
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA quantitative assessment of the dynamic modification of lipid–DNA probes on live cell membranes†
Yousef
Bagheri
,
Sara
Chedid
,
Fatemeh
Shafiei
,
Bin
Zhao
* and
Mingxu
You
 *
*
Department of Chemistry, University of Massachusetts, Amherst, MA 01003, USA. E-mail: mingxuyou@umass.edu; bzhao@umass.edu
First published on 24th October 2019
Abstract
Synthetic lipid–DNA probes have recently attracted much attention for cell membrane analysis, transmembrane signal transduction, and regulating intercellular networks. These lipid–DNA probes can spontaneously insert onto plasma membranes simply after incubation. The highly precise and controllable DNA interactions have further allowed the programmable manipulation of these membrane-anchored functional probes. However, we still have quite limited understanding of how these lipid–DNA probes interact with cell membranes and also what parameters determine this process. In this study, we have systematically studied the dynamic process of cell membrane modification with a group of lipid–DNA probes. Our results indicated that the hydrophobicity of the lipid–DNA probes is strongly correlated with their membrane insertion and departure rates. Most cell membrane insertion stems from the monomeric form of probes, rather than the aggregates. Lipid–DNA probes can be removed from cell membranes through either endocytosis or direct outflow into the solution. As a result, long-term probe modifications on cell membranes can be realized in the presence of excess probes in the solution and/or endocytosis inhibitors. For the first time, we have successfully improved the membrane persistence of lipid–DNA probes to more than 24 h. Our quantitative data have dramatically improved our understanding of how lipid–DNA probes dynamically interact with cell membranes. These results can be further used to allow a broad range of applications of lipid–DNA probes for cell membrane analysis and regulation.
Introduction
The cell membrane provides a physical barrier between the intracellular compartments and the extracellular environment. The cell membrane also plays important roles in the signal transmission between cells and the extracellular matrix or among neighbouring cells.1,2 A better understanding and modulation of the composition and function of cell membranes is critical for regulating cell signaling and interactions. Despite the tremendous progress that has been made, there are still many mysteries surrounding cell membranes, especially those pertaining to lipid-mediated membrane interactions and signalling pathways.1,3 On the other hand, these membrane lipids can be used as powerful building blocks for cell membrane modification and regulation.Synthetic lipid–DNA conjugates have emerged as powerful tools for cell membrane studies. These lipid–DNA conjugates can be self-assembled onto the outer surface of plasma membranes simply after incubation.4–6,9–11 In addition to this simplicity, highly precise and programmable DNA hybridizations can be used to fine-tune different cell membrane patterns and interactions.4,6–8 Lipid–DNA conjugates can be further modified with different functional moieties, including fluorophores, small molecule drugs, chemically reactive cross-linkers, photo-responsive groups, etc. These modifications have allowed the engineering of functional lipid–DNA probes to monitor cell membrane transportation of signalling molecules,12,13 to analyze membrane biophysics,14,15 to generate membrane nanopores,16,17 and to regulate intercellular interactions.18,19 In addition to their broad range of applications on plasma membrane surfaces, lipid–DNA probes can also be used for intracellular applications such as transmembrane cargo deliveries13,20–22 and membrane analysis of intracellular organelles.23
Lipid–DNA conjugates have provided a straightforward, rapid, and efficient approach to modify cell membranes. However, one major challenge is the limited persistence of these membrane modifications. Lipid–DNA probes can be internalized into cells, and as a result, they can only remain on cell membranes for ∼2–4 h.15,24,25 In order to extend the duration of these membrane modifications, it is critical to understand how these lipid–DNA probes are removed from the cell membranes. Moreover, a systematic understanding of how lipid–DNA probes insert into cell membranes is still missing. Previous studies have shown that lipid–DNA probes with different lipid structures, DNA and fatty acid lengths exhibit quite different cell membrane anchoring efficiencies.11,26,27 However, it is still not known how these variations happen and what parameters determine the membrane insertion kinetics, efficiencies, and durations of lipid–DNA probes.28
In this study, we aim to provide an in-depth understanding of how lipid–DNA probes interact with cell membranes. With quantitative assessment using a fluorescence microscope, we have determined the cell membrane insertion kinetics, magnitudes, and durations of different lipid–DNA probes. We have systematically studied the effect of lipid/DNA structure, hydrophobicity, concentration, temperature, and cell type on these membrane interactions. A kinetic model for the cell membrane modification process with these lipid–DNA probes has been proposed. In addition, based on this model, two approaches have been developed to improve the persistence of lipid–DNA probes on live cell membranes to at least 24 h.
Results and discussion
Design and cell membrane insertion kinetics of lipid–DNA probes
It is known that the cell membrane anchoring efficiency of lipid–DNA probes largely depends on their lipid structures.11,29–32 Here, we decided to synthesize a library of seven lipid structures containing one or two fatty acid chains of different lengths and saturation levels (Fig. 1a). As one of the most abundant natural components of plasma membranes, cholesterol (with a tetraethylene glycol linker) was also included in our library. We first conjugated each lipid with a 6-carboxyfluorescein (FAM)-labelled 20 nucleotide (nt)-long DNA strand. The sequence of this DNA strand was designed to have no secondary structure to avoid DNA interactions on the cell membranes (Table S1 and Fig. S1†). After purification, the successful synthesis of each lipid–DNA conjugate was confirmed by matrix-assisted laser desorption/ionization mass spectrometry and gel electrophoresis (Fig. S2a and Table S2†).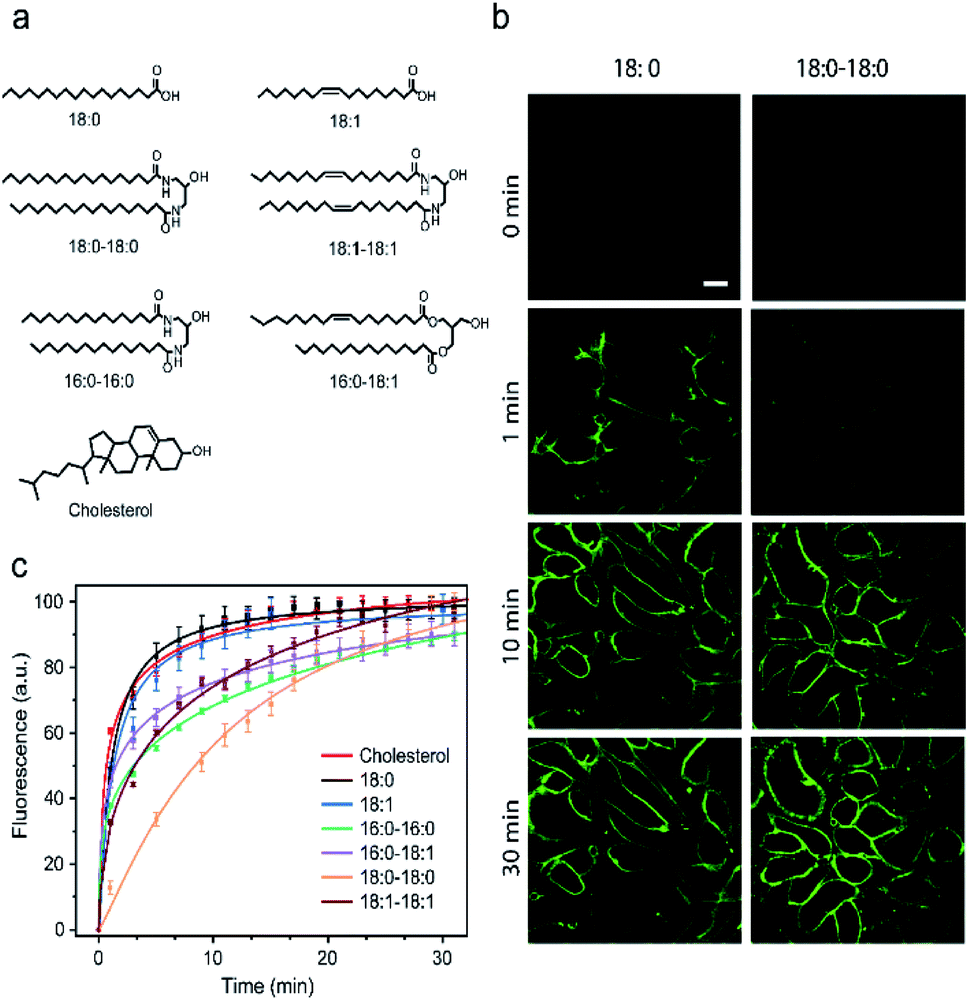 | ||
| Fig. 1 Probe structure and insertion kinetics on MDCK cell membranes. (a) Chemical structures of the lipid moieties used in this study. (b) Fluorescence imaging of the MDCK cell membrane insertion kinetics after adding 1 μM 18:0- and 18:0–18:0-modified 20 nt DNA probes at 0 min. Scale bar, 20 μm. (c) Normalized cell membrane insertion kinetics of different lipid–DNA probes. The same 20 nt single-stranded DNA was conjugated in each probe. At 0 min, 1 μM of each 20 nt lipid–DNA probe was added to the MDCK cells and incubated at room temperature. Shown are the mean and SEM values that were measured on 50–60 cell membranes. | ||
We next asked if we could monitor the insertion kinetics of these lipid–DNA probes on live cell membranes. We chose a commonly used Madin–Darby canine kidney (MDCK) epithelial cell line, added 1 μM of each probe at room temperature, and measured cellular fluorescence with a spinning disk confocal microscope. The imaging conditions were optimized to avoid photobleaching (Fig. S3a and b†). As shown in Fig. 1b, different lipid structures exhibit different insertion rates on MDCK cell membranes. For example, it only took 7 min for 90% of the 18:0-based probe (t0.9) to insert on the membranes, while in contrast, the same percentage of insertion was achieved in 25 min for the 18:0–18:0 probe (Fig. 1c). Indeed, the number of fatty acid chains will influence the insertion kinetics of the lipid–DNA probe. After further imaging all seven types of lipids, our results indicated that half-maximum cell membrane fluorescence could be observed in only 2–9 min, and it took 7–31 min to reach 90% maximum signal for each lipid–DNA probe (Fig. 1c). As a result, all the tested lipid–DNA probes can be rapidly inserted onto MDCK cell membranes.
Our next goal was to quantify the cell membrane insertion rate of each probe. To correlate the measured fluorescence signals with membrane probe densities, we prepared a lipid monolayer film using soybean polar extract (Fig. S3c†). Lipid–DNA probes can distribute homogeneously on this film.12,33 Under the same imaging conditions as those for the MDCK cells, a linear correlation between the monolayer fluorescence intensity and the probe density was observed for all the tested lipid–DNA probes (Fig. S3d†). This linear calibration curve was further used to determine probe densities on live cell membranes.
We next added different initial concentrations of lipid–DNA probes to the MDCK cells, and monitored their membrane fluorescence signals. Our results indicated that the cell membrane insertion of lipid–DNA probes followed a first-order reaction model (Fig. S3e†). Based on this model, under the experimental conditions, the apparent membrane insertion rate constant was determined to be in the range of 0.009–0.033 s−1 for each lipid–DNA probe (Fig. 2 and Table S3†). The cholesterol- and 18:0-based probes exhibited the fastest cell membrane insertion rate among the seven types of lipids.
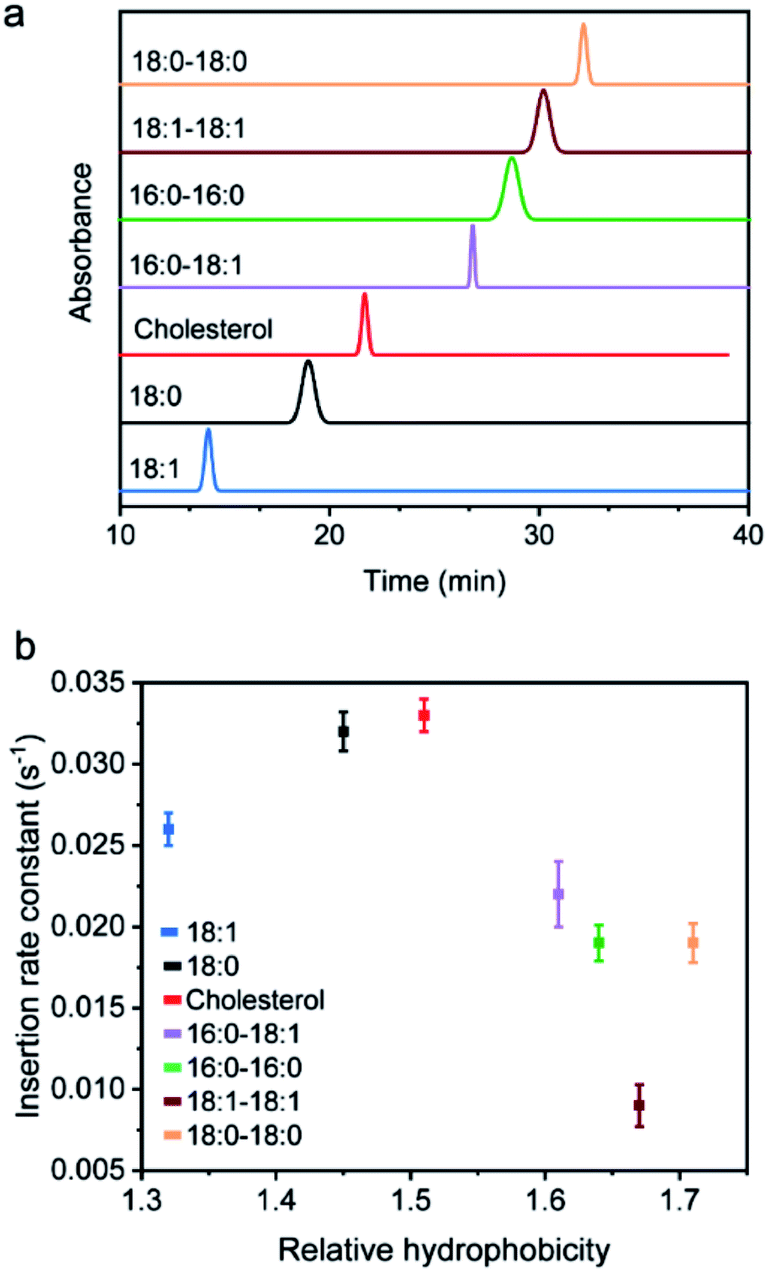 | ||
| Fig. 2 Hydrophobicity of the probes and its effect on membrane interaction kinetics. (a) The hydrophobicity value of each 20 nt lipid–DNA probe was determined with a reversed phase HPLC using a C4 column and a triethylammonium acetate/acetonitrile eluent. The retention time was further used to determine the relative hydrophobicity. (b) MDCK cell membrane insertion rate constant of each 20 nt lipid–DNA probe plotted against its corresponding hydrophobicity as measured in panel (a). Shown are the mean and SEM values that were measured on 50–60 cell membranes. | ||
Effect of hydrophobicity and aggregation status on the membrane insertion rates
We wondered what parameters of lipids could potentially lead to these different membrane insertion rates. We first asked if the insertion kinetics of the probe is correlated with its hydrophobicity. To determine the hydrophobicity value of each lipid–DNA probe, we developed an HPLC assay (Fig. 2a). Hydrophobicity can be quantified as the logarithm of the probe's distribution constant between a hydrophobic and a hydrophilic phase.34,35 In our HPLC assay, the retention time of each probe was used to calculate the corresponding capacity factor, which is directly proportional to the distribution constant of the probe between the stationary phase and mobile phase in a reversed-phase HPLC column. As a result, based on the retention time as measured using the same HPLC column and mobile phase, we can calculate the relative hydrophobicity value of each lipid–DNA probe.As shown in Fig. 2b, interestingly, the less hydrophobic cholesterol-, 18:0- and 18:1-based probes exhibited faster membrane insertion kinetics. This result is somehow unexpected considering the highly hydrophobic nature of plasma membranes. On the other hand, the amphiphilic nature of the lipid–DNA probe makes it easy to form nanometer- or micrometer-sized aggregates in an aqueous solution. We hypothesized that there could be a competing process between the formation of these aggregates and the cell membrane insertion of the probe.
To test this hypothesis, we first wanted to confirm the formation of aggregates using gel electrophoresis. Indeed, for all the tested probes, lipid-modified DNAs migrate significantly slower compared to unmodified DNAs (Fig. S2a†). Considering that lipid tails are relatively small in size, these band shifts are quite likely due to the assembly of multiple lipid–DNA probes. We further measured the sizes of these aggregates using dynamic light scattering. After adding 1 μM of each probe, except for the most hydrophilic 18:1-modified DNA probe, all other lipid–DNA probes could form aggregates with diameters in the range of 200–500 nm (Fig. S4†). The size distribution of the aggregates largely depends on the structure and hydrophobicity of the lipids. Large-sized aggregates could only be observed with less hydrophobic cholesterol- or 18:0-modified DNA probes, while highly hydrophobic lipid–DNA probes tended to form compact, small-sized nanoparticles. All these results demonstrated the existence of aggregates in the solution of lipid–DNA probes.
We next asked if the monomeric form and aggregated form of lipid–DNA probes exhibited similar membrane insertion kinetics. To determine the amount of monomers and aggregates in the tested lipid–DNA probes, we first measured the critical aggregation concentration (CAC) value of each probe. The CAC is the concentration at which amphiphilic monomers start to aggregate and all additionally added probes remain in the aggregated form. As a result, we can estimate the respective monomer and aggregate concentration of probes based on the CAC value. We measured the CAC value of each lipid–DNA probe using a Nile red dye, which is nearly non-fluorescent in aqueous solutions, but is highly fluorescent in a nonpolar environment (Fig. S5†). After adding different concentrations of each probe into a solution containing 30 mM Nile red, the CAC value of the probe was determined from a corresponding fluorescence response curve (Fig. S6†). Indeed, the less hydrophobic 18:1- and 18:0-modified probes exhibited large CAC values that are beyond the tested concentrations (Fig. S6†). A 2.2 μM CAC was observed for the cholesterol-modified probe, while all other highly hydrophobic lipid–DNA probes showed CAC values lower than 1 μM. Interestingly, the CAC values of the probes appeared to be positively correlated with their cell membrane insertion rates (Table S4†).
We next wanted to quantify the cell membrane insertion rates of probes in their aggregated and monomeric forms, respectively. We chose two representative lipid–DNA probes, cholesterol and 16:0–16:0. For each probe, we monitored the fluorescence signal change on the MDCK cell membrane after adding three different concentrations of the probe, 250 nM, 1 μM, and 5 μM. When 250 nM probes were used, i.e., far below the CAC value, the majority of the probes existed in their monomeric form. In comparison, both monomeric and aggregated forms of the probes existed at 5 μM concentration, which is above the CAC value. Based on the estimated initial monomer and aggregate concentrations in each case, our results indicated that the membrane insertion rate constant for the monomeric and aggregated forms of cholesterol–DNA was 0.041 s−1 and 0.0038 s−1, respectively (Fig. S7†). Similarly, a 10-fold higher membrane insertion rate was observed in monomeric 16:0–16:0 probe (0.074 s−1) compared to that in the aggregated form (0.0072 s−1). This result could be expected considering the structure of these monomers and aggregates. In the monomeric form, the lipid anchor is easily accessible and can be directly inserted onto cell membranes. In contrast, in the aggregated form, the lipid anchors will most likely be hidden within a negatively charged DNA corona, making interaction with cell membranes difficult. As a result, the membrane insertion rate is much faster with monomeric form lipid–DNA probes than the aggregated form (Fig. S7†).
As mentioned above, the less hydrophobic 18:0-, 18:1- and cholesterol-based probes exhibited larger CAC values (Fig. S6†). At a typical probe concentration (0.2–1 μM), most of these lipid–DNA probes stayed in the monomeric form, which could rapidly insert into cell membranes. In this case, the insertion kinetics could depend on the lipid structure and its tendency to interact with cell membranes. For example, cholesterol can efficiently interact with different lipids on the cell membrane,38–40 and as a result, exhibited fast insertion into the membranes. For probes with low CAC, both monomeric and aggregated forms existed. For each monomer, there will be a competition between aggregate formation and cell membrane insertion. As a result, the effective concentrations of the monomers were reduced. In other words, less hydrophobic lipid–DNA probes exhibited faster cell membrane insertion kinetics.
Effect of DNA on the membrane insertion kinetics of the probes
So far, we have focused on studying the effect of lipid structures on the membrane insertion kinetics of lipid–DNA probes. We next asked how different DNA structures could influence the membrane insertion rates. In each of the above-tested lipid–DNA probes, a 20 nt single-stranded DNA was used. We next studied the effect of DNA length by replacing the 20 nt DNA with 40 nt, 60 nt, and 80 nt single-stranded DNA. As expected from their larger hydrophilic-to-hydrophobic ratio, lipid–DNA probes containing these longer DNA strands exhibited larger CAC values (Table S4†). Even though a low diffusion rate constant and high membrane electrostatic repulsion were expected for longer lipid–DNA probes, we realized that these less hydrophobic lipid–DNA probes still exhibited faster insertion kinetics on MDCK cell membranes than 20 nt probes (Fig. S8a†). For example, it took 12.8 min, 5.7 min, 5.3 min, and 4.2 min to reach 90% maximum signal for cholesterol-modified 20 nt, 40 nt, 60 nt, and 80 nt DNA probes, respectively. This is another indication that less hydrophobic lipid–DNA probes exhibited faster cell membrane insertion kinetics.We next wanted to study the effect of double-stranded DNA on the insertion kinetics. To prepare lipid–DNA probes with double-stranded DNA, we incubated the lipid-modified 20 nt DNA probes with a 20 nt complementary DNA strand. Interestingly, the gel electrophoresis and dynamic light scattering result indicated that the formation of double strands actually reduced the size of the aggregates (Fig. S2b and S9†). This size reduction is probably due to the increased persistence length, steric hindrance and electrostatic repulsion in these double-stranded DNA-based lipid aggregates.36 We further measured the CAC values of these DNA duplex probes. As expected, the addition of complementary DNA only slightly increased the CAC values of the lipid–DNA probes (Fig. S10 and Table S4†).
When we further measured the membrane insertion kinetics of the probes, these less hydrophobic double-stranded DNAs actually slowed down the probe's membrane insertion compared to single-stranded DNA (Fig. S8b†). These reduced membrane insertion rates may have resulted from the increased persistence length37 and the large electrostatic repulsion between these highly negatively charged DNAs and the negatively charged cell membranes.25 It is worth mentioning that the opposite effect that less hydrophobic double-stranded DNAs exhibited slower membrane insertion may be explained by the similar CAC values and aggregation status of these double-stranded probes to those of single-stranded probes (Table S4†). These results indicated that not only the lipid hydrophobicity but also the structure of the DNA would influence the membrane insertion kinetics.
Cell membrane anchoring efficiency of lipid–DNA probes
Our next goal was to understand how the cell membrane insertion efficiency is determined in these lipid–DNA probes. We first asked if the amount of membrane-anchored probes is affected by the type of lipid anchor. To test that, we added 1 μM of each 20 nt lipid–DNA probe to the MDCK cells. Indeed, consistent with some previous studies,11 after 1 h of incubation at room temperature, we observed a large variation in the membrane insertion efficiency among different probes (Fig. 3a). We chose to add 1 μM of lipid–DNA probes because most of the tested probes have reached the maximum membrane insertion level at this concentration (Fig. S11a†).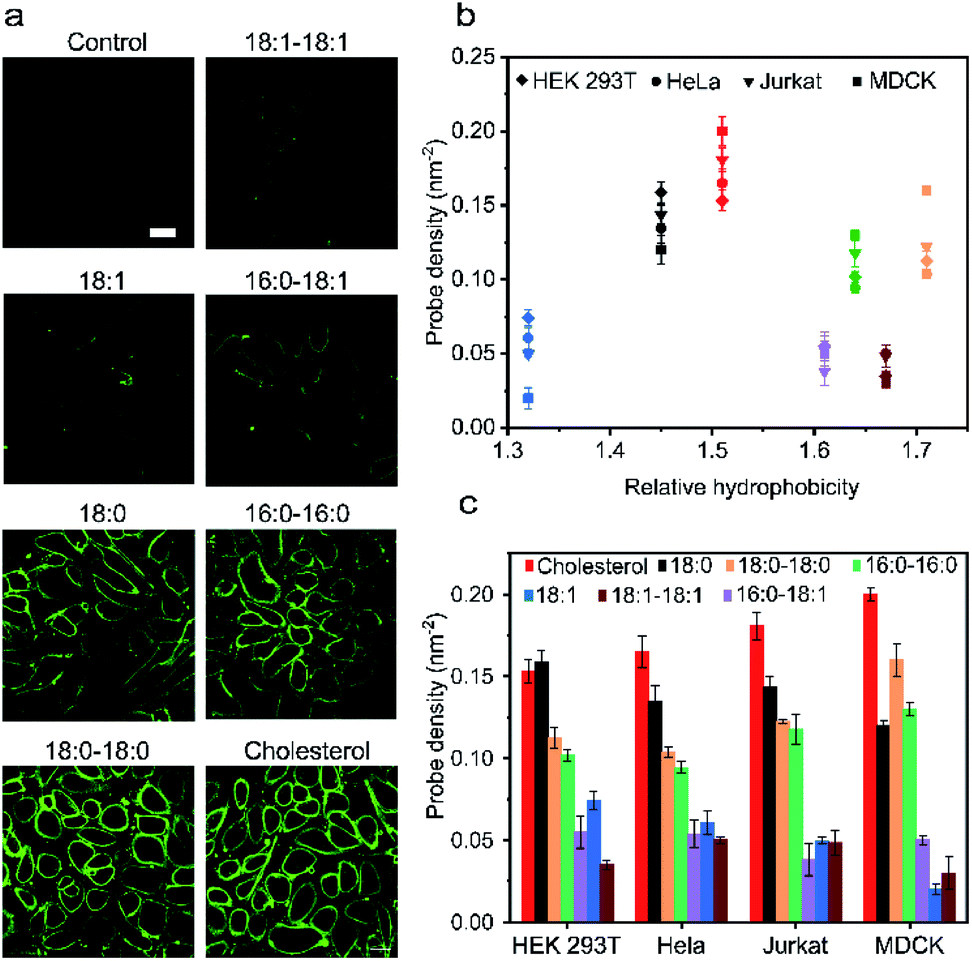 | ||
| Fig. 3 Cell membrane insertion efficiency of lipid–DNA probes. (a) Fluorescence imaging of the MDCK cell membrane insertion efficiency after incubation with 1 μM of each 20 nt lipid–DNA probe at room temperature for 1 h. Scale bar, 20 μm. (b) Membrane probe density on four different types of cell lines plotted against its corresponding hydrophobicity after 1 h of incubation with 1 μM of each 20 nt lipid–DNA probe. Each color represented a type of lipid, and the same color indication has been used as in panel (c). (c) Membrane probe densities on four different types of cell lines as measured after 1 h of incubation with 1 μM of each 20 nt lipid–DNA probe. Shown are the mean and SEM values that were measured on 50 cell membranes. | ||
Based on the observed fluorescence signal for each type of lipid–DNA probe, we further quantified the number of probes per unit area on the cell membrane (Fig. 3b). Our results indicated that there may be an optimal hydrophobicity of the probes to yield the maximum modification onto the membrane. For example, the cholesterol–DNA probe exhibited the highest membrane insertion efficiency despite its medium hydrophobicity compared to the other probes (Fig. 3b). Probes of low hydrophobicity may not efficiently insert on the membranes. Highly hydrophobic probes may tend to aggregate in the solution rather than insert onto the membrane. Furthermore, the structure of the lipid moiety could also influence the membrane anchoring efficiency. It is known that cholesterol is one of the most abundant lipids on mammalian plasma membranes. Cholesterol can fit into the flickering spaces between acyl chains and insert into different lipid domains on the cell membranes.38–40 This natural abundance of the cholesterol moiety may also explain the efficient membrane modification with cholesterol–DNA probes.
We wondered if other lipid anchors that mimic naturally abundant lipids on the plasma membranes would also result in highly efficient membrane insertion. We tested a 16:0–18:1-based probe for this purpose. Phospholipids containing a 16:0–18:1 fatty acid moiety are among the most abundant naturally existing lipids on the MDCK cell membranes.41 However, our result indicated that the membrane insertion efficiency of this probe is much lower than that of the cholesterol–DNA probe, and not even as high as that of the 16:0–16:0 probe (Fig. 3a). We asked if this low insertion efficiency could be due to the unsaturation on the 9th carbon of its acyl chain. Indeed, lipid anchors with saturated acyl chains (e.g., 18:0–18:0, 16:0–16:0, and 18:0) in general inserted more efficiently into MDCK cell membranes than lipids with unsaturated chains (e.g., 18:1–18:1, 16:0–18:1, and 18:1) (Fig. 3c). We cannot simply predict the insertion efficiency of a lipid–DNA probe based on the membrane abundance of the lipid moieties. For applications that require high probe densities on MDCK cells, cholesterol and 18:0–18:0 lipid anchors provide superior functionality.
We further quantified the membrane probe densities based on the above-mentioned calibration curve as measured in the lipid monolayer (Fig. S3d†). Under the experimental conditions, maximally 0.02–0.20 probe per nm2 membrane space can be efficiently achieved. These fluorescence microscopy results have been further validated by measuring the solution fluorescence intensities before and after incubating the probes with MDCK cells (Fig. S11b†).
We next asked if such probe insertion efficiency depends on the cell type. In addition to the above-mentioned MDCK cells, we further tested the membrane probe insertion with Jurkat, HEK 293T, and HeLa cells (Fig. 3b and c). Our results indicated that the probe insertion efficiency is generally consistent among these four types of cell lines. For example, the cholesterol–DNA probe normally exhibited the highest membrane insertion efficiency. Lipids with saturated acyl chains could anchor more into cell membranes than those with unsaturated chains, while at the same time, some probes behaved differently on different cell membranes. For example, single-acyl-chain 18:0 and 18:1 probes preferred to be inserted onto HEK 293T cells than MDCK cells, while double-acyl-chain 18:0–18:0 and 16:0–16:0 probes inserted more onto MDCK cells. These results might be correlated with different phospholipid compositions in these cell plasma membranes.42–45
We also studied the effect of double-stranded DNA on the probe insertion efficiency. After adding a complementary DNA strand, the probe insertion efficiency was slightly decreased on MDCK cell membranes (Fig. S11c†). This decreased membrane insertion may be due to the higher electrostatic repulsion between the double-stranded DNA with sialic acids and glycosaminoglycans on the cell membranes.25
Temperature could be another important factor in determining the probe insertion efficiency. We measured the MDCK cell membrane fluorescence after incubating each lipid–DNA probe for 1 h at room temperature or 37 °C. Our data indicated that the probe insertion efficiency was reduced at an elevated temperature (Fig. S11d†). This result is interesting because the probe insertion rate was faster at higher temperature (Fig. S8c†).
This difference between the membrane insertion kinetics and the efficiency led us to further study the process of probe departure from the cell membranes. Our hypothesis is that the membrane insertion efficiency depends on the net value of the difference between the probe's membrane insertion rate and its membrane departure rate. At 37 °C, the enhanced membrane fluidity and cellular functions may result in a larger membrane departure rate due to probe internalization or diffusion into the solution. As a result, to understand the membrane insertion efficiency, we need to further study the membrane departure or persistence of lipid–DNA probes.
Cell membrane persistence of lipid–DNA probes
One of the major limitations in applying lipid–DNA probes is their short persistence on the cell membranes. It is still challenging to use these probes for long-term cell membrane measurement or regulation. To solve this problem, our next goal is to understand the cell membrane persistence of lipid–DNA probes. Again, using 20 nt lipid–DNA probes, we monitored the probe intensities and distributions on the MDCK cell membranes. After 1 h of incubation, we washed away free probes in the solution. The cell membrane fluorescence was further tracked for another 2.5 h (Fig. 4a and b). The cell membrane fluorescence dramatically decreased over time for all the tested probes (Fig. 4b). For example, half of the membrane 18:0 probes disappeared within 45 min after removing the free probes. On the other hand, a longer membrane persistence was observed for 18:0–18:0 and cholesterol probes, with a half-life of 140 min and 100 min, respectively.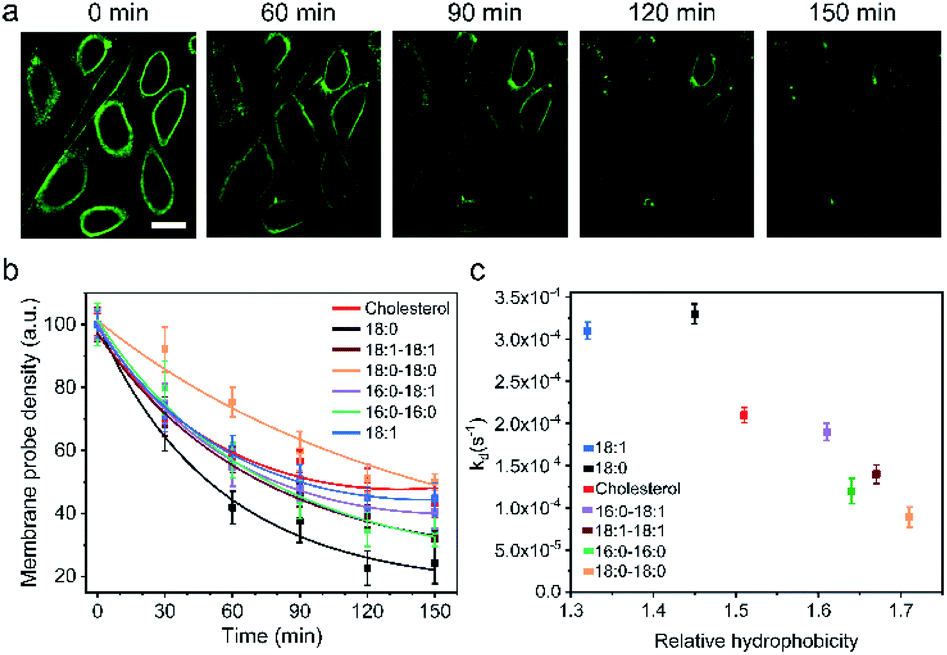 | ||
| Fig. 4 Cell membrane persistence of lipid–DNA probes. (a) Fluorescence imaging of the MDCK cell membrane fluorescence decay after removing the free unbound probes at 0 min. These cells were pre-incubated with 1 μM of 20 nt cholesterol–DNA probe at room temperature for 1 h. Scale bar, 20 μm. (b) MDCK cell membrane probe density decay kinetics as measured after removing the free unbound probes at 0 min. These cells were pre-incubated with 1 μM of the corresponding 20 nt lipid–DNA probe at room temperature for 1 h. (c) MDCK cell membrane decay rate constant of each 20 nt lipid–DNA probe plotted against its corresponding hydrophobicity. Shown are the mean and SEM values that were measured on 50–60 cell membranes. | ||
We further quantified the cell membrane fluorescence and probe density decay. Our data indicated a first-order reaction model for this observed membrane process (Fig. S12a†). After calculating the membrane decay rate constant (termed kd) for each lipid–DNA probe (Fig. 4c), interestingly, we realized that there was a similar correlation between the decay rate constant and the relative hydrophobicity value of the probe with the effect of hydrophobicity on the membrane insertion rate constant (Fig. 2b). In other words, less hydrophobic lipid–DNA probes exhibited both faster membrane insertion and departure, which may explain the relative independency of the membrane insertion efficiency from the hydrophobicity of lipid–DNA probes (Fig. 3b).
We realized that there could be two mechanisms for the loss of lipid–DNA probes from the cell membranes: internalization into the cells and direct outflow into the solution.21,24,26 We then asked if we could quantify the contribution of each mechanism in the membrane signal decay. Membrane-immobilized probes can be potentially internalized through different pathways such as transmembrane lipid flip-flop, endocytosis, etc. The negatively charged DNA strands will likely reduce the chance of lipid flip-flop. We hypothesized that the probe internalization probably mainly occurs through endocytosis. To test this hypothesis, we tracked the probe internalization with a lysosomal tracker, pHrodo red dextran (Fig. 5a). The fluorescence of pHrodo red dextran is pH-dependent, which can be used to stain lysosomes and late endosomes.46 Based on the Pearson correlation coefficient of the two fluorescent channels, 67% and 64% of the internalized 16:0–16:0 and cholesterol–DNA probes were localized in the endosome/lysosome (Fig. 5a). Indeed, endocytosis is one of the major mechanisms of the probe internalization.
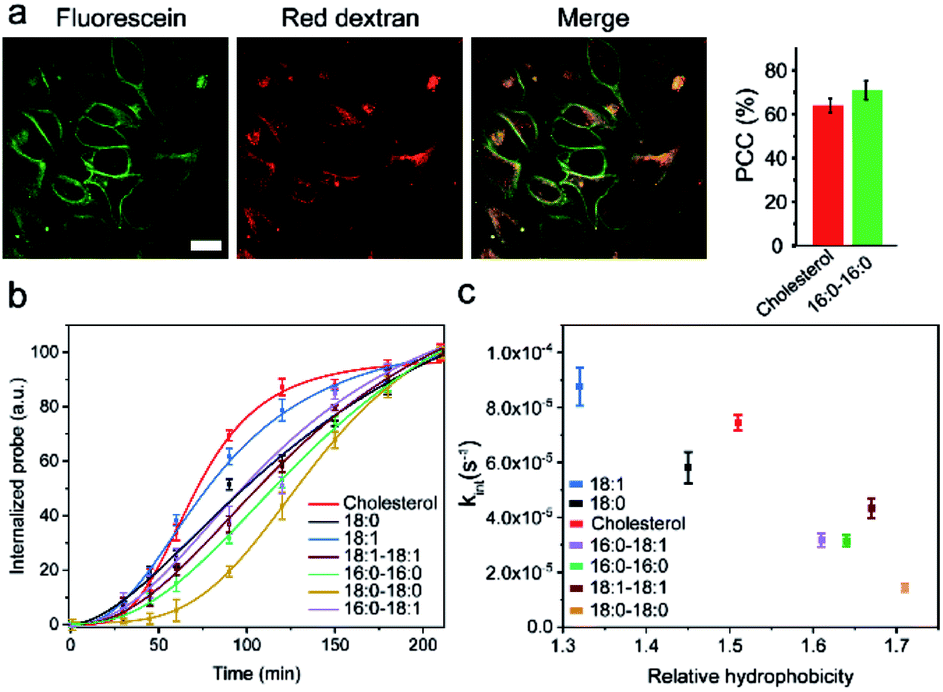 | ||
| Fig. 5 Cellular internalization of lipid–DNA probes. (a) Fluorescence imaging and colocalization of the 20 nt cholesterol–DNA probe (fluorescein channel) and a lysosomal tracker (red dextran channel). Scale bar, 20 μm. The Pearson correlation coefficient (PCC) of the two fluorescent channels was further quantified with both 16:0–16:0 and cholesterol probes. Shown are the mean and SEM values that were measured in 30 cells. (b) MDCK cellular internalization kinetics of each 20 nt lipid–DNA probe. The free unbound probes were removed at 60 min, after pre-incubating the cells with 1 μM of each probe at room temperature for 1 h. (c) MDCK cellular internalization rate constant of each 20 nt lipid–DNA probe plotted against its corresponding hydrophobicity. Shown are the mean and SEM values that were measured in 50 cells. | ||
In order to measure the internalization kinetics, we labelled our lipid–DNA probe with a pH-independent cyanine-5 (Cy5) dye rather than FAM, whose fluorescence signal is pH-dependent (Fig. S12b†). Our results indicated that the cellular internalization for all the tested probes followed a first-order reaction model toward the probe concentration in the solution (Fig. 5b and S12c and d†). The calculated internalization rate constant (kint) exhibited a similar hydrophobicity-dependent pattern to that for the overall membrane decay rate constant (kd) (Fig. 5c). Less hydrophobic lipid–DNA probes tended to internalize faster. For example, the 18:1 probe has the highest probe internalization rate, while the more hydrophobic 16:0–16:0 and 18:0–18:0 probes internalized the slowest. It appears that a stronger hydrophobic–hydrophobic interaction between the lipid anchor and the plasma membrane might cause more probes to be retained on the cell membranes.
In addition to the cellular internalization, membrane-anchored probes may also directly flow out into the solution. The hydrophilic DNA moieties and the probe concentration differences between the membrane and extracellular solution may result in this outflow. Here, we measured the kinetics of the outflow by monitoring the fluorescence signal of the probes that diffused out into the solution. For this purpose, we first incubated 1 μM 18:0 or cholesterol probe with MDCK cells for 1 h. After removing the excess probes, fresh HEPES buffer was added and the fluorescence of the probe in this buffer was measured over time with a plate reader (Fig. S13†). Our data indicated that the probe's outflow to the solution again followed a first-order reaction model, with a rate constant of 1.6 × 10−3 s−1 and 2.1 × 10−4 s−1, respectively, for the 18:0- and cholesterol-based probes. Indeed, the less hydrophobic 18:0 probe exhibited a faster outflow rate than the cholesterol probe. To further study the effect of hydrophobicity on the membrane outflow rate, we added the 20 nt or 80 nt cholesterol probe onto MDCK cell membranes. Indeed, the less hydrophobic 80 nt probe exhibited much faster membrane signal decay than the 20 nt probe (Fig. S12e†). The membrane outflow rate also appeared to be reversibly correlated with the hydrophobicity of the probe.
All these data indicated that the less hydrophobic lipid–DNA probes exhibited faster membrane insertion, internalization and outflow, which could explain the difference in the membrane anchoring efficiency of each lipid–DNA probe (Fig. 3b). For example, even though the 18:0 probe exhibited a similar membrane insertion rate to cholesterol, its membrane decay rate is much faster (Fig. 2b and 4b). As a result, the membrane modification efficiency of the 18:0 probe is obviously lower than that of cholesterol. Similarly, the 18:1 probe could insert into cell membranes at a high rate; however, it also exhibited a high membrane decay rate, which made 18:1-DNA not an ideal probe for efficient cell membrane modification. When choosing an appropriate lipid–DNA probe, both membrane insertion and departure kinetics should be considered. The net rate difference between these two processes determines the overall membrane modification efficiency.
Considering this dynamic membrane process of insertion and departure, we next asked if there is any correlation between the membrane lateral diffusion rate of the probe and its membrane insertion and departure. We measured the lateral diffusion rate of each lipid–DNA probe on MDCK cell membranes using a fluorescence recovery after photobleaching (FRAP) assay (Table S6 and Fig. S13†). Interestingly, the less hydrophobic 18:0, cholesterol, and 18:1 probes also exhibited faster membrane lateral diffusion rates than other lipids. There could be indeed a correlation between the probe membrane lateral diffusion rate and the dynamics of its membrane insertion and departure.
Based on these data and other literature studies,47–50 we proposed a pathway through which lipid–DNA probes could insert into, persist in, and depart from the cell membranes (Fig. 6). First, there is equilibrium between the monomeric and aggregated forms of lipid–DNA probes in the solution. Monomeric form lipid–DNA probes insert into the cell membranes. Then, some of the cell membrane-anchored probes are internalized into the cells through clathrin- or caveolae-mediated endocytosis. The majority of the internalized probes are then located inside the late endosomes or lysosomes where they are degraded or rejected out of the cells. Some probes are likely transferred to the Golgi apparatus or endoplasmic reticulum as well. On the other hand, the membrane-anchored probes can also directly flow out into the extracellular solution, which is the reverse process of the initial probe membrane insertion. It is worth mentioning that direct probe exchange between neighbouring cells can also occur at the cell–cell junctions. Our single-molecule tracking data indicated that lipid–DNA probes can be exchanged between neighbouring cells bi-directionally (Fig. S13e and movie S1†). Due to this bi-directional nature, we think that the membrane probe density will likely not be reduced because of these intercellular exchanges.
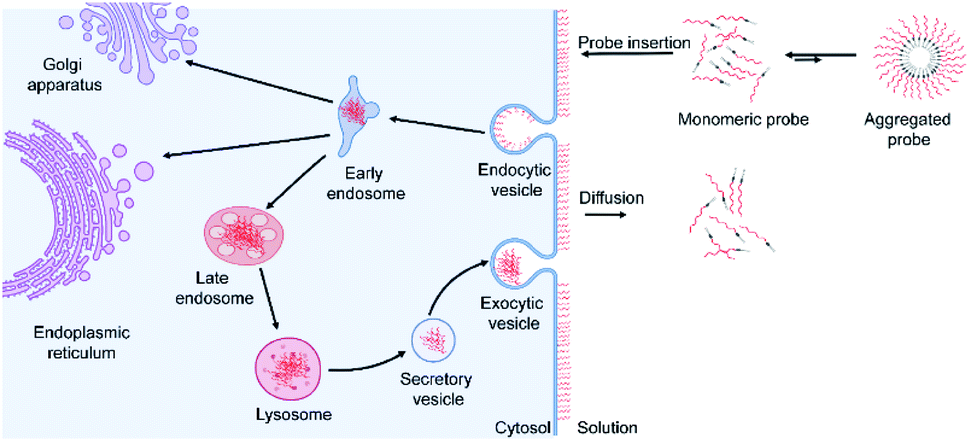 | ||
| Fig. 6 Schematic of the dynamic process of lipid–DNA probe modification on cell membranes. First, there is equilibrium between the monomeric and aggregated forms of lipid–DNA probes in the solution. Monomeric form lipid–DNA probes insert into the cell membranes. Then, some of the cell membrane-anchored probes are internalized into the cells through the endocytosis pathway. Some membrane-anchored probes can also directly diffuse out into the extracellular solution or neighbouring cell membranes. | ||
Improving the membrane persistence of lipid–DNA probes
Our next goal was to improve the probe persistence on the cell membranes. Based on our model of the dynamic membrane insertion and decay processes, our first hypothesis is that having excess probes in the solution can maintain high probe densities on the cell membranes. These excess probes in the solution (above the CAC value) could switch between the aggregated and monomeric forms, and maintain a high monomeric probe concentration to insert into cell membranes continuously. Indeed, our results showed that in the presence of excess probes, lipid–DNA probe densities on the MDCK cell membrane remained constant for 3–4 h (Fig. S14a†), while as shown above, during the same period of incubation, dramatic membrane signal decay was observed after removing the free unbound probes (Fig. 4b). We also measured the probe internalization rate in the presence of excess probes (Fig. S14b†). Interestingly, the obtained probe internalization rate is quite similar to that in the absence of excess probes (Fig. 5b). Indeed, these results indicated that the improved membrane persistence of the lipid–DNA probe is not due to the reduced internalization, but is instead because of the dynamic replacement of the membrane probes with fresh lipid–DNA from the solution. For applications that require long-term membrane modifications, having excess probes in the solution is thus useful.Considering the high background signal and interference from free probes in the solution, the existence of excess probes may not always be ideal. We next asked how we could further improve the probe persistence on the cell membrane after washing away free unbound probes. As mentioned above, endocytosis is one of the major pathways for the loss of membrane probes. We thus asked if we could retain a high membrane probe density by inhibiting endocytosis. We first tested methyl-beta-cyclodextrin (MβCD), which is known to inhibit caveolae-mediated endocytosis by removing cholesterols from the cell membrane.49 Unfortunately, the addition of MβCD dramatically reduced the number of probes on the cell membrane (Fig. S15a†). Meanwhile, MβCD exhibited a high toxicity towards MDCK cells, even at a reduced concentration (Fig. S16†).
We next tested hypertonic sucrose, which has been shown to block clathrin-mediated endocytosis by destroying clathrin lattices on the cell membranes. Indeed, we found that sucrose is effective in reducing probe internalization during the first 3 h (Fig. S15†). However, an unexpected fast probe internalization was observed afterwards. This fast internalization may be due to the limited tolerance of the cells towards long-term exposure to large amounts of sucrose.51
We next tested another well-studied endocytosis inhibitor, filipin. Filipin is known to inhibit caveolae-mediated endocytosis by interacting with cell membrane cholesterols to prevent their participation in the endocytosis.52 After optimizing the dosage and duration of inhibition, the inhibition of the endocytosis process could be achieved by first incubating MDCK cells with 0.5 μg mL−1 filipin for 1 h, and then adding a mixture of filipin and lipid–DNA probes (Fig. S15d†). As shown in Fig. 7a, the addition of filipin could significantly reduce the cellular internalization of all the tested lipid–DNA probes. We further quantified filipin-induced internalization inhibition using four representative probes including cholesterol, 18:0, 16:0–16:0 and 16:0–18:1. After 4 h of incubation, filipin could reduce the probe internalization by 40–60% (Fig. 7b). Moreover, the addition of filipin did not reduce the membrane insertion efficiency of all the tested probes (Fig. S15e†). All these results indicated that filipin could be used as an efficient endocytosis inhibitor to reduce the cellular internalization of lipid–DNA probes.
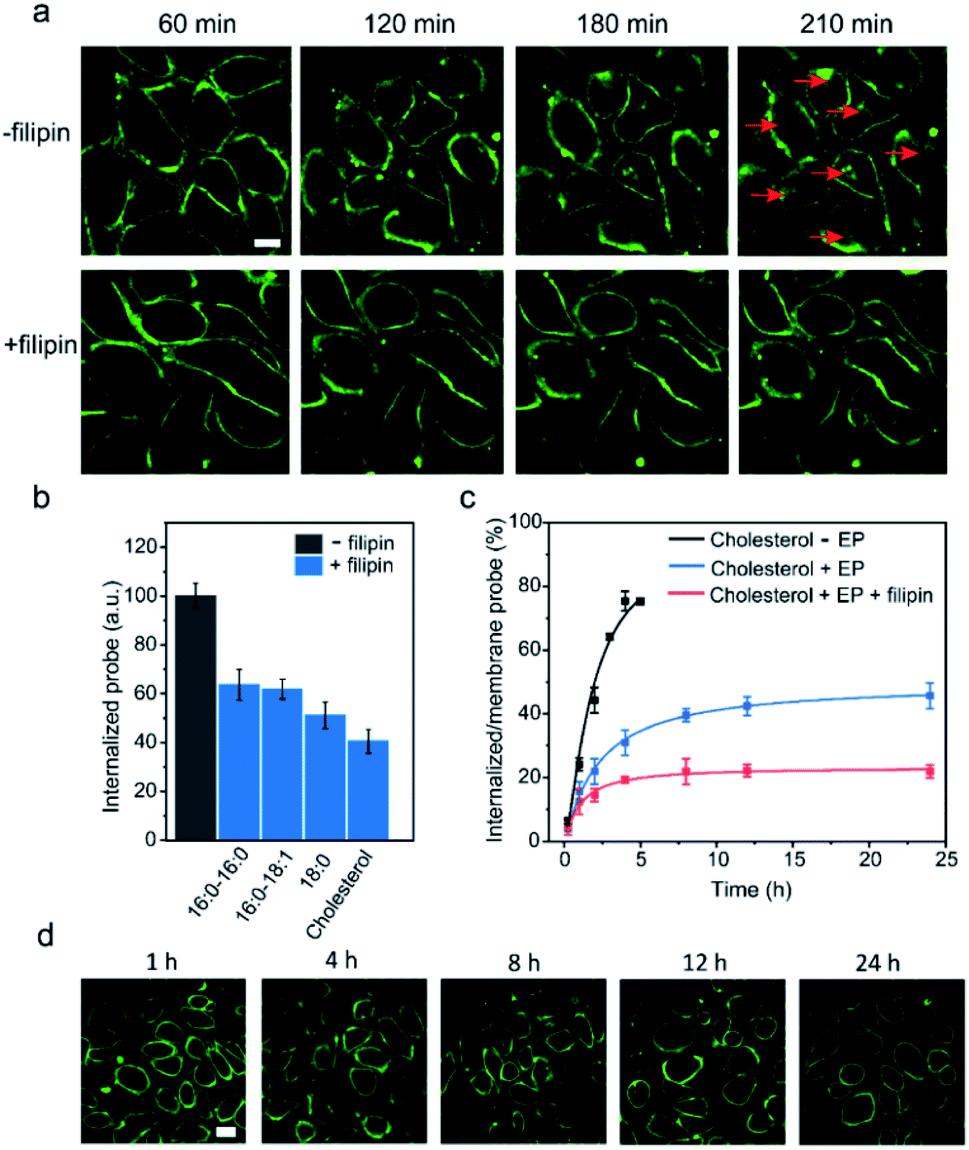 | ||
| Fig. 7 Improving the cell membrane persistence of lipid–DNA probes. (a) Fluorescence imaging of the MDCK cellular internalization efficiency after adding 1 μM of 20 nt cholesterol–DNA in the absence or presence of 0.5 μg mL−1 filipin. Scale bar, 20 μm. (b) Effect of filipin on the probe internalization as measured after 4 h of incubation of each lipid–DNA probe at room temperature. Shown are the mean and SEM values that were measured in 50 cells. (c) Long-term inhibitory effect of filipin on the cellular internalization of a cholesterol–DNA probe. The internalized probe density was divided by that on the cell membrane in the absence or presence of excess probes (EP) and/or filipin. Shown are the mean and SEM values that were measured in 50 cells. (d) Fluorescence imaging of the MDCK cells after adding 1 μM cholesterol–DNA probe in the presence of 0.5 μg mL−1 filipin. Scale bar, 20 μm. | ||
Our last goal was to test if we could use filipin for long-term inhibition of probe internalization. We chose the cholesterol–DNA probe as an example. As shown in Fig. 7c, we quantified the probe internalization efficiency through dividing the internalized probe density by that on the cell membrane. As expected, the addition of filipin dramatically reduced the internalization of the probes. Even after 24 h of incubation, the internalized probe concentration was only 22% of that on the cell membrane. We could now successfully monitor the lipid–DNA probe signal on cell membranes for at least 24 h, without interference from the internalized probes (Fig. 7d). These lipid–DNA probes could be potentially applied for various membrane studies that require long-term immobilization on the plasma membrane.
Conclusions
In this study, we have provided a quantitative assessment of the whole process of lipid–DNA probe modification on live cell membranes. We have now gained deeper insights into the structure–activity relationships of such dynamic interactions. These lipid–DNA probes could be broadly used for cell membrane analysis and regulation. Our results will have substantial impacts on the design of advanced cell membrane-anchored lipid–DNA probes.(1) We discovered that the membrane insertion of lipid–DNA probes is much faster with the monomeric form than the aggregated form. Generally, lipid–DNA probes with lower hydrophobicity insert faster onto cell membranes.
(2) The membrane insertion efficiency of lipid–DNA probes depends on the net value of the difference between their membrane insertion and departure rates. For example, cholesterol–DNA exhibited the highest insertion efficiency among the tested probes. Lipids with saturated acyl chains normally inserted more efficiently than the ones with unsaturated chains.
(3) Lipid–DNA probes can be removed from the cell membranes through either endocytosis or direct outflow into the solution or neighbouring cell membranes. Probes with lower hydrophobicity exhibited faster endocytosis, outflow, and overall decay from the cell membranes.
(4) Long-term probe modifications on the cell membrane can be realized with excess amounts of lipid–DNA probes in the solution and by adding endocytosis inhibitors, such as filipin. We are now able to dramatically improve the cell membrane persistence of the probes to at least 24 h.
Experimental section
Chemicals and reagents
Unless otherwise mentioned, all the chemicals were purchased from Sigma or Fisher and used without further purification. DNA strands including tetraethylene glycol (TEG)–cholesterol and 6-fluorescein-modified oligonucleotides were purchased from Integrated DNA Technologies or Keck Oligonucleotide Synthesis. 16:0–18:1-modified DNA was purchased from Bio-Synthesis, Inc. Soybean polar extract and 16:0–18:1 lipids were purchased from Avanti Polar Lipids.Cell culture
MDCK cells were cultured in a DMEM medium supplemented with 10% fetal bovine serum, penicillin (100 U) and streptomycin (0.1 mg mL−1) at 37 °C in a 5% CO2 atmosphere. MDCK cells were sub-cultured at 80% confluency and plated at a density of 50% following standard cell culture procedures. Cells were washed twice with a PBS buffer before adding the DNA probes.Cell imaging and data analysis
All the images were collected with NIS-Elements AR software using a Yokogawa spinning disk confocal unit on a Nikon Eclipse-TI inverted microscope. 6-Fluorescein amidite (FAM) was excited with a 488 nm laser line using 40× and 100× oil immersion objectives. All the data analysis was performed using the NIS-Elements AR Analysis software. Data were presented as mean ± SEM following the standard calculations for the mean value and the standard errors of the mean. A standard t-test was carried out to determine the differences between the mean values. If the p-value was lower than 0.05, it was considered as statistically significant.Critical aggregation concentration (CAC) determination
CAC values were measured by monitoring Nile red fluorescence in the presence of different concentrations of lipid–DNA conjugates. To determine the CAC of each conjugate, fluorescence spectra of 30 mM Nile red in HEPES buffer were measured after incubation with 10 nM to 15 μM of lipid–DNA conjugates at room temperature for 6 h. The fluorescence was measured using an excitation wavelength of 535 nm and emission at 630 nm.53Dynamic light scattering
Dynamic light scattering was performed on a Malvern Nano Zetasizer at room temperature to determine the size of aggregates. 1 μM of each lipid–DNA conjugate in HEPES buffer was used for these measurements. The HEPES buffer was filtered using a 0.2 μm syringe filter before the measurement.Lipid monolayer preparation
Soybean polar extract was mixed with different concentrations of lipid–DNA conjugates and incubated at 4 °C overnight to incorporate the conjugates into the lipid layer. 10 μL of the equilibrated lipid–DNA mixture was placed on a Teflon layer and dried under an air flow. Before imaging, the lipid monolayer was rehydrated with HEPES buffer. Fluorescence imaging was performed using the exact same parameters used for imaging the cells.Fluorescence recovery after photobleaching (FRAP) analysis
A Yokogawa spinning disk confocal unit on a Nikon Eclipse-TI inverted microscope was used for the FRAP experiment. The photobleaching was performed on a circular area of interest with a diameter of 2–3 μm using 50% laser power at 408 nm for 1 s. The fluorescence recovery was then monitored for another 4 min. The diffusion coefficient for each probe was determined following previous reports.54Cell viability test
A cell viability test was carried out with MDCK cells using an Alamar blue method.55 Briefly, cells were first incubated with lipid–DNA probes and endocytosis inhibitors for 4 h at room temperature. Afterwards, 400 μL of 10% Alamar blue solution was added and the cells were incubated for 2 h at 37 °C.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge NIH R35GM133507, a start-up grant from UMass Amherst and an IALS M2M seed grant. We are grateful to Dr James Chambers for assistance with fluorescence imaging. The authors also thank other members in the You Lab and Dr Craig Martin for useful discussion and valuable comments.Notes and references
- E. Sezgin, I. Levental, S. Mayor and C. Eggeling, Nat. Rev. Mol. Cell Biol., 2017, 18, 361–374 CrossRef CAS PubMed.
- H. E. Grecco, M. Schmick and P. I. H. Bastiaens, Cell, 2011, 144, 897–909 CrossRef CAS PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- M. E. Todhunter, N. Y. Jee, A. J. Hughes, M. C. Coyle, A. Cerchiari, J. Farlow, J. C. Garbe, M. A. LaBarge, T. A. Desai and Z. J. Gartner, Nat. Methods, 2015, 12, 975–981 CrossRef CAS PubMed.
- S. Chen, A. W. Bremer, O. J. Scheideler, Y. S. Na, M. E. Todhunter, S. Hsiao, P. R. Bomdica, M. M. Maharbiz, Z. J. Gartner and D. V. Schaffer, Nat. Commun., 2016, 7, 10309 CrossRef CAS PubMed.
- N. S. Selden, M. E. Todhunter, N. Y. Jee, J. S. Liu, K. E. Broaders and Z. J. Gartner, J. Am. Chem. Soc., 2012, 134, 765–768 CrossRef CAS PubMed.
- X. Qu, S. Wang, Z. Ge, J. Wang, G. Yao, J. Li, X. Zuo, J. Shi, S. Song, L. Wang, L. Li, H. Pei and C. Fan, J. Am. Chem. Soc., 2017, 139, 10176–10179 CrossRef CAS PubMed.
- M. Xiao, W. Lai, X. Wang, X. Qu, L. Li and H. Pei, Microphysiological Systems, 2018, 2, 1–17 CrossRef.
- M. J. Taylor, K. Husain, Z. J. Gartner, S. Mayor and R. D. Vale, Cell, 2017, 169, 108–119 CrossRef CAS PubMed.
- H. Liu, B. Kwong and D. J. Irvine, Angew. Chem., Int. Ed., 2011, 50, 7052–7055 CrossRef CAS PubMed.
- R. J. Weber, S. I. Liang, N. S. Selden, T. A. Desai and Z. J. Gartner, Biomacromolecules, 2014, 15, 4621–4626 CrossRef CAS PubMed.
- G. Feng, X. Luo, X. Lu, S. Xie, L. Deng, W. Kang, F. He, J. Zhang, C. Lei, B. Lin, Y. Huang, Z. Nie and S. Yao, Angew. Chem., Int. Ed., 2019, 58, 6590–6594 CrossRef CAS PubMed.
- L. Qiu, T. Zhang, J. Jiang, C. Wu, G. Zhu, M. You, X. Chen, L. Zhang, C. Cui, R. Yu and W. Tan, J. Am. Chem. Soc., 2014, 136, 13090–13093 CrossRef CAS PubMed.
- M. You, Y. Lyu, D. Han, L. Qiu, Q. Liu, T. Chen, C. Sam Wu, L. Peng, L. Zhang, G. Bao and W. Tan, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS PubMed.
- B. Zhao, C. O'Brien, A. P. K. K. K. Mudiyanselage, N. Li, Y. Bagheri, R. Wu, Y. Sun and M. You, J. Am. Chem. Soc., 2017, 139, 18182–18185 CrossRef CAS PubMed.
- J. R. Burns, N. Al-Juffali, S. M. Janes and S. Howorka, Angew. Chem., Int. Ed., 2014, 53, 12466–12470 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- M. E. Todhunter, R. J. Weber, J. Farlow, N. Y. Jee, A. E. Cerchiari and Z. J. Gartner, Curr. Protoc. Chem. Biol., 2016, 8, 147–178 Search PubMed.
- X. Xiong, H. Liu, Z. Zhao, M. B. Altman, D. Lopez-Colon, C. J. Yang, L.-J. Chang, C. Liu and W. Tan, Angew. Chem., Int. Ed., 2013, 52, 1472–1476 CrossRef CAS PubMed.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V Li, B. Huang, D. S. Van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS PubMed.
- Y. Wu, K. Sefah, H. Liu, R. Wang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5–10 CrossRef CAS PubMed.
- H. Liu, Z. Zhu, H. Kang, Y. Wu, K. Sefan and W. Tan, Chemistry, 2010, 16, 3791–3797 CrossRef CAS PubMed.
- A. Saminathan, J. Devany, K. S. Pillai, A. T. Veetil, M. Schwake and Y. Krishnan, bioRxiv, 2019, 523019.
- G. G. Borisenko, M. A. Zaitseva, A. N. Chuvilin and G. E. Pozmogova, Nucleic Acids Res., 2009, 37, e28 CrossRef PubMed.
- M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 6218–6223 CrossRef CAS PubMed.
- A. Patwa, A. Gissot, I. Bestel and P. Barthélémy, Chem. Soc. Rev., 2011, 40, 5844–5854 RSC.
- K. Börjesson, J. Tumpane, T. Ljungdahl, L. M. Wilhelmsson, B. Nordén, T. Brown, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2009, 131, 2831–2839 CrossRef PubMed.
- Y. Bagheri, F. Shafiei, S. Chedid, B. Zhao and M. You, Supramol. Chem., 2019, 31, 532–544 CrossRef CAS.
- S. C. Hsiao, B. J. Shum, H. Onoe, E. S. Douglas, Z. J. Gartner, R. A. Mathies, C. R. Bertozzi and M. B. Francis, Langmuir, 2009, 25, 6985–6991 CrossRef CAS PubMed.
- I. Pfeiffer and F. Höök, J. Am. Chem. Soc., 2004, 126, 10224–10225 CrossRef CAS PubMed.
- A. Lopez and J. Liu, Langmuir, 2018, 34, 15000–15013 CrossRef CAS PubMed.
- A. Bunge, M. Loew, P. Pescador, A. Arbuzova, N. Brodersen, J. Kang, L. Dähne, J. Liebscher, A. Herrmann, G. Stengel and D. Huster, J. Phys. Chem. B, 2009, 113, 16425–16434 CrossRef CAS PubMed.
- J. K. Hannestad, R. Brune, I. Czolkos, A. Jesorka, A. H. El-Sagheer, T. Brown, B. Albinsson and O. Orwar, ACS Nano, 2013, 7, 308–315 CrossRef CAS PubMed.
- K. N. Dejanovic and S. E. Cabaniss, Environ. Sci. Technol., 2004, 38, 1108–1114 CrossRef PubMed.
- K. Valkó, C. Bevan and D. Reynolds, Anal. Chem., 1997, 69, 2022–2029 CrossRef PubMed.
- M. P. Thompson, M.-P. Chien, T.-H. Ku, A. M. Rush and N. C. Gianneschi, Nano Lett., 2010, 10, 2690–2693 CrossRef CAS PubMed.
- P. Gross, N. Laurens, L. B. Oddershede, U. Bockelmann, E. J. G. Peterman and G. J. L. Wuite, Nat. Phys., 2011, 7, 731–736 Search PubMed.
- P. L. Yeagle, Biochim. Biophys. Acta, Rev. Biomembr., 1985, 822, 267–287 CrossRef CAS.
- H. Ohvo-Rekilä, B. Ramstedt, P. Leppimäki and J. Peter Slotte, Prog. Lipid Res., 2002, 41, 66–97 CrossRef.
- O. G. Mouritsen and K. Jørgensen, Chem. Phys. Lipids, 1994, 73, 3–25 CrossRef CAS.
- G. van Meer and K. Simons, EMBO J., 1982, 1, 847–852 CrossRef CAS PubMed.
- A. A. Spector and M. A. Yorek, J. Lipid Res., 1985, 26, 1015–1035 CAS.
- M. G. Lewis, T. L. Kaduce and A. A. Spector, Prostaglandins, 1981, 22, 747–760 CrossRef CAS.
- G. van Meer and K. Simons, EMBO J., 1982, 1, 847–852 CrossRef CAS PubMed.
- R. Dawaliby, C. Trubbia, C. Delporte, C. Noyon, J.-M. Ruysschaert, P. Van Antwerpen and C. Govaerts, J. Biol. Chem., 2016, 291, 3658–3667 CrossRef CAS PubMed.
- G. C. Kemmer, M. Urban, M. G. Palmgren, T. Vosch, J. Schillerd and T. G. Pomorsk, Analyst, 2015, 140, 6313–6320 RSC.
- G.-H. Chai, F.-Q. Hu, J. Sun, Y.-Z. Du, J. You and H. Yuan, Mol. Pharm., 2014, 11, 3716–3726 CrossRef CAS PubMed.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- A. Alshehri, A. Grabowska and S. Stolnik, Sci. Rep., 2018, 8, 3748–3757 CrossRef PubMed.
- F. Deng, H. Zhang, X. Wang, Y. Zhang, H. Hu, S. Song, W. Dai, B. He, Y. Zheng, X. Wang and Q. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 5803–5816 CrossRef CAS PubMed.
- D. Dutta and J. G. Donaldson, Cellular Logistics, 2012, 2, 203–208 CrossRef PubMed.
- Y. Kitajima, T. Sekiya and Y. Nozawa, Biochim. Biophys. Acta, Biomembr., 1976, 455, 452–465 CrossRef CAS.
- T. G. W. Edwardson, K. M. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2013, 5, 868–875 CrossRef CAS PubMed.
- M. Kang, C. A. Day, A. K. Kenworthy and E. DiBenedetto, Traffic, 2012, 13, 1589–1600 CrossRef CAS PubMed.
- B. P. Joshi, J. Hardie, M. A. Mingroni and M. E. Farkas, ACS Chem. Biol., 2018, 13, 2339–2346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S18, Tables S1–S6, and a description of materials and methods. See DOI: 10.1039/c9sc04251b |
| This journal is © The Royal Society of Chemistry 2019 |