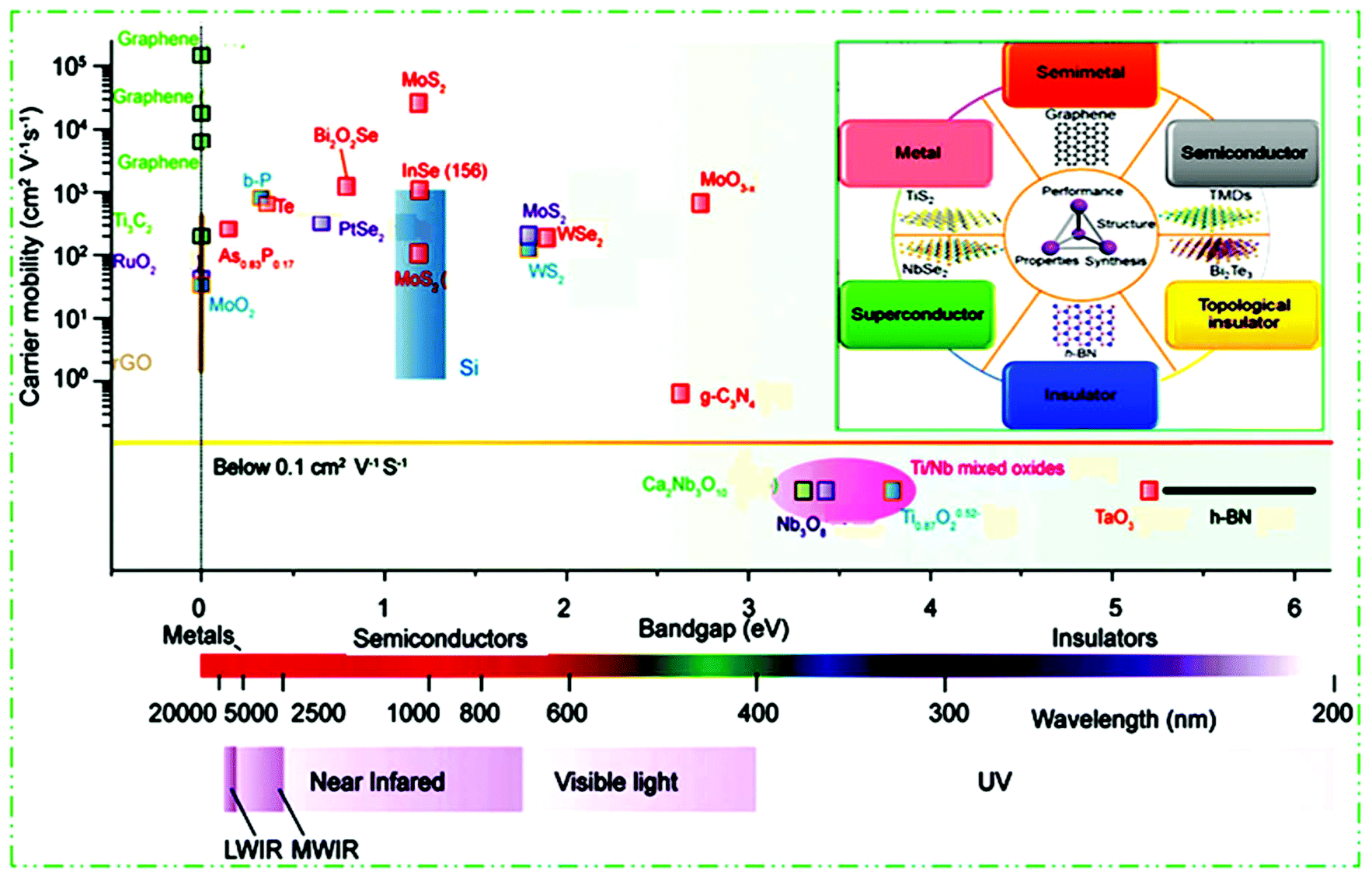 | ||
| Fig. 3 2D materials ranging from insulators and metals to semiconductors with diverse bandgaps, and electron/hole mobility summary scale diagram. This figure has been adapted/reproduced from ref. 104 with permission from Nature Publishing Group. | ||
2.2. 2D-nanomaterial based electrocatalysts
In all technologies for renewable energy production, the requirement of high-performing electrocatalysts is critical. Normally, an excellent electrocatalyst has high activity, high SSA, excellent electric conduction, and long-term stability.123,124 The catalyst reactivity mainly relies on the physio-chemical properties of the catalyst surface and electrode–electrolyte interface.124,125 To attain elevated activity, catalysts must be efficient at lowering the energy barriers of catalytic response and advancing the rate of surface electron charge transfer. Therefore, the improvement of distinct structure surfaces is beneficial to encourage electrochemical processes. Furthermore, high electrode SSAs and moderate conductivity can be obtain by organization of nanosized conductive supports/substrates.126,127 Catalyst stability demonstrates the working life of fuel cells, and is more significant for realistic use. In the ORR technique, PGM catalysts possess low tolerance for byproducts such as methanol/CO, resulting in a limited operational life, and thus highly active NPMs electrocatalysts are required. Thus, the synthesis of next-generation catalysts with improved reactivity, selectivity, stability and efficiency is a current challenge in energy conversion/storage. Thus, particular procedures to tailor the physical, chemical, and electronic surface characteristics of 2D nanomaterials (e.g., intermediary adsorption free energy, kinetics of charge transfer, etc.) are required, which can change their intrinsic nature. To recognize the result of these alterations, topical progress in surface classification allows the direct study of the active sites to disclose the reaction mechanism. Accordingly, quantum theoretical study, materials science, and surface electrochemistry have considerably helped in the development of 2D materials as highly active catalysts in energy conversion applications. Thus, 2D materials are presently one of the most up-to-date research fields in this direction.128Dai et al. in 2009 reported N-doped carbon nanotubes (N-CNT), and showed that they can replace Pt for the ORR in fuel cells. After this discovery, significant research progress was made to perfect the potential properties of 2D materials as metal-free electrocatalysts in various renewable energy formation processes. Among the these materials, the graphene family, graphdiyne, graphitic carbon nitride (g-C3N4), h-BN, and BP, will be discussed in detail in the following section. To date, among the 2D-based materials, graphene and graphene-based nanocomposite materials are promising electrode materials with high performances in the field of energy devices (sensors, electronics and optoelectronics, efficient catalysts, etc.) due to their high electrical conductivity and large SSA. The enormous achievement of graphene has been followed by remarkable search for further 2D materials at the atomic level for amazing feature/characteristics.1,6–9 The application of graphene in photovoltaic devices, supercapacitors (SCs), batteries, and fuel cells for hydrogen generation is an opportunity to deal with the increasing challenges globally in the energy field. The 2D graphene with a simulated ∼2600 m2 g−1 surface to mass ratio and high flexibility and electrical conductance show it can store electric ions, charge, or hydrogen. The benefit of using 2D crystals is the opportunity to generate and control layered synthetic structures with “customized” characteristics through their spins (spintronic). This results in the development of accessible materials with insulating, semiconducting, and metallic properties.
2.3. 2D material-based composites as electrocatalysts
The exceptional anisotropy and novel but flexible electronic properties of 2D nanosheets have boosted enormous significance in their broad range of applications and especially electrochemical use. Mounting 2D catalysts on supports with a high specific surface area is an efficient strategy to develop enhanced catalyst performances up to a limit, but synthesizing composites will further boost their performance. Carbon supports are attractive and widely utilized because they are suitable dispersion platforms that promote efficient electron transfer. Compared to NiFe-LDHs and NiFe-LDHs mixed with carbon nanotubes, the solvothermal synthesis of ultrathin NiFe-LDHs grown on oxidized carbon nanotubes resulted in higher OER activity. The strong interactions between NiFe-LDH and carbon nanotubes strengthen the electron transfer kinetics for the OER. Heteroatom doping and the addition of functional groups to carbon supports are also effective to facilitate interactions between the catalyst and support. A 3D catalyst with NiCo-LDH grown on N-doped graphene hydrogels outperformed that grown on undoped graphene. The enhanced HER performance of MoS2 nanoparticles prepared on rGO relative to the absence of rGO highlighted that the use of rGO provided good nanoparticle dispersion, which increased the accessibility of edge sites and enhanced the electron transport.3. 2D-material applications
The early application of 2D nanomaterials as lubricants was a result of their layered nature. Over the years, the applications of 2D nanomaterials have diversified, which are dictated by their electronic structures and intrinsic physical properties. Graphene is a zero band gap semimetal, where electrons are highly mobile, and it exhibits exceptional conductivity. Equipped with a varying band gap, other 2D nanomaterials, such as TMDCs, LDHs, metal oxides and MXenes present versatile opportunities. By tweaking the transition metal or chalcogen type, TMDCs can achieve a spectrum of properties, encompassing insulators (HfS2), semiconductors (MoS2), semimetals (PtSe2) and true metals (NbS2). Insulating h-BN and semiconducting g-C3N4 are prized for their thermal and chemical stability. Thanks to their vast array of properties, 2D nanomaterials hold promise in the fields of electronics, sensors and catalysis. Fig. 4 shows the different applications of 2D materials; however, in this review, we are mainly interested in their electrocatalytic applications in energy production, i.e., fuel cells. With the development in sustainable energy attracting global attention, it has become imperative to estimate the efficiency of different 2D nanomaterials in this area. Electrocatalysis is the main theme for clean energy conversion in future technologies via the HER, HOR, ORR, OER, ENR and ECR. Thus, 2D nanomaterials are pursued as economical alternatives to the expensive Pt-based catalysts for these reactions. The allure of 2D materials in catalysis can be mapped to three aspects, their SSA, mechanical properties and conductivity (thermal and electric). 2D materials have maximal surface to bulk ratios, providing a high density of surface active sites, which favor surface-active applications. Their excellent mechanical properties endow catalysts with durability and their thermal conductivity facilitates the diffusion of heat produced during exothermic reactions. Additionally, the tunable electronic properties of 2D materials tailor catalytic performances. The robust mechanical structures, wide SSA and high density of active sites in 2D nanomaterials are advantageous compared with bulk materials for catalyst stability and activity. Compared to bulk materials, 2D materials are the preferred building blocks for constructing hierarchical composite catalysts.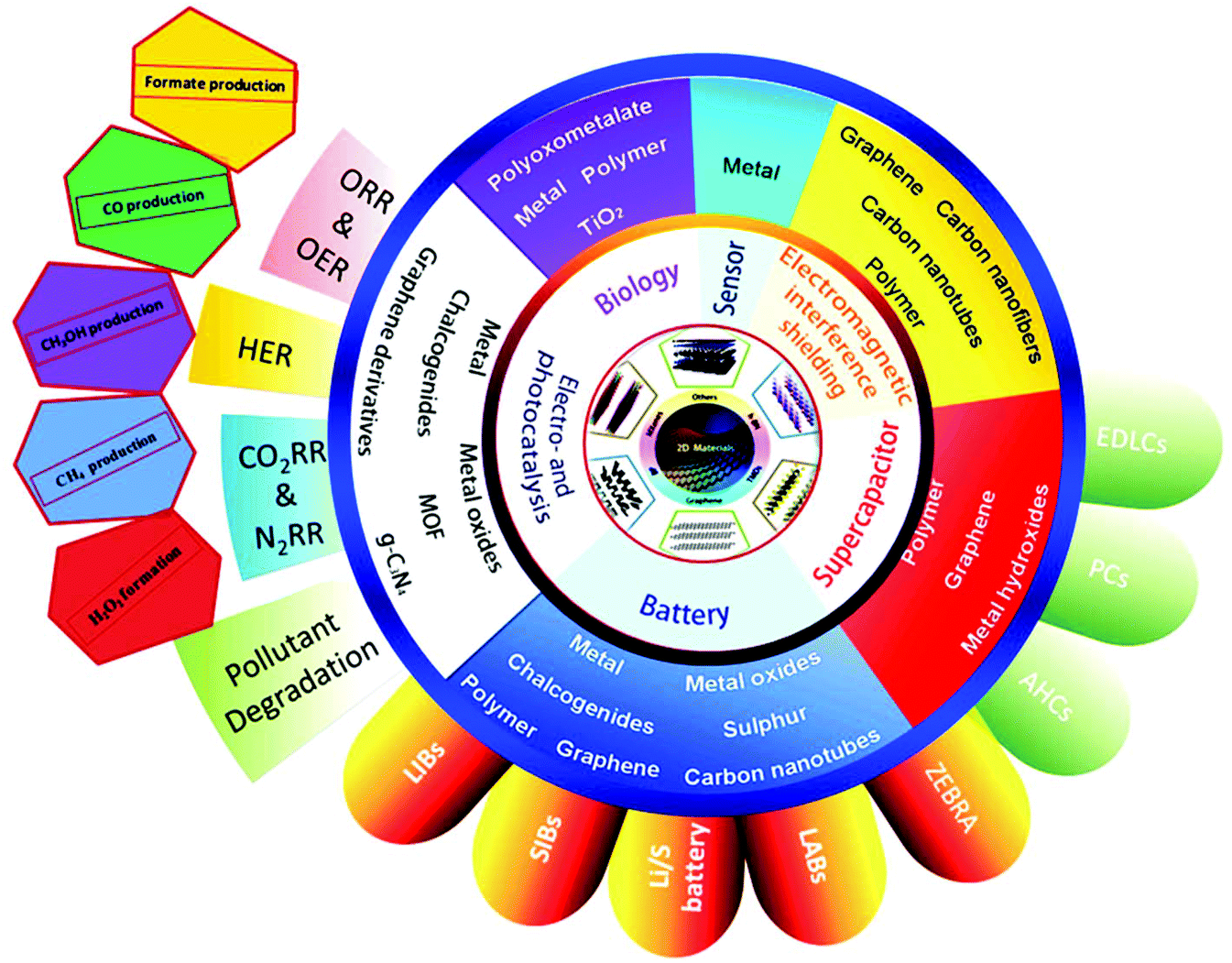 | ||
| Fig. 4 Electrocatalytic applications of 2D materials in energy production (fuel cells) and energy saving devices (batteries and superconductors). This figure has been adapted/reproduced from ref. 129 with permission from Elsevier Inc. | ||
4. Fuel cell technology
Fuel cells are devices that can switch chemical energy into electrical energy by reacting using O2 or other oxidizing agents.130 Compared to solar and wind energy, fuel cell energy is more reliable because of its unconventional manner of energy production. Usually, fuel cells are measured to be ideal for renewable energy as a result of their elevated efficiency, mild operation process, zero emission and most importantly, unlimited renewable source of reactants. The main areas of fuel cell technology include transportation, and stationary and portable power. It converts fuel chemical energy into electrical energy similarly to batteries. In the case of fuel cells, the combustion process does not follow the thermodynamic cycles as in combustion engines.131 The renewable energy production in fuel cells system is described by the Gibbs free energy. Regarding large-scale applications, one restriction of fuel cell-based technology from the commonly used technology is its high electrode activation energy barrier. This problem can be solved by the introduction of abundant, low cost, and stable electrocatalytic materials. Thus, catalytic properties are of supreme importance to overcome this barrier. Solar and electrical energy can be used and stored in chemical bonds to split water to generate hydrogen, which can also be oxidized to produce energy together with oxygen reduction. Clean energy production in devices via electrocatalysis involves two major energy cycles, the carbon cycle and water cycle.128 These cycles are mainly based on reactions such as the ORR, OER, and HER. The core reaction for energy production by fuel cells and metal–air batteries is the ORR.132 Similarly, reactions such as the HER and OER mainly occur in water electrolysis cells, in which ultrahigh pure hydrogen can be generated and supplied to fuel cells.133 More specifically, in the carbon cycle, direct methanol/ethanol participation is significant task for potential power, which can solve both storage and hydrogen transportation challenges.134Fuel cell-based energy production involves multi-electron charge transfer processes, for instance, the ORR is classified as a 2e− or 4e− system. These electrons though external circuit are channeled via the anode to cathode, which produces a current. Along with produced electricity, water is a by-product from its constituent atoms, i.e. H2 and O2. In the opposite case, H2 (fuel) and O2 are generated via the application of comparatively low energy. Thus, the combination of forward and reverse reactions, consisting of consecutive hydrogen- and oxygen-associated catalytic processes, makes a cycle, which is called the water cycle. Progress in this technology originated from the increasing hydrogen market, which has caused the electrocatalytic water cycle to be an important research topic. The perfect technical water cycle is composed of water electrolysis via the OER and HER for fuel production, followed by power production via the ORR and HOR. For electrolytic cells, the HER and OER occur at the cathode and anode, creating H2 gas and O2 molecules, respectively (Fig. 5(a)). The ORR and HOR occur at the cathode and anode for H2 and O2, respectively (Fig. 5(b)). In the last two decades, considerable investigations have addressed the progress in achieving high electrocatalytic properties for both the water and carbon cycles. Generally, the kinetics of the 2e− transfer method in half-cell HER and HOR is straightforward. However, the slow rates for the multi-electron transfer ORR and OER decrease the efficiency of the energy equipment. Conventionally, noble metals (Pt, Ir, and Ru), are used as a electrocatalysts for these reactions, but these expensive electrocatalysts suffer from poor stability, which significantly limits their large-scale, long-term industrial application. Thus, the development of low cost, high performance electrocatalysts has become important for clean and sustainable energy production.128 The water cycle was a novel achievement summed up in Grove's letter, On a gaseous voltaic battery, which was published in 1842.136 His working principle relied on the explanation by Christian Friedrich Schönbein (1838).137 Subsequently, two systems, discharging mode (fuel cell) and charging mode (capacitors and batteries), have been examined independently in the last few decades, in alkaline (anion conducting membranes (AEM)) and acid media (PEM) (Fig. 5(c and d), respectively). Moreover, interest in the development of H2/O2 energy storage systems led to the combination of both systems, the so-called URC, which was reported by Mitlitsk et al. (Fig. 5(e)).138–140
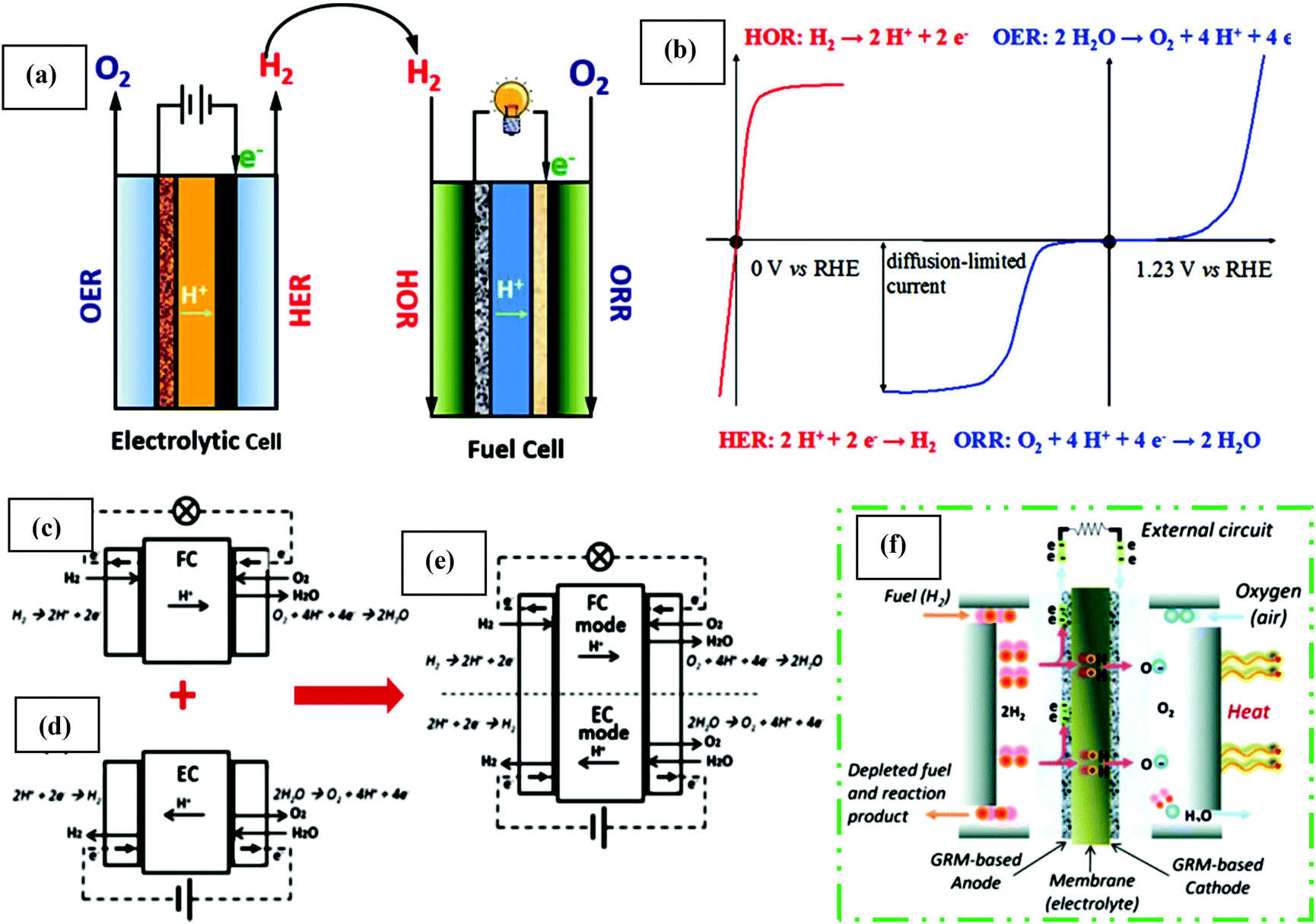 | ||
| Fig. 5 (a) Schematic illustration of an electrolytic/fuel cell. (b) Polarization curves of the H2- (red color curves) and O2-involved reactions (blue color curves), without a proper scale. This figure has been adapted/reproduced from ref. 128 with permission from the American Chemical Society. Innermost polymer electrolyte membrane (PEM) system (c) fuel cell (FC), (d) electrolyzer cell (EC), and (e) unitized regenerative cell (URC). Discharging mode: FC and recharging mode: EC. PEMFCs, where the fuel (e.g. H2) channel starting from one side, is split by the catalyst (e.g. graphene electrode) into H+ ions and e−. (f) More detail explanation of the current generation and combination of hydrogen ions and oxidant at the cathode, generating water and heat. This figure has been adapted/reproduced from ref. 135 with permission from the Royal Society of Chemistry. | ||
Electrochemical reactions play an essential role in energy conversion and energy storage systems, e.g., the ORR and HOR occurring on the cathode and anode of the H2–O2 fuel cell, whereas the OER and HER are the anodic and cathodic reactions in the electrolyzer, respectively.141 However, deep understanding of the basic principles and chemistry is required for understanding these future energy producing/storing devices. In the past few years, the advantages of fuel cell technology and its industrial application have drawn considerable attention especially in electric vehicles, and portable and residential power sources due to its high competence (specifically, 40–60% or up to 85% when dissipated heat is utilized), almost pollution free nature, minimal corrosion problems, and possible significant applications in many fields.142 The main advantages of fuel cells is that they are functionalized to produce electricity once a constant source of oxygen and fuel is applied. There are several types of fuel cells, including PEMFCs,143 solid oxide fuel cells (SOFCs),144 molten carbonate,145 and phosphoric acid,146 but they all share a cathode, anode, and electrolyte, which permit charges to travel in the fuel cell between two sides. A further detail explanation of the structure of the fuel cell, e.g., PEMFC, is shown in Fig. 5(f).
In fuel cells, rationally designed porous structures are the most advantageous electrocatalyst structures, especially hierarchical porous structures, where macro/meso-pores with a large number of electrocatalytically active sites make it the transportation of reactants and products (O2 and H2O) easy. However, major challenges exist regarding the synthesis of efficient electrocatalysts, and thus different synthetic methods, e.g., pyrolysis, are used. Another important factor regarding the efficiency of fuel cells is their durability, besides the activity of cathode catalysts, which can be enhanced either by synthesizing more stable electrocatalysts or anion exchange membranes (AEMs). Basically, AEMs consist of a polymer backbone with cationic ion-exchange groups that permit only the passage of OH−/H+ anions. The diffusion of OH− is considerably low compared to H+, which restricts the efficiency of fuel cells. An additional significant problem in fuel cells is their low durability, e.g., in AEMFCs due to the chemical degradation of OH− conducting ionomers. Therefore, suitable methods for membrane-electrode assembly (MEA) is of utmost importance for high performance. Despite the above-mentioned challenges, fuel cell technology holds great promise to reach the aim of developing affordable and sustainable power sources, and accordingly several breakthroughs have been made in fuel cell research in the last five years. Further increasing the stability, reducing cost, and elevating durability are hot topics in the development of a fuel cells, especially AEMFCs, which can work at a low-operating temperature.147 Similarly to batteries, fuel cells change fuel chemical energy in combination with oxidants into electric energy (Fig. 6).
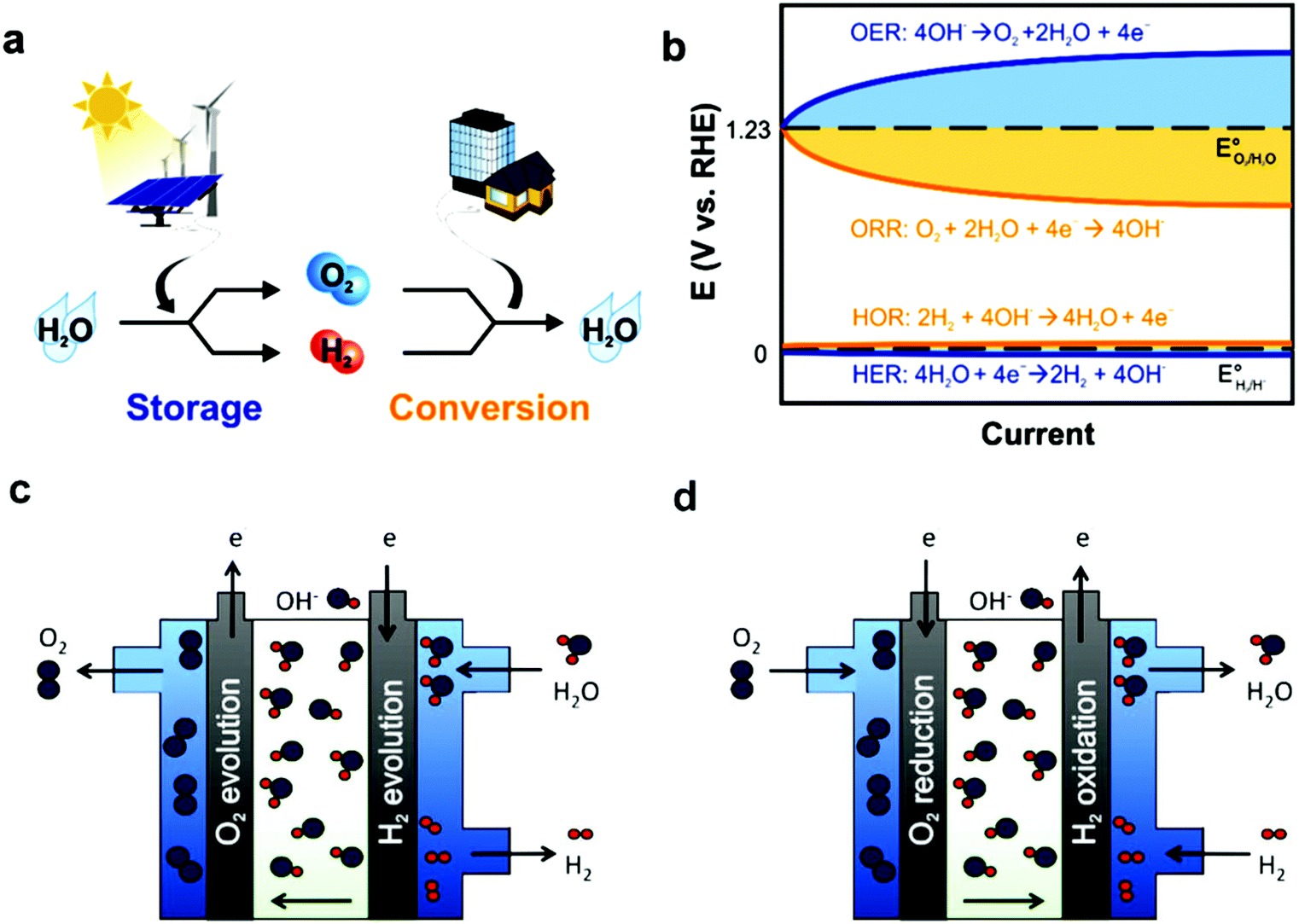 | ||
| Fig. 6 (a) Energy storage/conversion in the H2 and O2 cycle, where the energy storage two half-cell reactions in water splitting are the OER and HER. While for energy conversion, the half-cell reactions include the ORR and HOR. (b) Hydrogen electrocatalysis (HER, and HOR oxygen electrocatalysis (OER and ORR)) overpotential scheme. OER and ORR seriously limit the rate determination and energy efficiency in electrochemical energy devices because of the large η required to drive electrochemical reactions, even when utilizing benchmark catalysts in alkaline electrolytes. Anion-exchange membrane schematic representation (c) electrolyte and (d) alkaline electrolyte-based fuel cell. This figure has been adapted/reproduced from ref. 148 with permission from the Royal Society of Chemistry. | ||
The dissimilarity from batteries is that fuel cells do not require recharging if an oxidant and fuel are constantly provided. After the application of the fuel (hydrogen), the fuel cell produces mainly electricity, and some water and heat as a byproduct. In contrast to thermal engines, fuel cells have the advantages of enhanced effectiveness, no ecological contamination, and infinite availability of reactants. Thus, fuel cells are predicted to be applied in the future extensively for commercial uses in stationary, transportation and portable power production, and may facilitate overcoming the worldwide energy crisis with a clean atmosphere. Among the fuel cells, PEMFCs are actively utilized for moveable electronics, vehicles and combined heat and power (CHP) systems because of their elevated power density, low working temperature, and swift start-up. PEMFCs also have the advantages of low ecological impact and high energy density, and thus have gained worldwide attention. PEMFCs are particularly suitable as central power sources in automobiles and buses. Fuel cell vehicles (FCVs) are considered an absolute solution in the automotive trade and contain large scale benefits above battery-based electric vehicles (EVs). The first commercially produced FCVs, the Toyota Mirai, were commercially sold/leased in 2014, but with a high price due to the excessive Pt loading in the fuel cell.149 PEMFCs have already been widely recognized, and the commercialization of these devices is gradually progressing, but AEMFCs are also being intensively developed. As one of the subcategories of AEMFCs, alkaline direct methanol fuel cells (ADMFCs) are capable alternative power sources in portable electronics due to their easier storage and handling of alcohols compared to hydrogen.147 Fuel cells have many uses, such as in automobiles, backup power systems, smart phones, and smart textiles (implanted digital computing and electronics) since they present a long-lasting electricity supply. Hence, the worldwide increasing demand for clean energy has directed research efforts towards alternative and efficient energy conversion technologies. It is estimated that fuel cells will play an increasing role in future energy security. At peak times, it will be desirable to accumulate extra energy in the form of H2 gas (for example via water electrolysis), which allows one to store it for a longer time and then to convert the chemical energy into electricity in the fuel cell at any time.
Some automobile companies such as Toyota, Honda, and Hyundai have introduced fuel cell-based electrical vehicles (FCEVs). However, is a need to gain progress in this field for sophisticated energy exchange systems such as water electrolysis, metal–air batteries, and fuel cells. Considering the continuous decrease in fossil fuels and deterioration of the environment, it is significant and necessary to discover plentiful, environmentally friendly and renewable energy sources. The standard Nernst potential in the O2 half-cell reaction observed is 1.23 V versus RHE. This is determined by the standard H2 potential at a specific pH. At 0 pH, this potential is considered to be the standard hydrogen electrode (SHE) potential. The kinetically favourable ORR is preferentially below the half-cell potential since the OER lies above this potential. The ORR/OER kinetics is very poor due to immense deviation from the half-cell potential, which is called “η”, requiring considerable electricity. Thus, considering the physical response η is very important toward the further synthesis of proficient catalysts for the OER. The oxygen electrocatalysis benchmark catalysts in acidic solutions is Pt for the ORR (∼$50 per g) and ruthenium oxides for the OER (∼$2.50 per g). At practical i = 1.5 A cm−2geo, these benchmark electrocatalysts still have ∼0.4 V η. In contrast, in basic solution, the ORR is catalyzed using metal oxides that have earth abundant elements such as NPMCs (e.g. Fe: 0.0001 g−1; Co: 0.03 g−1; and Ni: ∼$0.02 g−1) and early rare-earth metals (e.g. La: ∼$0.02 g−1). Some good oxide catalysts contain MnOx for the ORR and Ni-Fe-Co oxides for the OER, needing an η of approximately 0.4 V at 10 mA cm−2geo, and recently our group introduced an rGO-coated abundant nature mayenite electride, 12CaO·7Al2O3 (C12A7:e−) for the ORR.1,148
Electrocatalysts have a major position in clean energy production, allowing a number of durable methods for potential pollution-free energy devices, such as fuel cells, electrolyzers and metal–air batteries. The basic reactions occurring in these devices are the ORR, OER and/or HER, which are multielectron-based electrochemical reactions with high η values and slow kinetics. Accordingly, many electrocatalysts are commercially available for these reactions, mostly based on PGMs; however, they are costly and unstable, and thus not feasible for common application. The requirements for an ideal type of electrocatalyst include easy preparation, very stable, inexpensive, and preferably have a bifunctional nature, which can simplify design protocols in rechargeable metal–air batteries and reproducible fuel cells/electrolyzers, and lead to the realization of more efficient but practical devices. The universal feedstock of abundant elements such as H2O, CO2, and N2 in the Earth's atmosphere provides a chance to convert the abovementioned products into energy by means of electrochemical methods coupled with inexhaustible energy if electrocatalysts with the required properties are developed. In the case of water splitting, hydrogen and oxygen evolution half-reactions occur, by which sustainable pure hydrogen can produced. Hydrogen is an energy carrier by which chemical energy can be transformed into clean electrical energy in fuel cells via the HOR and ORR reactions. Similarly, hydrogen peroxide can produced by the ORR, which is an important chemical used in the water treatment, pulp and paper bleaching industries.150 Similarly, atmospheric or open starting point sources of CO2 can also be used as fuel, commodity chemicals, final product chemicals, and starting materials for polymers through groundwork electric-reduction.151 Moreover, the electric reduction of N2 into NH3 can be utilized for the production of fertilizers and reduce distribution costs, which can be synthesized by the Haber–Bosch process and using the newly introduced C12A7:e− catalyst-based method.6 Based on these crucial ideas, the development of more active and stable electrocatalysts for further boosting the efficiency and selectivity in chemical conversions is important. Regarding improving activity and the reaction rate, increasing the number of active sites at a particular electrode or increasing the built-in activity of each active site are important to investigate. There are some physical limitations for catalysts loaded on the working electrode, which do not influence other significant factors, for example charge and mass transportation. In contrast, increasing inherent activity leads to a direct enhancement in electrode reactivity in a way that diminishes charge transport issues due to massive catalyst loadings, and with enhanced inherent reactivity, the catalyst components may be reduced, which can be cost-effective.2
4.1. Types of electrochemical reactions
The basic method for the manufacture of hydrogen gas is the HER. Water electrolysis is the electrochemical process traditionally used for the production of high-purity hydrogen. On the other hand, recently the HER acts as a connection between basic surface electrocatalysis and the behavior of recently reported materials. Thus, designing highly efficient HER electrocatalysts is necessary for clean energy production at the fundamental and utilization levels. Basically, the HER cathodic electrochemical reaction for water splitting is based on the 2e− transfer mechanism with one catalytic transitional species, H* (* = electrode active site), and it may occur via the Volmer–Heyrovsky or Volmer–Tafel mechanism.152,153 The overall HER rate is determined by the hydrogen adsorption free energy (ΔGH), which is strongly based on the electronic configuration of the electrocatalyst. The most favorable assessment of ΔGH = 0 eV, shows that the H2 bound on the catalyst surface may not be too strong nor too feeble. Noble metals, for instance, the benchmark Pt and Ru, have a ΔGH approaching 0 eV. In the case of earth-abundant metals, this value is either excessively high or excessively very low, resulting in insufficient catalytic activity, according to the prominent hydrogen volcano.154 Therefore, developing NPMCs that can replace Pt for the HER in acidic and basic electrolyte is an essential challenge. After catalyst activity, another important factor is lasting stability. The useful approaches to evaluate electrocatalyst durability include cyclic voltammetry (CV), stability tests that calculate the catalyst amount from drain to electrolyte, and utilizing thin-film-based catalyst configurations.153,155 Basically, in the HER, protons (acidic medium) or water molecules (alkaline medium) get reduced, and gaseous hydrogen is evaluated due to water splitting. Therefore, generally the HER process can be written as follows (catalyst active site “*”):
Acid medium
| 2H+ + 2e− → H2(g) | (1) |
Alkaline medium
| 2H2O + 2e− → H2(g) + 2OH− | (2) |
The HER process can also be explained further. The standard potentials (E°) depend on the nature of the active ions. Also, the HER consists of many electrochemical reactions, and the activation energy obstacle to advance the reaction is known as “η”. Consequently, materials having a lower η will have an enhanced reaction rate and high efficiency. The HER mechanisms are the Volmer–Heyrovsky and Volmer–Tafel mechanisms. The HER in an acid environment consists of the following steps:
1. Hydrogen atom adsorption (Had) due to the combination of electrons and protons on the electrode surface (proton release) via the Volmer reaction
| H+ + * + e− ⇔ Had* | (3) |
2. Had atom interaction with electrons and protons resulting in electrochemical desorption (Heyrovsky reaction)
| Had* + H+ + e− ⇔ H2 + * | (4) |
3. Two Had atoms coupling to result in the dissociative desorption of H2 vie the Tafel reaction
| 2*Had ⇔ H2 + 2* | (5) |
On the other hand, in alkaline electrolyte, owing to the OH− concentration, HER advances due to following steps:
1. Water molecule coupling to electrons, leading to the formation of Had atom at the electrode interface (Volmer reaction)
| H2O + e− ⇔ Had* + OH− | (6) |
2. Had atom combines with molecular water and electron, allowing electrochemical H2 desorption (Heyrovsky reaction):
| Had* + H2O + e− ⇔ H2 + * + OH− | (7) |
3. The Tafel reaction is similar to that previously discussed in acidic environment.
Hence, in alkaline and acid environment, for the HER, the formation of Had starts through the Volmer reaction (eqn (3) or (6)) while, the successive formation Had can continue through the Heyrovsky reaction (eqn (4) or (7)) or dissociative desorption through the Tafel reaction (eqn (5)). In contrast, the HER can be investigated via the Tafel slope obtained from the HER polarization curves. The Tafel slope analysis shows the native electrocatalyst properties, and the experiential magnitudes allow the mechanisms to be differentiated. In the Tafel rate-determining step (RDS), the Tafel slope is ∼30 mV dec−1, but for the Heyrovsky and Volmer RDS, the Tafel slopes are 40 and 120 mV dec−1, respectively. Co-based electrocatalysts follow the Volmer–Heyrovsky mechanism for the HER, where generally the Heyrovsky step is the RDS in an acid environment, whereas the Volmer step is the measured RDS in an alkaline environment.141,156,157
Acid medium
| 2H2O(aq) → 4H+(aq) + 4e− + O2(g) | (8) |
Alkaline medium
| 4OH−(aq) → 2H2O(aq) + 4e− + O2(g) | (9) |
In more detail, in the literature, various proposed OER mechanisms occur in acid (eqn (10)–(14)) and alkaline media (eqn (15)–(19)), involving MOH and MO intermediates. Fig. 7 shows two dissimilar pathways for the OER, where the black line shows that its progression involves the development of peroxide (M–OOH) intermediate and the green line shows the direct response for two nearby oxo (M–O) intermediates to produce an O2 molecule.141 In the green route, 2MO directly combines to make O2(g) (eqn (12)), including the generation of MOOH intermediate (eqn (13) and (18)), which later disintegrates via the black route to O2(g) (eqn (14) and (19)). In the heterogeneous OER process, all M–O bonding interactions in intermediates (MOH, MO and MOOH) are critical to the overall electrocatalytic activity.
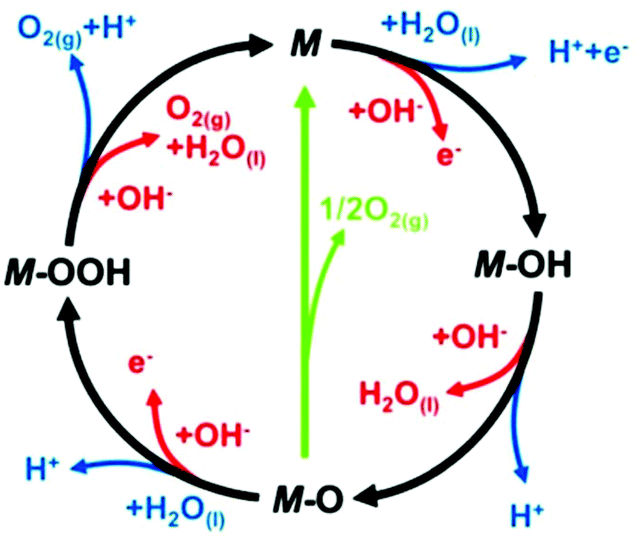 | ||
| Fig. 7 In loop blue line shows acidic and red line alkaline OER mechanism. This figure has been adapted/reproduced from ref. 141 with permission from the Royal Society of Chemistry. | ||
Acid medium
| H2O + M → MOH + H+ + e+ | (10) |
| MOH + OH− → MO + H2O + e− | (11) |
| MO → 2M + O2(g) | (12) |
| MO + HO → MOOH + H+ + e− | (13) |
| MOOH + H2O(l) → M + O2 + H+ + e− | (14) |
Alkaline medium
| OH + M− → MOH | (15) |
| MOH + OH− → MO + H2O | (16) |
| MO → 2M + O2(g) | (17) |
| MO + OH− → MOOH + e− | (18) |
| MOOH + OH− → M + O2(g) + H2O | (19) |
Thus far, the benchmark materials examined as potential NPMCs for the OER are Co- and Ni-based materials (both without support and with support on, e.g., carbon). Among the NPMCs for the OER, Co-based materials are only active in alkaline medium, e.g., CoOx and CoOOH.141 The limitation of the traditional OER process based on the four-step mechanism is that it is frequently restricted by its lethargic electron transfer kinetics. Fascinatingly, the progress of water oxidation catalysts with excellent performance and substantial stability is big challenge. Therefore, in the OER, noble-metal oxides, e.g., RuO2 and IrO2, are taken as the active catalysts, regardless of their tremendously high cost and shortage. Thus, the discovery of earth-abundant 2D materials as alternatives to replace the expensive Ir and Ru is required. Accordingly, substrate-free, transition metal-based catalysts and graphene show novel benefits due to their high charge transport, although the poor OER activity of graphene considerably prevents its general usage. Graphene-like TMDCs are also significantly active in the field of OER because of their novel structural-based electronic properties. However, previous studies showed that the most important shortcoming of layered TMDCs (WS2, MoS2, and MoSe2) is their low conductivity. Fortunately, BP is a metal-free layered semiconductor with comparatively elevated carrier transport (≥200 cm2 V−1 s−1) and substantial catalytic performances.
Direct 4e− pathway
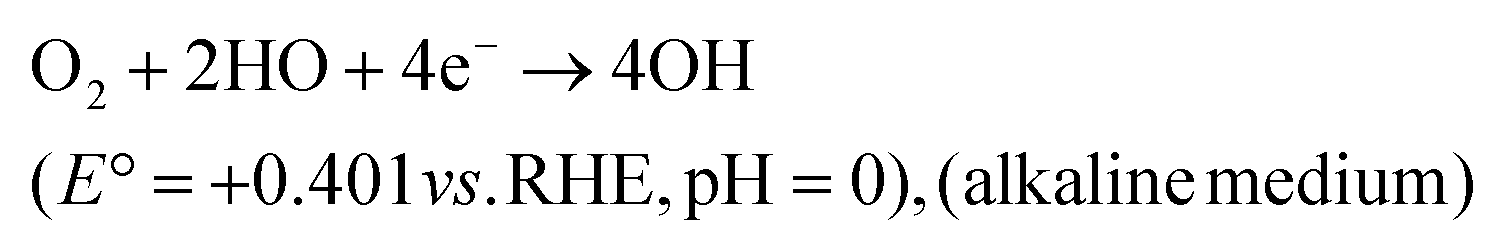 | (28) |
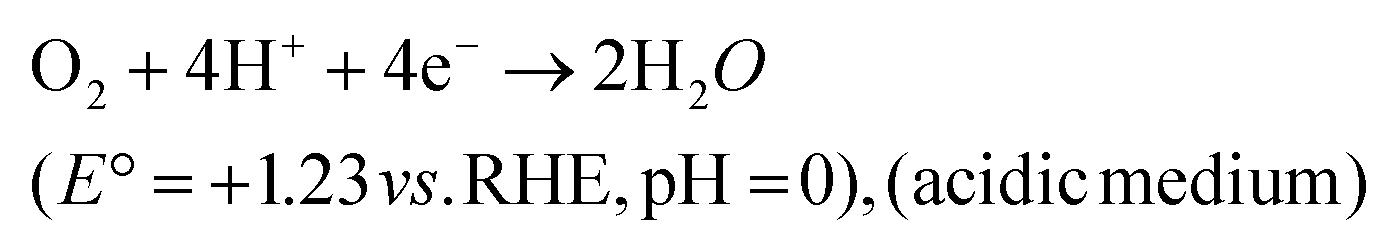 | (29) |
Two-step 2e− pathway
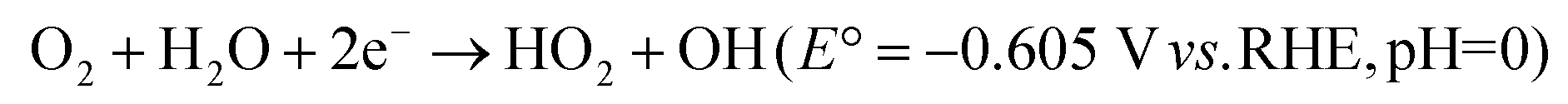 | (30) |
And
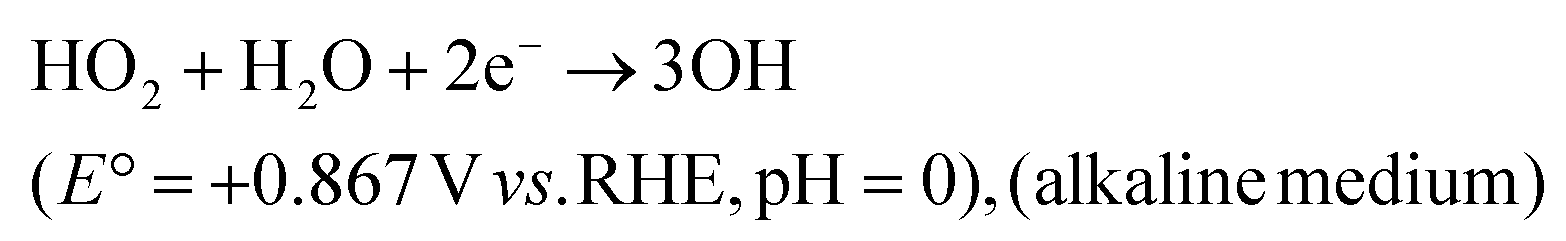 | (31) |
| O2 + 2H+ + 2e− → H2O2 (E° = +0.867 V vs. RHE, pH = 0) | (32) |
And
| H2O2 + 2H+ + 2e− → H2O \cr \tab \quad (E° = +1.76 V vs. RHE, pH = 0), (acidic medium) | (33) |
In fuel cells, the ORR with the 4e− pathway is an advantageous cathodic method due to its positive potential pass on a large open circuit potential. To gain maximum energy from the reaction, the production of peroxide should be avoided. Peroxide formation not only decreases the operating potential and current efficiencies, but also causes the degradation of fuel cell components, which results in an unstable performance. Thus, in more detail, the ORR mechanism can be explained graphically. The typical ORR polarization curves can be divided into three regions, i.e., kinetically controlled region, diffusion-controlled region, and mixed kinetic/diffusion-controlled region (Fig. 8). JL denotes the diffusion limited i. In the kinetically restricted region the O2 reduction rate is very slow with a small increase in i with a decrease in potential. Conversely, a considerable boost in i is observed in the mixed kinetic and diffusion interface area. In this region, acceleration of the reaction occurs with a marked drop in potential value. In the diffusion interface region, i is determined by the rate at which the diffusion of the reactants occur. Quantitative analysis of a catalyst in terms of its activity can be done considering two parameters, i.e. the onset potential (Eonset) and half-wave potential (E1/2). The more positive the potential, more active the ORR electrocatalyst.163 However, fast and more suitable screening techniques to describe the electrochemical nature of recently synthesized materials are necessary. Among them, the rotating disk electrode (RDE) is the widely applied method to exemplify the properties of catalysts/catalysts maintained in liquid electrolyte, which was reported by Stonehart and Ross in 1976.164
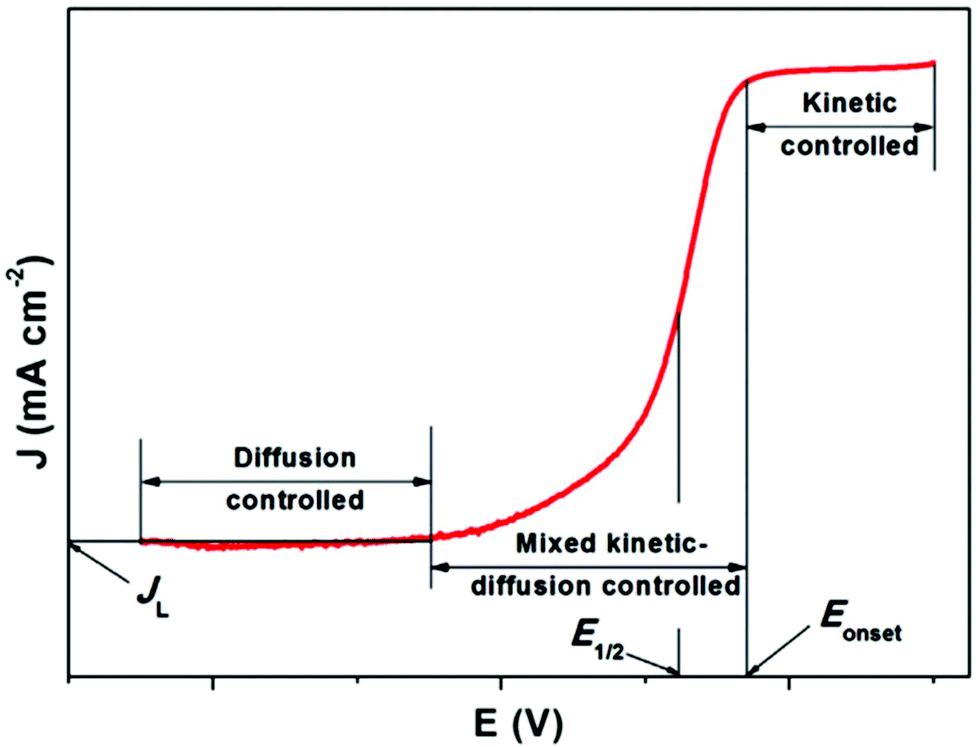 | ||
| Fig. 8 Characteristic ORR curve of a single catalyst. This figure has been adapted/reproduced from ref. 163 with permission from Elsevier. | ||
Generally, in RDE-based measurements, a thin film is fabricated, which was proposed by Gloaguen et al. (1994).165 Subsequently, the expansion of this method was reviewed by Schmidt and Gasteiger.166 Consequently, electrocatalysts are normally put in a water/alcohol/Nafion mixture to obtain a homogeneous ink. Actually, Nafion acts as a glue material to adhere the catalyst on the RDE during electrode rotation sets. There are two ways to add Nafion, either dilute Nafion solution is added directly to powder/solvent mixture at some stage in making the ink or just directly dropping Nafion on the catalyst film to make a Nafion coating. In either option, the quantity of Nafion must be very low to reduce extra dispersion, which limits the amount of O2 and decrease in IR content. Generally, the Nafion film thickness must be less the 0.2 μm when it is spread on the catalyst film or the quantity of solid Nafion must be less than 20 wt% of the catalyst weight when it is put in the ink. The obtained ink is deposited on glassy carbon electrode (GCE), which is known as the working electrode, to form catalyst films. To mollify the mass transfer in ORR activity measurements, the GCE is rotated at different rotation speeds to increase the O2 mass transfer rates at the electrode. The inherent ORR catalyst activity (kinetic current without mass transfer effect) can be calculated using the Koutecky–Levich (K–L) relation:
| 1/j = 1/jk + 1/jl,c = 1/jk + 1/0.62nFAC0 × D02/3v−1/6ω1/2 | (34) |
where, j, jk, and jl,c are the calculated i, kinetic i, and diffusion-limited i, respectively. The dispersion-limited current is calculated using the electron transfer (n) number, and Faraday constant (F), electrode geometric area (A), concentration of dissolved O2 in solution (C0*), diffusion coefficient of O2 (D0), kinetic viscosity of solution (v), and rotation speed of electrode (ω). jk is normally calculated using the K–L plot (j−1vs. ω−1/2) at different ration speeds. On the other hand, it can also be calculated using the calculated diffusion-limited current at fix rotation speed (normally, 1600 rpm). The K–L equation is ideally applied when the surface of the fabricated thin film on a GCE is smooth, under laminar flow hydrodynamics.
The catalyst thin film quality strongly affects RDE-based kinetic current measurements. Therefore, the fabricated catalyst film should be very thin, uniform, and very smooth. In case of thick films, mass transport is restricted throughout the film, which causes partial use of the catalyst. For PGM-based electrocatalysts, especially Pt, the thickness of the catalyst thin film on the GCE is generally calculated considering the quantity of carbon black. In the case of loading Pt electrocatalysts with different quantities, it must be considered that the weight of carbon black must remain the same on the working electrode because it will probably retain same the catalyst thin film thicknesses. This is noticeable in the case of a non-uniform, rough thin film surfaces for RDE calculations since the K–L relation is generally applied for a smooth film surface on the GCE. To get good and repeatable results by RDE calculations, the first thing is to make a well dispersed catalyst ink. However, due to the different surface natures of support materials used for catalytic measurements (such as carbon black or noncarbon) with electrocatalysts, it is not easy to develop a general method for all types of electrocatalysts. Besides, water and ethanol/isopropanol are generally use to make the ink because they help to keep the carbon surfaces wet. The water/alcohol ratios strongly depend on the type of carbon and electrocatalysts used, and also the pre-treatment conditions applied, such as annealing and acid treatment. Sometimes, even with a fine ink obtained using well-dispersed electrocatalyst and support material particles, a uniform catalyst film is not certain. The synthetic method, applied drying conditions, and environmental circumstances strongly affect the film quality. Therefore, Garsany et al.167,168 introduced new way to reproduce fine value electrocatalyst films by rotating the GCE at 700 rpm and adding the ink dropwise on top of the GCE surface. The normal variation was a lower electrochemical active area (ECA) and ORR value in the case when electrocatalyst films on GCE were prepared via the rotational drying process, and their activities were 70% greater than previously applied methods for the preparation of films. Similarly, Ke et al.169 also introduced the “intermittently micro-contact coating fine droplets” technique, in which the GCE can be uniformly covered with about 3 × 103 tiny droplets (3 nL per droplet). A solution for the reproducibility problem was also suggested by Shinozaki et al.,170 which when applied, they found that drying the electrocatalyst thin film in isopropanol gas atmosphere could help to make an excellent film with good ORR activity. The Nafion ionomer binder used for catalyst films has a negatively effect on the calculated catalytic activity. Therefore, due to the block effect using Nafion ionomer, the specific activity for Pt/C at 0.9 V was only 0.15 mA cm−2 Pt.149 However, most electrocatalysts have been evaluated using an RDE rather than real fuel-cell devices, i.e., MEA. It is well-known that good ORR activity from RDE tests cannot always transfer good MEA performance since the latter is in a much more complicated environment. Specifically, the RDE setup is more simplified and ideally designed to evaluate the intrinsic electrochemical property of electrocatalysts. Its limitation is also obvious, where it is not able to evaluate ion transfer, water management, architecture stability, and particularly mass transfer, which becomes even more severe for carbon-based metal-free cathodes due to their significantly thick catalyst layer. More importantly, there is very limited fundamental understanding of this gap because of much less effort placed on this.171
4.2. Fuel cell efficiency
The fuel cell operating efficiency by converting chemical energy of a fuel into electricity is the ratio between generated electrical energy and supplied fuel oxidation. The simulated thermodynamic efficiency, ηtherm, is defined as:| ηtherm = (Qreact − Qlat)/Qreact = ΔG/ΔH = Wel/Qreact | (35) |
| ηv = Ui/E° = UiEMF | (36) |
Hence, the η of fuel cells can be written as, η = ηtherm × ηv. For H2/O2 fuel cells, the rate of EMF = E° is 1.229 V. In an actual system, it is important to know the total efficiency of the fuel cell, “ηtotal”, not only depends ηtherm, and ηv, but also on two other features, e.g., efficiency because of reactant consumption or coulombic efficiency, ηcoul; and the efficiency of design, ηdesign (ηtotal = η × ηcoul × ηdesign). All these relations show that to improve the fuel cell efficiency, firstly, we should increase the following three factors, first ηv (via electrocatalysis), second ηcoul (through selectivity/tolerance of materials), and third ηdesign (through system and layer by layer structure design).172 Here, we discuss in detail about the electrochemical properties of 2D materials, the ORR, OER, HER, etc. The integration of fuel cells in the electronics community faces numerous challenges, and a few of them are as follows:
1. Synthesis of suitable electrodes materials, especially also applicable to flexible electronics.
2. Replacement of high cost noble metals and their alloys as electrocatalysts, to make the fuel cell-based technology accessible to common people.
3. The requirement of decreasing metal electrode poisoning and enhancing their long term stability.
To solve these issues, a new category of electrocatalytic materials that is inexpensive, highly efficient and high sustainable need to be synthesized considering fuel cell-based technology is the future of humanity, where common people can benefit from it. Regarding the solution to the aforementioned issues, 2D materials are perfect to tackle these problems and make electron transfer easier in both fuel oxidation and ORR. Also, 2D materials should have potential because of proton membranes and their high hydrogen ion conductivity. This, together with water impermeability, methanol, and H2 may resolve the setback in future fuel cells and electrode poisoning. In next section, we explain in detail the electrochemical applications of 2D materials for energy production and storage.
4.3. Engineering protocols for 2D electrocatalyst formation
In contrast to bulk materials, 2D materials are also preferred monomers for constructing ordered composite electrocatalysts. The 2D family also has enhanced electron and mass diffusion/transportation compared to other types of material structures such as their bulk counterparts, nanoparticles and nano-arrays. Moreover, when using normal electrocatalytic materials in the form of shells/hosts, the catalyst causes aggregation and/or detains them in small sizes or for a while does not homogeneously combine/coat. These problems limit the effective number of electrocatalytic active sites for a long period, and hence their efficiency decreases. Alternatively, the 2D family is perfect catalysts because of their simple and clear crystalline arrangement, well-matched to micro-fabrication encapsulation, and simple to manage using just one variable by just constructing a bridge between requirement and electrocatalyst design.173 Moreover, the atomic-scale thickness of 2D materials may have some structural defect symmetry or functionalization may increase the electron DOS and improve the activity. Significantly, in 2D materials, their electronic structures and DOS can mainly be adjusted via coupling with hetero-atoms or boundaries or varying their lattice constants by applying pressure. Thus, the future interest in 2D materials is because of their novel structure with moderate electronic nature, which is important for different devices applications, especially catalyst applications, comparatively resulting in other active sites and improved inherent activity than conventional electrocatalysts. The catalytic properties of some of 2D materials are due to three aspects, their SSA (e.g., 42600 m2 g−1 for monolayer graphene),174 mechanical properties and conductivity (thermal and electric).175 The maximum surface to bulk ratios maybe the reason for their high density of surface active sites, which favors surface-active applications. Also, their outstanding mechanical properties result in their durability and their thermal conductivity makes the diffusion of heat produced during exothermic reactions easy.
Additionally, the tunable electronic properties of 2D materials can tailor their catalytic performance.175 In 2D nanomaterials with moderate electrocatalytic applications, the most important parameters are the adsorption energy (microscopic property), number of active sites, reaction kinetics (macroscopic property) and moderate conductivity. The obvious rates of catalytic response are mainly reliant on extrinsic properties (crystalline morphology, etc.) and inherent structure (work function, d-band position, etc.) of the catalyst, followed by the calculated transitional adsorption energies, kinetics, and reaction energy barriers. Considerable progress has been achieved in experimental nanotechnologies based on DFT simulation, and particularly their combination, resulting in new approaches for catalytic methods at the atomic/molecular level. Therefore, different synthetic protocols based on the modification of electrocatalysts such as heteroatom doping, tuning band gaps via defects, and interfacial band gap tailoring, have been applied to further boost the electrocatalytic activity of 2D nanomaterials. Fig. 9 shows how the electrocatalytic properties of 2D materials can be enhanced, which can be separated into two groups, growing active surfaces (top semicircle) and increasing intrinsic catalyst activity (base semicircle). These methods will help with detail explanations of proposed electrocatalyst principles for specific catalytic process. For example, for OER electrocatalysis, most metal-free nanosheets and 2D MXenes have been found to be promising alternatives to the benchmark catalysts.95
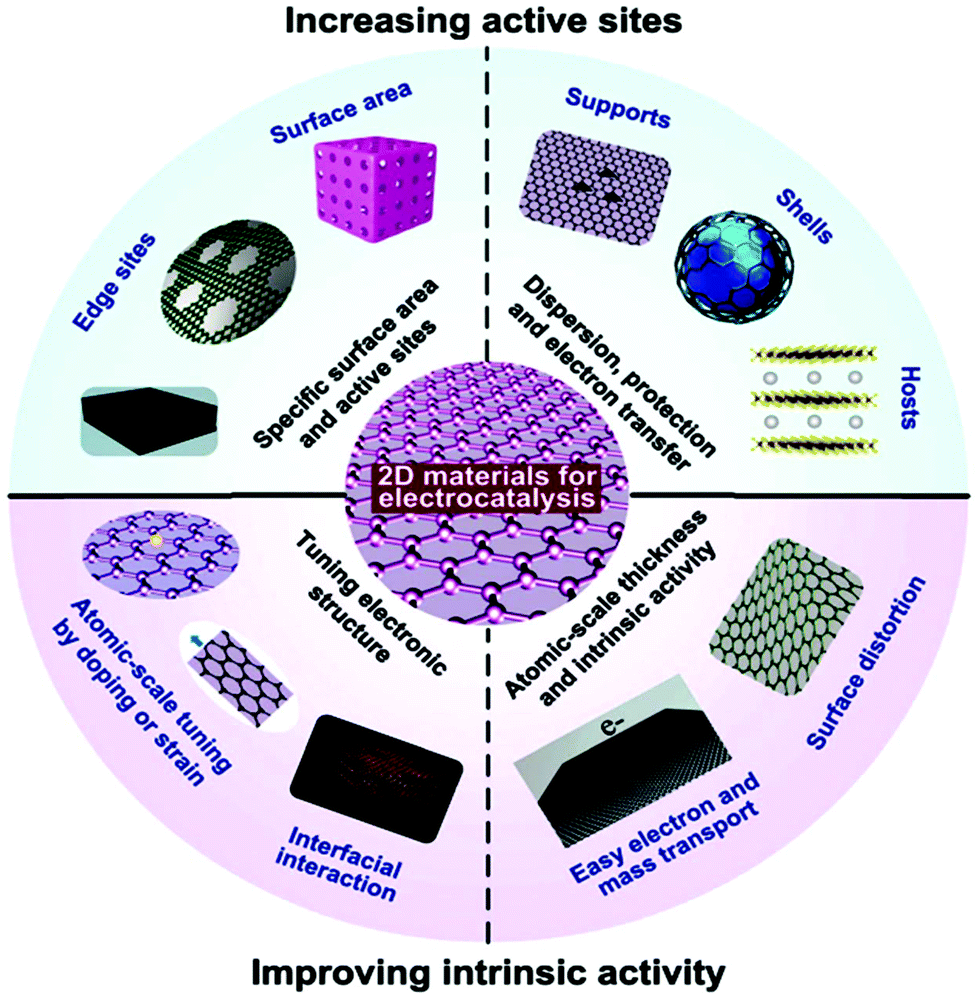 | ||
| Fig. 9 Electrocatalysis scheme showing the importance of 2D materials as electrocatalysts. This figure has been adapted/reproduced from ref. 173 with permission from the Royal Society of Chemistry. | ||
After the introduction of graphene in 2004, other 2D materials have been investigated and their potential applications studied in various research fields.80 Therefore, Jiao et al. carried out DFT simulations to establish an accurate structural description, and electrochemical experiments to determine the origin of the ORR and HER activity in heteroatom-doped graphene. The formed doped graphene with B, N, O, S, and P showed different physicochemical characteristics and was applied to study the comparison between morphology, SSA, and defects of the different heteroatom-doped samples (Fig. 10(a and b)). Also a theoretical-based study was conducted based on different molecular models to compare computational outcomes with experimental ones (Fig. 10(c)). The calculated free energy illustration for the HER, ORR and experimentally measured visible catalytic activity are shown in Fig. 10(d–g). By comparing the proper theoretical and experimental results (e.g., ΔG and i0, respectively), a volcano plot can be built, which shows the intrinsic activity trend and gives predictions about catalysts with superior performances (Fig. 10(h and i)). Basically, the volcano plot is associated with the Sabatier principle, according to which the highest catalytic activity can obtained when the catalyst surface has suitable binding energies for reactive intermediates. Therefore, in the case of loosely bonded intermediates, surface activation and reaction are very difficult. On the other hand, in the case of the too strongly bonded intermediates, they may engage all existing active sites and poison them. Fig. 10(h and i) show that all the samples are situated on the right side of the volcano, which represents that intermediate adsorption is comparatively weak. Thus, obviously, if a catalyst has a ΔG value in the center of volcano (i.e., small ΔG value, stronger adsorption in doped graphene), it will own a larger i0 value from both computational and experimental sides. By relating the molecular orbital theory (MOT), a catalyst design method to modify graphene-based materials can be proposed (nearer to the volcano tip). Besides graphene, several new types of 2D materials have also been studied for electrocatalytic applications.176 In comparison to other electrocatalytic materials, the two very significant structures of the 2D family in catalysis are their variable and consistently exposed lattice planes and exclusive electronic levels. 2D materials such as MoS2 exhibit great activity for the HER since their H2 adsorption Gibbs free energy (ΔGH*) is merely 0.08 eV, which is lower than that of the benchmark Pt (ΔGH* ≈ 0.09 eV).160 Here, we discuss in detail the electrocatalytic applications of 2D materials.
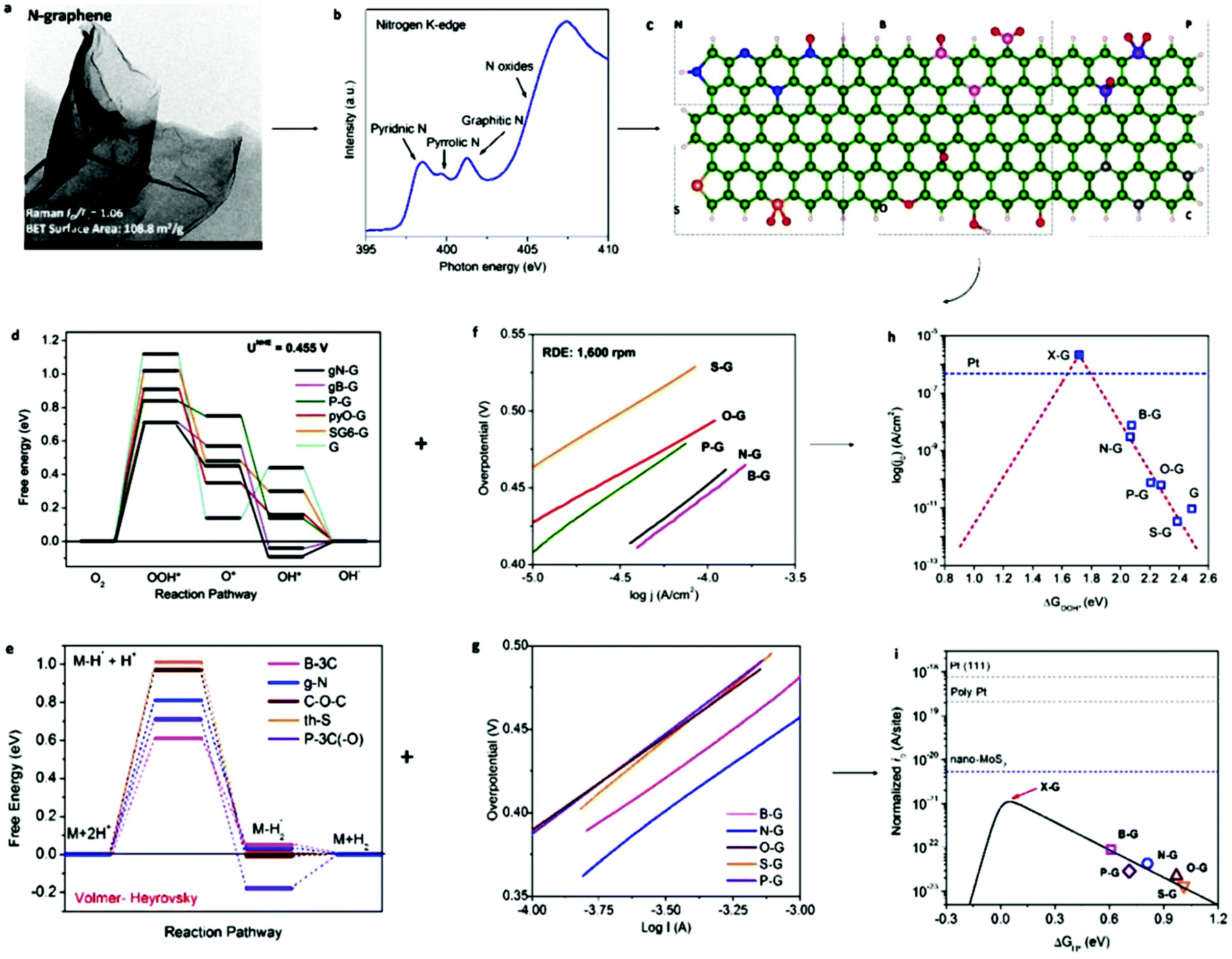 | ||
| Fig. 10 (a) TEM image of N2-graphene; with inset showing ID/IG and SSA values. (b) N2 K-edge NEXAFS spectrum. (c) Schematic heteroatom doping structure. (d) ORR and (e) HER free energy diagrams at equilibrium potential for diverse models. (f) ORR and (g) HER Tafel plots. (h) ORR and (i) HER volcano plots. This figure has been adapted/reproduced from ref. 128 with permission from the American Chemical Society. | ||
5. Oxygen reduction reaction (ORR) for 2D materials95
5.1. Prospects for replacing noble metals by graphene family
The graphene family presents active, low-cost, metal-free substitutes for PGM electrocatalysts for the ORR in sustainable energy production.177,178 This alternative choice is raised because of their novel structural types and numerous wonderful properties, e.g., large SSA, and elevated density of active sites.179 Significantly, they also have moderate conductivity and high mechanical strength, and under a moderate environment, thermal/mechanical/electrical stability (tolerance to harsh environments), which are beneficial for their applications as catalysts and catalyst support materials.95 Recently, much research has been performed to confirm the especially superior improvement by the 2D family towards replacing benchmark PGM-based materials for the ORR. High N-doped graphene180 and co-doping of B in N-graphene176 have verified steady development, which can get comparative performances to the benchmark materials. Similarly, in another study, defects were produced in graphene and the ORR results were almost similar to that of Pt/C.181 Very high N-doping in graphene is a bit challenging, but it can match its ORR results to that of Pt/C.182 However, codoping using S, Fe, and N hardly results in activity exceeding that of Pt/C.183 Here, we will discuss in detail the ORR properties of 2D materials, starting with the graphene family and will discuss their defect chemistry.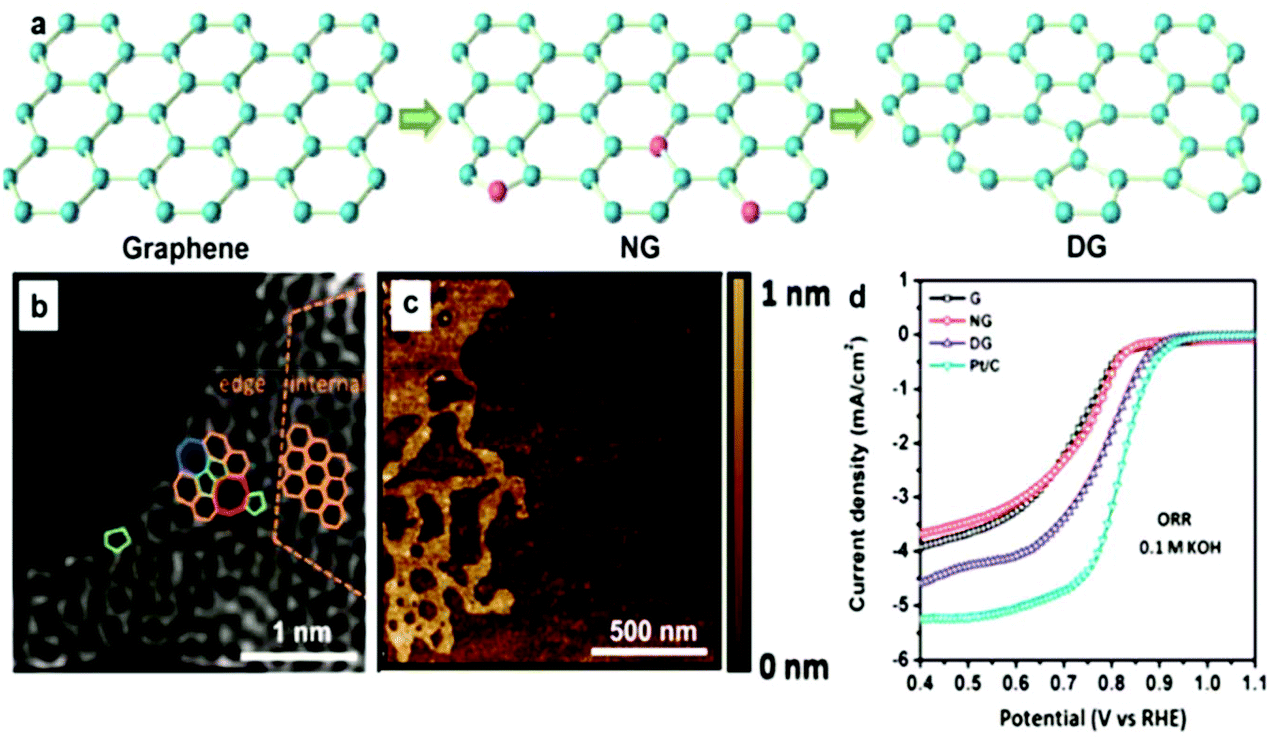 | ||
| Fig. 11 (a) Schematic representation of graphene with defects. (b) HAADF figure of defective graphene with hexagons, pentagons, heptagons, and octagons, shown by orange, green, blue, and red colors, respectively. (c) AFM image of graphene with defects. (d) LSVs for pristine, N-doped and defective graphene for the ORR. This figure has been adapted/reproduced from ref. 181 with permission from Wiley-Blackwell. | ||
Regarding ORR applications, the electron-deficient B atoms assist the chemisorption of negatively polarized O atoms and encourage O–O splitting. Recently, Qiao et al. doped B in graphene and showed that it exhibited the lowest ORR activation barrier free energy, resulting in its higher catalytic activity compared to P- and S-doped graphene.178 The O2 adsorption on B-doped graphene was calculated to continue via the end-on dioxygen intermediate, form elevated energy intermediate for dissociative system (I) and hydroperoxo species for associative method (II),192 which can largely activate the ORR process. On the other hand, the B content also had a limit, if the B content >140 ppm, then it causes a decrease in ORR activity in alkaline electrolyte due to an increase in the electrical resistivity.193
On the other hand, N2 doping provokes polarization in graphene sheets because of the higher electronegativity of N2 (3.04) than that of C. The N doping causes opening of the band gap, which makes it semiconducting.194 There are three types of primary N-bonding configurations observed, basal plane pyrrolic N, quaternary N (central graphitic), and edge pyridinic N. The sp2-coupled quaternary N and pyridinic N have a small influence on graphene, while sp3-hybridized pyrrolic N causes a distortion in the planar geometry of graphene. N-Doped graphene semiconductor is related to the N-bonding configuration. For quaternary N, the three (sp2) valence electrons of N2 form three bonds with adjacent carbon atoms, one (2pz) orbital occupied in p hybridization, and the fifth one is delocalized in the CB. The n-type doping in quaternary N leads to the donation of 0.5 electrons to the graphene lattice.195 On the other hand, pyridinic and pyrrolic N-atoms located at defective vacancies cause p-doping by withdrawing graphene electrons.196 The carrier concentration increased to 2.6 × 1013 cm2 (4 times that of pure graphene) using a very small doping concentration, i.e. 0.6% atom of quaternary N-doping.197 Dai et al. synthesized N-doped graphene and studied its ORR catalytic activity (2010),198,199 and showed that the quaternary N and pyridinic N fraction can make the ORR activity possible. For further explanations about these doping, Ruoff et al. further studied doped graphene in detail. They suggested that the quaternary N can only settle the limiting i, but on the other side, the pyridinic N can mainly develop the onset potential in the case of electrochemical ORR activity.200 Quaternary N may boost the density of electrons by p-orbital delocalization (n-type doping), causing improved nucleophile strength in the adjacent carbon, and hence will promote the O2 adsorption, which will further increase the ORR activation in alkaline electrolytes.200 In a more detailed study, it was suggested that the pyridinic N can further enhance the active surface for boosting the ORR activity in N-graphene.201 The importance of N-doping was also studied by Zhang et al., where they applied g-C3N4 as a template and source of N2 to synthesize highly porous N-doped graphene, with ORR catalytic activity analogous to the benchmark Pt/C in alkaline solution.182
Similarly to the previous studies on doping in graphene, the effect of P doping in graphene was also studied, which causes sp3 hybridization and tetrahedral analogue structure along three available carbon atoms. Due to the longer bond length (1.77 Å) of P–C than C–C, the P atoms stick out from the graphene plane by around 0.09 Å, where the P electronegativity (2.19) decreases compared to that of C (2.55), and the C–P polarity is opposite to the C–N bond. When contrasting to 0.5 electron, quaternary N relocates to the p orbitals, and P just donates about 0.2 electrons (Hirshfeld population analysis).202 Thus, due to the weaker electron contribution than N-doped graphene, P-doped bilayers show n-type behavior, and more stable in an O2 environment than n-type doping resultant from N-doping, and with higher mobility. P-Doped graphene has a high ORR performance with a more positive η and higher i.203 To explain this theoretically, Wu et al. demonstrated that the ORR occurs through the 4e− pathway.204 Similarly, recent DFT simulations by Yang et al. suggested that the ORR on P-doped graphene first occurs through the 2e− pathway, forming OOH transitional species, and then through the 4e− pathway via the O–O bond in OOH. The second step concerning OH reduction to H2O, with an energy barrier of 0.88 eV (very large), is suggested to be the rate-limiting step. The P atoms and their nearby C atoms are recommended to be more suitable as adsorption and active sites in the ORR. The P atoms may also function as a bridge, promoting electron transfer from C to adsorbates.205
Besides the aforementioned nonmetallic dopants in graphene, it is also very significant to study and discuss the doping of metals, e.g., Fe, Co, Ni, and Cu,206,207 and metal oxide (MnO2)208 in graphene, which may further boost the catalytic activity of doped graphene. Therefore, Pumera et al. doped a small quantity of Mn (18 ppm, 0.0018 wt%), which led to a potential shift by 80 mV in the ORR.208 Similarly, substitution of multiple hetero-atoms (B/N, S/N, P/N, I/N, B/N/P, N/P/S, N/Fe, and N/S/Fe) may modify the electronic structure of graphene.95 This can create a synergistic effect and engineer new properties. Fig. 12 shows Co-doped B in N-doped graphene, and Qiao et al. showed it will be very helpful in the ORR in alkaline solution. Fig. 12(a) shows that the η of B, and N-doped graphene is about 110 mV and E1/2 about 150 mV, and that Pt/C has a negative value.176 Co-Doping with S and N further affected the electron donor and acceptors,209 and enhanced the active sites.210 Zhou et al. demonstrated a codoping effect using aminothiazole-based Fe, S, O, and N co-doped graphene, AT-N/Fe-G,183 with a doping quantity of 6.5%, 6.8%, 1.5%, and 2.6% for N, O, S, and Fe, respectively. These co-doped graphene showed good ORR activity in both alkaline and acidic conditions compared to that of the benchmark 20% Pt/C. Regarding the stability in acidic solution, the potential of AT-N/Fe-G shifted positively by 12 mV after 10000 cycles, but up to date, the potential of Pt/C shifted by 58 mV.
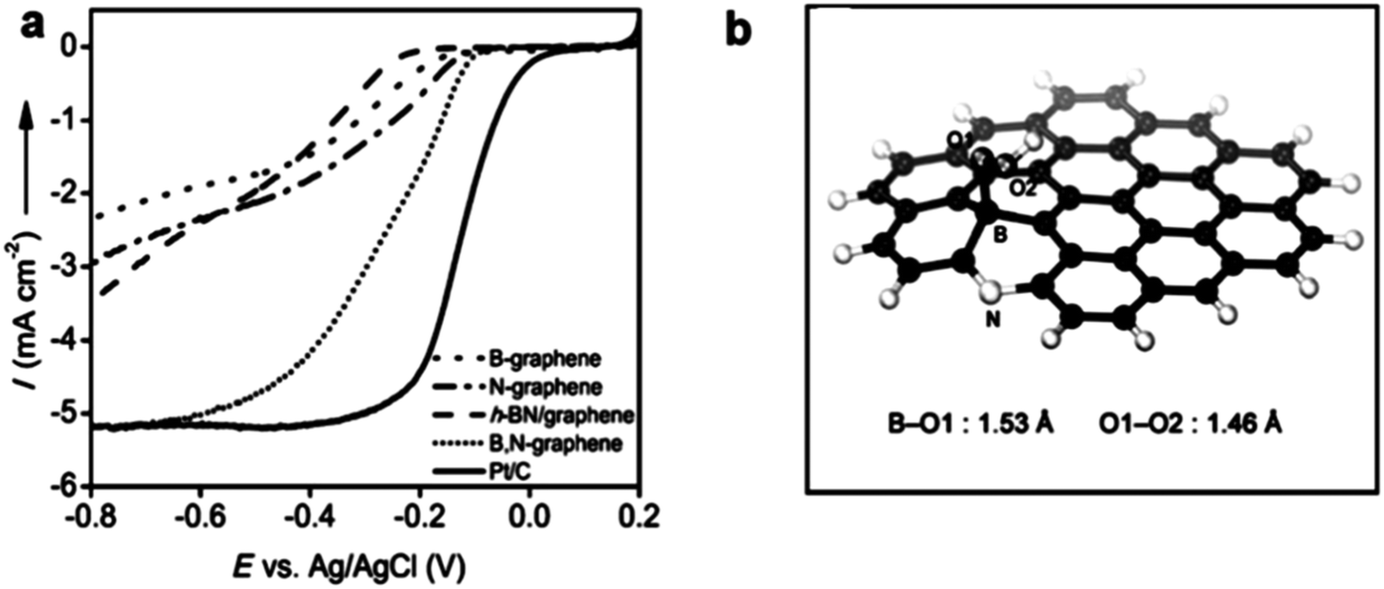 | ||
| Fig. 12 (a) LSVs curves at rotating rate of 1500 rpm, in O2-saturated alkaline solution (0.1 M KOH scan rate: 10 mV s−1). (b) HO2 adsorbed on B, N-graphene optimized arrangement structure. This figure has been adapted/reproduced from ref. 176 with permission from John Wiley and Sons Ltd. | ||
Theoretical studies based on DFT calculations show that dopant and doping vacancies strongly affect the ORR activity. A theoretical method based on free energy adsorption intermediates (O*, OH*, and OOH*) was introduced to calculate the ORR activity on graphene and its derivatives.178 Generally, DFT simulation-based experimental investigations for ORR activity of mono-element doping in graphene display the following trend: N-graphene ≥ B-graphene > P-graphene > S-graphene. In the case of di-atom-doped graphene, the activity changes as (P,N)-graphene > (B,N)-graphene > (S,N)-graphene. The best metal-free 2D catalyst demonstrated to date is N-doped porous graphene with a higher η of 0.076 V versus Ag/AgCl and limiting i of 5.79 mA cm−2 than that of the benchmark 20 wt% Pt/C (0.105 V vs. Ag/AgCl; 5.72 mA cm2). However, the ternary, Fe, S, and N-doped graphene still shows better ORR performances than N-doped catalysts. The E1/2 = 0.926 V vs. RHE for this ternary doped graphene is higher than that of Pt/C (0.907 V vs. RHE).95
Moreover, the addition of two or multiple components endows enhanced properties possibly due to the synergistic effect, and thereby improves the performances of MoS2-based composite materials. In addition, doping of MoS2 with other materials or atoms can alter the basal plane or enlarge the MoS2 interlayer spacing in nanosheets, and thus modify their d-band electronic properties. Here, first we discuss the most promising applications of the graphene family in composites with mayenite electride, C12A7:e−, for the ORR in fuel cells, which was introduced for the first time by our group.1 In this work, Karim Khan et al., for the first time, initiated a facile synthetic method,1,6–9 and the synthesized nanosized C12A7−xSnx:e−, where the doping level, x = 0.20 to 1 (thereafter, Sn-doped C12A7:e−) composite exhibited a high SSA of 244 m2 g−1 and increasing conductivity of 24 S cm−1, 68 S cm−1, 190 S cm−1 and 290 S cm−1, with an Sn doping level of x = 0.2, 0.4, 0.8, and 1, respectively. Fig. 13(a) show the CV curves, where the cathodic reduction currents revealed obvious diffusion-controlled ORR peaks at 0.78 V, and 0.8 V (vs. RHE) in O2-saturated 0.1 M KOH solution for Sn-doped C12A7:e− with and without rGO coating, respectively. The diffusion-limited i increased with an increase in the rotation rate, as usually observed (Fig. 13(b and c)). These results demonstrate the adequately high intrinsic activity without/with rGO coating for the Sn-doped C12A7:e− composite in the ORR.
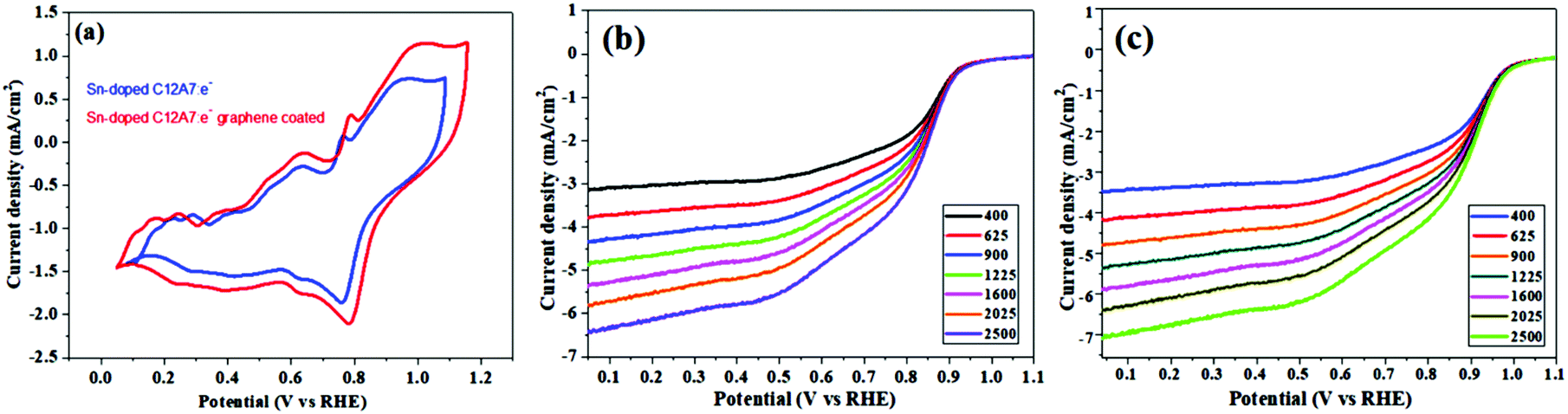 | ||
| Fig. 13 (a) Sn-doped C12A7:e− CVs curves (with and without rGO coated, with doping level, x = 1) at 50 mV s−1 scan rate in O2-saturated 0.1 M KOH electrolyte. Sn-doped C12A7e− (x = 1) (b) without rGO coating and (c) with rGO coating in O2-saturated 0.1 M KOH with various rotation rates at a scan rate of 10 mV s−1. This figure has been adapted/reproduced from ref. 1 with permission from the Royal Society of Chemistry. | ||
Fig. 14(a) shows that the i of Sn-doped C12A7:e− is approximately similar to that of the benchmark 20% Pt/C, but that of the rGO-coated sample is about 5.9 mA cm−2 higher. This may be because of the work-function difference between rGO and Sn-doped C12A7:e−, which is the reason for the transfer of electrons from the Sn-doped C12A7:e− to rGO.211 Consequently, the Sn-doped C12A7:e− composite with moderate conductivity, led to a low resistance and higher binding energy (BE), minimizing the unfavorable adsorption of intermediates, resulting in enhanced catalytic properties.212 These results demonstrate satisfactory high intrinsic activity with rGO-coated Sn-doped C12A7:e− in the ORR. The suggested number of exchanged electrons was 4, representing that the rGO-coated Sn-doped C12A7:e− composite followed the four-electron pathway, with water as the byproduct (Fig. 14(b)).213–215Fig. 14(c) shows that the relative current without and with rGO-coated Sn-doped C12A7:e− did not change after 11 h, and about a 59% decrease was observed for the 20% Pt/C electrode.213 Furthermore, the rGO-coated Sn-doped C12A7:e− exhibited no change in its LSV curve upon the addition of CH3OH.213,216
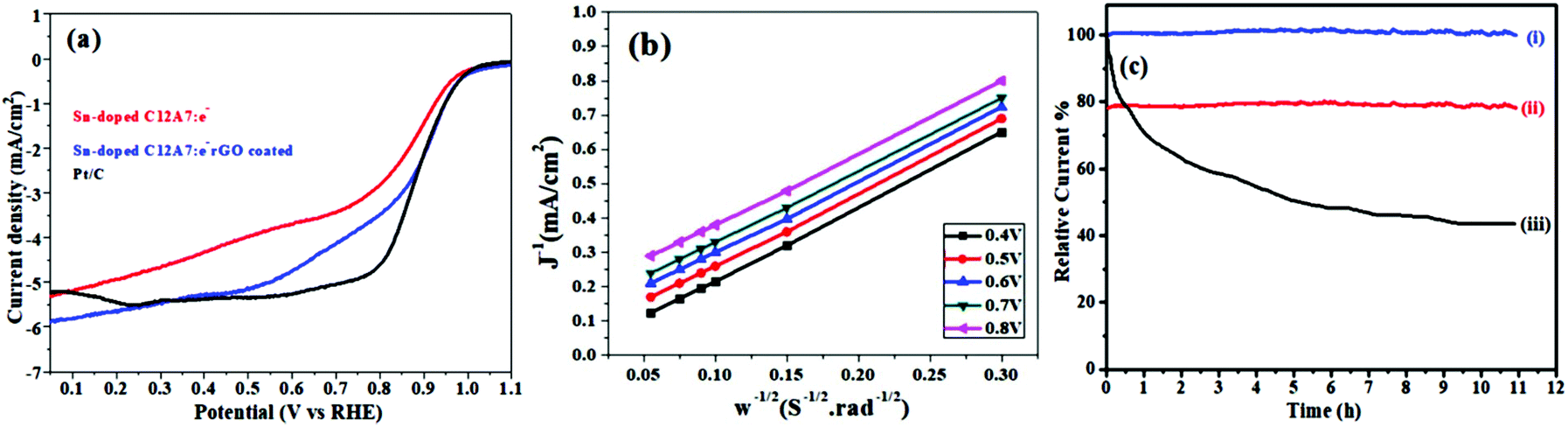 | ||
| Fig. 14 (a) ORR polarization curves of without/with rGO coated Sn-doped C12A7:e−, and Pt/C, in O2-saturated 0.1 M KOH, @1600 rmp, and sweep rate of 10 mV s−1. (b) rGO coated Sn-doped C12A7:e− K–L plots, and (c) Sn-doped C12A7:e− (with and without rGO coating, doping level, x = 1) chronoamperometry and Pt/C curves. This figure has been adapted/reproduced from ref. 1 with permission from the Royal Society of Chemistry. | ||
Although, a higher temperature generally supports the removal of free O2 species from C12A7 and improve the electrical conductivity of materials, in anticipation of certain edges.218 Zhou et al. theoretically calculated ORR measurements for N-doped graphene supported by MXenes (Ti2C, V2C, Nb2C, and Mo2C monolayers) (Fig. 15(A) and (B)). Graphitic sheets hybridized with V2C (G/V2C) and Mo2C (G/Mo2C) MXene monolayers had remarkable ORR catalytic activities. The most active site was the hollow-site C atoms located close to the pyridinic N dopants, giving rise to the low overpotentials of 0.36 V (G/V2C) and 0.39 V (G/Mo2C) (Fig. 15(C)), which are competitive to the benchmark Pt (0.45 V). The strong electronic coupling between MXene and graphitic materials as a potential multifunctional hybrid is promising for flexible energy devices. Additionally, Xie et al.219 experimentally calculated the ORR activity for Pt and Ti3C2Tx composite, which were stable even after 10000 cycles, and the half-wave potentials were constant (Fig. 15(D)), but Pt/C lost 21 mV (Fig. 15(E)). By compositing alkalization-intercalated MXene (Ti3C2(OH/ONa)2) with Ag composites, the urchin-structured MXene-Ag0.9Ti0.1 bimetallic nanowire featured pronounced electrocatalytic activity for the ORR (Fig. 15(F)),220 with onset- and half wave-potentials higher than that of the benchmark 20 wt% Ag/C and Ag-based catalysts.217Table 1 describes the details about the ORR properties of graphene-based 2D materials in alkaline medium. rGO-coated Sn-doped C12A7:e− exhibited increased catalytic activity for the ORR mostly due to the balanced relationship between the number of active sites and the electron transportation and stability. With increasing heat-treatment, the graphitization degree increased in the rGO-coated on Sn-doped C12A7:e− electride, which is beneficial for the transportation of electrons.
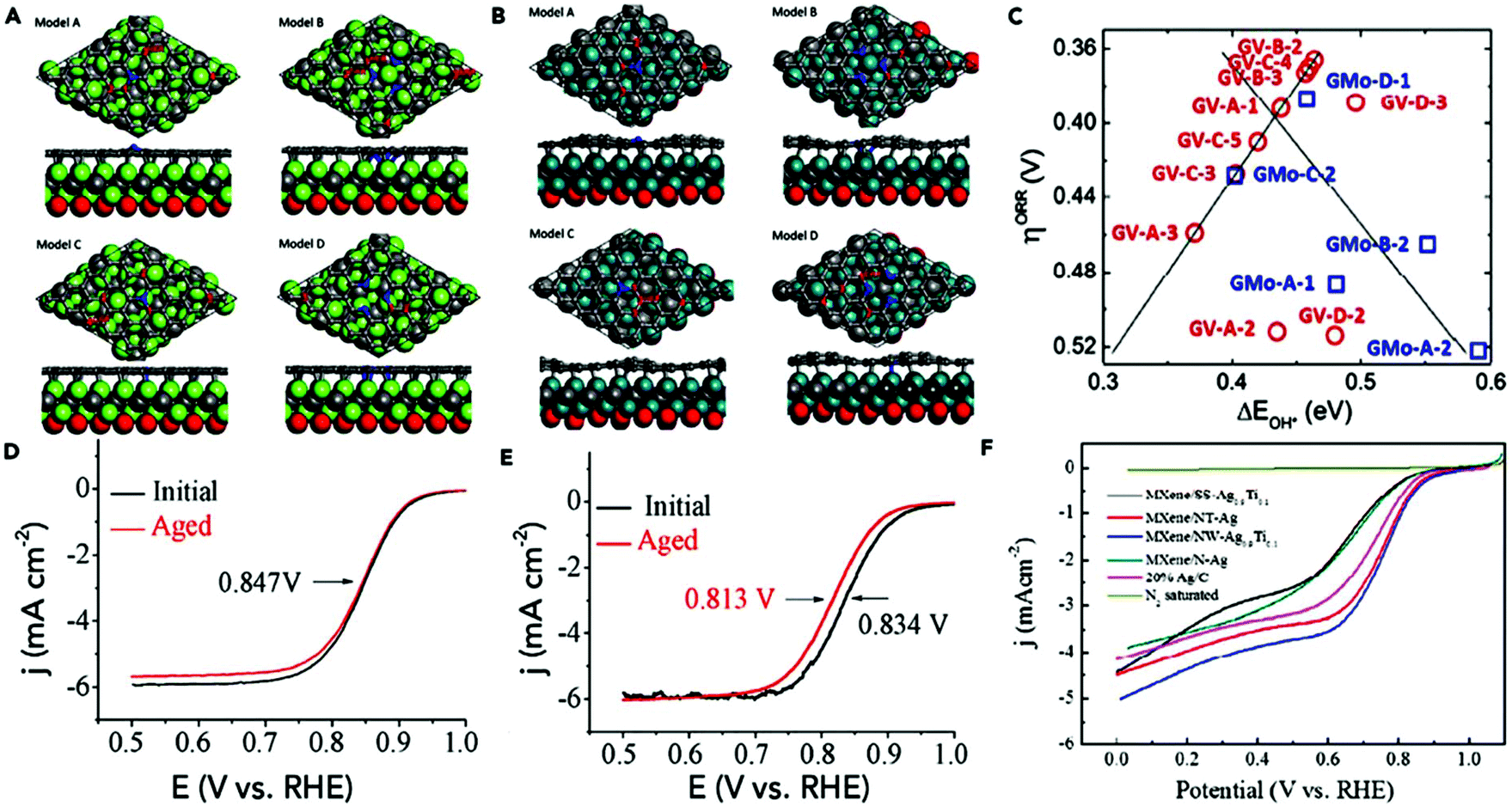 | ||
| Fig. 15 MXenes as catalysts for the ORR. (A–C) Four different models of N-doped graphene on (A) V2C and (B) Mo2C MXene monolayer. (C) Volcano plot of ORR overpotential and ηORRvs. OH* binding energy. The black box indicates the dimensions of the supercells. The red numbers label the C sites. C, N, O, V, and Mo atoms are denoted by gray, blue, red, green, and turquoise, respectively. ORR current on (D) Pt/Ti3C2X2 and (E) Pt/C catalysts before and after the accelerated durability tests. (F) ORR polarization curves of MXene-Ag0.9Ti0.1 nanowire and other control samples in 0.1 M KOH. This figure has been adapted/reproduced from ref. 217 with permission from Elsevier Inc. | ||
| Catalyst | Synthetic method (precursor) | Electrochemical performance | Ref. |
|---|---|---|---|
| Defect graphene | Ar plasma etching (GO) | Onset/half-wave potential: 0.912 V, 0.737 V, electrons transferred: 3.85 (0.4 V vs. RHE) | 221 |
| Annealing at 1150 °C in N2 (N-doped graphene) | Onset/Half-wave potential: 0.91 V, 0.76 V electrons transferred: 3.87 (0.50–0.65 V, vs. RHE) | 181 | |
| B-Doped graphene | Annealing at 900 °C in Ar (GO and B2O3) | Onset potential: 0.029 V i @ 0.3 V: 2.38 mA cm−2 (vs. NHE) | 178 |
| Annealing at 1200–1600 °C in N2 (B4C) | Overpotential difference relative to 20% Pt/C: 59 mV n: 3.6–4.2 (0.3 to 0.7 V vs. Ag/AgCl) Limiting current: 16.32 mA cm−2 (0.6 V vs. Ag/AgCl) | 222 | |
| N-Doped graphene | Fluorination and then annealing at 800 °C (reduced GO, XeF2, and NH3) | Onset potential: 0.05 V (vs. Ag/AgCl) i @ 1600 rpm: 4.99 mA cm−2 | 180 |
| Annealing at 700 °C (reduced GO and NH3) | Onset potential: 0.91 V, i @ 300 rpm: 1.4 mA cm−2 at 0.5 V (vs. RHE) | 223 | |
| Hydrothermal method (180 °C) (GO and NH3) | Onset potential: 0.08 V, i @ 0.3 V: 1.67 mA cm−2, electrons transferred: 3 (0.32 to 0.7 V) vs. Ag/AgCl | 224 | |
| Porous N-doped graphene | Annealing at 800–1000 °C in the mixture of H2 and Ar (porous Ni sheets and pyridine) | Onset potential: 0.08 V, i @ 0.4 V: 8.2 mA cm−2n: 3.9 (0.4 V) vs. Ag/AgCl | 225 |
| N-Doped porous carbon nanosheets | Hydrothermal treatment and then annealing at 900 °C in N2 (porous g-C3N4 and glucose) | Competitive with Pt/C catalysis Onset/Half-wave potential: 0.076 V, 0.21 V, electrons transferred: 3.8–3.92 (0.35 to 0.6 V vs. Ag/AgCl) Tafel slope: 143 mV dec−1 | 182 |
| P-Doped G | Pyrolysis at 1000 C in Ar (GO and triphenylphosphine) | Onset potential: 0.92 V, electrons transferred: 3.0–3.8 (0.5–0 V) vs. RHE | 203 |
| S-Doped G | Cycling of S-G composite-based Li–S batteries (S-G composite) | Onset/Half-wave potential: 0.15 V, 0.37, ORR reduction peak: 0.34 V, V (vs. SCE), electrons transferred: 3.13 (0.8 V) vs. SCE | 226 |
| Mn/reduced GO | Staudenmaier oxidation then hydrazine reduction | Onset potential: 183 mV (vs. Ag/AgCl) | 208 |
| B/N co-doped graphene | Ionic liquids in a Wurtz-type reductive coupling (CCl4, K, and C1C4ImBF4) | ORR reduction peak: 0.314 V, half-wave potential @ 1600 rpm: 86 mV relative to 40% Pt/C, i @ 1600 rpm: 5.2 mA cm−2, electrons transferred: 3.84–3.88 (0.8 to 0.4 V vs. SCE) | 227 |
| Two-step pyrolysis (dicyandiamide, GO, and boric acid) | Onset/Half-wave potential (@ 900 rpm) 0.86 V, 0.61 V, ORR reduction peak: 0.61 V, (vs. RHE), mass activity @ 0.75 V: 0.53 mA mg−1 | 209 | |
| S/N co-doped graphene | Pyrolysis at 800 °C in Ar (dopamine-cysteine-reduced GO) | Onset potential: 0.002 V, ORR reduction peak potential: 0.213 V, electrons transferred: 2.98 to 3.36 (0.30 to 0.60 V) vs. Hg/HgO | 228 |
| P/N co-doped graphene | Two-step pyrolysis (dicyandiamide, GO, and phosphoric acid) | Onset/Half-wave (@ 900 rpm) potential: 0.87 V, 0.64 V, ORR reduction peak: 0.64 V, mass activity @ 0.75 V: 0.80 mA mg−1 (vs. RHE) | 209 |
| (P/N co-doped graphene)/carbon nanosheets | Pyrolysis (GO, polyaniline and phytic acid) | Onset/Half-wave potential: 1.01 V, 20 mV, ORR reduction peak: 0.85 V, i @ 0.6 V: 5.56 mAcm−2; Tafel slope: 51 mV dec−1n: 3.96 | 229 |
| I/N co-doped graphene | Annealing at 900 °C in Ar (iodine, poly(aniline), and activated G) | Onset potential: 0.08 V, i @ 0.7 V: 11.76 mA cm−2, electrons transferred: 3.93 (0.7 V vs. Ag/AgCl) | 230 |
| I/P co-doped graphene | Reflux heating (GO and PI3) | Onset potential: 0.202 V (vs. Ag/AgCl) | 231 |
| N/P/S tri-doped graphene | Pyrolysis at 800 °C in N2 (GO, triphenylphosphine, and thiourea) | Onset potential: 0.03 V, ORR peak: 0.20 V (vs. Ag/AgCl) i: 6.41 mA cm−2 | 232 |
| N/P/F tri-doped graphene | Pyrolysis at 950 °C (polyaniline, GO, and NH4PF6) | Half-wave potential: 40 mV, electrons transferred: 3.85 (0–0.6 V vs. RHE) | 233 |
| N/Fe co-doped graphene | Pyrolysis and activation (Ferrocene, rGO, and NH3) | Onset/Half-wave potential: 0.09 V, 0.29 V, i: 15.7 mA cm−2, n: 3.83 (0.25 V) vs. Ag/AgCl | 234 |
| N/S/Fe tri-doped graphene | Polymerization and then annealing at 900 °C in Ar (carbon black, 2-aminothiazole, and FeCl3) | Competitive with Pt/C under acidic and alkaline conditions Half-wave potential: 0.926 V, mass activity @ 1.0 V: 0.56 A g−1n: 3.97(vs. RHE) | 183 |
| P-MoS2 | Thermolysis process (280 °C) (C3N4, MoO3, thiourea, and red phosphorus) | Onset/Half-wave potential: 0.96 V, 0.80 V, electrons transferred: 3.4–3.6 (0.6 to 0.8 V vs. RHE) | 235 |
| O-MoS2 | Pyrolysis synthesis followed by O incorporation (ammonium molybdate, thiourea, melamine, and H2O2) | Onset/Half-wave potential: 0.94 V, 0.80 V (vs. RHE), i: 3.49 mA cm−2 | 236 |
| MoS2/N-doped porous carbon nanosheets | Functionalization and then pyrolysis (4-iodophenylfunctionalized MoS2 and conjugated microporous polymer) | Half-wave potential: 0.14 V, diffusion-limited i @ 1600 rpm: 5.4 mA cm−2, i: 26.3 mA cm−2, electrons transferred: 2 4.0 (0.4 to 0.9 V) vs. Ag/AgCl | 237 |
| MoSe2/G | In situ solvothermal route (280 °C) (rGO, MoCl5, and Se) | Onset potential: 0.02 V, i @ 0.8 V: 5.35 mA cm−2, electrons transferred: 3.40 (0.2 V) vs. Ag/AgCl | 238 |
| WSe2/graphene | Thermolysis process (280 °C) (reduced GO, WCl6, and Se) | Onset potential: 0.02, i @ 0.8 V: 5.26 mA cm−2n: 3.57 (0.2 V) vs. Ag/AgCl | 235 |
| Co3O4/N-doped graphene | Hydrolysis and oxidation and then hydrothermal crystallization (GO, cobalt acetate, and NH4OH) | Half-wave potential: 0.83 V, electrons transferred: 4.0 (0.60 to 0.75 V vs. RHE) Tafel slope: 42 mV dec−1 | 239 |
| Carbon nanotubes/N-doped graphene | In situ doping during CVD growth (CH4 and NH3) | Onset potential: 0.88 V, electrons transferred: 3.22 (0.47 vs. RHE), limiting i: 132.8 mAcm−2 | 240 |
5.2. 2D-MXenes for ORR applications
MXenes based on earth-abundant elements are promising materials in the electrocatalytic field. The use of MXene materials, e.g., Ti3C2X2 (X = OH and F), as a support has two advantages in case of electrocatalytic applications. Firstly, as Ti-rich materials, they will show good durability under mechanical, acidic, and oxidative stress. Secondly, they are not just as good as carbon supports with effective conductive channels for electrons, but also efficiently avoid corrosion the support, which always happens in PGMCs/carbon-based electrocatalysts. For instance, Ag has a relatively high ORR performance and robustness in alkaline medium, but its reported electrocatalytic behavior is not satisfactory. Another brilliant plan to further boost the catalytic activity is to prepare more conductive supports coupled with some effective bimetallic catalysts. Thus, Peng et al. synthesized MXene-Ag composites by directly mixing AgNO3 and alkalization intercalated MXene (alk-MXene, Ti3C2(OH/ONa)2) solution containing polyvinylpyrrolidone (PVP) at room temperature. This unique alk-MXene-Ag0.9Ti0.1 nanowire composite with a width of 42 ± 5 nm demonstrated high activity toward the ORR with enhanced conductivity, increased abundant active sites and provided a synergistic effect. It possessed the best ORR activity with η = 0.921 V vs. RHE, E1/2 = 0.782 V vs. RHE at 1600 rpm, best structure stability and reversibility after 1000 cycles compared to the commercial Ag/C catalyst and pure Ag nanowires reported previously.6. 2D nanomaterials for oxygen evolution reaction (OER)
6.1. Prospects for replacing noble metals with graphene family for the OER
The particular attention to metal-free electrocatalysts for the OER is because of their high activity, abundance and inexpensive nature. Single-element doping such as N- and P-doping in graphene overcomes the OER obstacle, yielding η = 10 mA cm−2 (0.38 V for N-G241 and 0.33 V for P–G242) compared to that of the IrO2 benchmark (overpotential: 0.33 V). In the co-doping case, more interesting results and further improvement in OER activity were achieved.243 The activity based on electronegativity (EX) and electron affinity (AX) can be represented as ϕ = (EX/EC) × (AX/AC), where the subscript represents the element, and Xia et al. showed the binding strength and OER activity of p-block element-doped graphene.244 Plotting the lower limit of η as a function of ϕ resulted in a volcano shape for mono-elemental doping, which showed that P is the most excellent dopant in the highly active graphene 2D material for the OER.244 The OER is the reverse reaction of the ORR with 4e− transfer (Table 2).95 N-Doped graphene, TMs (Ni),83 metal oxides (Co3O4),239 composites with N-doped carbon nanotubes,245 and graphitic C, have been demostrated95 as very active catalysts in the OER process. Qiao et al. reported Ni nanoparticles co-doped in N-doped graphene (Ni/N-G)83 with OER η = 0.32 V, approaching that of the benchmark IrO2 (0.27 V). These amazingly high activities are credited to the dual active site system. Table 2 shows the 2D family with OER activities.| Catalyst | Electrochemical performance | Ref. |
|---|---|---|
| Defective graphene | Potential: 1.57 V (@ 10 mA cm−2vs. RHE) Tafel slope: 97 mV dec−1 | 181 |
| N-Doped graphene | i (@ 0.7 V vs. Ag/AgCl): 0.27 mA cm−2 | 224 |
| P-Doped graphene | Onset potential: 1.48 V, potential @ 10 mA cm−2: 1.56 V (vs. RHE) | 242 |
| (P/N co-doped graphene)/carbon nanosheets jORR, 3 mA cm−2: 0.71 V | Onset potential: 1.32 V, i: 70.75 mA cm−2(@ 0.9 V), potential: 1.57 V (@ 10 mA cm−2vs. RHE) Tafel slope: 70 mV dec−1, Difference in potential between jOER, 10 mA cm−2 and jORR, 3 mA cm−2: 0.71 V | 229 |
| N/P/F tri-doped graphene | Onset potential: 1.62 V (vs. RHE) Tafel slope: 136 mV dec−1 | 233 |
| La+ Ni-Fe LDH nanosheets | Over-potential: 0.26 V (@ 10 mAcm−2) | 246 |
| Ni–Fe LDH nanosheets | Overpotential: 0.305 V (@ 10 mA cm−2), i: 9.5 mA cm−2 (@ h = 300 mV), Tafel slope: 40 mV dec−1 | 247 |
| Ni–Co LDH nanosheets | Overpotential: 0.335 V (@ 10 mA cm−2), i: 1.5 mA cm−2(@ h = 300 mV), Tafel slope: 41 mV dec−1 | 247 |
| Co–Co LDH nanosheets | Overpotential: 0.355 V (@ 10 mA cm−2), i: 0.85 mA cm−2 (@ h = 300 mV), Tafel slope: 45 mV dec−1 | 247 |
| Ni–V LDH nanosheets | i: 27.0 1.6 mA cm−2 (@ h = 350 mV), Tafel slope: 50 mVdec−1, TOF: 0.054–0.003 s−1 | 248 |
| TiO2/NiO nanosheets | Overpotential (h): 0.32 V, potential: 1.55 V (@ 10 mA cm−2, vs. RHE), Tafel slope: 52 mV dec−1 | 249 |
| Porous b-Ni(OH)2 nanosheets | Overpotential: 0.415 V (@ 10 mA cm−2) Tafel slope: 60 mV dec−1 | 250 |
| Ni3N nanosheets | Mass activity: 572 A g−1, overpotential: 0.35 V (@ 52.3 mA cm−2), Tafel slope: 45 mV dec−1 | 251 |
| NiPS3@NiOOH nanosheets | Onset potential: 1.48 V (vs. RHE) Overpotential: 0.35 V (@ 10 mA cm−2), Tafel slope: 80 mV dec−1 | 95 |
| Porous Co3O4 nanosheets | Onset potential: 1.51 V (vs. RHE), i: 10 mA cm−2 at 1.56 V (vs. RHE) Tafel slope: 69 mV dec−1 | 240 |
| NiCo2O4 nano-sheets (O vacancies) | Overpotential: 0.32 V (@ 10 mA cm−2), i: 285 mA cm−2(@ 0.8 V, vs. Ag/AgCl), Tafel slope: 30 mV dec−1 | 252 |
| g-CoOOH nano-sheets | Onset potential: 1.47 V (vs. RHE), overpotential: 0.30 V(@ 10 mA cm−2), mass activity: 66.6 A g−1, (@ 0.3 V, vs. RHE) Tafel slope: 38 mV dec−1 | 253 |
| Ni/N-doped graphene | Overpotential: 0.32 V(@ 10 mA cm−2), i: 16.3 mA cm−2 (at an overpotential of 0.4 V) | 83 |
| Co3O4/N-doped graphene | Overpotential: 0.31 V (@ 10 mA cm−2), Tafel slope: 67 mV dec−1 | 239 |
| Carbon nano-tubes/N-doped graphene | Potential: 1.63 V (@ 10 mA cm−2, vs. RHE), Tafel slope: 83 mV dec−1 | 240 |
| g-C3N4/graphene | Onset potential: 0.58 V (vs. Ag/AgCl), over-potential: 0.539 V (@ 10 mA cm−2) | 254 |
| g-C3N4/Ti3C2 nanosheets | Onset potential: 1.44 V (vs. RHE), potential: 1.65 V (@ 10 mA cm−2, vs. RHE), i: 13 mA cm−2(@ 1.70 V, vs. RHE), Tafel slope: 74.6 mV dec−1 | 255 |
| Few-layer BP | Onset potential: 1.45 V (vs. RHE), Tafel slope: 88 mV dec−1 | 14 |
| BP bulk@Ti foil | Onset potential: 1.48 V (vs. RHE), Tafel slope: 91.5 mV dec−1 | 29 |
| IrO2 | Onset potential: 1.5 V (vs. RHE), Tafel slope: 49 mV dec−1 | 256 |
| N-Graphene/NiCo | Onset potential: 0.7 V (vs. Ag/AgCl), Tafel slope: 614 mV dec−1 | 257 |
| RuO2 | Onset potential: 1.42 V vs. RHE, Tafel slope: 76 mV dec−1 | 258 |
| Co3O4/N-graphene | Onset potential: 1.5 V (vs. RHE), Tafel slope: 67 mV dec−1 | 239 |
| N,O-Graphene/CNT | Onset potential: 0.315 (vs. Ag/AgCl), Tafel slope: 141 mV dec−1 | 259 |
| Co3O4 | Onset potential: 1.5 V (vs. RHE), Tafel slope: 234 mV dec−1 | 260 |
| Pt/C | Onset potential: 1.5 V (vs. RHE), Tafel slope: 317 mV dec−1 | 261 |
6.2. Other 2D material families for the OER
MXenes are highly active electrocatalysts due to their hydrophilic surface with good electrical conductance and durability. Metal-free nanosheet electrocatalysts do not achieve activity equal to or greater than that of the benchmark IrO2 electrocatalyst for the OER, but by mixing 2D g-N3C4 and Ti3C2, very active synergy was obtained, basically matching the activity of carbon-supported IrO2.255 The hybrid film of overlapping g-C3N4 and Ti3C2 2D sheets (TCCN), where in this film, Ti3C2 is coupled with g-C3N4 through Ti–N interaction, formed a porous free-standing film with a good OER performance, where at i = 10 mA cm−2, the potential value was 1.65 V, which is less than that of the IrO2/C benchmark (1.70 V) and comparable or even better than that of the benchmark PMCs. The catalytic nanosheets further showed high stability with only a slight anodic current decrease after 10 h reaction, accompanied by no change in morphology. Similarly, the activity of NiCo-LDH matched that of the state-of-the-art IrO2 electrocatalysts for the OER, and the activity of NiFe LDH 2D sheets was better than that of IrO2.247 The γ-CoOOH 2D sheets were approximately 20-fold more active than bulk γ-CoOOH and better than IrO2 by two-fold. Therefore, NNMC nanosheets are more active in the OER then the benchmark IrO2. 2D Ni-based composites are normally available in large quantities, containing 2D sheets with distinct 3d electrons and unique features e.g., orbitals, which make them superior OER electrocatalysts, e.g., Ni-based LDH 2D sheets (Ni–Fe and Ni–Co LDHs)246,262 are extremely active and used in water oxidation, reaching i = 10 mA cm−2 at a small overpotential (305 mV for Ni–Fe LDH and 335 mV for Ni–CO LDH). Ni–Fe and Ni–Co LDHs exhibit better OER activity than IrO2 and Co–Co LDHs.247 The Ni–V LDH monolayer also demonstrated i = 27 mA cm−2 (57 mA cm−2 after ohmic-drop correction) at an overpotential of 350 mV, similar to the most active Ni–Fe-LDHs for the OER in basic solution.248 The O2 vacancies confined in the 2D sheets of NiCo2O4, ZnCo2O4 and Co3O4 have a lower adsorption energy for H2O, causing improved OER activity (lower overpotential, smaller Tafel slopes, moderate current densities and larger TOF turn-over frequency).95 For example, porous 2D Co3O4 nanosheets showed an electrocatalytic current of up to 342 mA cm−2 at 1.0 V (vs. Ag/AgCl), about 50 and 30 times greater than that of bulk Co3O4 and the state-of-the-art 20 wt% Pt/C, respectively.263 A large amount of unsaturated coordination Co3+ ions on the surface provides reactive sites, which speed up the OER process.95Similarly, γ-CoOOH sheets with a thickness of 1.4 nm were used as an electrocatalyst for water oxidation, showing high mass activity (66.6 A g−1), which was 20-fold greater than that of bulk γ-CoOOH and 2.4-fold superior to that of the state-of-the-art IrO2,264 with the best OER activity among the NNMCs sheets published in the literature. This elevated activity is mainly due to their semi-metallic properties, resulting in improved H2O electrophilicity and quicker interfacial transfer of electrons between Co ions and adsorbed –OOH intermediate species to make O2. The NiOOH species resulting from the oxidation of Ni, and Ni–O–C or Ni–N–C (between Ni and N-doped graphene bonding) are active centers, which promote the oxidation of OH to O2. The porous g-C3N4 and MXene (Ti3C2) composite consists of double-sheets, which enhance the O2 electrochemistry, giving a sharp onset potential of 1.44 V (vs. RHE) and OER currents higher than that of the benchmark 20 wt% IrO2/C.255 This electrode offers a significant synergistic effect in comparison to the separate g-C3N4 and Ti3C2 electrodes. BP has gained significant attention due to its novel properties, which also result in enhanced electrocatalytic performances. Recently, Wang et al. synthesized bulk BP, which had an onset potential of 1.49 V (vs. RHE) and a low Tafel slope of 72.88 mV dec−1, showing comparative electrocatalytic characteristics to the RuO2 benchmark catalysts for the OER, but its bulk crystal structure still suffers from low active sites. Recently it was reported that a decrease in the number of layers in 2D materials will result in further exposed electrocatalytic active surface and amplified SSA than that of their bulk counterparts. These favorable results were first observed with the native ultrathin lamellar BP structure, which can open up alternative paths in using 2D layered materials with high electrocatalytic activity. Xiaohui Ren et al. used BP for the OER as a new rediscovered layered material (Fig. 16).14
 | ||
| Fig. 16 (a) Scheme of synthesis of BP nanosheets; (b) low- and (c) high-resolution SEM images of as-prepared BP nanosheets on SiO2/Si substrate; (d) TEM image (inset in (d): SAED pattern); (e) HR-TEM image; (f) Raman spectrum; (g) tapping-mode AFM image and comparative cross-sectional analysis (red, green, and blue solid line area). This figure has been adapted/reproduced from ref. 14 with permission from Wiley-VCH Verlag. | ||
Significantly, a decrease in the thickness of BP nanosheets will create extra active sites and boost their electrocatalytic activities.14Fig. 17 demonstrates the measured polarization curves of 2D BP under different C[OH−] electrolytes (1, 0.2, 0.1, and 0.05 M) in 1 M KOH, which showed improved i and lower η = 1.45 eV than the previously obtained 1.48 eV in bulk BP.14 Interestingly, the long-term robustness of the BP nanosheets was demonstrated in 1, 0.2, 0.1, and 0.05 M KOH. The i values of BP in 1 M KOH is shown in Fig. 17(a), which fluctuate near 6.2 mA cm−2, and becomes much larger than that in 0.2, 0.1, and 0.05 M KOH. After 10000 s, no noticeable decay in the i was observed in the 2D BP in the presence of 1, 0.2, and 0.1 M KOH, while a slight decline was detected in 0.05 m KOH electrolyte. In addition, linear sweep voltammetry (LSV) of the BP nanosheets in 1, 0.2, 0.1, and 0.05 M KOH was carried out. The stability before LSV and after LSV was studied for comparison, as shown in (Fig. 17(b)). The LSV curves demonstrate the high stability of the BP nanosheets in KOH solution, and comparatively, i in 1 M KOH is rather steady at 8 mA cm−2 (under a potential of 1.8 V) presenting no noticeable decay.14Fig. 17(c) shows the long-term stability of the BP nanosheets (Fig. 18).
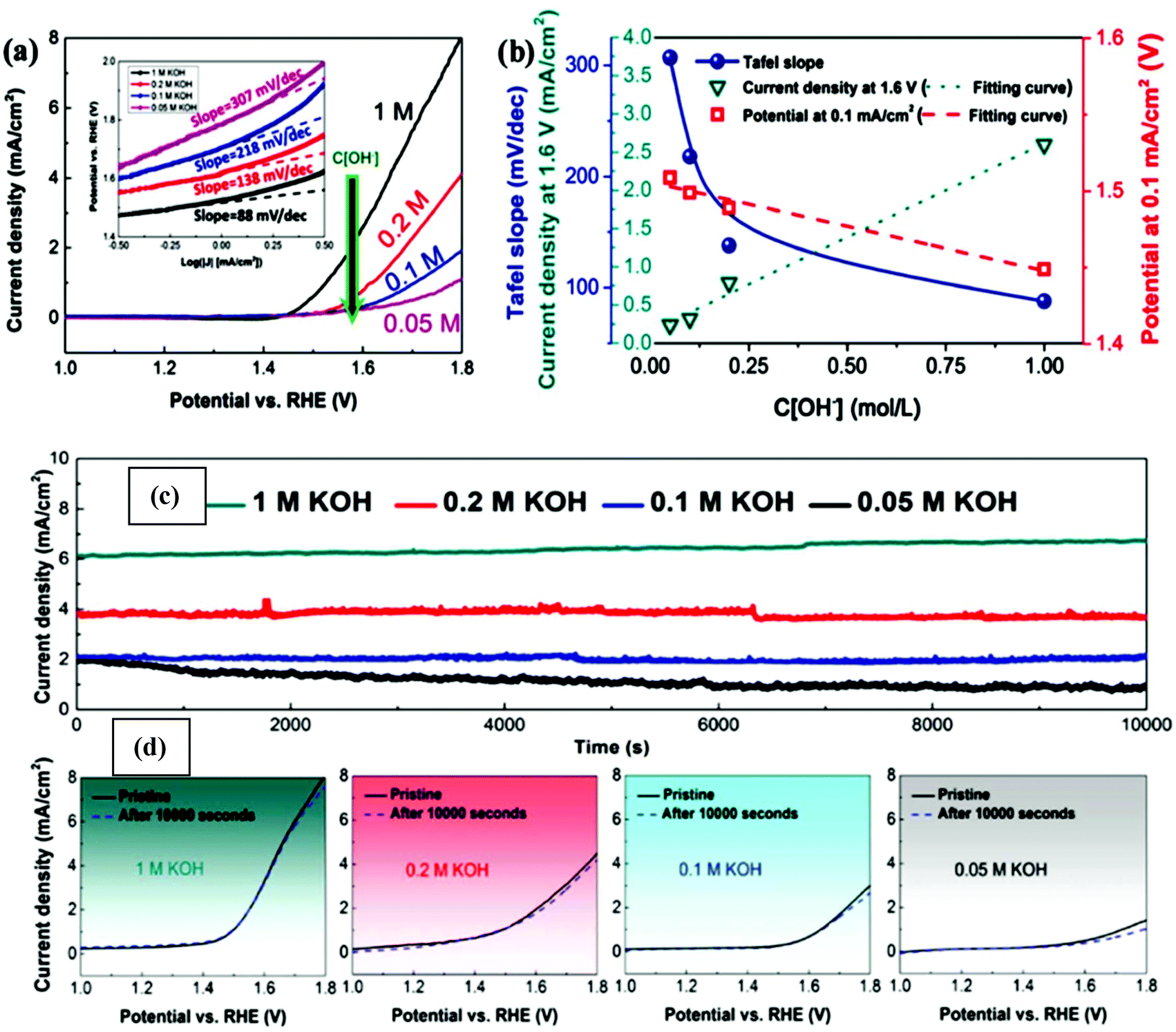 | ||
| Fig. 17 (a) Polarization curves (LSV) and inset corresponds to the Tafel plots of the BP nanosheets in KOH electrolyte with different concentrations (1, 0.2, 0.1, and 0.05 M). (b) Tafel slope at 10 mV cm−2 as a function of the concentration of OH− (C[OH−]).14 (c) Long-term stability of BP nanosheets in different basic electrolyte concentrations of 1, 0.2, 0.1, and 0.05 M KOH during the OER process. (d) LSVs of BP nanosheets in 1, 0.2, 0.1, and 0.05 M KOH before and after 10000 s measurement. This figure has been adapted/reproduced from ref. 14 with permission from Wiley-VCH Verlag. | ||
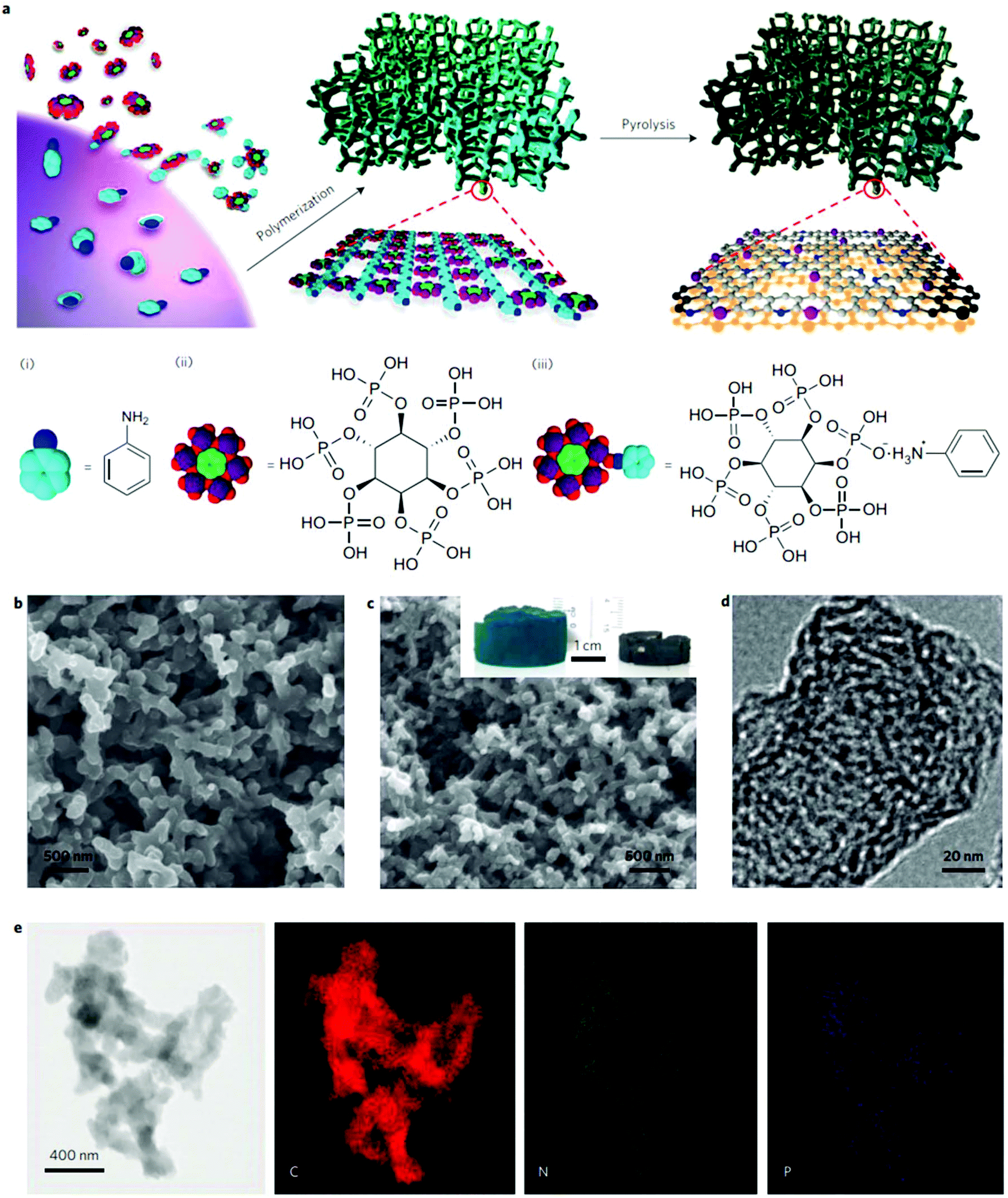 | ||
| Fig. 18 (a) Synthesis of N and P co-doped porous foam, (i) aniline, (ii) phytic acid, and (iii) complex formation, via oxidative polymerization to form a 3D PANi hydrogel cross-linked with phytic acid. SEM images of (b) PANi aerogel and (c) NPMC-1000. Inset in c: PANi aerogel digital photograph before (left) and after (right) pyrolysis at 1000 °C. (d) HR-TEM image and (e) TEM image (e, left), with equivalent element NPMC-1000 mapping images. The element mapping images in the TEM analysis for C, N and P show a uniform element distribution. This figure has been adapted/reproduced from ref. 261 with permission from Nature Publishing Group. | ||
Jingfang Zhang et al. studied the OER activity of transition metal-based electrocatalysts, especially for single precious metal atoms supported on LDHs. The single-atom Au supported on NiFe LDH (sAu/NiFe LDH) had overpotential of 0.21 V in contrast to the calculated result (0.18 V).265 We ascribe the excellent OER activity of sAu/NiFe LDH to the charge redistribution of active Fe and its surrounding atoms caused by the neighboring sAu on NiFe oxyhydroxide stabilized by interfacial CO32− and H2O interfacing with LDH (Fig. 19).265
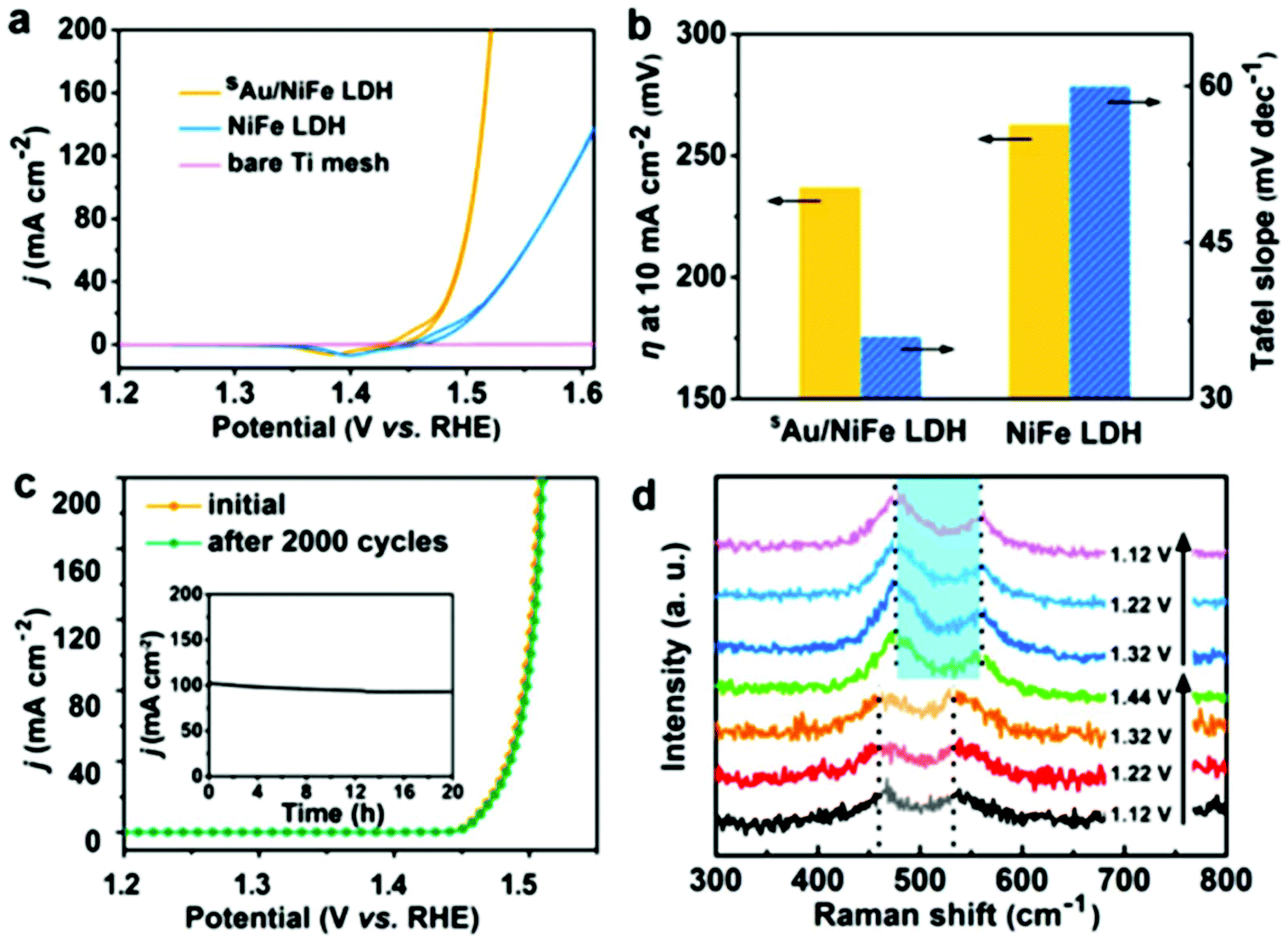 | ||
| Fig. 19 (a) CV curves, (b) Overpotential (η) at 10 mA cm−2 (left) and Tafel slope (right) for sAu/NiFe LDH and pure NiFe LDH. (c) Polarization curves of sAu/NiFe LDH before and after 2000 cycles. (d) Raman spectra of sAu/NiFe LDH at different potentials during the anodic and cathodic sweep in a CV cycle. This figure has been adapted/reproduced from ref. 265 with permission from the American Chemical Society. | ||
7. 2D-bifunctional OER/ORR electrocatalysts
The proposed principles based on theoretical calculations for ORR/OER bi-functional catalysts focus on the adsorption free energy (ΔGOH*) of OH*. Thus, Zheng et al. proposed a dual volcano plot for oxygen electrode catalyst activity. According to their calculations, they illustrated that 2D electrocatalysts with OH* adsorption inside the top region of the dual volcano plot are not good ORR or OER catalysts, but have good bifunctional activity in recoverable OER/ORR. The OER and ORR activity occur at the edges of similar structures but with dissimilar active sites. In addition, P/N co-doped carbon/graphene sheets are reported to be good bi-functional active catalysts for reversible oxygen electrodes (ROE),229 with a low overvoltage of 0.71 V for the OER, achieving 10 mA cm−2 in the OER and ORR, which is defined as the potential required to reach an i of 3 mA cm−2.261p-Valence element (N222 or B266) doped graphene has been used as a bifunctional electrocatalyst in both the ORR and OER. Theoretical-based evaluation suggests that nitrogen substitutes the carbon at the graphene edges, which results in OER and ORR low overpotentials (0.41 V OER; 0.45 V ORR), almost equal to that of the benchmark Pt electrocatalysts.224 Jintao Zhang et al. described the co-doping of N and P into mesoporous carbon foam, resulting in a great SSA ∼ 1663 m2 g−1 and good ORR and OER catalytic activity (Fig. 20 and 21). The N,P-doped carbon foam acts as air electrode in Zn-air batteries, as verified by its 1.48 V open-circuit potential, with 735 mA h gZn−1 (835 W h kgZn−1) specific capacity, 55 mW cm−2 peak power density, and 240 h stability after mechanical recharging. Two-electrode-based system rechargeable batteries could be cycled for 180 cycles at 2 mA cm−2 with satisfactory stability. DFT calculations show that the graphene periphery effects and N, P co-doping are indispensable in the catalytic performance of bi-functional catalysts.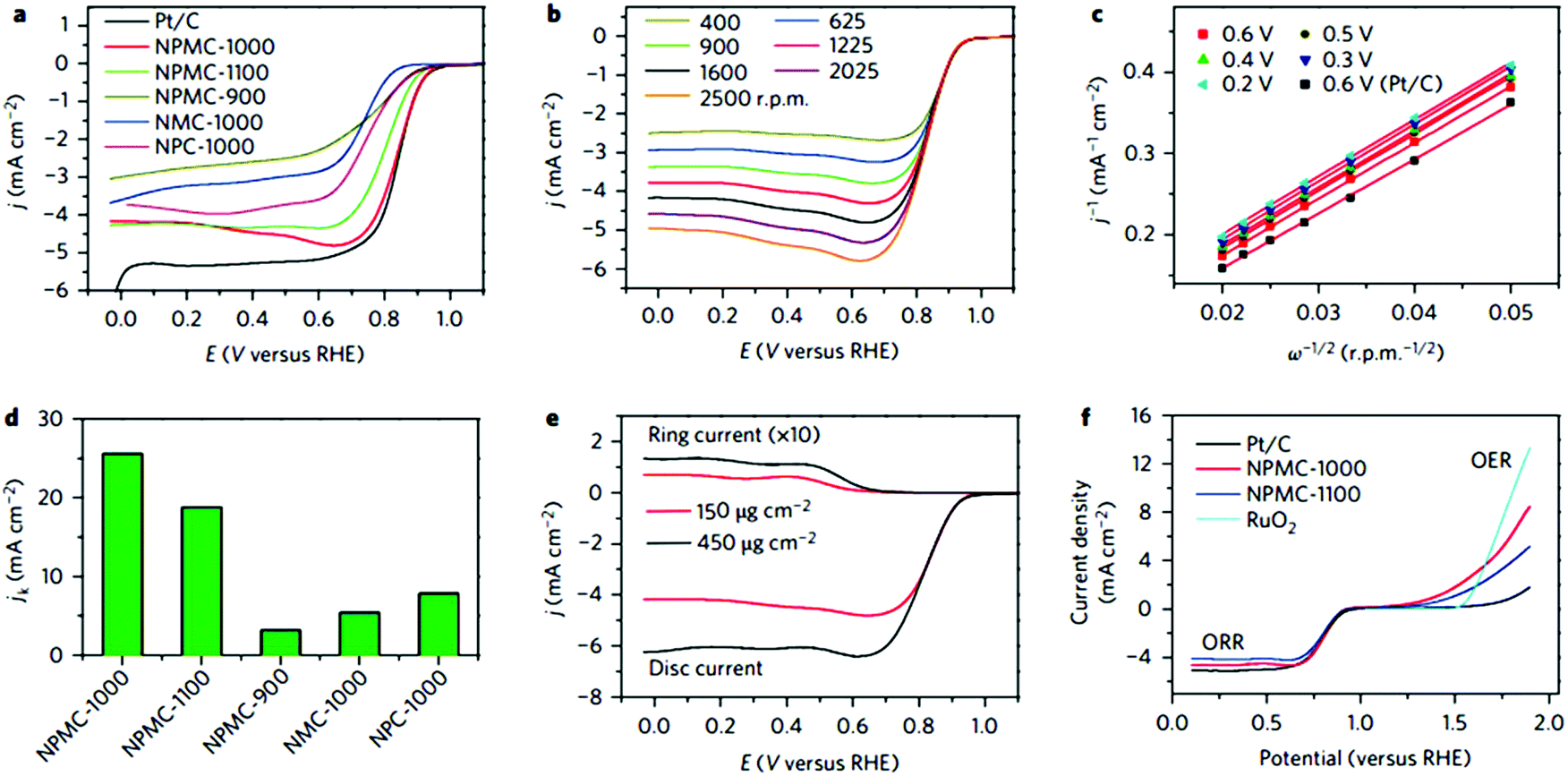 | ||
| Fig. 20 ORR and OER activity. (a) LSV curves for NPMC-900, 1000, 1100, 1000, NPC-1000 and benchmark Pt/C catalyst (1600 rpm.) in 0.1 M KOH O2-saturated solution at a scan rate of 5 mV s−1. (b) LSV curves of NPMC-1000 in 0.1 M KOH at different rotations. (c) NPMC-1000 and Pt/C K–L plots at different potentials. (d) Kinetic current of different samples at 0.65 V for O2 reduction. (e) Different catalyst loading RRDE measurements (1600 rpm.) for the ORR at the NPMC-1000 electrode. (f) LSV curves of NPMC1000, NPMC-1100, RuO2 and Pt/C catalyst (1600 rpm.) in 0.1 M KOH (5 mV s−1 scan rate). This figure has been adapted/reproduced from ref. 261 with permission from Nature Publishing Group. | ||
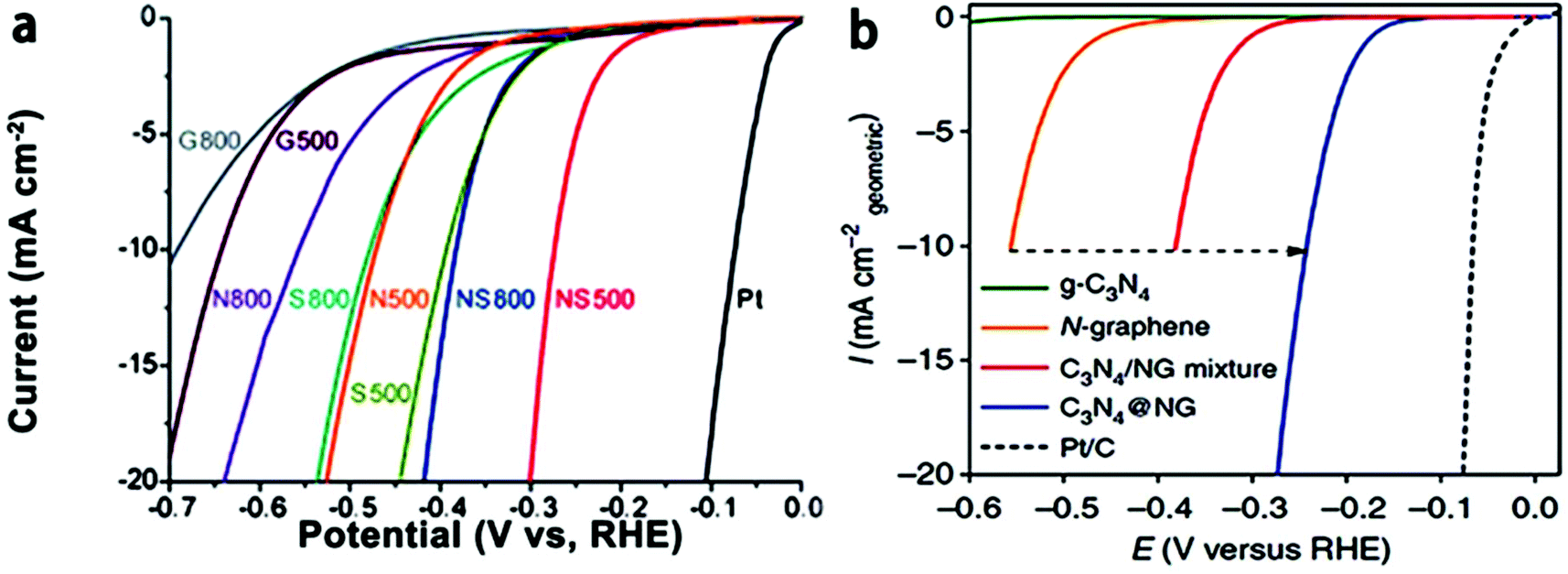 | ||
| Fig. 21 (a) CVs for doped and un-doped graphene nanoporous structure using diverse CVD heat treatments and unusual dopants. (b) HER polarization curves of the different materials in 0.5 M H2SO4, at a scan rate of 5 mV s−1. This figure has been adapted/reproduced from ref. 95 with permission from the Royal Society of Chemistry. | ||
8. 2D materials for hydrogen evolution reaction (HER)
8.1. Dependence on the characteristics of pristine 2D graphene and metallic TMDs materials for HER applications
Obviously, great progress has been achieved to discover benchmark Pt-substituting electrocatalysts for the HER, but a few problems still need to be resolved before their practical applications. Water splitting with high efficiency needs the use of electrocatalysts for minimizing the η value to drive the HER. Even though ultrathin metal nanosheets (Ru95 and Ni267) show onset potentials analogous to that of the benchmark Pt/C, the graphene family, TMDCs, and MXenes are excellent options because of their economical nature, easy availability with sufficient quantity, tailorable electronic structures, and good stability in acidic/alkaline solution. The interesting structural and potential electronic nature of 2D materials present new opportunities for discovering electrochemical reactions, particularly for the HER. Accordingly, several new studies have been reported for 2D electrocatalyst materials that can effectively catalyze the HER in both acidic and alkaline solution. The band structure and Gibbs free-energy of the transition state (|ΔGH*|) are critical issues in the HER. Thus, initially we will explain the characteristics of 2D graphene-based materials. Generally, |ΔGH*| calculations are very useful to give information about HER activity for metal and non-metal catalysts.268 Although more proficient PGM-based catalysts have a |ΔGH*| value of about 0.09 eV, pure graphene presents a very positive |ΔGH*| value (1.85 eV), showing that it is less active for the HER. However, the pioneer work completed by Qiao et al. demonstrated that B-, P-, or N-doped graphene could reduce the |ΔGH*|, and thus improve the preliminary activation state H* adsorption. In addition, pyridinic N/P dual-doped graphene was verified to exhibit a low |ΔGH*| value (0.08 eV). Comparable to the observed trend for the OER and ORR, in the HER, co-doping of graphene with N and P atoms results in a lower HER η (420 mV@10 mA cm−2) and smaller Tafel slope (91 mV dec−1) value than that of pure and singly doped graphene because of the downshift of the valence bands due to the presence of active carbons. Chen et al. also proposed that porous co-doped graphene especially by N/S, has considerably improved HER activity. The obtained overpotential was 280 mV, @ 10 mA cm−2, with 80.5 mV dec−1 Tafel slope (Fig. 21(a)).269,270 Co-doping with S may enhance the formation of graphitic-N due to the extremely low |ΔGH*| of 0.12 eV, which assists the HER. Nitrogen and sulfur co-doping was reported for carbon nanosheets, which also promoted the HER.271 Recent results from Qiao et al. showed that the combination of N-doped graphene with g-C3N4 resulted in a lower |ΔGH*| (0.19 eV) compared to that of g-C3N4 (|ΔGH*| = 0.54 eV) and N-G (|ΔGH*| = 0.57 eV). This combination of a 2D material with another 2D hybrid resulted in an estimated HER of η = 240 mV and Tafel slope = 51.5 mV dec−1, outperforming the individual components and their physical mixture, most MPMCs, and even approaching that of the benchmark MoS2 non-Pt HER catalyst (Fig. 21(b)).272 This improved HER activity was recognized due to the fundamental element electronic hybridization phenomenon, which synergistically enhances H2 ion adsorption and its ease to be reduced.160 Moreover, combining N-G with a small Co amount may create HER-active sites involving coupling effects between Co and N, resulting in considerably advanced H2-evolving activities, and outstanding performances in both N-G and Co-containing graphene.273Here, we first focus on 2D materials, especially the typical 2D graphene independent from metal and metal-based TMDs and explain their properties and applications based on combined DFT computations and experimental observations, especially for the HER.128 2D crystals have been further explored to boost their HER nature, by improving the electron transport efficiency.97 However, the HER mechanism strongly depends on the type of material; thus, it is significant to understand the fundamental properties of electrocatalysts to optimize their use in the HER. To advance the activity, we need to apply strategies to further adjust the electronic structure of 2D materials to afford abundant active sites because loss in resistance is one of the main setbacks in non-metal electrodes. Fig. 22 shows that there are four main way to enhance the HER performance of 2D electrocatalyst materials.
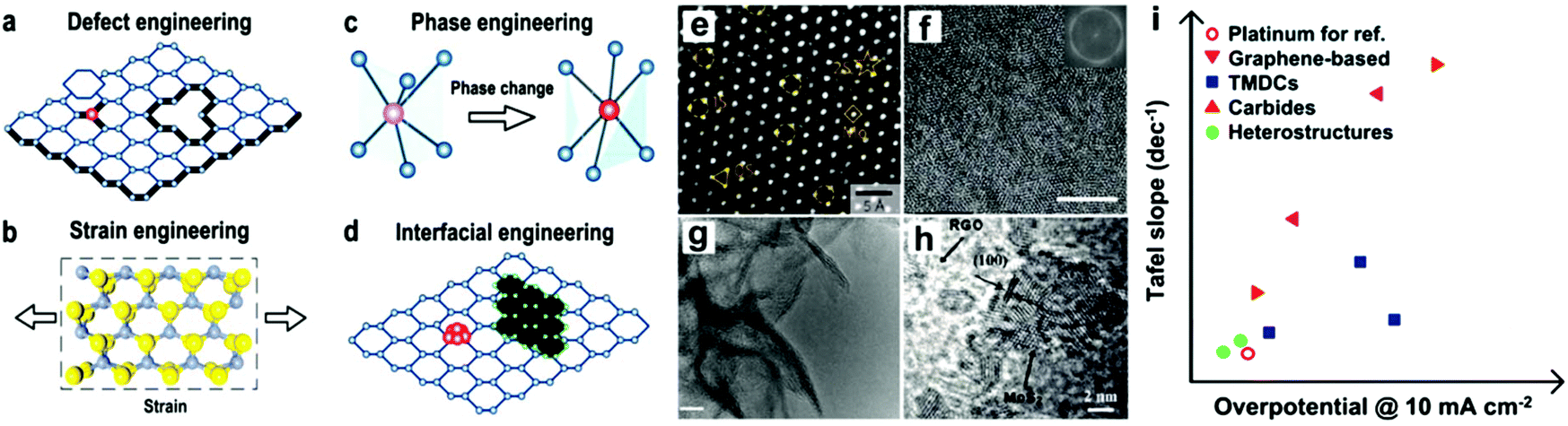 | ||
| Fig. 22 (a–d) Scheme for obtaining high efficiency 2D materials, (e) enhancing the catalytic efficiencies of MoS2 with S doping and strain, (f) O2 incorporation, (g) 1T0-MoS2, (h) Formation of heterostructures, and (i) summary of the overpotentials @10 mA cm2 with Tafel slopes of 2D HER electrocatalysts. Data points are broken down into two parameters, i.e., Volmer-Tafel route (purple) and Volmer–Heyrovsky route (blue). This figure has been adapted/reproduced from ref. 173 with permission from the Royal Society of Chemistry. | ||
1. Firstly, create surface defects (e.g., vacancies and edges) (Fig. 22(a)),274 which will cause high i with small coordinated active sites.
2. Secondly, create/introduced strain on the material (Fig. 22(b)), where strain and edge-site substitution with TMs (e.g., Fe, Ni, Cu, and Co) will further adjust the H bonding at the edges.275
3. The third is phase engineering (Fig. 22(c)).
4. Fourthly, interface modulation, by which the electronic structures and the DOS around the Fermi energy of 2D materials can be adjusted (Fig. 22(d)).
5. The fifth is the use of a highly conductive substrate or support coupling with 2D-materials.
In first case of surface defects, MoS2 is used as an example, which was initially known as a weak HER catalyst. Regarding improving the HER activity of MoS2 in acidic media, Jaramillo et al. explained the apparent contradictions160 that the HER normally occurs at the MoS2 edge sites with a less active basal plane. Thus, to get more potential use of the active edges, perpendicularly arranged MoS2 structures were fabricated, which exposed a huge quantity of edge sites, but the basal plane surface sites were not exploited and advance electron transportation to conductive substrates occurred. Using DFT simulation, Li et al.276 suggested that the fundamental MoS2 plane may become active by generating sulfur vacancies in 2D sheets (Fig. 22(e)).
Regarding exerting strain, Li et al.276 showed that the introduction of surface defects under applied strain will further boost the HER activity of MoS2 sheets (Fig. 22(e)). For example, in case of sulfur vacancies under applied strain, the fundamental plane ΔGH reached up to 0 eV and the DOS around the Fermi level increased, causing an enhancement in HER activity. Also, Xie et al.274 explained that the HER activity of 2D MoS2 was noticeably enhanced by simultaneously applying disorder manufacturing and oxygen doping (Fig. 22(f)), where, the disordered structure presented a large quantity of active unsaturated sulfur atoms, and O2 doping successfully modified the electronic structure, improving conductivity. The third strategy is phase engineering, for example, metallic 1T-phase MoS2 exhibited an enhanced HER performance compared to the 2H semiconducting phase. The main reason for this maybe that the electron transfer in metallic 1T phase MoS2 is greater at the fundamental planes than that in the 2H phase equivalent (Fig. 22(g)). The final plan related to interface modulation is to tune the electronic structures of 2D materials and their DOS near the Fermi energy. Li et al.277 observed that the HER performance in 2D MoS2 could further enhanced by synthesizing MoS2/rGO hetero-structures (Fig. 22(h)). Luo et al.278 also proved that the HER activity of MoS2 in alkaline medium can be enhanced by attaching OH nanoclusters in 2D MoS2. Moreover, besides MoS2, other 2D materials and their hetero-structures illustrate good HER activity, such as graphene, b-Mo2C, TaS2, and NbS2. The HER activities of different types of graphene, carbides, TMDCs, and 2D hetero-structures in acid solution are presented in Fig. 22(i).
8.2. Other 2D materials for HER
Graphene is a typical example of a layered material, which was first synthesized via simply exfoliation from its parent structure. Other than intrinsic (vdW gapped) 2D materials, particular types of non-layered 2D materials are emerging. Compositionally, these new types of 2D materials can be metallic, oxides, chalcogenides, or any other material with 3D-structured atoms that are made thin through gentle engineering via bottom-up synthesis. Since then, numerous 2D materials in both categories have been successfully prepared including metals, oxides, dichalcogenides, nitrides, and thiophosphates. Recent advancements in manufacturing and engineering in 2D materials show their high efficiency in HER applications. For example, naturally layered 2D C3N4 demonstrates extraordinary activity and stability for generating photo-carriers in the photocatalytic HER process, while edge-exposed 2D MoS2 has great future potential for electrocatalytic HER. Moreover, through various strategies such as adding co-catalysts and doping, defect engineering, increasing active sites, and hybridization, HER activities can be significantly enhanced.279Besides graphene, nanosheets of (N, S dual-doped) Mo2C,280 GaS,281 and TMDs e.g., MoS2, WS2, WSe2, ReS2,MoTe2, MoSe2, VS2, VSe2, and WTe2, also show HER activity.160,282,283 The layer-dependent electrocatalytic activity of MoS2 for the HER has been observed.284 The decrease in exchange i with a 4.47 factor was observed with an increase in the number of layers. Similarly, TM and chalcogen edges (MoSe2 and WS2) also show potential for the HER, while only the Se and Mo edges are active sites for MoS2 and WSe2, respectively. The W-edge sites of WSe2 and S-edge sites of MoS2, where the H binding is very weak and too strong, respectively, are unsuitable for the HER to occur.282 The experimental285 and theoretical studies based on the comprehensive performance of six TMDCs by Gholamvand et al.286 clearly showed the activity follows the trend selenides > sulphides > tellurides, with MoSe2 outperforming other materials.282,287 Monolayer VS2 is probably more important than all the above 6 TMDCs in catalyzing the HER with a performance analogous to the benchmark Pt,288 @ 10 mA cm−2i with an almost 30 mV more negative potential, and a Tafel slope of 34 mV dec−1 compared to 30 mV dec−1 for Pt.289 The TMDC edge planes are expected to support faster mixed electron transport than basal planes, and hence show higher HER activity. For example, Mo-edges with sulfide-end in 2H-MoS2 (trigonal prismatic phase) are HER active sites, but the basal planes are not.290 However, defects in MoS2 may significantly develop the HER activity of its “inert” in-plane domains.291 Furthermore electron hopping can affect the conductivity of TMDCs, which will also improve their HER properties. The meta-stable octahedral 1 T phase, relative to the trigonal prismatic 2H phase is thermodynamically stable, has intrinsically lower charge transfer resistance with elevated active sites due to strained lattice distortion, and causes a quicker HER.292 In 1T-TMDCs compared to 2H-TMDCs, their catalytic activity is independent of the edge sites.293 Especially, elevated fundamental HER activity is obtained on porous 1T-MoS2 nanosheets, with an overpotential of 153 mV@ 10 mA cm−2, 43 mV dec−1 Tafel slope, and a large electrochemical active SSA (Fig. 23).294
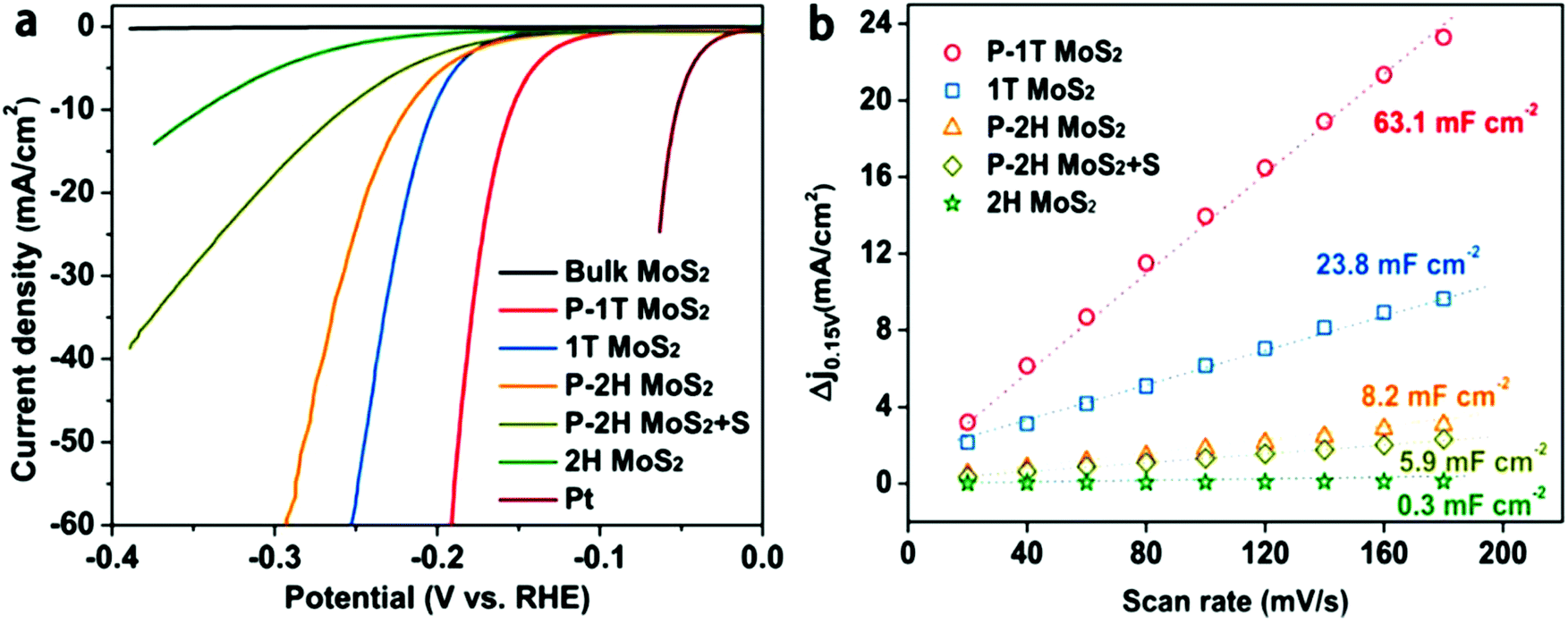 | ||
| Fig. 23 (a) iR-corrected J–V curves of porous 1T MoS2 (P-1T MoS2), 1T MoS2, porous 2H MoS2 (P-2H MoS2), 2H MoS2, porous 2H MoS2 after S compensation (P-2H MoS2 + S), bulk MoS2 and a Pt wire. (b) Extraction of the double-layer capacitance in MoS2. This figure has been adapted/reproduced from ref. 95 with permission from the Royal Society of Chemistry. | ||
Metal dopants such as Co, Mn, Cu, Ta, Ru, Fe, Rh and Ni can alter the potency of sulfur binding on edges and modify the hydrogen adsorption free energy to move toward the most favorable ΔGH*.295 However, in HER applications, Pumera et al. illustrated that bulk doped TMDCs (MoS2 and WS2) with p-doped TMDCs (Nb and Ta) showed relatively lower activity than their un-doped counterparts.296 Theoretical computation showed that strain notably altered the free energy of H2 ion adsorption at the interface of defected 1T WS2.297 The ΔGH* value was about 0.28 eV without strain and 0 eV at a strain of 2.7%. Disorder engineering permits the number of low-coordinated atoms to be enhanced, which act as active sites in the HER. The introduction of a few disorders can hinder electron transfer, resulting in a decrease in electrical conductance and catalytic activity, but some suitable disorders are favorites for an improvement in catalytic activity. Oxygen plasma treatment and hydrogen heat treatment are effective and simple experiential strategies to improve the HER activity of MoS2 single sheets.298 In this case, when the MoS2 nanosheets contain 34–40% disorder, then they can replace Pt catalysis, with an η = 160 mV more negative than that of the benchmark 20% Pt/C.274 TMDCs and graphene composites, and also doping in graphene can further boost their conductivity, which will enhance the HER kinetics.299 For example, MoS2/N-GO needed only η = 56 mV @ 10 mA cm−2, which shows good HER activity as an NPM nanosheet catalyst.299 The calculated Tafel slope for 2D/2D hybrid materials was 41.3 mV dec−1, almost similar to benchmark 20% Pt/C (30 mV dec−1). Very thin non-lamellar TMCs (CoSe2,300 NiSe2,301 and Fe–Ni sulfide302) nanosheets, having a metallic nature, are potential NPMCs for the HER. Additionally, doping pyrite CoSe2 with metal atoms, i.e., Mn, will generate more opportunities for active sites for the HER. Furthermore, Mn doping decreases the kinetic energy (K.E) barrier, promoting H–H bond formation among two neighboring adsorbed H atoms and enhancing H2 gas evolution, resulting in comparable catalytic properties for HER with benchmark NPMCs. The reported best TMDC HER catalyst is porous NiSe2 nanosheets, which are obtained from the transformation of LDH precursors,303 with a lower onset by η ∼ 90 mV. The η value required to obtain current densities of 10, 100, and 200 mA cm−2 are 135, 183, and 202 mV, respectively. In addition, well-known 2D HER electrocatalysts contain inexpensive NPMNs, e.g., NiMoNx,304 and NPMHOs, e.g., Ni(OH)2.95
Yanmei Shi et al. demonstrated the synthesis of 1T′-MoS2 crystals and studied different electrochemical microcells with individual MoS2 nanosheets, which revealed that the basal plane of 1T′-MoS2 is highly catalytically active.306 Recently, Yifu Yu et al. studied the phase arrangement, which plays a significant role in the specific formation of inorganic materials (Fig. 24).305 They reported the large-scale formation of micrometre-sized metallic-phase 1T′-MoX2 (X = S and Se)-layered massive crystals with elevated purity. The electrochemical study revealed that the basal plane of 1T′-MoS2 is much more active than that of 2H-MoS2 for the HER in an acid environment, with an onset overpotential of 65 mV and current density of 607 mA cm−2 at 400 mV, which is superior among the MoS2-based catalysts. This outstanding HER activity of the synthesized 2D materials is due to more active nature of their basal plane and improved charge transport ability of 1T′-MoS2 compared to 2H-MoS2.305 Chao Zhang et al.307 experimentally and theoretically investigated the HER performance of Co3S4 porous nanosheets with sulfur vacancies (Co3S4 PNSvac) in alkaline medium, with an onset overpotential of 18 mV and an overpotential of 63 mV for 10 mA cm−2, and large mass activity of 1056.6 A g−1 at an overpotential of 200 mV.307
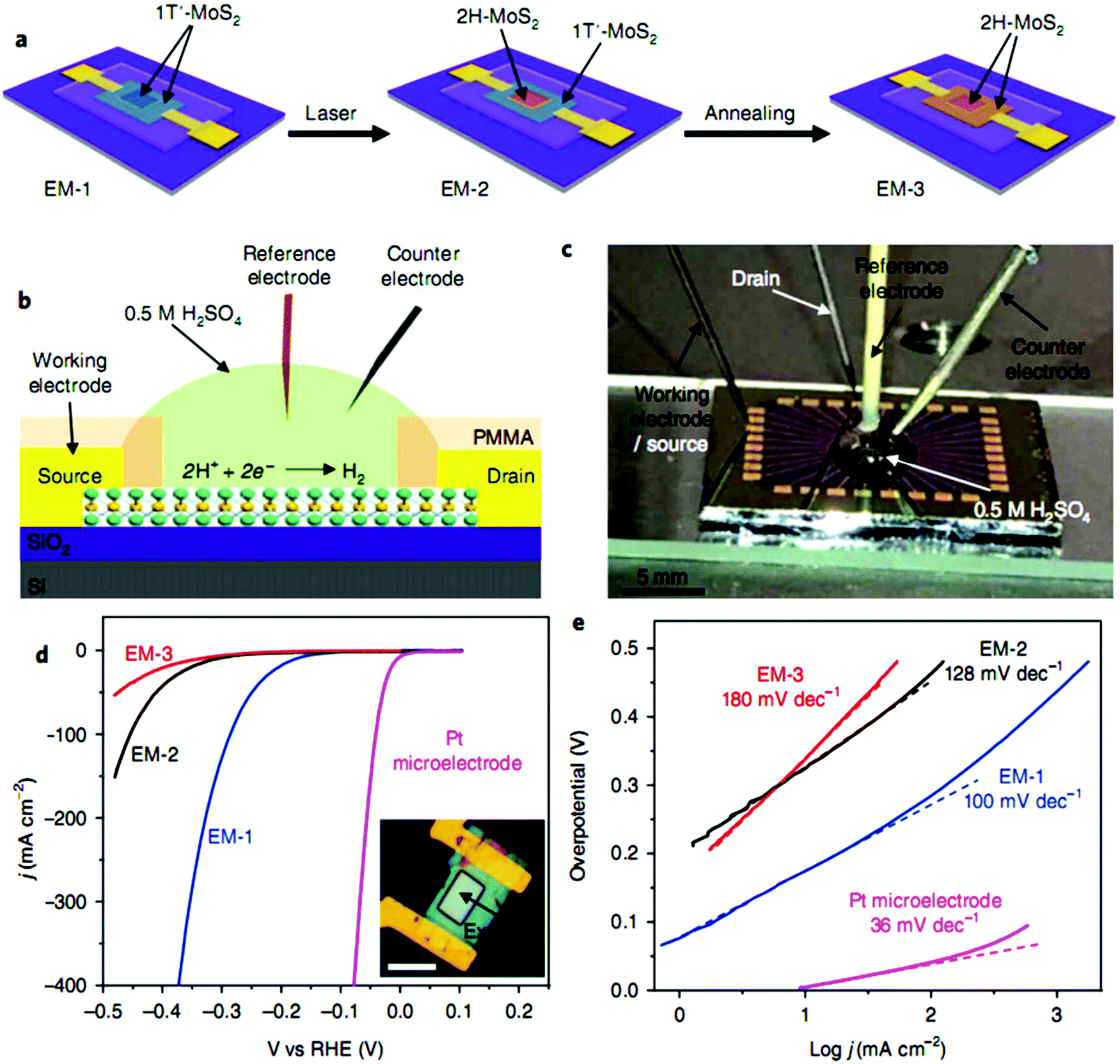 | ||
| Fig. 24 (a) Scheme showing the fabrication of 3 types of electrochemical microcells. (b) HER measurements and (c) optical view of the electrochemical set-up. (d and e) Polarization curves obtained and Tafel-slops. This figure has been adapted/reproduced from ref. 305 with permission from Nature Publishing Group. | ||
The main key to designing materials with improved HER activity is based on understanding how to organize their reactive intermediate binding energies on surfaces.2Fig. 25(a) shows the volcano plot, where each element has a different electrochemical reaction representation and show a wider structure, by which catalysts cross a broad selection of chemical reactions. Besides this volcano plot, further aspects are not present in the straightforward description model, which are necessary to quantitatively confirm the complete reaction rates. For example, deviations in kinetic barrier dimensions from one material class to the next, and therefore MoS2 has lower exchange current densities than PMCs, although it has ΔGH close to a moderate value. The kinetic barrier variations are also a function of pH for a given potential vs. RHE, which shows the pH dependence of i.159 Although feasible alterations in this process are challenging (e.g., regarding kinetic barriers or pH), it is noticeable that the activity volcano does not shift toward the right or left, and somehow only up and down, showing that the descriptor still provides information on the bonding nature for the most selective HER catalysts.308 However, for complete explanations and understanding, additional detailed and capable techniques for measuring electrochemical barriers are necessary for hydrogen ion transfer reactions containing OH and hydronium ions.162,309,310 Since Pt sits near the top of the hydrogen volcano, with an approximately thermo-neutral ΔGH, and is the most active catalyst for the HER, it needs small η values for elevated response times in acidic solutions (Fig. 25(b) and (c)). On the other hand, scarcity and high cost of Pt may decrease its extensive technological utilizations. Thus, the boosted earth-abundant NPMCs can probably replace Pt.153 For decades, MoS2 was considered an inactive catalyst for the HER,311 but motivated by hydrogen-producing enzymes (hydrogenases and nitrogenases), DFT-based simulations were performed to study the Mo(1010) edge of MoS2, showing about 50% hydrogen coverage, and a ΔGH = 0.08 eV, near 0 eV (optimum value) (Fig. 25(a))312 but the basal plane shows poor activity (ΔGH = 1.92 eV) in bulk MoS2 crystals.161 Therefore, due to these calculations, MoS2 was synthesized on a carbon black support to increase its edge sites and it was successfully tested in a membrane electrode setup. The statistical area-normalized @ 10 mA cm−2geoi was attained at η = ∼175 mV, which at that time was the most active reported NPMC for the HER in acidic electrolyte.2 Shortly after, it was experimentally shown that the MoS2 edges are certainly catalytic active sites in the HER.160 An MoS2 monolayer was deposited on the surface of Au(111) nanoparticles, which showed that the HER activity linearly increased with MoS2 perimeter length, but independent of its SSA (Fig. 25(b) and (d)). To explain this, simulations and experimental observations showed that the periphery is more active, thus motivating the development of MoS2 catalysts with considerable bare edge sites. Therefore, the 3D mesoporous MoS2 nanostructure with a double-gyroid morphology was discovered (Fig. 25(d)), which minimized the formation of basal planes, and exposed the maximum active edge sites. Thus, average turnover frequency (TOFavg) increased from 2 to 4 from that of MoO3–MoS2 nanowires synthesized via the same sulfide method (Fig. 25(d)).290,313 A limitation of the double-gyroid structure is the elongated electron transport distance from the active site to conductive substrate, which enhances resistive loss since the electron mobility perpendicular to the MoS2 basal planes is lower than the in-plane charge mobility by almost thrice in magnitude. Thus, synthesized vertically aligned MoS2 nanostructures bare numerous edge sites at the surface and also enable facile electron transfer to the conducting substrate (Fig. 25(d)).314,315 Another smart method in catalyst growth is to disperse high SSA nanoparticles on supports.2 For example, MoS2 nanoparticles were prepared on rGO,277 which resulted in better dispersion and decreased MoS2 aggregation, leadin to superior activity because of the improved edge sites and superior ion transport (Fig. 25(b)). Lithium ion intercalation into the vdW gap of MoS2 has also been examined to increase the HER activity by changing the electronic properties of MoS2.293,316 This showed that MoS2 chemical exfoliation and phase change from 2H semiconducting phase into metallic phase 1T are other ways to tailor the catalytic activity Fig. 25(d).
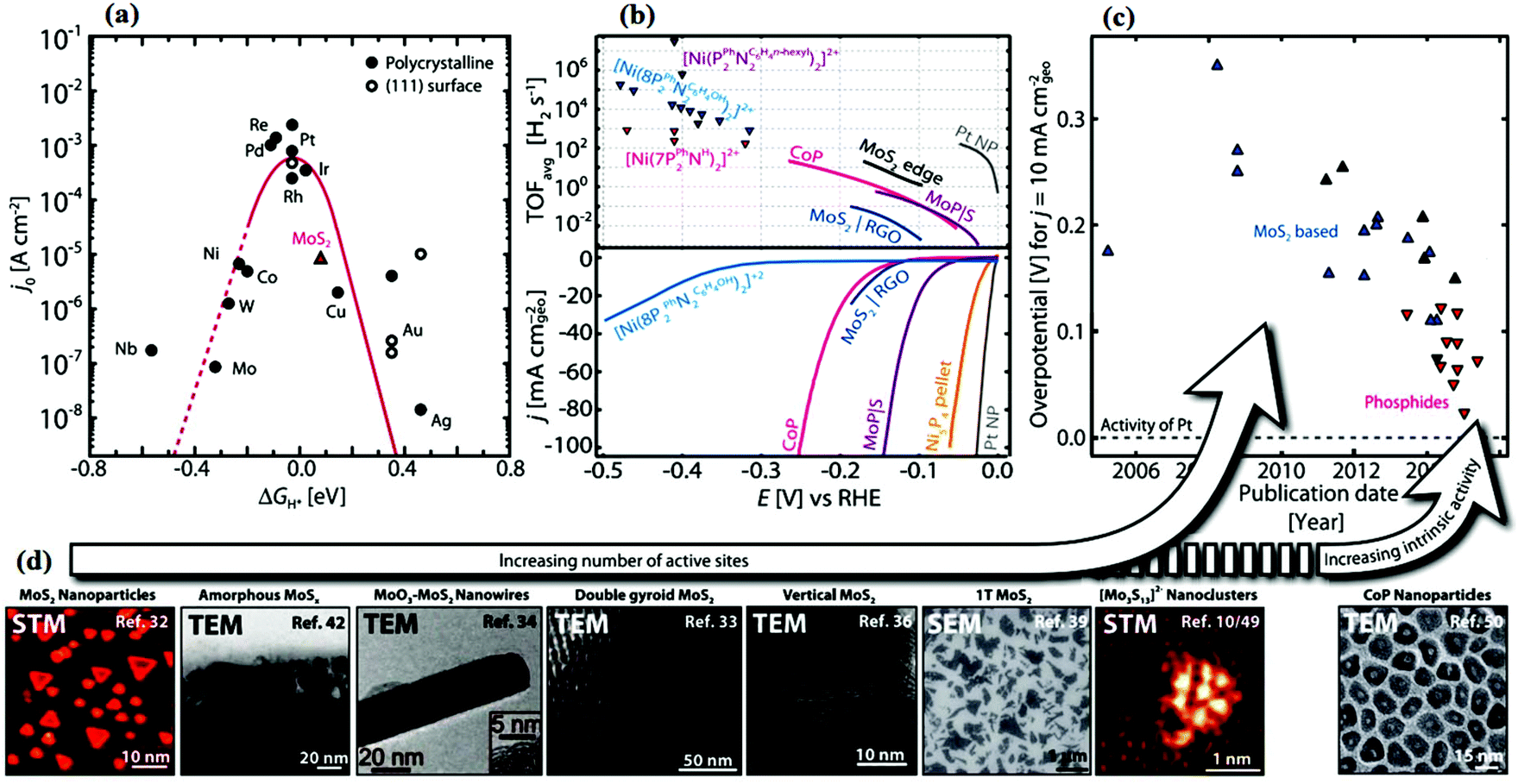 | ||
| Fig. 25 (a) HER volcano plot, (b) TOFavg plots with LSV, (c) order development in η of MoS2-based and phosphide HER catalysts, and (d) microscopy images of HER catalysts. This figure has been adapted/reproduced from ref. 2 with permission from the American Association for the Advancement of Science. | ||
It is suggested that the improved activity of 1T-MoS2 over similarly prepared 2H-MoS2 is due to the increase in active edge sites, and decrease in charge transfer resistance.316 Based on another study, the edge 1T-MoS2 is not the only major active sites, but the basal plane can also be possible catalytically active sites.293 Thus, recently, it was exposed that the vacancies in the MoS2 basal plane also show activity, which can also be modified by strain.276 Similarly, in the case of molybdenum sulfide, its high HER activity was studied, which was shown to be mainly because of its high SSA (Fig. 25(d)). The calculated as-synthesized amorphous MoS2 composition was close to MoS3, but considering its reduction potentials in an electrochemical cell, it was MoS2, which was also supported by in situ studies.317,318 Further doping with TMs (Ni, Fe, and Co), the catalyst performance is considerably improved in acidic medium319 because of the increase in SSA, while the TOFavg is enhanced in neutral environment. Different methods to tailor MoS2-based catalysts actually increase the active sites numbers; however, their overall activity is still limited, which may be due to the small portion of edge sites contributing to the rate of the reaction (Fig. 25(b–d)). This requires us to further modify our strategy and design molecular clusters with under coordinated S at the surface analogous to MoS2 edges, such as [Mo3S4]4+ cubanes, Mo-IV-disulfide, and thiomolybdate [Mo3S13]2− complexes (Fig. 25(d)). Another strategy to increase the number of active sites is proposed based on increasing the catalyst loading, but it results in the restriction of charge and mass transport. This will encourage advancement of further catalysts with higher intrinsic activity through experimental and simulation work on descriptors based on ΔGH ≈ 0. Hence, some TM phosphides, selenides, borides, carbides, and nitrides show HER activities closer to Pt in the form of η @ 10 mA cm−2geo. Although, due to high catalyst loadings and large SSAs, NPMCs lag behind the benchmark Pt regarding the TOFavg in acidic medium (Fig. 25(b–d)). In alkaline media, to date, NPMCs have also developed, e.g., Ni–Mo systems showed low η values @ 10 mA cm−2geo. However, they also exhibit lower TOFavg values than the benchmark Pt, similarly to NPMCs in acidic media. Homogeneous catalysts with high TOFavg have also been developed, even though they classically need a large η value to reach substantial current densities, thus leaving much room for improvement (Fig. 25(b)).2,320Table 3 shows recent studies on ultrathin 2D electrocatalysts for the HER.
| 2D material electrocatalyst | Electrochemical performance | Ref. |
|---|---|---|
| 2H-phase MoS2 | Onset over-potential: ca. 100 mV, Tafel slope: ca. 50 mV dec−1 | 321 |
| Defect- and S-rich MoS2 | Overpotential: 135 mV(@ 10 mA cm−2), Tafel slope: 48 mV dec−1 | 322 |
| 1T metallic MoS2 | Overpotential: 187 mV(@ 10 mA cm−2), Tafel slope: 43 mV dec−1 | 316 |
| Defect-rich MoS2 | Onset overpotential: ca. 120 mV, overpotential: 190 mV (@ 10 mA cm−2), Tafel slope: 50 mV dec−1 | 323 |
| MoS2 | Onset over-potential: ca. 100 mV, Tafel slope: 73 mV dec−1 | 324 |
| O-Doped MoS2 | Onset over-potential: ca. 120 mV, Tafel slope: 55 mV dec−1 | 274 |
| 1T-VS2 | Overpotential: 68 mV (@ 10 mA cm−2), Tafel slope: 34 mV dec−1 | 325 |
| WS2 | Onset over-potential: ca. 80 mV, overpotential: 240 mV (@ 10 mA cm−2), Tafel slope: 55 mV dec−1 | 297 |
| WS2 | Onset over-potential: ca. 100 mV, Tafel slope: 48 mV dec−1 | 326 |
| 1T-WS2 nanosheets | Overpotential: 142 mV (@ 10 mA cm−2), Tafel slope: 70 mV dec−1 | 327 |
| VSe2 | Onset over-potential: ca. 108 mV, overpotential: 206 mV (@ 10 mA cm−2), Tafel slope: 88 mV dec−1 | 328 |
| 1T-MoSe2 | Onset over-potential: ca. 60 mV, overpotential: 179 mV (@ 10 mA cm−2), Tafel slope: 78 mV dec−1 | 329 |
| 2H–1T MoSe2 mixture | Overpotential: 152 mV (@ 10 mA cm−2), Tafel slope: 52 mV dec−1 | 330 |
| MoS2(1−x)Se2x | Onset over-potential: ca. 80 mV, overpotential: 164 mV (@ 10 mA cm−2), Tafel slope: 48 mV dec−1 | 331 |
| Supramolecular polymer | Onset over-potential: ca. 110 mV, overpotential @ 10 mA cm−2: 333 mV, Tafel slope: 80.5 mV dec−1 | 332 |
| Pt-MoS2 | Overpotential: 55 mV (@ 10 mA cm−2), Tafel slope: 40 mV dec−1 | 333 |
| Single-atom Pt-doped MoS2 | Overpotential: 145 mV (@ 10 mA cm−2), Tafel slope: 96 mV dec−1 | 334 |
| Ru@C2N | Onset over-potential: ca. 95 mV, overpotential: 22 mV (@ 10 mA cm−2), Tafel slope: 30 mV dec−1 | 335 |
| NiCo2S4 | Onset over-potential: ca. 17 mV, overpotential: 65 mV (@ 10 mA cm−2), Tafel slope: 84.5 mV dec−1 | 336 |
| Mn-doped CoSe2 | Onset over-potential: ca. 174 mV, overpotential: 195 mV (@ 10 mA cm−2), Tafel slope: 36 mV dec−1 | 337 |
| Ultra-thin Fe–Ni–S | Overpotential @ 10 mA cm−2: 105–117 mV, Tafel slope: 40–48 mV dec−1 | 302 |
| NiSe2 | Overpotential: 135 mV (@ 10 mA cm−2), Tafel slope: 37 mV dec−1 | 250 |
| CoP | Onset over-potential: ca. 40 mV, overpotential: 90 mV (@ 10 mA cm−2), Tafel slope: 43 mV dec−1 | 338 |
| Ni5P4–Ni2P | Onset over-potential: ca. 54 mV, overpotential: 120 mV (@ 10 mA cm−2), Tafel slope: 79.1 mV dec−1 | 339 |
| Mo–W–P | Overpotential: 93 mV (@ 20 mA cm−2), Tafel slope: 52 mV dec−1 | 340 |
| MoP | Onset over-potential: ca. 50 mV, overpotential: 124 mV (@ 10 mA cm−2), Tafel slope: 58 mV dec−1 | 341 |
| CoP | Overpotential: 56 mV (@ 10 mA cm−2), Tafel slope: 44 mV dec−1 | 342 |
| C@Ni8P3 | Overpotential: 110 mV (@ 10 mA cm−2), Tafel slope: 46 mV dec−1 | 343 |
| Ni0.9Fe0.1PS3 | Overpotential: 72 mV (@ 10 mA cm−2), Tafel slope: 73 mV dec−1 | 344 |
| NiMoNx | Onset overpotential: ca. 78 mV, Tafel slope: 35.9 mV dec−1 | 304 |
| MoN | Onset over-potential: ca. 100 mV, overpotential: 220 mV (@ 10 mA cm−2), Tafel slope: 90 mV dec−1 | 221 |
| N/S co-doped G | Onset potential: 0.13 V (vs. RHE) Operating potential: 0.28 V (@ 10 mA cm−2, vs. RHE) Tafel slope: 80.5 mV dec−1 | 270 |
| (Co/N co-doped mesoporous carbon spheres)/N-doped carbon nanosheets | Over-potential: 0.22 V (@ 10 mA cm−2) Tafel slope: 81 mV dec−1 | 345 |
| CoS2/CNT/G | Overpotential: 0.142 V (@ 10 mA cm−2) Tafel slope: 51 mV dec−1 Exchange i: 6.26 105 mA cm2 | 346 |
| CoSe2 nanosheets | Overpotential: 0.27 V (@ 10 mA cm−2), TOF: 745 h−1, Tafel slope: 64 mV dec−1 | 300 |
| CoxW(1−x)S2 nanosheets | Over-potential: 0.121 V (@ 10 mA cm−2) Tafel slope: 67 mV dec−1 | 275 |
| Fe–Ni sulfide nanosheets | Overpotential: 0.105 V (@ 10 mA cm−2) Tafel slope: 40.0 mV dec1 | 302 |
| g-C3N4/N-doped G | Overpotential: 0.24 V (@ 10 mA cm−2) Exchange i: 3.5 107 A cm−2 | 272 |
| Co/N-Doped G | Overpotential: 0.03 V (@ 0.3 mA cm−2) Overpotential: 0.147 V (@ 10 mA cm−2), Tafel slope: 82 mV dec−1 | 273 |
| GaS nanosheets | Onset potential: 0.48 V (@ 1 mA cm−2), i @ 0.6 V (vs. RHE): 22 mA cm−2, Tafel slope: 85 mV dec−1 | 281 |
| Mn-CoSe2 nanosheets | Overpotential: 0.174 V (@ 10 mA cm−2), Tafel slope: 36 mV dec−1 Exchange i: 0.0683 mA cm−2 | 337 |
| N/S co-doped Mo2C nanosheets | Onset potential: 0.046 V (vs. RHE) Operating potential: 0.086 V (@ 10 mA cm−2, vs. RHE), exchange i: 3.8 102 mA cm−2, Tafel slope: 47 mV dec−1 | 347 |
| Strained MoS2 nanosheets (S vacancies) | Formal potential: 0.53 V (vs. Ag/AgCl), electron-transfer coefficient: 0.4, electron-transfer rate constant: 2.3 104 cm s−1 | 283 |
| Hierarchical MoS2 nanosheets | Over-potential: 0.167 V (@ 10 mAcm−2), Tafel slope: 70 mV dec−1, turnover frequency (TOF):0.41 S1 at an over-potential of 0.125 V | 291 |
| Metallic-phase MoS2 nanosheets | i:10 mA cm−2 (@ 0.175 V) Tafel slope: 41 mV dec−1 | 292 |
| MoS2/mesoporous G | Onset overpotential: 0.14 V, Tafel slope: 42 mV dec−1 | 348 |
| MoS2/N-doped G | Onset potential: 0.005 V (vs. RHE), over-potential: 0.056 V (@ 10 mA cm−2), Tafel slope: 41.3 mV dec−1, exchange i: 0.74 mA cm−2 | 299 |
| MoS2/G | Onset overpotential: 0.03 V, i @ over-potential of 0.11 V: 10 mA cm−2 | 349 |
| C/MoS2@G | Onset potential: 0.16 V (vs. RHE) Onset overpotential: 0. 165 V, Tafel slope: 46 mV dec1 | 350 |
| N-CNT/MoS2+x nanosheets | Onset potential: 0.135 V (vs. RHE), overpotential: 0.08 V (@ 10 mA cm−2) | 351 |
| MoS2/CoSe2 nanosheets | Onset potential: 0.011 V (vs. RHE), overpotential: 0.068 V(@ 10 mA cm−2), exchange i: 0.073 mA cm−2 | 352 |
| MoSe2 nanosheets | Exchange i: 1.9 × 10−3 mA cm−2, Tafel slope: 76 mV dec−1 | 286 |
| Ni nanosheets | Onset potential: 0.034 V (vs. RHE), Tafel slope: 114 mV dec−1 | 267 |
| NiMoNx/C nanosheets | Onset potential: 0.078 V (vs. RHE), Tafel slope: 35.9 mV dec−1 Exchange i: 0.24 mA cm−2 | 304 |
| NiSe2 nanosheets | Over-potential: 0.117 V (@ 10 mA cm−2), Tafel slope: 32 mV dec−1 | 353 |
| Vertically oriented ReS2 nanosheets/Au | Over-potential: 0.2 V (@ 10 mA cm−2), Tafel slope: 85 mV dec−1 | 354 |
| Ru nanosheets | Onset potential: 0 V (vs. RHE), overpotential: 0.02 V (@ 10 mA cm−2), Tafel slope: 46 mV dec−1 | 95 |
| SnS/N-doped G | Overpotential: 0.125 V (@ 10 mA cm−2), TOF: 0.23 S−1 at an over-potential of 0.125 V, Tafel slope: 38 mV dec−1 | 355 |
| VS2 nanosheets | Overpotential: 0.068 V (@ 10 mA cm−2), overpotential: 0.34 V (@ 100 mA cm−2), Tafel slope: 34 mV dec−1 | 289 |
| VSe2 monolayers | Overpotential: 0.206 V (@ 10 mA cm2), Tafel slope: 88 mV dec−1 | 328 |
| Hierarchical WS2 nanosheets | Overpotential: 0.16 V (@ 10 mA cm−2), Tafel slope: 60 mV dec−1,TOF: 1.24 S−1 at and over-potential of 0.15 V | |
| Vertically aligned WS2 nanosheets | Onset overpotential: 0.03 V IR-corrected kinetic i: 48 mA cm−2 at and overpotential of 0.221 V, Tafel slope: 61 mV dec−1 | 356 |
| WS2/G | Overpotential: 0.17 V (@ 10 mA cm−2), Tafel slope: 52 mV dec−1 | 357 |
| 2H-phase MoS2 | Onset overpotential: ca. 100 mV, Tafel slope: ca. 50 mV dec−1 | 321 |
| Mo2CTx (rich-O termination group) | Overpotential: 189 V (@ 10 mA cm−2), Tafel slope: 75 mV dec−1 Stability: >1000 CV | 358 |
| 0.5W-MoxC | Overpotential: 148 V (@ 20 mA cm−2), Tafel slope: 56 mV dec−1, stability: >24 h | 359 |
| Mo/Mo2C-HNS-750 | Onset overpotential: ca. 16 mV, overpotential: 89 V (@ 10 mA cm−2), Tafel slope: 70.72 mV dec−1, stability: >20 h | 360 |
| N-Doped Mo2C | Onset overpotential: ca. 48.3 mV, overpotential: 99 V (@ 10 mA cm−2), Tafel slope: 44.5 mV dec−1, stability: >12 h | 361 |
| Ti2CTx (rich-O termination group) | Onset overpotential: ca. 75 mV, overpotential: 170 V (@ 10 mA cm−2), Tafel slope: 100 mV dec−1, stability: >5 h | 362 |
| MoC@2D-NPCs | Onset overpotential: ca. 0 mV, overpotential: 45 V (@ 10 mA cm−2), Tafel slope: 46 mV dec−1, stability: >20 h | 363 |
| Mo2CTx (MXene with-O termination) | Overpotential: 189 V (@ 10 mA cm−2), Tafel slope: 70 mV dec−1, stability: >2 h | 364 |
| MoS2/Ti3C2-MXene@C | Onset overpotential: ca. 20 mV, overpotential: 135 V (@ 10 mA cm−2), Tafel slope: 45 mV dec−1, stability: >20 h | 365 |
| a-Mo2C | Overpotential: 198 V (@ 10 mA cm−2), Tafel slope: 56 mV dec−1, stability: >1000 CV | 366 |
| NS-Doped Mo2C | Onset overpotential: ca. 46 mV, overpotential: 86 V (@ 10 mA cm−2), Tafel slope: 47 mV dec−1, stability: >25 h | 367 |
8.3. Challenges with 2D-material HER catalysts in alkaline electrolyte
As aforementioned, the HER activity on a variety of 2D materials may be considerably improved using different methods to enhance the exposed active sites and their conductivity. However, although significant developments have been made, some unsettled questions are still unsolved. The majority of electrochemical experimentalists showed the very active phase is the 1T phase of MoS2, but some theoretical calculations also show that the 1T′ phase is more stable than the 1T phase because of local strain. This uncertainty obstructs the further development of HER activity through extensively applied phase engineering. Secondly, there is also some focus on scientifically examining the involvement of numerous other features such as strain, defects, S-vacancy, and phase conversion. Regarding graphene-related materials for the ORR, these features mostly operate cumulatively and add to essentially altering the MoS2 electronic structure, and consequently its HER activity. For instance, Yin et al. calculated S-vacancies, 1T-MoS2 edges, and their HER activity phase.294 Thirdly, analogous to the nanostructure manufacturing approach, the balance between the introduction of additional active sites, vacancies and defects with TMD crystallinity can be measured for getting desirable ΔGH* and charge transfer rates. Fourthly, catalyst stability and activity may be concurrently greater in a range of electrolyte pH. Such as, Staszak-Jirkovský et al. explained the stability and activity of TMDCs may be balanced by a suitable change in their composition.368 As a result, amorphous composites were combined, i.e., CoSx (high activity) with MoSx (high stability), and considered inexpensive and pH universal alternatives to PMCs for the HER. Additionally, compared to the numerous studies on 2D materials in acidic HER, studies on 2D materials for application in alkaline HER are scarce. However, electrolyzers working by means of alkaline anion exchange membranes may give mild conditions that NPMCs can withstand, where in alkaline solution, the HER rate is ∼2–3 orders less than that in acidic solution. Moreover, although the acidic HER is well recognized, mechanistic information in alkaline HER still has to be established.1289. 2D-bifunctional OER/HER electrocatalysts
Electrocatalytic H2O splitting represents a promising way to produce clean H2. However, the sluggish kinetics of the OER often limits the overall water splitting efficiency. Furthermore, although some well-designed electrocatalysts have been developed to promote the kinetics of the OER, a large potential is still required to match the HER, leading to a low energy conversion efficiency. Thus, alternative strategies to replace the anodic OER with the electrooxidation of thermodynamically more favorable species are of increasing importance to boost the HER. Chenqi Huang et al. explored an alternative anodic reaction with accelerating kinetics to produce value-added chemicals with high selectivity, especially integrated with promoted hydrogen (H2) generation, which is highly desirable (Fig. 26).369 Here, the selective semi-dehydrogenation of tetrahydroisoquinolines (THIQs) was demonstrated to replace the oxygen evolution reaction (OER) for boosting the HER in H2O over an Ni2P nanosheet electrode. The value-added semi-dehydrogenation products, dihydroisoquinolines (DHIQs), could be selectively obtained with high yields at the anode. The controllable semi-dehydrogenation is attributed to the in situ formed NiII/NiIII redox active species. This strategy can deliver a variety of DHIQs bearing electron-withdrawing/donating groups in good yields and excellent selectivity, and can be facilely applied to gram-scale synthesis. Furthermore, a two-electrode Ni2P bifunctional electrolyzer can produce both H2 and DHIQs with robust stability and high faradaic efficiencies at a much lower cell voltage than that of overall water splitting.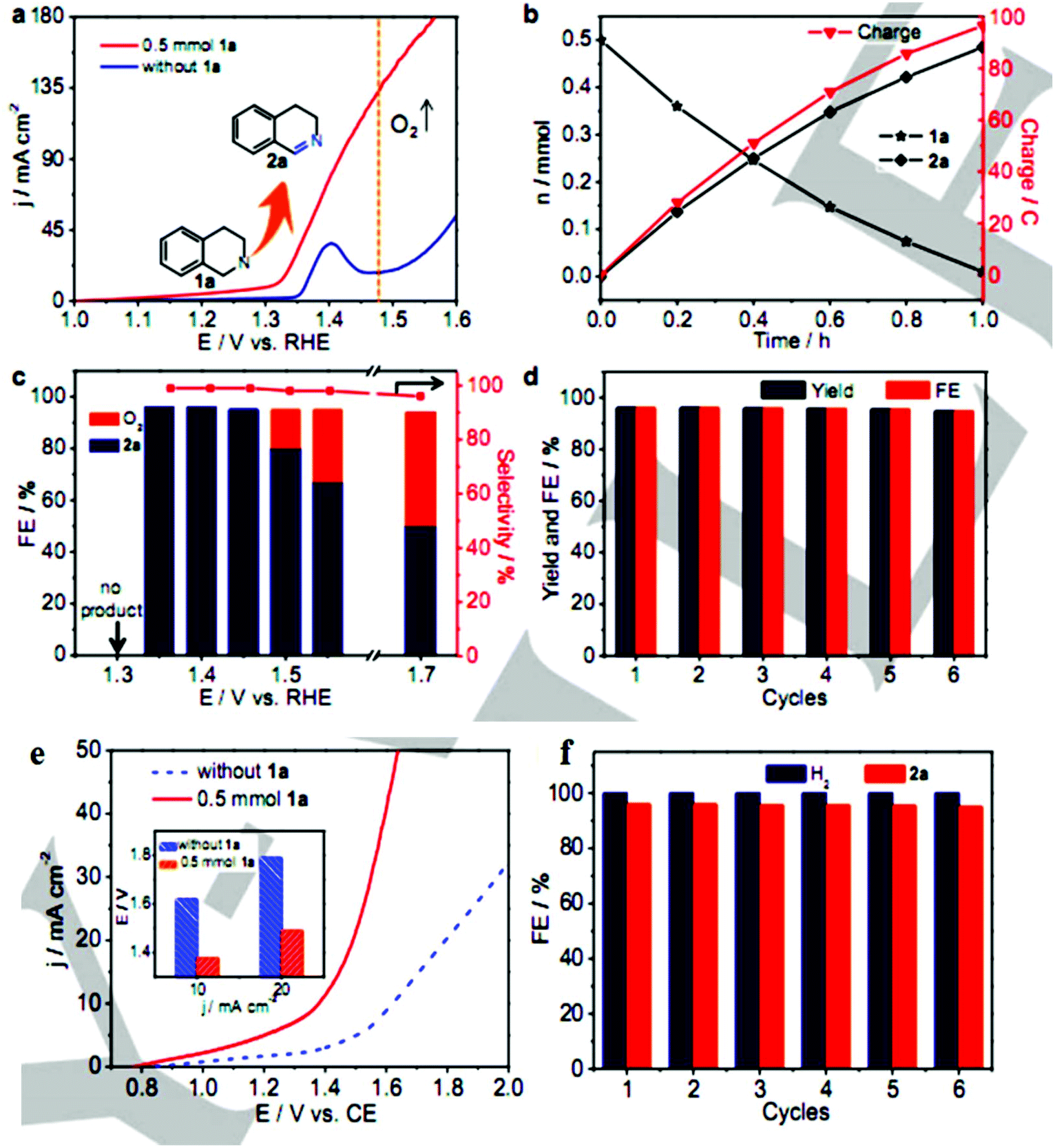 | ||
| Fig. 26 (a) LSV curves of Ni2P at a scan rate of 5 mV s−1 in 40 mL of 1.0 M KOH with and without 0.5 mmol 1a. (b) Time-dependent evolution of 1a and its semi-dehydrogenation product 2a. (c) FEs and selectivity of 2a at different potentials. (d) Cycle-dependent yields and FEs of 2a. (e) LSV curves over an Ni2P∥Ni2P electrolyzer. (f) Cycle-dependent FEs of an Ni2P∥Ni2P electrolyzer for both H2 and 2a production. This figure has been adapted/reproduced from ref. 369 with permission from John Wiley and Sons Ltd. | ||
10. Other important reactions of electrocatalysts
Besides the aforementioned methods relating to the OER, ORR, HOR, and HER, there are large number of additional promising energy conversion reactions that are comparatively less studied. Some of them can possibly be game-changing, where electrocatalysts can be applied with precise properties. These reactions may have different intermediates, number of electrons transferred, descriptors and volcanoes to judge selectivity, and activity, which will also be a first significant step in knowledge to accelerate the development of catalysts.10.1. Electrochemical carbon dioxide reduction reaction (ECR)
Due to the increase in the exhaustion of fossil fuels and nonstop CO2 release, using ECR and sustainable energy resources for fuel and chemical manufacture is extremely desirable for future energy sustainability and continuous supply. Even though the ECR is feasible from a thermodynamic viewpoint, due to the high stability of CO2, this reaction is kinetically very slow, and thus potential catalysts are required to enhance the reaction. In the few decades, broad efforts have been dedicated to the synthesis of proficient CO2 catalysts. Conventional bulk metals are usually divided into three categories based on their binding power of different intermediates and final products. Group I consists of, Sn, Hg, Pb, and In as formate manufacturing metals, group II consists of Au, Ag, Zn, and Pd as CO-forming metals, and group III contains Cu, which is capable of making hydrocarbons and alcohols. Although they are active for the ECR, bulk metals have extremely restricted active sites and can be deactivated from impurity poisoning or some tightly bound intermediates. Accordingly, recently, a variety of nanostructured materials have appeared as superior ECR catalysts. 2D materials, particularly atomically thin sheets, have plentiful unsaturated surface atoms, and thus possess an enhanced number of active sites.128 Similarly to the ORR, the ECR is a multielectron reduction reaction involving diverse surface-bound reaction intermediates. However, in contrast to the ORR, there are many probable CO2 reduction products, e.g., carbon monoxide, formate, formaldehyde, methane, methanol, and C2+ hydrocarbons and oxygenates, and several of these products need huge number of electrons and protons removed/assigned, and may have diverse intermediates as well.2 The ECR based on a ‘clean’ and competent method for CO2-fuels conversion proceeds via the 2e− (formate and CO), 6e− (methanol), 8e− (methane), and 12e− (ethylene) mechanisms. The applied conditions such as electrocatalyst, type and concentration of electrolyte, pressure, and temperature can influence this reduction process and product distributions. The HER often improves the CO2 reduction in aqueous electrolytes via metal electrocatalysts, and thus the synthesis of novel and further selective catalysts for CO2 transformation into useful products is necessary. There are three main steps to consider when synthesizing heterogeneous electrocatalysts for CO2 reduction, which are1. The chemical adsorption of CO2 molecules on the cathode catalyst active sites.
2. Transfer of electron and/or migration of protons to split C–O bonds and convert them into C–H bonds.
3. Products rearrangement followed by desorption from the electrode surface and discharge to the electrolyte.
However, the ECR is a complex process, and it is better to continue through proton-coupled multi-electron shift. Nevertheless, the definite full reaction mechanism mainly relies on numerous factors (Table 4).
| Electron transfer | Reaction | E° (V vs. SHE) |
|---|---|---|
| e− | CO2 + e− → CO2− | −1.9 |
| 2e− | CO2 + 2H+ + 2e− → CO + H2O | −0.53 |
| 2CO2 + 2H+ + 2e− → HCOOH | −0.61 | |
| 2CO2 + 2H+ + 2e− → H2C2O4 | −0.913 | |
| 4e− | CO2 + 4H+ + 4e− → HCHO + H2O | −0.48 |
| 6e− | CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 |
| 8e− | CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
| 12e− | 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | −0.349 |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | −0.329 | |
| 14e− | 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | −0.27 |
| 18e− | 3CO2 + 18H+ + 18e− → C3H7OH + H2O | −0.31 |
As shown in Fig. 27, the chemical features manage the selectivity, for example redox properties, binding affinity, and acid/base character. Structural and material properties can affect the activity, for example, crystallinity, particle size, and surface area. Lastly, the efficiency is identified by optical and electrical properties, i.e., quantum FE, photonic, and morphology. In this perspective, nature gives very active and selective catalysts, but less competence. In contrast, inorganic catalysts, e.g., metals and metal oxides, exhibit elevated efficiency and activity. However, alteration of selectivity at the molecular level is still very complicated. Homogeneous molecular catalysts are capable of achieving good selectivity and efficiency, but are intrinsically inadequate regarding activity. The view of understanding all three features in a single system may only be attained by reticular chemistry. The Nernst equation shows that the ECR mechanism is more thermodynamically favorable for processes having very positive standard redox potentials (E°). However, the ECR rate depends on several factors, e.g., pressure, electrolyte, electrocatalyst, and temperature, despite the position of thermodynamic equilibrium. Catalysts particularly possess a vital character in the ECR kinetic process, e.g., bulk TMs are categorized in three divisions based on their capability of producing BE-definite response transition species. Also, previous studies proposed that electrocatalyst size/structure, morphology, and chemical state have a considerable impact on reaction kinetics. Additionally, the electrolyte has a significant effect on the ECR kinetics. Generally, aqueous solutions having salts of inorganic metals (e.g., KHCO3 and NaHCO3) are used as the electrolyte, and thus the HER (E° = −0.42 V vs. SHE) is not the preferred challenging reaction. Although non-aqueous organic electrolytes can efficiently restrain the competing HER and advance the dissolubility of CO2, their comparatively low viscosity are responsible for poor electrolyte diffusion, and their elevated price and toxic nature restrict their large-scale use.
 | ||
| Fig. 27 Strategy for improving the emerging ECR. This figure has been adapted/reproduced from ref. 370 with permission from the Nature Publishing Group. | ||
The ECR kinetics is also influenced by temperature and pressure because of the insolubility of CO2 in electrolyte. Usually, a high CO2 partial pressure and low temperature are helpful in increasing the CO2 dissolution. Generally, a high CO2 concentration causes high kinetics, and thus more energy is necessary to generate this circumstance. Thus, ECR studies are mostly performed in CO2-saturated aqueous electrolyte at room temperature. Also, faradaic efficiency (FE), η, and energy efficiency, j, are two additional parameters that are significant for assessing ECR efficiency because of the range of products created. The FE can calculated as follows:
| FE = αnF/Q | (37) |
| EE = (E°/E° + η) × FE | (38) |
The ECR has potential as a main approach for the renewable manufacture of fuels and chemicals through the use of renewable CO2 free energy sources. However, the development of efficient catalysts is critical since currently, there are no industrial scale operations that use this technology due to its low energetic efficiency. For ECR on a large scale, electrocatalysts must have high activity and selectivity to some product.
In the 1980s, carbon ECR was calculated on many heterogeneous elemental surfaces.371,372 The electrocatalyst results were mainly divided based on their selectivity toward their major reaction product, CO (e.g., Au and Ag), formate (e.g., Pb and Sn), hydrocarbons (e.g., Cu), and hydrogen (e.g., Pt and Ni). Here, we show some non-nanosheet state-of-the-art values and compare them with that of some 2D materials. During the past few decades, many TMs (e.g., first transition series, coinage/precious metals, and few normal metals such as Bi and In), TMOs (TiO2, Cu2O, and SnO2), and TMDCs as well as metal-free carbon materials have been discovered as electrodes for the ECR. It was planned that the starting step in ECR involved CO2 anion radical formation after one-electron transfer in a CO2 molecule, which needs satisfactory energy to rearrange a linear CO2 molecule into a non-linear anion at 1.9 V (vs. SHE), and in most cases this is considered rate-determining. Electrochemical conversion obstacles such as large η (difference between applied potential and equilibrium), sluggish kinetics, unsatisfactory selectivity and low efficiency must be tackled in proposed catalysts, where the recent research on inexpensive electrocatalysts with high activity/selectivity is shown in Table 5.
| Catalyst | Synthetic method (precursor) | Electrolyte | Electrochemical performance | Ref. |
|---|---|---|---|---|
| Bismuth nanosheets | Electrochemical reduction (BiOCl nano-sheets) | 0.5 M KHCO3 | Onset potential: 1.3 V Max FE of formate: 92% at 1.5 V (vs. SCE) | 373 |
| N-Doped graphene like material | Carbonation at 1000 °C in Ar (3-pyridine carbonitrile) | 97 wt% [Bmim] BF4, 3 wt% water | Max CH4 FE: 93.5% at 1.4 V (vs. SHE) CH4 partial i (@ 1.4 V, vs. SHE): 3.26 mA cm−2 | 374 |
| 4-Atomthick Co3O4/Co sheet | Ligand-confined growth (Co(iii) acetyl-acetonate) | 0.1 M Na2SO4 | i (@ 0.85 V): 10.59 mA cm−2, FE for formate:90.1% (@ 0.85 V, vs. SCE) Overpotential @ 10.59 mA cm−2: 0.24 V | 303 |
| Mo-Bi bimetallic chalcogenide nanosheets | Solvothermal method (230 °C) (Bi(NO3)3·5H2O and (NH4)2MoS4) dec−1 | [Bmim]BF4/MeCN | Max FE of methanol: 71.2% at 0.7 V (vs. SHE), i at maximum FE of methanol: 12.1 mA cm−2 Tafel slope: 124.4 mV (@ overpotential range 0.16–0.26 V) | 375 |
| Vertically aligned MoS2 nanoflakes | Mechanical exfoliation (bulk MoS2) | 96 mol% water, 4 mol% EMIM-BF4 | Onset potential: 0.164 V (vs. RHE), i: 30 mA cm−2 (@ 0.764 V, vs. RHE) | 376 |
| WSe2 nanoflakes | CVD | 50 vol% EMIMBF4, 50 vol% deionized water | Onset potential: 0.164 V (overpotential of 54 mV), i: 18.95 mA cm−2(@ 0.164 V), i: 330 mA cm−2 (@ 0.764 V, vs. RHE), CO formation FE: 24% CO formation TOF: 0.28 s−1 | 377 |
| SnO2/G | Hydrothermal method (SnCl2, ethylene glycol, and water) | 0.1 M NaHCO3 | i: 13.1 mA cm−2 (@ 1.8 V), specific i: 266 A g−1 FE for formate: 93.6% (@ 1.8 V, vs. SCE) | 378 |
| Sn quantum sheets in G | Spatially confined reduction then the hydrothermal process (180 °C), calcinations at 500 °C and then 1000 °C (SnO2 layer and glucose) | 0.1 M NaHCO3 | i: 21.1 mA cm−2 (@ 1.8 V), onset potential: 0.85 V, Max FE for formate @ 1.8 V (vs. SCE): 89%, Tafel slope: 83 mV dec−1 | 379 |
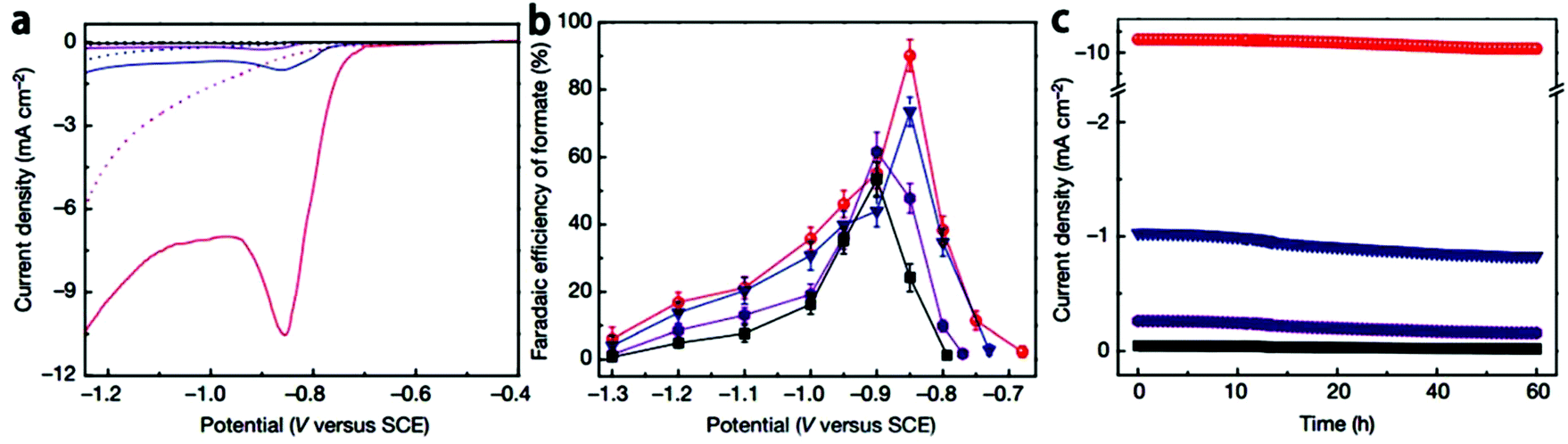 | ||
| Fig. 28 ECR to formate on four-atom thick partially oxidized Co (red) layers, four-atom thick pristine Co (blue), partially oxidized bulk Co (violet) and intact bulk Co (black). (a) LSVs in CO2-saturated (solid line) and N2-saturated (dashed line) 0.1 M Na2SO4 aqueous solution. (b) FE of formate for 4 h. (c) Chronoamperometry results at the equivalent potentials (in b) with the maximum FE. This figure has been adapted/reproduced from ref. 95 with permission from the Royal Society of Chemistry. | ||
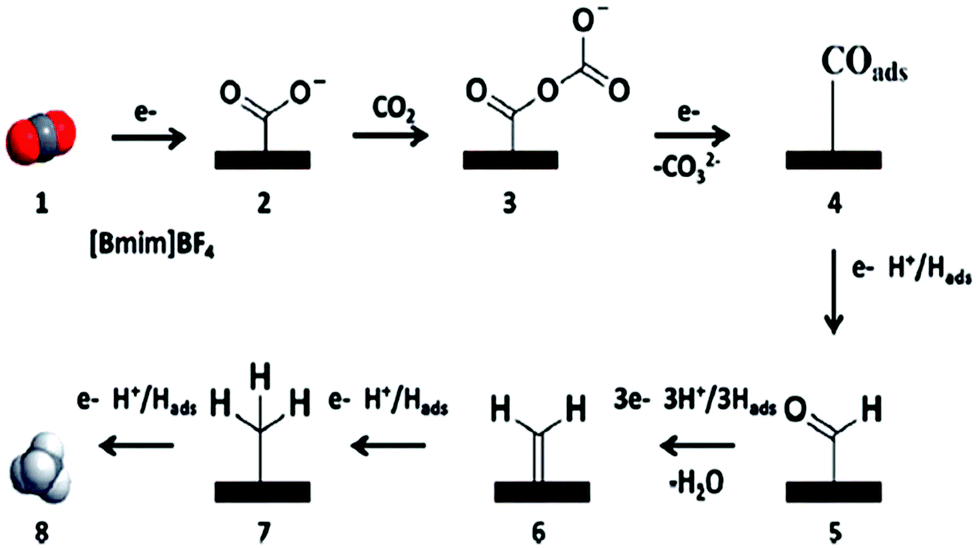 | ||
| Fig. 29 N2-Doped graphene catalysis method for CO2 (1) reduction to methane (8). This figure has been adapted/reproduced from ref. 95 with permission from the Royal Society of Chemistry. | ||
This established procedure involves maintaining the selectivity after the addition of a small quantity of H2O (3%) to the IL, with an increase in i from 1.4 to 3.3 mA cm2, which is about six-fold superior to that of Cu foil. The N2-doped electrocatalyst was significant since only CO and H2 were formed in its absence. This metal-free 2D material shows considerable success in improving metal-based electrocatalysis involving complex multi-electron methods. Many researchers use ILs, which exhibit electrolyte properties, in addition to coordinating and solvating CO2 (provided that pre-concentration occurs).
| H+ + * + O2 + e− → OOH* | (39) |
| OOH* + H+ + e− → H2O2 + * | (40) |
Thus, it is necessary to design electrocatalysts theoretically with almost zero η, which may have an excellent ΔGOOH and moderate OOH* intermediate BE.150 Although numerous catalysts, e.g., Pt, Ag, Au, Pd–Au alloys, N2-doped C, and hierarchically porous carbon, have been discovered, they were created to show merely reasonable efficiency in manufacturing H2O2. Appropriate catalysts are required to have high selectivity toward the 2e− in contrast to the 4e− pathway. DFT-based simulations were used to draw a volcano framework, which showed the theoretical η value for ΔGOOH for the 2e− reduction in O2 conversion into H2O2 and experimental η value at 1 mA cm−2 are overlaid in this plot (Fig. 30(a)).382 For metals that bind OOH* strongly, the 4e− ORR will follow the 2e− pathway. Alternatively, in the case of weak OOH* binding, the 2e− and 4e− volcano plots overlay on each another, which shows a decrease in activity for H2O2 selectivity with weaker OOH* binding. Thus, the very promising electrocatalysts having elevated activity/selectivity to H2O2 will lie on the top of the 2e− volcano plot. Simulation-based calculated Pt–Hg, Pd–Hg, and Ag–Hg alloys showed not only notable mass activity but also high selectivity (>95%).150,382 The normal design approach has exposed significant ideas to begin showing and recognizing very good electrocatalytic materials for H2O2 formation, especially to avoid the toxicity of Hg. Thus, the thermodynamic framework and expanding it to know the kinetic barriers and interfacial procedure along a wider array of materials and applied circumstances, will assist to give more information for the development of electrocatalysts that can operate at low overpotentials for the selective production of H2O2.2
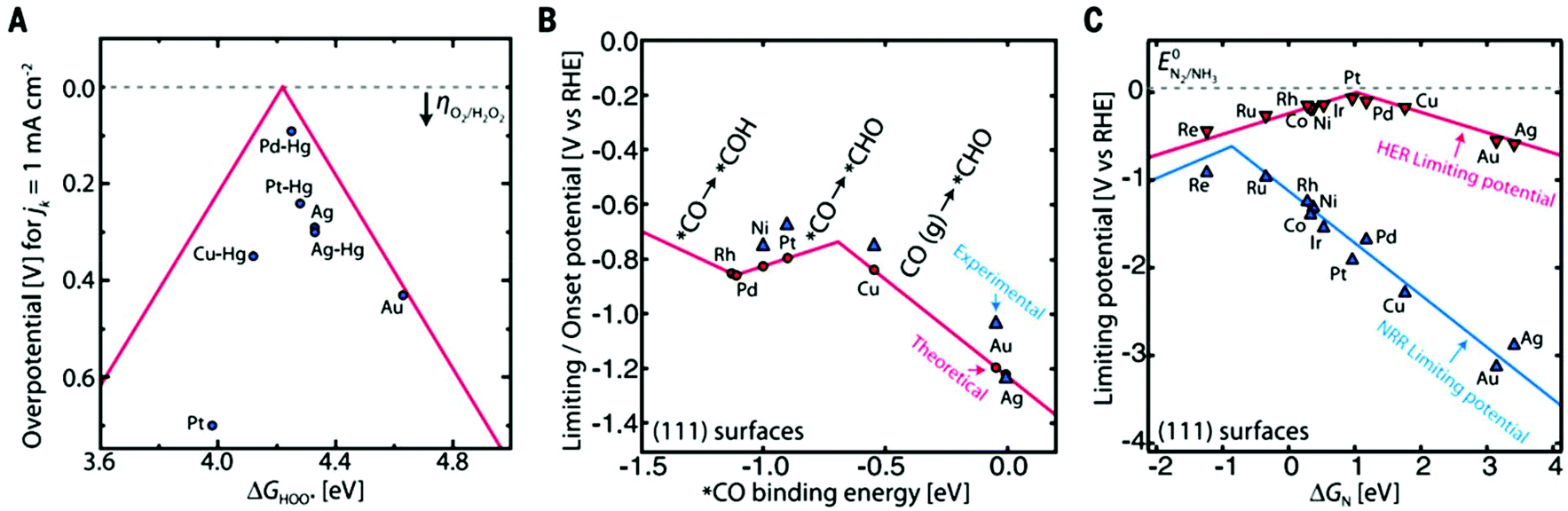 | ||
| Fig. 30 (a) Volcano plot for H2O2 production. (b) Volcano plot for ECR. (c) Volcano plot for N2 reduction. This figure has been adapted/reproduced from ref. 2 with permission from the American Association for the Advancement of Science. | ||
10.2. Nitrogen reduction reaction (N2RR)
Motivation for the electroreduction of N2 (N2 + 6H+ + 6e− → 2NH3) can be obtained from nitrogenase bacteria for N2 fixation under normal environmental conditions. Previous studies verified the possibility of this procedure in artificial circumstances, serving to inspire the development of catalysts e.g., Pt, Rh, and Ru. Similarly to the ECR, the N2RR also consists of many intermediates, and the HER is the main mechanism, making selectivity a big challenge. Experimental investigations demonstrate the extremely poor performance of catalysts for this reaction; therefore, there is much room for development.383 Thus, based on theoretical calculations, a volcano plot was built based on the theoretical limiting potential to DEN on an array of metal surfaces (Fig. 30(c)). Metals that bind N2 too loosely are imperfect due to the adsorption of N2 as N2H* in the initial reaction, but tightly-bound metals are imperfect by either NH* protonation to NH2* (flat surfaces) or elimination of NH2* as NH3 (stepped surfaces). Metals, e.g., Ru, Rh, Mo, and Fe, lie near the top of the volcano plot, which show moderate N2 binding. Although, these metals are located near the top of volcano plot, they still show large theoretical overpotentials of 0.5 V. This may be due to the non-ideal scaling relations between their intermediates. Thus, the condition for improved N2RR activity can be possibly achieved by stabilizing N2H* relative to NH2* or NH*. It was suggested that the flat surfaces of Sc, Ti, Zr, Re, and Y are competent for N2RR at −1 to −1.5 V vs. RHE with considerable containment of the HER contributing to their strong N2 binding than H2; however, methods to restrain the HER are required to achieve realistic results.384,385 DFT-based calculations also suggested that TMNs, such as ZrN and VN, are extremely active for the N2RR at small onset potentials and will suppress the HER.386 Thus, further work is needed to elucidate the atomic-scale processes at electrode–electrolyte interface, including the role of solvents, cations, and anions in addition to the kinetics of proton–electron conductance and N–N scission.2,38711. Dependence of different factors on electrocatalyst performance
11.1. Electrochemical stability of 2D materials as electrodes
Although there are increasing investigations on 2D materials for electrochemical applications, their electrode stability is insufficient. Electrode stability is clarified in terms of its intrinsic electrochemistry and catalytic reactions depending on the choice of electrolyte and applied potential window. In electrocatalysis, intrinsic electrochemistry explains the intrinsic redox performance of electrode materials when an electrochemical potential is applied.388 The electrochemical potentials are described vs. Ag/AgCl electrode. Since pristine graphene demonstrates no intrinsic electrochemistry over a stable and large window, chemically modified graphene, which has broad carbon–oxygen bonds, obviously has diverse electrochemical reductions created from its electro-active oxygen functionalities. Oxygen functionalities such as peroxyl and aldehyde groups are reduced under mild conditions, whereas carboxyl groups become reduced only at extreme negative potentials of ∼2.0 V.389 After the reduction of GO, O2 functional groups are chemically permanent. In the primary sweep, the intense cathodic wave begins at −0.7 V with peaks in the range −0.9 V to −1.5 V.390 Binary elemental components (TM and chalcogen) and the construction of predictable surface oxides determine the intrinsic electrochemistry of TMDCs, which are more multifaceted than graphene. Bonde et al. first investigated the intrinsic anodic signals of MoS2 and WS2 in acidic conditions.391 The XPS studies revealed that MoS2 was oxidized to yield MoO3, SO42− and S22− species and similar clarification was also observed for WS2. Recently, a series of electrochemical studies under neutral circumstances verified that bulk and exfoliated group VIB TMDCs show oxidative waves ranging from 1.0 V to 1.2 V, which originate from the oxidation of the metal centre from +4 to +6, as established via comparison to the intrinsic electrochemistry of associated TMOs.392 Unlike diselenides and disulfides, bulk and exfoliated MoTe2 and WTe2 share a distinct anodic wave at 0.5 V, which is traced to the tellurium electrochemistry, causing oxidation to TeO2.393 The tendency of TMDCs towards oxidation also adds to their intrinsic electrochemistry. The oxidation peak potential of group VIB TMDCs unveils an increasing trend in the order of WSe2 < MoSe2 < WS2 < MoS2.394 These findings are consistent with previous studies that show tungsten dichalcogenides oxidize easier than molybdenum dichalcogenides and diselenides over disulfides.395 Furthermore, ditellurides are known to be mainly prone to oxidation. In liquid-phase exfoliated TMDCs, Coleman et al. showed the lack of oxide impurities except for MoTe2 and WTe2.286 Raman spectra indicated the presence of TeO2 in ditellurides, where WTe2 contained a larger amount of oxides. Chalcogen-dependence of the inherent electro-oxidative waves was also observed in vanadium and platinum dichalcogenides, where the intrinsic oxidation potentials decrease going down the chalcogen group.396 Under acidic conditions for the HER, the oxidation tendency of TMDs is not be a key reflection since the onset of the HER occurs in the negative potential region. In the pnictogen family, multilayered phosphorene was the first to be examined for its electrochemistry, and found to have a single intrinsic anodic indicator. Its anodic peak stems from the oxidation of P(0) to P(V), resulting in P2O5 or H3PO4 species.397 Exposure to ambient light, moisture and air readily oxidizes its surface to PxOy, which reacts further to form H3PO4. Due to the quantum confinement effect, Martel et al. revealed that the degradation of BP is thickness-dependent, where the extent of passivation increases as the number of layers decreases. Other than it proclivity to oxidation, multi-layered phosphorene also displays a prominent ORR signal at −0.5 V.397 Subsequently, further electrochemical study of layered arsenene, antimonene and bismuthene was performed. The voltammograms of bulk pnictogens are devoid of electrochemical signals. Upon shear exfoliation, native redox signals of layered pnictogens appear. Going down the group, the anodic peak potentials of exfoliated pnictogens become increasingly negative from −0.10 V for arsenene to −0.19 V for bismuthene. In comparison to other 2D materials, g-C3N4 displays negligible intrinsic electroactivity. Regardless of the presence of nitrogen atoms, the CVs of g-C3N4 formed from diverse methods are featureless. Therefore, no electroactive surface functional groups exist on as-formed g-C3N4. Akin to pristine graphene, g-C3N4 shows a wide operational window that eliminates inherent interference in electrochemical measurement. The innate electrochemistry of the prototype MXene Ti3C2Tx has been observed recently. In electrolyte of pH 7.0, Ti3C2Tx showed a strong irreversible oxidation signal at 430 mV, which disappeared in successive sweeps. This signal is useful for the electrochemical oxidation of nicotinamide adenine dinucleotide (NADH).398 Electrochemically oxidized Ti3C2Tx that cannot be reproduced presents a stable window of operation in electrochemical applications.11.2. Electron transfer at 2D materials
After revealing the intrinsic electrochemistry of 2D materials, which experience inherent oxidation or reduction, another important aspect to argue is their electrode kinetics. The electrode kinetics fundamentally connotes the heterogeneous electron transfer (HET) between electroactive molecular probes in solution and solid-state electrode materials. Anisotropic, electronic and surface properties have been recognized to show an important effect on electron transfer kinetics. The anisotropy of the graphene family is obvious in the practical HET rates occurring on their edge and basal planes. On the edge planes of graphite-based highly pyrolytic graphite electrodes, the HET rates of the [Fe(CN)6]3−/4− redox couple are quick.399 In contrast, the HET rates are significantly slower on the basal planes. The same trend was observed for a single graphene layer, whereby the HET rates of molecular probes such as [Fe(CN)6]3−/4−, ascorbic acid and NADH were more rapid on the edge than the basal plane.400 In addition, defects in the graphene family produce quicker HET rates because of their superior electronic DOS. The energy level of defect states lies between the CBs and VBs in the disordered pristine sp2 structure. This brings the DOS close to the Fermi level.401 On the contrary, the minute overlap of the VB and CB of pristine graphene results in a low DOS at the Fermi level. The HET rates at the defect sites of CVD graphene, which had mechanically and chemically persuade defects, were an order of magnitude larger than at the basal plane.402 The O2-containing functional groups on graphene strongly influence the HET rates towards specific electroactive probes. In a study involving GO and rGO materials, the results demonstrated that the HET rate increased as the carbon-to-oxygen ratio increased.403 rGO, where oxygen groups are eliminated, showed cases of an elevated HET rate and minor charge transfer resistance (Rct) in the [Fe(CN)6]3−/4− redox probe related to GO.404 The electrostatic repulsion between the O2-containing groups in GO and negatively charged [Fe(CN)6]3−/4− redox probe hinders electron transfer across the electrode–electrolyte interface, causing slower HET rates in GO. Specific to surface-sensitive redox probes, such as [Fe(CN)6]3−/4−, the effects of oxygen-containing moieties on HET rates do not influence surface-insensitive probes such as [Ru(NH3)6]3+/2+.405Being anisotropic, TMDCs are similar to graphene in that discernible electron transfer characteristic occurs from their edge and basal planes. Gerischer et al. showed the method of electron relocation at these two orthogonal planes of MoS2 using [Fe(CN)6]3−/4−, Fe3+/2+ and Cu2+/+ redox probes and recognized a connection to the electronic structure of MoS2.407 As one may anticipate, the HET rate of the MoS2 edge plane surpasses that of its basal surface, as also seen in the case of graphene. This fact is recognized due to the considerable overlap between the dxy and dxy22 orbitals of Mo CB and orbitals of the redox probes. In agreement with this, it was determined that the edge plane of macroscopic MoS2 crystals (Fig. 31) demonstrates a fast HET rate and k0obs = 4.96 × 10−5 cm s−1 for [Fe(CN)6]3−/4− and 1.1 × 10−3 cm s−1 for [Ru(NH3)6]3+/2+ redox probes.406 On the other hand, the pristine basal plane showed slow HET rates close to zero for both redox probes. Exfoliation of bulk TMDCs may either enlarge or deteriorate the HET rates towards [Fe(CN)6]3−/4−. In comparison to their individual bulk counterparts, faster HET rates were observed for exfoliated MoSe2 and WS2 across conventional organolithium reagents and aromatic intercalants, but exfoliated MoS2 and WSe2 showed diverse properties, where the rate was quicker in some cases and slower in others relying on the intercalant.408 Sometimes, exfoliation may initiate oxides that obstruct electron movement at the electrode–electrolyte interface. Electrochemical treatment results in structural and electronic alteration, which modify the HET rates of TMDs. Zhang et al. formed exfoliated 2D MoS2 from molybdenite crystals via electrochemical Li-intercalation. Electroreduction of exfoliated MoS2 resulted in enhanced conductivity, with faster HET rates for redox probes than before treatment. On the other hand, oxidation weakens the HET rates of both bulk and exfoliated MoS2. The DFT-based computational study showed that the electron doping during electroreduction is the primary factor stabilizing the 1T phase, which enhances electron transfer, and hence catalytic properties for the HER.396 It has become a general characteristic for TMDCs that electro-reduction enhances their HET rates for [Fe(CN)6]3−/4−, but electrooxidation may impede electron transfer (Fig. 32). Dopants and impurities are known to modify their electron transfer characteristics. N-G, with electron-donating nitrogen dopants set in by thermal exfoliation of graphite oxide in NH3-saturated atmosphere, shows faster HET rates for the [Fe(CN)6]3−/4− redox couple than undoped graphene.409 Also, N-G prepared through N2 plasma treatment of graphene shows superior catalytic activity in the reduction of H2O2 to the untreated graphene.410 It is also reported that metal-based contaminants of Ni, Fe and Co in parts per billion concentrations in graphene also improve its electrocatalytic activity for analytes for example hydrazine and NaHS.411 Also, the occurrence of TM dopants in TMDCs impacts their final HET performance for the [Fe(CN)6]3−/4− redox probe. Nb- and Ta-doped MoS2 showed slightly a lower HET rate than undoped MoS2, whereas improved HET rate upon doping WS2 with Nb or Ta.296 The anisotropic nature of BP also adds to the diverse electron transfer rates on the edge and basal planes. For both the [Fe(CN)6]3−/4− and [Ru(NH3)6]3+/2+ redox probes, quicker electron transfer was observed for the edge but slow electron transfer rates on the basal plane, with poorly defined redox signals for the basal plane. Similarly, the edge plane of BP is very responsive to the oxidation of ascorbic acid analyte, as evident by its larger current compared to the mild current of the basal plane.412 Other members of the pnictogen family show that shear exfoliation enhance their electron transfer nature. There is a noticeable boost in the HET rate upon shear exfoliation of bulk pnictogens, wherein the most accentuated increase is shown in bismuthene contrast with its bulk state. All shear-exfoliated pnictogens exhibit improved catalytic nature towards the oxidation of ascorbic acid. Mainly, antimonene showed a dramatic decrease in potential for the oxidation of ascorbic acid by 0.1 V in contrast with bulk Sb.413 The effect of surface nature on the electron transfer characteristics of h-BN is evident by the different current signals of h-BN immobilized on various carbon-based substrates. Using the [Ru(NH3)2]3+/2+ redox probe, the HET rate decreases with an increase in h-BN mass loading, which denotes slower electrode kinetics occurring on the h-BN surface relative to the underlying carbon-based substrates.44 When h-BN is tailored on smooth substrates, such as a glassy carbon electrode, the cathodic current signal exhibited only a minor increase in intensity. However, there is a considerable rise in cathodic current upon the use of h-BN tailored on a screen-printed electrode, which offered a rough and ridged surface. Recently, the HET rates of MXenes have also been reviewed. Ti3C2, the archetypal MXene, was modified with fluorine and oxygen, resulting in slower electron transfer kinetics for [Fe(CN)6]3−/4– (ref. 414) and sluggish electron transfer on a halogen-terminated diamond electrode because of the feeble interface between the anionic [Fe(CN)6]3−/4− and electronegative fluorine and hydrophobic surface.415 Upon alkalization of Ti3C2, its electronegative fluorine functional groups were replaced with hydroxyl groups, which are less electronegative. Thus, alk-Ti3C2 showed quicker electron movement than before, together with a decline in Rct.414
 | ||
| Fig. 31 Anisotropic effects that influence electron transfer in 2D materials. (a) Schematic illustration of the edge and basal planes of MoS2. Inset: macroscopic molybdenite crystal (approximately 190 mm × 280 mm). Surface morphologies of the (b) edge and (c) basal planes of MoS2 by optical microscopy. This figure has been adapted/reproduced from ref. 406 with permission from John Wiley & Sons, Inc. | ||
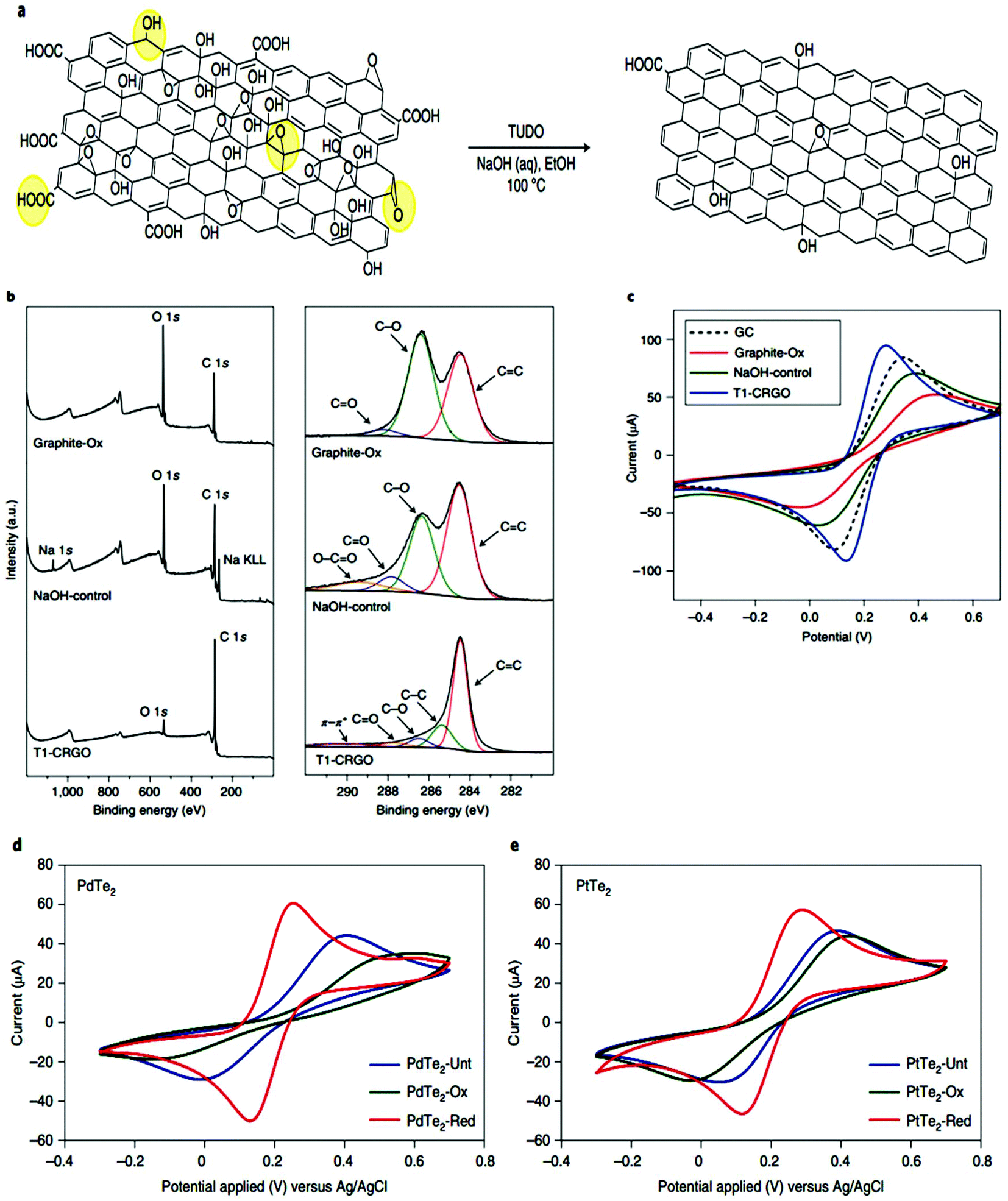 | ||
| Fig. 32 Surface characteristics that influence electron transfer in 2D materials. (a) Scheme of the reduction of functional groups on GO by thiourea dioxide (TUDO). (b), XPS spectra of graphite oxide after reduction with NaOH in the absence of TUDO for 2 h at 90 °C (NaOH control), and after reduction with NaOH and TUDO for 1 h at 90 °C (chemically modified rGO, T1-CRGO), and their (c) CV recorded in [Fe(CN)6]3−/4− probe. Voltammograms depicting the changes in electron transfer in the treated (d) PdTe2 and (e) PtTe2 towards the [Fe(CN)6]3−/4− probe. This figure has been adapted/reproduced from ref. 175 with permission from John Wiley and Sons Ltd. | ||
11.3. Mass transport
Mass transportation is critical in highly active catalysts since the fast reduction of interfacial reactant species (H+ or OH−) and the formation of gaseous products hamper response rates. Therefore, an incessant reactant supply and fast gas discharge are necessary to preserve elevated response effectiveness. In 2D catalysts, the interstitial spaces between neighboring sheets have been accepted as 2D channels to facilitate the mass transport in the liquid and gaseous phases. Including spacers in 2D MoS2 yielded open, vigorous and connective channels to the SSA and enhanced ion diffusion, with largely improved HER catalyst activity (Fig. 33).416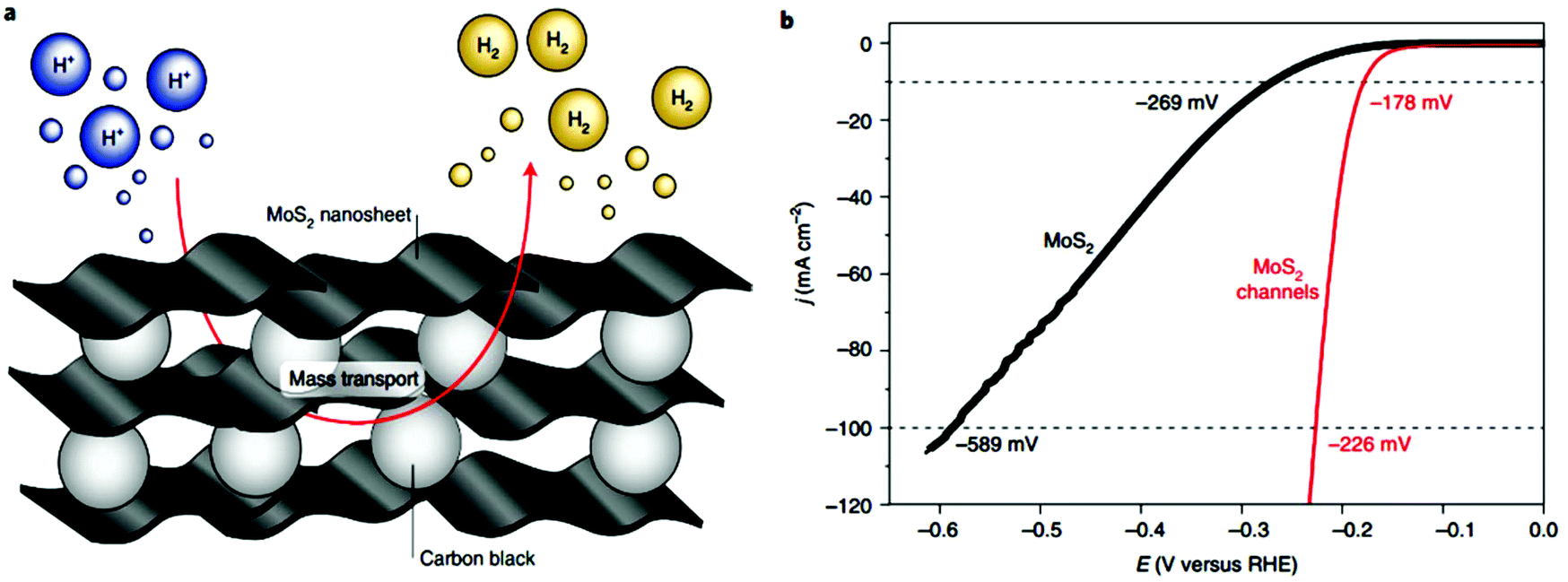 | ||
| Fig. 33 Mass transport affects 2D electrocatalytic materials. (a) Schematic illustration of engineered channels in MoS2. (b) Catalytic HER presentation of MoS2 nanosheets and MoS2 with engineered channels. This figure has been adapted/reproduced from ref. 175 with permission from John Wiley and Sons Ltd. | ||
11.4. Activity at the edges
The anisotropic factor of 2D materials for their catalytic nature is verified in their active edges and inert basal planes. Atoms residing on the edges of 2D materials are shown to have a diverse chemical setting than other parts of materials with better tendency for coordination compared with basal planes, which are normally of saturated coordination. Since the edge sites are responsible for the catalytic activity of 2D materials, it is vital to optimize their edge structure to improve their properties. The edges of graphene were experimentally exposed to have superior ORR activity compared to its basal plane.400 Graphene edges frequently have oxygen groups at their end points because of oxidation or the parent graphite material. Since these oxygen groups may facilitate ORR activity, the liquid-assisted mechanical exfoliation of graphite yielded graphene with a low oxygen content to minimize the effect of oxygen groups on the ORR.417 Therefore, the abundant edge sites of graphene were established as the main cause for its improved ORR activity. Zigzag edges are the thermodynamically favorable active sites and armchair edges are considered as inactive because of the strong adsorption of OH− species, which restrain the active sites for oxygen binding.418 To reduce the effect of dopants, edge-rich and dopant-free, graphene with competent ORR nature was formed via Ar plasma etching.419 The edge-rich graphene outperformed pure graphene with a lower onset potential and higher i for the ORR. The pristine graphene formed via plasma etching is ideal model to determine the role of edges as the ORR active sites. Similarly to graphene, h-BN on an Au electrode had less edges as active sites for the ORR, which showed the lower ORR activity for 2D h-BN sheets than BN nanotubes.420 The BN nanotubes showed few B- and N-edge structures, while well-known edge structures were noted in the BN nanosheets. In the HER, the edges of TMDCs are the catalytic sites, as shown for 2H-MoS2. By investigating a monolayer of MoS2 nanoparticles of various sizes deposited on the Au(111) surface, the HER activity demonstrated a linear relation with the number of edge sites of MoS2.160 For example, achieving an elevated fraction of exposed edge sites resulted in the formation of the mesoporous double-gyroid MoS2 structure.290 The curvature of the double-gyroid MoS2 catalyst gives a high density of edge sites, and thus improved HER property. The orientation of TMDCs on a specific substrate also boost their edge sites, e.g., when MoS2, MoSe2 and WSe2 films were vertically aligned on substrates, maximize edge-termination, and hence enhanced HER properties were observed.421It was also investigated that the basal planes of the metallic 1T-phase of TMDCs, similar to the edges, also have HER active nature.161 Exfoliated 1T-phase group VIB TMDC nanosheets showed better catalytic HER activity to that of the 2H-phase, with the low Tafel slope of 1T-MoS2 of 43 mV dec−1 because the increase in active sites led to higher conductivity in the 1T-phase of TMDCs.316 Besides TMDCs as HER electrocatalysts, MXenes equipped with O* or OH* termination are also promising candidates, although pure MXenes are seldomly used as electrocatalysts because of their low activity. It is exciting to show that the O* basal planes of the majority of MXenes are active for the HER. Especially, the delamination of Mo2CTx enhanced its HER activity despite of having a greater part of exposed basal planes.364 Unexpectedly, the edges of TMDCs are also active for the ECR in the presence of ionic liquids as co-catalysts, regardless of being active sites for the HER, an opposing reaction to the ECR in acidic conditions. In the ECR, CO2 reduction to CO occurs at the Mo-terminated edges of MoS2 due to their metallic nature and high d-electron density.376 The ionic liquid, 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIM-BF4), stabilizes CO2via complex arrangement and prevents the HER. A series of TMDCs nano-flakes including WS2 and WSe2 showing a superior density of edges than their bulk counterparts showed improved ECR catalysis.377 This was the most prominent for the WSe2 nano-flakes terminated with W atoms. Among the 2D materials, layered metal oxides and LDHs are the most exploited OER catalysts recognized due to their wonderful OER nature, where edges account for their OER activity. Liquid-phase exfoliation of bulk LDHs (NiFe, NiCo, and CoCo) into monolayers resulted in up to 4.5 times higher OER catalysis than before, similar to IrO2 catalysts.247 Furthermore, the OER activity was further enhanced upon fracturing single-layered NiFe LDHs into ultrafine nanosheets with a thickness of <3 nm and maximum exposed edges.422 When BP is thinned into nanosheets by liquid exfoliation followed by centrifugation, additional OER active sites are formed. These produced sites are considered to be edges.14 Compared with bulk phosphorus, nanosheets exhibit improved OER performance with an onset potential of 1.45 V and Tafel slope of 88 mV dec−1.14,175
12. Conclusions, challenges and future directions
The rich electrochemistry of 2D materials presents opportunities to discover their applications in energy production and storage. Despite the diversity of 2D materials, their resultant electrocatalytic and charge transfer properties are attributed to anisotropy and surface characteristics. We shed light on the inherent electrochemistry arising from intrinsic elemental composition, surface oxides and oxygen groups. In most 2D materials, edges display faster electron transfer and higher electrocatalytic activity against the sluggish electron transfer of basal planes, which improved by some treatments. The role of surface impurities or additional functional groups may also modify electron transfer properties. 2D materials have garnered much success in ORR, HER, OER, N2RR and ECR electrocatalysis, where mostly their edges are the catalytic active centers, together with the some other alterations in 2D sheets, e.g., doping. Nanostructure materials with optimal morphologies or exfoliation of materials into thinner and smaller nanoflakes are often implemented to increase the active DOS edge sites. Using catalyst supports will further improve the accessibility of these active sites. The integration of dopants and functional groups also enhances intrinsic catalytic activity. Generally, graphene is a promising choice as ORR catalyst, TMDCs take the helm as intrinsic HER electrocatalysts, while LDHs and associated oxides are best for catalyzing the OER. The burgeoning research into MXenes also show their potential as HER catalysts. The use of 2D materials for ECR has room for expansion as works to date mainly report TMDCs and graphene-based materials. The challenge in the ECR also lies in fabricating catalysts with high product selectivity. Employing 2D materials such as h-BN, g-C3N4 and pnictogens as energy-related electrocatalysts is limited and further investigations can elucidate their catalytic potential. Beyond TMDCs and graphene, MXenes are the next promising electrocatalysts, which are predicted to outperform TMDCs because the basal planes of MXenes upon functionalization may become catalytic and function as additional active sites. Published works on h-BN, g-C3N4 and pnictogens as electrocatalysts are currently scarce, though efforts have been undertaken either by doping or developing hybrids with better catalytic attributes. Moving forward, the field of 2D nanomaterials is brimming with possibilities. The development of hybrid 2D materials by integrating two or more materials can create novel composite structures that display unique functionality and tailored properties for renewable energy device applications. Anisotropy and surface characteristics can serve as guidelines when designing different compounds. To date, increasing research has resulted in the design of new materials (MXenes) and memorizing prosperity in graphene era. Although noteworthy progress has been made in electrocatalysts, this is still a growing area for developing new-type MXene materials for commercially applications, which is still in its infancy, and needs to be radically improved. The interfacial features of MXene cause the electrolyte to have crucial effects on its electrochemical activity. Currently, interrelated fundamental research on liquid/solid interfaces is totally lacking, which is necessary.12.1. 2D-materials as model materials in electrochemistry
2D-materials present a straightforward model in theoretical predictions because of their plane surface and uncomplicated crystal, and therefore may gave chance in elementary electrochemical study. Combining 2D material-based electrochemical reactions with microcircuit techniques may be incredibly fundamental for quantitative studies on mechanisms and the effect of discrete factors on their activity. These perceptions will guide the design of good electrocatalysts for electrodes. In addition, during electrochemical reactions, because of the high potential of electrons and high metal salt concentration surrounding electrodes, valency, surface chemistry and material chemical composition are not constant. These changes need to be characterized and understood to determine where the reactivity originates and how to tailor it. Thus, combining electrochemical 2D materials and in situ characterization techniques is critical to determine the relationship between reactants and materials in electrochemical reactions at the atomic level or nanoscale. Such studies will shed light on surface structure changes, valency, and 2D material composition at the nanoscale or atomic level for electrochemical materials.12.2. Metallic 2D materials for electrocatalysis
Metallic 2D materials, i.e., MXenes, are very hopeful substitutes for PMCs. They have improved electronic conductance and more catalytic active sites than semiconductor 2D-Xene electrocatalysts, which are similar to graphene, phosphorene, silicene etc., where just their edges are active. Nanosheets in contrast to nonlamellar structures, such as Co3O4 and Co9S8, have plentiful active sites arising from basal plane un-saturation coordination. Metallic 2D materials have a lamellar structure, which is robust and offer long-term stability in electrochemical reactions. Also, synthetic methods for 2D-materials besides CVD are required. Third, for electrocatalysts based on 2D metallic materials, possessing analogous properties with metals, there is a need to focus on their in situ surface characterization during electrochemical measurement, which is especially imperative for electrocatalytic mechanisms.12.3. Understanding and commercialization of catalysis
Most electrocatalyst studies reported to date for 2D materials are still at the trial and error stage. This is due to the lack of effective characterization methods and theories, which facilitate the direct design of efficient catalysts using 2D materials. During the last few years, although theoretical predictions have been introduced, there is still a large gap between predictions and realistic approach. Thus, in situ characterizations and calculations close to practical conditions are required. The investigation of 2D materials as electrocatalysts and their heterostructures cover approximately all kinds of heterogeneous catalysis, including traditional heterogeneous catalysis, biocatalysis, photocatalysis, and electrocatalysis. Pristine 2D materials and their heterostructures may be utilized as catalysts or catalyst support (donor or acceptor depending on interaction with metals). However, besides technical considerations, there are several technological issues that are still unsolved. 2D materials as large-area films and small nanosheets are the two main structures used in different applications and their massive production are frequently requisite for electrocatalyst applications. Recent graphene production expenditure is very high due to the shortcomings in accessible synthetic methods. Consequently, it is significant to extend further efficient synthetic approaches for the large-scale synthesis of 2D layers. Another urgent matter is the molding process or stacking 2D materials and their heterostructures into a valid commercial electrocatalyst. Generally, the 2D nanosheet structure is unfastened and inclined to aggregate at elevated temperatures due to functional group or adsorbed species desorption, which critically limit their industrial application. A promising method to resolve this dilemma is to fasten 2D-nanosheets on suitable supports. It was observed that by assembling 2D materials into 3D structures, e.g., foam-supporting materials such as Ni foam or carbon fiber paper, catalytic performances were enhanced. Thus, by understanding the catalytic nature of materials through in situ characterizations and theoretical simulations, supported with appropriate preparation method, then real catalytic applications will be achievable.13. Limitations and outlook for future work
The commercialization of2D materials is still early-to-mid stage technology in the Gartner Hype cycle, and probable a decade away starting from first technology production. Thus, industrialization with the development of the small-scale production for 2D materials from small start-up companies will push advancements toward a wide spectrum of possibilities. The achievements with 2D materials originated from basic technology factors, passed quickly through an inflated peak, joined proficiently with early adopters, and shifted efficiently towards the height of production. The time periods above in which the development of materials fluctuate greatly depend on technology complications. For example, technologies that rely on 2D inks will grow faster compared to that involving high-speed electronic switching. Cost-efficient synthesis, superior mobility, massive production and long-term chemical robustness may be basic factors that drive 2D materials into the upcoming photovoltaics marketplace.An exciting set of synergistic effects were exposed by graphene co-doped with elements close to carbon in the periodic table, e.g., P and S, besides B and N. N-Doped graphene supplied paramount non-metallic catalysts in the ORR to date, but the impacts of N on the quaternary basal plane against pyridinic N at the edges are still not established and demand additional critical analysis. The mixed metal/graphene composite approach has resulted in electrocatalysts comparable with IrO2, the benchmark OER electrocatalyst. Carbon/nitride MXene (e.g., TiCx carbide) has also been obtained as a better alternative than carbon-supported IrO2. Metal independent P-G, N-G and B/N-G have demonstrated encouraging OER activity. The codoping of graphene with N/S and N/P materials has shown very promising metal independent HER catalysts, approaching the Pt catalyst. The carbon nitrides supported on N-G emerged as comparable with the bulk MoS2 catalyst. In HER catalysis, TMDC nanosheets are being extensively explored, and to date, selenides have been proven to be the most efficient HER electrocatalysts. At the edge sites, the insertion of different metals actually offers bimetallic synergistic effects and enhanced HER rates. TMDCs/rGO combinations also show promise. The ECR is useful for obtaining useful chemicals products (methanol, ethanol, methane etc.). Moreover its reduction is also a key factor to minimize the carbon footprint from the combustion energy route because it is continually dependent on carbon oxidation. Converting CO2 back into fuel gas using 2D-Mxene-based electrocatalysts is in the early stages, and initial subsidiary achievements have been achieved with ultrathin metal and metal sulfide nanosheets. Metal-free carbon/graphene electrodes are also utilized in ionic liquid environment for CO2 reduction. The polymerized ionic liquids show outstanding CO2 adsorption capability, and thus it is probability that their synergism matching with graphene will create a number of thrilling outputs in CO2 reduction. The scalable utilization of 2D materials as electrocatalysts needs some consideration.
1. Understanding the nature of 2D material electrocatalysts through theoretical analysis and in situ characterizations will help in the design and development of customized diverse new nanosheets and atomic level electrocatalysts.
2. The mass production of 2D materials is key factor for their application. There is a need for the controllable production of a number of layers and flakes with dimension confinement for 2D-Mxene materials.
3. The modification of 2D materials in a controllable way, such as doping to regulate their electronic structures and surface engineering to bring in functional groups, is highly recommended for sustainable applications.
4. Manufacturing and functionalization of nanosheet heterostructures are required for tuning electronic properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from Natural Science Foundation of China (NSFC) (Grant No.: 61275043, 61307048, and 61171006).References
- K. Khan, A. K. Tareen, L. Jia, U. Khan, A. Nairan, Y. Yuan, X. Zhang, M. Yang and Z. Ouyang, Dalton Trans., 2018, 13498–13506 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- E. Mobil, Google Scholar, 2015.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- S. Jewell and S. M. Kimball, US Geological Survey, 2015, 9, 196.
- K. Khan, A. K. Tareen, M. Aslam, K. H. Thebo, U. Khan, R. Wang, S. S. Shams, Z. Han and Z. Ouyang, Prog. Solid State Chem., 2018, 1–19 CAS.
- K. Khan, A. K. Tareen, S. Elshahat, N. Muhammad, J. Li, I. Aboodd, L. Bibbò, A. Yadav, R. U. R. Sagar, U. Khan and Z. Ouyang, RSC Adv., 2018, 8, 24276–24285 RSC.
- K. Khan, A. K. Tareen, S. Elshahat, A. Yadav, U. Khan, M. Yang, L. Bibbò and Z. Ouyang, Dalton Trans., 2018, 3819–3830 RSC.
- K. Khan, A. k. Tareen, U. Khan, A. Nairan, S. Elshahat, N. Muhammad, M. Saeed, A. Yadav, L. Bibbò and Z. Ouyang, Sci. Rep., 2018, SREP-18–33491B Search PubMed.
- K. Khan, L. Jia, Z. Wenwei, X. Wei, Y. Ye and S. Weijie, J. Wuhan Univ. Technol.Mater. Sci. Ed, 2016, 31, 1201–1205 CrossRef CAS.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 41–58 RSC.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- J. Ma, S. Lu, Z. Guo, X. Xu, H. Zhang, D. Tang and D. Fan, Opt. Express, 2015, 23, 22643–22648 CrossRef PubMed.
- X. Ren, J. Zhou, X. Qi, Y. Liu, Z. Huang, Z. Li, Y. Ge, S. C. Dhanabalan, J. S. Ponraj and S. Wang, Adv. Energy Mater., 2017, 7, 1700396 CrossRef.
- H. Gleiter, Acta Mater., 2000, 48, 1–29 CrossRef CAS.
- V. Skorokhod, I. Uvarova and A. Ragulya, Google Scholar, 180.
- J. Lu, K. Zhang, X. F. Liu, H. Zhang, T. C. Sum, A. H. C. Neto and K. P. Loh, Nat. Commun., 2013, 4, 2681 CrossRef PubMed.
- L. Zhao, D. Tang, X. Wu and H. Zhang, Opt. Lett., 2010, 35, 2756–2758 CrossRef CAS PubMed.
- Z.-g. Dai, X.-h. Xiao, W. Wu, Y.-p. Zhang, L. Liao, S.-s. Guo, J.-j. Ying, C.-x. Shan, M.-t. Sun and C.-z. Jiang, Light: Sci. Appl., 2015, 4, e342 CrossRef CAS.
- W. Li, Y. Zhang, L. Liu, D. Li, L. Liao and C. Pan, Nanoscale, 2015, 7, 18147–18151 RSC.
- A. Gupta, T. Sakthivel and S. Seal, Prog. Mater. Sci., 2015, 73, 44–126 CrossRef CAS.
- K. Shehzad, Y. Xu, C. Gao and X. Duan, Chem. Soc. Rev., 2016, 45, 5541–5588 RSC.
- C. Zhang, Y. Li, P. Zhang, M. Qiu, X. Jiang and H. Zhang, Sol. Energy Mater. Sol. Cells, 2019, 189, 11–20 CrossRef CAS.
- W. Tao, X. Ji, X. Xu, M. A. Islam, Z. Li, S. Chen, P. E. Saw, H. Zhang, Z. Bharwani and Z. Guo, Angew. Chem., 2017, 129, 12058–12062 CrossRef.
- T. Wang, Y. Guo, P. Wan, H. Zhang, X. Chen and X. Sun, Small, 2016, 12, 3748–3756 CrossRef CAS PubMed.
- S. Yang, Y. Liu, W. Chen, W. Jin, J. Zhou, H. Zhang and G. S. Zakharova, Sens. Actuators, B, 2016, 226, 478–485 CrossRef CAS.
- J. Li, H. Luo, B. Zhai, R. Lu, Z. Guo, H. Zhang and Y. Liu, Sci. Rep., 2016, 6, 30361 CrossRef CAS PubMed.
- P. Wan, X. Wen, C. Sun, B. K. Chandran, H. Zhang, X. Sun and X. Chen, Small, 2015, 11, 5409–5415 CrossRef CAS PubMed.
- Q. Jiang, L. Xu, N. Chen, H. Zhang, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2016, 55, 13849–13853 CrossRef CAS PubMed.
- X. Ren, Z. Li, Z. Huang, D. Sang, H. Qiao, X. Qi, J. Li, J. Zhong and H. Zhang, Adv. Funct. Mater., 2017, 27, 1606834 CrossRef.
- Z. Zhang, Y. Liu, L. Ren, H. Zhang, Z. Huang, X. Qi, X. Wei and J. Zhong, Electrochim. Acta, 2016, 200, 142–151 CrossRef CAS.
- J. Shao, L. Tong, S. Tang, Z. Guo, H. Zhang, P. Li, H. Wang, C. Du and X.-F. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 5391–5399 CrossRef CAS PubMed.
- Z. Huang, Z. Zhang, X. Qi, X. Ren, G. Xu, P. Wan, X. Sun and H. Zhang, Nanoscale, 2016, 8, 13273–13279 RSC.
- J. Song, L. Zhang, Y. Xue, Q. Y. S. Wu, F. Xia, C. Zhang, Y.-L. Zhong, Y. Zhang, J. Teng and M. Premaratne, ACS Photonics, 2016, 3, 1986–1992 CrossRef CAS.
- J. Liu, Y. Xue, Z. Wang, Z.-Q. Xu, C. Zheng, B. Weber, J. Song, Y. Wang, Y. Lu and Y. Zhang, ACS Nano, 2016, 10, 3536–3542 CrossRef CAS PubMed.
- Q. Zhang, Z. Chang, G. Xu, Z. Wang, Y. Zhang, Z. Q. Xu, S. Chen, Q. Bao, J. Z. Liu and Y. W. Mai, Adv. Funct. Mater., 2016, 26, 8707–8714 CrossRef CAS.
- Y. Wang, E. Della Gaspera, B. J. Carey, P. Atkin, K. J. Berean, R. M. Clark, I. S. Cole, Z.-Q. Xu, Y. Zhang and Q. Bao, Nanoscale, 2016, 8, 12258–12266 RSC.
- Z.-Q. Xu, Y. Zhang, Z. Wang, Y. Shen, W. Huang, X. Xia, W. Yu, Y. Xue, L. Sun and C. Zheng, 2D Mater., 2016, 3, 041001 CrossRef.
- Y. Wang, Z. Xia, L. Liu, W. Xu, Z. Yuan, Y. Zhang, H. Sirringhaus, Y. Lifshitz, S. T. Lee and Q. Bao, Adv. Mater., 2017, 29, 1606370 CrossRef PubMed.
- Z.-Q. Xu, Y. Zhang, S. Lin, C. Zheng, Y. L. Zhong, X. Xia, Z. Li, P. J. Sophia, M. S. Fuhrer and Y.-B. Cheng, ACS Nano, 2015, 9, 6178–6187 CrossRef CAS PubMed.
- Y. Zhang, D. Li, X. Tan, B. Zhang, X. Ruan, H. Liu, C. Pan, L. Liao, T. Zhai and Y. Bando, Carbon, 2013, 54, 143–148 CrossRef CAS.
- Y. Zhang, C. Luo, W. Li and C. Pan, Nanoscale, 2013, 5, 2616–2619 RSC.
- C. Xirouchaki and R. Palmer, Philos. Trans. R. Soc., A, 2003, 362, 117–124 CrossRef PubMed.
- A. F. Khan, E. P. Randviir, D. A. Brownson, X. Ji, G. C. Smith and C. E. Banks, Electroanalysis, 2017, 29, 622–634 CrossRef CAS.
- M. A. Khan and Y.-S. Ho, Environ. Eng. Manage. J., 2015, 14, 2163–2168 CrossRef.
- W. Zou, K. Khan, X. Zhao, C. Zhu, J. Huang, J. Li, Y. Yang and W. Song, Mater. Res. Express, 2017, 4, 1–20 Search PubMed.
- W. Lee, S. H. Kang, J.-Y. Kim, G. B. Kolekar, Y.-E. Sung and S.-H. Han, Nanotechnology, 2009, 20, 335706 CrossRef PubMed.
- J. W. Stouwdam and R. A. Janssen, J. Mater. Chem., 2008, 18, 1889–1894 RSC.
- L. Lu, Z. Liang, L. Wu, Y. Chen, Y. Song, S. C. Dhanabalan, J. S. Ponraj, B. Dong, Y. Xiang and F. Xing, Laser Photonics Rev., 2018, 12, 1700221 CrossRef.
- X.-Y. Feng, B.-Y. Ding, W.-Y. Liang, F. Zhang, T.-Y. Ning, J. Liu and H. Zhang, Laser Phys. Lett., 2018, 15, 085805 CrossRef.
- V. Mokerov, Y. V. Fedorov, L. Velikovski and M. Y. Scherbakova, Nanotechnology, 2001, 12, 552 CrossRef CAS.
- Z. Guo, S. Chen, Z. Wang, Z. Yang, F. Liu, Y. Xu, J. Wang, Y. Yi, H. Zhang and L. Liao, Adv. Mater., 2017, 29, 1703811 CrossRef PubMed.
- J. Zheng, H. Zhang, S. Dong, Y. Liu, C. T. Nai, H. S. Shin, H. Y. Jeong, B. Liu and K. P. Loh, Nat. Commun., 2014, 5, 2995 CrossRef PubMed.
- Q. Bao, H. Zhang, B. Wang, Z. Ni, C. H. Y. X. Lim, Y. Wang, D. Y. Tang and K. P. Loh, Nat. Photonics, 2011, 5, 411 CrossRef CAS.
- K. Novoselov, D. Jiang, F. Schedin, T. Booth, V. Khotkevich, S. Morozov and A. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, I. V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147 CrossRef CAS PubMed.
- Q. Bao, H. Zhang, Y. Wang, Z. Ni, Y. Yan, Z. X. Shen, K. P. Loh and D. Y. Tang, Adv. Funct. Mater., 2009, 19, 3077–3083 CrossRef CAS.
- W. Tao, X. Zhu, X. Yu, X. Zeng, Q. Xiao, X. Zhang, X. Ji, X. Wang, J. Shi and H. Zhang, Adv. Mater., 2017, 29, 1603276 CrossRef PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- M. M. Alsaif, A. F. Chrimes, T. Daeneke, S. Balendhran, D. O. Bellisario, Y. Son, M. R. Field, W. Zhang, H. Nili and E. P. Nguyen, Adv. Funct. Mater., 2016, 26, 91–100 CrossRef CAS.
- T. Wang, D. Qi, H. Yang, Z. Liu, M. Wang, W. R. Leow, G. Chen, J. Yu, K. He and H. Cheng, Adv. Mater., 2019, 31, 1803883 CrossRef PubMed.
- D. K. Sang, H. Wang, M. Qiu, R. Cao, Z. Guo, J. Zhao, Y. Li, Q. Xiao, D. Fan and H. Zhang, Nanomaterials, 2019, 9, 82 CrossRef PubMed.
- T. Fan, Z. Xie, W. Huang, Z. Li and H. Zhang, Nanotechnology, 2019, 6186–6192 Search PubMed.
- X. Bao, Q. Ou, Z. Q. Xu, Y. Zhang, Q. Bao and H. Zhang, Adv. Mater. Technol., 2018, 3, 1800072 CrossRef.
- Z. Wang, Q. Ou, Y. Zhang, Q. Zhang, H. Y. Hoh and Q. Bao, ACS Appl. Mater. Interfaces, 2018, 10, 24258–24265 CrossRef CAS PubMed.
- X. Qi, Y. Zhang, Q. Ou, S. T. Ha, C. W. Qiu, H. Zhang, Y. B. Cheng, Q. Xiong and Q. Bao, Small, 2018, 14, 1800682 CrossRef PubMed.
- Q. Zhang, J. Lu, Z. Wang, Z. Dai, Y. Zhang, F. Huang, Q. Bao, W. Duan, M. S. Fuhrer and C. Zheng, Adv. Opt. Mater., 2018, 1701347 CrossRef.
- C. Zheng, Q. Zhang, B. Weber, H. Ilatikhameneh, F. Chen, H. Sahasrabudhe, R. Rahman, S. Li, Z. Chen and J. Hellerstedt, ACS Nano, 2017, 11, 2785–2793 CrossRef CAS PubMed.
- Y. Ping, Y. Zhang, Y. Gong, B. Cao, Q. Fu and C. Pan, Electrochim. Acta, 2017, 250, 84–90 CrossRef CAS.
- Y. Liu, B. N. Shivananju, Y. Wang, Y. Zhang, W. Yu, S. Xiao, T. Sun, W. Ma, H. Mu and S. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 36137–36145 CrossRef CAS PubMed.
- W. Li, M. U. Rothmann, A. Liu, Z. Wang, Y. Zhang, A. R. Pascoe, J. Lu, L. Jiang, Y. Chen and F. Huang, Adv. Energy Mater., 2017, 7, 1700946 CrossRef.
- G. Zhang, X. Tang, X. Fu, W. Chen, B. Shabbir, H. Zhang, Q. Liu and M. Gong, Nanoscale, 2019, 1762–1769 RSC.
- H. Xie, Z. Li, Z. Sun, J. Shao, X. F. Yu, Z. Guo, J. Wang, Q. Xiao, H. Wang and Q. Q. Wang, Small, 2016, 12, 4136–4145 CrossRef CAS PubMed.
- H. Mu, Z. Wang, J. Yuan, S. Xiao, C. Chen, Y. Chen, Y. Chen, J. Song, Y. Wang and Y. Xue, ACS Photonics, 2015, 2, 832–841 CrossRef CAS.
- S. C. Dhanabalan, J. S. Ponraj, Z. Guo, S. Li, Q. Bao and H. Zhang, Adv. Sci., 2017, 4, 1600305 CrossRef PubMed.
- P. Yan, R. Lin, H. Chen, H. Zhang, A. Liu, H. Yang and S. Ruan, IEEE Photonics Technol. Lett., 2015, 27, 264–267 CAS.
- S. Bai, C. Sun, H. Yan, X. Sun, H. Zhang, L. Luo, X. Lei, P. Wan and X. Chen, Small, 2015, 11, 5807–5813 CrossRef CAS PubMed.
- Z. Sun, Y. Zhao, Z. Li, H. Cui, Y. Zhou, W. Li, W. Tao, H. Zhang, H. Wang and P. K. Chu, Small, 2017, 13, 1602896 CrossRef PubMed.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- C. Xie, C. Mak, X. Tao and F. Yan, Adv. Funct. Mater., 2017, 27, 1603886 CrossRef.
- R. Wang, X. Li, Z. Wang and H. Zhang, Nano Energy, 2017, 34, 131–140 CrossRef CAS.
- S. Chen, J. Duan, J. Ran, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2013, 6, 3693–3699 RSC.
- D. Chimene, D. L. Alge and A. K. Gaharwar, Adv. Mater., 2015, 27, 7261–7284 CrossRef CAS PubMed.
- F. Torrisi and J. N. Coleman, Nat. Nanotechnol., 2014, 9, 738 CrossRef CAS PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372 CrossRef CAS PubMed.
- Z. Zhu, J. Guan, D. Liu and D. Tománek, ACS Nano, 2015, 9, 8284–8290 CrossRef CAS PubMed.
- C. Coleman, H. Goldwhite and W. Tikkanen, Chem. Mater., 1998, 10, 2794–2800 CrossRef CAS.
- Z. Sun, T. Liao, Y. Dou, S. M. Hwang, M.-S. Park, L. Jiang, J. H. Kim and S. X. Dou, Nat. Commun., 2014, 5, 3813 CrossRef CAS PubMed.
- S. Ida, C. Ogata, M. Eguchi, W. J. Youngblood, T. E. Mallouk and Y. Matsumoto, J. Am. Chem. Soc., 2008, 130, 7052–7059 CrossRef CAS PubMed.
- L. Liu, J. Park, D. A. Siegel, K. F. McCarty, K. W. Clark, W. Deng, L. Basile, J. C. Idrobo, A.-P. Li and G. Gu, Science, 2014, 343, 163–167 CrossRef CAS PubMed.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- C. E. Boott, A. Nazemi and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 13876–13894 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55(23), 6771–6775 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- W. Choi, N. Choudhary, G. H. Han, J. Park, D. Akinwande and Y. H. Lee, Mater. Today, 2017, 20, 116–130 CrossRef CAS.
- H. Şahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R. T. Senger and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155453 CrossRef.
- T. Ling, J. J. Wang, H. Zhang, S. T. Song, Y. Z. Zhou, J. Zhao and X. W. Du, Adv. Mater., 2015, 27, 5396–5402 CrossRef CAS PubMed.
- S. Zhang, Z. Yan, Y. Li, Z. Chen and H. Zeng, Angew. Chem., 2015, 127, 3155–3158 CrossRef.
- A. Mannix, Science, 2015, 350, 1513 CrossRef CAS PubMed.
- H. Yin and Z. Tang, Chem. Soc. Rev., 2016, 45, 4873–4891 RSC.
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim and K. L. Shepard, Nat. Nanotechnol., 2010, 5, 722 CrossRef CAS PubMed.
- Y. Ebina, T. Sasaki and M. Watanabe, Solid State Ionics, 2002, 151, 177–182 CrossRef CAS.
- X. Xu, K. Takada, K. Fukuda, T. Ohnishi, K. Akatsuka, M. Osada, B. T. Hang, K. Kumagai, T. Sekiguchi and T. Sasaki, Energy Environ. Sci., 2011, 4, 3509–3512 RSC.
- K. Akatsuka, G. Takanashi, Y. Ebina, M.-a. Haga and T. Sasaki, J. Phys. Chem. C, 2012, 116, 12426–12433 CrossRef CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- X. Zhang, Z. Lai, C. Tan and H. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8816–8838 CrossRef CAS PubMed.
- Y. Zhao, J. Qiao, Z. Yu, P. Yu, K. Xu, S. P. Lau, W. Zhou, Z. Liu, X. Wang and W. Ji, Adv. Mater., 2017, 29, 1604230 CrossRef PubMed.
- S. Kim, A. Konar, W.-S. Hwang, J. H. Lee, J. Lee, J. Yang, C. Jung, H. Kim, J.-B. Yoo and J.-Y. Choi, Nat. Commun., 2012, 3, ncomms2018 Search PubMed.
- H. Fang, S. Chuang, T. C. Chang, K. Takei, T. Takahashi and A. Javey, Nano Lett., 2012, 12, 3788–3792 CrossRef CAS PubMed.
- D. Ovchinnikov, A. Allain, Y.-S. Huang, D. Dumcenco and A. Kis, ACS Nano, 2014, 8, 8174–8181 CrossRef CAS PubMed.
- B. Liu, M. Köpf, A. N. Abbas, X. Wang, Q. Guo, Y. Jia, F. Xia, R. Weihrich, F. Bachhuber and F. Pielnhofer, Adv. Mater., 2015, 27, 4423–4429 CrossRef CAS PubMed.
- M. Long, A. Gao, P. Wang, H. Xia, C. Ott, C. Pan, Y. Fu, E. Liu, X. Chen and W. Lu, Sci. Adv., 2017, 3, e1700589 CrossRef PubMed.
- M. Yoshida, J. Ye, T. Nishizaki, N. Kobayashi and Y. Iwasa, Appl. Phys. Lett., 2016, 108, 202602 CrossRef.
- D. Bandurin, A. Tyurnina, G. Yu, A. Mishchenko, V. Zólyomi, S. Morozov, R. Krishna, R. Kumar, Z. Kudrynskyi and S. Pezzini, Electron Transport In Atomically Thin Crystals, 2018, p. 53 Search PubMed.
- J. Wu, H. Yuan, M. Meng, C. Chen, Y. Sun, Z. Chen, W. Dang, C. Tan, Y. Liu and J. Yin, Nat. Nanotechnol., 2017, 12, 530 CrossRef CAS PubMed.
- Y. Wang, G. Qiu, R. Wang, S. Huang, Q. Wang, Y. Liu, Y. Du, W. A. Goddard, M. J. Kim and X. Xu, Nat. Electron., 2018, 1, 228 CrossRef.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- J. Sato, H. Kato, M. Kimura, K. Fukuda and W. Sugimoto, Langmuir, 2010, 26, 18049–18054 CrossRef CAS PubMed.
- D. S. Kim, T. C. Ozawa, K. Fukuda, S. Ohshima, I. Nakai and T. Sasaki, Chem. Mater., 2011, 23, 2700–2702 CrossRef CAS.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- N. M. Markovic, Nat. Mater., 2013, 12, 101 CrossRef CAS PubMed.
- Y. Kang, P. Yang, N. M. Markovic and V. R. Stamenkovic, Nano Today, 2016, 11, 587–600 CrossRef CAS.
- G. A. Somorjai, Chem. Rev., 1996, 96, 1223–1236 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao and S. Z. Qiao, Adv. Mater., 2015, 27, 5372–5378 CrossRef CAS PubMed.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- J. Peng, X. Chen, W.-J. Ong, X. Zhao and N. Li, Chem, 2018, 5, 1–33 Search PubMed.
- G. G. Scherer, Solid State Ionics, 1997, 94, 249–257 CrossRef CAS.
- J. Kunze and U. Stimming, Angew. Chem., Int. Ed., 2009, 48, 9230–9237 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- S. K. Kamarudin, F. Achmad and W. R. W. Daud, Int. J. Hydrogen Energy, 2009, 34, 6902–6916 CrossRef CAS.
- A. C. Ferrari, F. Bonaccorso, V. Fal'Ko, K. S. Novoselov, S. Roche, P. Bøggild, S. Borini, F. H. Koppens, V. Palermo and N. Pugno, Nanoscale, 2015, 7, 4598–4810 RSC.
- W. R. Grove, London, Edinburgh, and Dublin Philos. Magaz. J. Sci., 1842, 21, 417–420 CrossRef.
- R. Meldola, Christian Friedrich Schönbein, 1799–1868. Ein Blatt zur Geschichte des 19. Jahrhunderts, Nature, 1900, vol. 62, p. 97 Search PubMed.
- N. Alonso-Vante, in Chalcogenide Materials for Energy Conversion, Springer, 2018, pp. 27–60 Search PubMed.
- F. Mitlitsky, B. Myers, A. H. Weisberg, T. M. Molter and W. F. Smith, Fuel Cells Bull., 1999, 2, 6–11 CrossRef.
- Y. Wang, D. Y. Leung, J. Xuan and H. Wang, Renewable Sustainable Energy Rev., 2016, 65, 961–977 CrossRef CAS.
- H. Zhong, C. Campos-Roldán, Y. Zhao, S. Zhang, Y. Feng and N. Alonso-Vante, Catalysts, 2018, 8, 559 CrossRef.
- S. Xu and P. Wu, J. Mater. Chem. A, 2014, 2, 13682–13690 RSC.
- V. Mehta and J. S. Cooper, J. Power Sources, 2003, 114, 32–53 CrossRef CAS.
- R. M. Ormerod, Chem. Soc. Rev., 2003, 32, 17–28 RSC.
- L. Plomp, J. Veldhuis, E. Sitters and S. Van der Molen, J. Power Sources, 1992, 39, 369–373 CrossRef CAS.
- M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura and P. Stonehart, J. Electrochem. Soc., 1994, 141, 2659–2668 CrossRef CAS.
- A. Sarapuu, E. Kibena-Põldsepp, M. Borghei and K. Tammeveski, J. Mater. Chem. A, 2018, 6, 776–804 RSC.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal and T. W. Hansen, Nat. Mater., 2013, 12, 1137 CrossRef CAS PubMed.
- D. T. Whipple and P. J. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- R. Parsons, Trans. Faraday Soc., 1958, 54, 1053–1063 RSC.
- T. R. Hellstern, J. D. Benck, J. Kibsgaard, C. Hahn and T. F. Jaramillo, Adv. Energy Mater., 2016, 6, 1501758 CrossRef.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 RSC.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. Van Der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Surf. Sci., 2015, 640, 133–140 CrossRef CAS.
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. d. Gudmundsdóttir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef.
- C. Goswami, K. K. Hazarika and P. Bharali, Mater. Sci. Energy Technol., 2018, 117–128 Search PubMed.
- P. Stonehart and P. N. Ross Jr., Electrochim. Acta, 1976, 21, 441–445 CrossRef CAS.
- F. Gloaguen, F. Andolfatto, R. Durand and P. Ozil, J. Appl. Electrochem., 1994, 24, 863–869 CrossRef CAS.
- T. Schmidt and H. Gasteiger, Handbook of fuel cells, 2010 Search PubMed.
- Y. Garsany, J. Ge, J. St-Pierre, R. Rocheleau and K. Swider-Lyons, ECS Trans., 2013, 58, 1233–1241 CrossRef.
- Y. Garsany, J. Ge, J. St-Pierre, R. Rocheleau and K. Swider-Lyons, ECS Trans., 2013, 58, 3–14 CrossRef.
- K. Ke, K. Hiroshima, Y. Kamitaka, T. Hatanaka and Y. Morimoto, Electrochim. Acta, 2012, 72, 120–128 CrossRef CAS.
- K. Shinozaki, J. W. Zack, R. M. Richards, B. S. Pivovar and S. S. Kocha, J. Electrochem. Soc., 2015, 162, F1144–F1158 CrossRef CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 1804799 CrossRef PubMed.
- N. Alonso-Vante, Fuel Cell Electrocatalysis, Springer, Cham, 2018, pp. 27–60 Search PubMed.
- X. Cai, Y. Luo, B. Liu and H.-M. Cheng, Chem. Soc. Rev., 2018, 6224–6266 RSC.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS PubMed.
- X. Chia and M. Pumera, Nat. Catal., 2018, 1 Search PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2013, 125, 3192–3198 CrossRef.
- X. Wang, G. Sun, P. Routh, D.-H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- F. Schedin, A. Geim, S. Morozov, E. Hill, P. Blake, M. Katsnelson and K. Novoselov, Nat. Mater., 2007, 6, 652 CrossRef CAS PubMed.
- Y. Liu, Y. Shen, L. Sun, J. Li, C. Liu, W. Ren, F. Li, L. Gao, J. Chen and F. Liu, Nat. Commun., 2016, 7, 10921 CrossRef CAS PubMed.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. Waterhouse, Y. Zhao, C. Zhou, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- C. Chen, X.-D. Yang, Z.-Y. Zhou, Y.-J. Lai, M. Rauf, Y. Wang, J. Pan, L. Zhuang, Q. Wang and Y.-C. Wang, Chem. Commun., 2015, 51, 17092–17095 RSC.
- Z. Shi, A. Kutana and B. I. Yakobson, J. Phys. Chem. Lett., 2014, 6, 106–112 CrossRef PubMed.
- P. Dai, T.-t. Yan, X.-x. Yu, Z.-m. Bai and M.-z. Wu, Nanoscale Res. Lett., 2016, 11, 226 CrossRef PubMed.
- X. Luo, J. Yang, H. Liu, X. Wu, Y. Wang, Y. Ma, S.-H. Wei, X. Gong and H. Xiang, J. Am. Chem. Soc., 2011, 133, 16285–16290 CrossRef CAS PubMed.
- Y.-B. Tang, L.-C. Yin, Y. Yang, X.-H. Bo, Y.-L. Cao, H.-E. Wang, W.-J. Zhang, I. Bello, S.-T. Lee and H.-M. Cheng, ACS Nano, 2012, 6, 1970–1978 CrossRef CAS PubMed.
- R. Faccio, L. Fernández-Werner, H. Pardo, C. Goyenola, O. N. Ventura and Á. W. Mombrú, J. Phys. Chem. C, 2010, 114, 18961–18971 CrossRef CAS.
- R. Cloke, J. Am. Chem. Soc., 2015, 137, 8872 CrossRef CAS PubMed.
- S. Kawai, Nat. Commun., 2015, 6, 8098 CrossRef CAS PubMed.
- J. Zhou, Q. Sun, Q. Wang and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 064505 CrossRef.
- G. Fazio, L. Ferrighi and C. Di Valentin, J. Catal., 2014, 318, 203–210 CrossRef CAS.
- L. Wang, Z. k. Sofer, P. Šimek, I. Tomandl and M. Pumera, J. Phys. Chem. C, 2013, 117(44), 23251–23257 CrossRef CAS.
- C. Zhang, L. Fu, N. Liu, M. Liu, Y. Wang and Z. Liu, Adv. Mater., 2011, 23, 1020–1024 CrossRef CAS PubMed.
- F. Joucken, Y. Tison, P. Le Fèvre, A. Tejeda, A. Taleb-Ibrahimi, E. Conrad, V. Repain, C. Chacon, A. Bellec and Y. Girard, Sci. Rep., 2015, 5, 14564 CrossRef CAS PubMed.
- T. Schiros, Nano Lett., 2012, 12, 4025 CrossRef CAS PubMed.
- E. Velez-Fort, C. Mathieu, E. Pallecchi, M. Pigneur, M. G. Silly, R. Belkhou, M. Marangolo, A. Shukla, F. Sirotti and A. Ouerghi, ACS Nano, 2012, 6, 10893–10900 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4(3), 1321–1326 CrossRef CAS PubMed.
- Z. Sun, J. Masa, P. Weide, S. M. Fairclough, A. W. Robertson, P. Ebbinghaus, J. H. Warner, S. E. Tsang, M. Muhler and W. Schuhmann, J. Mater. Chem. A, 2015, 3, 15444–15450 RSC.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 RSC.
- R. Ma, X. Ren, B. Y. Xia, Y. Zhou, C. Sun, Q. Liu, J. Liu and J. Wang, Nano Res., 2016, 9, 808–819 CrossRef CAS.
- H.-m. Wang, H.-x. Wang, Y. Chen, Y.-j. Liu, J.-x. Zhao, Q.-h. Cai and X.-z. Wang, Appl. Surf. Sci., 2013, 273, 302–309 CrossRef CAS.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- J. Wu, Z. Yang, Q. Sun, X. Li, P. Strasser and R. Yang, Electrochim. Acta, 2014, 127, 53–60 CrossRef CAS.
- X. Zhang, Z. Lu, Z. Fu, Y. Tang, D. Ma and Z. Yang, J. Power Sources, 2015, 276, 222–229 CrossRef CAS.
- G. Lu, Y. Zhu, K. Xu, Y. Jin, Z. J. Ren, Z. Liu and W. Zhang, Nanoscale, 2015, 7, 18271–18277 RSC.
- J. Masa, A. Zhao, W. Xia, Z. Sun, B. Mei, M. Muhler and W. Schuhmann, Electrochem. Commun., 2013, 34, 113–116 CrossRef CAS.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., 2013, 125, 14063–14066 CrossRef.
- C. H. Choi, M. W. Chung, H. C. Kwon, S. H. Park and S. I. Woo, J. Mater. Chem. A, 2013, 1, 3694–3699 RSC.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2012, 124, 11664–11668 CrossRef.
- M. M. Menamparambath, J.-H. Park, H.-S. Yoo, S. P. Patole, J.-B. Yoo, S. W. Kim and S. Baik, Nanoscale, 2014, 6, 8844–8851 RSC.
- Z. Li, Q. Gao, H. Zhang, W. Tian, Y. Tan, W. Qian and Z. Liu, Sci. Rep., 2017, 7, 43352 CrossRef PubMed.
- Y. Yuan, L. Yang, B. He, E. Pervaiz, Z. Shao and M. Yang, Nanoscale, 2017, 6259–6263 RSC.
- Z. Li, Q. Gao, H. Zhang, W. Tian, Y. Tan, W. Qian and Z. Liu, Sci. Rep., 2017, 7, 43352 CrossRef PubMed.
- J. Wei, Y. Hu, Y. Liang, B. Kong, Z. Zheng, J. Zhang, S. P. Jiang, Y. Zhao and H. Wang, J. Mater. Chem. A, 2017, 5, 10182–10189 RSC.
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- J. Peng, X. Chen, W.-J. Ong, X. Zhao and N. Li, Chem, 2018, 18–50 Search PubMed.
- K. Khan, A. K. Tareen, S. Elshahat, A. Yadav, U. Khan, M. Yang, L. Bibbò and Z. Ouyang, Dalton Trans., 2018, 47, 3819–3830 RSC.
- X. Xie, S. Chen, W. Ding, Y. Nie and Z. Wei, Chem. Commun., 2013, 49, 10112–10114 RSC.
- Z. Zhang, H. Li, G. Zou, C. Fernandez, B. Liu, Q. Zhang, J. Hu and Q. Peng, ACS Sustainable Chem. Eng., 2016, 4, 6763–6771 CrossRef CAS.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- T. V. Vineesh, M. P. Kumar, C. Takahashi, G. Kalita, S. Alwarappan, D. K. Pattanayak and T. N. Narayanan, Adv. Energy Mater., 2015, 5, 1500658 CrossRef.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- L. Wang, F. Yin and C. Yao, Int. J. Hydrogen Energy, 2014, 39, 15913–15919 CrossRef CAS.
- Y. Ito, H. J. Qiu, T. Fujita, Y. Tanabe, K. Tanigaki and M. Chen, Adv. Mater., 2014, 26, 4145–4150 CrossRef CAS PubMed.
- Z. Ma, S. Dou, A. Shen, L. Tao, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2015, 54, 1888–1892 CrossRef CAS PubMed.
- R. Ma, B. Y. Xia, Y. Zhou, P. Li, Y. Chen, Q. Liu and J. Wang, Carbon, 2016, 102, 58–65 CrossRef CAS.
- H. Zhang, X. Liu, G. He, X. Zhang, S. Bao and W. Hu, J. Power Sources, 2015, 279, 252–258 CrossRef CAS.
- R. Li, Z. Wei and X. Gou, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS.
- M. Hassan, E. Haque, A. I. Minett and V. G. Gomes, ChemSusChem, 2015, 8, 4040–4048 CrossRef CAS PubMed.
- L. Wang, Z. Sofer, R. Zboril, K. Cepe and M. Pumera, Chem. – Eur. J., 2016, 22, 15444–15450 CrossRef CAS PubMed.
- F. Razmjooei, K. P. Singh, M. Y. Song and J.-S. Yu, Carbon, 2014, 78, 257–267 CrossRef CAS.
- J. Zhang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 13296–13300 CrossRef CAS PubMed.
- Q. Dong, X. Zhuang, Z. Li, B. Li, B. Fang, C. Yang, H. Xie, F. Zhang and X. Feng, J. Mater. Chem. A, 2015, 3, 7767–7772 RSC.
- H. Huang, X. Feng, C. Du and W. Song, Chem. Commun., 2015, 51, 7903–7906 RSC.
- H. Huang, X. Feng, C. Du, S. Wu and W. Song, J. Mater. Chem. A, 2015, 3, 16050–16056 RSC.
- K. Yuan, X. Zhuang, H. Fu, G. Brunklaus, M. Forster, Y. Chen, X. Feng and U. Scherf, Angew. Chem., Int. Ed., 2016, 55, 6858–6863 CrossRef CAS PubMed.
- J. Guo, Y. Shi, X. Bai, X. Wang and T. Ma, J. Mater. Chem. A, 2015, 3, 24397–24404 RSC.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- G. L. Tian, M. Q. Zhao, D. Yu, X. Y. Kong, J. Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251–2259 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- Z. Xiao, X. Huang, L. Xu, D. Yan, J. Huo and S. Wang, Chem. Commun., 2016, 52, 13008–13011 RSC.
- X. Ren, B. Wang, J. Zhu, J. Liu, W. Zhang and Z. Wen, Phys. Chem. Chem. Phys., 2015, 17, 14605–14612 RSC.
- Z. Zhao, M. Li, L. Zhang, L. Dai and Z. Xia, Adv. Mater., 2015, 27, 6834–6840 CrossRef CAS PubMed.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 21138–21149 CrossRef CAS PubMed.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, J. R. Winkler and A. M. Müller, J. Am. Chem. Soc., 2014, 136, 13118–13121 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li and Y. Luo, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef CAS PubMed.
- H. Liang, L. Li, F. Meng, L. Dang, J. Zhuo, A. Forticaux, Z. Wang and S. Jin, Chem. Mater., 2015, 27, 5702–5711 CrossRef CAS.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., 2015, 127, 7507–7512 CrossRef.
- T. Skaltsas, G. Mountrichas, S. Zhao, H. Shinohara, N. Tagmatarchis and S. Pispas, Chem. – Eur. J., 2015, 21, 18841–18846 CrossRef CAS PubMed.
- J. Tian, Q. Liu, A. M. Asiri, K. A. Alamry and X. Sun, ChemSusChem, 2014, 7, 2125–2130 CrossRef CAS PubMed.
- T. Y. Ma, J. L. Cao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 1138–1142 CrossRef CAS PubMed.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 13567–13570 CrossRef CAS PubMed.
- T. Sun, L. Xu, Y. Yan, A. A. Zakhidov, R. H. Baughman and J. Chen, ACS Catal., 2016, 6, 1446–1450 CrossRef CAS.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2014, 26, 2925–2930 CrossRef CAS PubMed.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- H. Liang, F. Meng, M. Cabán-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976–3982 RSC.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu and Y. Jiang, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed.
- Z. Lin, G. H. Waller, Y. Liu, M. Liu and C.-p. Wong, Carbon, 2013, 53, 130–136 CrossRef CAS.
- Y. Kuang, G. Feng, P. Li, Y. Bi, Y. Li and X. Sun, Angew. Chem., Int. Ed., 2016, 55, 693–697 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, in Materials For Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World Scientific, 2011, pp. 280–284 Search PubMed.
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., 2015, 127, 2159–2164 CrossRef.
- Y. Zhou, Y. Leng, W. Zhou, J. Huang, M. Zhao, J. Zhan, C. Feng, Z. Tang, S. Chen and H. Liu, Nano Energy, 2015, 16, 357–366 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. D. Kim, E. L. Samuel, Z. Peng, Z. Zhu, F. Qin and J. Bao, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- T. A. Shifa, F. Wang, K. Liu, K. Xu, Z. Wang, X. Zhan, C. Jiang and J. He, Small, 2016, 12, 3802–3809 CrossRef CAS PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan and F. Abild-Pedersen, Nat. Mater., 2016, 15, 48 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- Y. Luo, X. Li, X. Cai, X. Zou, F. Kang, H.-M. Cheng and B. Liu, ACS Nano, 2018, 12, 4565–4573 CrossRef CAS PubMed.
- J. Di, C. Yan, A. D. Handoko, Z. W. Seh, H. Li and Z. Liu, Mater. Today, 2018, 749–770 CrossRef CAS.
- B. Conway and B. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- A. Harvey, C. Backes, Z. Gholamvand, D. Hanlon, D. McAteer, H. C. Nerl, E. McGuire, A. s. Seral-Ascaso, Q. M. Ramasse and N. McEvoy, Chem. Mater., 2015, 27, 3483–3493 CrossRef CAS.
- C. Tsai, K. Chan, F. Abild-Pedersen and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 13156–13164 RSC.
- H. Li, M. Du, M. J. Mleczko, A. L. Koh, Y. Nishi, E. Pop, A. J. Bard and X. Zheng, J. Am. Chem. Soc., 2016, 138, 5123–5129 CrossRef CAS PubMed.
- Y. Yu, S.-Y. Huang, Y. Li, S. N. Steinmann, W. Yang and L. Cao, Nano Lett., 2014, 14, 553–558 CrossRef CAS PubMed.
- X. Chia, A. Ambrosi, Z. k. Sofer, J. Luxa, D. Sedmidubský and M. Pumera, ACS Nano, 2015, 10, 112–123 CrossRef PubMed.
- Z. Gholamvand, D. McAteer, C. Backes, N. McEvoy, A. Harvey, N. C. Berner, D. Hanlon, C. Bradley, I. Godwin and A. Rovetta, Nanoscale, 2016, 8, 5737–5749 RSC.
- A. Ambrosi, Z. Sofer and M. Pumera, Chem. Commun., 2015, 51, 8450–8453 RSC.
- X. Fan, S. Wang, Y. An and W. Lau, J. Phys. Chem. C, 2016, 120, 1623–1632 CrossRef CAS.
- J. Yuan, J. Wu, W. J. Hardy, P. Loya, M. Lou, Y. Yang, S. Najmaei, M. Jiang, F. Qin and K. Keyshar, Adv. Mater., 2015, 27, 5605–5609 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- J. Zhang, S. Liu, H. Liang, R. Dong and X. Feng, Adv. Mater., 2015, 27, 7426–7431 CrossRef CAS PubMed.
- X. Geng, W. Sun, W. Wu, B. Chen, A. Al-Hilo, M. Benamara, H. Zhu, F. Watanabe, J. Cui and T.-p. Chen, Nat. Commun., 2016, 7, 10672 CrossRef CAS PubMed.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang and Z. Zhang, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- K. Wang, C. Zhou, D. Xi, Z. Shi, C. He, H. Xia, G. Liu and G. Qiao, Nano Energy, 2015, 18, 1–11 CrossRef CAS.
- X. J. Chua, J. Luxa, A. Y. S. Eng, S. M. Tan, Z. k. Sofer and M. Pumera, ACS Catal., 2016, 6, 5724–5734 CrossRef CAS.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy and G. Eda, Nat. Mater., 2013, 12, 850 CrossRef CAS PubMed.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed.
- Y. J. Tang, Y. Wang, X. L. Wang, S. L. Li, W. Huang, L. Z. Dong, C. H. Liu, Y. F. Li and Y. Q. Lan, Adv. Energy Mater., 2016, 6, 1600116 CrossRef.
- L. Liang, H. Cheng, F. Lei, J. Han, S. Gao, C. Wang, Y. Sun, S. Qamar, S. Wei and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 12004–12008 CrossRef CAS PubMed.
- E. J. Juarez-Perez, M. R. Leyden, S. Wang, L. K. Ono, Z. Hawash and Y. Qi, Chem. Mater., 2016, 28, 5702–5709 CrossRef CAS.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68 CrossRef CAS PubMed.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- Y. Yu, G.-H. Nam, Q. He, X.-J. Wu, K. Zhang, Z. Yang, J. Chen, Q. Ma, M. Zhao and Z. Liu, Nat. Chem., 2018, 10, 638 CrossRef CAS PubMed.
- Y. Shi and B. Zhang, Inorg. Chem. Front., 2018, 5, 1490–1492 RSC.
- C. Zhang, Y. Shi, Y. Yu, Y. Du and B. Zhang, ACS Catal., 2018, 8, 8077–8083 CrossRef CAS.
- N. Danilovic, R. Subbaraman, D. Strmcnik, V. Stamenkovic and N. Markovic, J. Serb. Chem. Soc., 2013, 78, 2007–2015 CrossRef CAS.
- Y. Huang, R. J. Nielsen, W. A. Goddard III and M. P. Soriaga, J. Am. Chem. Soc., 2015, 137, 6692–6698 CrossRef CAS PubMed.
- K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2016, 7, 1686–1690 CrossRef CAS PubMed.
- H. Tributsch and J. Bennett, J. Electroanal. Chem. Interfacial Electrochem., 1977, 81, 97–111 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- Z. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168–4175 CrossRef CAS PubMed.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- H. Wang, Z. Lu, S. Xu, D. Kong, J. J. Cha, G. Zheng, P.-C. Hsu, K. Yan, D. Bradshaw and F. B. Prinz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19701–19706 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- H. G. S. Casalongue, J. D. Benck, C. Tsai, R. K. Karlsson, S. Kaya, M. L. Ng, L. G. Pettersson, F. Abild-Pedersen, J. Nørskov and H. Ogasawara, J. Phys. Chem. C, 2014, 118, 29252–29259 CrossRef CAS.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515–2525 RSC.
- H. J. Brown, S. Wiese, J. A. Roberts, R. M. Bullock and M. L. Helm, ACS Catal., 2015, 5, 2116–2123 CrossRef CAS.
- D. Voiry, R. Fullon, J. Yang, C. d. C. C. e Silva, R. Kappera, I. Bozkurt, D. Kaplan, M. J. Lagos, P. E. Batson and G. Gupta, Nat. Mater., 2016, 15, 1003 CrossRef CAS PubMed.
- Y. Guo, X. Zhang, X. Zhang and T. You, J. Mater. Chem. A, 2015, 3, 15927–15934 RSC.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- H. Liang, H. Shi, D. Zhang, F. Ming, R. Wang, J. Zhuo and Z. Wang, Chem. Mater., 2016, 28, 5587–5591 CrossRef CAS.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, C. R. English, F. Meng, A. Forticaux, R. J. Hamers and S. Jin, Energy Environ. Sci., 2014, 7, 2608–2613 RSC.
- W. Zhao, B. Dong, Z. Guo, G. Su, R. Gao, W. Wang and L. Cao, Chem. Commun., 2016, 52, 9228–9231 RSC.
- M. Jiang, J. Zhang, M. Wu, W. Jian, H. Xue, T.-W. Ng, C.-S. Lee and J. Xu, J. Mater. Chem. A, 2016, 4, 14949–14953 RSC.
- Y. Yin, Y. Zhang, T. Gao, T. Yao, X. Zhang, J. Han, X. Wang, Z. Zhang, P. Xu and P. Zhang, Adv. Mater., 2017, 29, 1700311 CrossRef PubMed.
- H. Zhou, F. Yu, Y. Huang, J. Sun, Z. Zhu, R. J. Nielsen, R. He, J. Bao, W. A. Goddard III and S. Chen, Nat. Commun., 2016, 7, 12765 CrossRef CAS PubMed.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058–12063 CrossRef CAS PubMed.
- X. Huang, Z. Zeng, S. Bao, M. Wang, X. Qi, Z. Fan and H. Zhang, Nat. Commun., 2013, 4, 1444 CrossRef PubMed.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, Nat. Nanotechnol., 2017, 12, 441 CrossRef CAS PubMed.
- L. Ma, Y. Hu, R. Chen, G. Zhu, T. Chen, H. Lv, Y. Wang, J. Liang, H. Liu and C. Yan, Nano Energy, 2016, 24, 139–147 CrossRef CAS.
- Y. Liu, X. Hua, C. Xiao, T. Zhou, P. Huang, Z. Guo, B. Pan and Y. Xie, J. Am. Chem. Soc., 2016, 138, 5087–5092 CrossRef CAS PubMed.
- Z. Pu, Q. Liu, P. Jiang, A. M. Asiri, A. Y. Obaid and X. Sun, Chem. Mater., 2014, 26, 4326–4329 CrossRef CAS.
- X. Wang, Y. V. Kolen'ko, X. Q. Bao, K. Kovnir and L. Liu, Angew. Chem., 2015, 127, 8306–8310 CrossRef.
- X.-D. Wang, Y.-F. Xu, H.-S. Rao, W.-J. Xu, H.-Y. Chen, W.-X. Zhang, D.-B. Kuang and C.-Y. Su, Energy Environ. Sci., 2016, 9, 1468–1475 RSC.
- Z. Pu, S. Wei, Z. Chen and S. Mu, Appl. Catal., B, 2016, 196, 193–198 CrossRef CAS.
- C. Zhang, Y. Huang, Y. Yu, J. Zhang, S. Zhuo and B. Zhang, Chem. Sci., 2017, 8, 2769–2775 RSC.
- J. Su, J. Zhou, L. Wang, C. Liu and Y. Chen, Sci. Bull., 2017, 62, 633–644 CrossRef CAS.
- V. B. Saptal and B. M. Bhanage, ChemSusChem, 2016, 9, 1980–1985 CrossRef CAS PubMed.
- B. Bayatsarmadi, Y. Zheng, Y. Tang, M. Jaroniec and S. Z. Qiao, Small, 2016, 12, 3703–3711 CrossRef CAS PubMed.
- S. Peng, L. Li, X. Han, W. Sun, M. Srinivasan, S. G. Mhaisalkar, F. Cheng, Q. Yan, J. Chen and S. Ramakrishna, Angew. Chem., 2014, 126, 12802–12807 CrossRef.
- H. Ang, H. T. Tan, Z. M. Luo, Y. Zhang, Y. Y. Guo, G. Guo, H. Zhang and Q. Yan, Small, 2015, 11, 6278–6284 CrossRef CAS PubMed.
- L. Liao, J. Zhu, X. Bian, L. Zhu, M. D. Scanlon, H. H. Girault and B. Liu, Adv. Funct. Mater., 2013, 23, 5326–5333 CrossRef CAS.
- L. Ma, Y. Hu, G. Zhu, R. Chen, T. Chen, H. Lu, Y. Wang, J. Liang, H. Liu and C. Yan, Chem. Mater., 2016, 28, 5733–5742 CrossRef CAS.
- Y. Li, J. Wang, X. Tian, L. Ma, C. Dai, C. Yang and Z. Zhou, Nanoscale, 2016, 8, 1676–1683 RSC.
- J. Ekspong, T. Sharifi, A. Shchukarev, A. Klechikov, T. Wågberg and E. Gracia-Espino, Adv. Funct. Mater., 2016, 26, 6766–6776 CrossRef CAS.
- M.-R. Gao, J.-X. Liang, Y.-R. Zheng, Y.-F. Xu, J. Jiang, Q. Gao, J. Li and S.-H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed.
- F. Wang, Y. Li, T. A. Shifa, K. Liu, F. Wang, Z. Wang, P. Xu, Q. Wang and J. He, Angew. Chem., Int. Ed., 2016, 55, 6919–6924 CrossRef CAS PubMed.
- J. Gao, L. Li, J. Tan, H. Sun, B. Li, J. C. Idrobo, C. V. Singh, T.-M. Lu and N. Koratkar, Nano Lett., 2016, 16, 3780–3787 CrossRef CAS PubMed.
- S. Shinde, A. Sami, D.-H. Kim and J.-H. Lee, Chem. Commun., 2015, 51, 15716–15719 RSC.
- Y. Yang, H. Fei, G. Ruan, Y. Li and J. M. Tour, Adv. Funct. Mater., 2015, 25, 6199–6204 CrossRef CAS.
- J. Zhang, Q. Wang, L. Wang, X. a. Li and W. Huang, Nanoscale, 2015, 7, 10391–10397 RSC.
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2017, 1, 173–180 CrossRef.
- K. Zhang, G. Zhang, J. Qu and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 2451–2459 CrossRef CAS PubMed.
- J. Xiong, J. Li, J. Shi, X. Zhang, N.-T. Suen, Z. Liu, Y. Huang, G. Xu, W. Cai and X. Lei, ACS Energy Lett., 2018, 3, 341–348 CrossRef CAS.
- J. Jia, T. Xiong, L. Zhao, F. Wang, H. Liu, R. Hu, J. Zhou, W. Zhou and S. Chen, ACS Nano, 2017, 11, 12509–12518 CrossRef CAS PubMed.
- S. Li, P. Tuo, J. Xie, X. Zhang, J. Xu, J. Bao, B. Pan and Y. Xie, Nano Energy, 2018, 47, 512–518 CrossRef CAS.
- C. Lu, D. Tranca, J. Zhang, F. n. Rodríguez Hernández, Y. Su, X. Zhuang, F. Zhang, G. Seifert and X. Feng, ACS Nano, 2017, 11, 3933–3942 CrossRef CAS PubMed.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- X. Wu, Z. Wang, M. Yu, L. Xiu and J. Qiu, Adv. Mater., 2017, 29, 1607017 CrossRef PubMed.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
- Y. Zhao, S. Chen, B. Sun, D. Su, X. Huang, H. Liu, Y. Yan, K. Sun and G. Wang, Sci. Rep., 2015, 5, 7629 CrossRef CAS PubMed.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic and M. G. Kanatzidis, Nat. Mater., 2016, 15, 197 CrossRef PubMed.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 1090–1094 Search PubMed.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 1 Search PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- C. Sun, F. Li, C. Ma, Y. Wang, Y. Ren, W. Yang, Z. Ma, J. Li, Y. Chen and Y. Kim, J. Mater. Chem. A, 2014, 2, 7188–7196 RSC.
- X. Sun, X. Kang, Q. Zhu, J. Ma, G. Yang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 2883–2887 RSC.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu and R. Haasch, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato and R. Haasch, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed.
- D. H. Won, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Angew. Chem., Int. Ed., 2016, 55, 9297–9300 CrossRef CAS PubMed.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed.
- C. J. Van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Björgvinsdóttir, H. Ólafsdóttir and E. Skúlason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- X. Chia, A. Y. S. Eng, A. Ambrosi, S. M. Tan and M. Pumera, Chem. Rev., 2015, 115, 11941–11966 CrossRef CAS PubMed.
- C. K. Chua, Z. Sofer and M. Pumera, Chem. – Eur. J., 2012, 18, 13453–13459 CrossRef CAS PubMed.
- M. Zhou, Y. Wang, Y. Zhai, J. Zhai, W. Ren, F. Wang and S. Dong, Chem. – Eur. J., 2009, 15, 6116–6120 CrossRef CAS PubMed.
- J. Bonde, P. G. Moses, T. F. Jaramillo, J. K. Nørskov and I. Chorkendorff, Faraday Discuss., 2009, 140, 219–231 RSC.
- X. Chia, A. Ambrosi, Z. Sofer, J. Luxa and M. Pumera, ACS Nano, 2015, 9, 5164–5179 CrossRef CAS PubMed.
- J. Luxa, P. Vosecky, V. Mazánek, D. Sedmidubsky, M. Pumera, P. Lazar and Z. Sofer, ACS Catal., 2017, 7, 5706–5716 CrossRef CAS.
- A. Y. S. Eng, A. Ambrosi, Z. Sofer, P. Simek and M. Pumera, ACS Nano, 2014, 8, 12185–12198 CrossRef CAS PubMed.
- W. Jaegermann and D. Schmeisser, Surf. Sci., 1986, 165, 143–160 CrossRef CAS.
- X. Chia, A. Ambrosi, P. Lazar, Z. Sofer and M. Pumera, J. Mater. Chem. A, 2016, 4, 14241–14253 RSC.
- L. Wang, Z. Sofer and M. Pumera, ChemElectroChem, 2015, 2, 324–327 CrossRef CAS.
- L. Lorencová, T. Bertok, E. Dosekova, A. Holazová, D. Paprckova, A. Vikartovská, V. Sasinková, J. Filip, P. Kasák and M. Jerigová, Electrochim. Acta, 2017, 235, 471–479 CrossRef PubMed.
- T. J. Davies, M. E. Hyde and R. G. Compton, Angew. Chem., Int. Ed., 2005, 44, 5121–5126 CrossRef CAS PubMed.
- W. Yuan, Y. Zhou, Y. Li, C. Li, H. Peng, J. Zhang, Z. Liu, L. Dai and G. Shi, Sci. Rep., 2013, 3, 2248 CrossRef PubMed.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- C. Tan, J. n. Rodríguez-López, J. J. Parks, N. L. Ritzert, D. C. Ralph and H. D. Abruna, ACS Nano, 2012, 6, 3070–3079 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150–7188 CrossRef CAS PubMed.
- C. K. Chua, A. Ambrosi and M. Pumera, J. Mater. Chem., 2012, 22, 11054–11061 RSC.
- X. Ji, C. E. Banks, A. Crossley and R. G. Compton, ChemPhysChem, 2006, 7, 1337–1344 CrossRef CAS PubMed.
- S. M. Tan, A. Ambrosi, Z. Sofer, Š. Huber, D. Sedmidubský and M. Pumera, Chem. – Eur. J., 2015, 21, 7170–7178 CrossRef CAS PubMed.
- S. M. Ahmed and H. Gerischer, Electrochim. Acta, 1979, 24, 705–711 CrossRef CAS.
- S. M. Tan, Z. k. Sofer, J. Luxa and M. Pumera, ACS Catal., 2016, 6, 4594–4607 CrossRef CAS.
- H. L. Poh, P. Šimek, Z. Sofer, I. Tomandl and M. Pumera, J. Mater. Chem. A, 2013, 1, 13146–13153 RSC.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- S. Y. Chee and M. Pumera, Analyst, 2012, 137, 2039–2041 RSC.
- Z. Sofer, D. Sedmidubský, Š. Huber, J. Luxa, D. Bouša, C. Boothroyd and M. Pumera, Angew. Chem., Int. Ed., 2016, 55, 3382–3386 CrossRef CAS PubMed.
- R. Gusmão, Z. Sofer, D. Bouša and M. Pumera, Angew. Chem., 2017, 129, 14609–14614 CrossRef.
- X. Zhu, B. Liu, H. Hou, Z. Huang, K. M. Zeinu, L. Huang, X. Yuan, D. Guo, J. Hu and J. Yang, Electrochim. Acta, 2017, 248, 46–57 CrossRef CAS.
- T. Kondo, H. Ito, K. Kusakabe, K. Ohkawa, Y. Einaga, A. Fujishima and T. Kawai, Electrochim. Acta, 2007, 52, 3841–3848 CrossRef CAS.
- G. Wang, J. Tao, Y. Zhang, S. Wang, X. Yan, C. Liu, F. Hu, Z. He, Z. Zuo and X. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 25409–25414 CrossRef CAS PubMed.
- J. Benson, Q. Xu, P. Wang, Y. Shen, L. Sun, T. Wang, M. Li and P. Papakonstantinou, ACS Appl. Mater. Interfaces, 2014, 6, 19726–19736 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Pan, S. Wang, X. Chen, P. Hu, L. Sun and X. Bao, Chem. Commun., 2011, 47, 10016–10018 RSC.
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC.
- K. Uosaki, G. Elumalai, H. Noguchi, T. Masuda, A. Lyalin, A. Nakayama and T. Taketsugu, J. Am. Chem. Soc., 2014, 136, 6542–6545 CrossRef CAS PubMed.
- H. Wang, D. Kong, P. Johanes, J. J. Cha, G. Zheng, K. Yan, N. Liu and Y. Cui, Nano Lett., 2013, 13, 3426–3433 CrossRef CAS PubMed.
- Y. Zhao, X. Zhang, X. Jia, G. I. Waterhouse, R. Shi, X. Zhang, F. Zhan, Y. Tao, L. Z. Wu and C. H. Tung, Adv. Energy Mater., 2018, 1703585 CrossRef.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2019 |