
The supramolecular assemblies based on heteroatom-containing triangulenes
Andreas
Winter
 ab and
Ulrich S.
Schubert
*ab
ab and
Ulrich S.
Schubert
*ab
aLaboratory for Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de; Fax: +49 3641 948200; Tel: +49 3641 948200
bCenter for Energy and Environmental Chemistry Jena (CEEC Jena), Philosophenweg 7a, 07743 Jena, Germany
First published on 10th September 2019
Abstract
The heteroatom-containing derivatives of triangulene feature a rich supramolecular chemistry which results from their (mostly) planar π-conjugated structure – predestined for the self-assembly via ππ-stacking interactions into 1D or 2D architectures. Herein, this behavior will be discussed – also taking the potential applications of these assemblies into account.
 Andreas Winter | Andreas Winter was born in Herne (Germany) in 1972 and studied chemistry at the University of Dortmund (Germany) where he graduated in organic chemistry in 1999. In 2003 he received his PhD in chemistry (University of Paderborn, Germany) for work on applications of the Mannich reaction in the synthesis of pyridine derivatives under supervision of Prof. N. Risch, and stayed on as a postdoc. Subsequently, in 2005 he joined the group of Prof. U. S. Schubert at the TU Eindhoven, the Netherlands. In 2010, he moved with group from there to the Friedrich Schiller University Jena, Germany. His research is focused on the synthesis of metallo-supramolecular assemblies for opto-electronic and energy-conversion applications. |
 Ulrich S. Schubert | Ulrich S. Schubert was born in Tübingen (Germany) in 1969. He studied chemistry in Frankfurt and Bayreuth (both Germany) and at the Virginia Commonwealth University, Richmond (USA). His PhD studies were performed at the Universities of Bayreuth and South Florida. After a postdoctoral training with J.-M. Lehn at the University of Strasbourg (France), he moved to the TU Munich (Germany) and obtained his Habilitation in 1999. During 1999–2000 he was Professor at the University of Munich, and during 2000–2007 Full-Professor at the TU Eindhoven (the Netherlands). Since 2007, he is a Full-Professor at the Friedrich Schiller University Jena, Germany. |
Introduction
Parent triangulene (1), also referred to as Clar's hydrocarbon, represents the structurally simplest non-Kekulé-type polycyclic aromatic hydrocarbon (PAH) featuring a biradical triplet ground state.1 Though already hypothesized by Clar in 1953,2 the on-surface synthesis and structural characterization of the highly reactive compound 1 was accomplished by Pavliček et al. very recently (Fig. 1);3 a number of substituted, thus stabilized derivatives was reported already some years earlier.4 Open-shell non-Kekulé PAHs, such as 1, are essentially of academic interest, though applications in the field of organic magnetics5 and opto-electronics1 have been suggested. The magnetic and photophysical properties are expected to result from the self-assembly of the planar π-conjugated molecules into 1D columnar architectures, driven by ππ-stacking interactions. In such assemblies, coupling of electrons should give rise, e.g., to ferromagnetism or low-energy absorption.6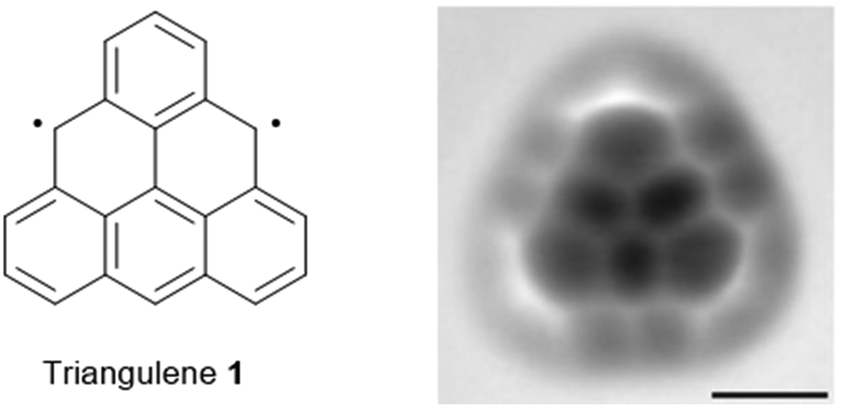 | ||
| Fig. 1 Schematic representation of the chemical structure of triangulene 1. A representative AFM image of 1 deposited onto a Cu(111) surface is also shown (scale bar: 5 Å).3 Figure reproduced with kind permission; © 2017 Springer Nature. | ||
The molecular structure of 1 can be varied basically by replacing carbon atoms – in the center and/or the in outer positions – by heteroatoms, thus configuring heteroatom-containing triangulene derivatives. A broad range of such structures has already been realized and one can distinguish between two subclasses: the cationic carbon-centered and the neutral heteroatom-centered derivatives (Fig. 2). In general, these types of compounds are much better understood than parent 1 and their fundamental chemistry as well as their photophysical and electrochemical properties will, before focusing on their supramolecular chemistry, briefly be discussed in the following.
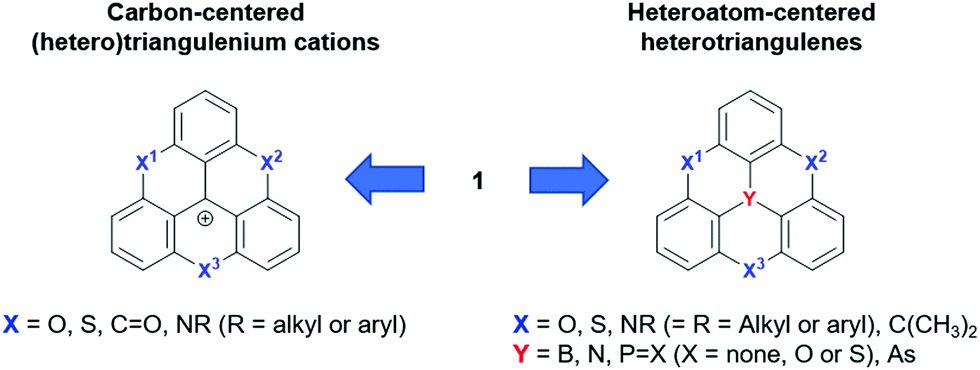 | ||
| Fig. 2 Schematic representation of the carbon-centered triangulenium and heteroatom-centered triangulene derivatives deduced from the parent compound 1. | ||
Preamble on heteroatom-containing triangulenes
Triangulenium cations are formed when the central C-atom of 1 is practically replaced by an sp2-hybridized C+ center. Thereby, heteroatom-bridged and planarized dyes of the triarylmethane family are formed (2–5, Fig. 3).7 The first example for this, planar sesquixanthylium cation (2, TOTA+) was already reported in 1964,8 but this compound was somehow disregarded until the late 1990s when a number of functionalized derivatives9 or, even more important, the nitrogen-bridged counterparts to 2 were reported (3 to 5).10,11 Special credit has to be given to Laursen and his co-workers for establishing reliable protocols to prepare these triangulenium cations. All these derivatives are stable and efficient dyes which exhibit UV/vis absorption bands in the region from ca. 450 to 650 nm.7 ADOTA+ (3) and DAOTA+ (4) are strong emitters with fluorescence quantum yields (ϕ) above 40% in acetonitrile solutions (emission lifetimes (τ) longer than 20 ns). These particular dyes have been considered in the context of bio-imaging applications, in vitro as well as in vivo.12 With respect to the electrochemical properties, a distinct trend can be observed for 2–5: briefly, with increasing number of nitrogen atoms, the oxidation becomes easier. This behavior can be attributed to the stronger donor-character of the nitrogen atoms; consequently, the more oxygen-rich derivatives are much easier to be reduced (reduction potentials of −0.385 and −1.40 V for 2 and 5, respectively). The ease of oxidation, in the case of TATA+ (5), was used by Gueret et al. for photosensitizing the light-driven hydrogen evolution reaction (HER) in the presence of a Co-based catalyst (remarkably, the TATA+ cation turned out to be a better photosensitizer than the benchmark Ru(bpy)32+ under the experiments’ conditions).13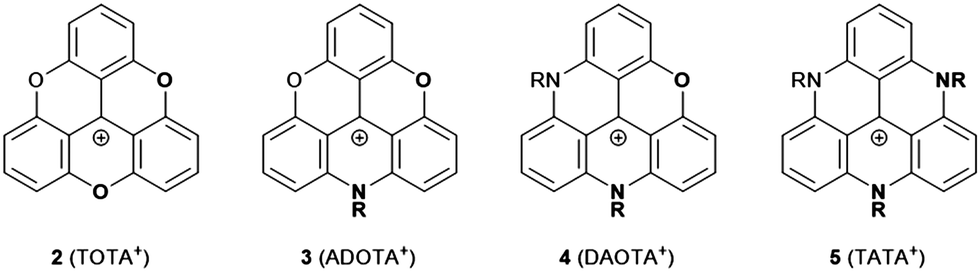 | ||
| Fig. 3 Schematic representation of O- and/or N-bridged triangulenium cations 2–5 (R denotes an alkyl or aryl residue, the counterions are omitted for clarity). | ||
For the neutral heteroatom-centered triangulenes a broad structural diversity is known. Concerning their synthesis two alternative strategies can basically be applied – the range of linear syntheses and the more universal divergent synthesis which was introduced recently by Hatakeyama and co-workers.14
The nitrogen-centered derivatives, either with isopropenyl (6) or carbonyl bridges (7), are already known since Hellwinkel's pioneering work in the 1970s.15–17 Whereas, their counterparts with higher group-15 elements in the center, i.e., phosphorous or arsenic, were published much later.18–22 As detailed below, the latter ones possess concave, bowl-shaped molecular structures and, therefore, their self-assembly behavior will significantly differ from that of the planar N-centered molecules. These, in turn, resemble planarized derivatives of the propeller-shaped triarylamines which offer a beneficial property: extension of the π-conjugation through the central N-atom and, as a result from, improved materials properties when compared to analogous non-bridged triarylamines.23 The photophysical and electrochemical properties of the core structures mainly depend on the nature of the bridging unit – ranging from a strongly electron-withdrawing carbonyl- (7) to a highly electron-donating oxygen-moiety (8, Fig. 4).24–26 It has been shown that these properties can be tuned over a wide range by appropriate modification of the parent structures.27 The Venkataraman group focused on the photophysical properties and the self-assembly behavior of 728 (vide infra) and it was realized that, after functionalization in the phenyl- and/or carbonyl-positions, materials with even semiconductor-like properties can be obtained.23 The same also holds true for isopropenyl-bridged 6: the incorporation into more complex architectures afforded materials with high ϕ values and large two-photon absorption (TPA) cross-sections.29 A more detailed discussion of functionalized N-centered triangulenes can be found in Kivala's personal account on this topic.23
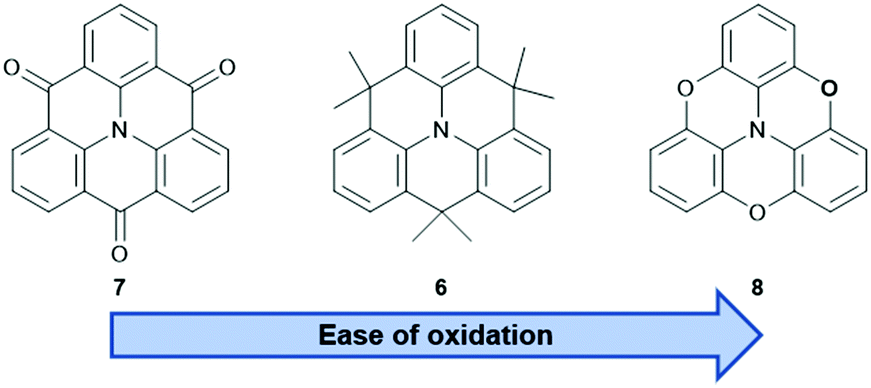 | ||
| Fig. 4 Schematic representation of the N-centered triangulenes 6–8. | ||
Compared to their N-centered counterparts, the boron-centered derivatives are relatively young members of the heteroatom-containing triangulene family with their syntheses being reported within the last decade, in particular by Yamaguchi and co-workers.14,30–34 Basically, triarylboranes represent the electron-acceptor counterparts to the electron-donating triarylamines and, thus, the play an important role in various opto-electronic applications: organic-light emitting diodes (OLEDs), TPA materials or sensing of anions.35–37 The planarization of the triarylborne framework, as in 9 and 10 (Fig. 5) strongly effects the solid-state properties of the material by enabling intermolecular interactions to occur much more efficiently. It was shown, for example, that π-extended derivatives of 9 functioned as electron-transport materials in OLEDs.31 The packing of such discotic species into columnar architectures via ππ-stacking interactions afforded a liquid crystalline (LC) assembly with ambipolar charge-carrier characteristics.32
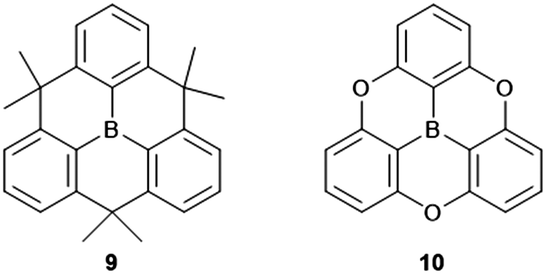 | ||
| Fig. 5 Schematic representation of the B-centered triangulenes 9 and 10. | ||
To sum up this section, the various types of heteroatom-containing triangulenes represent important structures to be employed in opto-electronic applications. This can be attributed to their rich photophysical and electrochemical properties which, in turn, are strongly associated with their intermolecular interactions, i.e., their supramolecular chemistry. The self-assembly behavior of triangulene derivatives, also in the context of potential applications, is in the spotlight of the subsequent sections.
Supramolecular assemblies based on C-centered triangulenium cations
The π-electronic triangulenium cations exhibit a self-assembly behavior that is strongly influenced by the accompanying anions. Thus, the supramolecular interaction of these compounds cannot be rationalized by the concepts of ππ-stacking interactions alone38 – ion-pairing interactions between the π-electronic ions significantly contribute to the supramolecular ordering/packing of the components. Recently, Haketa and Maeda pointed out that three different assembly modes have to be considered in this context (Fig. 6).39 First of all, the two most extreme scenarios have to be named: the charge-by-charge and the charge-segregated ordering. In the former case, columnar heterostacks with alternating cationic and anionic molecules are formed (here, the ππ-stacking is facilitated by electronic attraction of opposite charges); whereas the latter one involves the stacking of molecules of identical charges (in such assemblies, which are rare, the electronic repulsion must be surpassed by even stronger attractive forces). The third (and most common) mode, which is the intermediate one, involves contributions from the other two modes.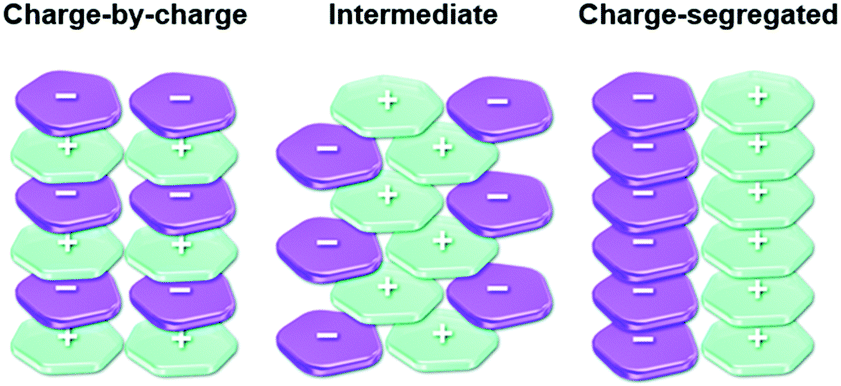 | ||
| Fig. 6 Schematic representation of the three different packing modes known for π-electronic ionic species.39 Figure reproduced with kind permission; © 2017 The Royal Chemical Society. | ||
In an initial study, Krebs et al. investigated the solid-state structures of the TOTA+ cations 2 and 11 as a function of their counterions.40 Notable differences with respect to solid-state ordering of the molecules were observed: in detail, for the monovalent anions (i.e., I−, BF4−, AsF6−, PF6−, NO3−, CF3SO3−), a zigzag-type packing of cations and anions was revealed by X-ray diffraction (XRD) analysis (Fig. 7). On the other hand, the bulky divalent anions (i.e., Mo6Cl142−, S2O62−) were found to be “sandwiched” between two TOTA+ sites. However, in both cases, the anions were detected in close proximity to the cationic centers of the TOTA+ molecules. The situation was somehow different in the case of (12)PF6:9 this salt featured a pseudohexagonal-type of stacking with strong contributions from the charge-segregated mode in which the PF6− anions were located close to the amino-substituents.
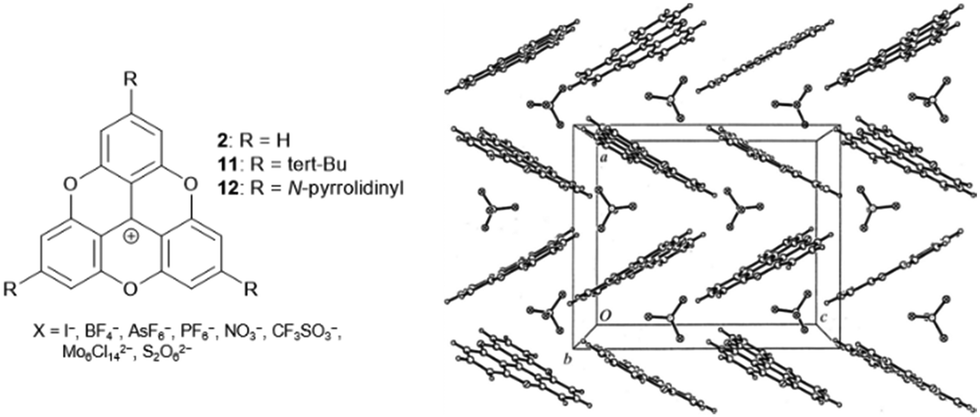 | ||
| Fig. 7 Schematic representation of the TOTA+ cations 2, 11 and 12. The solid-state structure of (2)BF4 is shown exemplarily (view along the b-axis of the Pbca unit cell).40 Figure reproduced with kind permission; © 1999 International Union of Crystallography. | ||
Laursen and co-workers investigated the formation of densely columnar packed Langmuir films at the water–air interface.41 For this purpose the amphiphilic TOTA+ derivatives 13, comprising a hydrophilic part, a rigid hydrophobic π-system and flexible hydrophobic alkyl chains were utilized. According to gracing incidence X-ray diffraction (GIXD) analysis, 13a and 13b yielded 2D-crystalline monolayers. In these structures, columnar stacks of the rigid-planar TOTA+ moieties were found, whereby these aggregates were well segregated from the accompanying PF6− counterions (a packing mode that was reminiscent of the solid-state structure of (12)PF6,9Fig. 8). The analytical data indicated an average column width which was almost identical to the diameter of the used molecules, i.e., about 17 Å; further, a remarkably short repeat distance of 3.45 Å along the columns was found, thus suggesting an almost cofacial alignment of the TOTA+ cations with only a marginal tilting of the molecules relative to the columnar axis. Later, the same group also prepared multilayer films from 13a by applying the Langmuir–Blodgett (LB) technique.42 These films featured the same type of supramolecular stacking as in the previous case. In-depth UV/vis absorption and emission studies gave evidence for an H-type aggregation of the molecules. Moreover, polarized fluorescence measurements revealed even a macroscopic alignment of the molecules resulting from the strong ππ-stacking interactions; thereby, the counterions were believed to be located between the single layers.
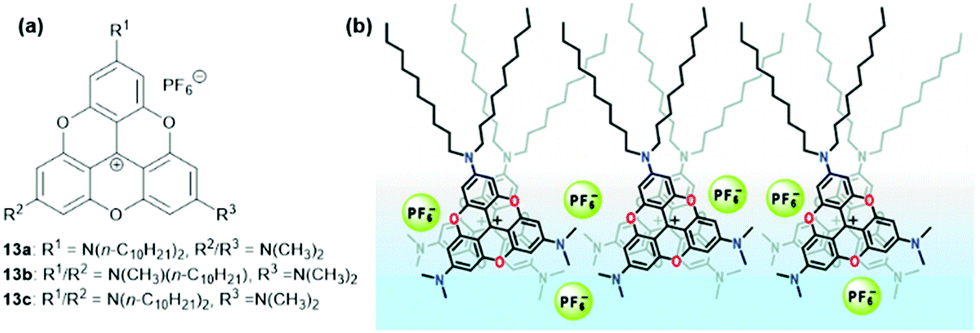 | ||
| Fig. 8 (a) Schematic representation of the amphiphilic TOTA+ derivatives 13. (b) Representation of the molecular packing of 13a within the assembled LB films.41 Figure reproduced with kind permission; © 2009 American Chemical Society. | ||
A different type of assembly was found in the case of the TATA+ cations 14. Due to the presence of the aliphatic or etheric chains, smectic-type bilayer assemblies were found in the solid state.43 Thereby, the layer-to-layer distance could be adjusted via the lengths of the pending chains which were oriented strictly perpendicular to the planar TATA+ moieties; the layers, in turn, were interwoven due to hydrophobic interactions of the chains. As a result, two TATA+ units were forced to stack in a face-to-face-type fashion, thereby overcoming their intrinsic electrostatic repulsion, eventually establishing a hexagonally ordered bilayer structure (Fig. 9). The authors processed the compounds into thin-solid films via spin-coating from solution. A high anisotropic polycrystallinity was observed in all these films, even on the cm length scale. The photophysical properties of the materials were investigated and suggested a potential use in opto-electronic applications due to their efficient exciton-transport behavior.44 Noteworthy, the corresponding triaryl-substituted TATA+ derivatives did not show this self-assembly behavior due to the perpendicular orientation of the substituents relative to the planar molecular core.45
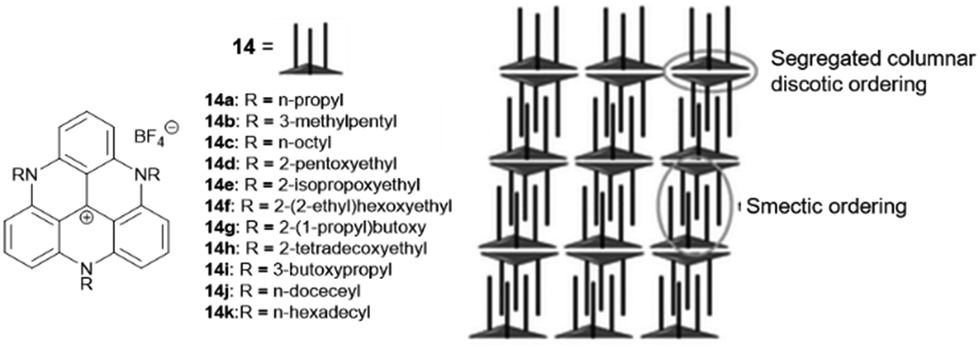 | ||
| Fig. 9 Schematic representation of the TATA+ derivatives 14 that assembled into a smectic-type structure. A representation of the supramolecular packing as evidenced from XRD analyses is also shown.43 Figure reproduced with kind permission; © 2012 The Royal Chemical Society. | ||
In all previous examples “conventional” small-molecule anions were accompanying the triangulenium cations. In addition to these, also the assembly of genuine π-electronic ion pairs, i.e., both the cation and the anion representing π-conjugated molecules, has been studied. Such species can, in principle, be obtained in a salt-metathesis reaction according to Fig. 10.39,46 It has to be noted that stable π-electronic anions are rare (compared to their cationic counterparts), since they prone to react with electrophiles. As one of the few exceptions, the cyclopentadienyl anions (Cp−) equipped with five strongly electron-withdrawing substituents (as the weak conjugated bases of superstrong organic acids47) have to be named in this context.
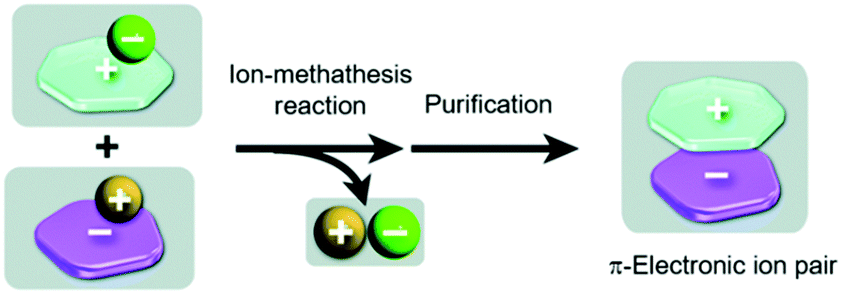 | ||
| Fig. 10 Schematic representation of the synthesis of π-electronic ion pairs via a salt-metathesis reaction of appropriate couples of precursor salts.46 Figure reproduced with kind permission; © 2013 The Royal Chemical Society. | ||
For example, Maeda and co-workers investigated the self-assembly properties of (14a)(PCCp) in the solid state (PCCp: 1,2,3,4,5-pentacyanocyclopentadienyl).48 X-ray crystal structure analysis revealed a regular packing of two pairs of 14a+ and PCCp− ions in an alternating fashion (Fig. 11); whereas, the (CV)PCCp ion salt (CV: crystal-violet cation) exhibited a strictly alternating stacking of the CV+ and PCCp− ions. The authors concluded that the highly ordered charge-by-charge assembly is disfavored when the π-electronic cation and anion strongly differ in their unit areas (as for the combination of 14a+ and PCCp−). Furthermore, the systems (14j)PCCp and (14k)PCCp, featuring long alkyl chains attached to the TATA+ cation, were investigated. These side chains were introduced to facilitate the dimension-controlled self-assembly of the ion pair due to additional van der Waals interactions.48 Micrometer-scaled prisms were obtained in both cases from CH2Cl2/methanol solutions. These materials were characterized by (polarized) optical microscopy ((P)OM), atomic force microscopy (AFM) imaging and synchrotron XRD analysis. The latter one gave evidence for the presence of 2D anisotropically ordered lamellar phases within the macroscopic structures. The mesophases were identified by differential scanning calorimetry (DSC) measurements and POM imaging revealed textures with crystalline behavior. The samples melted into isotropic liquids at 169 °C and 176 °C, respectively.
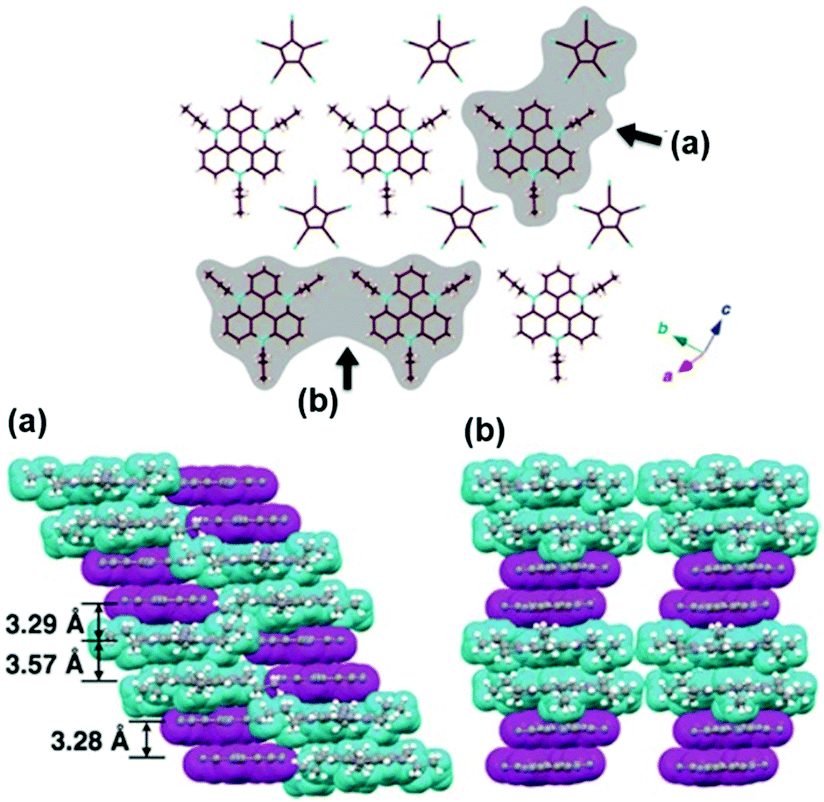 | ||
| Fig. 11 X-ray single-crystal structure (14a)PCCp. Top: Representative packing mode as top view of one layer. Bottom: Two different space-filling model side views (cyan and magenta units represent the cations and anions, respectively).48 Figure reproduced with kind permission; © 2016 Wiley-VCH. | ||
The binding of anions to appropriate π-electronic receptor molecules represents a versatile alternative to obtain planar π-electronic anions.39 Against this background, Maeda et al. introduced the BF2-complexes of 1,3-bis(pyrrol-2-yl)-1,3-diketones as efficient anion receptors for the formation of such anions and employed them in the context of supramolecular ion-pairing assembly (see e.g.ref. 39 and the literature cited therein). Among others, also the TATA+ cations 14a+, 14c+, 14j+ and 14k+ were combined with the anions (15·Cl)− and (15·Br)− (Fig. 12) to yield supramolecular gels in alkane solvents and thermotropic LC materials.49 The X-ray single crystal analysis of (14a)(15b·X) (X = Cl and Br) confirmed the expected packing of the planar π-electronic ions in a highly ordered charge-by-charge-type fashion (Fig. 13a). In all cases, supramolecular organogels with LC properties, comprising sub-μm-sized fibers, were observed (representative microscopy images are shown in Fig. 13b–d).
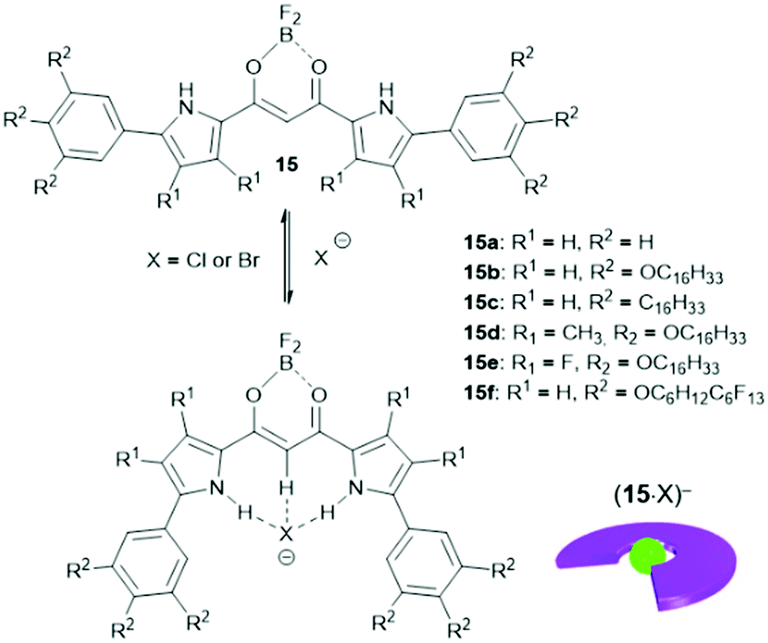 | ||
| Fig. 12 Schematic representation of the formation of π-electronic anions by binding of a halide to the anion receptor 15. | ||
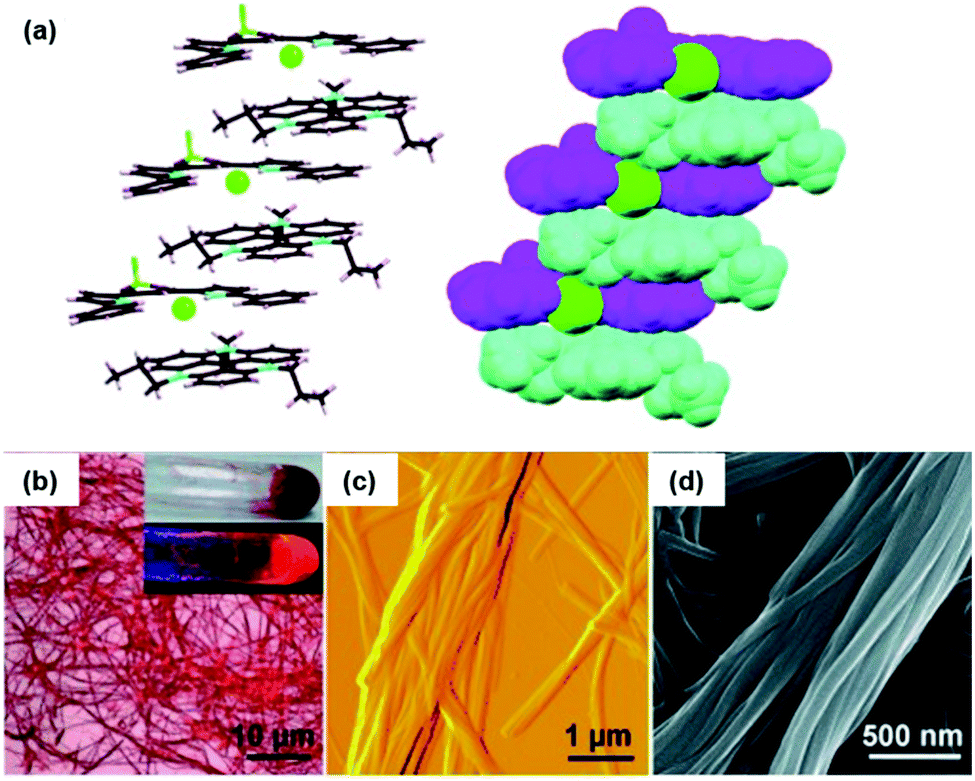 | ||
| Fig. 13 (a) Solid-state packing of (14a)(15a·Cl) in the stick-type (left) and space-filling representation (right; 14a, 15a and Cl− are depicted in cyan, magenta and green, respectively). (b–d) Microscopy images of the octane xerogel of (14a)(15b·Cl). (b) OM image (the inset shows pictures of the bulk sample upon visible (top) and UV light (bottom)), (c and d) representative AFM and SEM images, respectively.49 Figure reproduced with kind permission; © 2010 Wiley-VCH. | ||
The LC behavior of ion-pairing assemblies based on TATA+ cations was investigated further in particular focusing on the influence of the pending chains of the accompanying π-electronic anion. For (14a)(15b·Cl), the mesophase featured a focal-conic texture for which a triplet of transition temperatures of 94, 88 and 42 °C upon cooling from the isotropic liquid state (Iso) was identified by POM imaging (Fig. 14a).49 The structure of the LC phase was studied by synchrotron XRD measurements and the observed peaks suggested a Colh phase (Fig. 14b); the proposed stacking mode, i.e., the formation of tetramers which assembled in a charge-by-charge-type fashion, was also present in the crystalline material (Fig. 13a).
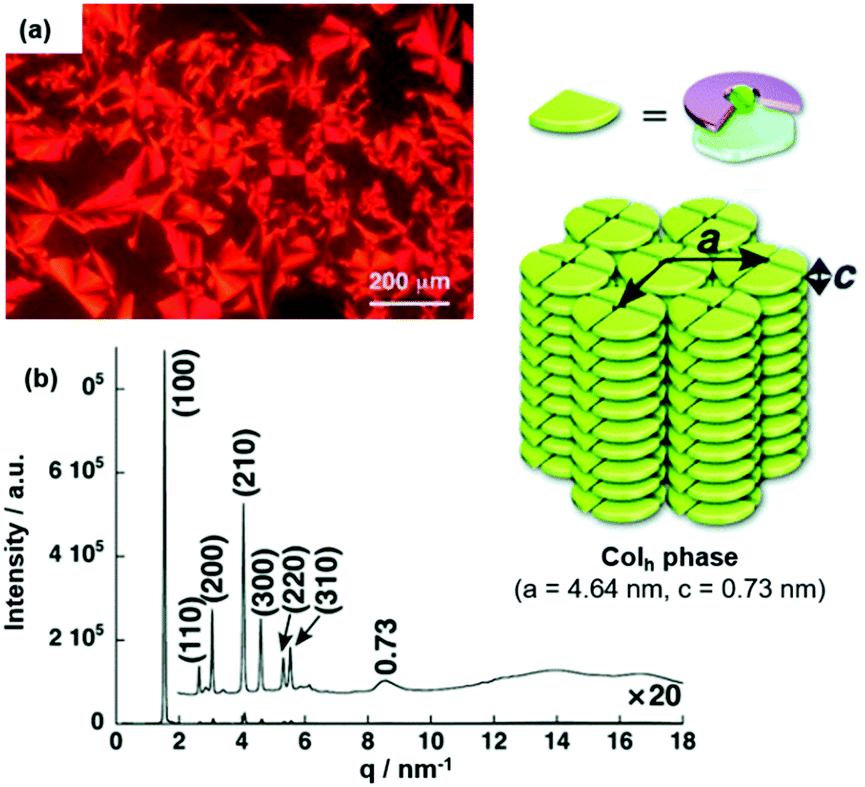 | ||
Fig. 14 (a) POM image of (14a)(15b·Cl) at 87 °C. (b) XRD pattern of (14a)(15b·Cl) in the mesophase at 70 °C (for both measurements the Iso was cooled down with a rate of 2 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) min−1). The proposed supramolecular stacking mode is also shown.49 Figure reproduced with kind permission; © 2010 Wiley-VCH. min−1). The proposed supramolecular stacking mode is also shown.49 Figure reproduced with kind permission; © 2010 Wiley-VCH. | ||
The morphology of the assembly could be modified simply via the nature of substituents on the anion-receptor molecule. For example, the π-electronic salt (14a)(15c·Cl) yielded a Colr mesophase at 50 °C upon cooling from the Iso instead of the Colh one as in the previous case (Fig. 15a).50 The XRD pattern featured a broad peak at which was assigned to a contribution of a charge-segregated packing mode to the overall assembly (Fig. 15b). This behavior was even more pronounced in the systems (14a)(15d·Cl) and (14a)(15e·Cl), where the stacking of π-electronic ions of identical charges was even dominant.51 The authors rationalized the strong contributions by charge-segregated stacking to the pronounced distortion of aryl rings due to the presence of the pyrrolic β-substituents (i.e., CH3 and F, respectively). As a consequence of this particular stacking behavior, ambipolar charge-transport (CT) properties with high mobilities were observed (e.g., (14a)(15e·Cl) exhibited a mobility of negative charges in the range of 5 × 10−3 cm2 V−1 s−1 at 4 × 103 V). It was concluded that the structure of the countercation strongly influences the conductivity of the ion-paired assemblies and, in particular, the aggregation in a charge-segregated mode is of importance to achieve high conductivity values (for examples see ref. 52).
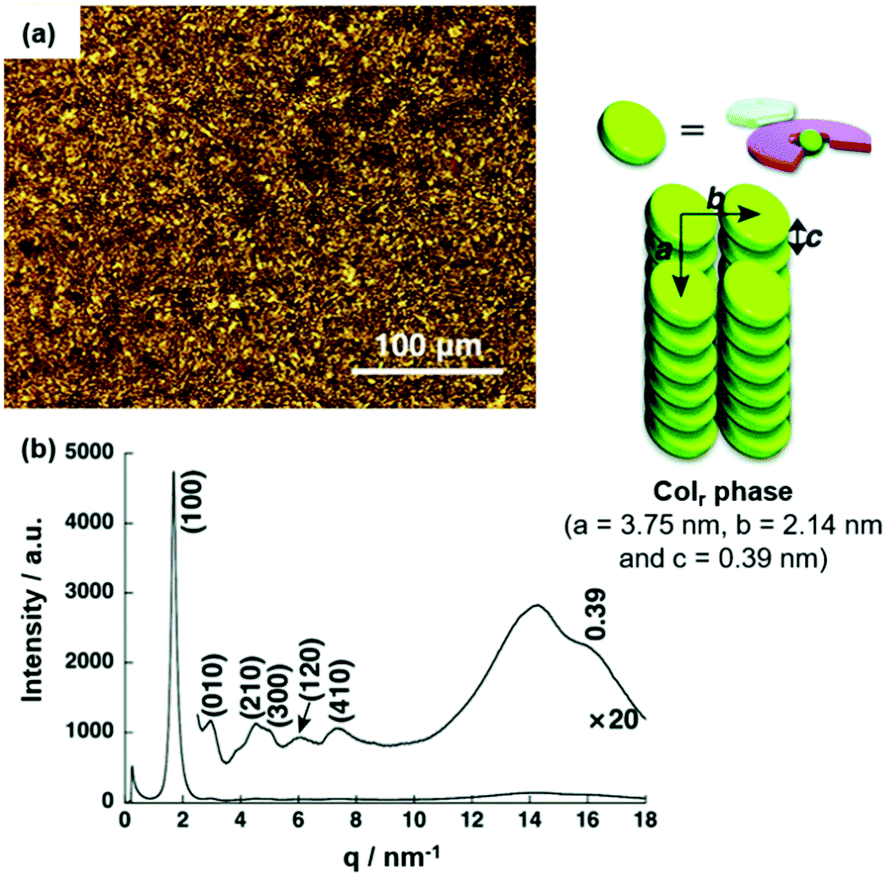 | ||
| Fig. 15 (a) POM image of (14a)(15c·Cl) at 62 °C. (b) XRD pattern of (14a)(15c·Cl) in the mesophase at 62 °C (for both measurements the Iso was cooled with a rate of 2 °C min−1). The proposed supramolecular stacking mode is also shown.50 Figure reproduced with kind permission; © 2016 Wiley-VCH. | ||
For the π-electronic salt (14a)(15f·Cl) in which partially fluorinated side chains were present in the anion-receptor molecule, the mesophase was formed below a much higher temperature as for the analogous non-fluorinated system (14a)(15b·Cl) (i.e., 153 °C vs. 80 °C); the increased stability of the mesophase was attributed to the much stronger interaction of the semi-fluorinated aliphatic chains.53 In these assemblies, a transient conductivity of 2.7 × 10−5 cm2 V−1 s−1 was observed.
In another study, also the π-conjugated system of the anion-receptor species was extended. For example, the benzo[c]-fused analogues of 15 were studied in this context.54 The π-electronic salts (14a)(16·Cl) were prepared and studied with respect to their self-assembly behavior. In the case of (14a)(16b·Cl), a tetragonal Colt mesophase was identified by POM imaging and XRD measurements (Fig. 16). In more detail, each disk of the column was formed by four sets of the ion pair; thereby, the flat cation and anion molecules formed the disks with the out-facing pending alkyl chains forming a hydrophobic shell along the columns.
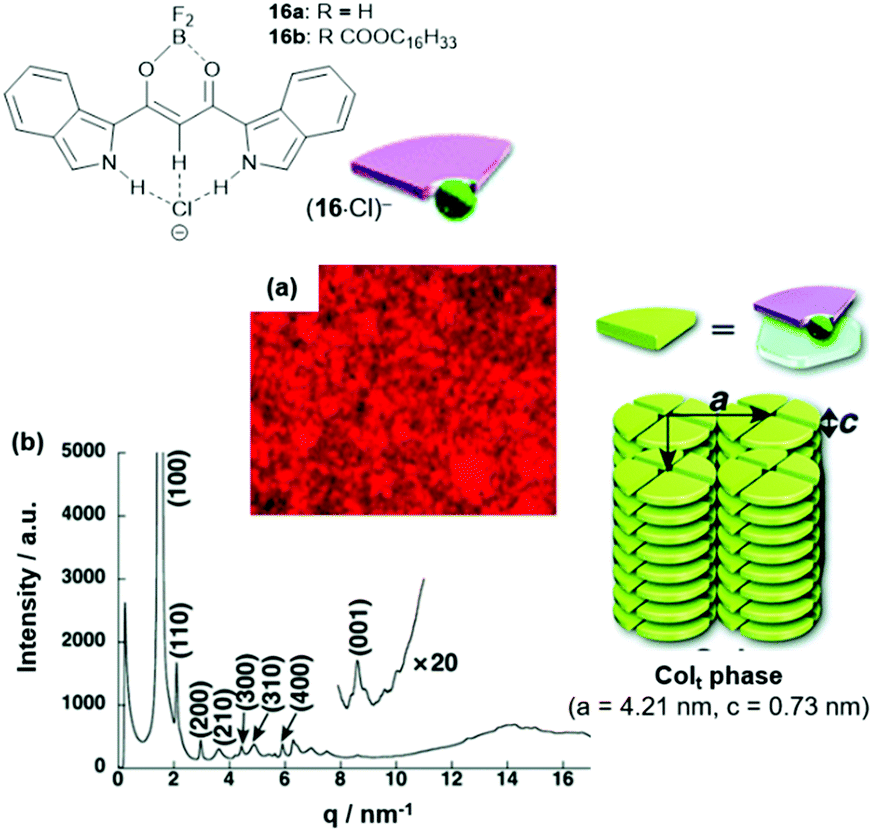 | ||
| Fig. 16 Top: Schematic representation of the π-electronic anions (16·Cl)−. Bottom: (a and b) POM image and synchrotron XRD pattern of (14a)(16b·Cl), respectively (both measurements were conducted at 80 °C); the proposed supramolecular stacking within the Colt structure is also shown.54 Figure reproduced with kind permission; © 2012 The Royal Chemical Society. | ||
The anion-receptor molecules were also equipped with bowl-shaped corannulene moieties, thereby expanding the π-electronic surface of the resulting anions.55 In particular, the ion salt (14a)(17·Cl), in which an unsymmetrical π-electronic anion was used, exhibited a rich self-assembly behavior. In 1,4-dioxane, for example, spherical aggregates with diameters in the range of 30 to 50 μm were identified by SEM imaging (Fig. 17a and b). However, a distinct mesophase could not be identified upon cooling of the Iso phase; instead, XRD measurements at 80 °C revealed a lamellar-type morphology with a much lower degree of ordering (compared to the aforementioned examples, Fig. 17c). Nonetheless, the LC materials showed a significantly increased charge-carrier mobility of up to 0.2 cm2 V−1 s−1; this behavior was attributed to the presence of the fused corannulene units which offered a better overlap of the π-conjugated systems. In a similar fashion, also the phenanthro[b]-fused derivatives 18 were employed for the complexation of chloride ions and, subsequently, the formation of π-electronic salts with TATA+ cations.56 The anion-receptor molecules featured an increased affinity towards halide anions (attributed to additional C–H⋯Cl interactions with the phenanthroline unit) and, as result, the (17·Cl)− were of highly planar structure. Accordingly, their π-electronic salts (14a)(18·Cl) yielded hexagonal columnar structures with liquid-crystalline mesophases which were analyzed by POM imaging and synchrotron XRD measurements (Fig. 29). For some of the systems (14a)(18·Cl), high photoconductivities were found in their anisotropically aligned mesophases (Fig. 18).
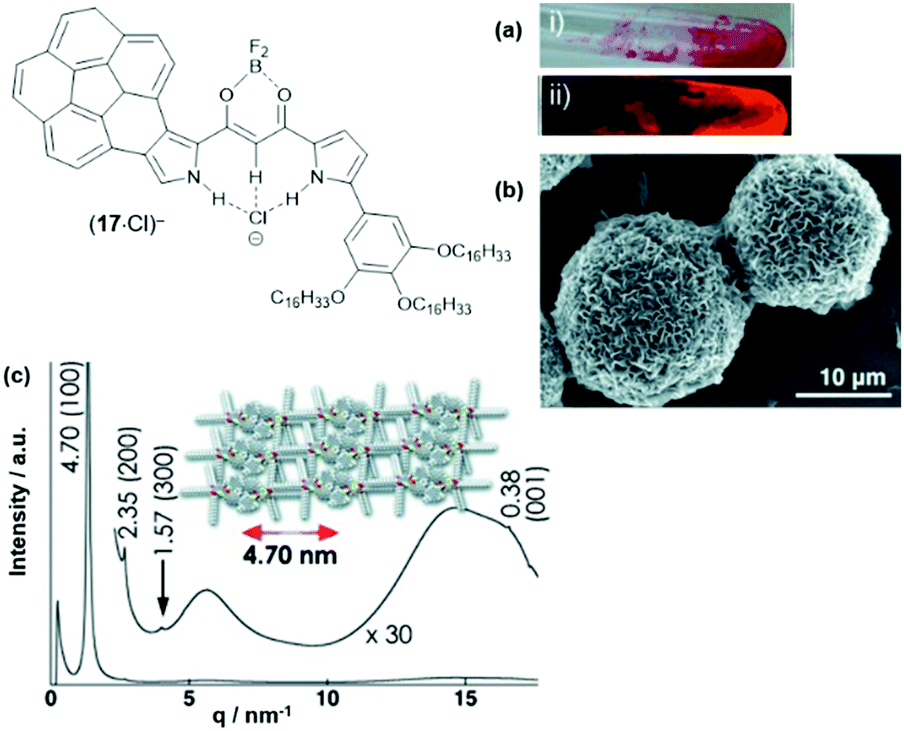 | ||
| Fig. 17 Schematic representation of the π-electronic anion (17·Cl)−. (a) Photographs of (14a)(17·Cl) in 1,4-dioxane under visible (i) and UV light (ii). (b) SEM image of the precipitated (14a) (17·Cl).(c) XRD pattern of (14a)(17·Cl) at 80 °C on cooling from the Iso phase; the proposed supramolecular structure of the assembly is also shown.55 Figure reproduced with kind permission; © 2013 Wiley-VCH. | ||
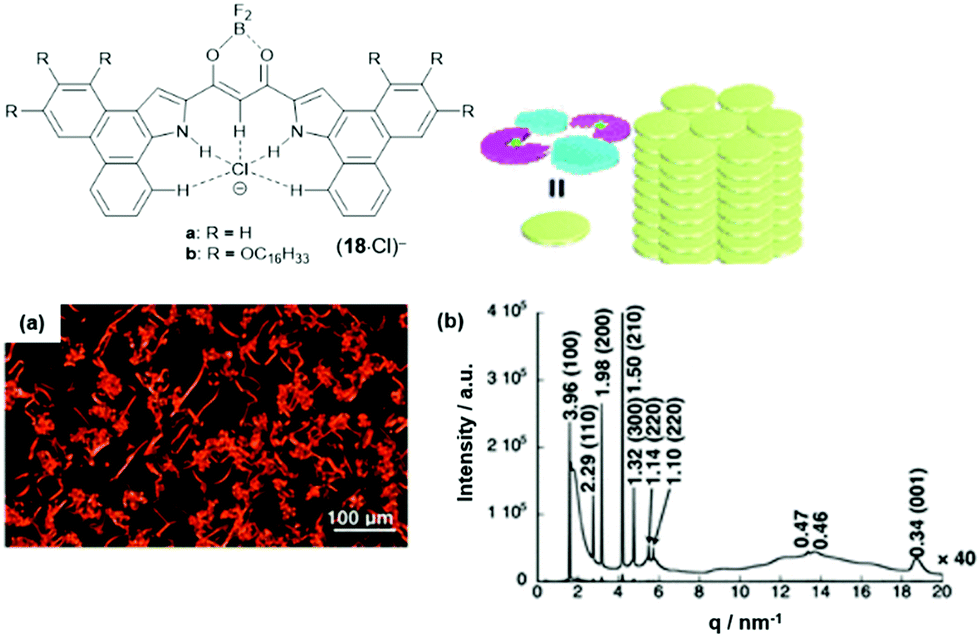 | ||
| Fig. 18 Schematic representation of the phenanthro[b]-fused π-electronic anion (18·Cl)−. (a) POM image of (14a)(18b·Cl) at 180 °C upon cooling. (b) Synchrotron XRD pattern of (14a)(18b·Cl) at 150 °C upon cooling; the proposed supramolecular packing mode of the assembly is also shown.56 Figure reproduced with kind permission; © 2019 American Chemical Society. | ||
Flood and co-workers pointed out that the aforementioned anion-receptor molecules were able to bind only halide ions; whereas, bulky anions, e.g., BF4− or PF6−, did not yield planar π-electronic anions due to a mismatch with the receptors’ binding sites (thus, planar stacking of the chromophores was prevented). To overcome this limitation, these authors used the so-called “cyanostar” receptor (CS, 19) which forms highly stable 2:1 sandwich-type complexes with large anions, e.g., BF4 (logβ2 ≈ 12).57 When combined with TOTA+ (2), the hierarchical self-assembly of the components gave a red-emitting material – both in solution as well as in the solid state, i.e., single crystals and drop-cast films; note, that (2)BF4 and 19 alone are green and blue emitting, respectively (Fig. 19a).58 A four-step assembly mechanism was identified by NMR as well as UV/vis absorption and emission spectroscopy: binding of the anion to 19, formation of an ion-paired salt, 1D columnar ππ-stacking and, eventually, crystallization (Fig. 19b). The photophysical studies revealed further that the supramolecular species {(2)[(19)2BF4]}n possessed its own excited-state manifold, significantly different from that of the TOTA+ cation. This CT state originated from the ππ-stacking interactions between the π-electronic ions. Thus, hierarchical ion-pairing assembly cannot only be applied to fabricate materials with LC behavior but also to form supramolecular architectures with rich opto-electronic properties. In both cases, the photophysical characteristic arise from the mixing of the ionic components and, therefore, represent a versatile tool in designing optical materials.
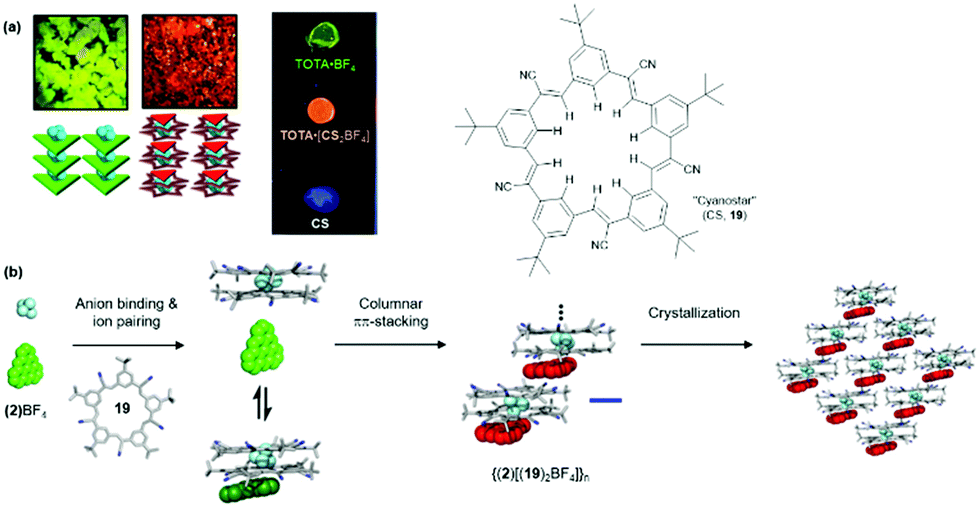 | ||
| Fig. 19 (a) Photographs of the single crystals (left) and drop-cast films (right) showing the characteristic emission of the individual components or of the supramolecular assembly. (b) Schematic representation of the proposed self-assembly mechanism of (2)(BF)4 and 19.58 Figure reproduced with kind permission; © 2017 American Chemical Society. | ||
Supramolecular assemblies based on nitrogen-centered triangulenes
Due to their neutral structure, the N-heterotriangulenes exhibit a self-assembly behavior that basically results from ππ-stacking interactions. The solid-structure of 7 was reported by Field and Venkataraman in 2002.28 The planar molecules densely packed into 1D columns which reported a herringbone-type ordering between the planes (plane-to-plane distance of 3.46 Å, Fig. 20). This packing mode already suggested the potential applicability of such planar building blocks to assemble nanostructures with opto-electronic and LC properties.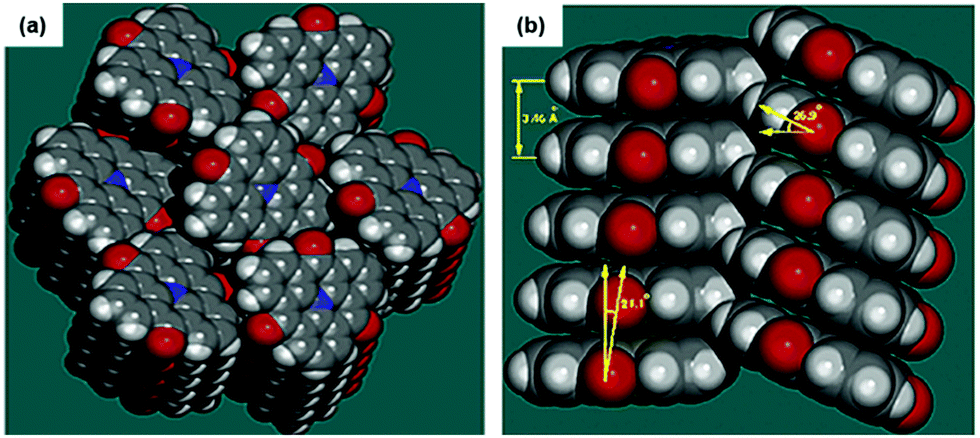 | ||
| Fig. 20 Illustration of the solid-state structure of 7 (a: top view; b: side view).28 Figure reproduced with kind permission; © 2017 American Chemical Society. | ||
In order to improve the solubility and, by the same time, modulate the photophysical properties as well as the aggregation behavior, the parent N-heterotriangulene 7 was modified by introducing substituents on the phenyl moieties.27 In one of the earliest examples in this respect, Zhang et al. attached carbazole-based dendrons (1st (G1) and 2nd generation (G2)) to the core structure of 7.59 These donor–acceptor-type featured an intramolecular CT transitions and aggregation-induced emission (AIE) was observed. The latter one is indicative for the planarization of the molecules due to intermolecular non-covalent interactions at high concentrations or in thin-solid films. For the derivatives with G1-substituents, a head-to-tail interaction (the so-called J-type aggregation) and not a columnar packing due to ππ-stacking interactions was concluded from UV/vis absorption and emission spectroscopy results, the derivatives with the large G2-substituents did not show this behavior, since the bulky dendrons prevented any non-covalent intermolecular interactions. Thus, placing sterically demanding moieties onto the core of 7 is beneficial for increasing the solubility and enables to adjust the photophysical and electrochemical properties, but is rather counterproductive with respect to the ability of the molecules to assemble into highly ordered arrays via non-covalent interactions.
Chen and co-workers introduced slim ethynylphenyl moieties by the Sonogashira-type cross-coupling reaction and investigated the self-assembly behavior of the resultant triangulene 20a in a binary methanol/dichloromethane solvent mixture.60 A two-level supramolecular polymerization of 20a, driven by concerted ππ-stacking and hydrophobic interactions, yielded nanowires which eventually afforded well-organized microrods (Fig. 21). XRD and X-ray scattering measurements confirmed the long-range ordering within the assemblies. The diameter of the nanowires, as determined by TEM imaging, was in good agreement with the one for the 1D columnar stack as derived from the XRD analysis (ca. 3 nm).
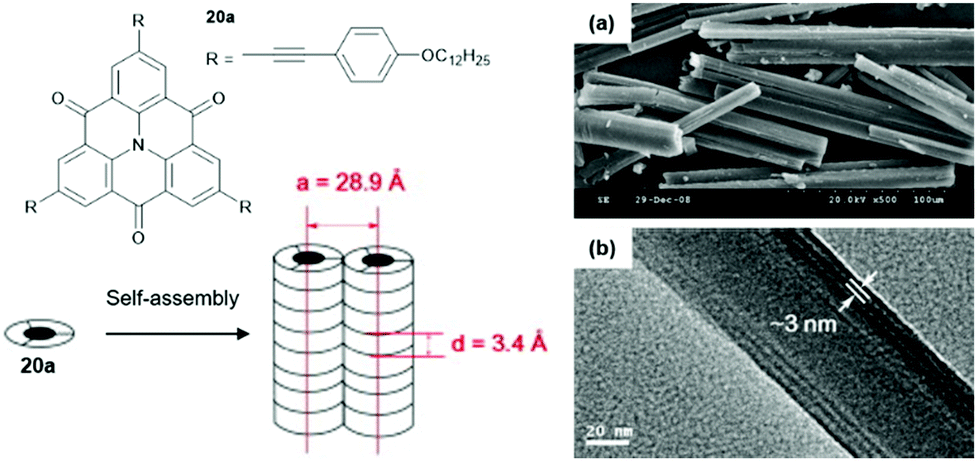 | ||
| Fig. 21 Schematic representation of the two-step self-assembly of 20 into microrods. A representative SEM (a) and a TEM image (b) are also shown.60 Figure reproduced with kind permission; © 2009 Elsevier B.V. | ||
Against the same background, Müllen and co-workers synthesized a small library of carbonyl-bridged N-centered triangulenes 20 in which long alkyl chains were attached to the core via different types of spacer units (Fig. 22).24 The aliphatic chains enabled a good solubility of all derivatives in broad range of organic solvents; however, the chemical structure and bulkiness of the substituents as well as the processing conditions had a profound influence on the supramolecular self-assembly of 20. In detail, the phenyl-bridged derivative 20c yielded columnar disc-like assemblies in solution and in the solid state; the derivatives with ethynylphenyl spacers (20d/e), however, revealed such a behavior only in the solid state. Within the series, 20b showed the lowest degree of aggregation in solution; whereas, well-aligned fibers could be fabricated by dip-coating from solution (Fig. 23a).24,61 As concluded from the electron-diffraction pattern, the highly crystalline fibers were formed by 1D columnar arrays of ππ-stacked molecules. In case of 20g, the solid-state structure could be visualized by XRD analysis of single crystals. The cofacial ππ-stacking of the molecules gave rise to an overall columnar arrangement (Fig. 23b).24 Thermotropic LC behavior was observed for the ethynylphenyl-equipped derivatives 20e/f which ordered themselves in homeotropic fashion (Fig. 24, no LC properties were found for the crystalline materials 20b–d).
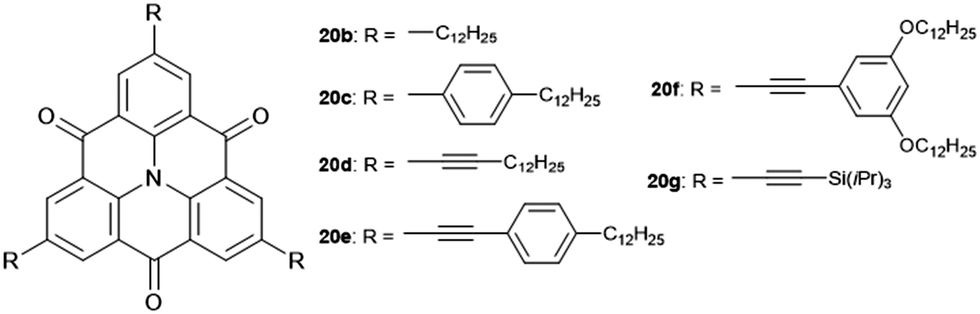 | ||
| Fig. 22 Schematic representation of the two-step self-assembly of 20 into microrods. A representative SEM (a) and a TEM image (b) are also shown.60 Figure reproduced with kind permission; © 2009 Elsevier B.V. | ||
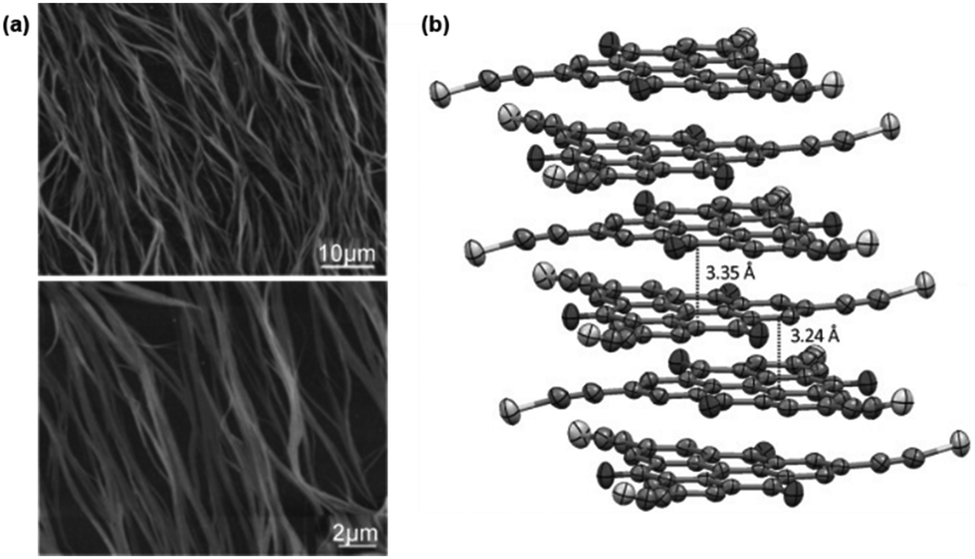 | ||
| Fig. 23 (a) Representative tapping-mode AFM images of the fibers fabricated from 21b.61 (b) Representation of the solid-state structure of 21g (side view).24 Schematic representation of the two-step self-assembly of 20 into microrods. A representative SEM (a) and a TEM image (b) are also shown.60 Figure reproduced with kind permission; © 2011 and 2009 Wiley-VCH, respectively. | ||
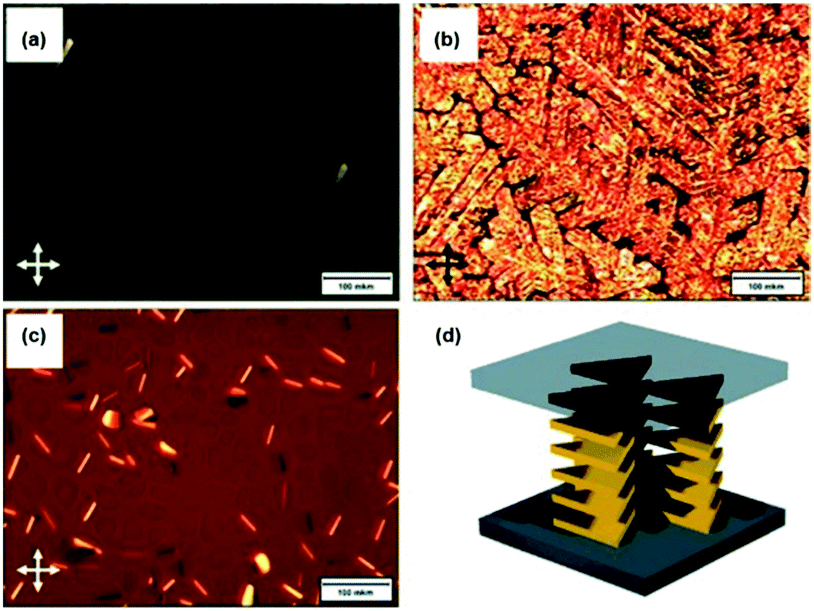 | ||
| Fig. 24 POM images of 20e (a: at 140 °C; b: at 30 °C) and 20f (c: at 30 °C). (d) Illustration of the homeotropic packing of the discotic molecules.24 Figure reproduced with kind permission; © 2009 Wiley-VCH. | ||
The multichromophoric compounds 21 were synthesized by Haedler et al. and the photophysical investigations revealed intramolecular CT processes (the direction of this depended on the chromophores in the periphery).62 The derivative 21b formed a fluorescent organo-gel even in the sub-mM concentration regime; thereby, the CT characteristics, as observed in solution, were retained. According to AFM and TEM imaging, this gel comprised 1D nanofibers with molecular-scale diameters (Fig. 25a).63 The fiber formation was attributed to a highly ordered cofacial ππ-stacking of the molecules, supported by hydrogen-bonding interactions involving the amide moieties of the pending groups. Remarkably, the fibers exhibited the ability to efficiently transport energy over μm distances, even at room temperature (i.e., basically limited only by the fiber lengths). Photoluminescence imaging was applied to quantify this long-range energy transport (Fig. 25b and c).
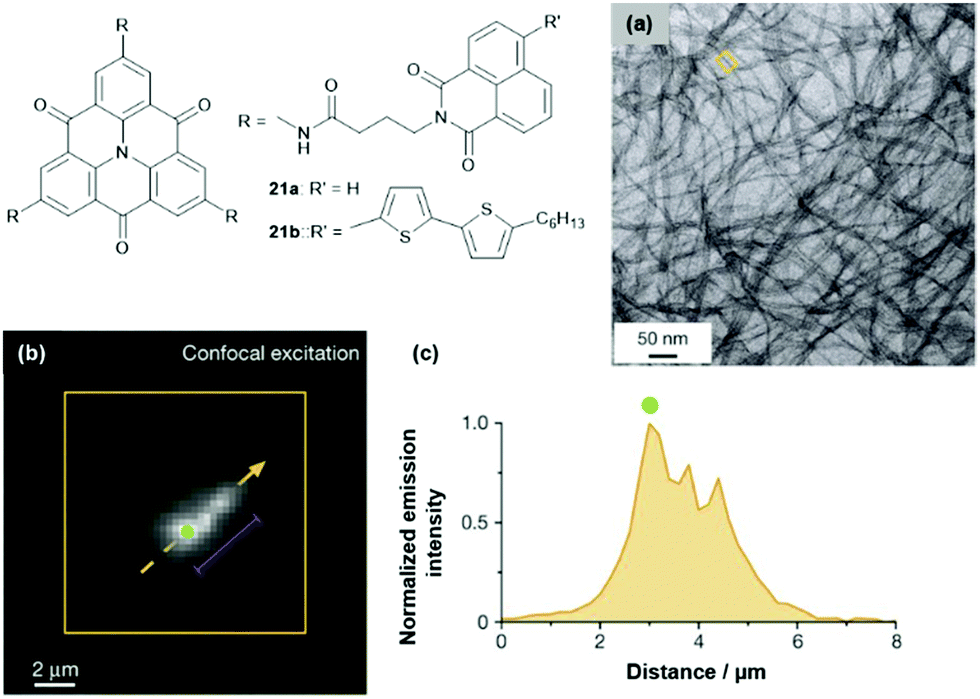 | ||
| Fig. 25 Schematic representation of the multichromophoric derivatives 21. (a) Representative TEM image of the organo-gel formed by 21b. (b) Representative photoluminescence image of a selected fiber. (c) Intensity profile along the orange dashed arrow in (b); the distance corresponds to the length along the arrow.63 Figure reproduced with kind permission; © 2015 Springer-Nature. | ||
The supramolecular polymerization of the chiral carbonyl-bridged N-heterotriangulenes 22 was studied by the Schmidt and the Meijer group.64 For both enantiomers, two distinctly different self-assembly pathways were identified (Fig. 26). The authors proposed an isodesmic supramolecular process, which could be trapped by cooling to 7 °C (state A). Moreover, a cooperative nucleation–elongation gave rise to a thermodynamically stable state B at the same temperature (induced by first undercooling to −5 °C). An in-depth discussion of the mechanisms of these two prominent types of supramolecular polymerizations can be found elsewhere.65 In both these states, a helical superstructure – arising from the same type of aggregation as for 21b – was formed. Further, the supramolecular polymerization proceeded in a controlled fashion when two samples of the same enantiomer, one in state A and the other in state B, were combined; thereby, the kinetically trapped state evolved into the thermodynamically favored one. Remarkably, this process was found to be highly enantioselective – no such transition was identified if the states formed of different enantiomers were mixed (e.g., combining (R)-22 in the state A with (S)-22 in the state B). It was concluded that a fundamental understanding of the self-assembly mechanism is as important as the structural design of the molecular components in order to create highly ordered functional supramolecular materials.
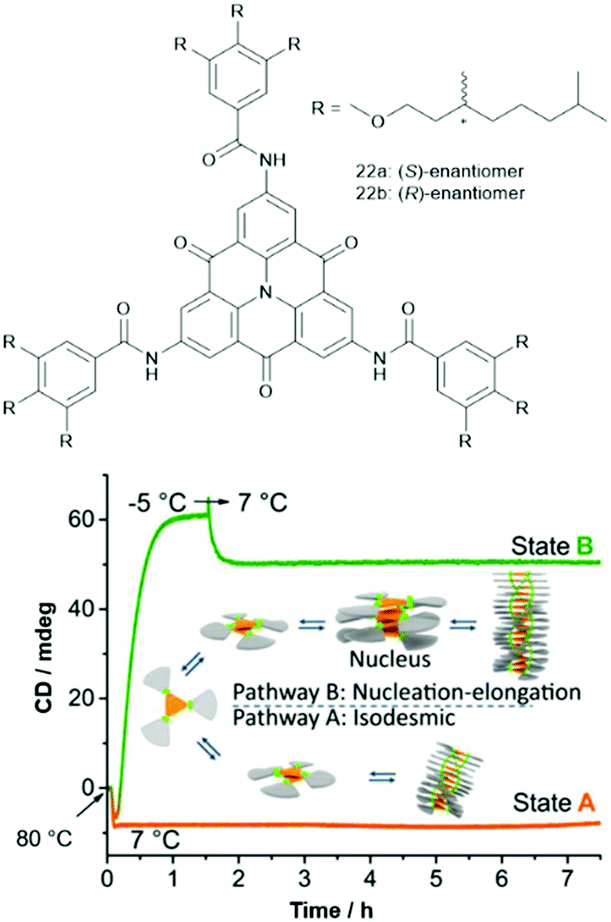 | ||
| Fig. 26 Top: Schematic representation of the chiral N-heterotriangulenes 21. Bottom: Illustration of the supramolecular polymerization of 21 which could be trapped in two different states by cooling.64 Figure reproduced with kind permission; © 2015 Springer-Nature. | ||
In 2014, Yamaguchi and co-workers reported that π-extension of parent 7 can also be achieved via the bridging positions.66 This finding enabled the synthesis of π-extended derivatives featuring a rich photophysical and electrochemical behaviour.27 In view of the supramolecular chemistry of these materials, the steric crowding around the exocyclic alkenyl moieties often prevents a planarization and, thus, the self-assembly into highly ordered architectures is obsolete. For example, the structure of the derivatives 23 and 24 was found to be slightly bowl-shaped and not planar (as for their aforementioned carbonyl-bridged analogues 20b and 20g, Fig. 27).67
 | ||
| Fig. 27 Schematic representation of the N-heterotriangulenes 23 and 24 featuring a π-extension via their bridging positions. | ||
Very recently, Dorca et al. demonstrated the pathway complexity of the 1D supramolecular polymerization of a chiral, dicyanovinyl-substituted derivative of 22.68 The authors claimed that their study represented the first example where the conformational change of a monomer enabled the pathway complexity of a supramolecular polymerization, i.e., the formation of two different aggregates from one type of molecular unit. The bowl-shaped monomer 22 existed also in a planar, yet metastable conformation. Both of these conformers were found to assemble into different types of aggregates – a kinetic as well as a thermodynamic one (Fig. 28). The stability of these aggregates was high enough to enable a co-existence in solution at room temperature (the interconversion between the different aggregates was energetically unfavored due to the steric hindrance arising from the substituents in the bridging positions). However, heating of the mixture resulted in the disassembly of the aggregates and 22 adopted only the thermodynamically favored C3-symmetric conformation. Upon cooling down to room temperature, the conformer afforded exclusively helically shaped columnar assemblies with distinct circular dichroism (CD) signatures which could be correlated to the chiral monomers.
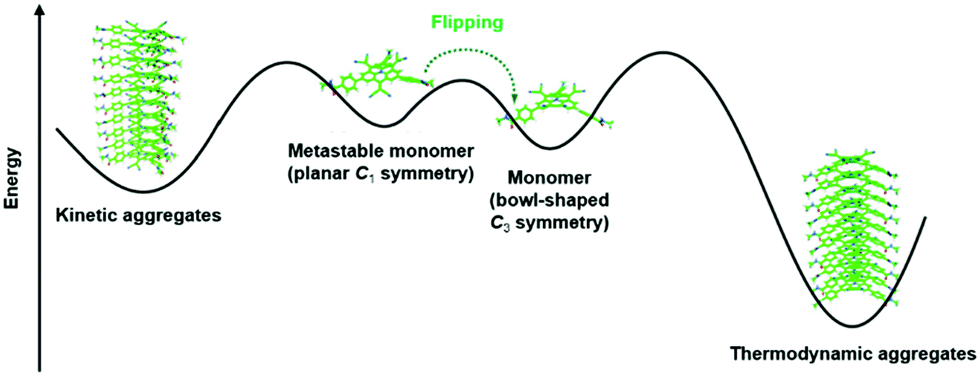 | ||
| Fig. 28 Illustration of the pathway complexity of a dicyanovinyl-equipped N-centered triangulene – two different types of supramolecular aggregates are accessible from the two conformers.68 Figure reproduced with kind permission; © 2019 American Chemical Society. | ||
In contrast to the rigid-planar carbonyl-bridged N-heterotriangulene 7, its dimethylmethylene-bridged counterpart 6 is slightly more flexible and a shallow bowl-shaped structure was verified by X-ray single crystal analysis.29 Moreover, the sp3-hybridized bridge atoms introduce an out-of-plane steric effect which basically excludes any cofacial ππ-stacking interactions (at least in the solid state). On the other hand, 6 is much more soluble and represents, due to the electron-rich nature, a useful precursor for introducing substituents in the para-positions of the phenyl rings. Consequently, a broad range of derivatives has been synthesized in the last decade and were, due to their planarized structure, found to exhibited a much higher degree of π-conjugation when compared to the members of the related triarylamine family.27 The π-extended derivatives of 6 have, for instance been used as hole-transport material in organic light-emitting devices (OLEDs), as photosensitizers in solar-cell devices or in non-linear optics due to their two-photon absorption behavior.
The dimethylmethylene-bridged N-heterotriangulenes do not enable a supramolecular self-assembly into 1D columnar materials. However, the straightforward threefold symmetrical functionalization of parent 6 yields derivatives suited for an assembly into 2D structures, i.e., sheet-like motives. This approach can be realized on the basis of covalent as well as supramolecular chemistry. As an example for the formation of covalent 2D structures derived from small-molecule N-heterotriangulene precursor molecules, the work of Bieri et al. has to be mentioned: the formation of such a sheet-like structure was achieved by assembling the symmetrical tribromo-derivative of 6 onto a Ag(111) surface, followed by thermal treatment at 300 °C.69 The same strategy was followed by Steiner et al. to prepare a covalent 2D network of carbonyl-bridged N-heterotriangulenes on an Au(111) surface.70 In order to utilize supramolecular chemistry for the very same purpose, the substituents attached to 6 need to function as supramolecular binding sites to force the molecular components into highly-ordered arrays on the basis of strong and directional, yet reversible non-covalent interactions (Fig. 29). In such a supramolecular process, the non-covalent bonds are formed and cleaved until eventually an equilibrium is reached in which the assembly features a high degree of order and – ideally – only a small number of structural defects can be found.71
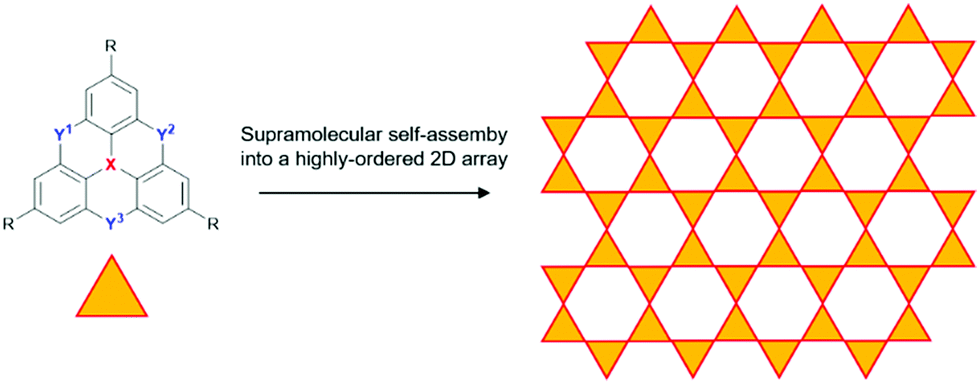 | ||
| Fig. 29 Idealized schematic representation of the self-assembly of heteroatom-containing triangulenes into a supramolecular sheet-like array. | ||
In this respect, the cyano group represents, despite its chemically simple structure, already a highly versatile functionality due to the highly asymmetric charge distribution that can be utilized to form ordered structures via intermolecular dipole–dipole72 or hydrogen-bonding interactions73 as well as, even more efficiently, by metal-to-ligand coordination.74 Against this background, Kivala et al. prepared the threefold cyano-functionalized N-centered triangulene 25 and studied its adsorption onto coinage-metal surfaces, i.e., Cu(111), Ag(111) and Au(111).75,76 Scanning tunneling microscopy (STM), low-energy electron diffraction, X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) calculations allowed the authors to identify all three above-mentioned bonding motifs. A hexagonally densely packed pattern of 25, stabilized by hydrogen-bonding interactions, was observed in the case of Ag(111). In contrast to this, two distinctly different partially porous phases were identified in the submonolayer coverage of Au(111) – the densely-packed α-phase originated from an interplay between dipolar-coupling and hydrogen-bonding interactions; whereas, the formation of the (filled) β-phase was explained by additional metal-to-ligand coordination involving metal centers of the Au(111) substrate (Fig. 30). However, on a Cu(111) surface only small areas of hexagonally close-packed sheets stabilized by metal-to-ligand coordination and areas of rather disordered molecules could be found. Thus, the role of the substrate also needs to be considered carefully when aiming for the assembly of supramolecular sheets on solid supports. The particular relevance of such porous sheets with respect to materials science, in particular when aiming for nanoelectronic devices, is highlighted by the ferromagnetic half-metallicity reported for a covalent material composed of a related N-centered triangulene monomer.77
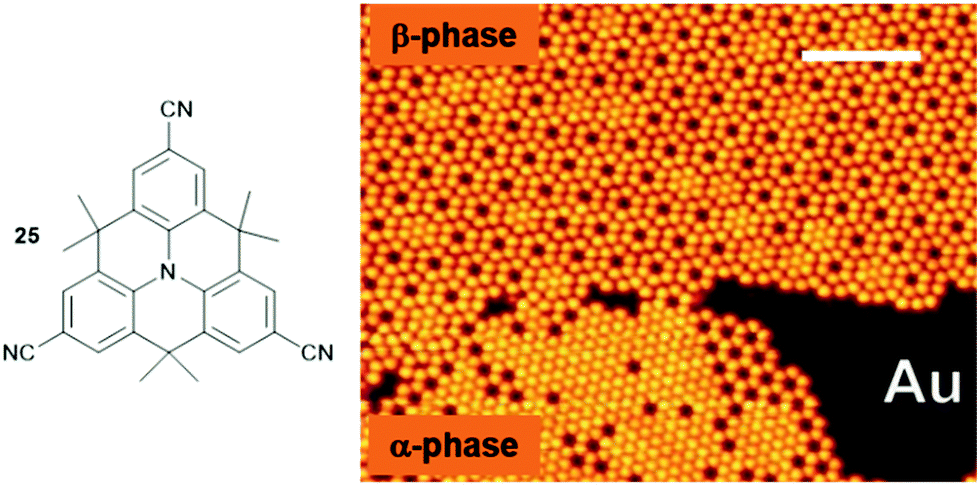 | ||
| Fig. 30 Schematic representation of the CN-functionalized N-centered triangulene 25. The adsorption onto an Au(111) substrate yielded two different phases as visualized by STM imaging (scale bar: 12.5 nm).75,76 Figure reproduced with kind permission; © 2014 Wiley-VCH. | ||
The Kivala group also transformed the CN-groups into carboxylic acids (26a) and 2,4-diamino-1,3,5-triazin-6-yl (DAT) units (26b), thereby introducing moieties designated for the self-assembly into 2D structures via self-complementary hydrogen-bonding interactions.23 Porous 2D and 3D structures formed by hydrogen-bonding interactions have attracted considerable attention and have recently been reviewed elsewhere.78 The solid-state structure of 26b was investigated by X-ray crystallographic analysis and confirmed the formation of regular planar sheets in which the molecules were linked by DAT-dimers (supported by hydrogen-bonding interactions involving co-crystallized solvent molecules). Besides the formation of supramolecular sheets on Au(111) of either of the monomers, also the co-adsorbance of 26a and 26b, representing a donor- and an acceptor-type molecule, was reported (Fig. 31).79 Presumably due to the maximization of the overall number of hydrogen bonds on the cost of the optimization of the hydrogen-bonding strength a mixed network was established. DFT calculations revealed that the band gap between the highest occupied and the lowest unoccupied molecular orbitals (i.e., the HOMO and the LUMO) was defined by both molecules, a fundamental characteristic of donor–acceptor assemblies.
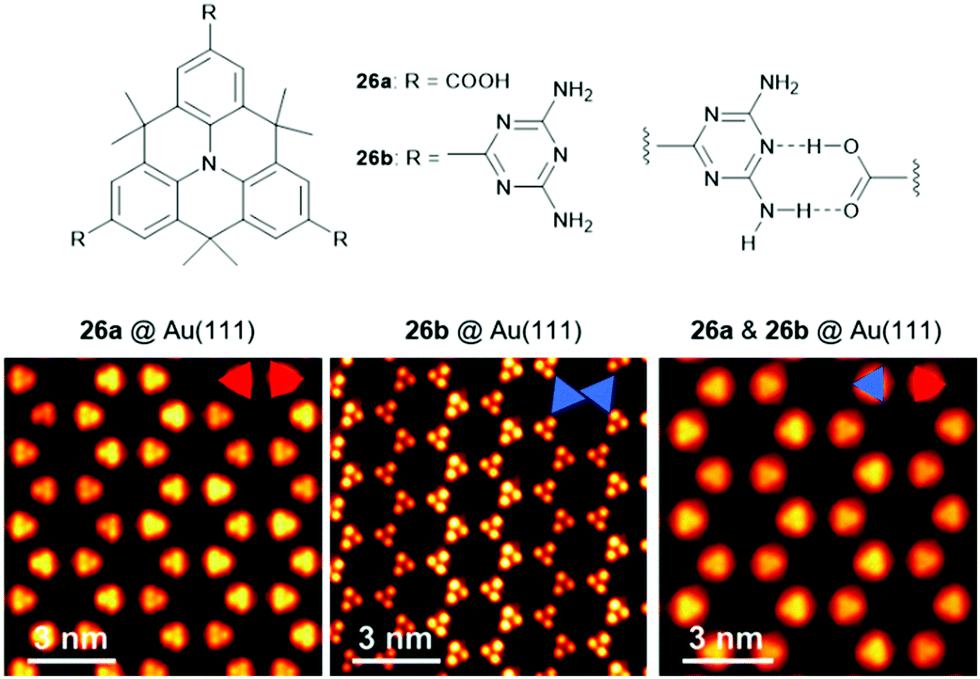 | ||
| Fig. 31 Top: Schematic representation of the functionalized N-centered heterotriangulenes 26; the proposed hydrogen-bonding interaction between the two entities is also shown. Bottom: STM images of the supramolecular sheets formed by 26a, 26b as well as a 1.1 mixture of 26a and 26b (from left to right).79 Figure reproduced with kind permission; © 2018 The Royal Chemical Society. | ||
Supramolecular assemblies based on boron-centered triangulenes
In 2012, Yamaguchi and co-workers reported on the synthesis of the alkyl-bridged boron-centered triangulene 9; the proposed planar structure of this bridged triphenylborane molecules could be verified by single-crystal XRD analysis (Fig. 32).33 However, intermolecular ππ-stacking interactions were prevented due to steric hindrance of the dimethylmethylene bridges (similar to the N-centered analogue 6); instead, a face-to-edge-type orientation of the molecules was found. The B-centered derivative 9 was highly stable against air, moisture, amines and heat. This behavior was rationalized by the structural constraint in 9 (which is absent in the parent triphenylborane), not requiring any further stabilizing bulky substituents. Moreover, the electron-deficient boron atoms retained their Lewis acidity and could reversibly coordinate fluoride ions; thereby, the molecular structure of 9 experienced a plane-to-bowl interconversion.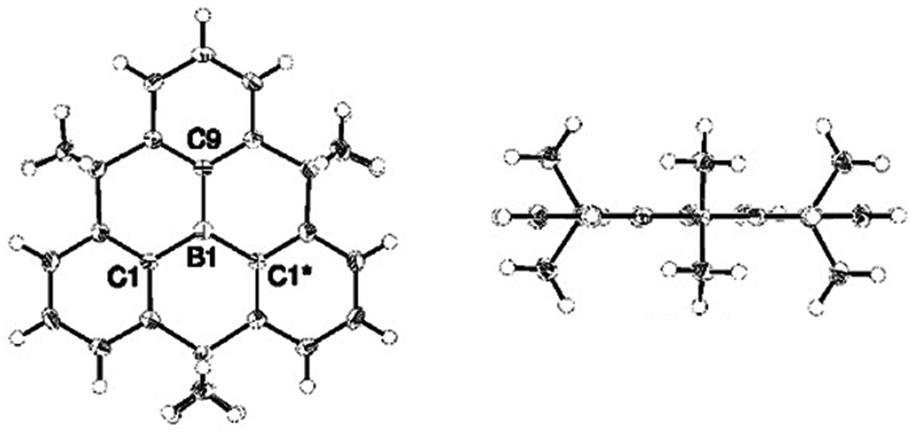 | ||
| Fig. 32 Top (left) and side view (right) of the solid-state structure of 9.33 Figure reproduced with kind permission; © 2012 American chemical Society. | ||
Decreasing the steric demand of the bridges allows for a much stronger intermolecular interaction and columnar assemblies can be obtained. This was shown by Yamaguchi et al. who studied the unsymmetrical derivative 27.32 According to DSC and XRD measurements, a ππ-stacked hexagonal columnar LC phase was assembled from the discotic compound at room temperature (disc-to-disc distance of ca. 3.6 Å, Fig. 33). The photophysical properties were investigated and revealed a blue-shifted and reduced fluorescence of 27 in the LC phase relative to that found in THF solution (LC phase: λem = 427 nm (ϕF = 0.61); THF solution: λem = 457 nm (ϕF = 0.99)). This finding was attributed to the dense packing of 27 in the LC phase, where the interconversion between the planar and the bowl-shaped conformation was impeded. The ordered columnar stacking and the short intermolecular distances in the LC assembly resulted in an ambipolar charge-carrier behavior: electron and hole mobilities of 10−3 and 3 × 10−5 cm2 V−1 s−1, respectively, were determined by time-of-flight measurements.
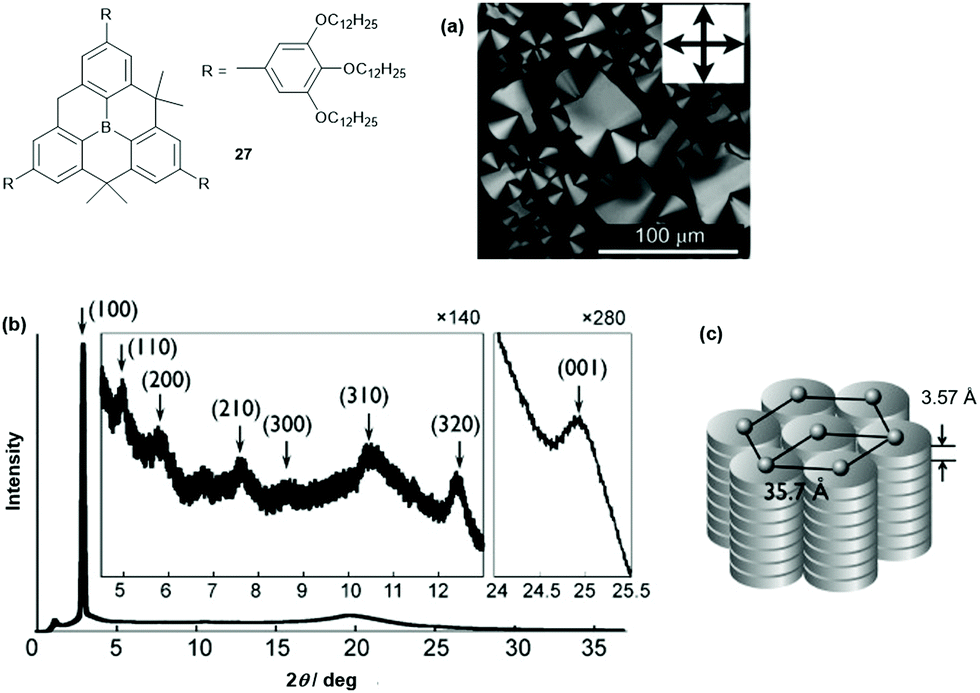 | ||
| Fig. 33 Schematic representation of 27. (a) POM image of 27 (at 70 °C upon cooling from the isotropic liquid; the inset shows the directions of the polarizer and analyzer). (b) XRD pattern of 27 (at room temperature after cooling from the isotropic liquid). (c) Schematic representation of the proposed hexagonal columnar phase of 27. Figure reproduced with kind permission; © 2012 American chemical Society. | ||
The planar heteroatom-bridged B-centered derivatives 28 and 29 were both reported very recently (Fig. 34).14,34 In contrast to the carbon-bridged analogue 9, the oxygen-bridged triangulene 28 exhibited intermolecular interactions in the solid state: due to the absence of steric hindrance, a ππ-stacking motive with a parallel offset and a plane-to-plane distance of ca. 3.38 Å was formed (Fig. 35).34 However, the phenyl substituents of 29, oriented perpendicular to the planar triangulene unit, prevented such a packing for steric reasons.
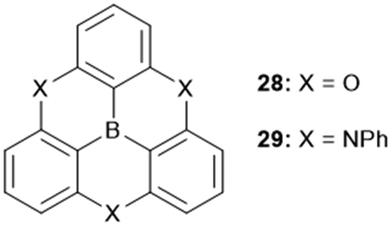 | ||
| Fig. 34 Schematic representation of the heteroatom-bridged boron-centered triangulenes 28 and 29. | ||
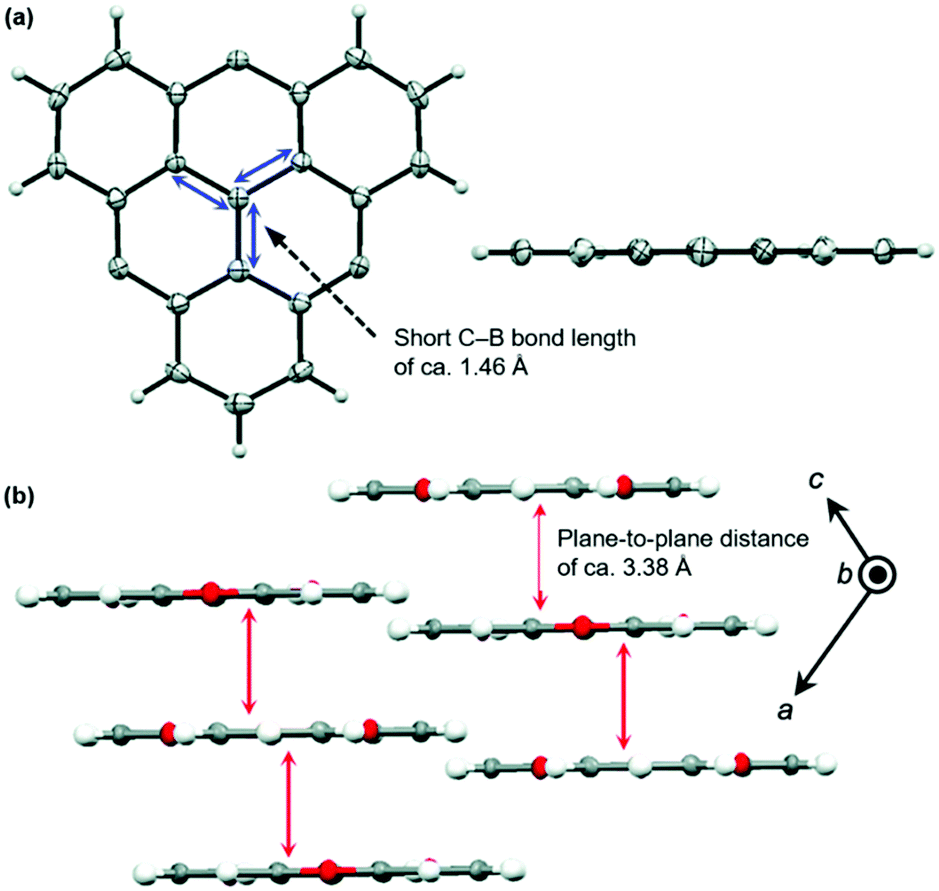 | ||
| Fig. 35 (a) Solid-state structure of 28 (left: top view; right: side view). (b) Cross section of the solid-state packing.34 Figure reproduced with kind permission; © 2016 The Royal Chemical Society. | ||
Supramolecular assemblies based on bowl-shaped heterotriangulenes
In contrast to the planar N-centered triangulenes, their phosphorous-centered counterparts were persistent bowl-shaped or convex/concave structures. This difference results from the presence of the P-atom in a threefold respect: firstly, the significantly larger covalent radius of phosphorous (when compared to B, C and N) urges the atom out of plane even when incorporated into a π-conjugated system.80 Secondly, the molecular structure is configurationally rather stable due to the large energy barrier for the pyramidal inversion.81 Finally, Bent's rule prevents phosphorous from being hybridized easily (P is by far more electropositive as C or N).82 The dimensions of the bowl-shaped P-centered triangulenes are defined by two parameters – the bowl depth and the cone angle (d and θ, respectively, Fig. 36). These parameters, in turn, are of relevance when considering the binding of appropriate guest molecules within the bowl (e.g., fullerenes, vide infra). Moreover, the structural and electronic features of such molecules also depend on the oxidation state of the central phosphorous atom: the lone electron pair of a trivalent P-center gives rise to a Lewis basic character; whereas, electronegative substituents in pentavalent species are known to induce Lewis acidity.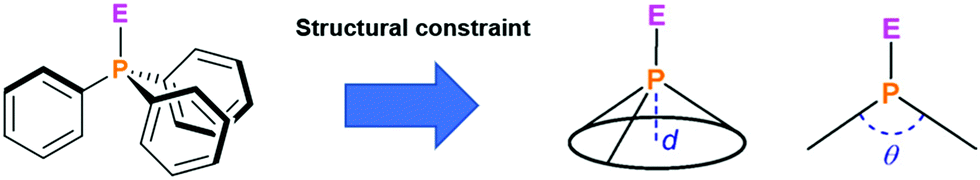 | ||
| Fig. 36 Schematic representation of triphenylphosphine and its axially occupied derivatives. The bowl depth (d) and the cone angle (θ) represent the key parameters when structural constraining the parent structure.27 Figure preproduced with kind permission; © 2019 American Chemical Society. | ||
Yamamura et al. pointed out that the molecular structure has a profound impact on the packing behavior and two different types of packing modes can basically be obtained (Fig. 37): ordered 1D concave/convex stacks (A and B) and less ordered assemblies (e.g., C).22
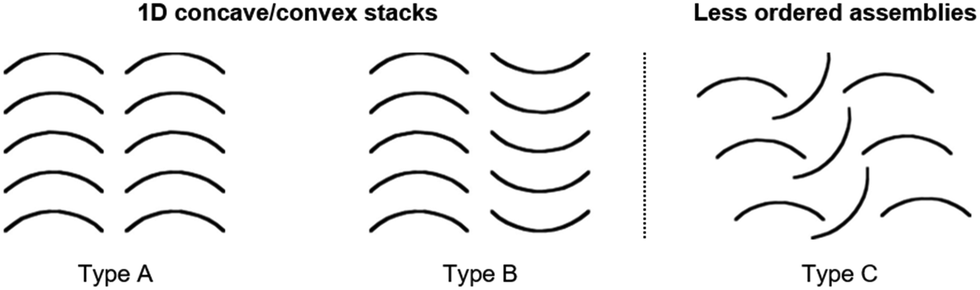 | ||
| Fig. 37 Schematic representation of the fundamental packing modes of bowl-shaped molecules.22 Figure reproduced with kind permission; © 2016 American Chemical Society. | ||
A variety of O- and/or S-bridged P-centered triangulenes 30 was prepared and their self-assembly behavior in solution as well as in the solid state was investigated (Fig. 38). The C3v-symmetric O-bridged derivative 30a with pyroelectric properties was reported by Krebs et al. already in 1997.21 The low sublimation temperature of 30a suggested only weak intermolecular interactions; however, a highly ordered packing mode due to ππ-stacking interactions was observed in the crystalline state (Fig. 39). The authors proposed a sufficiently high dipole moment (μ) as the main driving force for this self-assembly behavior. This assumption was verified later by applying multipolar modelling studies: the dipole moment of 30a was enhanced by ca. 42% when going from the solubilized to the crystalline state (μ = 4.7 D).83
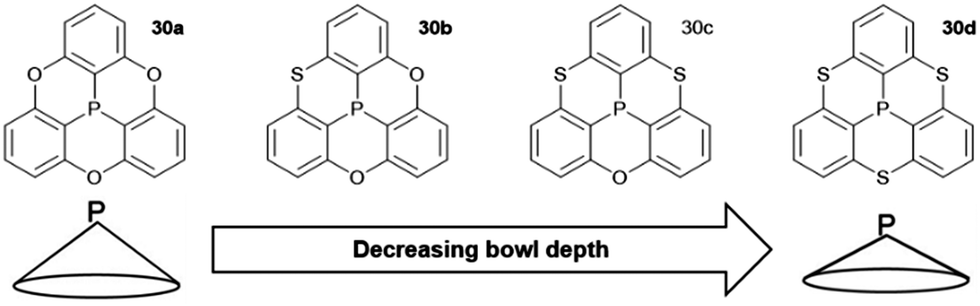 | ||
| Fig. 38 Schematic representation of the O- and/or S-bridged P-centered triangulenes 30. | ||
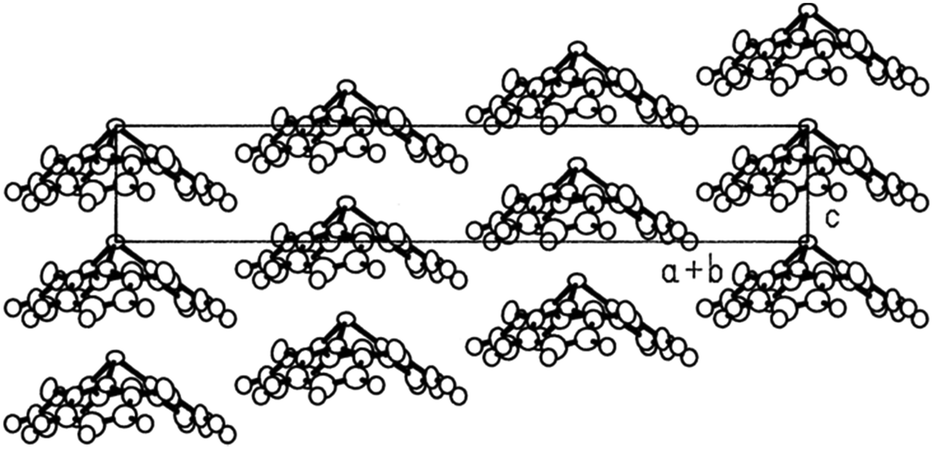 | ||
| Fig. 39 Representation of the molecular packing of 30a in the crystalline state. Figure reproduced with kind permission; © 1997 American Chemical Society. | ||
The S-containing derivatives 30b–d were reported by Yamamura et al.22 With increasing number of S-bridges, the cone angles increased and, consequently, the bowl depths decreased (30a: d = 2.43 Å; 30d: 2.03 Å). Not only the molecular parameters, but also the packing behavior was affected by the nature of the bridges. The C3v-symmetric derivative 30d again assembled into highly ordered 1D columnar stacks of type B (according to Fig. 37). On the contrary, the Cs-symmetric representatives 30b and 30c stacked in a less ordered zig-zag-type mode.
The same authors also investigated the self-assembly behavior of various “bowl chiral” derivatives of 30a (Fig. 40). Basically, the self-assembly of such chiral bowl-shaped molecules yields materials with supramolecular chirality which, in turn, might be used to create materials applicable in polarization-related photonics or chiral organic conducting systems (the chiroptical switching represents the base in this context).84
 | ||
| Fig. 40 Schematic representation of the chiral bowl-shaped P-centered triangulenes 31. | ||
In the crystalline state, rac-31a featured an ordered hexagonal array of anisotropic 1D columns of either enantiopure (M)-31a or (P)-31a molecules ππ-stacked in a convex/concave-type fashion.20 Thereby, the alignment of the columns corresponded to the type-B packing (Fig. 37) in which each enantiopure column was directly opposed to the neighboring one, which was formed by molecules of the second enantiomer (Fig. 41a). The XRD analysis of single-crystalline (M)-31a, however, revealed a different, i.e., an orthorhombic packing mode: each convex/concave columnar ππ-stack was surrounded by five columns – four of these aligned in the opposite and one aligned in the same direction (Fig. 41b).
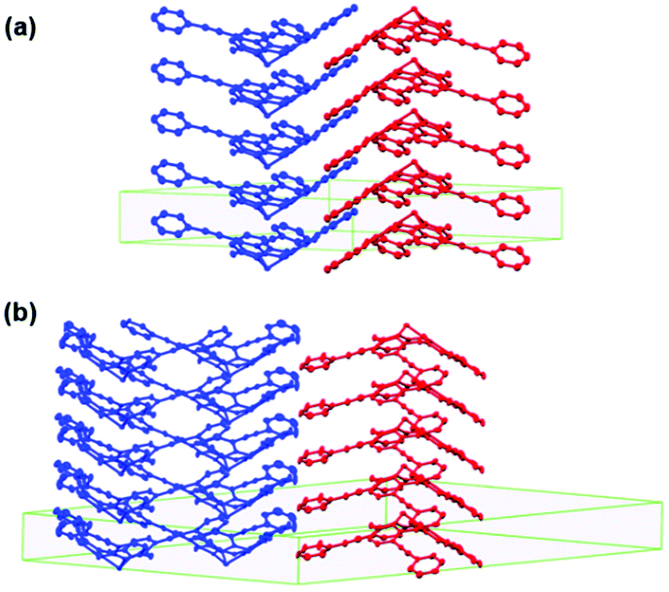 | ||
| Fig. 41 Representation of the packing of rac-31a (a) and (M)-31a (b) in the crystalline state.20 Figure reproduced with kind permission; © 2015 The Royal Chemical Society. | ||
The derivative 31b possessing alkoxy side chains assembled into columnar LC phases.20 According to DSC and XRD measurements, the LC phases derived from the enantiopure compounds were more ordered than the one formed by racemic 31b. Remarkably, the circular dichroism (CD) signal of the thin-solid film of enantiopure 31b had the opposite signature as in solution. This difference could directly be attributed to the self-assembly of the molecules in the condensed state. The reversibility of the chiroptical switching of enantiopure 31b was demonstrated by the multiple transition between the anisotropic liquid and the LC phases.
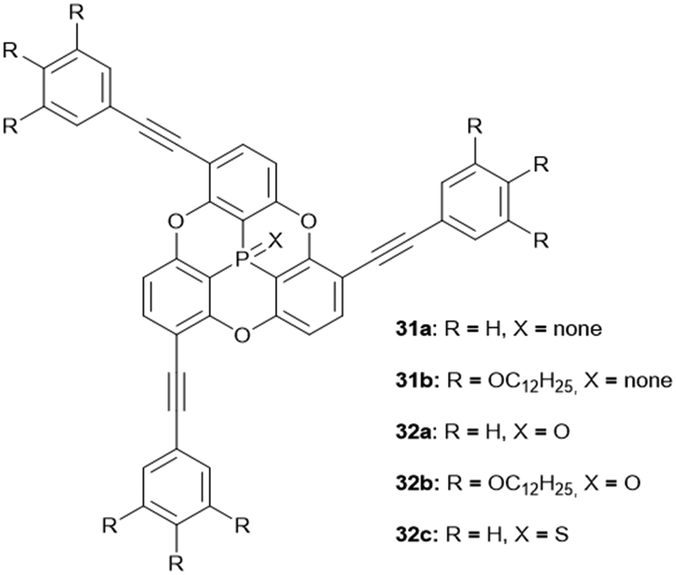
The N-bridged heterotriangulene 32 was reported only recently and did, due to steric hindrance of the pending phenyl moieties, not yield any ordered assemblies.14 The arsenic-centered triangulenes possess similar molecular structures as their phosphorous-containing counterparts;18 however, their self-assembly behavior in solution or in the solid state has not been discussed in the scattered reports on these compounds. The first example for a silicon-centered triangulene derivative was published by Hatakeyama and co-workers; the values of bowl parameters were in the same range as those reported for the similar P-centered derivatives.14 Thus, an application of these young members of the heterotriangulene family in the field of supramolecular host–guest complexation appears feasible. On the contrary, the steric hindrance arising from the pending substituents in concert with the presence of an axial substituent on the heteroatom is expected to prevent the formation of ordered supramolecular convex/concave ππ-stacks. The same also holds true for the various examples for axially-substituted phosphorous-containing triangulenes.27
Beyond 1D and 2D assemblies: miscellaneous supramolecular architectures based on heteroatom-containing triangulenes
Bowl-shaped molecules have attracted considerable attention in the field of supramolecular chemistry due to their ability to bind spherical (convex) guest molecules to their concave face via van der Waals, ππ-electronic and charge-transfer interactions.85 In particular, the recognition of pristine fullerene C60 represents an intensive field of research in this respect.86 For example, the bowl-shaped hydrocarbons corannulene87 and sumanene88 have been reported as potent host molecules, since their curved structures resemble fullerene subunits and, thus, exactly match C60's convex surface.Within this context, Yamamura et al. utilized the chiral P-centered triangulene P-oxide (P)-32a as bowl-shaped host for the complexation of C60.19 In the crystalline state four host molecules were found to almost perfectly cover the guest's surface (Fig. 42). Remarkably, the host–guest interaction was strong enough to withstand ionization: a species assigned to the (P)-32a⊃C60 1.1 complex was observed in the matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrum of the crystalline sample. Since the P-sulfide 32c featured almost the same bowl dimensions as 32a, (P)-32c yielded a similar host–guest complex of 4:1 stoichiometry with C60 in the solid state.20 Due to the lone electron pair, 31a possessed a deeper bowl and, consequently, a sandwich-type 2:1 complex was formed with C60 (Fig. 43). This type of architecture is reminiscent of Haino's biscalix[5]arene–C60 adducts; the CT interaction between biscalix[5]arene-based molecular tweezers and fullerene derivatives was utilized by these authors to assemble discrete supramolecular architectures as well as linear, side-chain or cross-linked supramolecular polymers.89 It may, thus, be concluded that also the ((P)-31a)2⊃C60 motive can be adapted for the very same purpose. However, reports targeting this application are not yet at hand.
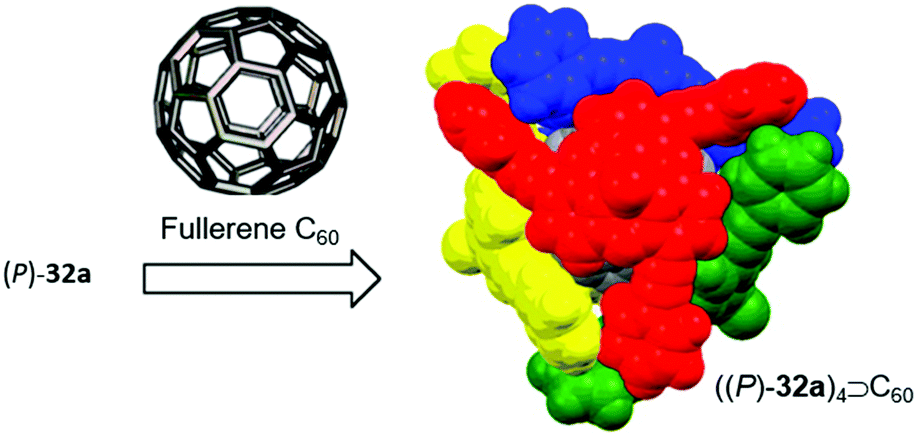 | ||
| Fig. 42 Schematic representation of the formation of a host–guest complex by coordination of four chiral (P)-32a host molecules to a fullerene C60 guest.19 Figure reproduced with kind permission; © 2014 American Chemical Society. | ||
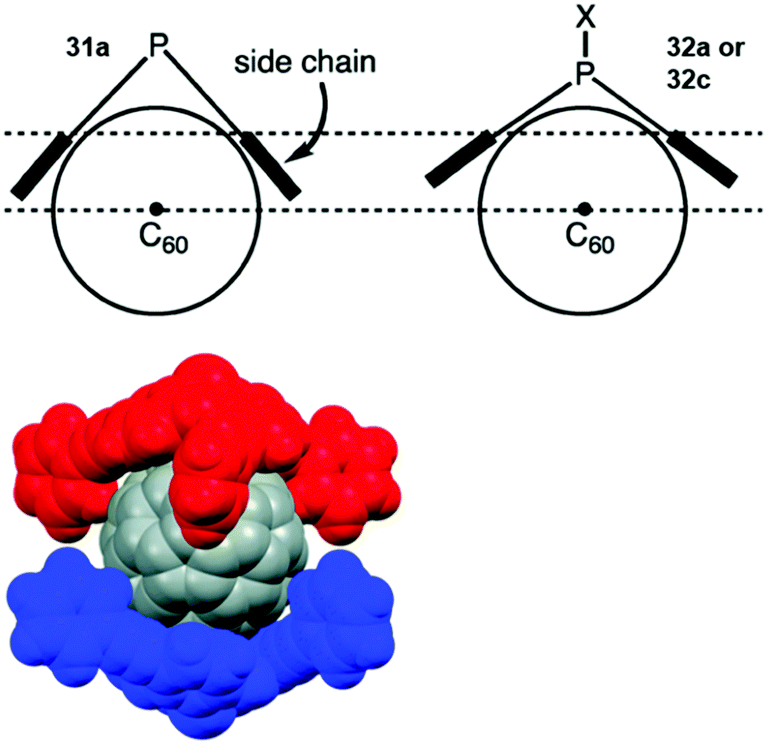 | ||
| Fig. 43 Top: Illustration of the coordination of a P-centered triangulene with a deep (31a) and a shallow bowl (32a/c) to C60. Bottom: Space-filling model of the ((P)-31a)2⊃C60 complex.20 Figure reproduced with kind permission; © 2015 The Royal Chemical Society. | ||
From the highly directional types of non-covalent interactions, halogen bonding (XB) has emerged as a powerful tool in various fields of chemical research – ranging from medical or catalytic applications to crystal engineering or materials science.90,91 Though being still under scientific debate, XB interactions are generally described by Clark's σ-hole concept: basically, the attractive interaction results from an electrophilic region on a molecule's halogen atom (the so-called σ-hole) and an electron-rich sites in another (or the same) molecule.92,93 1,2,4,5-Tetrafluoro-3,6-diiodobenzene represents a prominent ditopic XB donor which was employed by Schaub et al. for the co-crystallization with 33 in a 1:2 ratio.94 X-Ray crystallographic analysis of the single crystals revealed a packing mode in which XB-donor molecules were surrounded by in total six molecules of 33 (Fig. 44). Two of these were coordinated in a linear fashion to the aryl iodide by strong P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯I XB-type interactions (the strength of this interaction was reflected by the very short bonding length of ca. 2.683 Å). The other four molecules of 33 contributed by ππ-stacking, H⋯F as and Ar–H⋯π interactions to the overall packing mode. This (up to now) exclusive example shows how non-covalent interactions, beyond the ππ-stacking of planar or bowl-shaped molecules, can be utilized to force triangulene derivatives into ordered arrays.
O⋯I XB-type interactions (the strength of this interaction was reflected by the very short bonding length of ca. 2.683 Å). The other four molecules of 33 contributed by ππ-stacking, H⋯F as and Ar–H⋯π interactions to the overall packing mode. This (up to now) exclusive example shows how non-covalent interactions, beyond the ππ-stacking of planar or bowl-shaped molecules, can be utilized to force triangulene derivatives into ordered arrays.
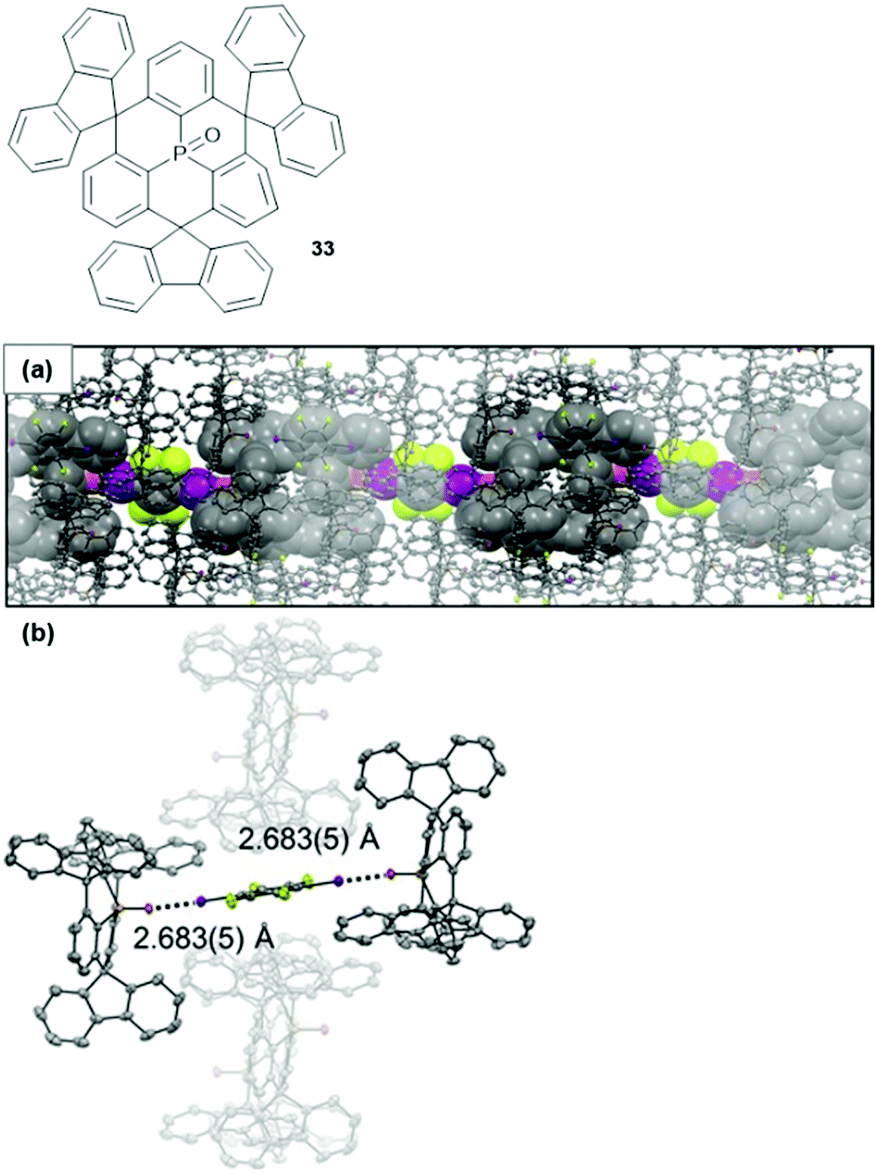 | ||
| Fig. 44 Top: Schematic representation of the P-centered triangulene 33. Bottom: X-ray single-crystal structure of the 2:1 complex of 33 and 1,2,4,5-tetrafluoro-3,6-diiodobenzene (thermal ellipsoids at 50% probability, hydrogen atoms omitted for clarity). (a) Illustration of the packing into chain-like assemblies. (b) Illustration of the linear halogen-bonding interaction contributing to the overall packing mode.94 Figure reproduced with kind permission; © 2017 Wiley-VCH. | ||
Finally, the planar triangulene scaffold has been used as template for the formation of metal complexes. Lacour and co-workers designed a tripodal ligand for the coordination of lanthanide(III) ions in which three tridentate binding sites were attached to the planar TATA+ unit (34, Fig. 45).95 The resultant Eu(III) complex of 34 was characterized by 1H NMR spectroscopy, mass spectrometry and single-crystal XRD analysis. Molecular-modelling studies were performed and supported the experimental data (Fig. 45). Further, [Eu(34)]4+ exhibited the emission behavior as 34 itself; thus, energy transfer from the excitated TATA+ core to the Eu(III) center could be excluded.
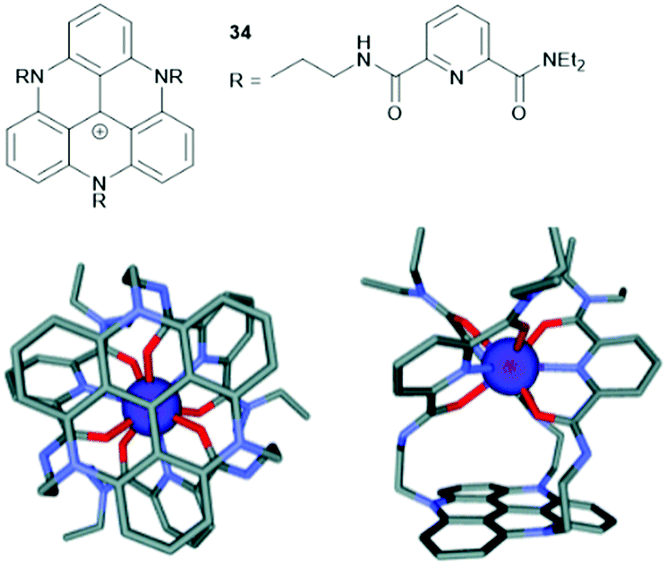 | ||
| Fig. 45 Top: Schematic representation of the tripodal ligand 34. Bottom: Illustration of the structural model [Eu(34)]4+ (Scigress software, hydrogen atoms are omitted for clarity); a top view (left) and a side view (right) are shown.95 Figure reproduced with kind permission; © 2012 The Royal Chemical Society. | ||
The last examples show how hydrogen-/halogen-bonding interactions and metal-to-ligand coordination can be combined with heteroatom-containing triangulene moieties. It can be speculated that the proper combination of these non-covalent forces with the ππ-stacking interactions offered by π-conjugated molecular structure might be of high virtue – not only in crystal engineering but also for the controlled supramolecular assembly of triangulene-based hierarchical 2D and even 3D supramolecular architectures.96 Such (metal-) organic frameworks might be employed in various opto-electronic applications, such as the harvesting of light or photocatalysis.97
Conclusions and outlook
The basic structures of the various types of heteroatom-containing triangulenes have already been known since some decades; however, applying their supramolecular chemistry as a tool to fabricate materials with tunable liquid-crystalline or opto-electronic properties has emerged as field of research rather recently. In particular, the planar boron- or nitrogen-centered representatives have been utilized for the formation dimension-controlled columnar assemblies.27 The same also holds true for the triangulenium cations which have been combined with appropriate anions to afford π-electronic salts.39 For these assemblies opto-electronic applications have already been envisioned. Moreover, the stacking of concave/convex or bowl-shaped derivatives offers one further opportunity to obtain 1D columnar assemblies. The C3-symmetric derivatives equipped with appropriate functionalities have been utilized for the self-assembly into 2D nanosheets via directional non-covalent interactions (e.g., dipole–dipole and hydrogen-bonding interactions, metal-to-ligand coordination). In addition to this, various non-covalent interactions play a pivotal role in the solid-state packing of functionalized triangulene derivatives, an aspect which is of relevance with respect to crystal engineering (i.e., tuning the structural or photophysical properties in the solid state).A broad range of coordination nanosheets (CONASHs) has been prepared from symmetrical planar building blocks and (transition) metal ions.97 Such materials are known to exhibit electron or proton conductivities, electrochroism as well as photo- or (photo-)electrocatalytic activities. Surprisingly, triangulene (or triangulenium) dyes have not yet been discussed in this context despite their rich photophysical properties predestined for such purposes.13 The same holds true for metal–organic frameworks, the 3D analogues to CONASHs, which are currently of particular interest regarding light-harvesting98 and water-splitting applications.99 We expect that such functional materials can also be derived from heteroatom-containing triangulene (or triangulenium) derivatives. However, the assembly of such hierarchically structured supramolecular 2D and 3D assemblies is non-trivial and requires a cautious control over the interplay between the ππ-stacking interactions and the other involved non-covalent forces (i.e., hydrogen-/halogen bonding interactions, metal-to-ligand coordination). We look forward to see such complex supramolecular assemblies to be realized and, even more, to be brought into action.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (CRC-TRR 234 “CataLight”, projects B01 and B02, as well as PP 2102, grant number SCHU1229-16/1) is kindly acknowledged.References
- Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- E. Clar and D. G. Stewart, J. Am. Chem. Soc., 1953, 75, 2667–2672 CrossRef CAS.
- N. Pavliček, A. Mistry, Z. Majzik, N. Moll, G. Meyer, D. J. Fox and L. Gross, Nat. Nanotechnol., 2017, 12, 308 CrossRef PubMed.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197 CrossRef CAS PubMed.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- V. Sharma, N. Som, S. D. Dabhi and P. K. Jha, ChemistrySelect, 2018, 3, 2390–2397 CrossRef CAS.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC.
- J. C. Martin and R. G. Smith, J. Am. Chem. Soc., 1964, 86, 2252–2256 CrossRef CAS.
- B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255–12263 CrossRef CAS.
- B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS PubMed.
- B. W. Laursen and F. C. Krebs, Chem. – Eur. J., 2001, 7, 1773–1783 CrossRef CAS.
- B. P. Maliwal, R. Fudala, S. Raut, R. Kokate, T. J. Sørensen, B. W. Laursen, Z. Gryczynski and I. Gryczynski, PLoS One, 2013, 8, e63043 CrossRef CAS PubMed.
- R. Gueret, L. Poulard, M. Oshinowo, J. Chauvin, M. Dahmane, G. Dupeyre, P. P. Lainé, J. Fortage and M.-N. Collomb, ACS Catal., 2018, 8, 3792–3802 CrossRef CAS.
- S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087–5090 CrossRef CAS PubMed.
- S. Bamberger, D. Hellwinkel and F. A. Neugebauer, Chem. Ber., 1975, 108, 2416–2421 CrossRef CAS.
- D. Hellwinkel and M. Melan, Chem. Ber., 1971, 104, 1001–1016 CrossRef CAS.
- D. Hellwinkel and M. Melan, Chem. Ber., 1974, 107, 616–626 CrossRef CAS.
- D. Hellwinkel, A. Wiel, G. Sattler and B. Nuber, Angew. Chem., 1990, 102, 677–680 CrossRef.
- M. Yamamura, T. Saito and T. Nabeshima, J. Am. Chem. Soc., 2014, 136, 14299–14306 CrossRef CAS PubMed.
- M. Yamamura, K. Sukegawa and T. Nabeshima, Chem. Commun., 2015, 51, 12080–12083 RSC.
- F. C. Krebs, P. S. Larsen, J. Larsen, C. S. Jacobsen, C. Boutton and N. Thorup, J. Am. Chem. Soc., 1997, 119, 1208–1216 CrossRef CAS.
- M. Yamamura, T. Hasegawa and T. Nabeshima, Org. Lett., 2016, 18, 816–819 CrossRef CAS PubMed.
- N. Hammer, T. A. Schaub, U. Meinhardt and M. Kivala, Chem. Rec., 2015, 15, 1119–1131 CrossRef CAS PubMed.
- M. Kivala, W. Pisula, S. Wang, A. Mavrinskiy, J.-P. Gisselbrecht, X. Feng and K. Müllen, Chem. – Eur. J., 2013, 19, 8117–8128 CrossRef CAS PubMed.
- F. Schlütter, F. Rossel, M. Kivala, V. Enkelmann, J.-P. Gisselbrecht, P. Ruffieux, R. Fasel and K. Müllen, J. Am. Chem. Soc., 2013, 135, 4550–4557 CrossRef PubMed.
- M. Kuratsu, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2005, 44, 4056–4058 CrossRef CAS PubMed.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed.
- J. E. Field and D. Venkataraman, Chem. Mater., 2002, 14, 962–964 CrossRef CAS.
- Z. Fang, T.-L. Teo, L. Cai, Y.-H. Lai, A. Samoc and M. Samoc, Org. Lett., 2009, 11, 1–4 CrossRef CAS PubMed.
- T. Kushida and S. Yamaguchi, Organometallics, 2013, 32, 6654–6657 CrossRef CAS.
- A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234–6237 CrossRef CAS PubMed.
- T. Kushida, A. Shuto, M. Yoshio, T. Kato and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 6922–6925 CrossRef CAS PubMed.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- Y. Kitamoto, T. Suzuki, Y. Miyata, H. Kita, K. Funaki and S. Oi, Chem. Commun., 2016, 52, 7098–7101 RSC.
- G. Turkoglu, M. E. Cinar and T. Ozturk, Molecules, 2017, 22, 1522 CrossRef PubMed.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- Y. Haketa and H. Maeda, Chem. Commun., 2017, 53, 2894–2909 RSC.
- F. C. Krebs, B. W. Laursen, I. Johannsen, A. Faldt, K. Bechgaard, C. S. Jacobsen, N. Thorup and K. Boubekeur, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 410–423 CrossRef PubMed.
- J. B. Simonsen, K. Kjær, P. Howes, K. Nørgaard, T. Bjørnholm, N. Harrit and B. W. Laursen, Langmuir, 2009, 25, 3584–3592 CrossRef CAS PubMed.
- J. B. Simonsen, F. Westerlund, D. W. Breiby, N. Harrit and B. W. Laursen, Langmuir, 2011, 27, 792–799 CrossRef CAS PubMed.
- T. J. Sørensen, C. B. Hildebrandt, J. Elm, J. W. Andreasen, A. Ø. Madsen, F. Westerlund and B. W. Laursen, J. Mater. Chem., 2012, 22, 4797–4805 RSC.
- T. J. Sørensen, C. B. Hildebrandt, M. Glyvradal and B. W. Laursen, Dyes Pigm., 2013, 98, 297–303 CrossRef.
- P. Hammershøj, T. J. Sørensen, B.-H. Han and B. W. Laursen, J. Org. Chem., 2012, 77, 5606–5612 CrossRef PubMed.
- B. Dong and H. Maeda, Chem. Commun., 2013, 49, 4085–4099 RSC.
- A. Kütt, T. Rodima, J. Saame, E. Raamat, V. Mäemets, I. Kaljurand, I. A. Koppel, R. Y. Garlyauskayte, Y. L. Yagupolskii, L. M. Yagupolskii, E. Bernhardt, H. Willner and I. Leito, J. Org. Chem., 2011, 76, 391–395 CrossRef PubMed.
- Y. Bando, Y. Haketa, T. Sakurai, W. Matsuda, S. Seki, H. Takaya and H. Maeda, Chem. – Eur. J., 2016, 22, 7843–7850 CrossRef CAS PubMed.
- Y. Haketa, S. Sasaki, N. Ohta, H. Masunaga, H. Ogawa, N. Mizuno, F. Araoka, H. Takezoe and H. Maeda, Angew. Chem., Int. Ed., 2010, 49, 10079–10083 CrossRef CAS PubMed.
- Y. Haketa, D. Katayama, S. Fukunaga, Y. Bando, T. Sakurai, W. Matsuda, S. Seki and H. Maeda, Chem. – Asian J., 2016, 11, 2025–2029 CrossRef CAS PubMed.
- Y. Haketa, Y. Honsho, S. Seki and H. Maeda, Chem. – Eur. J., 2012, 18, 7016–7020 CrossRef CAS PubMed.
- R. Yamakado, T. Sakurai, W. Matsuda, S. Seki, N. Yasuda, S. Akine and H. Maeda, Chem. – Eur. J., 2016, 22, 626–638 CrossRef CAS PubMed.
- Y. Terashima, T. Sakurai, Y. Bando, S. Seki and H. Maeda, Chem. Mater., 2013, 25, 2656–2662 CrossRef CAS.
- Y. Bando, S. Sakamoto, I. Yamada, Y. Haketa and H. Maeda, Chem. Commun., 2012, 48, 2301–2303 RSC.
- Y. Bando, T. Sakurai, S. Seki and H. Maeda, Chem. – Asian J., 2013, 8, 2088–2095 CrossRef CAS PubMed.
- S. Sugiura, W. Matsuda, W. Zhang, S. Seki, N. Yasuda and H. Maeda, J. Org. Chem., 2019, 8886–8898, DOI:10.1021/acs.joc.9b00754.
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704 CrossRef CAS PubMed.
- B. Qiao, B. E. Hirsch, S. Lee, M. Pink, C.-H. Chen, B. W. Laursen and A. H. Flood, J. Am. Chem. Soc., 2017, 139, 6226–6233 CrossRef CAS PubMed.
- H. Zhang, S. Wang, Y. Li, B. Zhang, C. Du, X. Wan and Y. Chen, Tetrahedron, 2009, 65, 4455–4463 CrossRef CAS.
- H. Zhang, Y. Li, X. Wan and Y. Chen, Chem. Phys. Lett., 2009, 479, 117–119 CrossRef CAS.
- S. Wang, M. Kivala, I. Lieberwirth, K. Kirchhoff, X. Feng, W. Pisula and K. Müllen, ChemPhysChem, 2011, 12, 1648–1651 CrossRef CAS PubMed.
- A. T. Haedler, S. R. Beyer, N. Hammer, R. Hildner, M. Kivala, J. Köhler and H.-W. Schmidt, Chem. – Eur. J., 2014, 20, 11708–11718 CrossRef CAS PubMed.
- A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Köhler, H.-W. Schmidt and R. Hildner, Nature, 2015, 523, 196 CrossRef CAS PubMed.
- A. T. Haedler, S. C. J. Meskers, R. H. Zha, M. Kivala, H.-W. Schmidt and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 10539–10545 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- C.-M. Chou, S. Saito and S. Yamaguchi, Org. Lett., 2014, 16, 2868–2871 CrossRef CAS PubMed.
- N. Hammer, T. E. Shubina, J.-P. Gisselbrecht, F. Hampel and M. Kivala, J. Org. Chem., 2015, 80, 2418–2424 CrossRef CAS PubMed.
- Y. Dorca, J. Cerdá, J. Aragó, E. Ortí and L. Sánchez, Chem. Mater., 2019, 31, 7024–7032 CrossRef CAS.
- M. Bieri, S. Blankenburg, M. Kivala, C. A. Pignedoli, P. Ruffieux, K. Müllen and R. Fasel, Chem. Commun., 2011, 47, 10239–10241 RSC.
- C. Steiner, J. Gebhardt, M. Ammon, Z. Yang, A. Heidenreich, N. Hammer, A. Görling, M. Kivala and S. Maier, Nat. Commun., 2017, 8, 14765 CrossRef CAS PubMed.
- H. Liang, Y. He, Y. Ye, X. Xu, F. Cheng, W. Sun, X. Shao, Y. Wang, J. Li and K. Wu, Coord. Chem. Rev., 2009, 253, 2959–2979 CrossRef CAS.
- P. A. Wood, S. J. Borwick, D. J. Watkin, W. D. S. Motherwell and F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 393–396 CrossRef CAS PubMed.
- S. Klyatskaya, F. Klappenberger, U. Schlickum, D. Kühne, M. Marschall, J. Reichert, R. Decker, W. Krenner, G. Zoppellaro, H. Brune, J. V. Barth and M. Ruben, Adv. Funct. Mater., 2011, 21, 1230–1240 CrossRef CAS.
- U. Schlickum, R. Decker, F. Klappenberger, G. Zoppellaro, S. Klyatskaya, M. Ruben, I. Silanes, A. Arnau, K. Kern, H. Brune and J. V. Barth, Nano Lett., 2007, 7, 3813–3817 CrossRef CAS PubMed.
- S. Gottardi, K. Müller, J. C. Moreno-López, H. Yildirim, U. Meinhardt, M. Kivala, A. Kara and M. Stöhr, Adv. Mater. Interfaces, 2014, 1, 1300025 CrossRef.
- K. Müller, J. C. Moreno-López, S. Gottardi, U. Meinhardt, H. Yildirim, A. Kara, M. Kivala and M. Stöhr, Chem. – Eur. J., 2016, 22, 581–589 CrossRef PubMed.
- E. Kan, W. Hu, C. Xiao, R. Lu, K. Deng, J. Yang and H. Su, J. Am. Chem. Soc., 2012, 134, 5718–5721 CrossRef CAS.
- Y.-F. Han, Y.-X. Yuan and H.-B. Wang, Molecules, 2017, 22, 266 CrossRef PubMed.
- C. Steiner, Z. Yang, B. D. Gliemann, U. Meinhardt, M. Gurrath, M. Ammon, B. Meyer, M. Kivala and S. Maier, Chem. Commun., 2018, 54, 11554–11557 RSC.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838, 10.1039/B801115J.
- R. D. Baechler and K. Mislow, J. Am. Chem. Soc., 1970, 92, 3090–3093 CrossRef CAS.
- I. V. Alabugin, S. Bresch and M. Manoharan, J. Phys. Chem. A, 2014, 118, 3663–3677 CrossRef CAS PubMed.
- G. K. H. Madsen, F. C. Krebs, B. Lebech and F. K. Larsen, Chem. – Eur. J., 2000, 6, 1797–1804 CrossRef CAS.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635–661 RSC.
- T. Kawase, in Supramolecular Chemistry of Fullerenes and Carbon Nanotubes, ed. N. Martín and J.-F. Nierengarten, VCH-Wiley, Weinheim, 2012, pp. 55–78 Search PubMed.
- L. T. Scott, M. M. Hashemi and M. S. Bratcher, J. Am. Chem. Soc., 1992, 114, 1920–1921 CrossRef CAS.
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed.
- T. Hirao, M. Tosaka, S. Yamago and T. Haino, Chem. – Eur. J., 2014, 20, 16138–16146 CrossRef CAS PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- T. A. Schaub, R. Sure, F. Hampel, S. Grimme and M. Kivala, Chem. – Eur. J., 2017, 23, 5687–5691 CrossRef CAS PubMed.
- J. Hamacek, C. Besnard, N. Mehanna and J. Lacour, Dalton Trans., 2012, 41, 6777–6782 RSC.
- R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523–528 CrossRef CAS.
- R. Sakamoto, K. Takada, T. Pal, H. Maeda, T. Kambe and H. Nishihara, Chem. Commun., 2017, 53, 5781–5801 RSC.
- M. C. So, G. P. Wiederrecht, J. E. Mondloch, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 3501–3510 RSC.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937 RSC.
| This journal is © the Partner Organisations 2019 |