
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer-supported metal catalysts for the heterogeneous polymerisation of lactones†
Ioli C.
Howard
a,
Ceri
Hammond
 b and
Antoine
Buchard
*a
b and
Antoine
Buchard
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
bCardiff Catalysis Institute, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 22nd October 2019
Abstract
A series of metal complexes were immobilised onto an inert poly(styrene) (PS) support and utilised in the solvent free ring-opening polymerisation (ROP) of various lactones. PS-LHZnOAc, PS-LHSnOct and PS-LClSnOct were identified as the most successful heterogeneous catalysts for the ROP of L-lactide. Investigations by in situ ATR-FT-IR revealed conversions reaching ca. 90% in 6, 2.3 hours and 55 minutes, respectively, with excellent molecular weight control and dispersities (ĐM 1.15–1.17). Catalyst loadings as low as 15 ppm metal and TOF values of up to 810 h−1 could also be achieved. Higher molecular weights could be targeted (ca. 35 kDa) whilst maintaining low dispersities in comparison to the industrial standard. Catalyst reuse was also possible, with up to 7 reuse cycles, albeit accompanied by a progressive reduction in conversion. Energy-Dispersive X-ray (EDX) spectroscopy and Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) showed low metal content in the unpurified polymer (as low as 335 ppm, similar to what is found in polymer purified by classical methods), suggesting these systems as promising reusable catalysts for the industrial production of metal-free renewable polymers.
Introduction
Polymers are some of the world's most widely used materials, due to their favourable properties such as high strength, low weight and low cost. However, although polymers are essential to modern society, several major challenges plague their use. For example, almost all of the world's supply of polymeric materials is derived from fossil resources, which is a resource that is depleting rapidly and exhibits significant negative environmental impact. Marine pollution is also a major source of concern; the leakage of waste plastics into the environment, and their subsequent breakdown into microplastics, has resulted in both the release of toxic chemicals into the environment (such as plasticisers and BPA), and the uptake of plastic by many aquatic organisms.1A potential solution to both of these challenges is to replace fossil fuel-derived polymers with those produced from renewable resources.2 In addition to being derived from a more sustainable feedstock, most renewable resources form oxygenated polymers (e.g. polyesters or polycarbonates) that are more susceptible to hydrolytic degradation, and thus could offer significant benefits over current plastics in terms of degradability.3 The synthesis of some of these polyesters occurs industrially via ring-opening polymerisation (ROP) of lactones such as lactide, glycolide, or ε-caprolactone. Among renewable polymers, poly(lactide) (PLA), is one of the most widely studied:4 it is a thermoplastic polymer with a versatile range of applications, including packaging and uses in the biomedical sector,3,5,6 which can also be composted industrially.
At present, industrial production of PLA is achieved in molten monomer by catalytic ROP using homogeneous tin(II) 2-ethylhexanoate, Sn(Oct)2.7 Although this catalyst has been approved by the US FDA,8 its homogeneous nature means that complete removal of the catalyst from the final product is not feasible, resulting in incorporation of the catalyst into the final product. In addition to increasing the cost of the polymer and decreasing the atom efficiency of the process, the presence of the catalyst in the final polymer is also problematic due to the potential toxicity of tin, which has been reported to inhibit cell growth by 50% in low doses.7–9 As such, difficulties arise in using PLA, and other polyesters made in similar way, in biomedical applications such as sutures, as the breakdown of the polymer could release residual metals into the body.
Since one of the draws of PLA is its potential to be used in biomedical applications, reduction of the metal content in the final polymer is vital. While much of the research into PLA has been focused on optimising the properties of the polymer by modifying homogeneous metal catalysts, very few examples have investigated minimising the toxic metal content within the polymer. Replacement of Sn(Oct)2 with organocatalysts such as organic bases,10–12 (thio)ureas13,14 and other bifunctional catalysts,15–17 has been investigated, however the toxicity of these catalysts has also been brought into question.18 A potential solution to this challenge involves the replacement of homogeneous Sn(Oct)2 with a heterogeneous i.e. solid, catalysts. Use of such a catalyst could prevent the metal from being incorporated into the polymer, thereby producing polymers of high purity and increased atom efficiency. Moreover, solid catalysts offer possibilities for the catalyst to be recycled, thus decreasing the cost of the process further.19–23
Heterogeneous catalysts which have been employed in the ROP of lactones have typically involved silica supports.24–31 One example showcased the ROP of L-LA using immobilised Ti(OiPr)3/SiO2 catalysts, which achieved 74% conversion over 12 hours at 70 °C in toluene (Mn 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, ĐM 1.20).32 Lee et al. also demonstrated the use of immobilised Sn(II) methoxide catalysts,27 however no attempt to reuse either of these catalysts or quantify final metal content in the polymer was made. Only a couple of examples have used SiO2-immobilised complexes in the melt ROP of lactide, which is the industrial preferred route. Jones et al. tethered a Zn(II) half-salen complex to a silica support for ROP of rac-LA in the melt at 130 °C ([LA]:[Cat] = 300:1). The heterogeneous catalyst achieved low conversions and modest Mn, although dispersities were improved due to confinement of the polymer within the catalyst (40%, 19300 Da, ĐM 1.34 compared to 1.42–1.83 for the homogeneous examples).26
000 Da, ĐM 1.20).32 Lee et al. also demonstrated the use of immobilised Sn(II) methoxide catalysts,27 however no attempt to reuse either of these catalysts or quantify final metal content in the polymer was made. Only a couple of examples have used SiO2-immobilised complexes in the melt ROP of lactide, which is the industrial preferred route. Jones et al. tethered a Zn(II) half-salen complex to a silica support for ROP of rac-LA in the melt at 130 °C ([LA]:[Cat] = 300:1). The heterogeneous catalyst achieved low conversions and modest Mn, although dispersities were improved due to confinement of the polymer within the catalyst (40%, 19300 Da, ĐM 1.34 compared to 1.42–1.83 for the homogeneous examples).26
However, acidic silica supports have often been cited to increase the rate of chain transfer and thus affect ROP, so that alternatives are needed.33 Amongst potential supports, polymers themselves represent an interesting option as usually non-acidic and non-porous. Polymer supported heterogeneous catalysts have previously been explored for small molecule organic synthesis,21,34–39 CO2 capture,40 and olefin and CO2 polymerisations.41–44 However, the use of such catalysts in ROP of cyclic esters is not so common, and these have, to the best of our knowledge, been exclusively used in solution.45,46
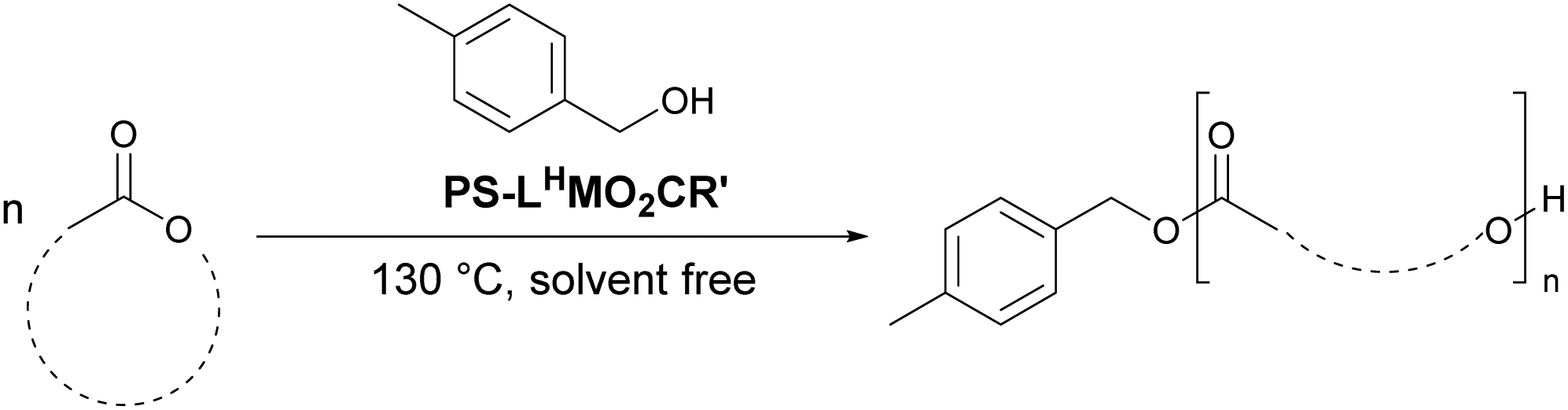
Herein, we report a series of single site metal complexes immobilised onto an inert poly(styrene) (PS) support, and their application for the bulk polymerisation of various lactones (L- and rac-LA, ε-caprolactone, ε-decalactone). The use of PLA as a model substrate in preliminary studies has enabled the optimisation of ROP and comparison of these catalysts to the homogeneous catalysts. Industrial conditions were targeted, with a final aim of Mn of 50000 Da. Particular focus has been placed on the metal leaching into the polymers, and catalyst recovery and reuse are also explored.
Results and discussion
Complex synthesis
A series of ligands and their complexes were immobilised onto poly(styrene) beads (PS), as depicted in Scheme 1. The complexes were synthesised in a simple 3-step pathway, in a procedure adapted from the literature.36,47,48 Commercial PS-CH2Cl, or “Merrifield's resin”, readily underwent a Kornblum oxidation of the chlorine functional group to form aldehyde PS-CHO. A new, sharp absorption band at 1697 cm−1 in the IR spectrum was consistent with an aldehyde carbonyl stretch (Fig. 1). A small peak at 2746 cm−1 corresponded to the aldehyde C–H stretching vibration,47 and the disappearance of the C–Cl stretch at 673 cm−1 further confirmed the conversion to the aldehyde. Calculations based on elemental analysis of PS-CHO (ESI†) indicated quantitative conversion to the aldehyde.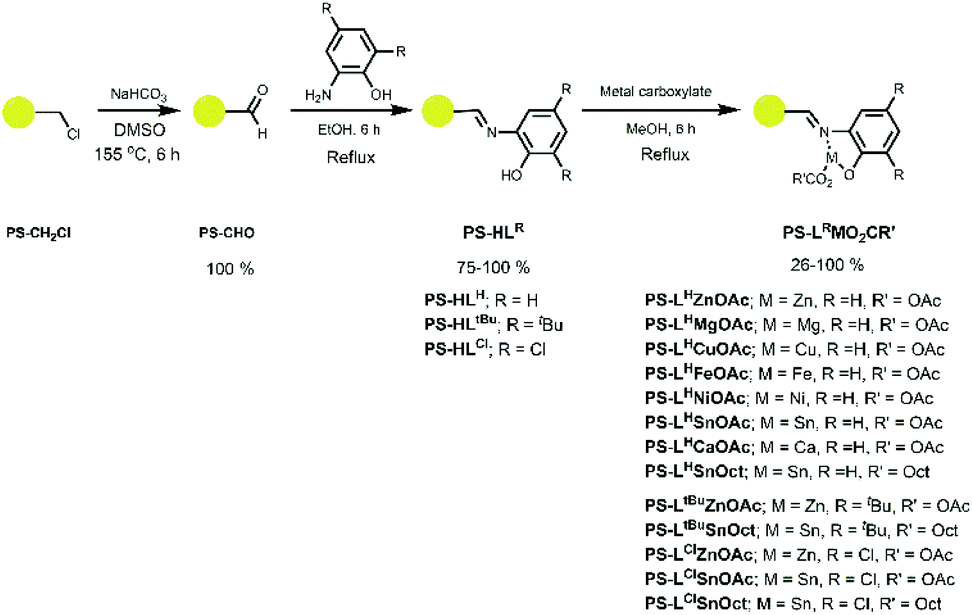 | ||
| Scheme 1 Synthetic route to the immobilised imine complexes with the chemical notation PS-LRMO2CR′, where M = metal, O2CR′ = carboxylate (OAc = acetate; Oct = 2-ethylhexanoate) and LR = ligand. | ||
 | ||
| Fig. 1 IR spectra of the PS-CH2Cl starting material (black), PS-CHO (red), the immobilised ligand PS-HLH (blue) and the complex PS-LHSnOct (green). | ||
The aldehyde was condensed with 2-aminophenol to yield the immobilised Schiff-base, PS-HLH. The intensity of the carbonyl peak was significantly reduced in the spectrum of PS-HLH, indicating incomplete conversion. At least partial formation of the ligand was confirmed by the presence of a hydroxyl vibration at 3381 cm−1; the aromatic C–N stretch was also observed at 1285 cm−1. Imine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretches have been reported at 1627 cm−1,49 consistent with the new peak seen in the IR spectrum of PS-HLH 1624 cm−1. PS-CHO was also condensed with two other amines: 2,4-di-tert-butyl-6-(methylamino)phenol, 2,4-dichloro-6-amino-phenol, to create ligands PS-HLtBu and PS-HLCl, respectively. Imine stretches at 1623 cm−1 appeared in all ligands, as well as a C–O vibration at 1247 cm−1. Elemental analysis of the ligands confirmed conversion to the ligands (>75%).
N stretches have been reported at 1627 cm−1,49 consistent with the new peak seen in the IR spectrum of PS-HLH 1624 cm−1. PS-CHO was also condensed with two other amines: 2,4-di-tert-butyl-6-(methylamino)phenol, 2,4-dichloro-6-amino-phenol, to create ligands PS-HLtBu and PS-HLCl, respectively. Imine stretches at 1623 cm−1 appeared in all ligands, as well as a C–O vibration at 1247 cm−1. Elemental analysis of the ligands confirmed conversion to the ligands (>75%).
Complexation of all ligands was achieved via reflux with the appropriate metal acetate source, as is typical of a salen ligand complexation, yielding similar structures to the 5-membered cycles reported in literature.48,50 A variety of metals were complexed to PS-HLH; disappearance of the –OH group at 3381 cm−1 indicated deprotonation of the hydroxyl group followed by covalent complexation to the metal, whilst the CN group shifted to 1604 cm−1.49,51 The acetate methyl C–H, COO−asymm and CO vibrations appeared at 1453, 1574 and 1698 cm−1, respectively. Morphological composition of the samples at each stage was obtained using Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDX). Triplicate EDX measurements of the samples showed some Cl in PS-CHO, suggesting either incomplete conversion to the aldehyde, or residual salt deposits (along with the presence of Na) on the surface from the work up (Fig. S3A†). EDX spectra also confirmed the presence of Zn and Sn in PS-LHZnOAc and PS-LHSnOct, respectively (Fig. S4 and S5†).
SEM imaging of the parent chlorinated resin revealed a highly porous matrix with pores of up to 5 μm (Fig. 2A). The porosity decreased gradually after each functionalisation step, with the surface appearing visibly smaller as the pores were coated by the complex (Fig. 2C).
 | ||
| Fig. 2 SEM images of (A) PS-CH2Cl, (B) PS-CHO, (C) PS-HLH and (D) PS-LHSnOct at ∼×100 (top), ×1000 (middle) and ×2000 (bottom) magnification. | ||
Further surface area analysis by Brunauer–Emmett–Teller (BET), using N2 sorption at 77.3 K, was carried out to investigate the porosity of catalysts PS-LHZnOAc and PS-LHSnOct (Fig. S6†). Low relative pressures (P/P0) of nitrogen surface adsorption implied that the pore sizes were too large to be effectively measured by BET.
Thermogravimetric analysis (TGA) of PS-HLH and its complexes (PS-LHSnOct and PS-LHZnOAc) revealed that the ligand and complexes possessed similar major degradation temperatures (300–380 °C, Fig. 3 and Fig. S7,† respectively). However, the first derivative of the TGA trace revealed that the ligand underwent two distinct degradation steps at 332 and 509 °C with weights approximately corresponding to the degradation of ligand followed by poly(styrene), respectively (Table S1†). The complexes had far more convoluted degradation pathways; PS-LHSnOct displayed weight losses occurring at 280, 388 and 562 °C, in line with the more intricate architecture of the complex.
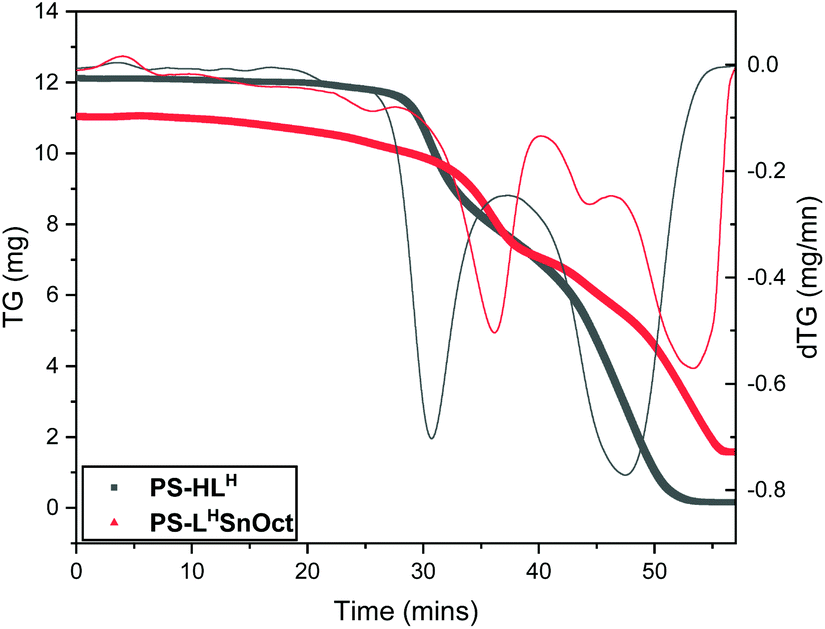 | ||
| Fig. 3 Thermogravimetric analysis (TGA) of ligand PS-HLH (grey) and its metal complex, PS-LHSnOct (red), and their first derivative traces (thinner lines). | ||
Polymerisation studies
To begin, the immobilised metal complexes were employed in the ring-opening polymerisation (ROP) of L-lactide (LA), which was conducted in the melt at 130 °C, with [LA]:[Cat]:[I] = 50:1:1, using p-methylbenzyl alcohol as co-initiator (I). Initial investigations into the activity of the catalysts focused on complexes of imine PS-HLH, coordinated to a metal acetate (PS-LHMOAc, entries 1–6, Table 1). Over 24 hours, clear differences between metal centres were seen. PS-LHZnOAc was by far the most promising catalyst, giving high conversion (84%) into PLA with controlled molecular weight close to the theoretical value (Mn 6450 and 6050 Da, respectively), and narrow dispersity (ĐM 1.23, entry 1, Table 1). The immobilised 5-membered metallacycle compared favourably with its 6-membered half-salen equivalent (40% conversion was achieved with a Zn/SiO2 catalyst prepared by Jones et al.).26 The effect of the carboxylate ligand was then studied. PS-LHSnOct was formed by reacting PS-HLH with Sn(Oct)2. While PS-LHSnOAc was inactive (entry 5, Table 1), switching the acetate to a 2-ethylhexanoate (Oct) ligand, increased the conversion to 93% (Mn 5900 Da, ĐM 1.49, entry 7, Table 1). The well-studied mechanism of homogeneous Sn(Oct)2 involves an initiation step where the pre-catalyst carboxylate exchanges with the p-MeBnO− co-initiator to generate an active metal alkoxide in situ.53 From our results, it is suggested that the rate of this exchange affects the rate and success of ROP. Our attempts to transpose these results to Zn catalysts were unfortunately limited due to difficulties in synthesising a pure Zn(Oct)2 precursor from ZnEt2.
:[Cat]:[I] = 50:1:1 in the melt at 130 °C for 24 hours
| Entry | Catalyst | Conv.a (%) | M n, Theo | M n, NMR | M n | M w | Đ M |
|---|---|---|---|---|---|---|---|
a Determined from the 1H NMR spectrum.
b
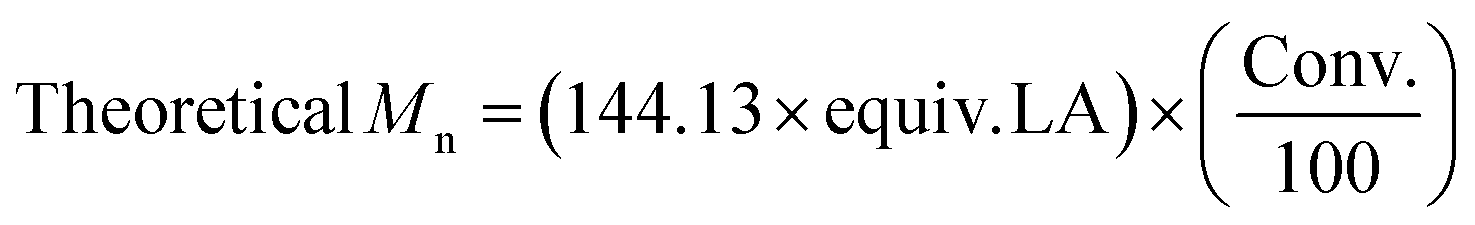 .
c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (multiplied by a factor of 0.58, rounded to the nearest 50).52
d Reaction quenched after 6 hours. .
c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (multiplied by a factor of 0.58, rounded to the nearest 50).52
d Reaction quenched after 6 hours.
|
|||||||
| 1 | PS-LHZnOAc | 84 | 6050 | 5800 | 6450 | 6600 | 1.23 |
| 2 | PS-LHMgOAc | 30 | 2150 | 1350 | 1150 | 1200 | 1.05 |
| 3 | PS-LHCuOAc | 7 | 500 | — | — | — | — |
| 4 | PS-LHNiOAc | 6 | 450 | 500 | — | — | — |
| 5 | PS-LHSnOAc | 8 | 600 | 550 | — | — | — |
| 6 | PS-LHCaOAc | 30 | 2150 | 950 | 1750 | 1950 | 1.11 |
| 7 | PS-LHSnOct | 93 | 6700 | 3150 | 5900 | 8800 | 1.49 |
| 8 | PS-HLH | 7 | 500 | 450 | — | — | — |
| 9 | PS-CHO | 7 | 500 | 450 | — | — | — |
| 10 | PS-CH2Cl | 4 | 300 | — | — | — | — |
| 11d | Sn(Oct)2 | 94 | 6800 | 1550 | 8750 | 15350 |
1.76 |
| 12 | Zn(OAc)2·2H2O | 94 | 6800 | 5600 | 3000 | 5220 | 1.73 |
A control reaction with homogeneous Sn(Oct)2 revealed comparable conversions after 24 hours, but with narrower dispersities for the heterogeneous PS-LHSnOct (ĐM 1.49 compared to 1.76, entries 7 and 11, Table 1). Due to its high activity, Sn(Oct)2 can participate in further transesterification once complete conversion is reached, thus increasing the range of Mn and ĐM.54 The same was observed for PS-LHZnOAc and its precursor, Zn(OAc)2·H2O (entries 1 and 12, Table 1) indicating that immobilisation generally leads to improved ROP control. Both PS-LHZnOAc and PS-LHSnOct produced white polymers (Fig. S13†), with no evidence of lactide epimerisation in the proton NMR (Fig. S9†) – both added benefits of the catalysts.
Polymers produced with PS-LHSnOct resulted in a melting point of 175 °C (Fig. S8†), close to that of fully isotactic PLLA (Tm 180 °C),55,56 whilst the polymer produced with Sn(Oct)2 resulted in a larger decrease to Tm 171 °C (Fig. S9†); this is indicative of a greater degree of epimerisation in PLA produced with Sn(Oct)2. No activity was seen in control reactions with any of the immobilised ligands or precursors (entries 8–10, Table 1).
A Sheldon test was carried out to investigate the heterogeneity of the catalysts.57PS-LHSnOct was removed after 45 minutes (62% conversion), and when compared to a reaction performed with a solid catalyst present throughout the time course, the hot filtration experiment showed a much smaller level of conversion after removal of the catalyst (72% and 94% after 2.5 hours, respectively, see Fig. S12†). Given that several forms of homogeneous Sn(II) are known to catalyse this reaction, the lack of a major increase in conversion likely indicates that either the species leached from the catalyst are inactive, or that only limited leaching occurs. However, in both scenarios, the lack of major increases in conversion confirm catalysis to be truly heterogeneous. Following these promising preliminary results, PS-LHZnOAc and PS-LHSnOct were taken forward for further studies.
Effect of altering the ligand structure
The ligand backbone of these two successful catalysts was modified in order to optimise their activity. In the first instance, a reduced form of the Schiff-base tether of ligand PS-HLH, PS-HLHred, was formed, featuring a secondary amine (Schemes S3 and S4†). It was thought that increasing the flexibility of the linker would improve access of the lactide to the active centre of the catalyst, by allowing the catalyst to adopt the optimal geometry of the coordination sphere – an observation which has previously been reported in the literature.48,50 Results from a 24-hour screen (Table S3†) showed the opposite, with activity decreasing dramatically. In fact, reduction of ligand PS-HLH to PS-HLHred caused large drops in conversion and Mn (down to 500 Da), although low dispersities were still achieved (ĐM 1.08–1.53). A maximum of 75% (Zn) and 22% conversion (Sn) for the unsubstituted amine PS-LHredMOR′ complexes were observed, after 24 hours (M = Zn or Sn, R′ = OAc or Oct). The same was observed with the complexed PS-HLtBured ligand.In the second instance, ligand PS-HLH was modified at positions ortho and para to the phenolic donor, to vary the electron density on the metal: ligands PS-HLtBu and PS-HLCl were synthesised to increase and decrease the electron donating ability of the ligand, respectively (Scheme 1). Increasing the steric bulk of the ligands using electron donating tBu groups to make ligand PS-HLtBu, caused a decrease in conversion within 24 hours for both PS-LtBuZnOAc (84 to 23%) and PS-LtBuSnOct (93 to 69%, entries 1 and 6, Table S3†). This is consistent with work by White and co-workers on salen ligands, who suggested that the larger substituents could block access of the monomer to the metal, resulting in a decrease in activity.58 The increase in electron density on the metal resulting from the inductive effect of the tBu groups could also have contributed to the decrease in conversion. This was made more evident when substituting PS-HLH with smaller, electron withdrawing Cl groups to form PS-HLCl. PS-LClSnOct required only 55 minutes to reach completion, as determined by in situ Attenuated Total Reflectance Fourier Transform Infra-Red (ATR-FT-IR) spectroscopy (entry 7, Table S3†). Comparatively, the unsubstituted complex (PS-LHSnOct) required 2.3 hours. The same increase in conversion over 24 hours was observed when using PS-LClSnOAc instead of PS-LHSnOAc: substitution of the ligand with Cl groups increased the conversion from only 8% to 96% (entry 5, Table S3†). Substituting the ligand with electron withdrawing groups seemingly reduces the electron density on the metal, increasing its Lewis acidity and thus facilitating the approach of the lactide to the metal centre.
In situ kinetic studies
Kinetic analysis of PS-LHZnOAc, PS-LHSnOct and PS-LClSnOct was conducted by monitoring the ether stretches of the lactide and PLA by in situ ATR-FT-IR at 1240 and 1185 cm−1, respectively, during ROP catalysis (Fig. S14†). A calibration using different concentration ratios of PLA and LA was used to directly convert the peak areas to concentration to get quantitative data (Fig. S1 and S2†).59 For each of the three catalysts, a [LA]:[Cat]:[I] ratio of 200:1:4 was used to reduce mechanical interference of the catalyst with the IR probe. Whilst a control reaction using Sn(Oct)2 reached full conversion within 7 minutes, the heterogeneous catalysts displayed excellent rates: PS-LClSnOct and PS-LHSnOct only took 55 minutes and 2.3 hours to reach completion, respectively (Fig. 4), while PS-LHZnOAc was complete within 6 hours (Fig. S15†). Turnover frequencies (TOF) of up to 181 h−1 were possible with the heterogeneous catalysts (entry 3, Table 2). A comparison of the substituent effect confirmed that reaction kinetics improve with EWG groups ortho and para to the phenoxy group: the Cl-substituted complex, PS-LClSnOct, was complete within 55 minutes (Fig. 4B), while the tBu substituted complex, PS-LtBuSnOct, had only reached 69% conversion after 24 hours (entry 6, Table S3†). Kinetic analysis confirmed that reaction was first order with respect to the monomer using each of the catalysts. However, the acquired data deviated from the first order kinetics towards the end due to viscosity, and an induction period was also seen. The latter could be attributed to both mass transfer limitations through the bulk to the active sites, and potentially to the exchange between the initiator and the carboxylate pre-catalyst. Further studies are required for an in-depth investigation into the rate of this exchange.
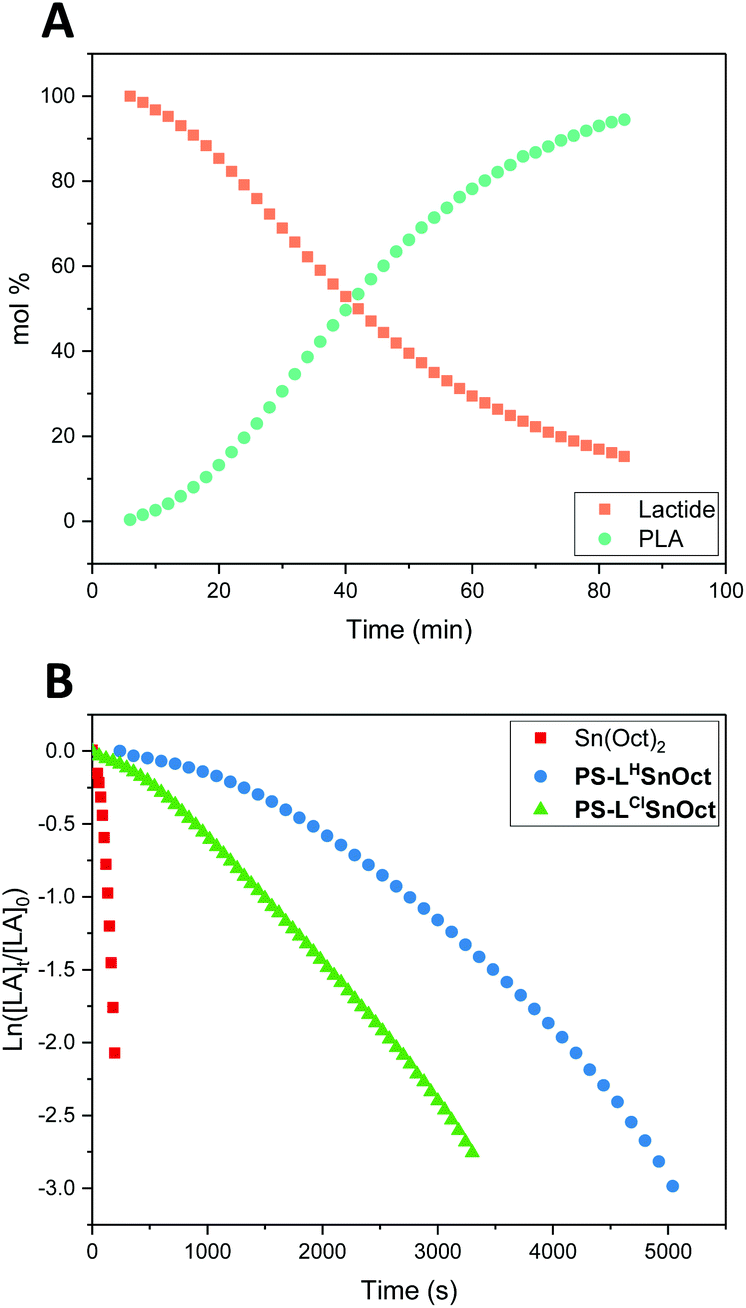 | ||
| Fig. 4 (A) Conversion of lactide to PLA over time for PS-LHSnOct, 2.3 hours in the melt, [LA]:[Cat]:[I] = 200:1:4, 130 °C. (B) Semi-logarithmic plot of the concentration of lactide, [LA], monitored by in situ ATR-FT-IR using PS-LXSnOct, where X = H or Cl. | ||
:[Cat]:[In] = 200:1:4 in the melt at 130 °C for 24 hours
| Entry | Catalyst | Time (h) | Conv.a (%) | M n, Theo | M n, NMR | M n | Đ M | k obs (×10−4 s−1)d | k obs (Sn(Oct)2)/kobs (Cat) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
a Determined from the 1H NMR spectrum.
b
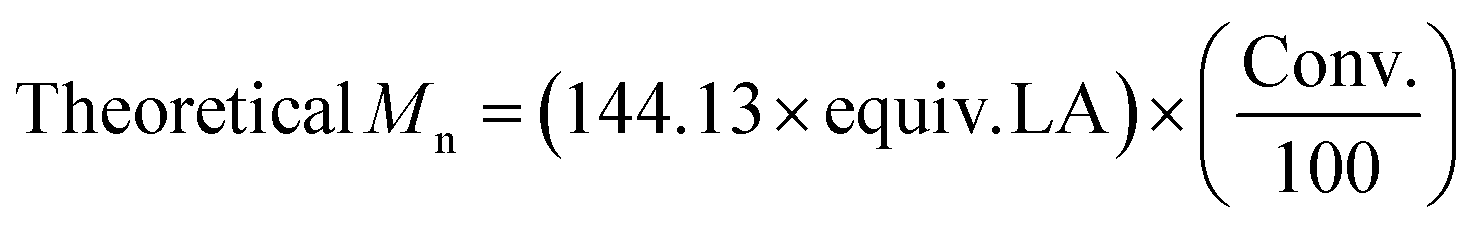 .
c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (multiplied by a factor of 0.58, rounded to the nearest 50).52
d Determined from in situ ATR-FT-IR kinetics. .
c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (multiplied by a factor of 0.58, rounded to the nearest 50).52
d Determined from in situ ATR-FT-IR kinetics.
|
|||||||||||
| 1 | PS-LHZnOAc | 6 | 89 | 6400 | 6100 | 7050 | 1.15 | 1.38 | 113 | 170 | 28.4 |
| 2 | PS-LHSnOct | 2.3 | 87 | 6250 | 6300 | 7300 | 1.17 | 6.44 | 24 | 174 | 75.6 |
| 3 | PS-LClSnOct | 0.8 | 83 | 6000 | 5800 | 5650 | 1.05 | 8.72 | 18 | 166 | 181.0 |
| 4 | Sn(Oct)2 | 0.12 | 89 | 6400 | 2450 | 6750 | 1.21 | 155 | 1 | 144 | 1231.7 |
A comparison of the kobs relative to that of Sn(Oct)2 revealed that PS-LClSnOct was 18 times slower than the industrial catalyst, while PS-LHZnOAc was approximately 113 times slower, at the same monomer:catalyst:initiator ([M]:[Cat]:[I]) ratio (Table 2, Scheme 2). These results were shown to be reproducible across different batches of catalyst; two separate batches of PS-LHSnOct produced kobs values of 6.44 × 10−4 s−1 and 6.10 × 10−4 s−1. Further, when the concentration of PS-LHSnOct was halved ([LA]:[Cat]:[I] = 400:1:8), the rate also halved, dropping to 3.49 × 10−4 s−1 (Fig. S16†), which indicates a likely first order in catalyst, and further hints that there are no mass-transfer limitations and that all catalyst sites are active at these concentrations.
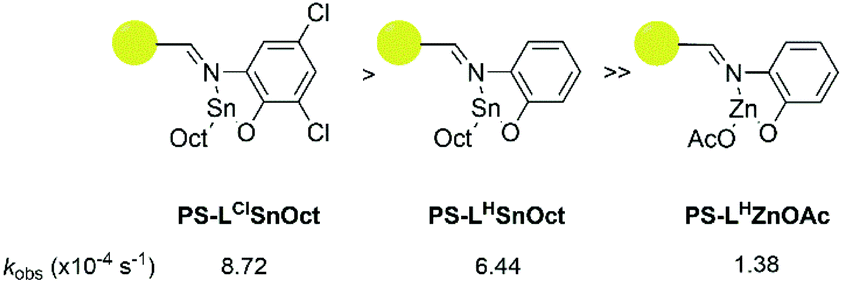 | ||
| Scheme 2 Order of reactivity of the three successful catalysts according to in situ ATR-FT-IR kinetic analysis: PS-LClSnOct (left), PS-LHSnOct (middle), PS-LHZnOAc (right). | ||
Analysis of the Matrix-Assisted Laser Desorption (MALDI-ToF) spectra of the catalysts revealed that all produced major series of MeBnO−/H+ end-capped PLA (Fig. 5, S17–S20†), with some transesterification. The transesterification was more apparent in PLA made using PS-LHZnOAc than with PS-LHSnOct, while PS-LClSnOct displays the best polymerisation control, with minimal transesterification and no evidence of cyclic species (Fig. 5). A comparison of the MALDI-ToF spectra and SEC traces of PS-LHSnOct produced PLA after 2.5 and 24 hours revealed that the dispersity increases drastically with longer reaction time, once the monomer is fully consumed (Fig. S18 and S21†). In the MALDI spectra, after 24 hours, the major series shifts to a lower molecular weight, and oligomers and minor series at higher Mn are now present, indicating that the catalyst can take part in side reactions at longer timescales.
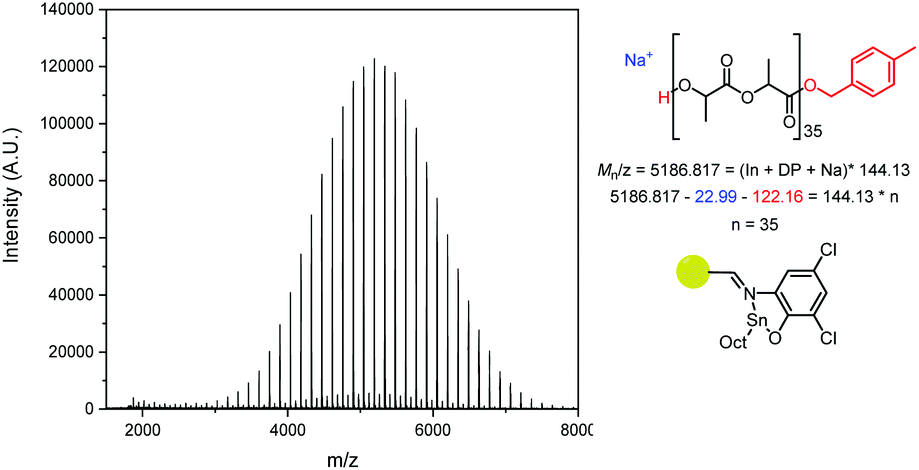 | ||
| Fig. 5 MALDI-ToF spectrum of PLA from PS-LClSnOct, showing a major series (MeBnO−/H+ end capped, Degree of Polymerisation (DP) ∼35) and a minor transesterification series. Conditions: [LA]:[Cat]:[I] = 50:1:1, 24 hours in the melt at 130 °C. | ||
As heterogeneous catalysts can be physically removed from the polymer, any potential side reactions can be suppressed without destroying the catalyst. Contrastingly, this problem cannot be remedied without destroying the catalyst when homogeneous catalysts are used, such as Sn(Oct)2. The dispersity for the latter was already slightly higher than with heterogeneous catalysts, despite the shorter timescale during in situ ATR-FT-IR monitored ROP, suggesting Sn(Oct)2 was already participating in transesterification reactions (ĐM 1.21, entry 4, Table 2). It is therefore evident that the ability to remove the heterogeneous catalysts from the polymer is a huge benefit of these catalysts: recovery of the catalysts allows for both greater control over the Mn whilst also creating the potential to reuse them.
Recovery and reuse
Both the polymer end groups and the effect of the carboxylate ligand on rates were highly suggestive of a coordination–insertion mechanism. In this mechanism, the PLA chain grows from the Lewis acidic metal centre, and participates in a dynamic exchange with the carboxylate, to yield the free PLA chain (Scheme S5†). It is hypothesised that the equilibrium can be driven towards the free polymer upon addition of a protic solvent (with acid, for example), which would then regenerate the pre-catalyst (vide infra).In all reactions, quenching the reaction was carried out with technical grade n-hexane to precipitate the free PLA, then re-dissolving the free polymer in dichloromethane (DCM) to filter out the catalyst. IR spectra were obtained of all recovered catalysts once the reaction had been quenched and compared to the original catalyst. The spectra of all the recovered catalysts contained peaks corresponding to the lactone CO and C–O at 1755 and 1089 cm−1, respectively (Fig. 6). This suggested that not all the PLA had been released from the active centres and supported the importance of the dynamic exchange process.
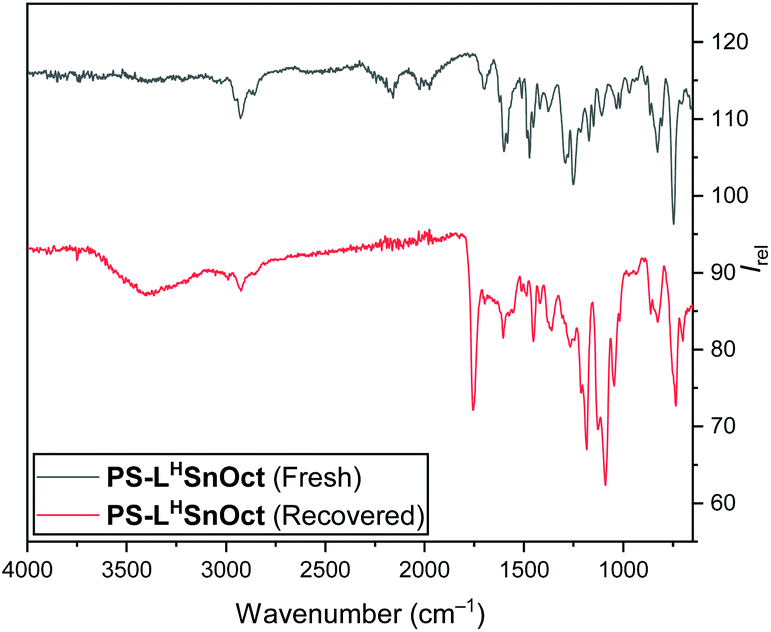 | ||
| Fig. 6 IR (ATR) spectra of PS-LHSnOct prior to ROP (top), and once recovered and dried in vacuo (bottom). | ||
To investigate the efficiency of the dynamic exchange, a reuse study was performed with several of the recovered catalysts (Fig. S23†). In all cases, recycling of the catalysts was carried out by quenching the reaction as described above, then drying the catalyst prior to reuse. Each reuse was accompanied by a significant drop in conversion: reuse of PS-LHZnOAc, for example, caused a drop from 84 to 33% (Mn 2750 Da, ĐM 1.25, entries 1 and 2, Table S4†). A more in depth reuse study of PS-LHSnOct was carried out over 7 reuse cycles; the same drop in conversion was observed throughout each cycle, stabilising out after the fourth cycle (Fig. 7 and S24†). The decline in conversion that occurred in the seventh cycle was attributed to the loss of catalyst mass through the various reuse procedures (Table S5†).
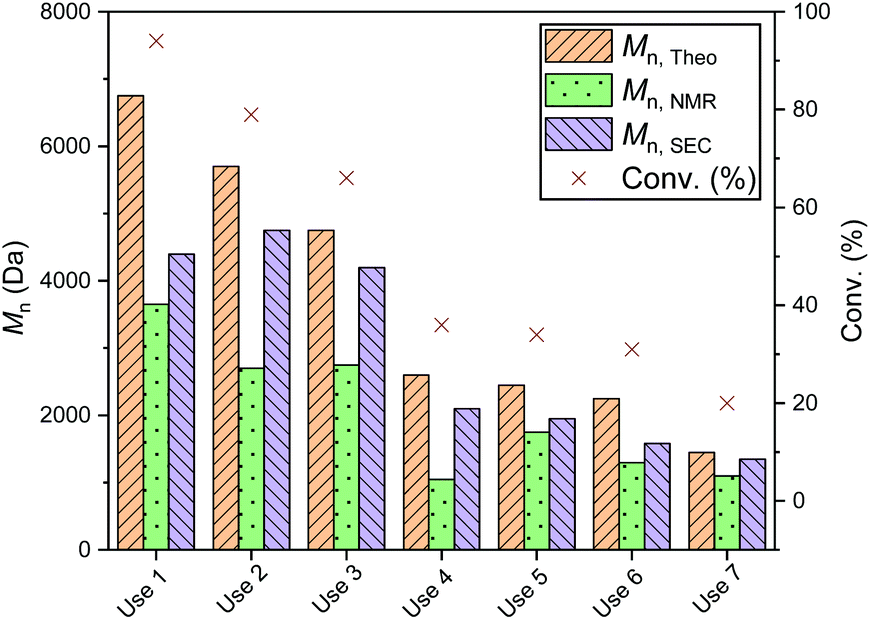 | ||
| Fig. 7 Polymerisation data from the ROP of L-LA with PS-LHSnOct over 6 reuse cycles. Conditions: [LA]:[Cat]:[I] = 50:1:1 in the melt at 130 °C, over 2.3 hours. | ||
The decrease in conversion can be either due to leaching of the metal, or from blocking of the active centres by the unreleased PLA.
Potential metal leaching was investigated by EDX, 119Sn NMR and Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES). 119Sn NMR was not sensitive enough to detect any Sn between 2000 and −2000 ppm at different numbers of scans, even in PLA samples produced from Sn(Oct)2 with a [LA]:[Cat]:[I] = 50:1:1 (Fig. S25†). However, Sn was detected in PLA produced with Sn(Oct)2 by EDX (Fig. 8A), yet no Sn was visible in PLA produced by the heterogeneous catalysts (Fig. 8B). This indicated that the decrease in conversion was unlikely to have been caused entirely by leaching of the metal into the PLA during reaction. In fact, EDX of the recovered catalyst still showed a distribution of Sn across the catalyst surface (Fig. 8C).
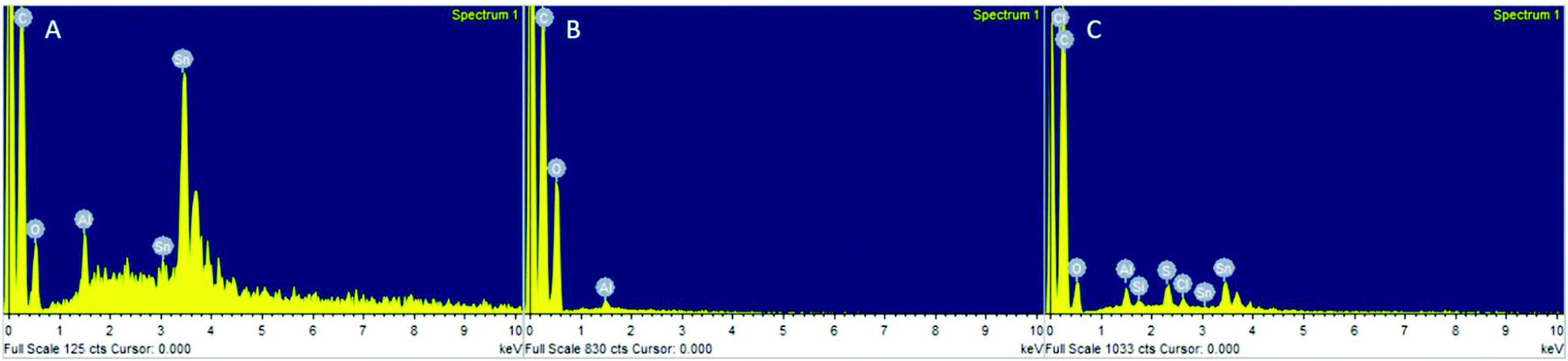 | ||
| Fig. 8 Energy Dispersive X-ray spectra of (A) PLA produced by Sn(Oct)2, showing the high Sn content in the polymer, (B) PLA from PS-LHSnOct, showing no evidence of Sn leaching, and (C) recovered PS-LHSnOct catalyst, with Sn still present in the catalyst. | ||
ICP-OES was used as a more quantitative tool to analyse the metal content of polymers before any purification was carried out (Table 3). PLA synthesised using Sn(Oct)2 contained 5573 ppm of Sn, similar to the theoretical maximum value ([LA]:[Cat]:[I] = 50:1:1, entry 4, Table 3). PLA made with the heterogeneous catalysts, on the other hand, contained significantly less metal content, of the same order of magnitude than polymers made using homogeneous systems and purified by classical methods involving dissolution and precipitation.60,61 The experimental metal content of PS-LHZnOAc was 385.5 ppm – a fraction of the theoretical maximum value calculated if 100% of the metal had leached into the polymer ([LA]:[Cat]:[I] = 50:1:1, entry 1, Table 3).
:catalyst ratios
| Entry | Cat. | [LA]:[Cat]:[I] |
Theoretical maximum metal ppm | Experimental metal ppm residue in crude polymer |
|---|---|---|---|---|
| 1 | PS-LHZnOAc | 50:1:1 |
17200 |
385.5 |
| 2 | PS-LHSnOct | 50:1:1 |
16551 |
1410.0 |
| 3 | PS-LHSnOct | 400:1:1 |
2060 | 335.0 |
| 4 | Sn(Oct)2 | 400:1:1 |
4100 | 5573.0 |
The same was observed for PS-LHSnOct, although the metal leaching was slightly higher with this catalyst (335 ppm ([LA]:[Cat]:[I] = 400:1:1, entry 3, Table 3). These findings could explain the slight increase in conversion observed in the supernatant during the Sheldon test. However, it has been shown to be practically impossible to entirely remove metal compounds from PLA, so the observation that the metal content in the final unpurified polymer is of the same order of magnitude than in purified polymers made with homogeneous catalysts already indicates that a major achievement has been made with the immobilised catalysts. These results suggest that purification of our polymer would result in a polymer of excellent purity, whose metal content would be far less than that of a polymer formed by classical methods. In addition, there is obvious potential for further ligand design to strengthen ligand–metal interactions and to suppress any metal leaching altogether.
A gradual increase in weight of catalyst over reuse cycles (Table S5†), further indicated that the PLA growing from the metal was increasing the mass of the catalyst. The decrease in mass after the fourth use was attributed to the degradation of the catalyst beads and subsequent loss of material during the recovery process. SEM imaging of the fresh and recovered catalyst showed that the catalyst was breaking up due to the mechanical stirring during reaction, exposing the unfunctionalised interior of the catalyst (Fig. S26†).
It was evident that the interior of the fresh catalyst could not be accessed by the PLA, as the interior remained highly porous, contrary to the exterior; the latter seemed to be blocked up with PLA (Fig. S26B†). A similar observation has been reported in the literature using SiO2 supported catalysts.27 Evidence from both SEM imaging and ICP-OES could partially explain the decrease in conversion over the various reuses. To maximise the conversion to PLA during each reuse cycle, regeneration of the pre-catalyst was attempted by quenching with either acetic acid or benzoic acid and dissolving the polymer in DCM. Both attempts were unsuccessful, as the acid tended to cause depolymerisation, evidenced by the lactic acid signals in the 1H NMR spectra (Fig. S27†). Washing the recovered catalyst in DCM over 72 hours recovered some more PLA, suggesting that polymer was still trapped on the catalyst surface. It would therefore be of interest to use a less porous catalyst in future studies.
Catalyst scope
To demonstrate the versatility of the catalysts, PS-LHSnOct and PS-LHZnOAc were used in the solution phase was attempted in THF at room temperature and toluene at 80 °C over 24 hours. The polymerisations in THF were unsuccessful, presumably due to the coordinating oxygen which blocked the access of lactide to the metal (entries 1 and 2, Table S6†). Polymerisation in toluene, however, proceeded with good conversions for PS-LHSnOct, and improved dispersity compared to the industrial standard (entries 3–5, Table S6†).The industrial relevance of the heterogeneous catalysts was then explored by altering the monomer to catalyst ratios up to 400:1:1 (Table 4). Whilst longer reaction times were required to achieve higher conversions comparable to the 50:1:1 system, it was possible to target higher molecular weights up to Mn 35250 Da (ĐM 1.27, entry 7, Table 4). A control using Sn(Oct)2 showed that even at these low loadings of catalyst ([LA]:[Cat]:[I] = 400:1:1), the homogeneous catalyst produced a polymer of lower Mn and higher dispersity due to increased transesterification (Mn 24000 Da, ĐM 1.88, entry 8, Table 4). Catalyst loadings down to 15 ppm of metal were possible ([LA]:[I] = 400:1), although higher temperatures or longer timescales were required to reach modest conversions (69% conversion at 1250 ppm catalyst, Mn 31350 Da, ĐM 1.13, entry 5, Table S4†). When the temperature was increased to 180 °C, a TOF of 810 h−1 was achieved with 15 ppm of metal using PS-LHSnOct (41% conversion, entry 2, Table S7†). When a [LA]:[Cat]:[I] ratio of 200:1:4 was used at 180 °C, PS-LHSnOct showed impressive activity, reaching full conversion within 20 minutes and displaying excellent control (kobs 1.25 × 10−3 s−1, Mn 7500 Da, ĐM 1.25, Fig. S26†), comparing well with Sn(Oct)2 under the same conditions (kobs 6.67 × 10−3 s−1, Mn 8250 Da, ĐM 1.53, Fig. S27†).
| Entry | [LA]:[Cat]:[I] |
Time (h) | Conv.a (%) | M n, Theo | M n | M w | Đ M | TON | TOF (h−1) |
|---|---|---|---|---|---|---|---|---|---|
a Determined from the 1H NMR spectrum.
b
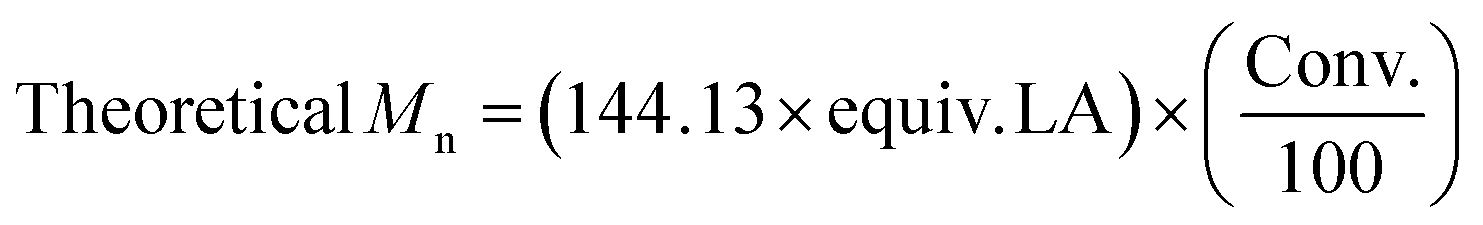 .
c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (multiplied by a factor of 0.58, rounded to the nearest 50).52
d Entry 8 used Sn(Oct)2 as the catalyst in a control reaction, to directly compare to entry 7. .
c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (multiplied by a factor of 0.58, rounded to the nearest 50).52
d Entry 8 used Sn(Oct)2 as the catalyst in a control reaction, to directly compare to entry 7.
|
|||||||||
| 1 | 100:1:1 |
2.5 | 86 | 12400 |
11100 |
12850 |
1.16 | 86 | 34.4 |
| 2 | 200:1:1 |
2.5 | 86 | 24800 |
23700 |
28300 |
1.19 | 172 | 68.8 |
| 3 | 300:1:1 |
2.5 | 40 | 17300 |
19600 |
21200 |
1.08 | 60 | 24.0 |
| 4 | 300:1:1 |
6 | 72 | 41500 |
25300 |
27900 |
1.10 | 215 | 35.9 |
| 5 | 400:1:1 |
6 | 38 | 21900 |
18800 |
20350 |
1.08 | 152 | 25.3 |
| 6 | 400:2:1 |
6 | 69 | 39800 |
28200 |
32700 |
1.16 | 138 | 23.0 |
| 7 | 400:1:1 |
24 | 90 | 51900 |
35250 |
44750 |
1.27 | 360 | 15.0 |
| 8d | 400:1:1 |
24 | 90 | 51900 |
24000 |
45200 |
1.88 | 179 | 7.5 |
Finally, the ROP of other rac-LA (a 50:50 mixture of D- and L-LA) and other lactones, commonly used in literature, was carried out to demonstrate the monomer scope of the catalysts (Table 5).62–70 Neither catalyst showed any isotactic bias when using rac-LA, with Pr (probability of racemic enchainment) around 0.50, characteristic of an atactic polymer (Fig. S10, Table S2†).59 When using less sterically hindered lactones such as ε-caprolactone (ε-CL), high conversions were achieved when using the optimised times from the L-LA study (up to 91%, entries 5–8, Table 5). Larger lactones such as ε-decalactone (ε-DL) struggled to reach high conversions within the L-LA optimised times (entries 9–12, Table 5). However, high conversions were still possible after 24 hours (entries 10 and 12, Table 5), and it is tentatively proposed that the sterically bulky alkane branch off ε-DL prevented the efficient approach of the monomer to the active site, although the activation of the monomer was not hindered completely. ε-DL was used without any purification prior to ROP, further proving the robustness of the heterogeneous catalysts.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | M | Time (h) | Conv.a (%) | M n, Theo | M n | M w | Đ M |
Conditions: [M]:[Cat]:[I] = 50:1:1 in the melt at 130 °C (M = monomer).a Determined from the 1H NMR spectrum.b 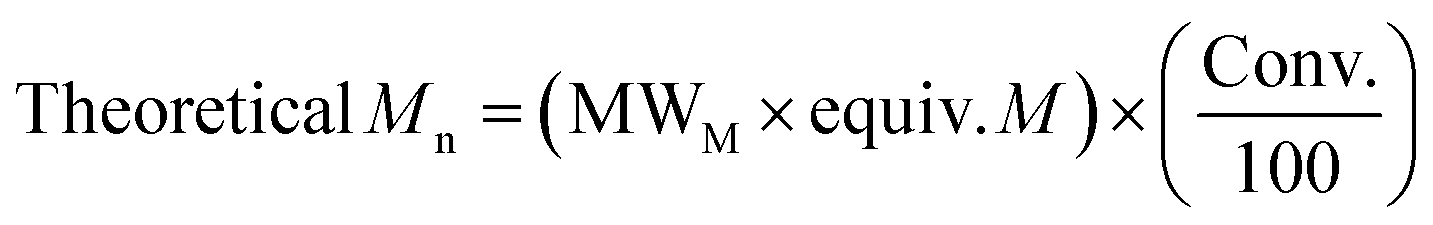 .c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (rounded to the nearest 50). Molecular weights multiplied by the respective correction factor for the polymer: PLA values multiplied by a factor of 0.58,52 PCL by 0.56.71 .c As determined by SEC (THF) using RI methods, relative to poly(styrene) standards (rounded to the nearest 50). Molecular weights multiplied by the respective correction factor for the polymer: PLA values multiplied by a factor of 0.58,52 PCL by 0.56.71 |
||||||||
| 1 | PS-LHZnOAc | rac-LA | 6 | 89 | 6400 | 6100 | 10200 |
1.67 |
| 2 | PS-LHSnOct | rac-LA | 2.5 | 91 | 6550 | 5450 | 6950 | 1.28 |
| 3 | Sn(Oct)2 | rac-LA | 10 min | 90 | 6500 | 6450 | 7850 | 1.22 |
| 4 | Zn(OAc)2·2H2O | rac-LA | 24 | 84 | 6050 | 2800 | 4550 | 1.63 |
| 5 | PS-LHZnOAc | ε-CL | 6 | 91 | 5200 | 6300 | 9400 | 1.49 |
| 6 | PS-LHSnOct | ε-CL | 2.5 | 89 | 5100 | 5350 | 7350 | 1.38 |
| 7 | Sn(Oct)2 | ε-CL | 10 min | 77 | 4400 | 5350 | 6150 | 1.15 |
| 8 | Zn(OAc)2·2H2O | ε-CL | 24 | 94 | 5350 | 3400 | 4800 | 1.42 |
| 9 | PS-LHZnOAc | ε-DL | 6 | 36 | 2050 | 2950 | 3500 | 1.18 |
| 10 | PS-LHZnOAc | ε-DL | 24 | 46 | 1550 | 4050 | 4600 | 1.13 |
| 11 | PS-LHSnOct | ε-DL | 2.5 | 0 | — | — | — | — |
| 12 | PS-LHSnOct | ε-DL | 24 | 74 | 2100 | 6300 | 7100 | 1.12 |
| 13 | Sn(Oct)2 | ε-DL | 10 min | 8 | 450 | — | — | — |
| 14 | Sn(Oct)2 | ε-DL | 24 | 92 | 5250 | 8400 | 11450 |
1.36 |
| 15 | Zn(OAc)2·2H2O | ε-DL | 24 | 64 | 5450 | 3500 | 4050 | 1.15 |
Conclusions
Several highly efficient heterogeneous catalysts based on SnII and ZnII were synthesised and employed in the ROP of L-LA, with PS-LClSnOct displaying the best rate and control. It was noted that both changing the carboxylate ligand from the acetate to 2-ethylhexanoate, and employing electron withdrawing groups on the catalyst (such as Cl-substituents ortho and para to the phenoxy donor) improved the rate of ROP drastically, producing white polymers of high purity. Catalyst loading as low as 15 ppm metal and TOF values of up to 810 h−1 could be achieved. Although polymerisation rates were slower than homogeneous industrial standard Sn(Oct)2, these catalysts were shown to reproducibly deliver excellent control under a wide range of reaction conditions and tolerate several lactone substrates, providing much improvement on the sparse number of known heterogeneous systems for ROP. The ability to use these catalysts in the melt and at industrially relevant ratios and temperatures is beneficial for industrial applications compared to other heterogeneous examples. Most significantly, ICP-OES showed that metal content in the final unpurified polymers was of the same order of magnitude than for purified polymers made with homogeneous catalysts. Recovery and reuse were possible, and these results suggest that the potential for achieving heterogeneous polymerisation with these systems is real. However, understanding the full mechanism of loss of activity, and hence improving the overall reusability of the catalyst towards an even greener process, will be the topic of following studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Analytical facilities were provided through the Material and Chemical Characterisation Facility (MC2) and at the University of Bath. IH would like to thank the EPSRC and the CDT in Catalysis for funding this project (EP/L016443/1). AB acknowledges Roger and Sue Whorrod (Whorrod Research Fellowship) and the Royal Society (UF/160021 fellowship) for research funding. CH acknowledges the Royal Society (UF/140207 fellowship). Dr Gerrit Gobius du Sart and Dr Matthew D. Jones are thanked for useful discussions when preparing the manuscript.References
- K. Law and R. C. Thompson, Science, 2014, 345, 144–145 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- W. H. Carothers, G. L. Borough and F. J. Natta, J. Am. Chem. Soc., 1932, 54, 761–772 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- S. Lambert and M. Wagner, Chem. Soc. Rev., 2017, 46, 6855–6871 RSC.
- R. Platel, L. Hodgson and C. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- S. P. Parwe, S. D. Warkad, M. V. Mane, P. S. Shedage and B. Garnaik, Polymer, 2017, 111, 244–251 CrossRef CAS.
- A. Arbor, Food Chem. Toxicol., 1995, 33, 273–283 CrossRef.
- M. C. Tanzi, P. Verderio, M. G. Lampugnani, M. Resnati, E. Dejana and E. Sturani, J. Mater. Sci. Mater. Med., 1994, 5, 393–396 CrossRef CAS.
- H. Li and L. Gu, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 968–976 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- X. Zhang, G. O. Jones, J. L. Hedrick and R. M. Waymouth, Nat. Chem., 2016, 8, 1047–1053 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- B. Lin and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 1645–1652 CrossRef CAS PubMed.
- C. Thomas, F. Peruch, A. Deffieux, A. Milet, J. P. Desvergne and B. Bibal, Adv. Synth. Catal., 2011, 353, 1049–1054 CrossRef CAS.
- B. Wang, L. Pan, Z. Ma and Y. Li, Macromolecules, 2018, 51, 836–845 CrossRef CAS.
- S. Naumann, P. B. V. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS PubMed.
- A. Nachtergael, O. Coulembier, P. Dubois, M. Helvenstein, P. Duez, B. Blankert and L. Mespouille, Biomacromolecules, 2015, 16, 507–514 CrossRef CAS PubMed.
- S. M. Islam, K. Ghosh, R. A. Molla, A. S. Roy, N. Salam and M. A. Iqubal, J. Organomet. Chem., 2014, 774, 61–69 CrossRef CAS.
- Y. Chauvin, D. Commereuc and F. Dawans, Prog. Polym. Sci., 1977, 5, 95–226 CrossRef CAS.
- A. Kirschning, H. Monenschein and R. Wittenberg, Angew. Chem., Int. Ed., 2001, 40, 650–679 CrossRef CAS PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- S. Hübner, J. G. De Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- M. D. Jones, M. G. Davidson, C. G. Keir, A. J. Wooles, M. F. Mahon and D. C. Apperley, Dalton Trans., 2008, 3655–3657 RSC.
- T. M. Abdel-Fattah and T. J. Pinnavaia, Chem. Commun., 1996, 665–666 RSC.
- M. D. Jones, M. G. Davidson, C. G. Keir, L. M. Hughes, M. F. Mahon and D. C. Apperley, Eur. J. Inorg. Chem., 2009, 635–642 CrossRef CAS.
- E. J. Lee, K. M. Lee, J. Jang, E. Kim, J. S. Chung, Y. Do, S. C. Yoon and S. Y. Park, J. Mol. Catal. A: Chem., 2014, 385, 68–72 CrossRef CAS.
- E. Martin, P. Dubois and R. Jérôme, Macromolecules, 2003, 36, 7094–7099 CrossRef CAS.
- K. Tortosa, T. Hamaide, C. Boisson and R. Spitz, Macromol. Chem. Phys., 2001, 3, 1156–1160 CrossRef.
- J. H. Khan, F. Schue and G. A. George, Polym. Int., 2009, 58, 296–301 CrossRef CAS.
- B. C. Wilson and C. W. Jones, Macromolecules, 2004, 37, 9709–9714 CrossRef CAS.
- E. Kim, E. W. Shin, I. K. Yoo and J. S. Chung, J. Mol. Catal. A: Chem., 2009, 298, 36–39 CrossRef CAS.
- K. Yu and C. W. Jones, J. Catal., 2004, 222, 558–564 CrossRef CAS.
- J. M. J. Fréchet, M. Jean Farrall and G. Darling, React. Polym., Ion Exch., Sorbents, 1982, 1, 27–34 CrossRef.
- W. Xu, R. Mohan and M. M. Morrissey, Tetrahedron Lett., 1997, 38, 7337–7340 CrossRef CAS.
- S. M. Islam, K. Ghosh, A. S. Roy and R. A. Molla, RSC Adv., 2014, 4, 38986–38999 RSC.
- F. Wu, Y. Feng and C. W. Jones, ACS Catal., 2014, 4, 1365–1375 CrossRef CAS.
- S. Canellas, C. Ayats, A. H. Henseler and M. A. Pericàs, ACS Catal., 2017, 7, 1383–1391 CrossRef CAS.
- Y. Feng, M. E. Lydon and C. W. Jones, ChemCatChem, 2013, 5, 3636–3643 CrossRef CAS.
- J. J. Lee, C. Yoo, C. Chen, S. E. Hayes, C. Sievers and C. W. Jones, Langmuir, 2018, 34, 12279–12292 CrossRef CAS PubMed.
- M. W. Mckittrick and C. W. Jones, J. Am. Chem. Soc., 2004, 126, 3052–3053 CrossRef CAS PubMed.
- J. Wang, X. Shan, S. Shan, H. Su, S. Wu and Q. Jia, Catal. Commun., 2015, 59, 116–121 CrossRef CAS.
- K. Yu and C. W. Jones, Organometallics, 2003, 22, 2571–2580 CrossRef CAS.
- G. G. Hlatky, Chem. Rev., 2000, 100, 1347–1376 CrossRef CAS PubMed.
- W. Long, C. S. Gill and C. W. Jones, Dalton Trans., 2010, 39, 1470–1472 RSC.
- M. Oshimura, A. Takasu and K. Nagata, Macromolecules, 2009, 42, 3086–3091 CrossRef CAS.
- C. Schuerch and J. M. J. Fréchet, J. Am. Chem. Soc., 1971, 93, 492–496 CrossRef CAS.
- C. L. Lee, Y. F. Lin, M. T. Jiang, W. Y. Lu, J. K. Vandavasi, L. F. Wang, Y. C. Lai, M. Y. Chiang and H. Y. Chen, Organometallics, 2017, 36, 1936–1945 CrossRef CAS.
- S. Jain and O. Reiser, ChemSusChem, 2008, 1, 534–541 CrossRef CAS PubMed.
- S. Gesslbauer, H. Cheek, A. J. P. White and C. Romain, Dalton Trans., 2018, 47, 10410–10414 RSC.
- G. G. Mohamed, M. M. Omar and A. M. M. Hindy, Spectrochim. Acta, Part A, 2005, 62, 1140–1150 CrossRef PubMed.
- J. Baran, A. Duda, A. Kowalski, R. Szymanski and S. Penczek, Macromol. Rapid Commun., 1997, 18, 325–333 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromol. Rapid Commun., 1998, 19, 567–572 CAS.
- P. Dubois, O. Coulembier and J.-M. Raquez, Handbook of Ring-Opening Polymerization, Wiley-VCH, Germany, 2009, pp. 255–286 Search PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- H. E. B. Lempers and R. A. Sheldon, J. Catal., 1998, 175, 62–69 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed.
- P. McKeown, S. N. McCormick, M. F. Mahon and M. D. Jones, Polym. Chem., 2018, 9, 5339–5347 RSC.
- A. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- G. Schwach, J. Coudane, R. Engel and M. Vert, Polym. Bull., 1996, 37, 771–776 CrossRef CAS.
- S. Lee, K. Lee, J. Jang, J. Sun, W. Jin, G. Kim, Y. Kim and J. Shin, Polymer, 2017, 112, 306–317 CrossRef CAS.
- J. Bai, J. Wang, Y. Wang and L. Zhang, Polym. Chem., 2018, 9, 4875–4881 RSC.
- J. Lin, W. Chen, Z. Shen and J. Ling, Macromolecules, 2013, 46, 7769–7776 CrossRef CAS.
- Y. Huang, W. Wang, C. Hsu, W. Lu, W. Chuang, M. Y. Chiang, Y. Lai and H. Chen, Polym. Chem., 2016, 7, 4367–4377 RSC.
- W. M. Stevels, M. J. K. Ankone, P. J. Dijkstra and J. Feijen, Macromolecules, 1996, 29, 8296–8303 CrossRef CAS.
- M. Tarnacka, A. Dzienia, P. Maksym, A. Talik, A. Zi, R. Bielas, K. Kaminski and M. Paluch, Macromolecules, 2018, 51, 4588–4597 CrossRef CAS.
- R. Polymerization, L. Using, C. Huang, F. Wang, B. Ko, T. Yu and C. Lin, Macromolecules, 2001, 34, 356–361 CrossRef.
- T. Borke, K. Odelius and A. Albertsson, Biomacromolecules, 2013, 14, 2883–2890 CrossRef PubMed.
- M. T. Martello, A. Burns and M. Hillmyer, ACS Macro Lett., 2012, 1, 131–135 CrossRef CAS.
- G. Gontard, A. Amgoune and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2016, 3253–3256 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, metal loading calculations, NMR spectra of monomer and polymers, plots of Mn and ĐMvs. conversion, polymerisation data and associated kinetic data, images of SEC traces, MALDI-ToF mass spectra, TGA and DSC traces. See DOI: 10.1039/c9py01472a |
| This journal is © The Royal Society of Chemistry 2019 |