
Push–pull type quinoidal perylene showing solvent polarity dependent diradical character and negative solvatochromism†
Wangdong
Zeng
a and
Jishan
Wu
 *ab
*ab
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmwuj@nus.edu.sg
bJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
First published on 18th October 2019
Abstract
Synthesis and physical properties of a new push–pull type quinoidal perylene (DA-Per) are reported. Three resonance forms, a closed-shell quinoid, an open-shell diradicaloid and a closed-shell zwitterion, all contribute to the ground-state electronic structure. The molecule shows solvent polarity dependent diradical character and negative solvatochromism, which can be explained by the subtle balance among the three resonance forms in different solvent environments.
Recent studies demonstrated that π-conjugated molecules with intermediate π-bonding could show open-shell diradical character in the ground state.1–6 For example, quinoidal oligo-para-phenylenes,7–12 quinoidal oligothiophenes,13,14 and polycyclic hydrocarbons embedded with a quinodimethane unit,15–20 have an intrinsic tendency to recover aromaticity and become open-shell diradicals. This type of molecule usually possesses a small energy gap and shows amphoteric redox behaviour. They show paramagnetic properties at increased temperatures due to the thermal population from the singlet ground state to the triplet excited state. In addition, they exhibit enhanced third-order non-linear optical (NLO) properties (e.g., two-photon absorption) at an intermediate diradicaloid state.21–25 However, so far, most of the studies have been conducted for symmetric molecules, while quinoidal molecules with asymmetric push–pull type structure are rarely investigated.26,27 There are very limited examples of molecules showing both open-shell diradical character and dipolar zwitterionic character because they are counterparts to each other.28,29 It was also demonstrated that π-conjugated molecules with a zwitterionic character usually show interesting solvatochromism and second-order NLO properties.30–32 In particular, T. Marks et al. showed that highly twisted π-zwitterions were good second-order NLO chromophores with ultrahigh hyperpolarizability.33–38 Herein, we report a new push–pull type quinoidal perylene molecule DA-Per, which contains a dicyanomethanide unit as an electron donor and a pyridinium moiety as an electron acceptor (Fig. 1a). The molecule can be drawn in three major resonance forms: a closed-shell quinoidal form, a closed-shell zwitterionic form, and an open-shell diradical form (Fig. 1a). The latter two forms can be stabilized by aromatic stabilization energy of the aromatic perylene unit. Our detailed studies revealed interesting solvent polarity dependent open-shell diradical character and unusual negative solvatochromism for DA-Per in different environments.
 | ||
| Fig. 1 (a) Chemical structure and three representative resonance forms of DA-Per. (b) Synthesis of DA-Per. Reagents and conditions: (a) 4-pyridinylboronic acid, Pd2(dba)3, Sphos, K3PO4, toluene, 110 °C, 12 h; (b) (i) malononitrile, NaH, Pd2(dba)3, dppf, THF; (ii) MeOTf, DCM, r.t., 20 min; (iii) NaHCO3 (aq). | ||
Compound DA-Per was synthesized according to the scheme shown in Fig. 1b. One-fold Suzuki coupling reaction between the 3,10-dibromo-1,1-bis(4-tetradecylphenyl)-1H-cyclopenta[ghi]perylene 139 and 4-pyridinylboronic acid (1 equivalent) gave the compound 2 in 70% yield. The Pd-catalyzed Takahashi coupling reaction between 2 and malononitrile in the presence of sodium hydride in anhydrous THF provided the aryl malononitrile intermediate, which was treated with methyl trifluoromethanesulfonate (MeOTf) followed by deprotonation with aqueous NaHCO3 solution to afford the final target compound DA-Per in an overall 75% yield from 2. Compound DA-Per is stable and can be purified by silica gel column chromatography on trimethylamine deactivated silica gel. It is soluble in normal organic solvents because the 4-tetradecylphenyl groups attached to the sp3-carbon at the perylene bay region can help to suppress aggregation.
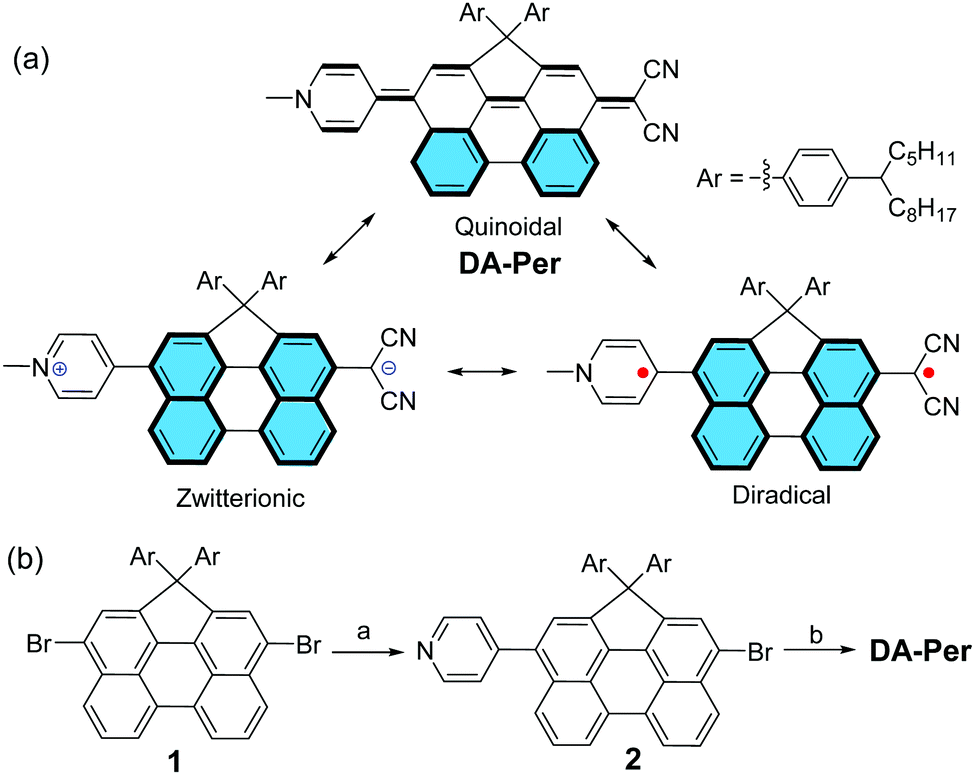
The electronic absorption spectra of DA-Per in different solvents show an unusual hyperchromic shift upon increase of the solvent polarity, that is, negative solvatochromism (Fig. 2a). The absorption maximum (λmax) in chloroform, tetrahydrofuran (THF), dichloromethane (DCM), acetone, DMSO and methanol appears at 875, 822, 809, 702, 654 and 634 nm, respectively (Table 1). The peak at 716 nm observed in chloroform is similar to that of an H-aggregated perylene dimer.27 However, concentration dependent UV-vis measurements indicate that there is almost no aggregation at low concentration (5 × 10−6–5 × 10−5 M) (Fig. S1 in the ESI†). This band may originate from some solvent polarity dependent higher-energy electronic transitions. The peak at about 480 nm observed in methanol could be correlated to the absorption of the aromatic perylene bridge due to the large stabilization of the zwitterionic form in the very polar solvent. The observed negative solvatochromism indicates that the molecule has larger dipole moment (μ) in the singlet ground state (S0) than that in the first singlet excited state (S1). The ground-state and the first singlet excited-state structures of DA-Per are optimized at the B3LYP/6-31G(d,p) level of theory (the long aliphatic chains are replaced by methyl groups during the calculations). Environment effects on S0 and S1 geometries are included by using the polarizable continuum model (PCM). Under gas phase, a large ground-state dipole moment (μg = 23.49 Debye) is calculated, and the negative/positive charges are mainly distributed at the dicyanomethide/pyridinium moiety, respectively, according to the calculated electrostatic potential map (Fig. 2b). The μg becomes larger with the increase of the solvent dielectric constant (εr) (Table 1). The calculated dipole moments at the first singlet excited state (μe) are slightly larger than that μg when the environment is gas phase and chloroform. However, when the solvent becomes more polar (in DCM, THF, acetone, DMSO and methanol), the μe is smaller than μg (Table 1), which explains the observed negative solvatochromism. Time-dependent (TD) DFT calculations (B3LYP/6-31G(d,p) combined with PCM) indicate that the longest wavelength absorption band is originated from the HOMO → LUMO electronic transition. The HOMO and LUMO are partially segregated, but have large spatial overlap, leading to a large oscillator strength (f = 1.068 under gas phase) and intense absorption (Fig. S8 and Table S1, ESI†). TD DFT calculations in different solvents also properly predict that the absorption maximum will show hyperchromic shift with increase of the solvent polarity (Table 1), and the trend is in agreement with the experimental observation, although there is still quite large deviation from the absolute wavelengths. We should also note that the observed solvent-dependent spectral change cannot be simply correlated with a single solvent polarity parameter (neither ET(30) nor εr). Hydrogen bonding between the methanol molecules with the CN group, or between THF molecules with the acidic protons in the pyridinium unit, may explain the abnormality in methanol and THF.
 | ||
| Fig. 2 (a) Electronic absorption spectra of DA-Per in different solvents. (b) Optimized geometry with dipole moment (left) and the calculated electrostatic potential map (right) of DA-Per (gas phase). | ||
| Solvents | Dipole moment (μ) (D) | λ max (nm) | ε (M−1 cm−1)/f | y 0 | ΔES–T (kcal mol−1) | Solvent polarity | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| μ g (S0) | μ e (S1) | Δμeg | Obs. | Calcd | Obs. (ε) | Calcd (f) | E T(30) | ε r | |||
| Gas | 23.49 | 27.47 | +3.98 | — | 627.0 | — | 1.0913 | 0.4261 | −7.17 | — | — |
| Chloroform | 39.70 | 39.78 | +0.08 | 875 | 717.0 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 490 490 |
1.2723 | 0.3545 | −9.38 | 39.1 | 4.7 |
| DCM | 43.85 | 42.34 | −1.51 | 822 | 712.8 | 25860 |
1.1695 | 0.3388 | −9.91 | 40.7 | 8.9 |
| THF | 42.85 | 41.70 | −1.15 | 809 | 711.9 | 22720 |
1.1858 | 0.3426 | −9.78 | 37.4 | 7.4 |
| Acetone | 46.84 | 44.41 | −2.43 | 703 | 700.4 | 18730 |
1.0677 | 0.3286 | −10.26 | 42.2 | 20.7 |
| DMSO | 48.67 | 45.47 | −3.20 | 654 | 702 | 15030 |
1.0446 | 0.3243 | −10.40 | 45.1 | 48.9 |
| Methanol | 47.75 | 45.10 | −2.65 | 634 | 694.8 | 7630 | 1.0312 | 0.3262 | −10.35 | 55.4 | 32.6 |
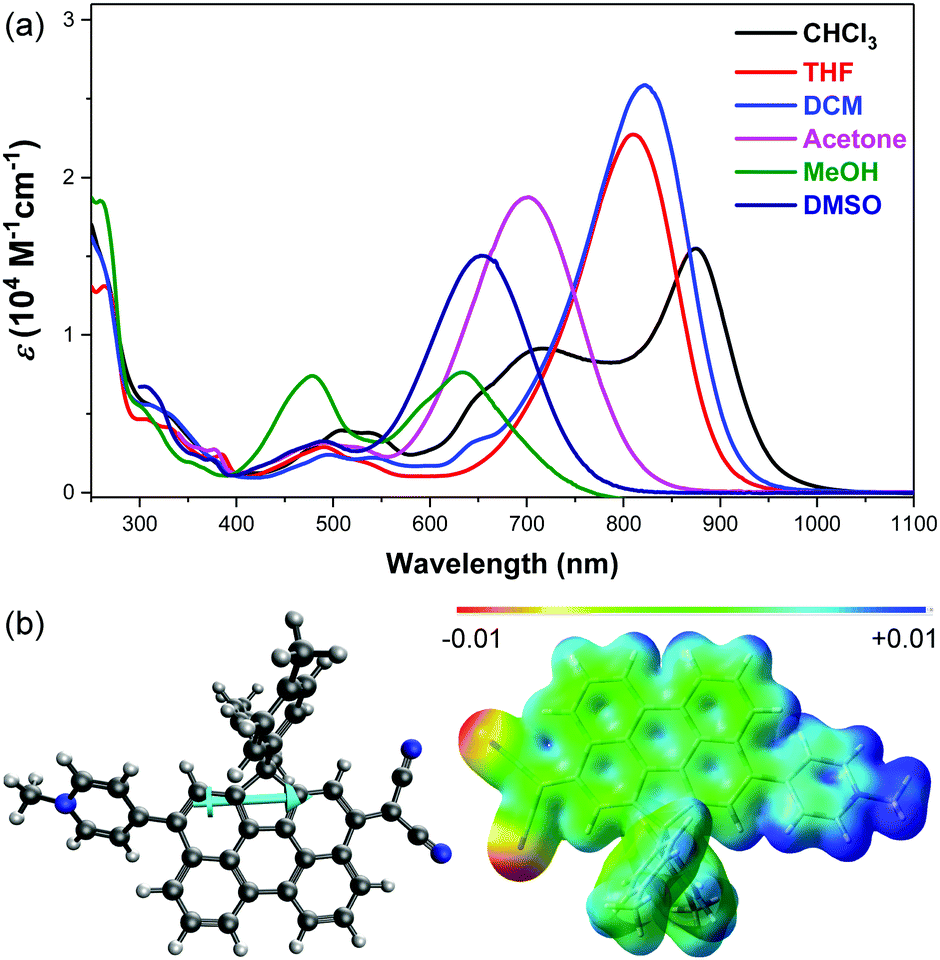
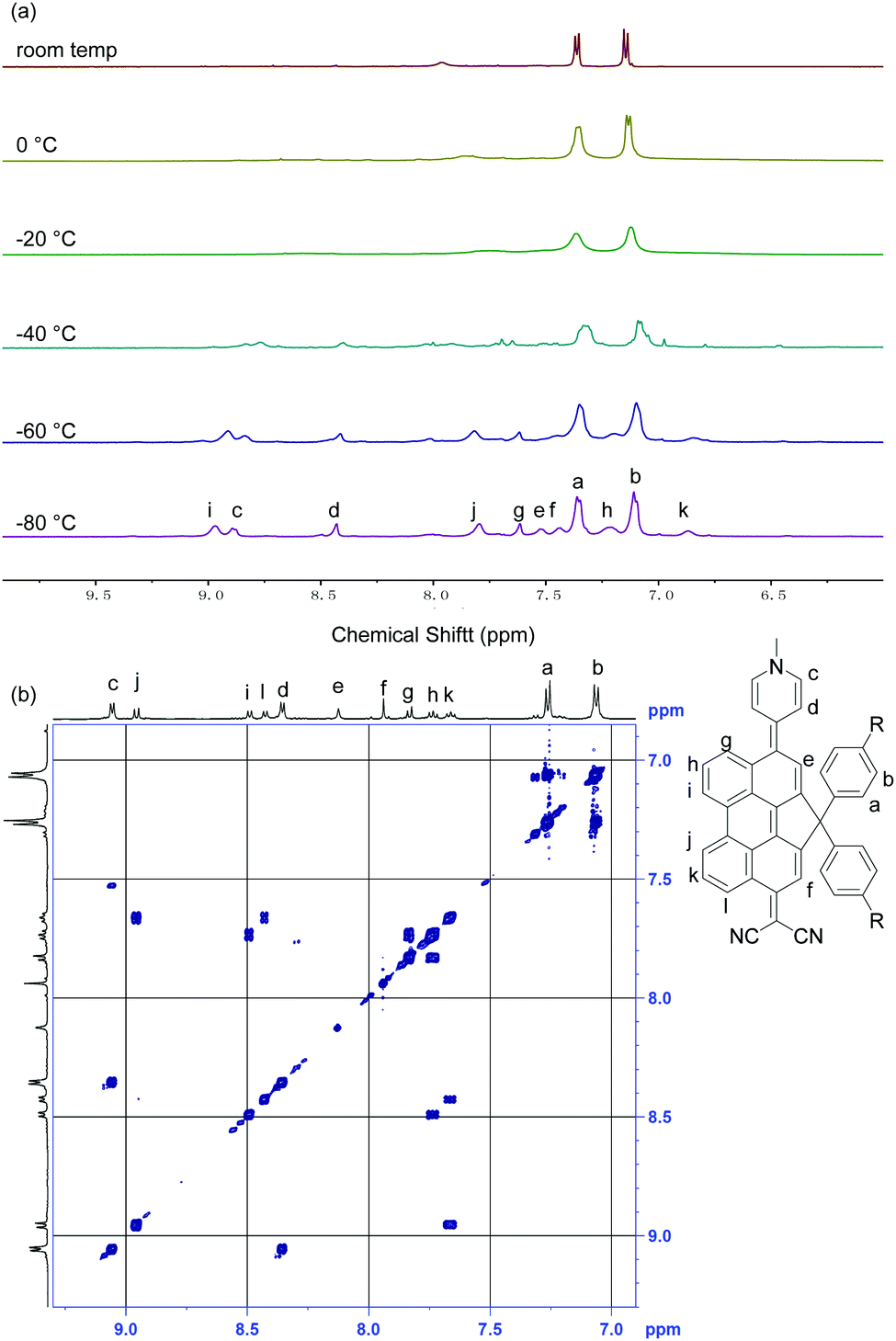
The 1H NMR spectrum of DA-Per in CD2Cl2 shows that the resonances for the protons on the π-conjugated backbone are significantly broadened and cooling the solution down to −80 °C leads to sharpening of the peaks (Fig. 3a). Such observation indicates that DA-Per in CD2Cl2 may display open-shell diradical character and the NMR broadening is due to the existence of thermally populated paramagnetic triplet diradical species, a typical phenomenon for many open-shell singlet diradicaloids.1–6 A broad ESR signal (ge = 2.0029) was determined (Fig. 4a). The ESR intensity (I) decreased with decreasing temperature (T) for the solid sample (Fig. S2 in the ESI†) and fitting of the IT–T data by Bleaney–Bows equation40 gave a singlet–triplet energy gap (ΔES–T) of −2.26 kcal mol−1 (Fig. 4b). The spin densities of the triplet diradical are mainly distributed along the quinoidal conjugated backbone (Fig. 4b). Similar NMR broadening was observed in CDCl3 and THF-d8 at room temperature (Fig. S3 and S4 in the ESI†). On the other hand, the 1H NMR spectra of DA-Per in polar DMSO-d6, acetone-d6, and methanol-d4 all exhibit sharp peaks (Fig. 3b and Fig. S5 and S6 in the ESI†), indicating a closed-shell nature. The resonances in DMSO-d6 are clearly assigned by using a 2D COSY technique (Fig. 3b). In agreement with the NMR spectra, ESR measurements of the DA-Per in different solvents with the same concentration (c = 1.66 × 10−4 M) show that the ESR intensity decreases in the order of CHCl3, DCM, THF, acetone, methanol and DMSO. There are only very weak signals in the latter three polar solvents (Fig. 4a). These observations suggest that the diradical character is also dependent on the polarity of the solvent, that is, in the more polar solvents, the zwitterionic form is more stabilized, leading to smaller diradical character.
 | ||
| Fig. 3 (a) VT 1H NMR spectra of DA-Per (500 MHz, aromatic region) in CD2Cl2. (b) 2D-COSY NMR spectrum of DA-Per (500 MHz, rt) in DMSO-d6. | ||
 | ||
| Fig. 4 (a) ESR spectra of DA-Per in different solvents at the same concentration (c = 1.66 × 10−4 M). (b) Fitted IT–T curve for DA-Per based on the VT ESR data measured in powder; I: integrated ESR intensity; T: temperature. Inset is the calculated spin density distribution map of the triplet state. (c) Calculated (RAS-SF/6-31G*) natural orbital profiles (isovalue: 0.04) and occupation numbers (in the parentheses). | ||
To deeply understand this trend, we conducted multi-reference restricted active space spin flip (RAS-SF)41,42 calculations based on the optimized triplet state geometries in different solvents. The diradical character index (y0) is defined as the occupation number of the lowest unoccupied natural orbital (LUNO). According to the calculations, the diradical character decreases with the increase of solvent polarity in the order of CHCl3, DCM, THF, acetone, methanol and DMSO (Table 1), which is the same as that of the ESR experiment. The unpaired electron densities are distributed along the π-conjugated backbone and the diradical character is calculated to be 0.426 in the gas phase (Fig. 4c). Similarly, the calculated vertical singlet–triplet excitation energy (ΔES–T) increases with increasing solvent polarity in the same order (Table 1). It should be noted that the calculated data only provide a reference to understand the trend, and it is not meaningful to compare the absolute values between the experiment and calculation (e.g., ΔES–T).
Compound DA-Per showed different electrochemical behavior in DCM and DMSO (Fig. 5). In DCM, two oxidation waves with half-wave potential Eox1/2 at 0.47 and 0.86 V (vs. Fc+/Fc) and two overlapped reduction waves with half-wave potential Ered1/2 at −1.36 and −1.46 V (vs. Fc+/Fc). In DMSO, one oxidative wave with Eox1/2 at 0.87 V and one reduction wave with Ered1/2 at −1.50 V were observed. Therefore, the electrochemical energy gap in DCM (1.83 eV) is smaller than that in DMSO (2.37 eV), in agreement with the optical energy gaps, which can be explained by the large contribution of open-shell electronic structure in DCM and the large zwitterionic contribution in DMSO.
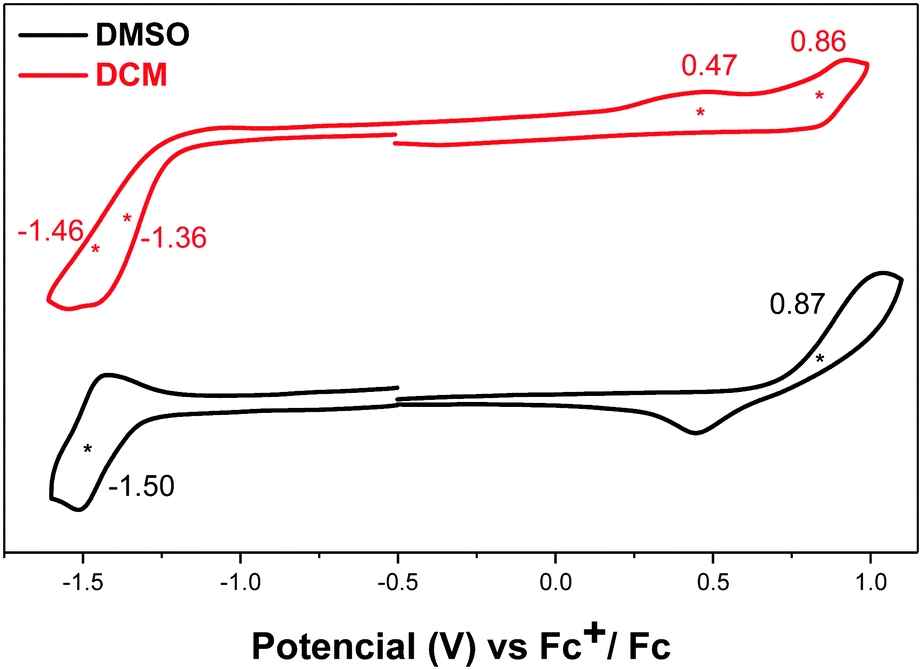 | ||
| Fig. 5 Cyclic voltammograms of DA-Per in DMSO and DCM with n-Bu4N·PF6 as a supporting electrolyte, and scan rate at 50 mV s−1. | ||
In conclusion, a new push–pull type quinoidal perylene based diradicaloid DA-Per was synthesized, which shows a negative solvatochromic effect due to a larger dipole moment at the first singlet excited state compared to that in the ground state. Notably, the open-shell diradical character also exhibits solvent dependence, and it displays closed-shell nature in the very polar solvents (DMSO, MeOH, acetone), while open-shell singlet diradical character in the less polar solvents (CHCl3, DCM, THF). Therefore, a subtle balance between the three resonance forms is tunable by solvent polarity. This work represents a rare report on diradicaloids showing solvent polarity dependent radical character. Investigation on the solvent dependent NLO properties is in our plan.
Experimental
Synthesis of compound 2
A mixture of compound 1 (0.48 g, 0.5 mmol), pyridine-4-boronic acid (61.5 mg, 0.5 mmol, 1.0 equiv.), Pd2(dba)3 (22.9 mg, 5 mol%), SPhos (40.1 mg, 20 mol%), and K3PO4 (425 mg, 2.5 equiv.) in toluene (100 mL) was degassed and heated at 110 °C for 12 h. After cooling to room temperature, water was added, and the reaction mixture was extracted with chloroform (50 mL). The organic layer was dried over sodium sulfate and the solvent was removed under vacuum. The residue was subjected to column chromatography (silica gel, hexane/DCM = 1/1) to afford the title product 2 (0.33 g) in 70% yield. 1H NMR (CDCl3, 500 MHz): δ ppm 8.77 (br, 2H), 8.27 (m, 2H), 8.09 (d, J = 8.50 Hz, 1H), 7.99 (br, 2H), 7.92 (s, 1H), 7.77–7.66 (m, 3H), 7.61 (s, 1H), 7.19 (d, J = 8.10 Hz, 4H), 7.00 (d, J = 8.10 Hz, 4H), 2.42 (m, 2H), 1.58–1.25 (m, 8H), 1.25–1.18 (m, 36H), 0.88–0.78 (m, 12H); 13C NMR (CDCl3, 125 MHz): δ ppm 149.90, 149.05, 145.99, 145.76, 139.73, 136.09, 135.79, 132.71, 131.10, 128.31, 127.86, 127.43, 126.17, 125.93, 124.63, 121.34, 45.67, 36.78, 31.86, 29.74, 29.52, 27.66, 27.30, 22.66, 14.11. HR-MS (APCI): m/z = 964.5403, calcd for C66H79BrN (M + 1): m/z = 964.5390, error = −1.4 ppm.Synthesis of compound DA-Per
To an ice-cooled suspension of NaH (46 mg, 1.15 mmol, 4.8 equiv.) in anhydrous THF (10 mL) was added dropwise via syringe a solution of malononitrile (49.5 g, 0.57 mmol, 2.4 equiv.) in anhydrous THF (2.0 mL) under argon. The mixture was stirred for 1 h at room temperature and then compound 2 (231 mg, 0.24 mmol), Pd2(dba)3 (11.0 mg, 5 mol%), dppf (20.0 mg, 15 mol%), and THF (20 mL) were added subsequently under argon. The mixture was purged with argon for 0.5 h and was then heated at 85 °C for 12 h. The solvent was removed under reduced pressure, water was added and the reaction mixture was extracted with DCM (50 mL). The organic layer was dried over sodium sulfate and the solvent was removed under vacuum. The residue was dissolved in dry DCM (20 mL) and then a solution of methyl trifluoromethanesulfonate (39.8 mg, 0.24 mmol) in DCM (2 mL) was added. The mixture was stirred at room temperature for 20 min and then poured into ice water and neutralized with saturated aqueous NaHCO3 solution (2 M, 50 mL). The organic layer was separated, washed with water, and dried over anhydrous Na2SO4. The solvent was removed under vacuum and the residue was subjected to column chromatography (deactivate silica gel by Et3N, acetone/DCM = 1/1) to afford the title product DA-Per (0.17 g) in an overall 75% yield from 2. 1H NMR (DMSO-d6, 500 MHz): δ ppm 9.05 (d, J = 6.65 Hz, 2H), 8.95 (d, J = 8.65 Hz, 1H), 8.48 (d, J = 7.50 Hz, 1H), 8.42 (d, J = 7.50 Hz, 1H), 8.35 (d, J = 6.65 Hz, 2H), 8.12 (s, 1H), 7.94 (s, 1H), 7.83 (d, J = 8.50 Hz, 1H), 7.74 (m, 1H), 7.67 (m, 1H), 7.26 (d, J = 8.25 Hz, 4H), 7.06 (d, J = 8.25 Hz, 4H), 4.40 (s, 3H), 2.42 (m, 2H), 1.52–1.43 (br, 8H), 1.24–0.99 (m, 36H), 0.78–0.74 (m, 12H). HR-MS (APCI): m/z = 963.6567, calcd for C70H81N3: m/z = 963.6530, error = −3.8 ppm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MOE Tier 3 programme (MOE2014-T3-1-004), NRF Investigatorship (NRF-NRFI05-2019-0005), MOE Tier 2 grant (MOE2018-T2-2-094) and NSFC grant (21801074).Notes and references
- M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed.
- Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582 CrossRef CAS PubMed.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC.
- T. Kubo, Chem. Lett., 2015, 44, 111 CrossRef.
- T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186 RSC.
- C. Liu, Y. Ni, X. Lu, G. Li and J. Wu, Acc. Chem. Res., 2019, 52, 2309 CrossRef CAS PubMed.
- A. E. Tschitschibabin, Chem. Ber., 1907, 40, 1810 CrossRef CAS.
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004 CrossRef CAS PubMed.
- K. Dimroth, W. Umbach and K. H. Blocher, Angew. Chem., Int. Ed. Engl., 1963, 2, 620 CrossRef.
- R. West, J. A. Jorgenson, K. L. Stearly and J. C. Calabrese, J. Chem. Soc., Chem. Commun., 1991, 2, 1234 RSC.
- G. Li, H. Phan, T. S. Herng, T. Y. Gopalakrishna, C. Liu, W. Zeng, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 5012 CrossRef CAS PubMed.
- G. Li, Y. Han, Y. Zou, J. J. C. Lee, Y. Ni and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 14319–14326 CrossRef CAS PubMed.
- T. Takahashi, K. I. Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127, 8928 CrossRef CAS PubMed.
- R. P. Ortiz, J. Casado, V. Hernandez, J. T. López Navarrete, P. M. Viruela, E. Orti, K. Takimiya and T. Otsubo, Angew. Chem., Int. Ed., 2007, 46, 9057 CrossRef CAS PubMed.
- K. Ohashi, T. Kubo, T. Masui, K. Yamamoto, K. Nakasuji, T. Takui, Y. Kai and I. Murata, J. Am. Chem. Soc., 1998, 120, 2018 CrossRef CAS.
- A. Shimizu, T. Kubo, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Hirao, K. Matsumoto, H. Kurata, Y. Morita and K. Nakasuji, J. Am. Chem. Soc., 2010, 132, 14421 CrossRef CAS PubMed.
- A. Shimizu, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, M. Uruichi and K. Yakushi, Chem. Commun., 2012, 48, 5629 RSC.
- Z. Sun, K.-W. Huang and J. Wu, J. Am. Chem. Soc., 2011, 133, 11896 CrossRef CAS PubMed.
- Z. Sun, S. Lee, K. Park, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, S. Das, Y. Li, C. Chi, R. Li, K. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 18229 CrossRef CAS PubMed.
- R. Huang, H. Phan, T. S. Herng, P. Hu, W. Zeng, S. Dong, S. Das, Y. Shen, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 10323 CrossRef CAS PubMed.
- M. Nakano, H. Nagao and K. Yamaguchi, Phys. Rev. A: At., Mol., Opt. Phys., 1997, 55, 1503 CrossRef CAS.
- M. Nakano, R. Kishi, T. Nitta, T. Kubo, K. Nakasuji, K. Kamada, K. Ohta, B. Champagne, E. Botek and K. Yamaguchi, J. Phys. Chem. A, 2005, 109, 885 CrossRef CAS PubMed.
- M. Nakano, R. Kishi, S. Ohta, H. Takahashi, T. Kubo, K. Kamada, K. Ohta, E. Botek and B. Champagne, Phys. Rev. Lett., 2007, 99, 033001 CrossRef PubMed.
- S. Ohta, M. Nakano, K. Kubo, K. Kamada, K. Ohta, R. Kishi, N. Nakagawa, B. Champagne, E. Botek, A. Takebe, S. Umezaki, M. Nate, H. Takahashi, S. Furukawa, Y. Morita, K. Nakasuji and K. Yamaguchi, J. Phys. Chem. A, 2007, 111, 3633 CrossRef CAS PubMed.
- K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S. I. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 46, 3544 CrossRef CAS.
- S. Inoue, Y. Aso and T. Otsubo, Chem. Commun., 1997, 1105 RSC.
- Z. Zeng, S. Lee, M. Son, K. Fukuda, P. M. Burrezo, X. Zhu, Q. Qi, R.-W. Li, J. T. López Navarrete, J. Ding, J. Casado, M. Nakano, D. Kim and J. Wu, J. Am. Chem. Soc., 2015, 137, 8572 CrossRef CAS.
- Y. Hirao, N. Nagamachi, K. Hosoi and T. Kubo, Chem. – Asian J., 2018, 13, 510 CrossRef CAS PubMed.
- Y. Hirao, Y. Hamamoto, N. Nagamachi and T. Kubo, Phys. Chem. Chem. Phys., 2019, 21, 12209 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- L. Beverina and G. A. Pagani, Acc. Chem. Res., 2014, 47, 319 CrossRef CAS PubMed.
- A. J.-T. Lou and T. J. Marks, Acc. Chem. Res., 2019, 52, 1428 CAS.
- I. D. L. Albert, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1998, 120, 11174 CrossRef CAS.
- H. Kang, A. Facchetti, P. Zhu, H. Jiang, Y. Yang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S.-T. Ho and T. J. Marks, Angew. Chem., Int. Ed., 2005, 44, 7922 CrossRef CAS.
- H. Kang, A. Facchetti, H. Jiang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S.-T. Ho, E. C. Brown, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 3267 CrossRef CAS.
- G. S. He, J. Zhu, A. Baev, M. Samoc, D. L. Frattarelli, N. Watanabe, A. Facchetti, H. Ågren, T. J. Marks and P. N. Prasad, J. Am. Chem. Soc., 2011, 133, 6675 CrossRef CAS PubMed.
- Y. Shi, D. Frattarelli, N. Watanabe, A. Facchetti, E. Cariati, S. Righetto, E. Tordin, C. Zuccaccia, A. Macchioni, S. L. Wegener, C. L. Stern, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 12521 CrossRef CAS PubMed.
- A. J.-T. Lou, S. Righetto, C. Barger, C. Zuccaccia, E. Cariati, A. Macchioni and T. J. Marks, J. Am. Chem. Soc., 2018, 140, 8746 CrossRef CAS.
- W. Zeng, H. Phan, T. S. Herng, T. Y. Gopalakrishna, N. Aratani, Y. Ni, Z. Zeng, H. Yamada, J. Ding and J. Wu, Chem, 2017, 2, 81 CrossRef CAS.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
- M. Head-Gordon, Chem. Phys. Lett., 2003, 372, 508 CrossRef CAS.
- D. Casanova and M. Head-Gordon, Phys. Chem. Chem. Phys., 2009, 11, 9779 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectra and characterization data, and theoretical calculation details. See DOI: 10.1039/c9qm00566h |
| This journal is © the Partner Organisations 2019 |