
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarved nanoframes of cobalt–iron bimetal phosphide as a bifunctional electrocatalyst for efficient overall water splitting†
Yuebin
Lian
ab,
Hao
Sun
ab,
Xuebin
Wang
ab,
Pengwei
Qi
ab,
Qiaoqiao
Mu
ab,
Yujie
Chen
ab,
Jing
Ye
c,
Xiaohui
Zhao
ab,
Zhao
Deng
 *ab and
Yang
Peng
*ab
*ab and
Yang
Peng
*ab
aSoochow Institute for Energy and Materials Innovations, College of Energy, Soochow University, Suzhou 215006, P. R. China
bKey Laboratory of Advanced Carbon Materials and Wearable Energy Technologies of Jiangsu Province, Soochow University, Suzhou 215006, P. R. China. E-mail: zdeng@suda.edu.cn; ypeng@suda.edu.cn
cAnalysis and Testing Center, Soochow University, Suzhou 215123, China
First published on 15th October 2018
Abstract
Water electrolysis for hydrogen production has long been regarded as an ideal tactic for renewable energy conversion and storage, but is impeded by the sluggish kinetics of both the hydrogen and oxygen evolution reactions, which are therefore in urgent need for high-performance but low-cost electrocatalysts. Herein, nanoframes of transition metal phosphides (TMPs) with the 3D framework carved open have been demonstrated as highly potent bifunctional catalysts for overall water splitting, reaching the benchmark performance of the Pt/C‖RuO2 couple, and are much superior to their nanocubic counterparts. This excellent water splitting behavior can be attributed to the enlarged active surface area, less obstructed electrolyte infiltration, promoted charge transfer, and facilitated gas release. Further through in-depth activity analysis and post-electrocatalysis characterization, special attention has been paid to the fate and role of phosphorus in the electrocatalytic process, suggesting that despite the chemical instability of the TMPs (especially under OER conditions), excellent electrocatalytic stability can still be achieved through the amorphous bimetallic hydroxides/oxides formed in situ.
1. Introduction
Water electrolysis for hydrogen production offers a promising solution to convert and store intermittent clean energy resources such as wind, tidal, solar etc. in a scalable fashion.1,2 It involves a hydrogen evolution reaction (HER) at the cathode and an oxygen evolution reaction (OER) at the anode, both of which are kinetically sluggish and in urgent need for highly active electrocatalysts to expedite the redox electron transfer.3–7 So far, the state-of-the-art candidates in this regard have been catalysts based on noble metals (e.g. Pt for the HER and RuO2/IrO2 for the OER), but their exorbitant cost and scarcity greatly limit broader applications. Moreover, using different catalysts for each side of the water splitting reaction will unavoidably increase the material and processing costs, and therefore bifunctional catalysts based on earth-abundant elements with low fabrication costs are highly desirable. In such contexts, considerable efforts have been made to develop high-efficiency bifunctional electrocatalysts for overall water splitting, including metals,8,9 alloys,10,11 metal oxides,12,13 metal nitrides,14 metal chalcogenides,15,16 metal carbides,17–20 metal hydroxides,21–23etc. However, more advancement is imperative to further reduce the energy input and maximize the current density in the electrolysis process for practical implementation.Transition-metal phosphides (TMPs), such as Ni2P,24,25 CoP,26,27 FeP,28–30 RuP2,31 Cu3P32etc., emerge as a new class of noble-metal-free catalysts for electrochemical water splitting, owing to their low fabrication cost, good electrochemical stability, controllable valence of central metals, and suitable bonding energy for the intermediates of both the HER and OER.33,34 To further enhance the electrochemical performance, research has been progressing to extend from mono-metallic to multi-metallic phosphides by doping one or more metal atoms, aiming for optimizing both the electronic structure and surface potential of the catalysts. As a result, high-activity catalysts with reduced overpotential and enhanced exchange current have been achieved. Despite all that, further insights into the role and fate of phosphorus in such catalysts during electrolysis are still lacking and thus highly desired for fundamental understanding of the catalytic mechanism and electrolytic process. In addition, while most of the previous studies on TMPs mainly dealt with half reactions (either the HER or OER), their applications in overall water splitting have been still limited and need to be realized with extended stability.35,36
The structural framework and surface morphology of nanomaterials are important prerequisites for determining the exposure of active sites and the activity of individual catalytic sites, which ultimately impart the overall catalytic potency.37,38 TMP nanostructures with the morphology of nanoparticles,39 nanosheets,40 nanowires41etc., owing to their high porosity and specific surface area for promoting electrolyte infiltration and contact, have been demonstrated as efficient electrocatalysts for accelerating both the HER and OER. For example, Lou et al. fabricated Ni–Co–P nanobricks with oriented nanosheets as the bifunctional electrocatalyst for overall water splitting, and achieved a low cell voltage of 1.62 V for reaching a current density of 10 mA cm−2.42 Using MOF-74 as the structural precursor, Zhao et al. synthesized a series of Co–Ni–P catalysts, with the best composition of Co4Ni1P in the form of nanotubes affording a current density of 10 mA cm−2 at a cell voltage of 1.59 V.43 Recently, nanostructures with the open framework such as nanocages44 and nanoframes45 as electrocatalysts have drawn further interests because of the facilitated charge and mass transportation, and the increased defective surface sites, both leading to greatly enhanced reaction kinetics. Hence, it can be rationalized that open frameworks of TMPs, in comparison to their bulky counterparts, should exhibit superior catalytic performance towards efficient water splitting.
To testify this hypothesis, herein, we adopt bimetallic MOF – Co–Fe Prussian Blue Analogues (Co–Fe PBAs) as the structural template, followed by etching with urea and phosphorizing with sodium hypophosphite, and fabricate nanoframes of Co–Fe phosphides for use as the bifunctional electrocatalyst for overall water splitting. The obtained Co0.6Fe0.4P nanoframes inherit the uniform cubic structure of PBAs with open 3D frameworks, which effectively promote the electroactive surface area, as well as the charge and mass transportation. In addition, the utilization of MOFs as the structural precursor allows the homogeneous distribution of various metal centers in a conductive and protective carbon matrix. As a result, with an optimized etchant dosage excellent HER and OER activities were simultaneously achieved, outperforming most of the TMP-based catalysts reported to date.35,46–48 What's more, our bifunctional catalyst needs only 1.57 V to achieve 10 mA cm−2 for overall water splitting with excellent long-term stability beyond 120 h, benefitting from the structural merits including largely exposed active surface, unobstructed mass diffusion pathways, and improved charge transportation.
2. Results and discussion
Co0.6Fe0.4P nanoframes were prepared by a straightforward strategy containing three sequential steps including template growth, framework etching, and phosphorization. The detailed procedure and synthesis conditions are illustrated in Fig. 1 and detailed in the Experimental section (ESI†). Samples etched with different amounts of urea prior to phosphorization are respectively denoted as Co0.6Fe0.4-0, Co0.6Fe0.4-0.75, Co0.6Fe0.4-1.125 and Co0.6Fe0.4-1.5 according to the mass ratio of urea to the Co–Fe PBA template. After phosphorization, they are denoted as Co0.6Fe0.4P-0, Co0.6Fe0.4P-0.75, Co0.6Fe0.4P-1.125 and Co0.6Fe0.4P-1.5 accordingly. The Co/Fe ratio in all samples was verified using Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES), unanimously showing a ratio of approximately 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which reflects the feed ratio of the reactants (Table S1†). Fig. 2a presents the X-ray diffraction (XRD) spectra of the as-prepared Co0.6Fe0.4-0 (Co–Fe PBA), Co0.6Fe0.4-1.125 (before phosphorization), and Co0.6Fe0.4P-1.125 (after phosphorization). The XRD spectra of Co–Fe PBAs with and without etching exhibit similar patterns with prominent peaks at 2θ = 16.7°, 24.1° and 34.9° that are assignable to the (200), (220) and (400) planes of Co–Fe PBAs, respectively, matching well with its standard profile of PDF # 75-0039. Notably, while the Co–Fe PBA without etching displays broaden and skewed diffraction peaks, those of the etched samples appear sharper with a narrowed full width at half maximum (FWHM). This is likely due to the high temperature etching conditions that improve the crystallinity of PBAs. After phosphorization, the crystallinity of all samples drastically decreases, with a few broad peaks distinguishable at 2θ = 31.9°, 36.5°, 48.3°, and 56.8°, inferring a homogeneous solid solution of CoP (PDF # 29-0497) and FeP (PDF # 71-2262), both having similar crystallographic features to each other (Fig. 2a).39,43,48 This point of view was further corroborated by the observation of increased crystallinity with increasing phosphorization temperature (Fig. S1 and S2†). The resemblance of XRD patterns of both CoP and FeP legitimizes the notation of the bimetal phosphides as Co0.6Fe0.4P, instead of the M2P stoichiometry that was also observed previously for other TMPs.39
:1, which reflects the feed ratio of the reactants (Table S1†). Fig. 2a presents the X-ray diffraction (XRD) spectra of the as-prepared Co0.6Fe0.4-0 (Co–Fe PBA), Co0.6Fe0.4-1.125 (before phosphorization), and Co0.6Fe0.4P-1.125 (after phosphorization). The XRD spectra of Co–Fe PBAs with and without etching exhibit similar patterns with prominent peaks at 2θ = 16.7°, 24.1° and 34.9° that are assignable to the (200), (220) and (400) planes of Co–Fe PBAs, respectively, matching well with its standard profile of PDF # 75-0039. Notably, while the Co–Fe PBA without etching displays broaden and skewed diffraction peaks, those of the etched samples appear sharper with a narrowed full width at half maximum (FWHM). This is likely due to the high temperature etching conditions that improve the crystallinity of PBAs. After phosphorization, the crystallinity of all samples drastically decreases, with a few broad peaks distinguishable at 2θ = 31.9°, 36.5°, 48.3°, and 56.8°, inferring a homogeneous solid solution of CoP (PDF # 29-0497) and FeP (PDF # 71-2262), both having similar crystallographic features to each other (Fig. 2a).39,43,48 This point of view was further corroborated by the observation of increased crystallinity with increasing phosphorization temperature (Fig. S1 and S2†). The resemblance of XRD patterns of both CoP and FeP legitimizes the notation of the bimetal phosphides as Co0.6Fe0.4P, instead of the M2P stoichiometry that was also observed previously for other TMPs.39
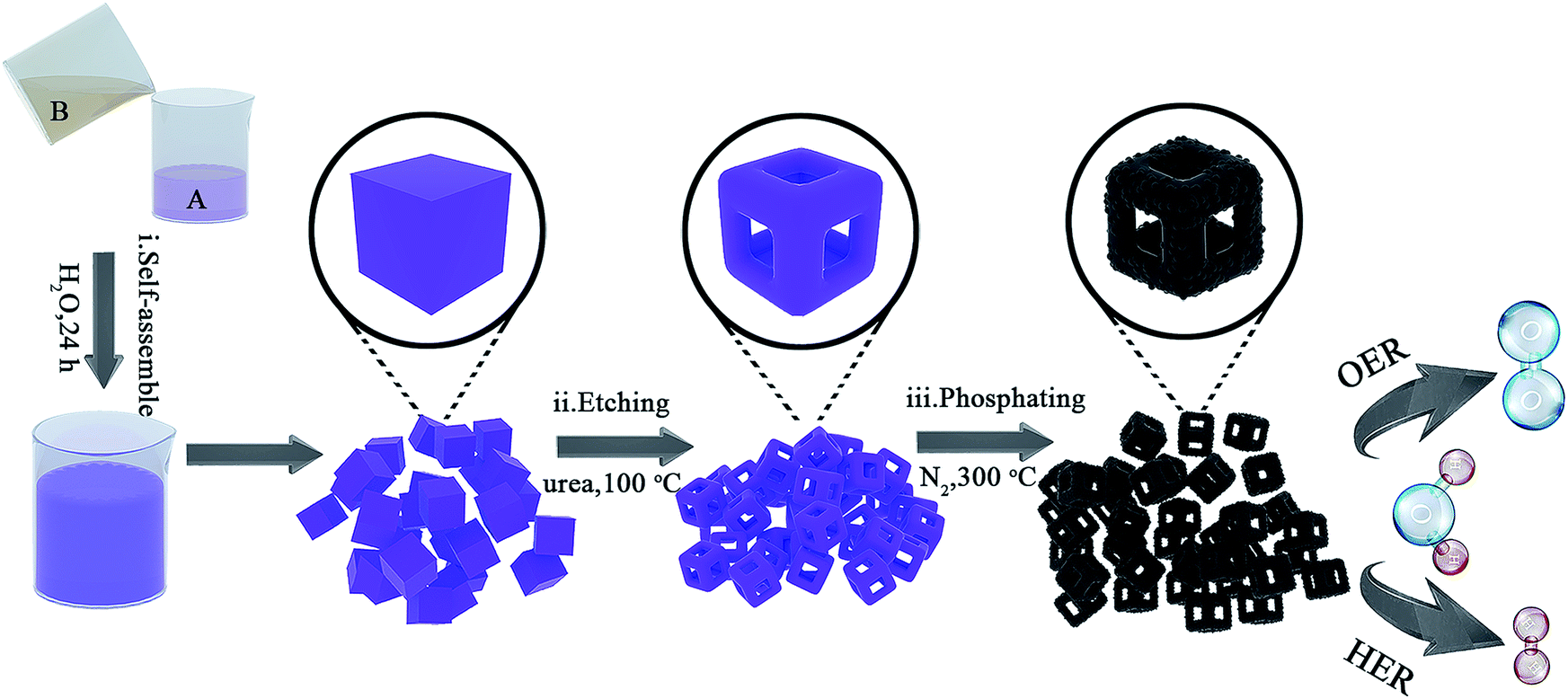 | ||
| Fig. 1 Schematic illustration of the preparation of the nanoframes composed of Co0.6Fe0.4P NPs coated with the carbon matrix. (i) Self-assembly of Co–Fe PBA; (ii) etching of the Co–Fe PBA to produce nanocages; (iii) phosphidation of the Co–Fe PBA nanocages to obtain subsequent Co0.6Fe0.4P NPs coated with the carbon matrix. | ||
 | ||
| Fig. 2 (a) Powder XRD patterns of the Co0.6Fe0.4 PBA before and after etching, and the Co0.6Fe0.4P-1.125 after phosphorization; high-resolution XPS (b) Co 2p, (c) Fe 2p, (d) P 2p spectra of the Co0.6Fe0.4P-1.125. | ||
X-ray photoelectron spectroscopy (XPS) analysis further confirmed the successful phosphorization of the Co–Fe PBA nanoframes. The survey spectra of the Co0.6Fe0.4P-1.125 reveal the presence of Co, Fe, P, C, N and O in the examined sample (Fig. S3a, ESI†), in which the O element may originate from the absorbed oxygen species and superficial oxidation due to air contact. In the high-resolution XPS Co 2p spectrum (Fig. 2b), the binding energies (BE) located at 778.6 eV and 793.5 eV are respectively the spin-orbitals of Co 2p3/2 and 2p1/2 that can be assigned to Co–P. The peaks centered at 781.8 eV and 797.8 eV can be respectively attributed to the 2p3/2 and 2p1/2 spin-orbitals of cobalt in Co–O, indicating partial oxidation of cobalt at the surface in contact with oxygen in air. Satellite peaks located at 785.4 and 802.9 eV are likely due to the shakeup excitation of the high-spin Co2+ ions.49–52 As for the high-resolution XPS spectra of Fe 2p (Fig. 2c), while the BE peaks at 706.7 eV and 718.1 eV are respectively assigned to the 2p3/2 and 2p1/2 spin-orbitals of Fe–P, the peaks at 710.4 eV and 723.7 eV can be attributed to the oxidation of Fe on the surface as well.53 Correspondingly, the P species with the binding energy of 129.0 and 130.1 eV are assigned to the P 2p1/2 and P 2p3/2 states of metal phosphides,54,55 and the peaks at about 133.8 eV represent the oxidized P species (P–O, P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, etc.) due to air exposure (Fig. 2d).56–59 It is worthwhile to note that when compared to the metallic Fe, Co, and elemental P species, the binding energies of Fe 2p and Co 2p observed here show apparently a positive shift and those of P 2p are negatively shifted, evidencing partial charge transfer between the metal and phosphorus atoms. In electrolysis, the positive metallic centers can act as the hydride acceptor whereas the negative P centers can play the role of a proton acceptor.60,61 Furthermore, the positive BE shift of the Co and Fe 2p orbitals indicates the enhanced ability of electron transfer, whereas the negative shift of P 2p suggests higher potency for electron-donation, paving the way for the use of Co0.6Fe0.4P as a bifunctional catalyst for both the HER and OER.62 Furthermore, the high-resolution XPS N 1s spectrum can be deconvoluted into three sub-peaks, corresponding to the pyridinic-N (398.51 eV), pyrrolic-N (399.80 eV) and quaternary-N (402.28 eV), respectively (Fig. S3b†). The abundant pyridinic-N is reportedly beneficial to the electrocatalytic performance by offering an electron-rich local environment surrounding the metal centers.63
O, etc.) due to air exposure (Fig. 2d).56–59 It is worthwhile to note that when compared to the metallic Fe, Co, and elemental P species, the binding energies of Fe 2p and Co 2p observed here show apparently a positive shift and those of P 2p are negatively shifted, evidencing partial charge transfer between the metal and phosphorus atoms. In electrolysis, the positive metallic centers can act as the hydride acceptor whereas the negative P centers can play the role of a proton acceptor.60,61 Furthermore, the positive BE shift of the Co and Fe 2p orbitals indicates the enhanced ability of electron transfer, whereas the negative shift of P 2p suggests higher potency for electron-donation, paving the way for the use of Co0.6Fe0.4P as a bifunctional catalyst for both the HER and OER.62 Furthermore, the high-resolution XPS N 1s spectrum can be deconvoluted into three sub-peaks, corresponding to the pyridinic-N (398.51 eV), pyrrolic-N (399.80 eV) and quaternary-N (402.28 eV), respectively (Fig. S3b†). The abundant pyridinic-N is reportedly beneficial to the electrocatalytic performance by offering an electron-rich local environment surrounding the metal centers.63
The Raman spectrum of the obtained Co0.6Fe0.4P nanoframes displays a D-band at 1338 cm−1 and G-band at 1580 cm−1, characteristic of the carbon matrix derived from MOF linkers (Fig. S3c†). The G-band corresponds to the E2g-vibration of graphitic carbon and the D-band arises from the defects within the carbon matrix. Specifically, for Co0.6Fe0.4P-1.125 the intensity ratio of ID/IG is 1.33, suggesting a low degree of graphitization and the existence of a large amount of defects in the carbon matrix.64 The thermogravimetric analysis (TGA) curves of Co–Fe PBA (Fig. S3d†) in air indicate that the MOF decomposition starts from 260 °C after the desorption of water, which is lower than the temperature of phosphorization. As for Co0.6Fe0.4P-1.125, TGA in air shows that after the loss of ligand content the sample regains weight at higher temperature due to the oxidation of Co, Fe and P elements (Fig. S3e†). The N2 adsorption–desorption measurements (Fig. S4†) indicate that etching with urea almost doubles the specific surface area for Co0.6Fe0.4-1.125 (vs. Co0.6Fe0.4-0), and the phosphorization treatment further promotes the specific surface area, resulting in a high specific surface area of 104.52 m2 g−1 and a total pore volume of 0.53 cm3 g−1 for Co0.6Fe0.4P-1.125, which are amongst the highest reported for TMP nanostructures.65
The micro-morphology and ultrafine structure of Co–Fe PBA nanoframes before and after phosphorization were characterized using scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM). The as-synthesized Co–Fe PBA presents highly uniform nanocubes with a smooth surface and an average size of 167 ± 22 nm (Fig. 3a). After etching, carved nanoframes that inherit the shape and size of the Co–Fe PBA prototype were obtained, as a result of NH4+ etching from the slowly decomposed urea and the different reaction and diffusion rates of Co/Fe ions between the edge and face center of the Co–Fe PBA nanocubes (Fig. 3b and S5b†). In contrast, direct etching with ammonium leads to irregular and collapsed nanostructures because of the strong solvent polarity and fast reaction rate (Fig. S6†).66 Unlike previously reported etching methods using either the thermal heating alone or ammonia solutions,67–71 here urea serves as both structure-directing and etching agents. Due to the high surface energy and unsaturated coordination of metal ions at the edge and corner sites of Co0.6Fe0.4 PBA, urea molecules are preferably and more strongly coordinated to these places and therefore slow down their subsequent etching by the NH4+. By contrast, the Co/Fe ions at the face center are more vulnerable to NH4+, resulting in nanocubes with concaved sides when a low etchant dosage was used (Fig. S7a and S7b†), nanoframes when the proper amount of etchant was employed (Fig. 3b), and collapsed nanoframes when the etchant was overdosed (Fig. S7c†). Additionally, when Co0.6Fe0.4 PBA nanocubes were subjected to the same heat treatment without adding urea (Fig. S5a†), no particle etching was visualized, indicating that with the reactant precursors and reaction conditions employed in this study, the thermal treatment alone was not capable of restructuring the PBA nanoparticles.
 | ||
| Fig. 3 (a1) and (a2) SEM and TEM images of the Co0.6Fe0.4 PBA nanocubes without etching; (b1) and (b2) SEM and TEM images of Co0.6Fe0.4-1.125 nanoframes before phosphorization; (c1) and (c2) SEM and TEM images of Co0.6Fe0.4P-1.125 nanoframes after phosphorization; (d) elemental mapping images of an individual Co0.6Fe0.4P-1.125 nanoframe showing the uniformly distributed Co, Fe, and P elements; (e) and (f) HR-TEM and SAD images of Co0.6Fe0.4P-1.125 showing polycrystalline lattice fringes. | ||
After phosphorization, the structure of nanoframes remains intact, with a slightly roughened surface (Fig. 3c and S5c†). To further investigate the lattice feature of the Co0.6Fe0.4P nanoframes and the surrounding environment of the metal phosphides, high-resolution TEM (HR-TEM) and energy dispersive X-ray spectroscopy (EDX) analyses were carried out, showing that Co0.6Fe0.4P nanoparticles are homogeneously embedded in the amorphous carbon matrix. Both HR-TEM images (Fig. 3f and S8a†) and XRD Debye–Scherrer calculations (Fig. S8b†) suggest that the average size of Co0.6Fe0.4P nanoparticles is about only 3.5 nm, thanks to the low annealing temperature and the slow Ostwald ripening. Clear lattice fringes with interplanar distances of 0.282, 0.231 and 0.188 nm, respectively, corresponding to the (110), (102), and (112) planes of Co0.6Fe0.4P, are measured in the HR-TEM images, further supporting that the bimetal phosphide is a solid solution of CoP and FeP, rather than a simple mix of phases. This argument is further verified by the selected area (electron) diffraction (SAD) image (Fig. 3f, inset) showing the polycrystalline nature with three major diffraction planes and the EDX mapping images (Fig. 3d) showing the uniform distribution of Co, Fe and P in the nanoframes. In addition, the EDX elemental analysis (Table S1†) further confirms that the atomic ratio of Co:Fe is approximately 1.5:1, in line with the theoretical content (6:4) and ICP measurements. All the above microscopic and spectroscopic characterizations unambiguously show that the low-temperature phosphorization process enables transformation of the bimetallic PBA precursors into homogeneous bimetallic phosphides without damaging the framework and morphology, providing a large specific surface area and porous structure of the catalyst with abundant active sites.72,73
The HER performance of the as-prepared Co0.6Fe0.4P nanoframes was investigated using a standard three-electrode system in 1.0 M KOH electrolyte. To minimize the capacitive current, the scan rate was set to 5 mV s−1 for acquiring all the linear sweep voltammetry (LSV) curves without iR compensation (Fig. 4a). Among all Co0.6Fe0.4P samples, Co0.6Fe0.4P-1.125 exhibits an overpotential of 133 mV at a current density of 10 mA cm−2, which is much smaller than those of Co0.6Fe0.4P-0 (164 mV), Co0.6Fe0.4P-0.75 (156 mV) and Co0.6Fe0.4P-1.5 (162 mV). Moreover, to reach a current density of 50 mA cm−2, the HER overpotential of the above samples are, respectively, 211 mV (Co0.6Fe0.4P-1.125), 262 mV (Co0.6Fe0.4P-0), 249 mV (Co0.6Fe0.4P-0.75), and 259 mV (Co0.6Fe0.4P-1.5). Obviously, upon etching with urea the carved Co0.6Fe0.4P nanoframes demonstrate better HER activities than the Co0.6Fe0.4P nanocubes (Co0.6Fe0.4P-0), thanks to the open 3D frameworks. Notably, the best HER activity obtained using Co0.6Fe0.4P-1.125 is only 102 mV away from the overpotential required by 20% Pt/C for achieving the current density of 50 mA cm−2. To further comprehend the HER kinetics, Tafel slopes were examined by linearly fitting the transformed polarization curves using the Tafel equation (η = blogj + a, where η is the overpotential, b is the Tafel slope, j is the current density, and a is a constant that equals the onset overpotential). Generally speaking, the Tafel slope can be regarded as the required overpotential for augmenting the exchange current by ten times, and therefore the lower the slope, the higher the reaction kinetics. Fig. 4b shows the linear Tafel slopes of all Co0.6Fe0.4P catalysts in comparison to the Pt/C benchmark. As expected, in the alkaline electrolyte the 20% Pt/C exhibits the lowest Tafel slope of only 41 mV dec−1. Co0.6Fe0.4P-1.125 presents a Tafel slope of 61 mV dec−1, lower than those of all other Co0.6Fe0.4P nanoframes, suggesting a rapid HER rate and a Volmer–Heyrovsky mechanism with the electrochemical desorption of hydrogen as the rate-limiting step.
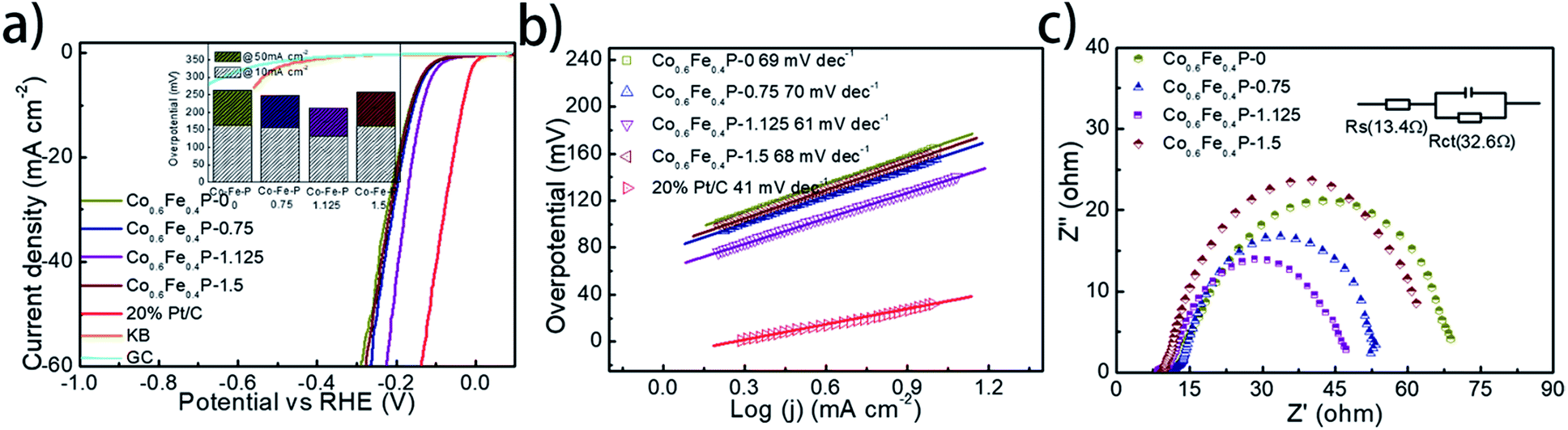 | ||
| Fig. 4 (a) Polarization curves of Co0.6Fe0.4P and control samples for the HER in 1.0 M KOH (inset: the column diagram of HER activities for all Co0.6Fe0.4P catalysts); (b) the corresponding Tafel plots of all the Co0.6Fe0.4P catalysts and 20% Pt/C; (c) Nyquist plots of all Co0.6Fe0.4P catalysts at −0.15 V over the frequency range from 100000 to 0.01 Hz. | ||
The electrochemically active surface area (ECSA) of Co0.6Fe0.4P nanoframes was estimated by calculating the double-layer capacitance from cyclic voltammetry (CV) curves at different scan rates in the voltage range between 0.22 and 0.42 V (vs. RHE) without any redox processes (Fig. S9†). A linear correlation can be observed when the current density at 0.32 V is plotted against the scan rate for all samples. It is clear that Co0.6Fe0.4P-1.125 possesses the largest Cdl (22.05 mF cm−2) amongst all Co0.6Fe0.4P samples, indicative of the highest surface area and exposure of active sites,74 which is in good agreement with the former BET measurements. In addition to the ECSA, the charge transfer resistance (Rct) at the electrode/electrolyte interface is another crucial parameter that reflects the kinetics of the redox reactions. The lower value of Rct suggests the enhanced charge transfer rate between the electrocatalyst and redox species, and thus the improved HER activity. Among all Co0.6Fe0.4P samples, Co0.6Fe0.4P-1.125 has the lowest Rct of 32.6 Ω (Fig. 4c), revealing a much faster electron-transfer rate than its counterpart nanocubes (Co0.6Fe0.4P-0). Consequently, both the enlarged ECSA and lowered Rct should help explain the enhanced HER activity of the Co0.6Fe0.4P-1.125 nanoframes.
To further study the pH versatility of the Co0.6Fe0.4P nanoframes regarding their HER activities, neutral and acidic electrolytes were also examined. In 1.0 M phosphate buffered saline (PBS) (pH = 7.0), all catalysts including Pt/C exhibit inferior HER activities (Fig. S10a1 and S10a2†) in comparison to those under alkaline conditions. To reach a current density of 10 mA cm−2, the 20% Pt/C needs an overpotential of 98 mV, whereas Co0.6Fe0.4P-1.125 requires 140 mV. However, at a higher current density of 30 mA cm−2, the HER overpotential of Co0.6Fe0.4P-1.125 (250 mV) is even lower than that of Pt/C (340 mV). In the acidic medium of 0.5 M H2SO4, the catalytic activities of all examined samples surpass those observed under both alkaline and neutral conditions, with Co0.6Fe0.4P-1.125 exhibiting overpotentials of only 97 and 170 mV to achieve the current densities of 10 and 50 mA cm−2, respectively (Fig. S10b1 and S10b2†). The high HER activities in the acidic medium are likely due to the readily available H+ ions and thus facilitated proton adsorption. It is worth noting that the HER activities observed here on the Co0.6Fe0.4P nanoframes surpass those of the majority of the non-noble-metal based TMP catalysts reported to date.49,75
To facilitate high-efficiency overall water splitting, the OER is the other equally important half reaction as the HER but might be even more kinetically sluggish due to the multiple electron transfer. Fig. 5a shows the polarization curves of all Co0.6Fe0.4P samples obtained by LSV without any iR compensation in the alkaline electrolyte (1.0 M KOH, pH = 14). Commercial RuO2 is used for benchmark comparison. Among all examined samples, Co0.6Fe0.4P-1.125 shows the best OER activity with a required overpotential of only 298 mV to reach a current density of 10 mA cm−2, even smaller than that of RuO2 (326 mV). The corresponding Tafel slope of Co0.6Fe0.4P-1.125 for the OER is 48 mV dec−1, also below that of RuO2 (62 mV dec−1). By contrast, the overpotentials to reach a current density of 10 mA cm−2 for Co0.6Fe0.4P-0, Co0.6Fe0.4P-0.75 and Co0.6Fe0.4P-1.5 are 308, 301, and 304 mV, respectively, with the corresponding Tafel slopes of 53, 54, and 55 mV dec−1. In addition, Nyquist plots (Fig. 5c) obtained at 1.53 V (vs. RHE) reveal a dramatically decreased Rct for Co0.6Fe0.4P-1.125 (24.2 Ω), much smaller than those of other Co0.6Fe0.4P samples under the same OER operating conditions. Such a good OER performance of Co0.6Fe0.4P-1.125 can be attributed to the same reasons in terms of the open 3D frameworks (thus facilitating electrolyte infiltration and gas release) and higher electrochemically active surface used to explain its excellent HER activity. However, when switched to a neutral or acidic electrolyte, the Co0.6Fe0.4P nanoframes exhibit less impressive OER activities, likely due to the dissolution of the in situ formed metal hydroxides/oxides, which will be discussed later.
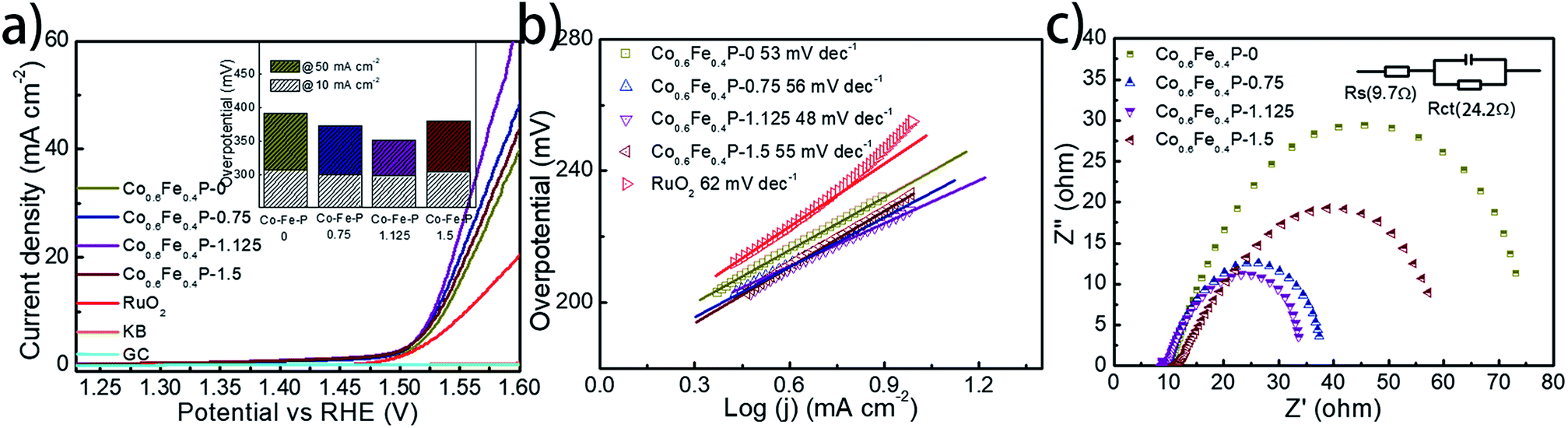 | ||
| Fig. 5 (a) Polarization curves of Co0.6Fe0.4P and control samples for the OER in 1.0 M KOH (inset: the column diagram of OER activities for all Co0.6Fe0.4P catalysts); (b) the corresponding Tafel plots of all the Co0.6Fe0.4P catalysts and RuO2; (c) Nyquist plots of all Co0.6Fe0.4P catalysts at −1.53 V over the frequency range from 100000 to 0.01 Hz. | ||
To further explore the intrinsic catalytic activity and unveil the percentage of effective active sites of the catalyst, a slow scan of CV from 0.72 to 1.42 V was acquired (Fig S11†) for Co0.6Fe0.4P-1.125. The voltage range was so chosen for the complete oxidation of the catalytically active Co0.6Fe0.4P species. The prominent oxidation peak between 0.9 V and 1.4 V is presumably ascribed to the one-step oxidation of active Co0.6Fe0.4P to Co–Fe oxide/hydroxide through an 8-electron process (eqn (1)). By integrating the faradaic charge of the oxidation peak, we surmise that about 21.1% of the Co0.6Fe0.4P is effective and exploited in electrocatalysis,18 again thanks to the open framework of Co0.6Fe0.4P-1.125 (see the ESI† for calculation details). Therefore, the turnover frequency (TOF) can be estimated by quantifying the H2/O2 conversion per unit active site per unit time (Fig. S12†), whose logarithmic transformation shows a semi-linear increment with the applied overpotential. Specifically, at a current density of 10 mA cm−2 the TOFs calculated for the HER and OER are 0.092 and 0.046 s−1, respectively, assuming the effective active sites work equally for both the HER and OER at a faradaic efficiency of 100%.
| Co0.6Fe0.4P + 11OH− → Co0.6Fe0.4OOH + PO43− + 8e− + 5H2O | (1) |
The remarkable HER and OER performance of the Co0.6Fe0.4P nanoframes in the alkaline electrolyte can be attributed to the open 3D frameworks, in addition to the high activity of bimetallic phosphides.76 On one hand, previous studies have shown that nanostructures of bimetallic phosphides exhibit superior water oxidation capability compared with their monometallic counterparts, owing to a synergic effect from the alloyed metals, as well as the increased density of states (DOS) at the Fermi level.77–79 This is further corroborated by our control studies on comparing the CoP, FeP and Co0.6Fe0.4P nanostructures (Fig. S13 and S14†). The thus optimized electronic structure of the electrocatalysts further enables more charge carriers to participate in the catalytic reactions, and allows optimizing the free energies of the reaction intermediates such as ΔG(H*) and ΔG(HO*), which are fundamental to their adsorption and desorption kinetics.80,81 On the other hand, the open 3D frameworks not only endow Co0.6Fe0.4P nanostructures with a higher electrochemically active surface area and thus promote the charge transfer among reaction species, but also effectively facilitate the electrolyte infiltration and release of the evolved gas bubbles by providing unobstructed mass diffusion pathways. Moreover, the Co0.6Fe0.4P nanoparticles are embedded in the nitrogen-doped carbon shell derived from the decomposition of the cyanide ligands, which should help maintain good conductivity and stabilize the nanoparticles by keeping them from aggregation.
With both remarkable HER and OER performances demonstrated above, a two-electrode cell using Co0.6Fe0.4P-1.125 as a bifunctional catalyst for both the anode and cathode was constructed in the alkaline electrolyte of 1.0 M KOH for overall water splitting. For comparison, an electrolyzer with RuO2 as the anode and Pt/C as the cathode was also inspected. All catalysts were coated onto a 5 × 5 mm2 nickel foam (NF) with a loading density of 2 mg cm−2. Fig. 6a shows that the Co0.6Fe0.4P-1.125 couple is able to deliver the overall water splitting with an exchange current density of 10 mA cm−2 at a cell voltage of 1.57 V, slightly lower than that of the simple addition of overpotentials from the HER (133 mV) and OER (298 mV). This is likely due to the larger specific surface area and better current-collecting ability of NF when compared to glassy carbon. The 1.57 V cell voltage is only 0.05 V higher than that of the Pt/C‖RuO2 couple (1.52 V @ 10 mA cm−2), and surpasses those of the majority of bifunctional electrocatalysts reported for overall water splitting (Table S2†).
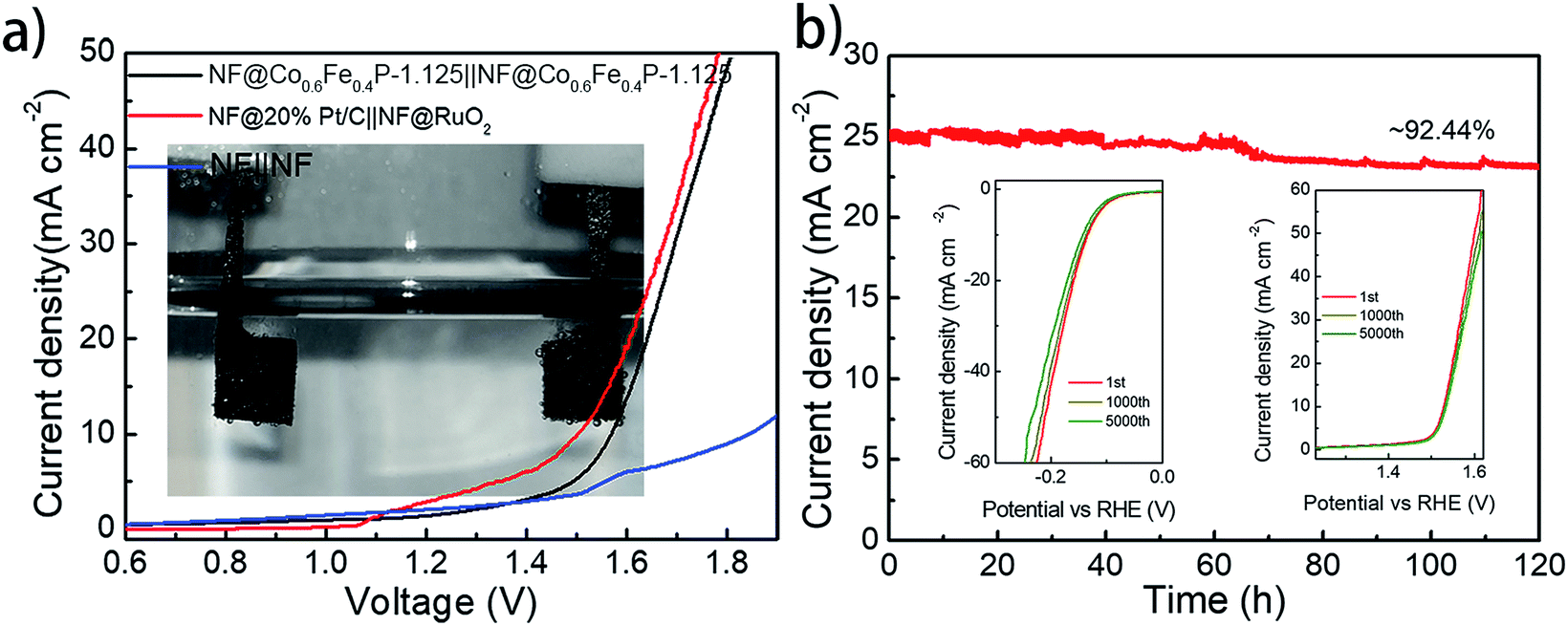 | ||
| Fig. 6 (a) The overall water splitting performance of the Co0.6Fe0.4P-1.125 and Pt/C‖RuO2 couple in 1.0 M KOH (inset: photo of the overall water splitting reaction in a two-electrode configuration); (b) the catalytic stability of the Co0.6Fe0.4P-1.125 for overall water splitting in 1.0 M KOH (inset: the durability of Co0.6Fe0.4P-1.125 for the HER and OER after 1000 and 5000 CV cycles). | ||
Durability and stability are vital criteria to assess electrocatalysts for practical applications. Herein, the durability of the Co0.6Fe0.4P-1.125 couple was evaluated through successive CV scanning for 5000 continuous cycles. For the HER, the scan potential range was set from 0.10 V to −0.25 V with a rate of 100 mV s−1, whereas for the OER, the scan range was from 1.20 to 1.65 V with the same scan rate. At the end of the CV measurements, both HER and OER polarization curves were acquired, showing negligible change in the overpotential and current density (insets in Fig. 6b). As for the electrochemical stability, a prolonged chronoamperometric test was conducted at a cell voltage of 1.65 V, resulting in an exchange current density of 26.11 mA cm−2. After 120 h of extensive testing, the current density dropped only about 7.6%, exhibiting excellent long-term stability (Fig. 6b). Such outstanding performance of Co0.6Fe0.4P-1.125 endorses its promising application for efficient overall water splitting.
To further examine whether there are any morphological and compositional changes to the catalyst after the prolonged water splitting reaction, XRD, SEM-EDX, TEM, and XPS analyses were performed, with special attention being paid to the role and fate of the phosphorus element throughout the electrolytic process. Both SEM and TEM images show no morphological changes for Co0.6Fe0.4P-1.125 on both the cathode and anode sides after the prolonged electrocatalytic reaction (Fig. S15†). EDX analysis (Fig. S16†), however, reveals that the loss of P and increase of O species after both the HER and OER. Specifically, for the P element, the atomic ratio drops from 32% in the pristine Co0.6Fe0.4P-1.125 sample to 16% and 1% in the post-HER and post-OER samples, respectively. In contrast, the atomic ratio of O increases from 17% to 35% and 46% for the pristine, post-HER and post-OER samples, respectively. This elemental change was further confirmed by the XPS analysis (Fig. 7a–c, for more detailed peak analysis see Fig. S17†). In the high-resolution XPS spectra of Co 2p, Fe 2p and P 2p spin-orbitals, all peaks associated with the metal phosphides decrease in intensity evidently after the HER, and almost completely disappear after the OER. Instead, both Co–O and Fe–O species increase after electrolysis. As for the P–O species, the highest intensity is observed on the pristine Co0.6Fe0.4P-1.125 sample, followed sequentially by the post-HER and post-OER samples. This is because at the high electric potential under OER conditions, almost all the active P species will be converted to PO43− and dissolved into the electrolyte (eqn (1)), and even at the HER potential there is still a partial oxidation of the surface P atoms (see the ESI† for possible phosphorus-involving reaction in the HER process). These observations suggest while partially oxidized Co0.6Fe0.4P is still HER active, CoFe metal oxides/hydroxides are the catalytic OER active sites formed in situ.31,82 As a result, in combination with the above CV study on effective active sites, these post-electrolysis characterizations unequivocally reveal the fate of phosphorus from the TMP catalysts in both the HER and OER sides of overall water splitting.
Furthermore, XRD analysis on the post-electrolysis samples shows that Co0.6Fe0.4P-1.125 exhibits a subtle change of the crystalline structure after the HER, but more seemingly oxide features after the OER (Fig. 7d) (Co–Fe oxide PDF # 22-1086). This, again, suggests that the Co0.6Fe0.4P nanoparticles are partially oxidized on the surface under the HER conditions but more severely oxidized in the OER process, resulting in mostly amorphous metal oxides/hydroxides. Lastly, to further interrogate if the metal phosphides are indeed contributing to the outstanding catalytic performance observed, we annealed the sample Co0.6Fe0.4-1.125 in air at 300 °C and inspected both the HER and OER activities of the obtained nanoframes of Co–Fe oxides (Fig. S18†). Much inferior electrocatalytic activities with the overpotentials of 373 and 382 mV at a current density of 10 mA cm−2 respectively for the HER and OER were witnessed, indicating that the nanostructures of TMPs, although easily oxidized in situ, are crucial for their high water splitting performance.
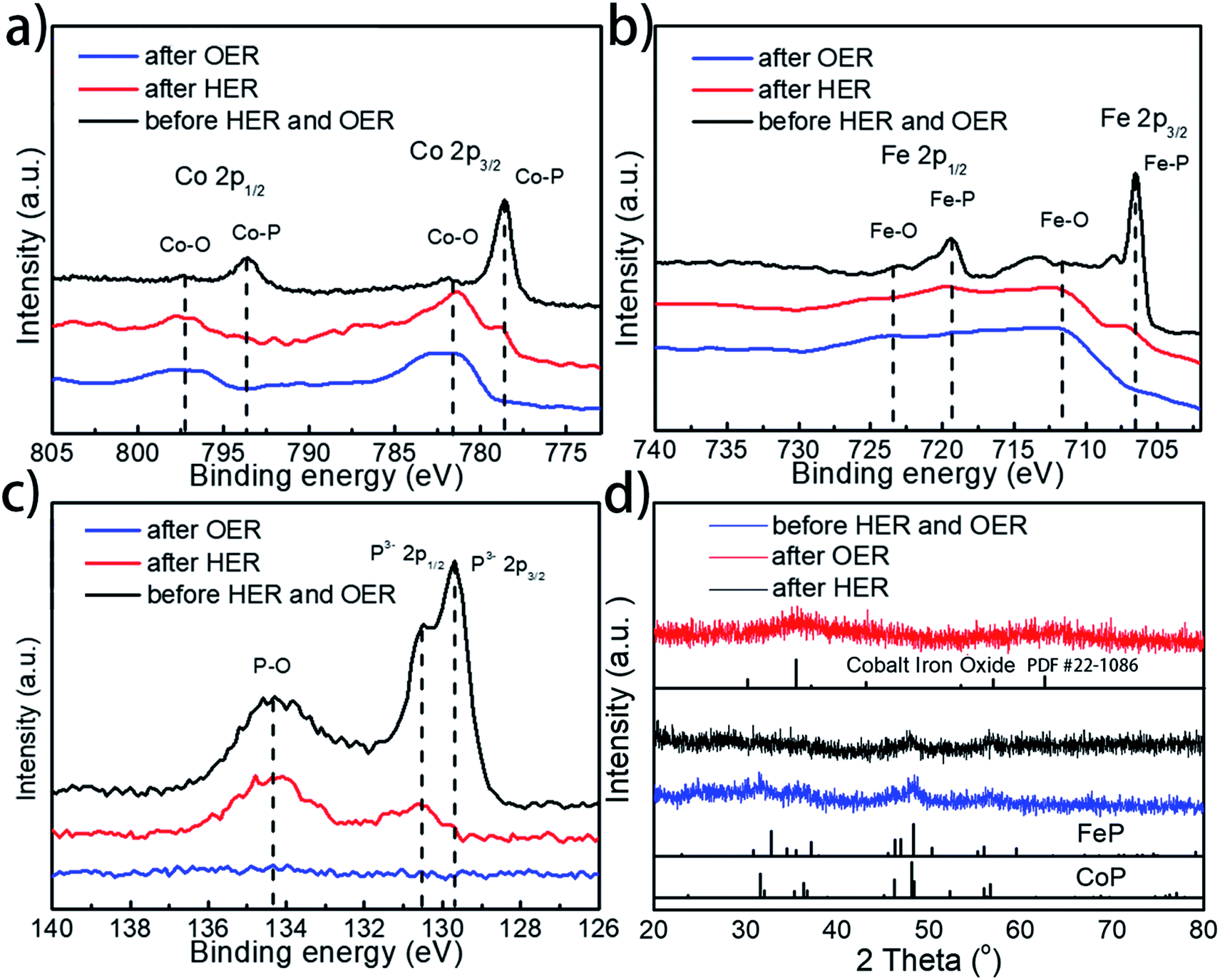 | ||
| Fig. 7 High-resolution (a) Co 2p, (b) Fe 2p, and (c) P 2p XPS spectra of the as-prepared, post-HER and post-OER Co0.6Fe0.4P-1.125 catalysts; (d) XRD patterns of the as-prepared, post-HER and post-OER Co0.6Fe0.4P-1.125 catalysts. | ||
3. Conclusions
In summary, we have demonstrated a facile route to prepare open 3D frameworks of Co–Fe bimetal phosphides as a superior catalyst for electrochemical water splitting. The structural and compositional advantages inherited from the Co–Fe PBA precursor account for the enhanced catalytic activities of the obtained nanoframes in both the HER and OER. Impressively, for the HER the Co0.6Fe0.4P-1.125 nanoframes only require an overpotential of 97 mV to achieve a 10 mA cm−2 current density in 0.5 M H2SO4 and 133 mV for 10 mA cm−2 in 1.0 M KOH. For the OER, the required overpotential to reach 10 mA cm−2 in the alkaline medium is only 297 mV. An electrolyzer employing the Co0.6Fe0.4P-1.125 nanoframes as a bifunctional catalyst for both the cathode and anode delivered a current density of 10 mA cm−2 at a cell voltage of 1.57 V with extended stability beyond 120 h, which is very close to those of integrated Pt/C and RuO2 counterparts. This outstanding performance is attributed to the enhanced electrochemically active surface area, and the promoted mass diffusion and charge transportation, thanks to the open 3D framework of the TMP nanostructures. Further in-depth activity analysis and post-electrolysis characterization revealed that while partially oxidized Co0.6Fe0.4P species are HER active, CoFe metal oxides/hydroxides are the catalytic OER active sites formed in situ. Through the carving of nanostructures for obtaining open 3D frameworks, this study demonstrates an effective tactic to promote both the HER and OER performance of bimetal phosphides towards efficient overall water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21701118), Natural Science Foundation of Jiangsu Province (No. BK20161209 and No. BK20160323) and Key Technology Initiative of Suzhou Municipal Science and Technology Bureau (SYG201748). We also extend our sincere appreciation to the support by Suzhou Key Laboratory for Advanced Carbon Materials and Wearable Energy Technologies, Suzhou 215006, China.References
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- H. Su, H. H. Wang, B. Zhang, K. X. Wang, X. H. Li and J. S. Chen, Nano Energy, 2016, 22, 79–86 CrossRef CAS.
- H. Sun, Y. Lian, C. Yang, L. Xiong, P. Qi, Q. Mu, X. Zhao, J. Guo, Z. Deng and Y. Peng, Energy Environ. Sci., 2018, 11, 2363–2371 RSC.
- Y. Xu, S. L. Yin, C. J. Li, K. Deng, H. R. Xue, X. N. Li, H. J. Wang and L. Wang, J. Mater. Chem. A, 2018, 6, 1376–1381 RSC.
- F. Qin, Z. H. Zhao, M. K. Alam, Y. Z. Ni, F. Robles-Hernandez, L. Yu, S. Chen, Z. F. Ren, Z. M. Wang and J. M. Bao, ACS Energy Lett., 2018, 3, 546–554 CrossRef CAS.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- Z. Wu, J. Wang, L. Han, R. Lin, H. Liu, H. L. Xin and D. Wang, Nanoscale, 2016, 8, 4681–4687 RSC.
- Z. Chen, Y. Ha, Y. Liu, H. Wang, H. Yang, H. Xu, Y. Li and R. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 7134–7144 CrossRef CAS PubMed.
- H. Hu, L. Han, M. Z. Yu, Z. Wang and D. Lou, Energy Environ. Sci., 2015, 9, 107–111 RSC.
- X. Shi, M. Fujitsuka, S. Kim and T. Majima, Small, 2018, 14, e1703277 CrossRef PubMed.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef CAS PubMed.
- Y. Liu, G. Yu, G. D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS PubMed.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS PubMed.
- X. J. Yang, X. J. Feng, H. Q. Tan, H. Y. Zang, X. L. Wang, Y. H. Wang, E. B. Wang and Y. G. Li, J. Mater. Chem. A, 2016, 4, 3947–3954 RSC.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed.
- L. Wang, Q. Zhou, Z. Pu, Q. Zhang, X. Mu, H. Jing, S. Liu, C. Chen and S. Mu, Nano Energy, 2018, 53, 270–276 CrossRef CAS.
- S. H. Bae, J. E. Kim, H. Randriamahazaka, S. Y. Moon, J. Y. Park and I. K. Oh, Adv. Energy Mater., 2017, 7, 1601492 CrossRef.
- M. Ledendecker, S. Krick Calderon, C. Papp, H. P. Steinruck, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- X. Xiao, L. Tao, M. Li, X. Lv, D. Huang, X. Jiang, H. Pan, M. Wang and Y. Shen, Chem. Sci., 2018, 9, 1970–1975 RSC.
- S. W. Li, Y. C. Wang, S. J. Peng, L. J. Zhang, A. M. Al-Enizi, H. Zhang, X. H. Sun and G. F. Zheng, Adv. Energy Mater., 2016, 6, 1501661 CrossRef.
- D. Y. Chung, S. W. Jun, G. Yoon, H. Kim, J. M. Yoo, K. S. Lee, T. Kim, H. Shin, A. K. Sinha, S. G. Kwon, K. Kang, T. Hyeon and Y. E. Sung, J. Am. Chem. Soc., 2017, 139, 6669–6674 CrossRef CAS PubMed.
- F. Yu, H. Q. Zhou, Y. F. Huang, J. Y. Sun, F. Qin, J. M. Bao, W. A. Goddardiii, S. Chen and Z. F. Ren, Nat. Commun., 2018, 9, 2551 CrossRef PubMed.
- Z. Pu, I. S. Amiinu, C. Zhang, M. Wang, Z. Kou and S. Mu, Nanoscale, 2017, 9, 3555–3560 RSC.
- Z. Pu, I. S. Amiinu, Z. Kou, W. Li and S. Mu, Angew. Chem., Int. Ed., 2017, 56, 11559–11564 CrossRef CAS PubMed.
- A. Han, H. Zhang, R. Yuan, H. Ji and P. Du, ACS Appl. Mater. Interfaces, 2017, 9, 2240–2248 CrossRef CAS PubMed.
- L. A. Stern, L. G. Feng, F. Song and X. L. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- H. H. Zou, C. Z. Yuan, H. Y. Zou, T. Y. Cheang, S. J. Zhao, U. Y. Qazi, S. L. Zhong, L. Wang and A. W. Xu, Catal. Sci. Technol., 2017, 7, 1549–1555 RSC.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- G.-F. Chen, T. Y. Ma, Z.-Q. Liu, N. Li, Y.-Z. Su, K. Davey and S.-Z. Qiao, Adv. Funct. Mater., 2016, 26, 3314–3323 CrossRef CAS.
- P. Zhang, B. Y. Guan, L. Yu and X. W. D. Lou, Angew. Chem., Int. Ed., 2017, 56, 7141–7145 CrossRef CAS PubMed.
- L. Yu, J. F. Yang, B. Y. Guan, Y. Lu and X. W. D. Lou, Angew. Chem., Int. Ed., 2018, 57, 172–176 CrossRef CAS PubMed.
- C. C. Du, M. X. Shang, J. X. Mao and W. B. Song, J. Mater. Chem. A, 2017, 5, 15940–15949 RSC.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2017, 56, 842 CrossRef CAS PubMed.
- H. J. Zhang, X. P. Li, A. Hahnel, V. Naumann, C. Lin, S. Azimi, S. L. Schweizer, A. W. Maijenburg and R. B. Wehrspohn, Adv. Funct. Mater., 2018, 28, 1706847 CrossRef.
- E. L. Hu, Y. F. Feng, J. W. Nai, D. Zhao, Y. Hu and X. W. Lou, Energy Environ. Sci., 2018, 11, 872–880 RSC.
- L. T. Yan, L. Cao, P. C. Dai, X. Gu, D. D. Liu, L. J. Li, Y. Wang and X. B. Zhao, Adv. Funct. Mater., 2017, 27, 1703455 CrossRef.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- Y. Bai, L. Fang, H. Xu, X. Gu, H. Zhang and Y. Wang, Small, 2017, 13, 1603718 CrossRef PubMed.
- Z. S. Cai, Y. Shi, S. S. Bao, Y. Shen, X. H. Xia and L. M. Zheng, ACS Catal., 2018, 8, 3895–3902 CrossRef CAS.
- Z. Cao, T. T. Zhou, W. Xi and Y. F. Zhao, Electrochim. Acta, 2018, 263, 576–584 CrossRef CAS.
- M. Zhuang, X. Ou, Y. Dou, L. Zhang, Q. Zhang, R. Wu, Y. Ding, M. Shao and Z. Luo, Nano Lett., 2016, 16, 4691–4698 CrossRef CAS PubMed.
- C. Xia, Q. Jiang, C. Zhao, M. N. Hedhili and H. N. Alshareef, Adv. Mater., 2016, 28, 77–85 CrossRef CAS PubMed.
- X. L. Ge, Z. Q. Li and L. W. Yin, Nano Energy, 2017, 32, 117–124 CrossRef CAS.
- Y. P. Li, J. D. Liu, C. Chen, X. H. Zhang and J. H. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 5982–5991 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- X. Y. Yu, Y. Feng, B. Y. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 RSC.
- B. Y. Guan, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56, 2386–2389 CrossRef CAS PubMed.
- M. Xu, L. Han, Y. Han, Y. Yu, J. Zhai and S. Dong, J. Mater. Chem. A, 2015, 3, 21471–21477 RSC.
- M. Liu and J. Li, ACS Appl. Mater. Interfaces, 2015, 8, 2158–2165 CrossRef PubMed.
- R. Zhang, X. Wang, S. Yu, T. Wen, X. Zhu, F. Yang, X. Sun, X. Wang and W. Hu, Adv. Mater., 2017, 29, 1605502 CrossRef PubMed.
- P. He, X. Y. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef CAS PubMed.
- Y. W. Tan, H. Wang, P. Liu, Y. H. Shen, C. Cheng, A. Hirata, T. Fujita, Z. Tang and M. W. Chen, Energy Environ. Sci., 2016, 9, 2257–2261 RSC.
- J. Yu, Q. Q. Li, Y. Li, C. Y. Xu, L. Zhen, V. P. Dravid and J. S. Wu, Adv. Funct. Mater., 2016, 26, 7644–7651 CrossRef CAS.
- X. D. Wang, Y. F. Xu, H. S. Rao, W. J. Xu, H. Y. Chen, W. X. Zhang, D. B. Kuang and C. Y. Su, Energy Environ. Sci., 2016, 9, 1468–1475 RSC.
- Z. Pu, C. Zhang, I. S. Amiinu, W. Li, L. Wu and S. Mu, ACS Appl. Mater. Interfaces, 2017, 9, 16187–16193 CrossRef CAS PubMed.
- T. H. Zhou, Y. H. Du, S. M. Yin, X. Z. Tian, H. B. Yang, X. Wang, B. Liu, H. M. Zheng, S. Z. Qiao and R. Xu, Energy Environ. Sci., 2016, 9, 2563–2570 RSC.
- J. Nai, Y. Lu, L. Yu, X. Wang and X. W. D. Lou, Adv. Mater., 2017, 29, 1703870 CrossRef PubMed.
- X. Y. Yu, L. Yu, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 5331–5335 CrossRef CAS PubMed.
- L. Han, X. Y. Yu and X. W. Lou, Adv. Mater., 2016, 28, 4601–4605 CrossRef CAS PubMed.
- J. Nai, B. Y. Guan, L. Yu and X. W. D. Lou, Sci. Adv., 2017, 3, e1700732 CrossRef PubMed.
- J. Nai, J. Zhang and X. W. Lou, Chem, 2018, 4, 1967–1982 CAS.
- J. Nai and X. W. D. Lou, Adv. Mater., 2018 DOI:10.1002/adma.201706825.
- J. Nai, Y. Lu, L. Yu, X. Wang and X. W. D. Lou, Adv. Mater., 2017, 29, 1703870 CrossRef PubMed.
- M. H. Sun, S. Z. Huang, L. H. Chen, Y. Li, X. Y. Yang, Z. Y. Yuan and B. L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
- C. M. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC.
- H. B. Wu, B. Y. Xia, L. Yu, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- Z. Pu, I. Saana Amiinu, M. Wang, Y. Yang and S. Mu, Nanoscale, 2016, 8, 8500–8504 RSC.
- J. Hao, W. Yang, Z. Zhang and J. Tang, Nanoscale, 2015, 7, 11055–11062 RSC.
- J. Kibsgaard, C. Tsai, K. Chan, J. D. Benck, J. K. Norskov, F. Abild-Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 RSC.
- C. Tang, L. Gan, R. Zhang, W. Lu, X. Jiang, A. M. Asiri, X. Sun, J. Wang and L. Chen, Nano Lett., 2016, 16, 6617–6621 CrossRef CAS PubMed.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1781 RSC.
- P. Liu and J. A. Rodriguez, J. Am. Chem. Soc., 2005, 127, 14871–14878 CrossRef CAS PubMed.
- P. Xiao, M. A. Sk, L. Thia, X. M. Ge, R. J. Lim, J. Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 RSC.
- J. Ryu, N. Jung, J. H. Jang, H. J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066–4074 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03877e |
| This journal is © The Royal Society of Chemistry 2019 |