
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of micrococcin P1 and thiocillin I enabled by Mo(VI) catalyst†
Siddhartha
Akasapu‡
,
Aaron B.
Hinds‡
 ,
Wyatt C.
Powell
and
Maciej A.
Walczak
*
,
Wyatt C.
Powell
and
Maciej A.
Walczak
*
Department of Chemistry, University of Colorado, Boulder, CO 80309, USA. E-mail: maciej.walczak@colorado.edu
First published on 3rd December 2018
Abstract
Thiopeptides are a class of potent antibiotics with promising therapeutic potential. We developed a novel Mo(VI)-oxide/picolinic acid catalyst for the cyclodehydration of cysteine peptides to form thiazoline heterocycles. With this powerful tool in hand, we completed the total syntheses of two representative thiopeptide antibiotics: micrococcin P1 and thiocillin I. These two concise syntheses (15 steps, longest linear sequence) feature a C–H activation strategy to install the trisubstituted pyridine core and thiazole groups. The synthetic material displays promising antimicrobial properties measured against a series of Gram-positive bacteria.
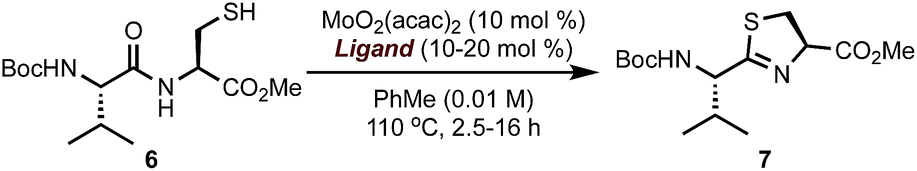
The surge in drug-resistant strains of bacteria poses a major global health threat and the need for expedient discovery of new therapeutics characterized by novel modes of action represents a high priority.1 The thiopeptide family of antibiotics contains ∼150 members of post-translationally modified peptides of ribosomal origin that display antibacterial activities against drug-resistant pathogens, including methicillin-resistant Staphylococcus aureus (MRSA).2 All thiopeptide antibiotics share a common molecular scaffold involving a nitrogen-containing heterocyclic core decorated with varying numbers of thiazol(in)e rings, assembled into macrocycles or acyclic chains of varying sizes and lengths.2c The macrocycle, whose size is correlated with molecular targets, is responsible for distinct modes of action, allowing thiopeptides to inhibit bacterial cell growth by means of blocking ribosomal protein synthesis.2d Because of their unique biological activities and intriguing structure, thiopeptides have received considerable attention from the synthetic community.3 However, the lack of concise, scalable, and practical syntheses amenable for rapid molecular modifications limits access to these valuable natural products. In this paper, we report a general approach to the synthesis of thiopeptide antibiotics featuring a novel Mo(VI) oxide catalyst to promote the cyclodehydration of cysteine residues under mild and neutral conditions and is demonstrated by the preparation of D series thiopeptides micrococcin P1 (1)3u,3w,4 and thiocillin I (2),3x,5 This practical and scalable route is suitable for analogue preparation and site-selective modifications.6
Our retrosynthetic analysis of the synthetic targets led to the dissection of the macrocycle into two “top” and “bottom” fragments, which could be united through a peptide coupling and then macrocyclised at the thiazole carboxylic acid adjacent to valine to furnish the final product(s) (Fig. 1). For construction of the two fragments, we envisioned a convergent approach involving the strategic introduction of selected thiazole rings through a catalytic cysteine cyclodehydration. Catalytic methods for cysteine cyclodehydrations are scarce despite the myriad obstacles encountered when employing common stoichiometric reagents due to their high Lewis acidity, likely incompatibility with acid-sensitive groups, possible epimerization, and generation of undesired by-products.7 We hypothesized that a MoO2(L)2 complex with the appropriate ligands might minimize epimerization of the endo- and exocyclic methylene groups since the cyclization event occurs under neutral conditions. Furthermore, bidentate ligands may extend the lifetime of the catalyst and prevent precipitation of heterogeneous molybdenum particles.8
 | ||
| Fig. 1 Selected thiopeptide antibiotics of the D series. | ||
To validate the feasibility of this proposal, we evaluated a series of Mo(VI) oxide complexes (Table 1). Initially, we studied the cyclodehydration reaction under conditions using only acac (acetylacetonate) as the ligand (entry 1). We observed that the molybdenum complex precipitated from the solution, forming insoluble particles, resulting in low yields of thiazoline product 7. In order to maximize the conversion of 6 into 7, catalyst loadings of MoO2(acac)2 had to be increased up to 60 mol% and the molybdenum complex had to be added portion-wise over a period of 12 h to ensure reproducible yields. We hypothesized that a bidentate ligand that (a) stabilizes Mo, preventing the formation of heterogeneous particles, and (b) modulates the electrophilic character of Mo presented a viable solution to these problems. To this end, we investigated a series of ligands based on the pyridine 2-carboxylate core (entries 2–11). These additives (20 mol%) were pre-mixed with MoO2(acac)2 (10 mol%) and used directly in the cyclodehydration reactions with 6. Addition of picolinic acid 8a was sufficient to improve the yield of 7 to 85% (entry 2) whereas carboxylates with electron-donating groups such as 8d, 9a, 9b gave 7 in <30% yield. Although still suboptimal, electron-poor ligands (entries 10 and 11) improved conversion of 6 into 7 with respect to the original conditions using only the acac ligand. The geometry of the Mo centre is also important as demonstrated by the reaction with bis-acid 8e forming a tridentate complex with Mo (entry 7). We found that a combination of sterics at C6 and a moderate electron-donating ability of the substituent resulted in excellent yields of 7 with <1% epimerization (entries 3–5). Although 6-methyl- and 6-ethylpyridine carboxylates gave comparable results (entries 2–5), we elected to use 8b as the ligand for further studies because of the significant cost difference between these two reagents. For comparison, a ligand with a basic sp3 nitrogen completely suppressed the reaction (entry 12). To exclude the possibility that the cyclodehydration reactions are catalyzed by a Brønsted acid, thiol 6 was exposed to catalytic amounts of pTSA (10 mol%) or 8b (10 mol%) under otherwise identical conditions from Table 1 and gave 7 in <1% after 16 h at 110 °C.
|
|
|||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Entry | L | Time | Yield | Entry | L | Time | Yield |
| 1 | None | 16 h | 18% | 7 | 8e | 16 h | 19% |
| 2 | 8a | 16 h | 85% | 8 | 9a | 16 h | 8% |
| 3 | 8b | 2.5 h | 87% | 9 | 9b | 16 h | 9% |
| 4 | 8b | 16 h | 97% | 10 | 9c | 16 h | 68% |
| 5 | 8c | 16 h | 98% | 11 | 10 | 16 h | 62% |
| 6 | 8d | 16 h | 26% | 12 | L-Proline | 16 h | <1% |

We rationalized the reactivity trend from Table 1 by a mechanism depicted in Scheme 1A: complex 11 undergoes a ligand exchange with amide 12, replacing one of the pyridine coordination sites. Picolinic acid-based ligands that contain electron-donating groups (e.g., 8d, 9a, 9b) retard the initial displacement step because the electron-rich heterocycle forms a tighter complex with the electrophilic Mo(VI), whereas electron-withdrawing groups at C4 and C6 of the pyridine ring (e.g., 9c) facilitate the displacement. Once the amide is bound to Mo (13), a proton transfer occurs from the amide NH to one of the oxo ligands. This step is likely promoted by a neighbouring nitrogen atom on the pyridine ring and may proceed intramolecularly. Ring closure from 14 followed by elimination of H2O and azoline product 16 regenerates catalyst 11. The highest rate of cyclodehydration of 6 with 8b can be rationalized by its electron-donating ability combined with steric hindrance of the methyl group that likely distorts the geometry of the MoO2(L2) complex, thus accelerating access of the amide substrate. This mechanistic proposal is consistent with DFT calculations (Scheme 1B). The most stable configuration of the MoO2(pic)2 complex is the one with pyridine nitrogen atoms in a cis configuration (e.g., 11). The Mo–N bond in the unsubstituted picolinic acid complex is 2.38 Å. This linkage is elongated in complexes substituted at C4 with an electron-withdrawing group (9c) and in sterically-congested complexes with the 6-Me group making the initial ligand exchange more facile. On the contrary, attachment of an electron-donating group at the para position increases the bond strength between Mo and N, demonstrated by a shortened Mo–N distance in a complex with 9a.
 | ||
| Scheme 1 (A) Proposed mechanism of Mo-catalysed cyclodehydration (second carboxylate ligand abbreviated for clarity). (B) Selected geometrical parameters of MoO2(X)2 complexes optimized at B3LYP/6-31G(d)/LANL2DZ level of theory. The optimized structure of the thermodynamic complex of MoO2(pic)2, pic = 2-picolinate is presented in the middle of the catalytic cycle. | ||
Having established a robust method for the catalytic installation of select thiazole groups, we proceeded towards the preparation of bottom fragments 25 (Scheme 2). The optimized cyclodehydration conditions were applied to two dipeptides 17 followed by oxidation to thiazoles 18 in excellent yields for both substrates. The remaining portion of the bottom fragment was prepared via a modified Hantzsch method using thioamide 19 and bromopyruvate 20 in 94% yield. Coupling of 21 with serine 22 followed by a stereoselective (dr > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) installation of the trisubstituted olefin with MsCl and a base (DABCO, Et2NH) resulted in thiazole 24. With this key precursor in hand, the amine and carboxylate groups in 18 and 24 were liberated and the subsequent amide couplings resulted in gram-scale syntheses of bottom fragments 25 in seven steps for both micrococcin P1 and thiocillin I in 47% and 51% overall yield, respectively.
:1) installation of the trisubstituted olefin with MsCl and a base (DABCO, Et2NH) resulted in thiazole 24. With this key precursor in hand, the amine and carboxylate groups in 18 and 24 were liberated and the subsequent amide couplings resulted in gram-scale syntheses of bottom fragments 25 in seven steps for both micrococcin P1 and thiocillin I in 47% and 51% overall yield, respectively.
 | ||
| Scheme 2 Synthesis of bottom fragments 25. (a) MoO2(acac)2 (10 mol%), 8b (20 mol%), PhMe, 110 °C, 2.5 h: 18a – 92%, 18b – 98%; (b) DBU, CBrCl3, CH2Cl2, 0 °C, 1 h: 18a – 97%, 18b – 91%; (c) 20, NaHCO3, THF, 0 °C to rt, 16 h then pyridine, TFAA, 0 °C, 20 min, 94%; (d) HCl, 1,4-dioxane, rt, 7 h then 22, HATU, DIPEA, DMF, 0 °C to rt, 1 h, 76%; (e) MsCl, Et3N, CH2Cl2, 0 °C, 1 h then DABCO, Et2NH, 0 °C to rt, CH2Cl2, 16 h, 91%; (f) HCl, 1,4-dioxane, 0 °C to rt, 45 min; (g) Bu2SnO, MeOH, 80 °C, 4 h then LiOH, H2O/THF, 90% (over two steps); (h) HATU, DIPEA, DMF, 0 °C to rt, 2 h: 25a – 72%, 25b – 78%. | ||
The synthesis of top fragment 37, common to D series thiopeptides, started from a Pd-catalyzed C–H activation of thiazole 26 (Scheme 3). We found that the yield of this reaction was critically dependent on the size of the ester group and the methyl group was incompatible with the cross-coupling conditions. The reaction of ethyl thiazole ester 26b with Pd(OAc)2 (5 mol%) and CyJohnPhos (15 mol%) gave, after conversion from 28a, methyl ester 28b9 which was then coupled with amine 30 following the two-step, selective chlorination of the pyridine core (97%). At this stage, the sulfur protective group was removed, and MoO2(acac)2-catalyzed cyclodehydration and oxidation afforded 32 in 86% yield. We found that the pyridine scaffold in 31 was sufficient to stabilize the Mo(VI) catalyst and prevented decomposition of the Mo complex in the cyclodehydration reaction thus alleviating the need of further stabilization by additional ligands. Next, the Stille coupling with ethyl vinyl ether 33 followed by bromination with NBS afforded bromoketone 34. Inspired by the prior work of Williams,10 we converted 34 into thiazole 36 in 86%. Because the exocyclic methylene group in the Hantzsch intermediate can undergo epimerization, we optimized the cyclization step by buffering the conditions with a bulky base (2,6-lutidine) whereas larger bases (e.g., 2,6-di-t-butyl-pyridine) were ineffective in promoting this transformation. To confirm the optical purity of the newly formed thiazole 36, the carbamate was removed, and the resultant amine was converted into a Mosher amide giving a 95:5 dr (determined by 19F NMR). Finally, site-selective hydrolysis of the methyl ester in 36 proceeded without concomitant removal of the i-Pr group to yield top fragment 37 in 73%.3l
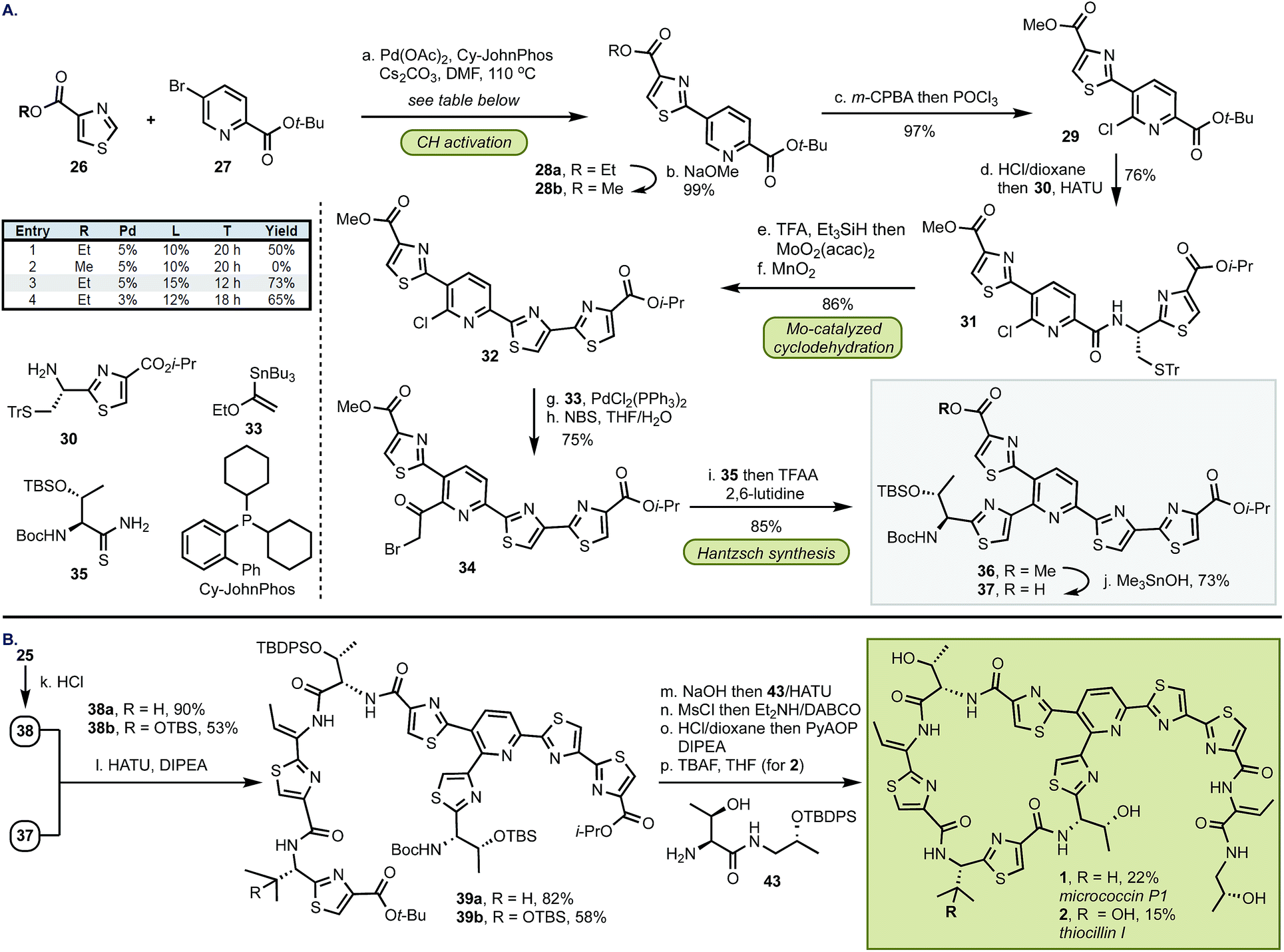 | ||
| Scheme 3 Preparation of the top fragment and completion of the syntheses. (a) Pd(OAc)2 (5 mol%), CyJohnPhos (15 mol%), Cs2CO3, DMF, 110 °C, 12 h, 73%; (b) NaOMe, MeOH, −20 to 0 °C, 99%; (c) i – m-CPBA, CH2Cl2, rt, 2 d; ii – POCl3, DMF, 0 °C, CH2Cl2, 97% (over two steps); (d) HCl, 1,4-dioxane, rt, 9 h then 30, HATU, DIPEA, DMF, 0 °C to rt, 2 h, 72%; (e) i – TFA, Et3SiH, CH2Cl2, 0 °C, 1 h then MoO2(acac)2 (10 mol%), PhMe, 110 °C, 13 h; (f) MnO2, CH2Cl2, rt, 24 h, 86% (from 31); (g) 33, PdCl2(PPh3)2 (10 mol%), 1,4-dioxane, 90 °C, 18 h, 97%; (h) NBS, THF, pH 7 phosphate buffer, rt, 1 h, 79%; (i) i – 35, KHCO3, dimethoxyethane, −40 °C to rt, 24 h then TFAA, 2,6-lutidine, dimethoxyethane, −20 °C, 20 h, 85%; (j) Me3SnOH, (CH2Cl)2, 80 °C, 20 h, 73%; (k) HCl, 1,4-dioxane, 0 °C to rt, 30–45 min: 38a – 90%, 38b – 53%; (l) HATU, DIPEA, DMF, 0 °C to rt, 2 h: 39a – 82%, 39b–53%; (m) NaOH, MeOH, 0 °C to rt, 2 h then 43, HATU, DIPEA, DMF, 0 °C to rt, 2 h; (n) MsCl, Et2NH, CH2Cl2 0 °C to rt, 15 min then DABCO, Et2NH, CH2Cl2, 0 °C to rt, 2.5 d; (o) HCl, 1,4-dioxane, rt, 16 h then PyAOP, DIPEA, DMF, −20 °C, 30 min, 22% (for 1 from 39a); (p) TBAF, THF, 0 °C, 1–2 h, 15% (for 2 from 39b). | ||
In preparation for the union of the two fragments, the Boc protective group was removed from bottom segments 25 to yield free amines 38 which were then coupled with top unit 37 to afford oligopeptides 39. Next, the i-Pr ester in 39 was removed under more forcing conditions (NaOH/H2O), and the resultant acid was merged with dipeptide 43. The installation of the olefin was accomplished by elimination of the hydroxyl group in threonine with MsCl in exclusive Z selectivity. The final steps of the synthesis involved global deprotection with HCl to cleave all alcohol, amine, and carboxylic acid protective groups in one step, followed by macrocyclization of the resulting amino acid intermediate with PyAOP furnished micrococcin P1 1 in 22% yield. The similarity of micrococcin P1 and thiocillin I, which differ only by a mutation of L-valine into 3-hydroxy-L-valine, led us to adopt identical conditions for both syntheses, with the exception of the deprotection for thiocillin I. We found that acidic conditions alone were insufficient and resulted in only partial cleavage of the silicon group at the hydroxyvaline position (ca. 30%); thus, following the final macrocyclization, the resultant precursor was subjected to TBAF to reveal thiocillin I (2) in 15%. Micrococcin P1 was completed in 14 steps and thiocillin I was completed in 15 steps from known commercial materials (longest linear sequence).
Having access to two representative members of the thiopeptide family, we evaluated the in vitro antimicrobial activity against a panel of recent Gram-positive bacterial strains (Table 2). Micrococcin P1 and thiocillin I exhibited similar potencies, showing 2- to 4-fold difference in their minimum inhibitory concentration (MIC) values in nearly all of the test bacterial isolates. An exception to this trend was observed for B. subtilis ATCC 6633, where micrococcin P1 was inactive at 16 μg mL−1 while thiocillin I displayed activity at concentrations as low as 4 μg mL−1, a result comparable to a previously published report showing thiocillin I as active at 1.56 μg mL−1 against B. subtilis PCI 219.5 Additionally, micrococcin P1 and thiocillin I were more potent than comparator antibiotics against vancomycin-resistant E. faecalis.
| Isolate | MIC (μg mL−1) | ||||
|---|---|---|---|---|---|
| 1 | 2 | Vancomycin | Ciprofloxacin | ||
| S. aureus | ATCC 29213 | 4 | 2 | 1 | 0.5 |
| 1974149 | 2 | 2 | 0.5 | 0.25 | |
| 1974148 | 8 | 2 | 1 | >16 | |
| E. faecalis | 1674621 | 1 | 0.5 | 0.5 | 1 |
| 1674614 | 1 | 0.5 | >16 | >16 | |
| B. subtilis | ATCC 6633 | >16 | 4 | 0.25 | 0.06 |
| S. pyogenes | 1744264 | 1 | 0.5 | 0.5 | 0.25 |
In summary, we described a general synthetic route toward thiopeptide antibiotics belonging to the D series. Given the structural commonalities shared by all family members of this class, the presented method is easily adaptable to suit the preparation of thiopeptides belonging to other series as well as derivatives. The novel molybdenum catalyst eliminates the need for stoichiometric reagents and generates water as the only by-product. Moreover, the modular approach lends itself into future SAR studies to attain compounds with desirable properties for pre-clinical and clinical development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Colorado at Boulder and in part by the NSF (CHE-1753225). Mass spectral analyses were recorded by the University of Colorado Boulder Central Analytical Laboratory Mass Spectrometry Core Facility (partially funded by NIH S10 RR026641). Dr Bimala Lama is acknowledged for the assistance with NMR experiments.References
- (a) M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089 CrossRef CAS PubMed; (b) C. f. D. C. a. Prevention, Antibiotic Resistance Threats in the United States, 2013 Search PubMed; (c) I. B. Seiple, Z. Zhang, P. Jakubec, A. Langlois-Mercier, P. M. Wright, D. T. Hog, K. Yabu, S. R. Allu, T. Fukuzaki, P. N. Carlsen, Y. Kitamura, X. Zhou, M. L. Condakes, F. T. Szczypiński, W. D. Green and A. G. Myers, Nature, 2016, 533, 338 CrossRef CAS PubMed; (d) M. F. Chellat, L. Raguž and R. Riedl, Angew. Chem., Int. Ed., 2016, 55, 6600 CrossRef CAS PubMed; (e) M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299 CrossRef CAS PubMed; (f) S. E. Rossiter, M. H. Fletcher and W. M. Wuest, Chem. Rev., 2017, 117, 12415 CrossRef CAS PubMed.
- (a) M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685 CrossRef CAS PubMed; (b) C. Li and W. L. Kelly, Nat. Prod. Rep., 2010, 27, 153 RSC; (c) X. Just-Baringo, F. Albericio and M. Álvarez, Angew. Chem., Int. Ed., 2014, 53, 6602 CrossRef CAS PubMed; (d) X. Just-Baringo, F. Albericio and M. Álvarez, Mar. Drugs, 2014, 12, 317 CrossRef CAS PubMed.
- (a) C. J. Moody and M. C. Bagley, Chem. Commun., 1998, 2049 RSC; (b) C. J. Moody and M. C. Bagley, Synlett, 1998, 361 CrossRef CAS; (c) K. C. Nicolaou, C. N. C. Boddy, S. Bräse and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096 CrossRef; (d) M. C. Bagley, K. E. Bashford, C. L. Hesketh and C. J. Moody, J. Am. Chem. Soc., 2000, 122, 3301 CrossRef CAS; (e) K. C. Nicolaou, M. Nevalainen, B. S. Safina, M. Zak and S. Bulat, Angew. Chem., Int. Ed., 2002, 41, 1941 CrossRef CAS; (f) C. J. Moody, R. A. Hughes, S. P. Thompson and L. Alcaraz, Chem. Commun., 2002, 1760 RSC; (g) K. C. Nicolaou, B. S. Safina, C. Funke, M. Zak and F. J. Zécri, Angew. Chem., Int. Ed., 2002, 41, 1937 CrossRef CAS; (h) K. C. Nicolaou, M. Nevalainen, M. Zak, S. Bulat, M. Bella and B. S. Safina, Angew. Chem., 2003, 115, 3540 CrossRef; (i) R. A. Hughes, S. P. Thompson, L. Alcaraz and C. J. Moody, Chem. Commun., 2004, 946 RSC; (j) K. C. Nicolaou, M. Zak, B. S. Safina, S. H. Lee and A. A. Estrada, Angew. Chem., Int. Ed., 2004, 43, 5092 CrossRef CAS PubMed; (k) K. C. Nicolaou, M. Zak, B. S. Safina, A. A. Estrada, S. H. Lee and M. Nevalainen, J. Am. Chem. Soc., 2005, 127, 11176 CrossRef CAS PubMed; (l) K. C. Nicolaou, A. A. Estrada, M. Zak, S. H. Lee and B. S. Safina, Angew. Chem., Int. Ed., 2005, 44, 1378 CrossRef CAS; (m) K. C. Nicolaou, B. S. Safina, M. Zak, S. H. Lee, M. Nevalainen, M. Bella, A. A. Estrada, C. Funke, F. J. Zécri and S. Bulat, J. Am. Chem. Soc., 2005, 127, 11159 CrossRef CAS PubMed; (n) K. C. Nicolaou, B. Zou, D. H. Dethe, D. B. Li and D. Y. K. Chen, Angew. Chem., Int. Ed., 2006, 45, 7786 CrossRef CAS PubMed; (o) H. M. Müller, O. Delgado and T. Bach, Angew. Chem., Int. Ed., 2007, 46, 4771 CrossRef PubMed; (p) R. A. Hughes and C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 7930 CrossRef CAS PubMed; (q) T. Mori, S. Higashibayashi, T. Goto, M. Kohno, Y. Satouchi, K. Shinko, K. Suzuki, S. Suzuki, H. Tohmiya, K. Hashimoto and M. Nakata, Chem.–Asian J., 2008, 3, 984 CrossRef CAS PubMed; (r) O. Delgado, H. M. Mueller and T. Bach, Chem.–Eur. J., 2008, 14, 2322 CrossRef CAS PubMed; (s) T. Mori, S. Higashibayashi, T. Goto, M. Kohno, Y. Satouchi, K. Shinko, K. Suzuki, S. Suzuki, H. Tohmiya, K. Hashimoto and M. Nakata, Chem.–Asian J., 2008, 3, 1013 CrossRef CAS PubMed; (t) K. C. Nicolaou, D. H. Dethe, G. Y. C. Leung, B. Zou and D. Y. K. Chen, Chem.–Asian J., 2008, 3, 413 CrossRef CAS PubMed; (u) D. Lefranc and M. A. Ciufolini, Angew. Chem., Int. Ed., 2009, 48, 4198 CrossRef CAS PubMed; (v) C. Ammer and T. Bach, Chem.–Eur. J., 2010, 16, 14083 CrossRef CAS PubMed; (w) M. A. Ciufolini and D. Lefranc, Nat. Prod. Rep., 2010, 27, 330 RSC; (x) V. S. Aulakh and M. A. Ciufolini, J. Am. Chem. Soc., 2011, 133, 5900 CrossRef CAS PubMed; (y) K. C. Nicolaou, Angew. Chem., Int. Ed., 2012, 51, 12414 CrossRef CAS; (z) X. Just-Baringo, P. Bruno, L. K. Ottesen, L. M. Cañedo, F. Albericio and M. Álvarez, Angew. Chem., Int. Ed., 2013, 52, 7818 CrossRef CAS PubMed; (a a) K. P. Wojtas, M. Riedrich, J.-Y. Lu, P. Winter, T. Winkler, S. Walter and H.-D. Arndt, Angew. Chem., Int. Ed., 2016, 55, 9772 CrossRef CAS PubMed.
- T. L. Su, Br. J. Exp. Pathol., 1948, 29, 473 CAS.
- J. Shoji, H. Hinoo, Y. Wakisaka, K. Koizumi and M. Mayama, J. Antibiot., 1976, 29, 366 CrossRef CAS PubMed.
- (a) W. J. Wever, J. W. Bogart and A. A. Bowers, J. Am. Chem. Soc., 2016, 138, 13461 CrossRef CAS PubMed; (b) X. Luo, C. Zambaldo, T. Liu, Y. Zhang, W. Xuan, C. Wang, S. A. Reed, P.-Y. Yang, R. E. Wang, T. Javahishvili, P. G. Schultz and T. S. Young, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3615 CrossRef CAS PubMed.
- K. C. Majumdar and S. K. Chattopadhyay, Heterocycles in Natural Product Synthesis, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- (a) A. Sakakura, R. Kondo and K. Ishihara, Org. Lett., 2005, 7, 1971 CrossRef CAS PubMed; (b) A. Sakakura, R. Kondo, S. Umemura and K. Ishihara, Adv. Synth. Catal., 2007, 349, 1641 CrossRef CAS; (c) A. Sakakura, S. Umemura, R. Kondo and K. Ishihara, Adv. Synth. Catal., 2007, 349, 551 CrossRef CAS; (d) A. Sakakura, S. Umemura and K. Ishihara, Chem. Commun., 2008, 3561 RSC; (e) A. Sakakura, R. Kondo, S. Umemura and K. Ishihara, Tetrahedron, 2009, 65, 2102 CrossRef CAS.
- T. Martin, C. Verrier, C. Hoarau and F. Marsais, Org. Lett., 2008, 10, 2909 CrossRef CAS PubMed.
- Q. Qiao, S.-S. So and R. A. Goodnow, Org. Lett., 2001, 3, 3655 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04885a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |