
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectro-activity and magnetic switching in lanthanide-based single-molecule magnets
Olivier
Cador
 ,
Boris
Le Guennic
and
Fabrice
Pointillart
*
,
Boris
Le Guennic
and
Fabrice
Pointillart
*
Univ Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) - UMR 6226, F-35000 Rennes, France. E-mail: fabrice.pointillart@univ-rennes1.fr
First published on 1st October 2019
Abstract
The control of the single-molecule magnet (SMM) behaviour is highly challenging but crucial to design molecular materials suitable for potential applications in molecular devices. To switch the SMM behaviour, a plethora of external stimuli can be employed such as electro-activity, solvents, protonation, magnetic and electric fields, and light. All these stimuli induce structural modifications and thus, to some degree, changes in the electronic properties. Lanthanide ions are elements extremely sensitive to crystal field effects i.e. they are sensitive to any structural changes making them ideal candidates for the design of molecular switches. This article reviews both the pool of electro-active lanthanide SMMs and the various switching modes such as redox-, solvato-, photo- and proton-switching lanthanide SMM behaviours. These examples highlight the importance of inducing electronic distribution changes close to the lanthanide centre to reach a complete ON–OFF switching but also the preponderance of further structural changes and intermolecular interactions to modulate the magnetic properties. The present review should convince scientific communities that the use of lanthanide ions in the design of molecular switches is a promising strategy.
 Oliver Cador | The research activities of Olivier Cador started in 1994 in Bordeaux (France). He mainly focused on the studies of the magnetic properties of systems with various dimensionalities, from nanoparticles to isolated molecules which show quantum behaviours, and finally to magnets with atypical behaviours. In Rennes, his aim was at first to develop the synergy between electronic properties, such as magnetism and electrical conductivity, in new molecular edifices based on organic/inorganic networks. He orients now his research field to Single Molecule Magnets (SMMs) based on lanthanides. |
 Boris Le Guennic | Boris Le Guennic received his Ph.D. in Chemistry from the University of Rennes in 2002. He then successively moved to the Universities of Erlangen, Buffalo, and Bonn for postdoctoral stays in the groups of Jochen Autschbach and Markus Reiher. In 2005, he was appointed as a CNRS researcher at ENS de Lyon (France). In 2011, he moved to the Chemical Sciences Institute of Rennes (University of Rennes, France) where he is currently a Senior CNRS researcher. His research is devoted mainly to the application of quantum chemical methodologies for the interpretation of magnetic and optical properties in transition metal and f-element complexes, with particular emphasis on lanthanide-based single molecule magnets. |
 Fabrice Pointillart | Fabrice Pointillart received his PhD from the Pierre et Marie University in 2005, supervised by Profs. Cyrille Train and Michel Verdaguer. After a two-year period of postdoctoral research at the University of Florence with Prof. Roberta Sessoli, he joined the University of Rennes 1 in 2007 as a CNRS researcher. His current research interests are focused on single-molecule magnets of lanthanides associated with other properties such as chirality, luminescence and spin-crossover. He was awarded the CNRS bronze medal in 2014 and the junior prize of the Coordination Chemistry Division of the French Chemical Society in 2017 and received the ERC Consolidator Grant in 2016. |
1. Introduction
Next generation devices such as hard disks would use molecular-scale components instead of the actual silicon-based electronics. Molecules able to retain their magnetization in a given direction were discovered in the early 90s with the famous Mn12 cluster.1 Such molecules behave as a tiny magnet at the molecular scale and are called single-molecule magnets (SMMs). While such objects were built from transition metals until the beginning of the 2000s, lanthanide ions became rapidly good candidates for the design of SMMs due to their high magnetic moment and strong magnetic anisotropy.2 Recently two crucial factors have been dramatically improved that allow lanthanide-based SMMs to be back in the race for potential applications in high-density data storage,3 and also in quantum computing4 and spintronics:5 the high blocking temperature6 and the level of understanding of the magnetic properties. Indeed the dynamics of a SMM can be modelled by the quantitative formulation of the relaxation time τ:7 | (1) |
This relaxation time is composed of thermally-dependent (first three terms in eqn (1)) and thermally-independent (last term in eqn (1)) regimes. The thermally assisted processes between two states |b〉 and |a〉 are illustrated in Scheme 1.
(i) The direct process involves a single phonon and needs the application of an external magnetic field. This process is slightly thermal dependent but strongly field dependent and characterized by the parameter A and the exponent m (eqn (1)) where m is fixed to 2 for non-Kramers ions and 4 for Kramers ions. (ii) The Raman process involves a virtual state and the simultaneous absorption/emission of two phonons. It is thermally-dependent and characterized by the parameter C and the exponent n (eqn (1)) which usually takes the value of 7 for non-Kramers ions and 9 for Kramers ions but could be lowered when both acoustic and optical phonons are involved in the relaxation process.8 (iii) The Orbach process involves real states and the simultaneous absorption/emission of two phonons. It gives the energy barrier (Δ) and has to be favoured to obtain high-performance SMMs. (iv) The QTM (not illustrated in Scheme 1) induces transitions between doublet states due to the distortion from the perfect axial symmetry9 and dipolar10 or hyperfine11 interactions.
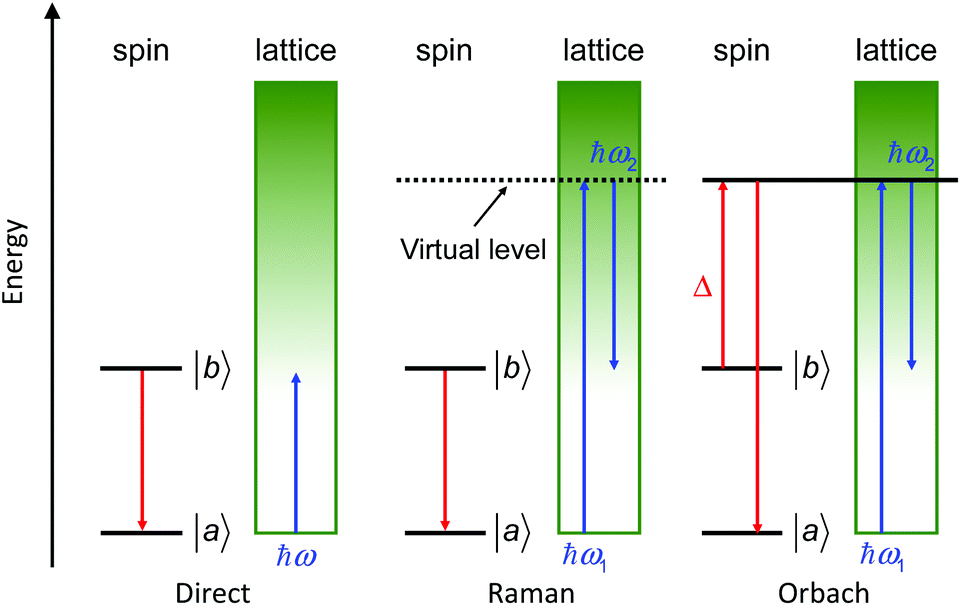 | ||
| Scheme 1 Illustration of the three thermally assisted relaxation processes between two states |b〉 and |a〉. | ||
Even more challenging is the design of molecular materials whose physical properties can be controlled by external stimuli such as temperature, pressure, light irradiation, and magnetic field (Scheme 2).12 In fact the future circuits and devices composed of molecular-scale components should be able to switch between distinct states at high speeds with the input of minimal energy.13 To date, most of the transition metal-based magnetic switches in which the SMM behavior is modulated under an external stimuli were obtained when the electronic properties are strongly affected. Thus photo-irradiation induced change of the spin state,14 while oxidizing or reducing agents induced change of the oxidation state of the organic ligand15 or the metal centre.16 Only recently, a few examples of solvato-switching of the SMM behavior were reported for transition metals.17
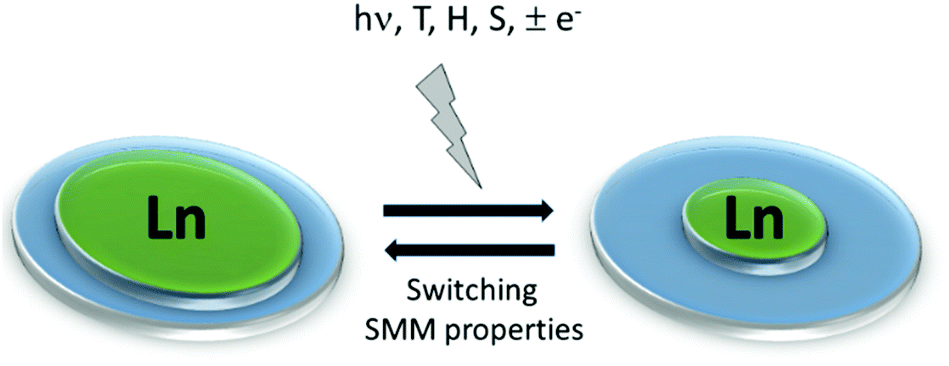 | ||
| Scheme 2 Lanthanide-based systems displaying switching of their SMM behavior under various stimuli such as light irradiation (hν), temperature (T), magnetic field (H), solvent (S) and oxidizing/reducing agent (±e−). | ||
Concerning lanthanide complexes, several examples of magnetic switching have appeared in the literature these last five years. This review aims to summarize these recent examples. The next section is devoted to the quite large pool of redox-active lanthanide SMMs while the third section shows how the redox activity of the ligand or the metal can be used to modulate the SMM behavior. The next two sections are focused on the solvato- and photo-switching SMMs. Finally the sixth section is devoted to proton-switching SMMs. Through reading this review, the goal is to detail the different possibilities of inducing magnetic modulations giving some clue about the degree as well as the origin of the observed magnetic modulations.
2. Electro-active single-molecule magnets
In this review the notion of electro-activity is attributed to species which can be easily oxidized or reduced i.e. with potentials (electro)chemically accessible |E| < 2.5 V vs. SCE. It is worth noting that the redox activity is exclusively centred on the organic ligands. Thus the most known electro-active ligands, i.e. tetraoxolene, phthalocyanine and tetrathiafulvalene, involved in the design of electro-active SMMs are described.2.1 Tetraoxolene based single-molecule magnets
The tetraoxolene-based ligands have a high potential interest level because they could be obtained in their closed shell dianionic and radical trianionic forms (Fig. 1a) giving potential access to radical bridging lanthanide species with possible interesting magnetic properties.18 Nevertheless among the several tetraoxolene-bridged dinuclear complexes of lanthanides which are reported in the literature, their magnetic properties are not studied.19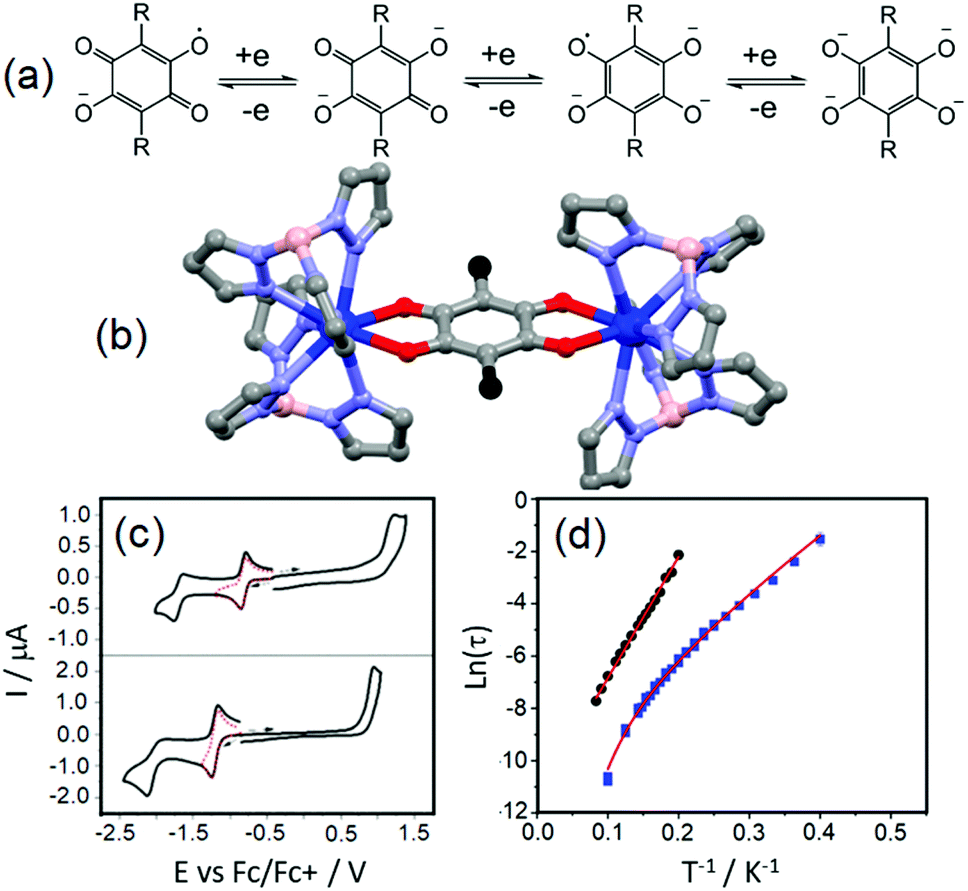 | ||
| Fig. 1 (a) Electrochemical processes for tetraoxolene ligands. (b) Molecular structures of the dinuclear complexes 1 (R = Cl in black) and 2 (R = CH3 in black). (c) Cyclic voltammograms of the yttrium analogues of 1 (up) and 2 (below). (d) Arrhenius plot of relaxation rates of 1 (blue squares) and 2 (black spots) in a 1600 Oe applied magnetic field with the best fits (red lines, see main text for descriptions). Colour code: carbon: grey, nitrogen: light blue, boron: pink, oxygen: red and dysprosium: blue. Adapted from ref. 20. | ||
The unprecedented deep magnetic study of a dinuclear complex involving lanthanide ions and tetraoxolene ligands was reported by C. Boskovic and collaborators in 2017.20 The Dy(III) derivatives were obtained by the one-pot reaction of hydrotris(pyrazolyl)borate (HB(pz)3−), the corresponding dysprosium salt and doubly deprotonated chloranilate (ca2−) ligands. The X-ray structures revealed the formation of dinuclear complexes in which the ca2− ligands (R = Cl (L1) and R = CH3 (L2)) bridged two lanthanide ions and the HB(pz)3− anions played the role of blocking ligands (Fig. 1b). The nature of the substituents on the ca2− bridges induced both the electronic effect and change from dodecahedral for [((HB(pz)3)2Dy)2(μ-L1)] (1) to square antiprismatic for [((HB(pz)3)2Dy)2(μ-L2)] (2) coordination environments which induced changes in the electrochemical and magnetic properties. The compounds with both L1 and L2 ligands displayed two quasi-reversible reduction processes (−0.96 to −0.81 V and −1.73 to −1.65 V vs. Fc/Fc+) corresponding to the formation of the radical tri-anionic and tetra-anionic species, and one irreversible oxidation process (0.89 to 1.18 V vs. Fc/Fc+) corresponding to the formation of the radical mono-anionic species (Fig. 1c). All the redox processes have been attributed to tetraoxolene-based ligands.21 The substitution of the chloride with methyl groups induced a negative shift of the reduction processes of 0.2–0.3 V because of the electron donating character of the methyl group making the oxidation of L2 more difficult compared to L1. The dynamic magnetic properties of 1 and 2 revealed a frequency dependence of the magnetic susceptibility at high frequency.
Once the QTM is suppressed by an optimal field (1600 Oe), the magnetic relaxation of 2 occurred through the sole Orbach regime (Δ = 47 K and τ0 = 1.0 × 10−5 s) while 1 displayed magnetic relaxation through both Orbach (Δ = 24 K and τ0 = 1.0 × 10−5 s) and Raman (C = 2.7 × 10−3 s K−n with n fixed to 7) regimes (eqn (1), Fig. 1d). The electrochemistry attested the possibility of isolating the radical tri-anionic oxidation state of the bridges as shown and discussed in section 3.1.
2.2 Phthalocyanine based single-molecule magnets
Phthalocyanine (Pc) ligands are well-known in molecular magnetism because the first lanthanide-based SMM discovered in 2003 was the Tb(Pc)2 double decker sample.22 These kinds of double decker compounds present a high anisotropy barrier23 making them potential candidates for applications in high density storage and spintronic devices. Heteroleptic lanthanide porphyrin (Por)-bisphthalocyanine (L3) triple decker complexes (L3)Ln(L3)Ln(Por) (Ln = Tb(III) (3) and Dy(III) (4)) were synthesized via microwave-assisted preparation.24 A complete electrochemical study showed that such complexes highlighted four one-electron oxidations and three one-electron reductions which are centred on the ligands. The oxidation and reduction potentials of the L3 ligand were identified at 0.50 and −0.65/−1.10 V vs. Fc/Fc+, respectively. The direct current (dc) magnetic properties of 3 and 4 showed ferromagnetic dipolar interactions at low temperature.The alternating current (ac) magnetic properties under an applied magnetic field of 1000 Oe (in order to suppress the QTM contribution from eqn (1)) demonstrated the SMM behaviour for 3 with two main magnetic relaxations i.e. an Orbach thermally dependent regime at high temperature (Δ = 132.0 K and τ0 = 5.0 × 10−9 s) and an under barrier direct mechanism at low temperature (A = 1.65 × 105 s−1 K−1 T−m with m fixed to 4).
However the advantages of the Pc ligands having accessible redox potentials25 can be problematic when attempting to deposit them on the Cu(111) surface26 because unexpected reduction can occur the same as for Mn12 complexes on the Au(111) surface.27 Thus Veciana's group designed new Pc derivatives in which the Pc is functionalized by phthalimides for stabilising the anionic double-decker complexes TBA[Tb(Ln)2] (5)–(7) (TBA is tetrabutylammonium).28 The success of the synthetic strategy was highlighted with electrochemical characterisation which presents strong shifts of 0.6–0.8 V depending on the nature of the phthalimide decorated group (R = −C15H31 (L4) (5), −C3H7 (L5) (6) and –CH(CH3)Ph (L6) (7)) (Fig. 2). The two oxidation potentials and the two reduction potentials are found at +0.3/+0.8 V and −0.9/−1.2 V vs. Fc/Fc+ compared to 0.0/−0.4 V and −1.6/−1.8 V vs. Fc/Fc+ for the unsubstituted Pc ligand in TBA[Tb(Pc)2].29
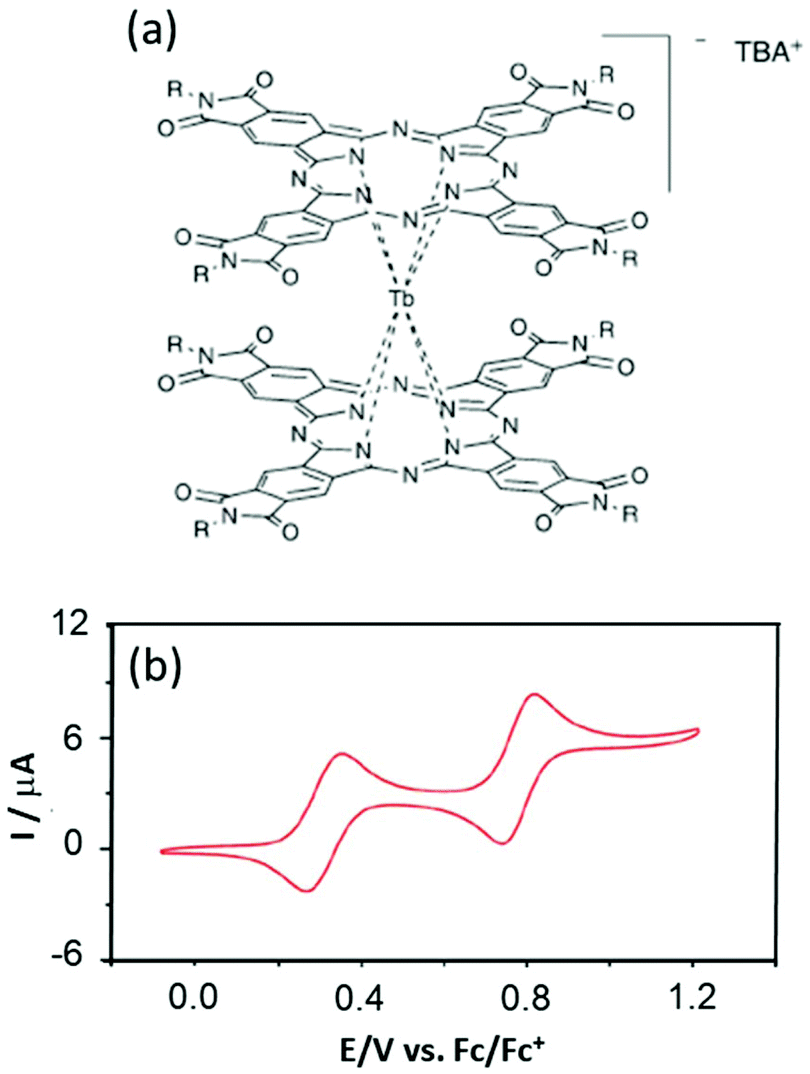 | ||
| Fig. 2 (a) Molecular structures of the TBA[Tb(Ln)2] complexes (R = –C15H31 (L4) (5), –C3H7 (L5) (6) and –CH(CH3)Ph (L6) (7)). (b) Cyclic voltammograms of 7. Adapted from ref. 28. | ||
The ac magnetic measurements showed classical SMM behaviour with both the thermally activated Orbach relaxation process and QTM at lower temperature.30 The fit of the Orbach regime led to energy barrier values with pre-exponential factors of 640 K and τ0 = 6.35 × 10−11 s for 5, 616 K and τ0 = 1.34 × 10−10 s for 6, and 666 K and τ0 = 2.22 × 10−11 s for 7. For the three derivatives a butterfly-shape hysteresis is observed at 2 K in accordance with the combination of Orbach and QTM magnetic relaxation processes.31 The strong shift in redox potentials has no effect on the magnetic properties and the nature of the substituent only slightly acted on the QTM regime.
2.3 Tetrathiafulvalene based single-molecule magnets
The tetrathiafulvalene (TTF) derivatives are probably one of the most expanded and used redox-active classes of compounds. They have been intensively employed to design molecular conductors and superconductors.32 One of their advantages is the fact that they can be easily decorated with one to four coordinated groups33 to design d–π or f–π systems. Such a synthetic strategy allowed the combination of both electro-activity and magnetic/luminescence properties.34J.-L. Zuo's group is very active in the field of electro-active lanthanide SMM. In 2013, they exploited the ability of a π-conjugated TTF-Schiff base ligand (L7 = 2,2′-((2-(4,5-bis(methylthio)-1,3-dithiol-2-ylidene)-1,3-benzodithiole-5,6-diyl)bis-(nitrilomethylidyne)bis(4-chlorophenolate))35 to coordinate lanthanides36 for obtaining a redox-active field-induced SMM.37 The X-ray structure showed that the system [(L7)Dy(LOEt)] (8) could be described as a mononuclear complex in which the Dy(III) ion is coordinated in the N2O2 pocket of the TTF-fused Schiff base ligand and capped by the tripodal metallo complex LOEt− (where LOEt− = [(η5-C5H5)Co(P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OEt)2)3]−) resulting in a N2O5 seven-coordinated Dy(III) centre (Fig. 3).
O)(OEt)2)3]−) resulting in a N2O5 seven-coordinated Dy(III) centre (Fig. 3).
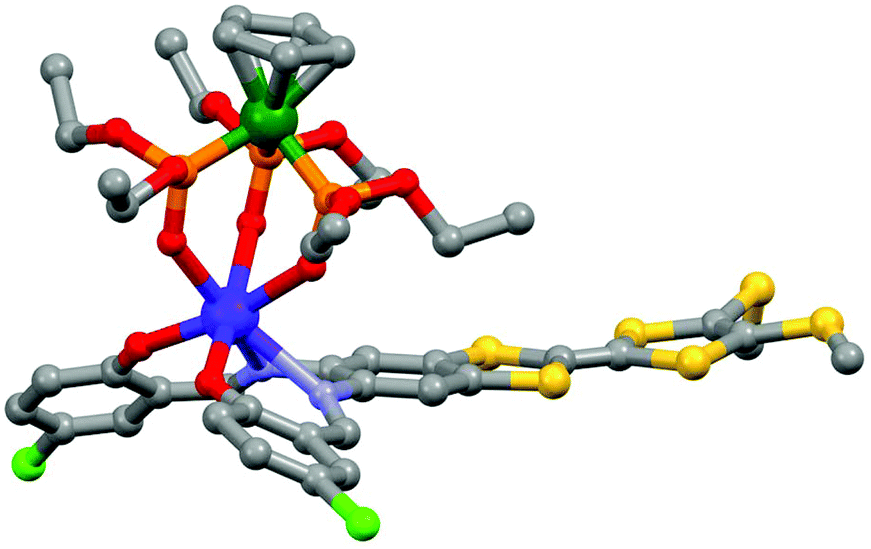 | ||
| Fig. 3 Molecular structure of 8. Hydrogen atoms and solvent of crystallization are omitted for clarity. Colour code: carbon: grey, nitrogen: light blue, phosphorus: orange, oxygen: red, sulphur: yellow, chloride: green, cobalt: dark green and dysprosium: blue. Adapted from ref. 37. | ||
The electrochemical properties of 8 exhibited two reversible single-electron oxidation waves localized at 0.87 V and 1.19 V vs. SCE corresponding respectively to the formation of the radical cationic and dicationic species as shown in Fig. 4a for the TTF core. These potentials are slightly cathodically shifted compared to the free ligand L7 (0.91 V and 1.25 V) which is the opposite trend to that expected through coordination. The authors attributed this negative shift to the competitive deprotonation effect (from phenol to phenolate) associated with the lanthanide coordination. The magnetization dynamic of 8 was investigated by alternating current magnetic measurements. Due to the efficient QTM, no maxima of the out-of-phase component of the magnetic susceptibility were observed and an optimal field of 4000 Oe was applied leading to thermally activated magnetic relaxation processes.
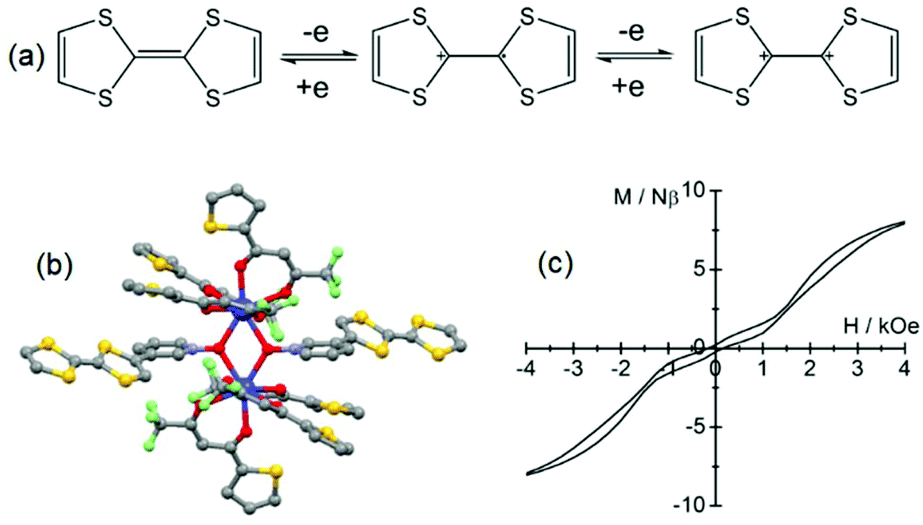 | ||
| Fig. 4 (a) Electrochemical processes for the TTF fragment. (b) Molecular structure of 9. (c) Hysteresis loop of 9 at 0.5 K. Colour code: fluoride: light green and sulphur: yellow. Adapted from ref. 37 and 38. | ||
The corresponding curve obtained by the Arrhenius law was fitted with an energy barrier of Δ = 41.6 K and τ0 = 9.2 × 10−8 s and 8 can be described as a field-induced electro-active single-ion magnet (SIM). The first observation of slow magnetic relaxation in a lanthanide coordination complex involving a TTF-based ligand was made two years earlier38 on a dinuclear complex in which significant magnetic interaction could operate. It is formed by the bridging of two Dy(tta)3 units (tta− = 2-thenoyltrifluoroacetonate) with two tetrathiafulvalene-3-pyridine-N-oxide ligands (L8). In the resulting complex [Dy(tta)3(L8)2]·0.5CH2Cl2 (9, Fig. 4b), the two Dy(III) ions are in an O8 square antiprismatic coordination environment. As expected, the cyclic voltammogram displayed two one-electron oxidation waves at potentials of 0.46 V and 0.88 V vs. SCE. 9 behaved as an SMM with frequency dependence of the magnetic susceptibility below 20 K. The temperature dependence of the relaxation time showed a thermally activated regime above 10 K which is characterized by Δ = 87(1) K and τ0 = 5.5 × 10−7 s and a deviation from linearity due to a non-thermally activated regime below 10 K with saturation occurring within 10 s. A double-butterfly shape hysteresis loop was observed at 2 K and 0.5 K (Fig. 4c).39 The fast magnetic relaxation at H = 0 Oe is due to QTM while at 1600 Oe it is due to the crossing of the antiferromagnetically coupled energy levels which opens a new tunnelling of magnetization. Consequently the 1600 Oe field value is directly connected to the intensity of the magnetic interactions between the two Dy(III) ions. It is worth noting that due to the antiferromagnetic interactions, the magnetic relaxation did not take place in the non-magnetic ground state but within the excited states. Such magnetic behaviour associated with this kind of hysteresis loop was also observed for other antiferromagnetically coupled Dy(III) ions in dinuclear SMMs.40
Changing the interaction from antiferromagnetic to ferromagnetic is of great interest to stabilize a magnetic ground state.
Such change in the nature of magnetic interaction for TTF-based SMMs was achieved by associating the two 4,5-bis(thiomethyl)-4′-carboxylictetrathiafulvalene (L9) and 4,5-bis(thiomethyl)-4′-ortho-pyridyl-N-oxide-carbamoyltetrathiafulvalene (L10) ligands with Ln(tta)3(H2O)2 units.41 Such association led to the formation of dinuclear complexes of formula [Ln(tta)2(L9)(L10)]2·2CH2Cl2 (Ln = Dy(III) (10) and Yb(III) (11)) in which the two lanthanide centres adopt a D2d O8 coordination sphere and are bridged by two μ(η1,η2) L9 while L10 is in a terminal coordination mode (Fig. 5a). Cyclic voltammetry of 10 (Fig. 5b) and 11 showed two single electron waves (at 0.42 V and 0.57 V vs. SCE) corresponding respectively to the formation of the radical cation form of L9 and L10 and one double electron oxidation (0.89 V vs. SCE) wave attributed to the simultaneous formation of the dicationic form of L9 and L10. The μ(η1,η2) coordination mode of L9 is suitable for observing magnetic interactions between the two lanthanide ions as shown in Fig. 5c. In fact the thermal variation of the χMT product increases at low temperature, a clear sign of significant ferromagnetic interactions. The Hamiltonian depicted in eqn (2) can be used to quantify both the crystal field effect through the extended Stevens operators’ technique and the intramolecular magnetic interaction:
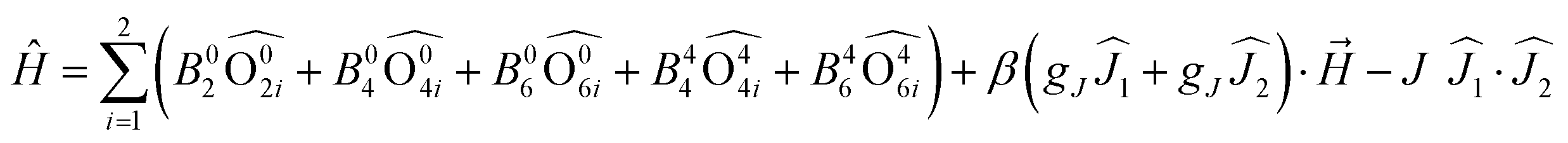 | (2) |
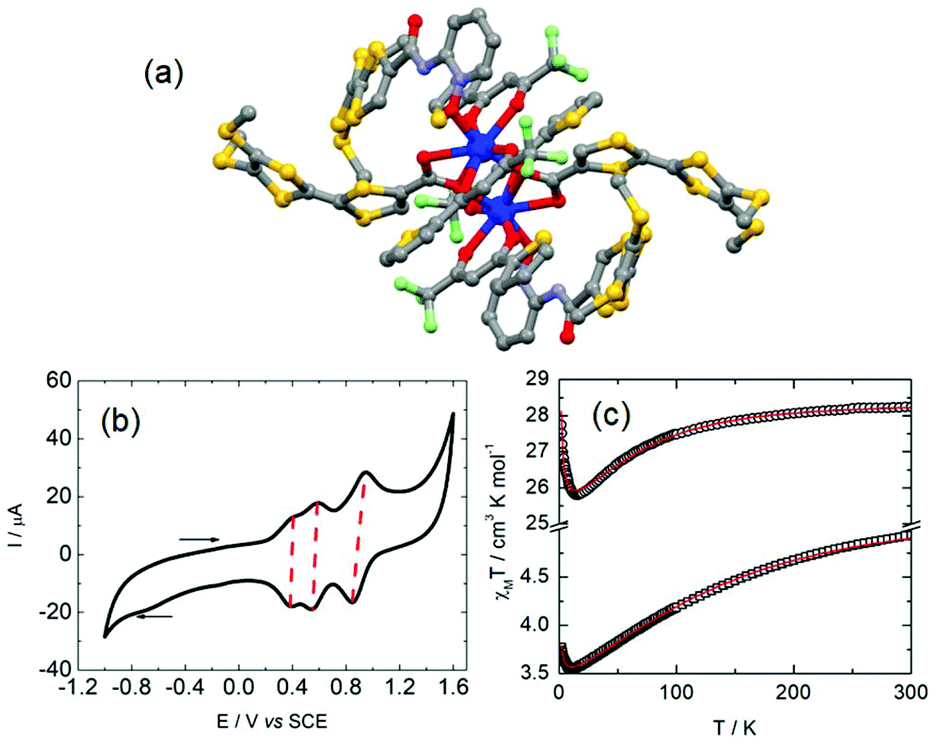 | ||
| Fig. 5 (a) Molecular structure of 11. (b) Cyclic voltammograms of 10. (c) Thermal dependence of the χMT products for 10 (circles) and 11 (squares) with best fit (red lines, see main text for details). Adapted from ref. 41. | ||
The best fit (Fig. 5c) for 10 was obtained with gJ = 4/3 (fixed), J = 4.29 × 10−3 K, B(0,2) = 2.32 K, B(0,4) = −5.66 × 10−3 K, B(4,4) = 4.44 × 10−2 K, B(0,6) = −3.46 × 10−6 K and B(4,6) = 1.03 × 10−4 K. The ground state is described by MJ = ±15/2 (99.9%) with an almost pure first excited state MJ = ±13/2 (95%) lying at 223 K. A simplified model considering a D4d symmetry and negligible magnetic interaction was used to obtain the best fit for 11 (Fig. 5c). The dynamic magnetic measurements revealed an SMM behaviour in zero applied magnetic field for 10 with Δ = 29 K and τ0 = 8.6 × 10−6 s while 11 is a field-induced SMM at 2000 Oe with Δ = 21 K and τ0 = 1.7 × 10−6 s. The latter ytterbium compound is remarkable because the energy splitting of the 2F7/2 multiplet state obtained from the magnetic properties could be compared to the Yb(III) luminescence which is experimental proof of such a splitting. 11 is one of the rare examples of redox active luminescent Yb(III) SMMs for which a magneto-structural correlation was done.42
A few years later, we reported another redox-active luminescent Yb(III) SMM.43 It was obtained by the association of the 4,5-ethylenedioxy-4′,5′-bis(2-pyridyl-N-oxidemethylthio)tetrathiafulvalene ligand (L11) and the Yb(tta)3·2H2O unit. The resulting mononuclear complex [Yb(tta)3(L11)]·2CH2Cl2 (12) presents redox-activity with reversible single-electron oxidation potentials at 0.42 V and 0.87 V vs. SCE. Since this system crystallised in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, rotating single crystal magnetometry was performed giving access to the g factors (gx = 5.84, gy = 2.00 and gz = 1.77) in the effective spin ½ framework. Ab initio calculations at the MS-CASPT2/RASSI-SO level corroborated the experimental values (gx = 5.96, gy = 0.73 and gz = 0.33). The resulting ground state is composed of a mixing of MJ = ±5/2 (50%) and MJ = ±7/2 (34%) which are well separated from the excited states MJ = ±7/2 (337 K), MJ = ±3/2 (429 K) and MJ = ±1/2 (672 K). Light irradiation at 22
space group, rotating single crystal magnetometry was performed giving access to the g factors (gx = 5.84, gy = 2.00 and gz = 1.77) in the effective spin ½ framework. Ab initio calculations at the MS-CASPT2/RASSI-SO level corroborated the experimental values (gx = 5.96, gy = 0.73 and gz = 0.33). The resulting ground state is composed of a mixing of MJ = ±5/2 (50%) and MJ = ±7/2 (34%) which are well separated from the excited states MJ = ±7/2 (337 K), MJ = ±3/2 (429 K) and MJ = ±1/2 (672 K). Light irradiation at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 of 12 led to an efficient sensitisation of the NIR Yb(III) emission composed of four contributions (0 K, 337 K, 432 K and 702 K) perfectly in agreement with the energy splitting given by the ab initio calculations. While 12 did not show any out-of-phase signal in a zero applied magnetic field (in agreement with the 50% MJ = ±5/2 doublet), under 1000 Oe field it showed frequency dependence of the magnetic susceptibility below 4 K. The temperature dependence of relaxation was fitted by an Orbach thermally activated process with Δ = 6 K and τ0 = 1.9 × 10−5 s.
000 cm−1 of 12 led to an efficient sensitisation of the NIR Yb(III) emission composed of four contributions (0 K, 337 K, 432 K and 702 K) perfectly in agreement with the energy splitting given by the ab initio calculations. While 12 did not show any out-of-phase signal in a zero applied magnetic field (in agreement with the 50% MJ = ±5/2 doublet), under 1000 Oe field it showed frequency dependence of the magnetic susceptibility below 4 K. The temperature dependence of relaxation was fitted by an Orbach thermally activated process with Δ = 6 K and τ0 = 1.9 × 10−5 s.
In previous lines, it was shown that redox-active lanthanide SMMs can be designed using various redox-active ligands such as tetraoxolene (section 2.1), phthalocyanine (section 2.2) and tetrathiafulvalene (section 2.3). Another strategy is to employ dyes (D–A)44 or triads (A–D–A)45 in which both donor (D) and acceptor (A) are electroactive. Thus in 2012, the 4,4′,7,7′-tetra-tert-butyl-2,2′-bi-1,3-benzo-dithiole-5,5′,6,6′-tetrone (L12) A–D–A triad46 was used as a connector between two Dy(hfac)3(H2O) terminal units to form the dinuclear compound [Dy2(hfac)6(H2O)2(L12)] (13) (Fig. 6).
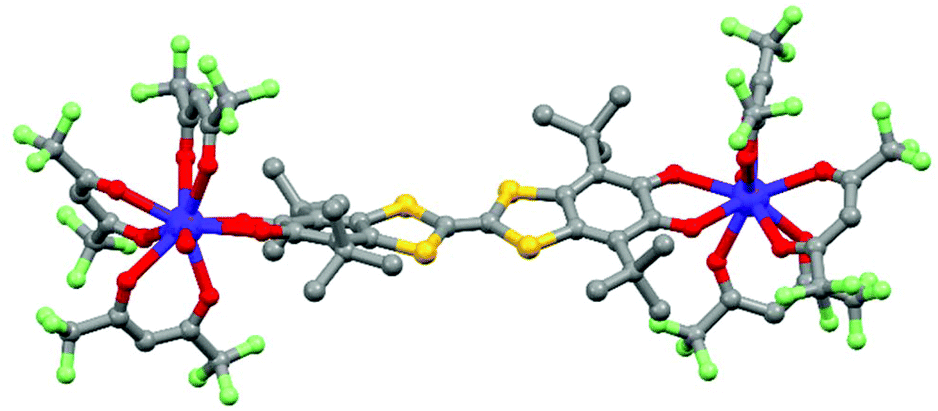 | ||
| Fig. 6 Molecular structure of 13. Hydrogen atoms are omitted for clarity. | ||
The triad L12 displayed a complex cyclic voltammogram with four reversible single-electron reduction waves at −0.40, −0.61, −1.04 and −2.56 V vs. Hg/HgCl2 in DMF and two oxidation waves at 1.62 V and 1.82 V vs. Hg/HgCl2 in chlorobenzene/MeCN. The four distinct reduction waves were attributed to the reduction of the two o-quinone acceptors. They are a good indication of an efficient electronic communication through the fused TTF. The two oxidation waves were attributed to the formation of the radical cationic and dicationic TTF species. Their large anodic shift is ascribed to the strong electron-withdrawing effect of the fused o-quinone acceptors. This multi electro-active compound behaved as an SMM in zero applied magnetic field with magnetic relaxation occurring through QTM (τQTM = 5.5 × 10−4 s) and Orbach (Δ = 15 K and τ0 = 2.4 × 10−6 s) regimes.
Very recently, a semiquinone acceptor instead of quinone was stabilised in a crystalline solid state by replacing the TTF donor with a stronger donor i.e. a p-phenylene-extended TTF.47 The resulting 2,2′-benzene-1,4-diylbis(6-hydroxy-4,7-di-tert-butyl-1,3-benzodithiol-2-ylium-5-olate) biradical triad (L13) presented three one-electron reduction waves at −0.43 V, −0.57 V and −0.89 V vs. SCE and four one-oxidation waves at 0.88 V, 1.10 V, 1.60 V and 1.80 V vs. SCE.48 The first two reduction processes were attributed to the two consecutive reductions from protonated semiquinone to catechol while the third reduction process was attributed to the reduction of the central p-quinoid spacer. The first two oxidation waves were attributed to the successive oxidation from protonated semiquinone to quinone moieties while the two highest oxidation potentials were attributed to the formation of the radical cationic and di-cationic TTF species. The multi-redox-active triad L13 can be involved in the formation of a one-dimensional polymer when it reacted with the Dy(hfac)3 precursor. The resulting polymer {[Dy(hfac)3(L13)]·2C6H14}n (14) could be described as an auto-assembly of field-induced SIM with an energy barrier of 20 K and τ0 = 2.1 × 10−5 s.49
Multi-redox-active lanthanide SMMs can be achieved combining two different electro-active ligands in the coordination sphere of the metal. For example J.-L. Zuo and collaborators designed a triple-decker sandwich dysprosium complex with both Pc and the Schiff base fused TTF (L7) ligands.50 The electrochemical properties of [Dy2(Pc)(L7)2(CH3OH)]·5(ClCH2CH2Cl) (15) were driven in the oxidation region showing the reversibility of the oxidation of L7 but no data on the electro-activity of the Pc were given. 15 displayed a field-induced SMM at 3000 Oe with Δ = 29 K and τ0 = 3.6 × 10−6 s.
Section 2 demonstrated that lanthanide SMMs can be achieved with reversible oxidation or/and reduction of the electro-active ligand(s). Nevertheless until this point of the review, no oxidized or reduced species were isolated in the solid state. The last example of this section relates, to the best of our knowledge, the unique example of a lanthanide coordination complex involving the radical cation oxidation state of the TTF-based ligand and SMM behaviour. This complex was isolated by the galvanostatic oxidation of a mixture of 4,5-bis(3-pyridyl-N-oxidemethylthio)-4′,5′-methyldithio-tetrathiafulvalene ligand (L14) and Dy(hfac)3·2H2O in the presence of the TBASO3CF3 electrolyte in CH2Cl2.51 This technique allowed the isolation of single crystals of a dinuclear sample of formula [Dy(hfac)2(SO3CF3)((L14)˙+)]2(CF3SO3)2·2CH2Cl2 (16) (Fig. 7). The X-ray structure of 16 consisted of a dinuclear complex in which the two Dy(III) ions are bridged by two 3-Py-N-oxide arms. The fully oxygenated coordination sphere is completed by one terminal 3-Py-N-oxide arm, two hfac− anions and one sulfonate anion. Two non-coordinated sulfonate anions were identified leading to a radical cation oxidation state for the two L14 ligands to assume the electroneutrality of the sample. The radical cation form of L14 was confirmed by both EPR measurements on a single crystal (g = 2.00798)52 and absorption spectroscopy with the appearance of an intra-ligand charge transfer band at very low energy.5316 behaved as a field-induced SMM at 800 Oe with Δ = 12 K. Unfortunately, the crystal packing did not allow electronic conductivity and 16 was described as an insulator.
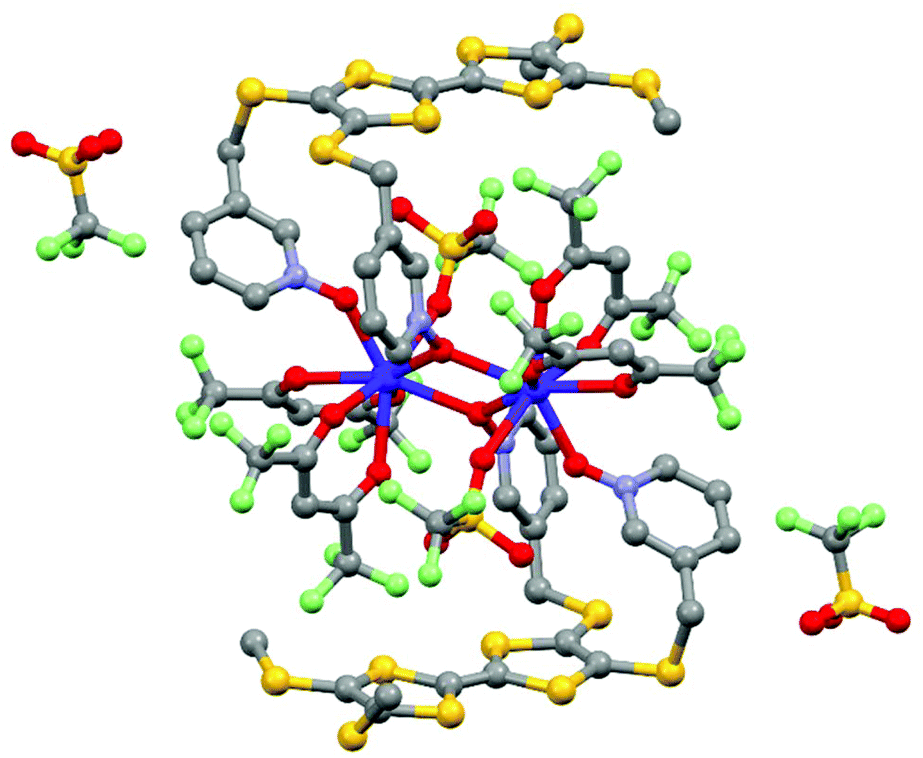 | ||
| Fig. 7 Molecular structure of 16. Hydrogen atoms and solvent molecule of crystallization are omitted for clarity. Adapted from ref. 51. | ||
3. Redox switching of single-molecule magnet behaviour
The aim of section 3 is to show the readers how the magnetic properties i.e. the SMM behaviour can be modulated in function of the oxidation state of the ligand or the metal.3.1 Ligand-centred redox-activity
The first examples presented in section 2 were based on the tetraoxolene ligand (complexes 1 and 2) but the SMM behaviour was studied only for the closed-shell bridges.54 Interest for studying open-shell ligand-based lanthanide compounds comes from the high-performance of the N23− radical-bridged lanthanide dinuclear complexes in which the exchange interactions vanished the efficiency of the QTM giving rise to a blocking temperature as high as 14 K.55 Then several lanthanide SMMs were successfully obtained with the implication of radical bridges.56 The first example of lanthanide complexes involving a tetraoxolene radical was published in 2018 by J. van Slageren and collaborators.57 It was obtained by the chemical reduction of 1 with cobaltocene leading to the dinuclear complex (1˙−)(CoCp2) in which tetraoxolene is in its tri-anionic radical form. While 1 did not show any out-of phase component of the magnetic susceptibility under a zero applied magnetic field, its reduced form (1˙−)(CoCp2) did (Table 1). The modification of the magnetic properties was mainly attributed to the presence of ferromagnetic interactions between the radical bridge and the Dy(III) ion (determined equal to 6 K in the Gd(III) analogue). In other words, the SMM behaviour in the zero magnetic field of 1 was switched on after single-electron reduction.| Stimuli | Compounds | SMM behaviour | Orbach parameters | Origin of the magnetic modulation | Ref. |
|---|---|---|---|---|---|
| Ligand-centered oxidation–reduction | 1 | OFF | 57 | ||
| ON (H = 1000 Oe) | Δ = 39 K, τ0 = 5.46 × 10−6 s | ||||
| 1(CoCp2) | ON | ||||
| ON (H = 1000 Oe) | Δ = 31 K, τ0 = 7.39 × 10−9 s | Magnetic interaction | |||
| 17 | OFF | — | 58 | ||
| (17)(CoCp2) | ON | Δ = 12.4 K, τ0 = 1.68 × 10−5 s | Magnetic interaction | ||
| (18)(n-Bu4N) | ON | Δ = 728 K | 8-Coordination | 60 | |
| 18[SbCl6] | ON | Δ = 791 K | Compressed 8-coordination | ||
| Metal-centered oxidation–reduction | 19 | ON (H = 1200 Oe) | Δ = 32.8 K, τ0 = 1.15 × 10−7 s | — | 63 |
| (19)(BF4) | ON (H = 800 Oe) | Δ = 43.5 K, τ0 = 0.46 × 10−7 s | |||
| 20 | ON | Δ = 39.4 K, τ0 = 1.63 × 10−6 s | Lower symmetry + magnetic interaction | 67 | |
| (20)K(THF)5 | OFF | — | |||
| 21 | OFF | — | |||
| (21)K(THF)5 | ON (H = 500 Oe) | Δ = 41.7 K | |||
| Solvation–desolvation | 22 | OFF | — | 7-Coordination | 72 |
| 23 | ON | Δ = 65.7 K, τ0 = 3.10 × 10−5 s | 6-Coordination | ||
| (24)(MeOH)·3MeOH | ON | Δ = 439 K | D 5h coordination symmetry | 73 | |
| 24 | OFF | — | O h coordination symmetry | ||
| 25 | OFF | — | 79 | ||
| 26 | ON | Δ = 45.9 K, τ0 = 2.43 × 10−6 s | |||
| (27)·2DMF | ON | No maxima for χ′′ | 80 | ||
| (27)·2CH3CN | ON | Δ = 97 K, τ0 = 1.81 × 10−11 s | Dipolar interaction | ||
| (28)·2C6H6 | ON | No maxima for χ′′ | 81 | ||
| (28)·2CH2Cl2 | ON | Δ = 49.4 K, τ0 = 2.55 × 10−9 s | Exchange interaction | ||
| Photo-irradiation | 29 | ON | Δ = 23.8 K, τ0 = 2.39 × 10−7 s | 88 | |
| 29 after irradiation | ON | No change | |||
| 30 | ON | Δ = 22.0 K, τ0 = 2.11 × 10−7 s | Dipolar interaction | ||
| 30 after irradiation | ON | No maxima for χ′′ | |||
| 31 | ON (H = 1500 Oe) | Δ = 14.2 K, τ0 = 1.90 × 10−8 s | 89 | ||
| 31 after irradiation | ON (H = 1500 Oe) | Δ = 14.7 K, τ0 = 7.25 × 10−9 s | |||
| 32 | ON | Δ = 14.5 K, τ0 = 5.33 × 10−6 s | 90 | ||
| 32 after irradiation | ON | No maxima for χ′′ | |||
| o-33 | ON | Δ = 19.1 K, τ0 = 5.20 × 10−7 s | D 2d coordination symmetry | 91 | |
| c-33 | ON | Δ = 28.3 K, τ0 = 1.02 × 10−6 s | D 4d coordination symmetry | ||
| 34@Y | ON | Δ = 55.1 K, τ0 = 9.81 × 10−9 s | 94 | ||
| 34@Y | OFF | — | |||
| trans-36 | ON | Δ = 35.4 K, τ0 = 4.41 × 10−6 s | 101 | ||
| cis-36 | ON | Δ = 32.9 K, τ0 = 5.76 × 10−7 s | |||
| 37 | OFF | — | 8-Coordination | 102 | |
| 37-UV | ON | Δ = 47.3 K, τ0 = 1.26 × 10−7 s | 9-Coordination | ||
| Protonation–deprotonation | H2-38 | ON (H = 1000 Oe) | Δ = 70.6 K, τ0 = 1.40 × 10−4 s | 105 | |
| H-38 | ON (H = 1000 Oe) | Δ = 56.1 K, τ0 = 1.25 × 10−8 s | |||
| 38 | ON (H = 0 Oe and H = 1000 Oe) | Δ = 34.7 K, τ0 = 2.51 × 10−7 s | |||
| — | |||||
| 39 | OFF | 7-Coordination | 106 | ||
| 40 | ON | Δ = 387.1 K, τ0 = 1.6 × 10−11 s | 8-Coordination | ||
Similar modulation of the SMM behaviour due to the reduction of a closed-shell ligand to a radical ligand was observed for the first time a year before by the group of K. R. Dunbar.58 3,6-Bis(2-pyridyl)-1,2,4,5-tetrazine (L15) was selected for its easily accessible reduction potential and to bridge two [Dy(tmhd)3] units (tmhd = 2,2,6,6-tetramethyl-3,5-heptane dionate) leading to the [(Dy(tmhd)3)2(L15)] (17) dinuclear compound. 17 displayed a thermally independent out-of-phase signal of magnetic susceptibility which has been attributed to magnetic relaxation through a QTM process. After the single reduction of 17 with Cp2Co, the resulting [Cp2Co][(Dy(tmhd)3)2(L15)˙−] (Cp2Co)(17) displayed a frequency dependence of the χ′′M component allowing the extraction of the energy barrier Δ = 12.4 K and time constant τ = 1.68 × 10−5 s for an Orbach process (Table 1). The presence of S = ½ centred on the bridging ligand led to magnetic interactions with the two Dy(III) ions that efficiently suppress the QTM observed for 17.
The most known redox-active ligands in molecular magnetism are the phthalocyanine (Pc) derivatives since the [PcTb]− anion was the first mononuclear lanthanide SMM reported. The Pc ligand can be easily reduced and oxidized as shown in section 2.2. Thus Ishikawa and collaborators compared the magnetic properties of the [(L16)2Tb]− anion (18)[nBu4N] (L16 = 2,3,9,10,16,17,23,24-octaethoxyphthalocyanine) with those of the [(L16)2Tb]+ cation (18)[SbCl6], both obtained from the reduction and oxidation of the neutral [(L16)2Tb],59 respectively.60 While the reduced form of 18 showed temperature dependence of the χ′′M component until 40 K, the oxidised form displayed it until 52 K.
Consequently the energy barrier of (18)[SbCl6] is found to be 8% greater than the one of (18)[nBu4N] (Δ = 791 K) (Table 1). This difference in the energy barrier was attributed to the compression of the coordination sphere after oxidation of the L16 ligand leading to a stronger crystal field.
Few years later, J. Veciana and collaborators probed the magnetic bistability of the three stable oxidation states of the same derivative (Fig. 8).61 For ease of isolation and stability of the samples, it appeared that magnetic circular dichroism (MCD) spectroscopy was the most powerful tool to probe the magnetic hysteresis, as already demonstrated for Mn12 SMMs.62 Both reduced and oxidised forms displayed butterfly-shape hysteresis with a greater coercive field for (18)+ than (18)− (Fig. 8) in agreement with what was observed in alternating current magnetic measurements. The neutral derivative (18) displayed an open hysteresis loop which remains open in the zero magnetic field (Fig. 8) due to a less efficient QTM. The observation of coercive field at H = 0 Oe is probably due to the association of S = ½ with the Tb(III) ion.
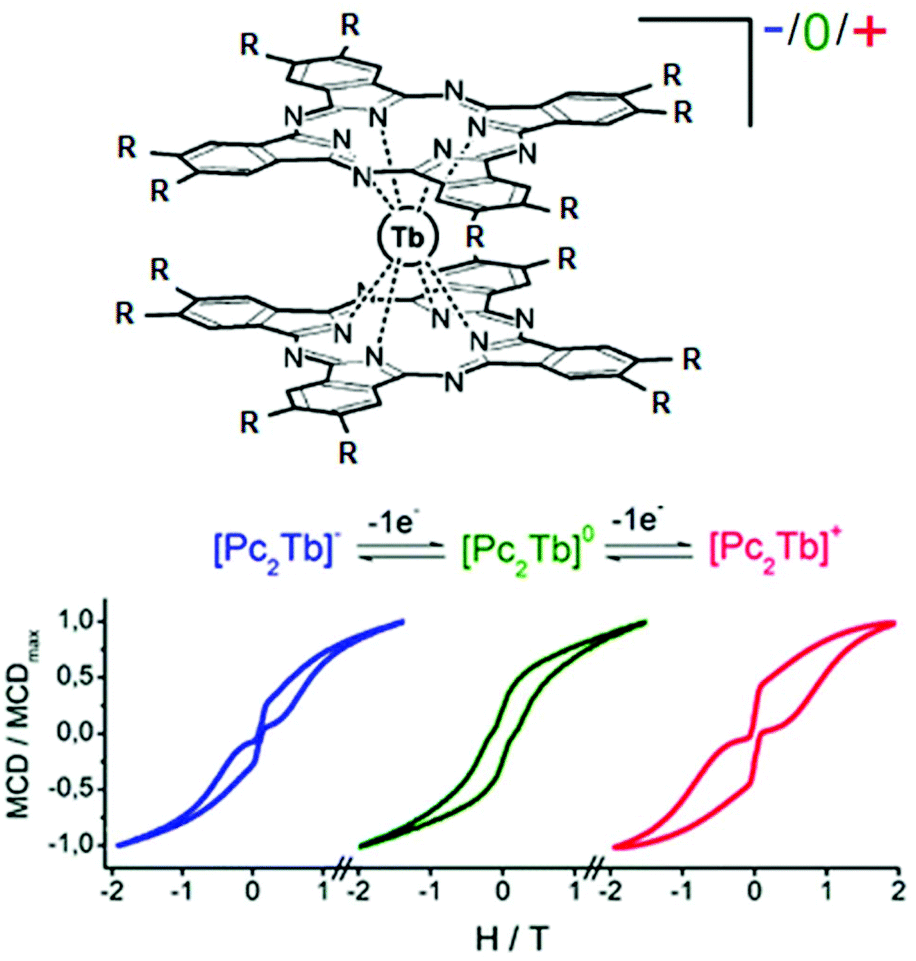 | ||
| Fig. 8 Molecular structure of 18. Hydrogen atoms and solvent molecule of crystallization are omitted for clarity. Adapted from ref. 60 and 61. | ||
3.2 Metal-centred redox-activity
The redox activity can also be carried by a 3d or 4d transition metal associated with the lanthanide. One example was obtained by the association of a ruthenium carbon-rich complex with the Dy(tta)3 unit.63 The ruthenium moiety brings the redox activity and it has already proved its ability to switch conductivity,64 non-linear optics65 or luminescence.66 It was decorated with 2,2′-bipyridine to coordinate the lanthanide centre. The cyclic voltammogram of trans-[(PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(dppe)2RuCC-bipyridyl)(Dy(tta)3)] (19) (dppe = 1,2-bis(diphenylphosphino)ethane) showed a reversible single-electron process at 0.062 V vs. Fc/Fc+ allowing the oxidation of Ru(II) in Ru(III) in the presence of acetylferrocenium tetrafluoroborate at −60 °C under argon leading to (19)(BF4). Both neutral 19 and radical cations (19)(BF4) behaved as field-induced SMMs with magnetic relaxation through an Orbach process which is characterized by Δ = 32.8 K and τ0 = 1.15 × 10−7 s (H = 1200 Oe) and Δ = 43.5 K and τ0 = 0.46 × 10−7 s (H = 800 Oe) respectively. The oxidation of the ruthenium carbon-rich complex induced an increase of the energy barrier of 30% and the magnetic relaxation of (19)(BF4) was measured forty times slower than 19 at 2 K (Table 1). The enhancement of the magnetic performances should be attributed to both the S = ½ carried by the ruthenium carbon-rich moiety and the structural changes around the Dy(III) ion after oxidation. The role of the intermolecular interaction was not evaluated for this system. It is worth noting that the reversibility of oxidation was proved by absorption spectroscopy.
C(dppe)2RuCC-bipyridyl)(Dy(tta)3)] (19) (dppe = 1,2-bis(diphenylphosphino)ethane) showed a reversible single-electron process at 0.062 V vs. Fc/Fc+ allowing the oxidation of Ru(II) in Ru(III) in the presence of acetylferrocenium tetrafluoroborate at −60 °C under argon leading to (19)(BF4). Both neutral 19 and radical cations (19)(BF4) behaved as field-induced SMMs with magnetic relaxation through an Orbach process which is characterized by Δ = 32.8 K and τ0 = 1.15 × 10−7 s (H = 1200 Oe) and Δ = 43.5 K and τ0 = 0.46 × 10−7 s (H = 800 Oe) respectively. The oxidation of the ruthenium carbon-rich complex induced an increase of the energy barrier of 30% and the magnetic relaxation of (19)(BF4) was measured forty times slower than 19 at 2 K (Table 1). The enhancement of the magnetic performances should be attributed to both the S = ½ carried by the ruthenium carbon-rich moiety and the structural changes around the Dy(III) ion after oxidation. The role of the intermolecular interaction was not evaluated for this system. It is worth noting that the reversibility of oxidation was proved by absorption spectroscopy.
The second example is made from the reaction between two equivalents of [K2(OEt2)]fc[NSi(t-Bu)Me2]2 and one equivalent of LnI3 leading to the trinuclear complexes K(THF)5[Ln(fc[NSi(t-Bu)Me2]2)2] (Ln(III) = Dy (20)K(THF)5 and Er (21)K(THF)5) (Fig. 8).67 These two compounds could be oxidised by iodine to generate the mixed valence Fe(III)/Fe(II) [Ln(fc[NSi(t-Bu)Me2]2)2] (Ln(III) = Dy (20) and Er (21)) (Fig. 9). Starting from the Dy(III) analogue, (20)K(THF)5 displayed a zero magnetic field SMM behaviour with a magnetic relaxation which followed QTM, Raman and Orbach processes. Eqn (1) was used to determine the parameters68 giving the best fit of the temperature dependence of the magnetic relaxation (τQTM = 5.03 × 10−4 s, C = 0.08 s−1 K−n with n = 5 and Δ = 39.3 K, τ0 = 1.63 × 10−6 s). Single oxidation of (20)K(THF)5 in 20 switched OFF the SMM behaviour in the zero applied magnetic field. Under an optimal applied magnetic field of 1000 Oe, both (20)K(THF)5 and 20 presented field-induced SMM behaviour but 20 highlighted a much faster magnetic relaxation than (20)K(THF)5. The magnetic relaxation involved a complex combination of relaxation processes i.e. direct, QTM, Raman and Orbach regimes.68 The energy barrier of (20)K(THF)5 was evaluated to be 66.2 K which is higher than that of the oxidised sample 20 (Δ = 39.1 K). Another difference is the presence of remaining QTM at 1000 Oe for 20 (τQTM = 8.77 × 10−4 s).
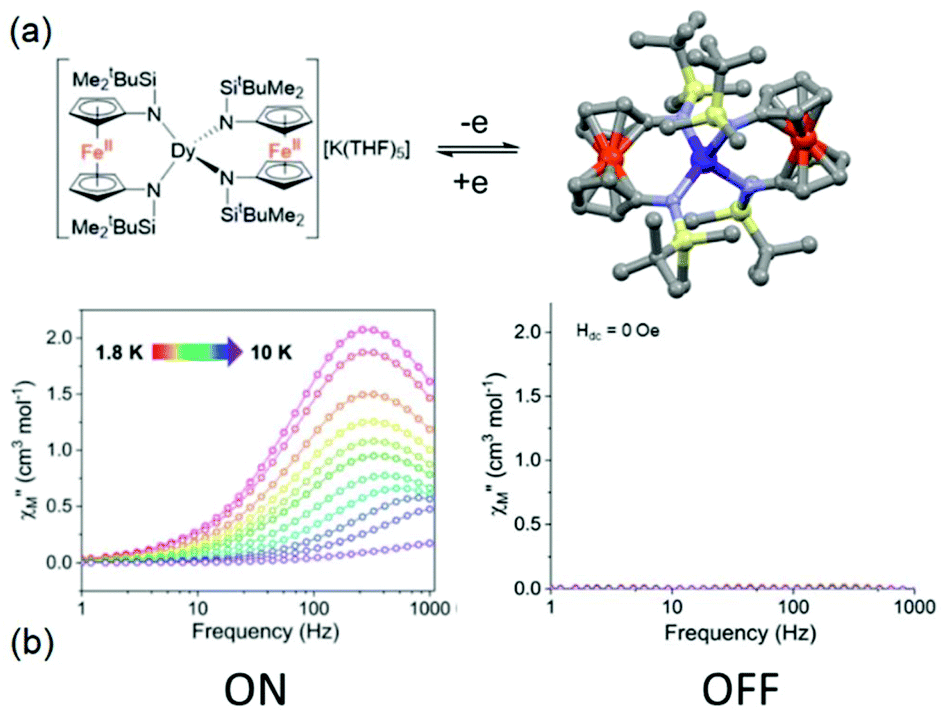 | ||
| Fig. 9 (a) Molecular structures of (20)K(THF)5 (left) and its oxidised form 20 (right). (b) Frequency dependence of the magnetic susceptibility for (20)K(THF)5 (left) and 20 (right) highlighting the ON and OFF SMM behaviour respectively. Colour code: silicon: light yellow and iron: orange. Adapted from ref. 67. | ||
Concerning the Er(III) analogue, any of the two (21)K(THF)5 and 21 samples behaving as SMMs in the zero magnetic field while applying a moderate field of 500 Oe to vanish the QTM allowed the observation of an out-of-phase component of the magnetic susceptibility for (21)K(THF)5 while 21 did not show any SMM behaviour. The dynamic parameters of the four processes involved in the magnetic relaxation were determined with among others an energy barrier of 41.7 K. As already observed for the Dy(III) analogue, the single-electron oxidation switched OFF the field-induced SMM for the Er(III) derivative. It is worth noting that this remains the unique example until now of redox-switching of an Er(III) SMM behaviour.
The authors attributed the significant deterioration of the magnetic performances after single electron oxidation to the faster QTM in 20/21 than for (20/21)K(THF)5 due to both the lower symmetry around the Ln(III) ion and the paramagnetic Fe(III) (S = ½). The strong electronic communication between the two Fe(II) and Fe(III) ions has been demonstrated by (i) a large comproportionation constant due to an important difference of Fe-centred oxidation potentials which is in agreement with a Robin and Day class II classification (mixed valence compound),69 (ii) Mössbauer spectroscopy because the Fe(III) site is quadrupole split instead of showing a singlet70 and (iii) UV-vis-NIR spectroscopy with the observation of an intervalence charge transfer transition at 1050 nm.71
4. Solvato switching of single-molecule magnet behaviour
Magnetic modifications can be induced by other stimuli such as solvation/desolvation, light irradiation, and protonation/deprotonation. The next sections will show the most representative examples of magnetic switches under these various stimuli.The year 2013 has been very rich on the topic of solvato-magnetic switches. The dinuclear compound {[Dy(H2O)2(CH3COCH3)]2(γ-SiW10O36)2}10− (22) in which the two Dy(III) ions were in a distorted monocapped trigonal prism can be reversibly transformed in the [Dy2(μ-OH)2(γ-SiW10O36)2]12− dinuclear compound (23) in the presence of the TBAOH base.72 In the latter compound, the two Dy(III) ions are bridged by two hydroxyl anions and now are in a distorted trigonal prism environment. The recovery of 22 from 23 was possible in the presence of HNO3 acid in acetone/acetonitrile/H2O. In the dynamic magnetic point of view, 22 did not show any significant SMM behaviour while 23 behaved as an SMM characterized by a thermally dependent regime at high temperature (Δ = 65.7 K and τ0 = 3.11 × 10−7 s) and QTM at low temperature. The Gd(III) analogue of 23 allowed the determination of the antiferromagnetic interaction between the lanthanide ions through the hydroxyl anion but since the diluted samples behaved as the non-diluted ones the authors concluded that the magnetic interaction plays a minor role in the magnetic switching between 22 and 23. Then the SMM switching was attributed to the change in coordination sphere symmetry when solvation/desolvation took place.
The second example of the same year was published by M.-L. Tong and collaborators. The ligand 2,2′,2′′-(((nitrile-tris(ethane-2,1-diyl))tris(azanediyl))tris(methylene))tris-(4-bromo-phenol) (L17) was used to react with both Zn(II) and Dy(III) (2:1) giving rise to [Zn2Dy(L17)2(MeOH)]NO3·3MeOH·H2O (24)(MeOH)·3MeOH (Fig. 10).73 Both coordinated and uncoordinated MeOH molecules can be removed by exposure to air in a single-crystal-to-single-crystal transformation giving the compound (24) (Fig. 10). (24)(MeOH)·3MeOH showed SMM behaviour without applied magnetic field while 24 did not. In the high-temperature regime, the relaxation of magnetization for 24 follows an Orbach process with Δ = 439 K in perfect agreement with the calculated ab initio value of 425 K.
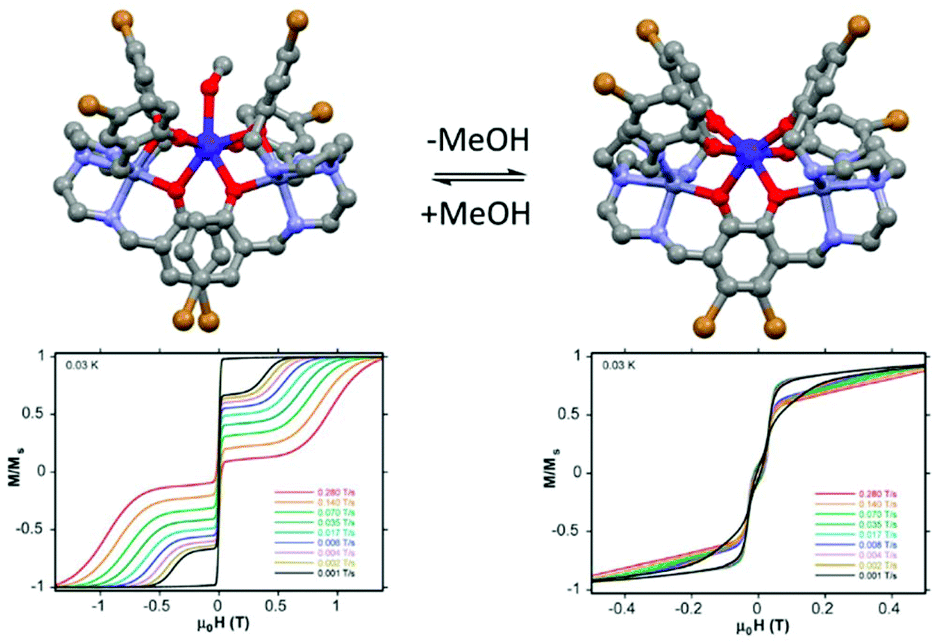 | ||
| Fig. 10 Molecular structures of (24)(MeOH)·3MeOH with its hysteresis loop at 0.03 K (left part) and its methanol free analogue (24) with its hysteresis loop at 0.03 K (right part). Colour code: bromide: brown. Adapted from ref. 73. | ||
The high energy barrier for (24)(MeOH)·3MeOH was attributed to both the quasi-D5h symmetry of the Dy(III) coordination environment and the two coordinating axial phenoxyl groups.74 In fact the computational approach showed that the ground state Kramers doublet in D5h symmetry is perfectly axial. After the single-crystal-to-single-crystal transformation the SMM behaviour is quenched due to the quasi-Oh symmetry of the Dy(III) coordination environment in 24 leading to an efficient QTM. Thus a magnetic field of 1200 Oe should be applied to observe an out-of-phase component of the magnetic susceptibility. The Arrhenius plot involved both direct and Raman processes while the Orbach process was determined to be negligible. Both previous similar examples in the literature75 and the discrepancy between the experimental (64 K) and calculated (>400 K) “energy barrier” are in agreement with a negligible Orbach contribution in the magnetic relaxation processes.76 Finally the difference in magnetic performances can be visualized by measuring the hysteresis loop at 0.03 K for single crystals of both (24)(MeOH)·3MeOH and 24 using a micro-SQUID magnetometer.77 While (24)(MeOH)·3MeOH showed an open hysteresis until 11 K, the remnant magnetization is negligible for 24 (Fig. 10).
The third example is based on a metal–organic framework (MOF) reported in 2006 by Qiu and collaborators.78 [Dy(L18)(H2O)]·DMF (25) (where H3L18 = 1,3,5-benzenetricarboxylic acid and DMF = N,N′-dimethylformamide) was obtained by mixing the nitrate salt of Dy(III) and H3L18 in DMF/EtOH followed by the successive addition of dibutylamine and nitric acid. The Dy(III) ion is seven-coordinated in a pentagonal-bipyramidal prismatic surrounding. The coordinated water and guest DMF molecules can be removed by calcination at 240 °C for 12 h in a single-crystal-to-single-crystal transformation. The resulting [Dy(L18)] (26) MOF revealed Dy(III) ions in a distorted trigonal prismatic environment after removing the coordinated water molecule. 25 can be reversibly recovered by single-crystal-to-single-crystal transformation by immersion of 26 in a H2O/DMF mixture.7925 did not show any SMM behaviour in zero and non-zero applied magnetic fields while after removing the guest solvent molecules, 26 behaved as an SMM in both zero and 1000 Oe applied magnetic fields. Both thermally dependent and independent magnetic relaxation processes were present. The high temperature Orbach regime fit gave an energy barrier of 45.9 K and τ0 = 2.43 × 10−6 s. The switch OFF of the SMM behaviour after desolvation was attributed to the change in the coordination surrounding symmetry.
In 2015, another system based on a MOF was published by J. R. Long and collaborators.80 This MOF of formula [Dy2(L19)4(NO3)2]·2DMF (27)·2DMF was elaborated by the solvothermal reaction of nitrate salt of Dy(III), L19 (L19 = isonicotinic acid N-oxide) in DMF. The X-ray structure determined from single-crystal revealed the formation of a 3D network in which the nods are formed by dinuclear Dy(III) units and the linkers between the nods are the L19 ligands. The pores are filled by the guest DMF molecules. (27)·2DMF could be converted into (27)·2CH3CN by single-crystal-to-single-crystal transformation soaking (27)·2DMF in CH3CN and vice versa. While (27)·2DMF displayed slow magnetic relaxation at very high frequencies (10 kHz) through a direct mechanism, (27)·2CH3CN displayed an SMM behaviour at much lower frequency through a multiple relaxation process. The plot of ln(τ) versus T−1 was fitted using eqn (2) with an Orbach regime at high temperature (Δ = 97 K and τ0 = 1.81 × 10−11 s) (Table 1), Raman regime at intermediate temperature (C = 0.0476 s−1 K−5.71, n = 5.71) and QTM at low temperature (τQTM−1 = 26.3 s−1). The most fundamental difference in the three previous examples of this section is that the guest solvent molecules are not coordinated to the Dy(III) centres and so the single-crystal-to-single-crystal transformation did not induce change in the nature of the lanthanide surrounding. In order to give an explanation to the observed magnetic modulation in function of the nature of the guest solvent molecules, the authors evaluated the dipolar interactions (which have been calculated much stronger than the exchange interactions) on the basis of ab initio calculations. Thus the tunnelling gap Δtun and dipolar applied magnetic field Hdip (Δtun = 1/2gx,yμBHdip) were found to be respectively equal to 10−3 K and 10−2 K, and 100 mT and 90 mT for (27)·2CH3CN (Jdip = 3.04 K) and (27)·2DMF (Jdip = 2.6 K) leading to magnetic relaxation 100 times slower for the former than for the latter (QTM ∼ Δtun2). This system is the first example of SMM behaviour modulation by tuning the dipolar interaction depending on the nature of the guest non-coordinated solvent molecules.
The second example of magnetic modulation by changing the lattice solvent nature was published in 2017 by J.-L. Zuo and collaborators.81 The benzene lattice of the [Er2(thd)4Pc]·2C6H6 (28)·2C6H6 dinuclear compound (Hthd = 2,2,6,6-tetramethylheptanedione) could be reversibly exchanged by immersion in dichloromethane to give [Er2(thd)4Pc]·2CH2Cl2 (28)·2CH2Cl2 by single-crystal-to-single-crystal transformation (Fig. 11).
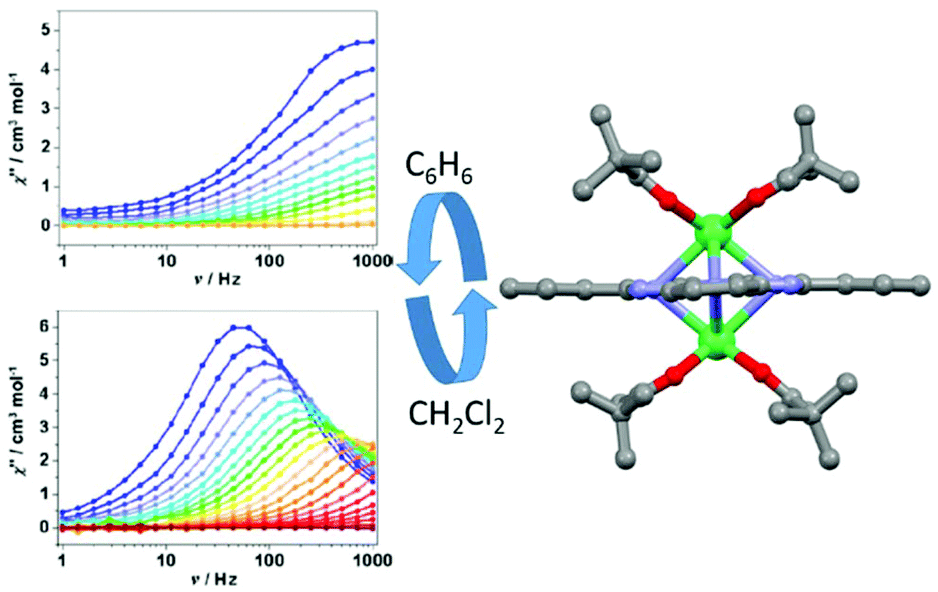 | ||
| Fig. 11 X-ray structure of complex 28 (right part) and out-of-phase component of magnetic susceptibility for both benzene (C6H6) and dichloromethane (CH2Cl2) solvent lattices. Colour code: erbium: green. Adapted from ref. 81. | ||
Except for the solvent lattice, the structure of the complex [Er2(thd)4Pc] is very similar in the two solvates. Ab initio calculations gave very similar axial anisotropy for both compounds. Nevertheless dynamic magnetic measurements clearly showed that magnetic relaxation for (28)·2C6H6 is faster than that for (28)·2CH2Cl2. In fact reasonable parameters could be obtained only for (28)·2CH2Cl2. The best fit of the thermal dependence of magnetic relaxation was obtained for a combination of Orbach (Δ = 49.4 K, τ0 = 2.55 × 10−9 s) (Table 1), direct (A = 6.01 × 10−4 s−2 Oe−2 with m = 2) and Raman (C = 1.88 s−1 K−n, n = 5.19) processes. The thermal variation of the magnetic susceptibilities was fitted using POLY-ANISO82 and considering the magnetic interaction as Jtotal = Jexchange + Jdipolar. The dipolar interactions were found to be ferromagnetic and very close in both solvates while the exchange interactions were found to be antiferromagnetic and stronger in (28)·2C6H6 than in (28)·2CH2Cl2. Consequently, Jtotal was calculated to be equal to 9.11 K and 2.92 K in (28)·2C6H6 and (28)·2CH2Cl2 respectively and used as the main reason for the drastic difference in magnetic relaxation.
5. Photo-switching of single-molecule magnet behaviour
Another efficient applied external stimulus is light irradiation when the systems incorporate photochromic moieties.5.1 Dithienylethene (DTE) based-photochromic ligands
One of the most famous photochromic moieties is undoubtedly dithienylethene (DTE). DTE derivatives are widely used because they are thermally stable and their fatigue-resistant performances are remarkable in both the solution and solid-state.83 UV and visible light irradiation led to the reversible isomerization of the DTE between ring-opened and ring-closed isomers, respectively.84 The DTE core can be functionalised by chemical groups able to coordinate metal ions.85One of the first examples incorporating the coordination of a DTE derivative with lanthanide ions was published in 2011 by M. Yamashita and M. Irie86 combining the well-known SMM [CuTb(L20)(NO3)3] (L20 = 1,3-bis((3-methoxysalicylidene)-amino)propane)87 and 1,2-bis(5-carboxyl-2-methyl-3-thienyl)perfluorocyclopentene (L21).88 Two different systems could be isolated depending on if the open form (o-L21) or closed form (c-L21) of the DTE ligand is used. o-L21 led to the formation of a tetranuclear ring-like structure of formula [Cu2Tb2(L20)2(o-L21)2(NO3)2]·2(n-BuOH) (29) while c-L21 led to the formation of a mono-dimensional ladder-type structure of formula {[(CuTb(L20)(n-BuOH)0.5)2(c-L21)3]·5DMF·4(n-BuOH)·2H2O)}n (30). The two compounds behaved as SMMs with an Orbach regime characterised by Δ = 23.8 K, τ0 = 2.39 × 10−7 s for 29 and Δ = 22.0 K, τ0 = 2.11 × 10−7 s for 30 (Table 1). The authors could demonstrate through absorption spectroscopy that the two compounds presented a photochromic activity of the o-L21 or c-L21 ligands under light irradiation. After irradiation, the ac magnetic behaviour of 29 did not change while 30 showed a shift of the χ′′M signal at higher frequencies. The modulation of the magnetic behaviour was attributed to the change in intermolecular interactions when the photochromic ligand is switched from one to the other form but unfortunately no X-ray structures of the systems after irradiation could be obtained.
Three years later, M. Yamashita and collaborators used the same ligand o-L21 to design a pure 4f system involving the most employed Dy(III) ion. A 2D-coordination network in which dinuclear Dy2-nodes are connected by a double o-L21 bridge in one direction and simple o-L21 bridge in the other direction was reported.89 The 2D polymer {[Dy2(L21)3(DMSO)3(MeOH)]·10MeOH}n (31) was exposed to UV irradiation (350 nm) and absorption spectroscopy undoubtedly showed the cyclisation of L21, nevertheless the diffraction pattern highlighted the loss of crystallinity. The cyclisation of L21 is reversible since the open form can be recovered by visible irradiation (480 nm) but powder X-ray diffraction clearly demonstrated that the structural transformation due to UV irradiation was not reversible. In a magnetic point of view, 31 behaved as a field-induced SMM (H = 1500 Oe) with a thermally dependent regime at high temperature characterized by Δ = 14.2 K and τ0 = 1.90 × 10−8 s and a remaining QTM at low temperature. After UV irradiation, the Orbach regime remained almost unchanged with Δ = 14.7 K and τ0 = 7.25 × 10−9 s while the QTM regime was affected since the system relaxed four times faster than before the irradiation. The authors attributed the changes of the QTM regime to geometrical variations in the Dy(III) surroundings after cyclisation of 31. After visible irradiation, the modulation of the magnetic behaviour was not significant. In other words, the closing and opening of L21 were reversible while the structural and so the magnetic modulations were not.
In 2015, the same group continued its investigation of photochromic SMMs based on the L21 ligand introducing the 2,2′-bipyridine (bpy) blocking ligand to induce a better crystal field around the Dy(III) centre. Once again a 2D coordination polymer was obtained with the formula [Dy2(L21)3(bpy)2(H2O)2]n (32).90 The Dy(III) ions lied in a N2O6 square anti-prism coordination sphere (D4d symmetry). 32 behaved as a SMM without applied magnetic field showing the effect of the introduction of the bpy ligand in the coordination sphere of the Dy(III) ion (Δ = 14.5 K, τ0 = 5.33 × 10−6 s and τQTM = 2.77 × 10−4 s) (Table 1). The QTM could be partially suppressed applying a field of 1000 Oe (Δ = 21.7 K, τ0 = 8.1 × 10−7 s and τQTM = 9.6 × 10−3 s). Absorption spectroscopy demonstrated that UV irradiation of 32 led to the cyclisation of L21 while visible irradiation led to its opening. Unfortunately this technique also showed the irreversibility of the structural changes because the π–π* of the bpy and intra-ligand transition for L21 were altered after visible irradiation compared to the starting 32 compound. After UV irradiation, the system still showed an out-of-phase signal of the magnetization but relaxed faster than before the irradiation and thus no maxima were observed. Under an applied field of 1000 Oe, the dynamic parameters could be extracted showing that the Orbach regime is only slightly modified while at low temperature the UV-irradiated sample relaxed faster than the starting sample.
Finally, the carboxylate groups of L21 were replaced by two 2,2′-bipyridine groups giving the new DTE derivative 1,2-bis(5-(2,2′-bipyridine)-2-methylthiophen-3-yl)cyclopent-1-ene (L22).91 The open form of this ligand (o-L22) was reacted with two equivalents of Dy(hfac)3·2H2O leading to the formation of a dinuclear complex of formula [(Dy(hfac)3)2(o-L22)] (o-33) (Fig. 12). The close form of the dinuclear complex was obtained after irradiation (385 nm) of the open form of the free ligand to give c-L22 that was reacted with two equivalents of Dy(hfac)3·2H2O leading to c-33. Since the cyclo-isomerisation is only active in solution, the conversion from o-33 to c-33 was not possible through single-crystal-to-single-crystal transformation.
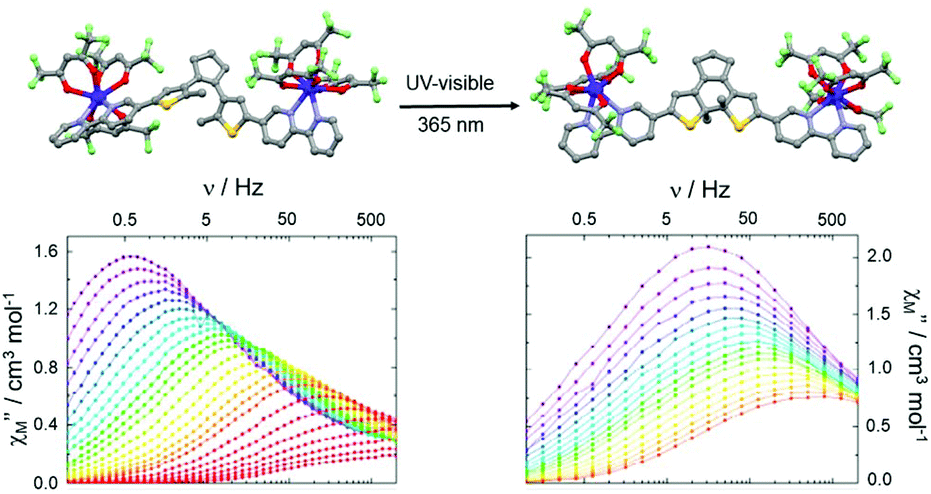 | ||
| Fig. 12 Molecular structures and out-of-phase component of magnetic susceptibility for o-33 (left) and c-33 (right) for the temperature range from 1.85 K to 10 K. Adapted from ref. 91. | ||
Both open and close forms displayed field-induced SMMs (H = 1000 Oe) as often observed in the case of the association between bipyridine derivatives or similar nitrogenated bischelating ligands and Dy(β-diketonate)3 unit.92 A combination of direct–Raman and direct–Orbach relaxation processes was determined for o-33 and c-33, respectively. In the bulk state, c-33 highlighted a slower magnetic relaxation than o-33 but the different coordination symmetries (D2d for o-33 and D4d for c-33) associated with the distinct crystal packing made the interpretation difficult. Thus the authors studied the dynamic magnetic properties of the two forms in frozen solution leading to an almost similar magnetic behaviour. In other words, the cyclo-isomerization of the photochromic ligand L22 induced only very small structural changes and thus the magnetic modulation remains weak at the molecular scale.
5.2 1,2-Bis(4-pyridyl)ethene (bpe) photo-reactive ligand
Other photoactive ligands can induce structural changes after irradiation. This is the case of the 1,2-bis(4-pyridyl)ethane ligand (L23) which is well-known to realize photochemical [2 + 2] cycloaddition with the formation of a cyclobutyl ring.93 Such photo-reaction was involved in the modulation of various physical properties but to the best of our knowledge only one article was devoted to SMM behaviour.94 The [Dy(L23)(H2O)4(NO3)2](NO3)·2L23 (34) compound can be converted into [Dy2(L24)(H2O)8(NO3)4](NO3)2·2L23·L24 (35) (where L24 = tetrakis(4-pyridyl)cyclobutane) by UV irradiation by a single-crystal-to-single-crystal transformation (Fig. 13). AC magnetic investigation concluded a SMM behaviour for 34 but with χ′′M maxima out from the experimental frequency window while 35 is not an SMM. The authors performed magnetic dilution to reduce the dipolar interactions, source of the efficiency of the QTM in these systems. 34@Y (dilution of 5.5% of 34 in a Y(III) matrix) showed clear SMM behaviour with an Orbach regime at high temperature (Δ = 55.1 K, τ0 = 9.81 × 10−9 s). To better cancel the QTM, a magnetic field of 300 Oe was applied leading, as expected, to a shift of χ′′M signals to a lower frequency range than those obtained in the zero magnetic field. Similar magnetic measurements were done for 35@Y (dilution of 11% of 35 in a Y(III) matrix). SMM behaviour was detected only under an applied magnetic field with the following dynamic magnetic parameters for the Orbach regime (Δ = 47.9 K, τ0 = 1.22 × 10−9 s) (Table 1).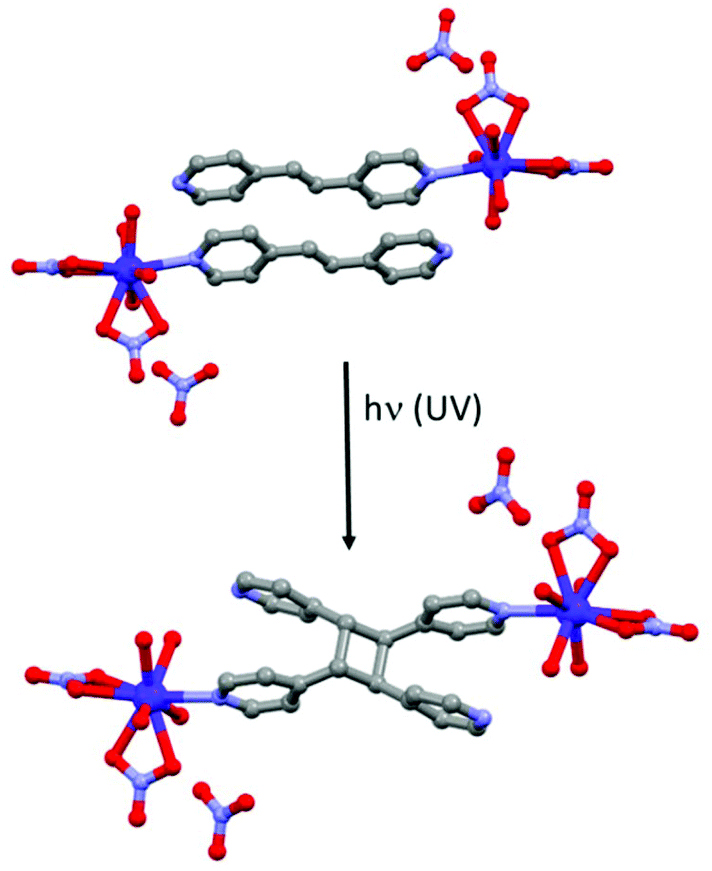 | ||
| Fig. 13 Photochemical [2 + 2] cycloaddition occurring in 34 (top) leading to the formation of 35 (bottom). | ||
The deviation from the linearity of the Arrhenius law could be fitted taking into account the remaining QTM, direct and Raman processes. At this point, it is clear that the photo-[2 + 2] cycloaddition induced a modulation of the magnetic properties from SMM behaviour for 34@Y to field-induced SMM for 35@Y. These observations were rationalized by ab initio calculations which concluded (i) an higher Ising character (gz = 19.09 vs. gz = 18.40), (ii) a greater energy gap between the ground state and the first excited state (112.8 K vs. 72.7 K)95 and (iii) less efficient QTM for 34@Y than for 35@Y.
5.3 Spiropyran-merocyanine based-photochromic ligands
Spiropyrans (SPs) are one of the best choices in the library of photochromic ligands.96 This class of ligands is very popular because of the cyclo-isomerization reaction to their merocyanine forms (trans-MC) upon light irradiation.97 Such reaction induces drastic structural and electronic modulations which have been exploited in several applications.98 The photochemical isomerization process is well-documented and it was clearly shown that the conversion of the trans-MC to SP form passes through a cis-conformation of the MC.99 Recently L. Norel and collaborators used the isomerization reaction from trans-MC to cis-MC to modulate the magnetic properties of a SMM.100 Thus a bis(pyridinemethyl)amine substituted spiropyran ligand101 was reacted with Dy(hfac)3·2H2O inducing the conversion of the spiropyran form into the trans-merocyanine form (trans-L25) of the ligand and the formation of the mononuclear complex of formula [Dy(trans-L25)(OTf)2(H2O)2]OTf (trans-36). Trans-36 behaved as an SMM in both solid and frozen solution states with a similar Orbach regime at high temperature (T > 5 K) (Δ = 32.9 K, τ0 = 1.24 × 10−5 s for solid state and Δ = 35.4 K, τ0 = 4.51 × 10−6 s for frozen solution state) (Table 1) while other under-barrier mechanisms are involved at low temperature (T < 5 K) (τQTM = 0.10 s for solid-state and 0.14 s for frozen solution state). Ab initio calculations corroborate the Ising character of magnetic anisotropy (gz = 19.69) as well as the presence of temperature-independent relaxation processes since the calculated energy difference between the ground state and the first excited state is overestimated (198.6 K). Under blue irradiation (450 nm) trans-36 was converted at 78% into the cis-36 form in solution. The ac magnetic measurements of cis-36 showed an almost unchanged Orbach regime (Δ = 32.9 K, τ0 = 5.76 × 10−7 s) compared to trans-36 while the low temperature regime was two times faster after irradiation (τQTM = 0.023 s). In other words, the trans–cis isomerization of 36 under irradiation induced slight magnetic modulation in frozen solution. Since the comparison of the magnetic behaviour was done in solution, the crystal packing effect should be neglected, and this might explain why the magnetic modulation is small. Similar observations were done for magnetic modulations of a DTE-based system after irradiation in solution (see section 5.1 with compounds o-33 and c-33).915.4 Solvato-switching of single-molecule magnet in a photo-reactive system
The last example of this section is one of the unique multi-switching of SMM behaviour with simultaneous photo- and solvato-switching phenomena. The concerned system was designed by reacting L26 (where L26 = N′,N′′,E,N′,N′′,E-N′,N′′-(ethane-1,2-diylidene)dipyrazine-2-carbohydrazide), L27 (where L27 = 9-anthrylmethylphosphonic acid), sodium oxalate and Dy(OAc)3·4H2O. The resulting complex of formula {[Dy5(L26)2(μ3-OAc)2(μ5-L27)(μ4-L27)(μ-OAc)2(OAc)2(H2O)(CH3OH)2]2(μ4-C2O4)} (37) can be described as two cyclic pentanuclear complexes of Dy(III) linked by a μ4-oxalate ligand.10237 did not behave as an SMM. After UV irradiation (365 nm) for 3 days, a single-crystal-to-single-crystal transformation led to the system 37-UV which displayed slow magnetic relaxation. The temperature dependence of the relaxation time was fitted using eqn (1).103 Three regimes were characterized i.e. the Orbach regime at high temperature (Δ = 47.3 K, τ0 = 1.26 × 10−7 s) (Table 1), a Raman process at intermediate temperature (C = 0.0011 s−1 K−n with n = 5.21) and a combination of Raman/direct processes at low temperature (A′ = 3.58 s−1 K−1 with A′ = AHm of eqn (1) and τQTM−1 = 0.0036 s−1).104 The switch ON of the SMM behaviour after irradiation was associated with the change of the coordination mode for L27 from μ4 to μ5 and the replacement of two methanol molecules with two water molecules giving the formula {[Dy5(L26)2(μ3-OAc)2(μ5-L27)2(μ-OAc)2(OAc)2(H2O)3]2(μ4-C2O4)} for 37-UV. Consequently the crystal field around the Dy(III) ions was modified as attested by ab initio calculations showing the increase of both the energy gap between the ground state and the first excited state from 102.4 K to 128.9 K and the axial character of the magnetic anisotropy from gz = 18.51 to gz = 19.41. Even more remarkable is that 37-UV could evolve to 37-Ar/N2 by a second single-crystal-to-single-crystal transformation by heating at 100 °C under an argon (37-Ar) or nitrogen (37-N2) atmosphere. This second transformation led to the switch OFF of the SMM behaviour due to the loss of two (37-N2) or three (37-Ar) coordinated water molecules. Nevertheless it was difficult to rationalize the magnetic behaviour after annealing with ab initio calculations because the average energy gap and Ising character for 37-Ar are close to those of 37-UV with Δ = 126.5 K and gz = 19.33. In addition, significant intramolecular interactions were calculated (J = −0.76 K for 37, J = −1.29 K for 37-UV and J = 0.46 K for 37-Ar) without the determination of their respective role in the observation or not of slow magnetic relaxation.6. Proton switching of single-molecule magnet behaviour
The previous section of this review showed that the dibenzotetraaza[14]annulenes like phthalocyanines or porphyrins are suitable macrocyclic ligands to design redox-active SMMs (see section 2.2), to observe redox-switching (see section 3.1) and solvato-switching (see section 4) SMM behaviour. The last section of the review shows that such ligands are also suitable for proton switching of the SMM behaviour.A recent example of such switching has been published by M. Yamashita and collaborators for a mononuclear double-decker complex made from a Dy(III) centre and an indolenine meso-substituted dibenzotetraaza[14]-annulene ligand (L28).105 In the absence of a base and acid in the reaction mixture the compound of formula H[Dy(L28)2]·solvents (H-38) was obtained. Both 1H-NMR and DFT methods concluded the position of the proton on the indolenine N-atom rather than on a N-atom of the annulene moiety. In the presence of an excess of acetic acid H-38 could be protonated to give H2-38 whereas with an excess of 1,8-diazabicyclo[5.4.0]-undec-7-ene base (DBU) H-38 could be deprotonated leading to 38. The three compounds have been magnetically studied in frozen solution and under an applied magnetic field of 1000 Oe. The neutral H-38 compound behaved as a field-induced SMM and its magnetization relaxed through both direct (A = 9.88 × 10−13 s−1 Oe−4 K−2 with direct process expressed as AT2H4) and Raman (C = 8.27 × 10−4 s−1 K−n with n = 9) processes (see eqn (1)).
After protonation of H-38, the magnetization of the cationic H2-38 compound relaxed faster through a complex association of mechanisms i.e. Orbach (Δ = 70.6 K, τ0 = 1.40 × 10−12 s), direct (A = 7.74 × 10−12 s−1 Oe−4 K−2) and Raman (C = 5.48 × 10−2 s−1 K−n with n = 9) processes. In contrast, after deprotonation of H-38, the magnetization of the anionic 38 compound relaxed slower through an Orbach regime (Δ = 56.1 K, τ0 = 1.25 × 10−8 s) and a Raman contribution (C = 3.33 × 10−4 s−1 K−n with n = 9). The relaxation of magnetization for 38 was slow enough to observe a SMM behaviour in zero applied magnetic field for which an Orbach regime (Δ = 34.7 K, τ0 = 2.51 × 10−7 s) and QTM (τQTM = 2.7 × 10−3 s) were identified. In conclusion the following trend for magnetic modulation was observed τ(H2-38) < τ(H-38) < τ(38) and it might be attributed to the increase of the under-barrier relaxation process contributions (direct and Raman) when protonation of the sample occurred. The second example of protonation/deprotonation of the first coordination sphere of the lanthanide is another double-decker complex made from the tetraphenylporphyrin (L29) macrocycle and Tb(III) ion.106 The protonated form has the formula [TbH(L29)2] (39) while the deprotonated form has the formula [Tb(L29)2](H-DBU) (40) (Fig. 14). The reversibility of the protonation/deprotonation process was verified by absorption spectroscopy. The presence of the protonated L29 was attested by electrochemistry with only four single-electron oxidation waves for 40 while six peaks were observed for 39. The two additional oxidation peaks in 39 were previously attributed to the protonated double-decker complex.107 The X-ray structures revealed a significant difference. Thus the Tb(III) centre is seven coordinated in 39 while it is eight coordinated in 40. The twisted angle value increases from 35° in 39 to 45° in 40. These structural modifications led to drastic changes in the magnetic properties since the SMM behaviour is switched on after deprotonation with an Orbach relaxation process in zero (Δ = 387.1 K, τ0 = 1.6 × 10−11 s) (Table 1) or applied magnetic field (2000 Oe) (Δ = 407.2 K, τ0 = 6.8 × 10−12 s) (Fig. 14). The proton switching of the SMM behaviour was associated with the loss of Ising-type magnetic anisotropy after protonation (seven-coordination instead of D4d eight-coordination) leading to a large off-diagonal ligand field symmetry and thus a loss of SMM behaviour.23a One more time, one could remark that the deprotonation led to an enhancement of the magnetic properties with much more drastic effect since the protonation/deprotonation process took place on the first coordination sphere of the lanthanide ion.
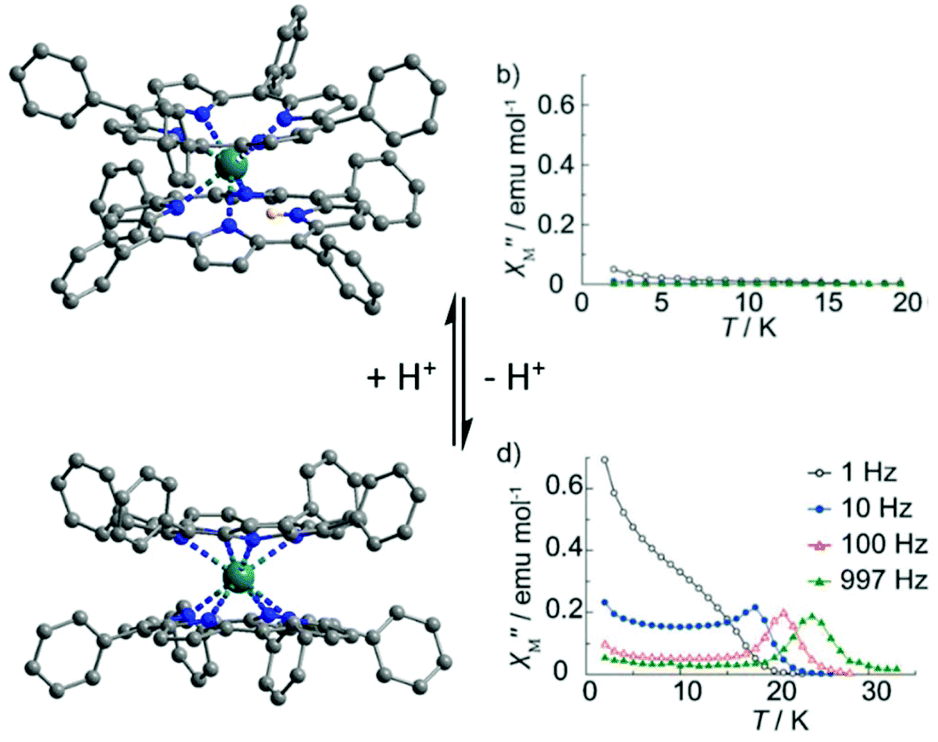 | ||
| Fig. 14 Crystal structure of 39 (top) and 40 (bottom) with their associated out-of-phase magnetic susceptibility contributions. Colour code: terbium: turquoise and hydrogen: light pink. Adapted from ref. 106. | ||
7. Conclusions and perspectives
The electro-activity and magnetic-switching of lanthanide SMMs are recent phenomena making such research fields grow quickly in less than 10 years. This article reviewed the main stimuli which can be used to modulate the SMM behaviour i.e. oxidation/reduction, solvation/desolvation and protonation/deprotonation processes, and light irradiation. Starting from the quite large pool of redox active lanthanide SMMs, the redox-switching of SMM behaviour is the most developed phenomena. It can be centred on the organic ligand or the metal ion. It is also probably the most complicated phenomena to understand because oxidation/reduction processes induced both change in the spin state and structural modifications. Thus magnetic interaction and electronic distribution could be simultaneously altered and need to be well understood to give a clear interpretation of the magnetic switching. For the solvato-, photo- and proton-switching of the SMM behaviour, the crucial parameter is the modification of the structural properties under the applied stimulus. The degree of change of the symmetry, electronic distribution around the lanthanide and crystal packing drives the importance of magnetic modulations. In this context, the phenomena of solvation/desolvation and protonation/deprotonation which took place in the first coordination sphere of the lanthanide ion have remarkable effects on the magnetic properties with ON–OFF switching of the SMM behaviour (Table 1). In contrast, structural changes which took place far from the metal centre such as change of solvent matrix or cyclo-isomerisation of DTE derivatives under light irradiation have only a slight effect on the magnetic properties. In these cases, the change of crystal packing i.e. change of dipolar interactions, took precedence over molecular structural changes. Lanthanide ions for the design of molecular switches are amazing suitable candidates since they are the metal ions most sensitive to any slight changes of structural properties especially compared to transition metal ions for which the switch of physical properties under external stimuli is mainly due to the change of their electronic configuration (metal-centred oxidation, change of spin-state etc.⋯).13–16 Thus examples of lanthanide molecular switches will expand undoubtedly in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Université de Rennes 1, CNRS France-Russia MULTISWITCH PRC grant (no. 199001) and the European Commission through the ERC-CoG 725184 MULTIPROSMM (project no. 725184).Notes and references
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Y. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford Univ. Press, 2006 Search PubMed.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- K. S. Pedersen, A.-M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed.
- (a) F.-S. Guo, B.-M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed; (b) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed; (c) K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492–8503 RSC; (d) F.-S. Guo, B.-M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- (a) J. M. Zadrozny, M. Atanasov, A. M. Bryan, C.-Y. Lin, B. D. Rekken, P. P. Power, F. Neese and J. R. Long, Chem. Sci., 2013, 4, 125–138 RSC; (b) R. L. Carlin, Magnetochemistry, Springer, Berlin, 1986 CrossRef.
- (a) A. Singh and K. N. Shrivastava, Phys. Status Solidi B, 1979, 95, 273–277 CrossRef CAS; (b) K. N. Shirivastava, Phys. Status Solidi B, 1983, 177, 437–458 CrossRef.
- T. Fukuda, K. Matsumura and N. Ishikawa, J. Phys. Chem. A, 2013, 117, 10447–10454 CrossRef CAS PubMed.
- (a) F. Pointillart, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2011, 17, 10397–10404 CrossRef CAS PubMed; (b) X. Yi, K. Bernot, O. Cador, J. Luzon, G. Calvez, C. Daiguebonne and O. Guillou, Dalton Trans., 2013, 42, 6728–6731 RSC; (c) F.-S. Guo and R. A. Layfield, Chem. Commun., 2017, 53, 3130–3133 RSC; (d) H. Tian, B.-L. Wang, J. Lu, H.-T. Liu, J. Su, D. Li and J. Dou, Chem. Commun., 2018, 54, 12105–12108 RSC; (e) L. Zhang, Y.-Q. Zhang, P. Zhang, L. Zhao, M. Guo and J. Tang, Inorg. Chem., 2017, 56, 7882–7889 CrossRef CAS PubMed; (f) M. J. Giansiracusa, E. Moreno-Pineda, R. Hussain, R. Marx, M. Martinez Prada, P. Neugebauer, S. Al-Badran, D. Collison, F. Tuna, J. van Slageren, S. Carretta, T. Guidi, E. J. L. McInnes, R. E. P. Winpenny and N. F. Chilton, J. Am. Chem. Soc., 2018, 140, 2504–2513 CrossRef CAS PubMed.
- (a) F. Luis, M. J. Martinez-Pérez, O. Montero, E. Coronado, S. Cardona-Serra, C. Marti-Gastaldo, J.-M. Clemente-Juan, J. Sesé, D. Drung and T. Schurig, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 060403 CrossRef; (b) F. Pointillart, K. Bernot, S. Golhen, B. Le Guennic, T. Guizouarn, L. Ouahab and O. Cador, Angew. Chem., Int. Ed., 2015, 54, 1504–1507 CrossRef CAS PubMed; (c) Y. Kishi, F. Pointillart, B. Lefeuvre, F. Riobé, B. Le Guennic, S. Golhen, O. Cador, O. Maury, H. Fujiwara and L. Ouahab, Chem. Commun., 2017, 53, 3575–3578 RSC; (d) E. Moreno-Pineda, M. Damjanovic, O. Fuhr, W. Wernsdorfer and M. Ruben, Angew. Chem., Int. Ed., 2017, 56, 9915–9919 CrossRef CAS PubMed; (e) Y.-C. Chen, J.-L. Liu, W. Wernsdorfer, D. Liu, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2017, 56, 4996–5000 CrossRef CAS PubMed; (f) J. Flores Gonzalez, F. Pointillart and O. Cador, Inorg. Chem. Front., 2019, 6, 1081–1086 RSC; O. Sato, Nat. Chem., 2016, 8, 644–656 Search PubMed.
- (a) O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed; (b) W.-B. Chen, Y.-C. Chen, J.-L. Liu, J.-H. Jia, L.-F. Wang, Q.-W. Li and M.-L. Tong, Inorg. Chem., 2017, 56, 8730–8734 CrossRef CAS PubMed.
- (a) M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M.-A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS PubMed; (b) R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357–359 CrossRef CAS PubMed; (c) A. Coskun, J. M. Spruell;, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- (a) X. Feng, C. Mathonière, I.-R. Jeon, M. Rouzières, A. Ozarowski, M. L. Aubrey, M. I. Gonzalez, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2013, 135, 15880–15884 CrossRef CAS PubMed; (b) C. Mathonière, H.-J. Lin, D. Siretanu, R. Clérac and J. M. Smith, J. Am. Chem. Soc., 2013, 135, 19083–19086 CrossRef PubMed.
- S. Fortier, J. J. Le Roy, C.-H. Chen, V. Vieru, M. Murugesu, L. F. Chibotaru, D. J. Mindiola and K. G. Caulton, J. Am. Chem. Soc., 2013, 135, 14670–14678 CrossRef CAS PubMed.
- (a) G. N. Newton, S. Yamashita, K. Hasumi, J. Matsuno, N. Yoshida, M. Nihei, T. Shiga, M. Nakano, H. Nojiri, W. Wernsdorfer and H. Oshio, Angew. Chem., Int. Ed., 2011, 50, 5716–5720 CrossRef CAS PubMed; (b) W. Jiang, C. Jiao, Y. Meng, L. Zhao, Q. Liu and T. Liu, Chem. Sci., 2018, 9, 617–622 RSC.
- (a) J. Vallejo, E. Pardo, M. Viviano-Chumillas, I. Castro, P. Amoros, M. Deniz, C. Ruiz-Perez, C. Yuste-Vivas, J. Krzystek, M. Julve, F. Lloret and J. Cano, Chem. Sci., 2017, 8, 3694–3702 RSC; (b) D. Q. Wu, D. Shao, X.-Q. Wei, F.-X. Shen, L. Shi, D. Kempe, Y.-Z. Zhang, K. R. Dunbar and X.-Y. Wang, J. Am. Chem. Soc., 2017, 139, 11714–11717 CrossRef CAS PubMed; (c) D. Shao, L. Shi, L. Yin, B.-L. Wang, Z.-X. Wang, Y.-Q. Zhang and X.-Y. Wang, Chem. Sci., 2018, 9, 7986–7991 RSC.
- (a) C. Carbonera, A. Dei, J.-F. Létard, C. Sangregorio and L. Sorace, Angew. Chem., Int. Ed., 2004, 43, 3136–3138 CrossRef CAS PubMed; (b) J. Tao, H. Maruyama and O. Sato, J. Am. Chem. Soc., 2006, 128, 1790–1791 CrossRef CAS PubMed.
- A. Md Subhan, R. Kawahata, H. Nakata, A. Fuyuhiro, T. Tsukuda and S. Kaizaki, Inorg. Chim. Acta, 2004, 357, 3139–3146 CrossRef.
- M. A. Dunstan, E. Rousset, M.-E. Boulon, R. W. Gable, L. Sorace and C. Boskovic, Dalton Trans., 2017, 46, 13756–13767 RSC.
- D. Guo and J. K. McCusker, Inorg. Chem., 2007, 46, 3257–3274 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- (a) N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., Int. Ed., 2005, 44, 2931–2935 CrossRef CAS PubMed; (b) N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Phys. Chem. B, 2004, 108, 11265–11271 CrossRef CAS; (c) N. Ishikawa, M. Sugita and W. Wernsdorfer, J. Am. Chem. Soc., 2005, 127, 3650–3651 CrossRef CAS PubMed; (d) N. Ishikawa, Polyhedron, 2007, 26, 2147–2153 CrossRef CAS.
- H.-G. Jin, X. Jiang, I. A. Kühne, S. Clair, V. Monnier, C. Chendo, G. Novitchi, A. K. Powell, K. M. Kadish and T. S. Balaban, Inorg. Chem., 2017, 56, 4864–4873 CrossRef CAS PubMed.
- (a) P. Zhu, F. Lu, N. Pan, D. P. Arnold, S. Zhang and J. Jiang, Eur. J. Inorg. Chem., 2004, 510–517 CrossRef CAS; (b) K. M. Kadish, T. Nakanishi, A. Gurek, V. Ahsen and I. Yilmaz, J. Phys. Chem. B, 2001, 105, 9817–9821 CrossRef CAS.
- L. Vitali, S. Fabris, A. M. Conte, S. Brink, M. Ruben, S. Baroni and K. Kern, Nano Lett., 2008, 8, 3364–3368 CrossRef CAS PubMed.
- M. Mannini, P. Sainctavit, R. Sessoli, C. Cartier dit Moulin, F. Pineider, M. A. Arrio, A. Cornia and D. Gatteschi, Chem. – Eur. J., 2008, 14, 7530–7535 CrossRef CAS PubMed.
- M. Gonidec, D. B. Amabilino and J. Veciana, Dalton Trans., 2012, 41, 13632–13639 RSC.
- D. P. Arnold and J. Jiang, J. Phys. Chem. A, 2001, 105, 7525–7533 CrossRef CAS.
- M. Gonidec, F. Luis, À. Vílchez, J. Esquena, D. B. Amabilino and J. Veciana, Angew. Chem., Int. Ed., 2010, 49, 1623–1626 CrossRef CAS PubMed.
- M. Waters, F. Moro, I. Krivokapic, J. McMaster and J. V. Slageren, Dalton Trans., 2012, 41, 1128–1130 RSC.
- (a) J. M. Williams, J. R. Ferraro, R. J. Thorn, K. D. Carlson, U. Geiser, H. H. Wang, A. M. Kini and M.-H. Whangbo, Organic Superconductors (Including Fullerenes), Synthesis, Structure, Properties and Theory, Prentice Hall, 1992 Search PubMed; (b) M. R. Bryce, J. Mater. Chem., 1995, 5, 1469–1479 RSC; (c) J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS; (d) P. Batail, Chem. Rev., 2004, 104, 4887–4890 CrossRef CAS (and the following articles of this thematic issue).
- D. Lorcy, N. Bellec, M. Fourmigué and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398–1438 CrossRef CAS.
- (a) F. Pointillart, S. Golhen, O. Cador and L. Ouahab, Dalton Trans., 2013, 42, 1949–1960 RSC; (b) F. Pointillart, B. Le Guennic, O. Cador, O. Maury and L. Ouahab, Acc. Chem. Res., 2015, 48, 2834–2842 CrossRef CAS PubMed.
- J.-C. Wu, S.-X. Liu, T. D. Keene, A. Neels, V. Mereacre, A. K. Powell and S. Decurtins, Inorg. Chem., 2008, 47, 3452–3459 CrossRef CAS PubMed.
- (a) G. Cosquer, F. Pointillart, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2011, 17, 12502–12511 CrossRef CAS PubMed; (b) G. Cosquer, F. Pointillart, B. Le Guennic, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2012, 51, 8488–8501 CrossRef CAS PubMed.
- F. Gao, L. Cui, W. Liu, L. Hu, Y.-W. Zhong, Y.-Z. Li and J.-L. Zuo, Inorg. Chem., 2013, 52, 11164–11172 CrossRef CAS PubMed.
- F. Pointillart, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2011, 17, 10397–10404 CrossRef CAS PubMed.
- F. Pointillart, B. Le Guennic, O. Maury, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2013, 52, 1398–1408 CrossRef CAS PubMed.
- (a) F. Habib, P.-H. Lin, J. Long, I. Korobkov, W. Wernsdorfer and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 8830–8833 CrossRef CAS PubMed; (b) X. Yi, K. Bernot, F. Pointillart, G. Poneti, G. Calvez, C. Daiguebonne, O. Guillou and R. Sessoli, Chem. – Eur. J., 2012, 18, 11379–11387 CrossRef CAS PubMed; (c) C. Y. Chow, H. Bolvin, V. E. Campbell, R. Guillot, J. W. Kampf, W. Wernsdorfer, F. Gendron, J. Autschbach, V. L. Pecoraro and T. Mallah, Chem. Sci., 2015, 6, 4148–4159 RSC; (d) S. Xue, Y.-N. Guo, L. Ungur, J. Tang and L. F. Chibotaru, Chem. – Eur. J., 2015, 21, 14099–14106 CrossRef CAS PubMed.
- (a) F. Pointillart, B. Le Guennic, S. Golhen, O. Cador, O. Maury and L. Ouahab, Chem. Commun., 2013, 49, 615–617 RSC; (b) F. Pointillart, S. Golhen, O. Cador and L. Ouahab, Eur. J. Inorg. Chem., 2014, 4558–4563 CrossRef CAS.
- F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150–175 CrossRef CAS.
- K. Soussi, J. Jung, F. Pointillart, B. Le Guennic, B. Lefeuvre, S. Golhen, O. Cador, Y. Guyot, O. Maury and L. Ouahab, Inorg. Chem. Front., 2015, 2, 1105–1117 RSC.
- C. Jia, S.-X. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804–3812 CrossRef CAS PubMed.
- B. Chen, Z.-P. Lv, C. Hua, C. F. Leong, F. Tuna, D. M. D'Alessandro, D. Collison and J.-L. Zuo, Inorg. Chem., 2016, 55, 4606–4615 CrossRef CAS PubMed.
- V. Kuropatov, S. Klementieva, G. Fukin, A. Mitin, S. Ketkov, Y. Budnikova, V. Cherkasoc and G. Abakumov, Tetrahedron, 2010, 66, 7605–7611 CrossRef CAS.
- Y. Yamashita, Y. Kobayashi and T. Miyashi, Angew. Chem., Int. Ed. Engl., 1989, 28, 1052–1053 CrossRef.
- N. O. Chalkov, V. K. Cherkasov, G. A. Abakumov, G. V. Romanenko, S. Y. Ketkov, I. V. Smolyaninov, A. G. Starikov and V. A. Kuropatov, Eur. J. Org. Chem., 2014, 4571–4576 CrossRef CAS.
- J. F. Gonzalez, O. Cador, L. Ouahab, S. Norkov, V. Kuropatov and F. Pointillart, Inorganics, 2018, 6, 45–54 CrossRef.
- F. Gao, X.-M. Zhang, L. Cui, K. Deng, Q.-D. Zeng and J.-L. Zuo, Sci. Rep., 2014, 4, 5928–5935 CrossRef CAS PubMed.
- F. Pointillart, B. Le Guennic, S. Golhen, O. Cador and L. Ouahab, Chem. Commun., 2013, 49, 11632–11634 RSC.
- J. S. Scout, Semiconductor and Semimetals, in Highly Conducting Quasi-One-Dimensional Organic Crystals, ed. E. Conswell, Academic Press, New York, 1988, vol. 27, p. 385 Search PubMed.
- C. Jia, S.-X. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804–3812 CrossRef CAS PubMed.
- (a) M. A. Dunstan, E. Rousset, M.-E. Boulon, R. W. Gable, L. Sorace and C. Boskovic, Dalton Trans., 2017, 46, 13756–13767 RSC; (b) J. O. Moilanen, A. Mansikkamaki, M. Lahtinen, F.-S. Guo, E. Kalenius, R. A. Layfield and L. F. Chibotaru, Dalton Trans., 2017, 46, 13582–13589 RSC.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- (a) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed; (b) S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS PubMed; (c) S. Demir, M. Nippe, M. I. Gonzalez and J. R. Long, Chem. Sci., 2014, 5, 4701–4711 RSC; (d) B. S. Dolinar, S. Gomez-Coca, D. I. Alexandropoulos and K. R. Dunbar, Chem. Commun., 2017, 53, 2283–2286 RSC; (e) C. A. Gould, L. E. Darago, M. I. Gonzalez, S. Demir and J. R. Long, Angew. Chem., Int. Ed., 2017, 56, 10103–10107 CrossRef CAS PubMed; (f) F.-S. Guo and R. A. Layfeld, Chem. Commun., 2017, 53, 3130–3133 RSC.
- P. Zhang, M. Perfetti, M. Kern, P. P. Hallmen, L. Ungur, S. Lenz, M. R. Ringenberg, W. Frey, H. Stoll, G. Rauhut and J. van Slageren, Chem. Sci., 2018, 9, 1221–1230 RSC.
- B. S. Dolinar, S. Gomez-Coca, D. I. Alexandropoulos and K. R. Dunbar, Chem. Commun., 2017, 53, 2283–2286 RSC.
- A. V. Ivanov, P. A. Zhukov, L. G. Tomilova and N. S. Zefirov, Russ. Chem. Bull., 2006, 55, 281–286 CrossRef CAS.
- S. Takamatsu, T. Isikawa, S.-Y. Koshihara and N. Ishikawa, Inorg. Chem., 2007, 46, 7250–7252 CrossRef CAS PubMed.
- M. Gonidec, E. S. Davies, J. McMaster, D. B. Amabinino and J. Veciana, J. Am. Chem. Soc., 2010, 132, 1756–1757 CrossRef CAS PubMed.
- (a) E. J. L. McInnes, E. Pidcock, V. S. Oganesyan, M. R. Cheesman, A. K. Powell and A. J. Thomson, J. Am. Chem. Soc., 2002, 124, 9219–9228 CrossRef CAS PubMed; (b) N. Domingo, B. E. Williamson, J. Gomez-Segura, P. Gerbier, D. Ruiz-Molina, D. B. Amabilino, J. Veciana and J. Tejada, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 052405 CrossRef; (c) P. Gerbier, N. Domingo, J. Gomez-Segura, D. Ruiz-Molina, D. B. Amabilino, J. Tejada, B. E. Williamson and J. Veciana, J. Mater. Chem., 2004, 14, 2455–2460 RSC; (d) M. R. Cheesman, V. S. Oganesyan, R. Sessoli, D. Gatteschi and A. J. Thomson, Chem. Commun., 1997, 1677–1678 RSC.
- L. Norel, M. Feng, K. Bernot, T. Roisnel, T. Guizouarn, K. Costuas and S. Rigaut, Inorg. Chem., 2014, 53, 2361–2363 CrossRef CAS PubMed.
- F. Meng, Y.-M. Hervault, Q. Shao, B. Hu, L. Norel, S. Rigaut and X. Che, Nat. Commun., 2014, 5, 3023–3303 CrossRef PubMed.
- K. A. Green, M. P. Cifuentes, M. Samoc and M. G. Humphrey, Coord. Chem. Rev., 2011, 255, 2530–2541 CrossRef CAS.
- E. Di Piazza, L. Norel, K. Costuas, A. Bourdolle, O. Maury and S. Rigaut, J. Am. Chem. Soc., 2011, 133, 6174–6176 CrossRef CAS PubMed.
- C. M. Dickie, A. L. Laughlin, J. D. Wofford, N. S. Bhuvanesh and M. Nippe, Chem. Sci., 2017, 8, 8039–8049 RSC.
- (a) V. V. Novikov, A. A. Pavlov, Y. V. Nelyubina, M.-E. Boulon, O. A. Varzatskii, Y. Z. Voloshin and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137, 9792–9795 CrossRef CAS PubMed; (b) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC.
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- E. M. Broderick, N. Guo, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278–9281 CrossRef CAS PubMed.
- S. Patra, T. A. Miller, B. Sarkar, M. Niemeyer, M. D. Ward and G. K. Lahiri, Inorg. Chem., 2003, 42, 4707–4713 CrossRef CAS PubMed.
- K. Suzuki, R. Sato and N. Mizuno, Chem. Sci., 2013, 4, 596–600 RSC.
- J.-L. Liu, Y.-C. Chen, Y.-Z. Zheng, W.-Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M.-L. Tong, Chem. Sci., 2013, 4, 3310–3316 RSC.
- (a) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC; (b) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (c) D. Gatteschi, Nat. Chem., 2011, 3, 830 CrossRef CAS PubMed; (d) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (e) H. L. C. Feltham, Y. Lan, F. Klöwer, L. Ungur, L. F. Chibotaru, A. K. Powell and S. Brooker, Chem. – Eur. J., 2011, 17, 4362–4365 CrossRef CAS PubMed; (f) A. Yamashita, A. Watanabe, S. Akine, T. Nabeshima, M. Nakano, T. Yamamura and T. Kajiwara, Angew. Chem., Int. Ed., 2011, 50, 4016–4019 CrossRef CAS PubMed.
- J.-L. Liu, K. Yuan, J.-D. Leng, L. Ungur, W. Wernsdorfer, F.-S. Guo, L. F. Chibotaru and M.-L. Tong, Inorg. Chem., 2012, 51, 8538–8544 CrossRef CAS PubMed.
- (a) A. Singh and K. N. Shrivastava, Phys. Status Solidi B, 1979, 95, 273–277 CrossRef CAS; (b) K. N. Shrivastava, Phys. Status Solidi B, 1983, 117, 437–458 CrossRef CAS.
- (a) W. Wernsdorfer, N. E. Chakov and G. Christou, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 132413 CrossRef; (b) W. Wernsdorfer, Supercond. Sci. Technol., 2009, 22, 064013 CrossRef.
- X. Guo, G. Zhu, Z. Li, F. Sun, Z. Yang and S. Qiu, Chem. Commun., 2006, 3172–3174 RSC.
- Q. Zhou, F. Yang, B. Xin, G. Zeng, X. Zhou, K. Liu, D. Ma, G. Li, Z. Shi and S. Feng, Chem. Commun., 2013, 49, 8244–8246 RSC.
- X. Zhang, V. Vieru, X. Feng, J.-L. Liu, Z. Zhang, B. Na, W. Shi, B.-W. Wag, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef CAS PubMed.
- J.-Y. Ge, L. Cui, J. Li, F. Yu, Y. Song, Y.-Q. Zhang, J.-L. Zuo and M. Kurmoo, Inorg. Chem., 2017, 56, 336–343 CrossRef CAS PubMed.
- (a) L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS PubMed; (b) L. Ungur, W. Van den Heuvel and L. F. Chibotaru, New J. Chem., 2009, 33, 1224–1230 RSC.
- (a) T. Yamada, S. Kobatake, K. Muto and M. Irie, J. Am. Chem. Soc., 2000, 122, 1589–1592 CrossRef CAS; (b) T. Yamada, S. Kobatake and M. Irie, Bull. Chem. Soc. Jpn., 2000, 73, 2179–2184 CrossRef CAS; (c) S. Kobatake, K. Uchida, E. Tsuchida and M. Irie, Chem. Commun., 2002, 2804–2805 RSC; (d) S. Kobatake and M. Irie, Bull. Chem. Soc. Jpn., 2004, 77, 195–210 CrossRef CAS; (e) M. Morimoto and M. Irie, Chem. Commun., 2005, 3895–3905 RSC; (f) S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed; (g) M. Irie, Bull. Chem. Soc. Jpn., 2008, 8, 917–926 CrossRef.
- (a) M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed; (b) H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- M. Morimoto, H. Miyasaka, M. Yamashita and M. Irie, J. Am. Chem. Soc., 2009, 131, 9823–9835 CrossRef CAS PubMed.
- T. Shiga, H. Miyasaka, M. Yamashita, M. Morimoto and M. Irie, Dalton Trans., 2011, 40, 2275–2282 RSC.
- (a) J.-P. Costes, F. Dahan, A. Dupuis and J.-P. Laurent, Inorg. Chem., 1996, 35, 2400–2402 CrossRef CAS PubMed; (b) J.-P. Costes, F. Dahan, A. Dupuis and J.-P. Laurent, Inorg. Chem., 1997, 36, 3429–3433 CrossRef CAS PubMed; (c) J.-P. Costes, F. Dahan and A. Dupuis, Inorg. Chem., 2000, 39, 165–168 CrossRef CAS PubMed; (d) T. Kajiwara, M. Nakano, S. Takaishi and M. Yamashita, Inorg. Chem., 2008, 47, 8604–8606 CrossRef CAS PubMed; (e) M. Sakamoto, K. Manseki and H. O. kawa, Coord. Chem. Rev., 2001, 219–221, 379–414 CrossRef CAS; (f) R. E. P. Winppeny, Chem. Soc. Rev., 1998, 27, 447–452 RSC; (g) S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420–421 CrossRef CAS PubMed; (h) F. He, M.-L. Tong and X.-M. Chen, Inorg. Chem., 2005, 44, 8285–8292 CrossRef CAS PubMed; (i) J.-P. Costes, F. Dahan and W. Wernsdorfer, Inorg. Chem., 2006, 45, 5–7 CrossRef CAS PubMed; (j) J.-P. Costes, M. Auchel, F. Dahan, V. Peyrou, S. Shova and W. Wernsdorfer, Inorg. Chem., 2006, 45, 1924–1934 CrossRef CAS PubMed; (k) R. Gheorghe, M. Andruh, J.-P. Costes and B. Donnadieu, Chem. Commun., 2003, 2778–2779 RSC; (l) R. Gheorghe, P. Cucos, M. Andruh, J.-P. Costes, B. Donnadieu and S. Shova, Chem. – Eur. J., 2006, 12, 187–203 CrossRef CAS PubMed; (m) J. Long, L.-M. Chamoreau and V. Marvaud, Dalton Trans., 2010, 39, 2188–2190 RSC; (n) M. Andruh, Chem. Commun., 2007, 2565–2567 RSC; (o) M. Andruh, J.-P. Costes, C. Diaz and S. Gao, Inorg. Chem., 2009, 48, 3342–3359 CrossRef CAS PubMed.
- M. Morimoto and M. Irie, Chem. – Eur. J., 2006, 12, 4275–4282 CrossRef CAS PubMed.
- D. Pinkowicz, M. Ren, L.-M. Zheng, S. Sato, M. Hasegawa, M. Morimoto, M. Irie, B. K. Breedlove, G. Cosquer, K. Katoh and M. Yamashita, Chem. – Eur. J., 2014, 20, 12502–11513 CrossRef CAS PubMed.
- G. Cosquer, M. Morimoto, M. Irie, A. Fetoh, B. K. Breedlove and M. Yamashita, Dalton Trans., 2015, 44, 5996–6002 RSC.
- G. Cosquer, M. Kamila, Z.-Y. Li, B. K. Breedlove and M. Yamashita, Inorganics, 2018, 6, 9–19 CrossRef.
- (a) D.-P. Li, T.-W. Wang, C.-H. Li, D.-S. Liu, Y.-S. Li and X.-Z. You, Chem. Commun., 2010, 46, 2929–2931 RSC; (b) L. Norel, K. Bernot, M. Feng, T. Roisnel, A. Caneschi, R. Sessoli and S. Rigaut, Chem. Commun., 2012, 48, 3948–3950 RSC; (c) G.-J. Chen, Y.-N. Guo, J.-L. Tian, J. Tang, W. Gu, X. Liu, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem. – Eur. J., 2012, 18, 2484–2487 CrossRef CAS PubMed; (d) T. T. Da Cunha, J. Jung, M.-E. Boulon, G. Campo, F. Pointillart, C. Pereira, B. Le Guennic, O. Cador, K. Bernot, F. Pineider, S. Golhen and L. Ouahab, J. Am. Chem. Soc., 2013, 135, 16332–16335 CrossRef CAS PubMed; (e) F. Pointillart, J. Jung, R. Berraud-Pache, B. Le Guennic, V. Dorcet, S. Golhen, O. Cador, O. Maury, Y. Guyot, S. Decurtins, S.-X. Liu and L. Ouahab, Inorg. Chem., 2015, 54, 5384–5397 CrossRef CAS PubMed; (f) Y. Bi, Y.-N. Guo, L. Zhao, Y. Guo, S.-Y. Lin, S.-D. Jiang, J. Tang, B.-W. Wang and S. Gao, Chem. – Eur. J., 2011, 17, 12476–12481 CrossRef CAS PubMed; (g) G. Cosquer, F. Pointillart, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2013, 19, 7895–7903 CrossRef CAS PubMed; (h) M. Perfetti, M. Gysler, Y. Rechkemmer-Patalen, P. Zhang, H. Tastan, F. Fischer, J. Netz, W. Frey, L. W. Zimmermann, T. Schleid, M. Hakl, M. Orlita, L. Ungur, L. Chibotaru, T. Brock-Nannestad, S. Piligkos and J. van Slageren, Chem. Sci., 2019, 10, 2101–2110 RSC; (i) B. Lefeuvre, O. Galangau, J. Flores Gonzalez, V. Montigaud, V. Dorcet, L. Ouahab, B. Le Guennic, O. Cador and F. Pointillart, Front. Chem., 2018, 6, 552–563 CrossRef CAS PubMed; (j) J. Flores Gonzalez, V. Montigaud, N. Saleh, O. Cador, J. Crassous, B. Le Guennic and F. Pointillart, Magnetochemistry, 2018, 4, 39–49 CrossRef.
- (a) Y.-C. Ou, W.-T. Liu, J.-Y. Li, G.-G. Zhang, J. Wang and M.-L. Tong, Chem. Commun., 2011, 47, 9384–9386 RSC; (b) M. Nagarathinam and J. J. Vittal, Chem. Commun., 2008, 438–440 RSC; (c) I.-H. Park, R. Medishetty, J.-Y. Kim, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5591–5595 CrossRef CAS PubMed; (d) K. M. Hutchins, T. P. Rupasinghe, L. R. Ditzler, D. C. Swenson, J. R. G. Sander, J. Baltrusaitis, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2014, 136, 6778–6781 CrossRef CAS PubMed; (e) X.-Y. Wang, Z.-M. Wang and S. Gao, Chem. Commun., 2007, 1127–1129 RSC; (f) H. Sato, R. Matsuda, M. H. Mira and S. Kitagawa, Chem. Commun., 2012, 48, 7919–7921 RSC.
- L.-F. Wang, J.-Z. Qiu, J.-L. Liu, Y.-C. Chen, J.-H. Jia, J. Jover, E. Ruiz and M.-L. Tong, Chem. Commun., 2015, 51, 15358–15361 RSC.
- J. Ruiz, A. J. Mota, S. Rodriguez-Dieguez, S. Titos, J. M. Herrera, E. Ruiz, E. Cremades, J.-P. Costes and E. Colacio, Chem. Commun., 2012, 48, 7916–7918 RSC.
- (a) R. J. Guglielmetti and J. C. Crano, in Topics in Applied Chemistry, Vol. 1, Main photochromic families, ed. A. R. Katritzky, Kluwer Academic Publisher, 2002 Search PubMed; (b) G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1753 CrossRef CAS PubMed; (c) M. M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed; (d) S. V. Paramonov, V. Lokshin and O. A. Fedorova, J. Photochem. Photobiol., C, 2011, 12, 209–236 CrossRef CAS; (e) J. J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed; (f) M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed.
- (a) Z. Y. Tian and A. D. Q. Li, Acc. Chem. Res., 2013, 46, 269–279 CrossRef CAS PubMed; (b) R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC; (c) Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed.
- P. Nuernberger, S. Ruetzel and T. Brixner, Angew. Chem., Int. Ed., 2015, 54, 11368–11386 CrossRef CAS PubMed.
- P. Selvanathan, V. Dorcet, T. Roisnel, K. Bernot, G. Huang, B. Le Guennic, L. Norel and S. Rigaut, Dalton Trans., 2018, 47, 4139–4148 RSC.
- P. Selvanathan, G. Huang, T. Guizouarn, T. Roisnel, G. Fernandez-Garcia, F. Totti, B. Le Guennic, G. Calvez, K. Bernot, L. Norel and S. Rigaut, Chem. – Eur. J., 2016, 22, 15222–15226 CrossRef CAS PubMed.
- H. Tian, J.-B. Su, S.-S. Bao, M. Kurmoo, X.-D. Huang, Y.-Q. Zhang and L.-M. Zheng, Chem. Sci., 2018, 9, 6424–6433 RSC.
- A. J. Brown, D. Pinkowicz, M. R. Saber and K. R. Dunbar, Angew. Chem., Int. Ed., 2015, 54, 5864–5868 CrossRef CAS PubMed.
- (a) I. J. Hewitt, J. Tang, N. T. Madhu, C. E. Anson, Y. Lan, J. Luzon, M. Etienne, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2010, 49, 6352–6356 CrossRef CAS PubMed; (b) J. J. Le Roy, M. Jeletic, S. I. Gorelsky, I. Korobkov, L. Ungur, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 3502–3510 CrossRef CAS PubMed; (c) K. L. M. Harriman, J. L. Brosmer, L. Ungur, P. L. Diaconescu and M. Murugesu, J. Am. Chem. Soc., 2017, 39, 1420–1423 CrossRef PubMed; (d) B. S. Dolinar, D. I. Alexandropoulos, K. R. Vignesh, T. James and K. R. Dunbar, J. Am. Chem. Soc., 2018, 140, 908–911 CrossRef CAS PubMed.
- Z. Liang, M. Damjanovic, M. Kamila, G. Cosquer, B. K. Breedlove, M. Enders and M. Yamashita, Inorg. Chem., 2017, 56, 6512–6521 CrossRef CAS PubMed.
- D. Tanaka, T. Inose, H. Tanaha, S. Lee, N. Ishikawa and T. Ogawa, Chem. Commun., 2012, 48, 7796–7798 RSC.
- G. A. Spyroulia, C. P. Raptopoulou, D. D. Montauzon, A. Mari, R. Poilblanc, A. Terzis and A. G. Coutsolelos, Inorg. Chem., 1999, 38, 1683–1699 CrossRef PubMed.
| This journal is © the Partner Organisations 2019 |