
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-controlled diastereodivergent, enantio- and regioselective copper-catalyzed hydroxyalkylboration of 1,3-dienes with ketones†
Jian-Jun
Feng‡
 ,
Yan
Xu‡
and
Martin
Oestreich
*
,
Yan
Xu‡
and
Martin
Oestreich
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: martin.oestreich@tu-berlin.de
First published on 20th August 2019
Abstract
A copper-catalyzed three-component coupling of 1,3-dienes, bis(pinacolato)diboron, and ketones allows for the chemo-, regio-, diastereo- and enantioselective assembly of densely functionalized tertiary homoallylic alcohols. The relative configuration of the vicinal stereocenters is controlled by the chiral ligand employed. Subsequent transformations illustrate the versatility of these valuable chiral building blocks.
Introduction
The enantioselective synthesis of tertiary homoallylic alcohols1 continues to attract attention as these are highly useful intermediates in complex molecule synthesis and for medicinal chemistry.2 An established way to access that motif is by ketone allylation3–7 where enantiofacial discrimination and low reactivity are the key challenges compared to aldehydes as electrophiles.8 Many methods are based on preformed allylmetal reagents.3–6 An alternative to these nucleophiles is their in situ formation by hydrometalation of 1,3-dienes9,10 and allenes,10 and examples of transition-metal-catalyzed reductive couplings with ketones were recently achieved.10–12 A powerful variation of this approach is the borylmetalation of 1,3-dienes in the presence of a carbon electrophile.13–17 These and related stereoselective borylative coupling reactions of other π-systems form a carbon–boron and a carbon–carbon bond in a single operation.13 However, reactions involving ketones as electrophiles are scarce.14,17a,d–g To the best of our knowledge, there are only three examples of the preparation of tertiary homoallylic alcohols by the borylative coupling strategy. Morken and co-workers reported a nickel-catalyzed three-component coupling of 1,3-dienes, bis(pinacolato)diboron, and ketones in racemic fashion (Scheme 1, top).14 The reaction outcome was dependent on the substitution pattern of the 1,3-diene; (E)-penta-1,3-diene converted into 4,3-hydroxyalkylboration products while isoprene (one example) afforded the 4,1-hydroxyalkylboration product. Starting from allenes as the precursor of the allylic nucleophiles, Hoveyda and co-workers realized enantioselective borylative couplings with carbonyl compounds with syn selectivity but enantiocontrol was lower for ketones than for aldehydes (Scheme 1, middle).17a Low enantioselectivity was found by Tian and Tao in an intramolecular borylative cyclization of allenes tethered to cyclohexanediones (not shown).17f Hence, there is a demand for the development of new enantioselective borylative coupling reactions of π-systems and ketones to access chiral tertiary homoallylic alcohols. We disclose here such a copper-catalyzed three-component reaction with 1,3-dienes as the allylic coupling partner where the diastereoselectivity is determined by the ligand (Scheme 1, bottom).9d,e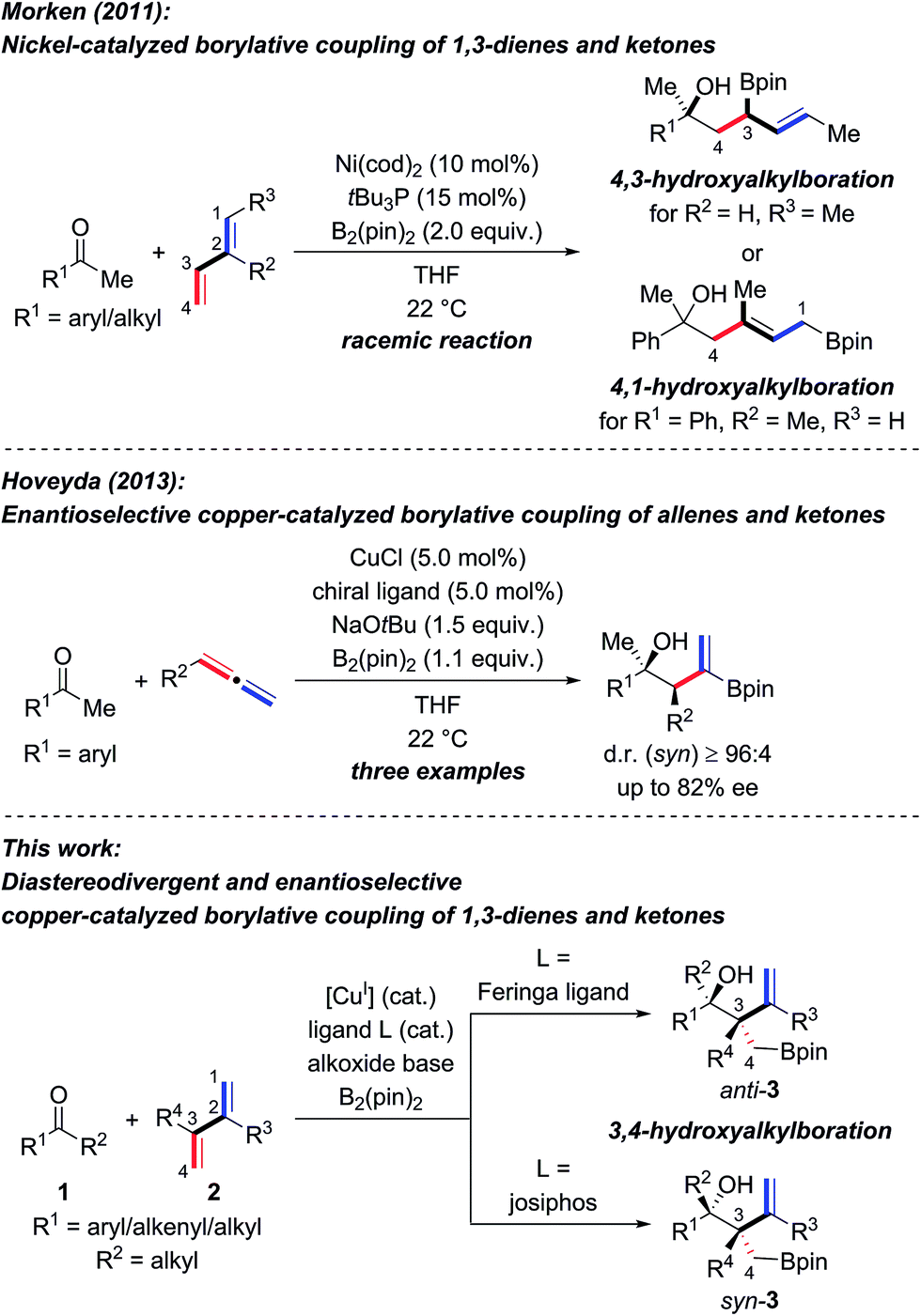 | ||
| Scheme 1 Transition-metal-catalyzed intermolecular borylative coupling reactions of ketones for the construction of tertiary homoallylic alcohols. cod = cycloocta-1,5-diene, pin = pinacolato. | ||
Results and discussion
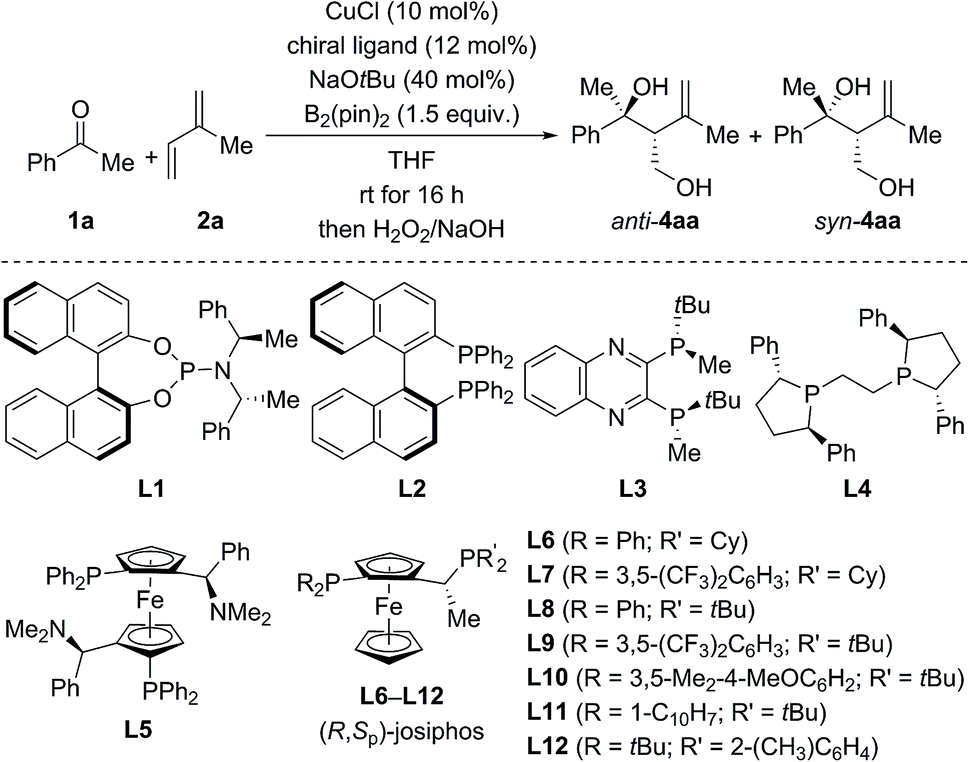
For optimization, the three-component reaction of acetophenone (1a), isoprene (2a), and B2(pin)2 was chosen as the model reaction. The ligand effects are summarized in Table 1. In general, the reaction catalyzed by CuCl and phosphoramidite ligands afforded anti-4aa as the major diastereomer after oxidative degradation of the carbon–boron bond (see the ESI† for the complete set of data).18 As an example, anti-4aa formed in decent yield and with moderate stereoselectivity at room temperature in the presence of CuCl/L1 and NaOtBu (entry 1). Further optimization of the copper source, solvent, and temperature led to a system which afforded the tertiary homoallylic alcohol anti-4aa as the major diastereomer in 94% NMR yield and with 90% ee (entries 2–4). In contrast to phosphoramidite ligands, bisphosphine ligands commonly used in copper catalysis such as L2 to L12 furnished syn-4aa as the major diastereomer at room temperature (entries 5–17), and commercially available josiphos derivative L9 was found to be optimal (entry 12). Lowering of reaction temperature from room temperature to −20 °C increased the enantiomeric excess and diastereoselectivity significantly but was detrimental to the yield (entry 13). Finally, high yield (98% NMR yield) and stereoselectivity (93% ee and d.r. = 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 in favor of syn) were restored in toluene/THF 8:2 with 5.0 mol% CuOAc and 6.0 mol% L9 as the catalyst–ligand combination (entry 14).
:13 in favor of syn) were restored in toluene/THF 8:2 with 5.0 mol% CuOAc and 6.0 mol% L9 as the catalyst–ligand combination (entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Yieldb (%) | d.r. (anti:syn) |
eec (%) | |
| anti-4aa | syn-4aa | ||||
|
a Unless otherwise noted, the reactions were performed with 1a (0.2 mmol), 2a (1 mmol), and B2(pin)2 (0.3 mmol) in THF (2 mL).
b Combined NMR yield determined by 1H NMR spectroscopy with CH2Br2 as an internal standard.
c Determined by HPLC analysis on chiral stationary phases.
d CuOAc instead of CuCl.
e Toluene instead of THF.
f Run at −30 °C.
g The other enantiomer was obtained.
h Run at −20 °C.
i 0.4 mmol scale, 5.0 mol% CuOAc and 6.0 mol% L9 were used and toluene/THF 8:2 instead of THF.
|
|||||
| 1 | L1 | 53 | 71:29 |
60 | 21 |
| 2d | L1 | 88 | 66:34 |
64 | 10 |
| 3d,e | L1 | 96 | 68:32 |
68 | 30 |
| 4d,e,f | L1 | 94 |
80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :20 :20
|
90 | 64 |
| 5 | L2 | 75 | 42:58 |
43 | 32g |
| 6 | L3 | 92 | 35:65 |
6 | 35 |
| 7 | L4 | 93 | 28:72 |
35g | 32g |
| 8e | L5 | 84 | 44:56 |
13 | 22 |
| 9 | L6 | 45 | 23:77 |
22g | 61 |
| 10 | L7 | 98 | 23:77 |
13g | 80 |
| 11 | L8 | 80 | 22:78 |
72g | 88 |
| 12 | L9 | 98 | 23:77 |
74g | 88 |
| 13h | L9 | 61 | 15:85 |
79g | 94 |
| 14h,i | L9 | 98 |
13:87
|
71 | 93 |
| 15 | L10 | 65 | 28:72 |
71g | 87 |
| 16 | L11 | 37 | 47:53 |
0 | 37 |
| 17 | L12 | 29 | 49:51 |
— | — |
We next investigated the scope of ketones using L1 in the anti-selective procedure and L9 in the syn-selective setup (Conditions A and B, Scheme 2). Acetophenones with various substituents in the para position, including electron-donating groups (as in 1b, c) and halogens (as in 1d–f), exhibited high reactivity and stereoselectivity. A carboxyl group was compatible (as in 1g), thus further emphasizing the functional-group tolerance of this reaction. 1h and i with meta substitution also gave satisfactory results. The reaction of ortho-methyl-substituted 1j was successful under Condition B and yielded syn-4ia with 98% ee (anti-4ja: 80% ee); conversely, poor stereoselectivity was obtained under Condition A. Pyridyl-substituted 1l reacted smoothly under Condition B and furnished syn-4la with good diastereoselectivity (d.r. = 90:10) and enantioselectivity (90% ee); in turn, the reaction of 1l under Condition A produced anti-4la with a moderate ee value. Aside from aromatic methyl ketones, propiophenone (1m), which had not been compatible with Morken's14 and Hoveyda's17a catalytic system (cf.Scheme 1), also furnished anti-4ma in excellent yield and good enantioselectivity with moderate diastereoselectivity under Condition A; B afforded the target compound in a similar yield yet with a high diastereomeric ratio and a markedly diminished ee value. Interestingly, α,β-unsaturated ketone 1n reacted regioselectively (1,2- over 1,4-addition) with good to excellent diastereoselectivity; syn-4na was the major product under both Condition A and B. Moreover, dialkyl ketone 1o converted into the corresponding products anti- and syn-4oa under A and B but with low diastereoselectivity likely due to the little steric differentiation between the methyl and methylene groups attached to the carbonyl carbon atom.
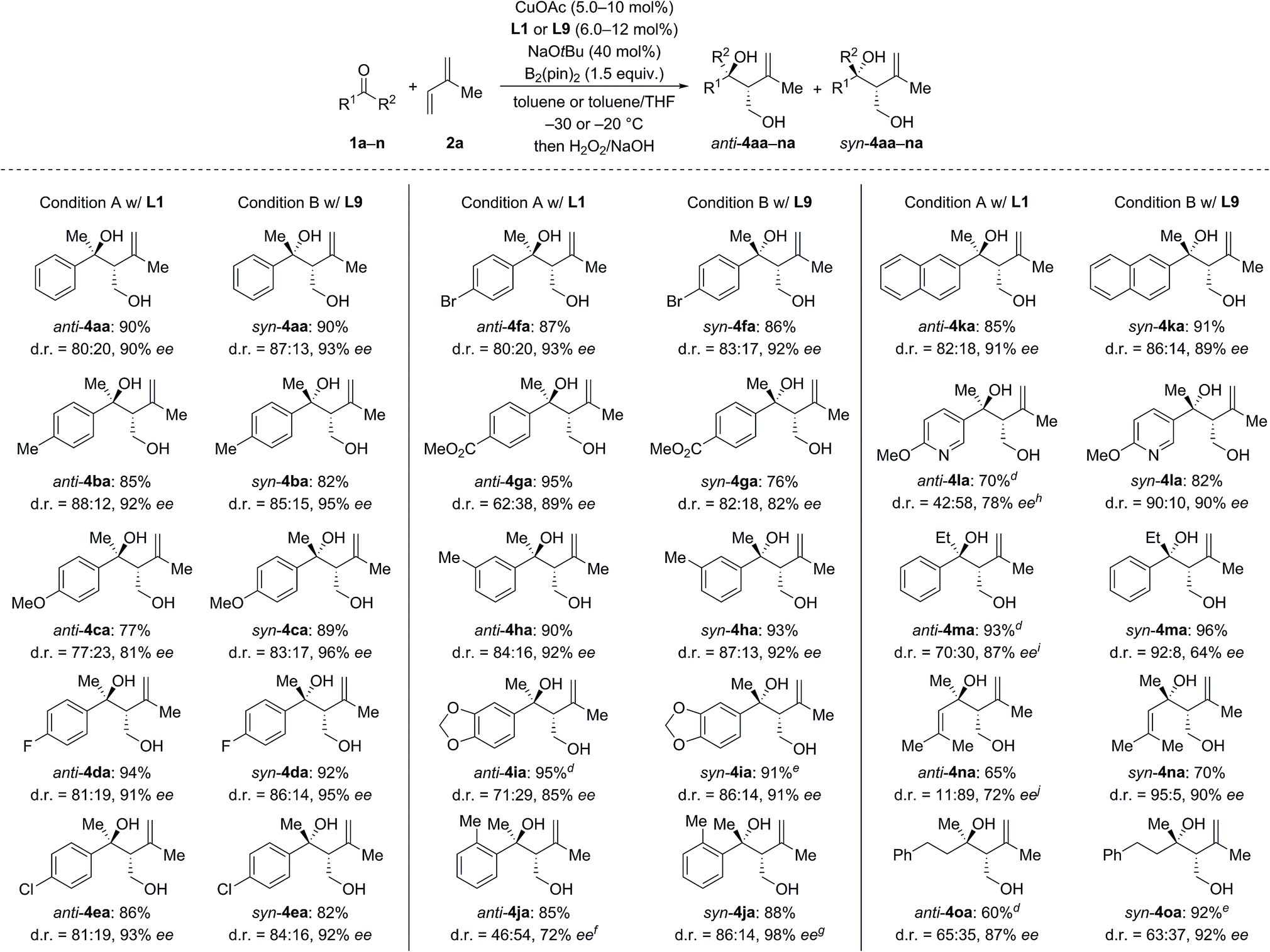 | ||
| Scheme 2 Scope I: variation the ketone.a–c aCondition A: CuOAc (10 mol%), L1 (12 mol%), NaOtBu (40 mol%), ketone 1 (0.20 mmol), isoprene (2a, 1.0 mmol), and B2(pin)2 (1.5 equiv.) in toluene (2 mL) at −30 °C. Condition B: CuOAc (5.0 mol%), L9 (6.0 mol%), NaOtBu (40 mol%), ketone 1 (0.40 mmol), isoprene (2a, 2.0 mmol), and B2(pin)2 (1.5 equiv.) in toluene/THF – 8:2 (3.5 mL) at −20 °C. bYields are combined isolated material; diastereomers are usually separable by flash chromatography on silica gel. cThe enantiomeric excess of the major diastereomer was determined by HPLC analysis on chiral stationary phases. dCuOAc (15 mol%) and L1 (18 mol%) were used. eCuOAc (10 mol%) and L9 (12 mol%) were used. fanti-4ja: 29% ee. ganti-4ja: 80% ee. hee value of anti-4la. isyn-4ma: 78% ee. jsyn-4na: 72% ee. | ||
We then examined the scope of 1,3-dienes (Scheme 3). Isoprene (2a) could be replaced by buta-1,3-diene (2b), myrcene (2c), its functionalized derivative 2d, and 2,3-dimethylbuta-1,3-diene (2e). Yields were generally good but stereoselectivities ranged from poor to good under Condition A. In contrast, good to excellent stereoselectivities were observed for these 1,3-dienes under Condition B, e.g., d.r. = 96:4 and 92% ee for 1n → syn-4nb and d.r. = 93:7 and 91% ee for 1a → syn-4ad. In the case of 2-aryl-substituted 1,3-diene 1f, diastereodivergency was not achieved. Subjecting 1f to Condition A afforded syn-4af in low yield as a single syn-isomer (not shown). However, applying Condition B at −5 °C significantly improved the yield and furnished the syn-4af with d.r. > 98:2 and 85% ee.
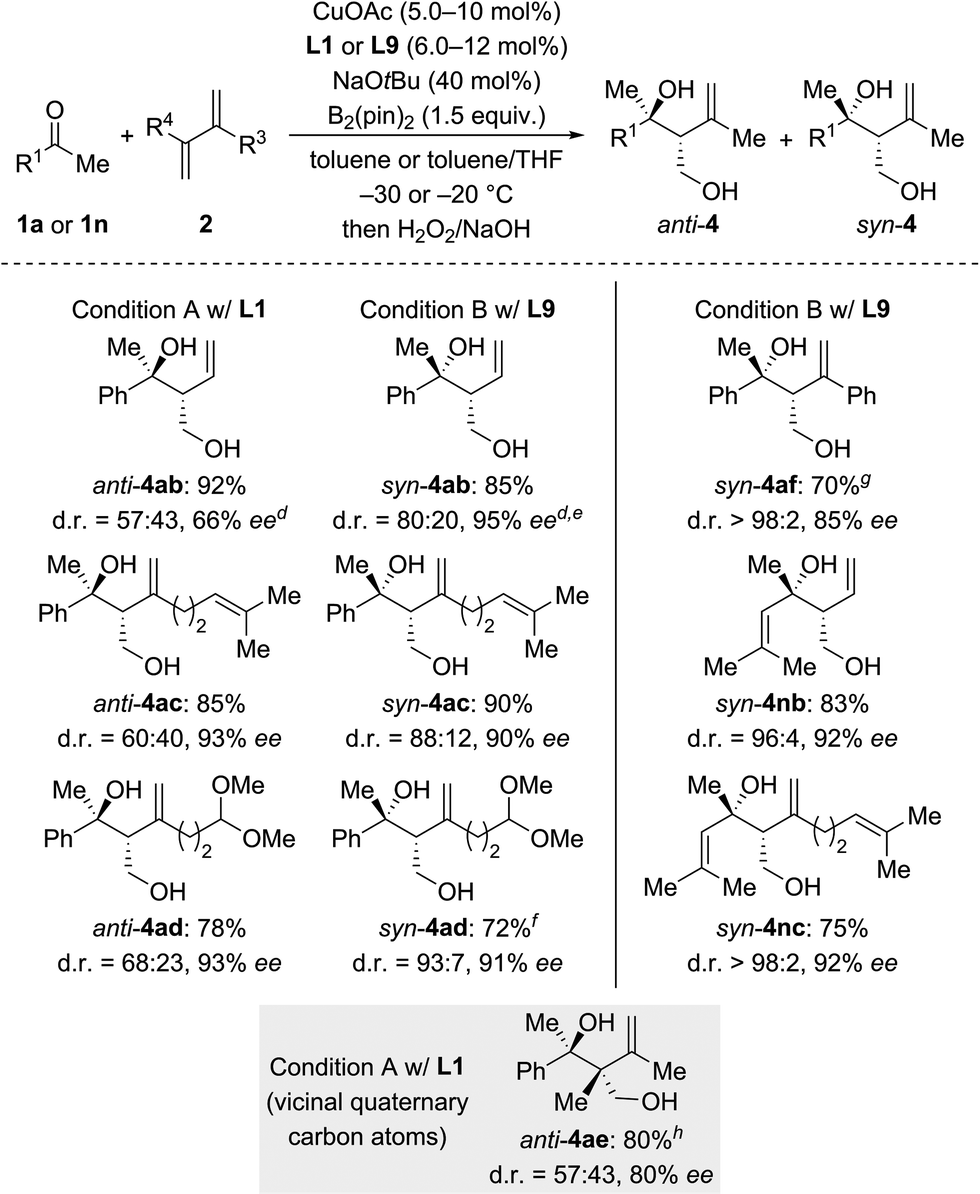 | ||
| Scheme 3 Scope II: variation of the 1,3-diene.a–c For footnotes a–c, see Scheme 2. dThe absolute configuration was assigned by chemical correlation after separation of the diastereomers by flash chromatography on silica gel (see the ESI†). eanti-4ab: 84% ee. fCuOAc (8.0 mol%) and L9 (10 mol%) were used. gRun at −5 °C with CuOAc (10 mol%), L9 (12 mol%), NaOtBu (50 mol%), and B2(pin)2 (2.0 equiv.). hCuOAc (15 mol%) and L1 (18 mol%) were used. | ||
To explore synthetic transformations of these tertiary homoallylic alcohols (Scheme 4), a scale-up synthesis of syn-4aa (1.0 mmol) under Condition B was done without any loss in efficiency and selectivity (see the ESI†). The primary alkyl borane generated by the multicomponent reaction was subjected to a Suzuki–Miyaura coupling to afford syn-5 in 83% yield (Scheme 4, top). The versatility of the diol products 4 is illustrated for several transformations (Scheme 4, bottom). The 1,1-disubstituted double bond in anti-4ha was hydrogenated over Pd/C to produce anti-6 in 87% yield. The hydroxy group in syn-4aa was replaced by an azide group through an SN2 reaction of an intermediate mesylate with NaN3 (syn-4aa → syn-7). Pyran syn-8 was synthesized from syn-4ab by sequential alcohol allylation and ring-closing metathesis. Of note, a chemoselective tosylation of the primary alcohol in syn-4aa followed by a 4-exo-tet ring closure allowed for the construction of enantioenriched, trisubstituted oxetane trans-9 in 86% yield.
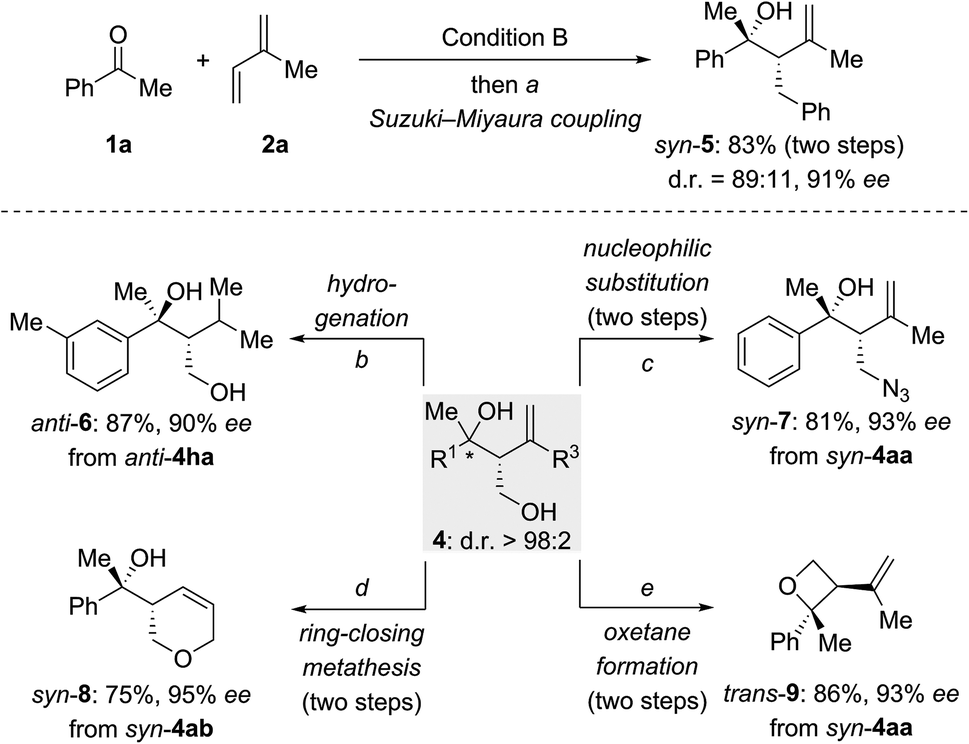 | ||
| Scheme 4 Tertiary homoallylic alcohols as versatile building blocks. (a) PhBr (1.8 equiv.), Pd(OAc)2 (5.0 mol%), RuPhos (10 mol%), KOtBu (3.0 equiv.), toluene/H2O (10/1), 80 °C, 24 h; (b) Pd/C (10%), H2 (1 atm), MeOH, rt, 26 h; (c) (i) MsCl (1.5 equiv.), Et3N (1.5 equiv.), CH2Cl2, 0 °C to rt, 50 min; (ii) NaN3 (2.0 equiv.), DMF/H2O (10/1), 80 °C, 12 h; (d) (i) NaH (2.0 equiv.), allyl bromide (1.1 equiv.), THF, 0 °C to rt, 14 h; (ii) Hoveyda–Grubbs II (5.0 mol%), CH2Cl2, Δ, 12 h; (e) (i) TsCl (2.4 equiv.), pyridine, 0 °C to rt, 24 h; (ii) nBuLi (1.1 equiv.), −25 °C to rt, 15 h. Ms = methanesulfonyl. | ||
Conclusion
In summary, we have developed an efficient copper-catalyzed diastereodivergent and enantioselective borylative coupling of 1,3-dienes and ketones. Using a Feringa-type ligand L1, the reaction yielded anti-configured tertiary homoallylic alcohols while a switch to josiphos ligand L9 resulted in syn selectivity (see the ESI† for a discussion of the reaction mechanism). This three-component coupling reaction represents a useful method for the preparation of stereochemically diverse tertiary alcohols bearing versatile alkenyl and boryl motifs from feedstock 1,3-dienes, ketones, and B2(pin)2. The synthetic utility of the reaction was showcased by several transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J.-J. F. gratefully acknowledges the Alexander von Humboldt Foundation for a postdoctoral fellowship (2017–2020), and Y. X. thanks the China Scholarship Council for a predoctoral fellowship (2018–2022). M. O. is indebted to the Einstein Foundation Berlin for an endowed professorship. Solvias AG is thanked for a generous gift of josiphos ligands.Notes and references
- For reviews of related asymmetric allylation of carbonyl compounds, see: (a) Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS; (b) M. Hatano and K. Ishihara, Synthesis, 2008, 1647–1675 CAS; (c) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2793 CrossRef CAS.
- M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS PubMed.
- With allylboron reagents: (a) R. Wada, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 8910–8911 CrossRef CAS; (b) S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2006, 128, 12660–12661 CrossRef CAS; (c) U. Schneider, M. Ueno and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13824–13825 CrossRef CAS; (d) S.-L. Shi, L.-W. Xu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 6638–6639 CrossRef CAS; (e) D. S. Barnett, P. N. Moquist and S. E. Schaus, Angew. Chem., Int. Ed., 2009, 48, 8679–8682 CrossRef CAS; (f) R. Alam, T. Vollgraff, L. Eriksson and K. J. Szabó, J. Am. Chem. Soc., 2015, 137, 11262–11265 CrossRef CAS; (g) D. W. Robbins, K. A. Lee, D. L. Silverio, A. Volkov, S. Torker and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 9610–9614 CrossRef CAS; (h) K. A. Lee, D. L. Silverio, S. Torker, D. W. Robbins, F. Haeffner, F. W. van der Mei and A. H. Hoveyda, Nat. Chem., 2016, 8, 768–777 CrossRef CAS; (i) F. W. van der Mei, C. Qin, R. J. Morrison and A. H. Hoveyda, J. Am. Chem. Soc., 2017, 139, 9053–9065 CrossRef CAS.
- With allylsilicon reagents: (a) S. Yamasaki, K. Fujii, R. Wada, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 6536–6537 CrossRef CAS; (b) M. Wadamoto and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 14556–14557 CrossRef CAS; (c) M. Wadamoto, M. Naodovic and H. Yamamoto, Eur. J. Org. Chem., 2009, 5132–5134 CrossRef CAS.
- With allyltin reagents: (a) S. Casolari, D. D'Addario and E. Tagliavini, Org. Lett., 1999, 1, 1061–1063 CrossRef CAS; (b) K. M. Waltz, J. Gavenonis and P. J. Walsh, Angew. Chem., Int. Ed., 2002, 41, 3697–3600 CrossRef CAS; (c) A. Cunningham and S. Woodward, Synthesis, 2002, 43–44 CAS; (d) J. G. Kim, K. M. Waltz, I. F. Garcia, D. Kwiatkowski and P. J. Walsh, J. Am. Chem. Soc., 2004, 126, 12580–12585 CrossRef CAS; (e) Y.-C. Teo, J.-D. Goh and T.-P. Loh, Org. Lett., 2005, 7, 2743–2745 CrossRef CAS; (f) J. G. Kim, E. H. Camp and P. J. Walsh, Org. Lett., 2006, 8, 4413–4416 CrossRef CAS; (g) X. Zhang, D. Chen, X. Liu and X. Feng, J. Org. Chem., 2007, 72, 5227–5233 CrossRef CAS.
- With allyl halides using a stoichiometric metal reductant: (a) J. J. Miller and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 2752–2753 CrossRef CAS; (b) X.-R. Huang, C. Chen, G.-H. Lee and S.-M. Peng, Adv. Synth. Catal., 2009, 351, 3089–3095 CrossRef CAS; (c) R.-Y. Chen, A. P. Dhondge, G.-H. Lee and C. Chen, Adv. Synth. Catal., 2015, 357, 961–966 CrossRef CAS ; for a review, see: ; (d) Z.-L. Shen, S.-Y. Wang, Y.-K. Chok, Y.-H. Xu and T.-P. Loh, Chem. Rev., 2013, 113, 271–401 CrossRef CAS.
- With hydrocarbon pronucleophiles: (a) R. Yazaki, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 3195–3197 CrossRef CAS; (b) R. Yazaki, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 5522–5531 CrossRef CAS; (c) X.-F. Wei, X.-W. Xie, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2017, 139, 4647–4650 CrossRef CAS.
- For authoritative reviews, see: (a) M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853–2873 CrossRef CAS; (b) C. García and V. S. Martín, Curr. Org. Chem., 2006, 10, 1849–1889 CrossRef.
- For key reviews, see: (a) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371–2380 CrossRef CAS; (b) K. D. Nguyen, B. Y. Park, T. Luong, H. Sato, V. J. Garza and M. J. Krische, Science, 2016, 354, aah5133 CrossRef ; for seminal work on the use of 1,3-dienes as allylmetal equivalents, see: ; (c) F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6338–6339 CrossRef CAS; (d) E. L. McInturff, E. Yamaguchi and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 20628–20631 CrossRef CAS; (e) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324–327 CrossRef CAS PubMed.
- M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS.
- For reviews of reductive couplings, see: (a) M. Kimura and Y. Tamaru, Top. Curr. Chem., 2007, 279, 173–207 CrossRef CAS; (b) J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890–3908 CrossRef CAS.
- Selected examples of reductive couplings of ketones and 1,3-dienes. Racemic reactions: (a) Y. Sato, M. Takimoto, K. Hayashi, T. Katsuhara, K. Takagi and M. Mori, J. Am. Chem. Soc., 1994, 116, 9771–9772 CrossRef CAS; (b) M. Kimura, H. Fujimatsu, A. Ezoe, K. Shibata, M. Shimizu, S. Matsumoto and Y. Tamaru, Angew. Chem., Int. Ed., 1999, 38, 397–400 CrossRef CAS; (c) M. Kimura, A. Ezoe, M. Mori, K. Iwata and Y. Tamaru, J. Am. Chem. Soc., 2006, 128, 8559–8568 CrossRef CAS; (d) J. C. Leung, L. M. Geary, T.-Y. Chen, J. R. Zbieg and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 15700–15703 CrossRef CAS; (e) B. Y. Park, T. P. Montgomery, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 16320–16323 CrossRef CAS PubMed . Asymmetric reactions: ; (f) Y. Yang, I. B. Perry, G. Lu, P. Liu and S. L. Buchwald, Science, 2016, 353, 144–150 CrossRef CAS; (g) C. Li, R. Y. Liu, L. T. Jesikiewicz, Y. Yang, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 5062–5070 CrossRef CAS.
- For authoritative reviews of borylative couplings, see: (a) K. Semba and Y. Nakao, Tetrahedron, 2019, 75, 709–719 CrossRef CAS; (b) D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC; (c) A. P. Pulis, K. Yeung and D. J. Procter, Chem. Sci., 2017, 8, 5240–5247 RSC; (d) H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS; (e) Y. Tsuji and T. Fujihara, Chem. Rec., 2016, 16, 2294–2313 CrossRef CAS; (f) K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Tetrahedron, 2015, 71, 2183–2197 CrossRef CAS; (g) H. Y. Cho and J. P. Morken, Chem. Soc. Rev., 2014, 43, 4368–4380 RSC.
- H. Y. Cho, Z. Yu and J. P. Morken, Org. Lett., 2011, 13, 5267–5269 CrossRef CAS.
- Examples of catalytic asymmetric platinum- and nickel-catalyzed borylative aldehyde–diene couplings: (a) J. B. Morgan and J. P. Morken, Org. Lett., 2003, 5, 2573–2575 CrossRef CAS; (b) H. E. Burks, L. T. Kliman and J. P. Morken, J. Am. Chem. Soc., 2009, 131, 9134–9135 CrossRef CAS; (c) L. T. Kliman, S. N. Mlynarski, G. E. Ferris and J. P. Morken, Angew. Chem., Int. Ed., 2012, 51, 521–524 CrossRef CAS.
- Selected examples of enantioselective copper-catalyzed borylative couplings of dienes. With α,β-unsaturated acceptors: (a) X. Li, F. Meng, S. Torker, Y. Shi and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 9997–10002 CrossRef CAS; with imines: (b) L. Jiang, P. Cao, M. Wang, B. Chen, B. Wang and J. Liao, Angew. Chem., Int. Ed., 2016, 55, 13854–13858 CrossRef CAS; with acylsilanes: (c) J.-J. Feng and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 8211–8215 CrossRef CAS; with electrophilic cyanating reagents: (d) T. Jia, M. J. Smith, A. P. Pulis, G. J. P. Perry and D. J. Procter, ACS Catal., 2019, 9, 6744–6750 CrossRef CAS; with aryl halides: (e) S. R. Sardini and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 9823–9826 CrossRef CAS; (f) K. B. Smith, Y. Huang and M. K. Brown, Angew. Chem., Int. Ed., 2018, 57, 6146–6149 CrossRef CAS.
- Selected examples of copper-catalyzed borylative couplings with carbonyl compounds. Intermolecular: (a) F. Meng, H. Jang, B. Jung and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 5046–5051 CrossRef CAS; (b) F. Meng, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 11304–11307 CrossRef CAS; (c) S. Gao and M. Chen, Chem. Sci., 2019, 10, 7554–7560 RSC; (d) X.-C. Gan and L. Yin, Org. Lett., 2019, 21, 931–936 CrossRef CAS ; intramolecular: ; (e) A. R. Burns, J. S. González and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 10827–10831 CrossRef CAS; (f) Y.-S. Zhao, X.-Q. Tang, J.-C. Tao, P. Tian and G.-Q. Lin, Org. Biomol. Chem., 2016, 14, 4400–4404 RSC; (g) E. Yamamoto, R. Kojima, K. Kubota and H. Ito, Synlett, 2016, 27, 272–276 CAS.
- For reports on the synthesis of chiral compounds by carbonyl (hydroxymethyl)allylations with anti-diastereoselectivity, see: (a) Y. J. Zhang, J. H. Yang, S. H. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 4562–4563 CrossRef CAS PubMed; (b) J. Feng, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2014, 136, 8911–8914 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03531a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |