
Bioinspired metal complexes for energy-related photocatalytic small molecule transformation
Hao-Lin
Wu
 ab,
Xu-Bing
Li
ab,
Chen-Ho
Tung
ab and
Li-Zhu
Wu
*ab
ab,
Xu-Bing
Li
ab,
Chen-Ho
Tung
ab and
Li-Zhu
Wu
*ab
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, the Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: lzwu@mail.ipc.ac.cn
bSchool of Future Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 16th November 2020
Abstract
Bioinspired transformation of small-molecules to energy-related feedstocks is an attractive research area to overcome both the environmental issues and the depletion of fossil fuels. The highly effective metalloenzymes in nature provide blueprints for the utilization of bioinspired metal complexes for artificial photosynthesis. Through simpler structural and functional mimics, the representative herein is the pivotal development of several critical small molecule conversions catalyzed by metal complexes, e.g., water oxidation, proton and CO2 reduction and organic chemical transformation of small molecules. Of great achievement is the establishment of bioinspired metal complexes as catalysts with high stability, specific selectivity and satisfactory efficiency to drive the multiple-electron and multiple-proton processes related to small molecule transformation. Also, potential opportunities and challenges for future development in these appealing areas are highlighted.
 Hao-Lin Wu | Dr Hao-Lin Wu received her BS degree in chemistry from Jilin University in 2014 and PhD degree in chemistry from the Technical Institute of Physics and Chemistry, Chinese Academy of Sciences in 2020. Currently, she is a postdoctor at Beijing Forestry University. Her research interest focuses on photoelectrochemical water splitting. |
 Xu-Bing Li | Dr Xu-Bing Li obtained his BS degree from Wuhan University in 2010 and his PhD degree at the Technical Institute of Physics and Chemistry of Chinese Academy of Sciences (CAS) under the supervision of Prof. Li-Zhu Wu and Prof. Chen-Ho Tung in 2015. Then, he continued his research work in the same group as an assistant researcher and was promoted as an associate professor in 2017. His research interests mainly focus on artificial photosynthesis, including water splitting, carbon dioxide reduction, and photoredox organic transformation. |
 Chen-Ho Tung | Prof. Chen-Ho Tung graduated from the University of Science and Technology of China in 1963 and was awarded his PhD degree in 1983 from Columbia University in New York City under the supervision of Prof. Nicholas J. Turro. He was elected as an academician of CAS in 1999 and is currently a professor at TIPC. His research interests include photochemical reactions, photoinduced electron transfer and energy transfer in supramolecular systems, and photocatalytic water splitting. |
 Li-Zhu Wu | Prof. Li-Zhu Wu received her BS degree from Lanzhou University in 1990 and PhD from the Institute of Photographic Chemistry of Chinese Academy of Sciences in 1995 under the supervision of Prof. Chen-Ho Tung. After a one-year postdoctoral stay at the University of Hong Kong with Prof. Chi-Ming Che, she was appointed as a professor at the Technical Institute of Physics and Chemistry, CAS. She was elected as an academician of CAS in 2019. Her research interests include artificial photosynthesis, photocatalytic organic transformation, and photoinduced electron transfer and energy transfer in supramolecular systems. |
Introduction
Because of their fascinating potential to alleviate problems related to energy, environment and economy, the transformation of inexhaustible or readily available small molecules like water (H2O), carbon dioxide (CO2), methane (CH4), nitrogen (N2) and other organic molecules into highly value-added chemicals is of great concern nowadays.1,2 However, the inherent features of these small molecules make their catalytic conversion quite challenging. One of the key problems is that their fairly inert bonds, such as C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, often require high thermodynamic potentials for molecular activation, and their conversion usually involves multi-electron and multiple-proton transfer processes, which make the selective chemical reaction rather difficult to either occur or control.3 It is encouraging that thermocatalysis and electrocatalysis have been realized to effectively activate their inert bonds and thus the conversion of these small molecules; however, they usually require an additional non-renewable energy input, which is limited by the needs of large-scale and sustainable development in the future.4
O bonds, often require high thermodynamic potentials for molecular activation, and their conversion usually involves multi-electron and multiple-proton transfer processes, which make the selective chemical reaction rather difficult to either occur or control.3 It is encouraging that thermocatalysis and electrocatalysis have been realized to effectively activate their inert bonds and thus the conversion of these small molecules; however, they usually require an additional non-renewable energy input, which is limited by the needs of large-scale and sustainable development in the future.4
Natural photosynthesis (NP) has shown us an excellent example to realize small molecule transformation.5 Upon light absorption by antenna systems, the harvested photon energy is transferred to the active site to power subsequent redox-reactions (Fig. 1). The water oxidation, for example, occurs by using the photo-generated oxidative holes to activate water molecules at the oxygen-evolving centre (OEC) in PSII.6 Then, the released four protons and four electrons are transferred via the electron transfer chains to the catalytic active center of PSI, so as to realize the corresponding reduction reactions via energetically enriched bio-reducing agents such as NADPH or ATP. In this regard, most green plants use high-energy electrons to reduce protons and CO2 to carbohydrate at the [NiFeS]-metalloenzyme catalytic centre for energy storage. In some green algae and photosynthetic bacteria, however, ferric redox proteins continue to transfer high-energy electrons to hydrogenases (H2ases) that reduce protons to produce H2 with remarkable activity.7 At this point, the proton source for H2 evolution of these anaerobic organisms comes from the carbon metabolism of organic matters. On the whole, plants and organisms can produce O2 and carbohydrates (or H2) from CO2 and H2O through the cooperation of PSII and PSI under sunlight irradiation to complete the transformation of solar energy into chemical energy.
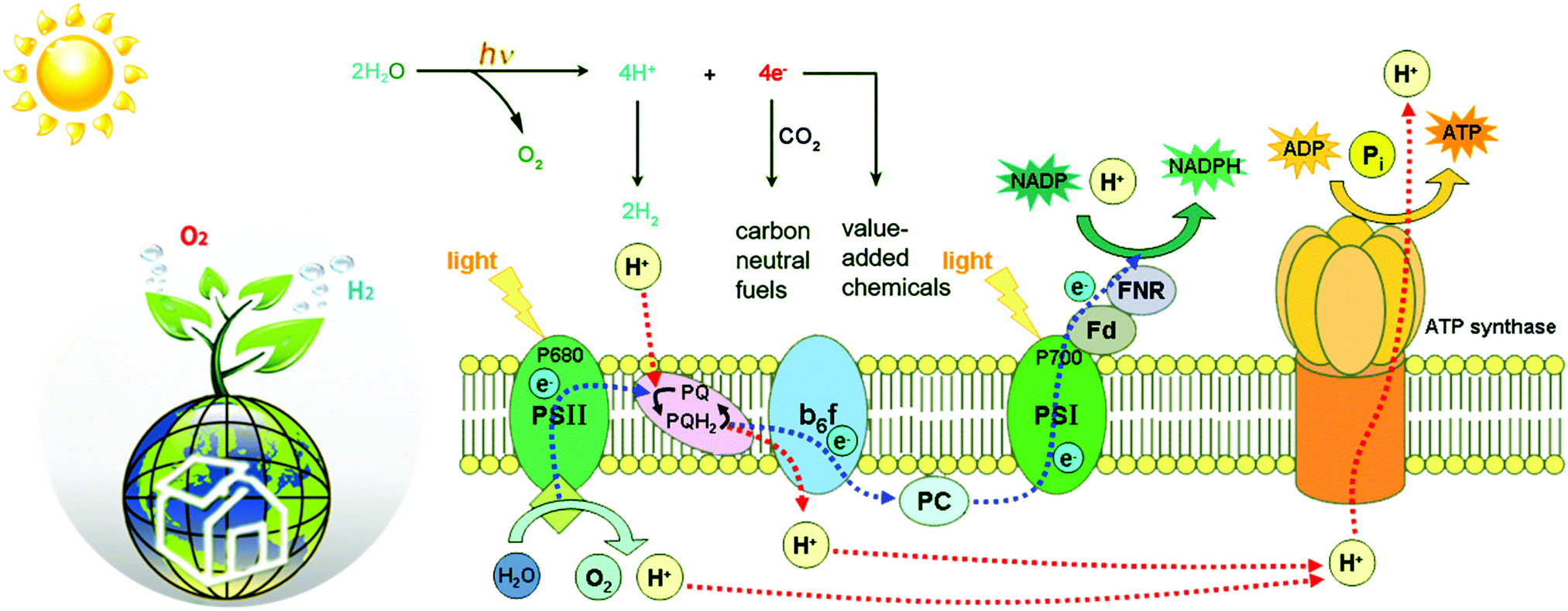 | ||
| Fig. 1 Schematic representation of the natural photosynthetic chain in photosystems. | ||
Inspired by NP, it was promising to realize small molecule transformation by reproducing the structure and functions of natural photosynthesis, that is, artificial photosynthesis.8 An ideal artificial photosynthetic system is usually based on a typical Z-scheme consisting of both oxidation and reduction catalysts (Fig. 2). Since most small molecules do not have light-absorbing capacity, appropriate structural design allows the catalyst molecules to match well with light harvesters to achieve efficient electron transfer, such as an appropriate driving force as well as flexible interface interaction.9 Following the capture of exciton carriers from photosensitizers, effective catalysts can reduce the activation energy and accelerate the reaction rate, which are undoubtedly important prerequisites for the construction of efficient artificial systems.10,11
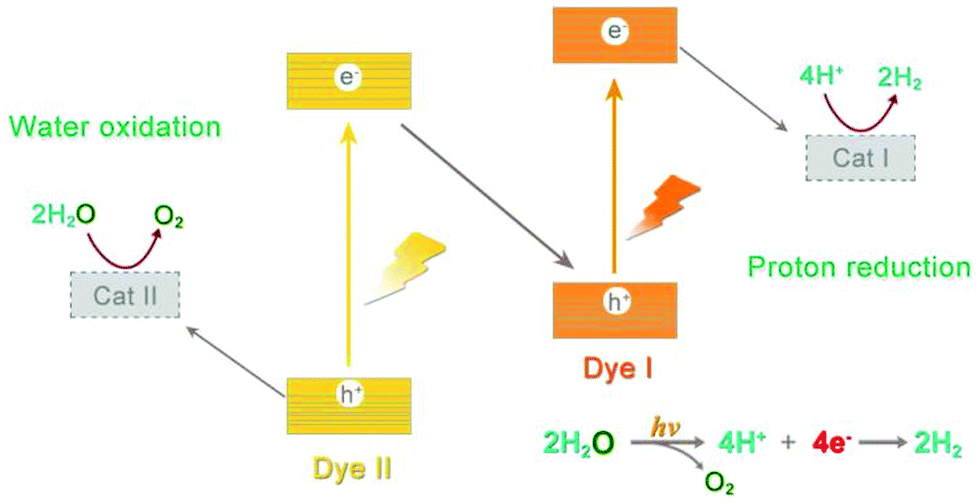 | ||
| Fig. 2 Schematic representation of the general concept of artificial photosynthesis. | ||
The archetypal biological systems have represented promising blueprints for the design of biomimetic catalysts. In light of the hydrogenase for reversible H2 production, the carbon monoxide (CO) dehydrogenase for reversible CO2 fixation, or the oxygen evolving complex for water oxidation, these delicate biocatalysts usually consist of a relatively small metal-containing active site.12 Though the fine structure and catalytic mechanism remain controversial, it is found that the precise control of metals and ligands has led to the design and synthesis of metal complex catalysts with rich photophysical, electrochemical, and catalytic properties to mimic the metalloenzymes.13
As the active sites, the metal centers within the complexes possess partially filled d-orbitals, which can reversibly store electrons and protons from small molecules via multiple redox states as well as provide binding sites for small molecule transformation. For example, Re(I)-based catalysts enable the conversion of CO2 to CO, partly due to the selective coordination of CO2 molecules to the reduced metal centres of the complexes.14 The individual metal center of a typical metal complex catalyst is usually coordinated by organic ligands, which expose each metal atom to the system to catalyze the reaction and provide higher coefficient of atomic utilization. The highly controllable ligands within metal complexes can be readily and systematically functionalized using different moieties to provide multiple functions, including electron-donating and electron-withdrawing substituents, accessional redox sites, intramolecular proton relays, sterically bulky groups with either hydrophobic or hydrophilic characteristics and so on.15,16 For instance, the CO-displacement of the all-CO [FeFe]-H2ase mimics by a poorly σ-donating phosphine ligand, tris(N-pyrrolyl)phosphine (P(pyr)3), could change the reduction potentials of diiron complexes with small cathodic shifts, ca. 30 mV for single replacement and 60 mV for dual replacement.17 Ligand modification can also facilitate the interaction between the catalyst and other components in the system. By introducing four dangling phosphonic acid groups to the outer sphere of a DuBois-type catalyst, the photocatalytic experiments can be carried out in both homogeneous and heterogeneous systems with a phosphonated bipyridine-based Ru(II) photosensitizer, and the outer sphere provides the catalyst with good solubility in aqueous solution as well as anchors for the immobilization on metal oxide semiconductors.18
It's no exaggeration to say that the unique properties of metal complexes offer infinite possibilities for photocatalytic small molecule transformation. Through the coordination with photosensitizers, the entire artificial photocatalytic process can be viewed as consisting of three consecutive subprocesses: capture of photonic energy by a variety of chromophores giving rise to the formation of excited states, conversion of these excited states into redox power by a charge separation process and finally catalysis using stored redox equivalents to drive the corresponding small molecule reduction or oxidation reactions at the active sites.19 In this respect, water can be oxidized to O2, while the corresponding electrons can be used to drive the reduction-conversion reactions, including proton reduction to produce H2 or reducing CO2 to carbon-neutral fuels. Due to the complexity of the catalytic conversion processes, at present, a more effective strategy is to divide the overall process into two half reactions as occurring in natural photosynthesis, which is conducive for further investigation and optimization of the catalysts. For any oxidation or reduction half reaction, the corresponding sacrificial reagents (sacrificial electron donor, SED or sacrificial electron acceptor, SEA) need to be added to provide the required reductive or oxidative equivalents. Once optimized, these half reactions can be integrated, such as introducing them into the PEC systems, to realize the coupling of oxidation and reduction processes.20 Furthermore, the approach can also be extended to couple the abovementioned reduction reactions with oxidative transformations of small organic molecules to produce high value-added organic products, mimicking the coupled redox reactions occurring in NP. A typical example is cross-coupling hydrogen evolution (CCHE), which will be discussed later.21
Over the past few decades, numerous homogeneous and heterogeneous molecular systems based on metal catalysts have been developed. This Feature Article provides a comprehensive account of several photochemical transformation reactions of critical small molecules that are catalyzed by metal complexes, e.g., water oxidation, proton and CO2 reduction and catalytic oxidation of some small organic molecules. All these fields are involved in energy-related small molecule activation and are also the hot spots of chemistry, materials, energy and environmental science. Herein, we do not intend to provide an exhaustive report of the works in these areas, but rather to pay attention to what researchers have learnt from NP for small molecule activation using bioinspired metal complexes as catalysts. Representative works, current challenges and promising perspectives will be highlighted.
Molecular water-oxidation catalysts (WOCs)
In NP, water oxidation occurs with a low overpotential (a thermodynamic parameter reflecting the difficulty of water oxidation) of ∼160 mV and a high reaction rate of 100–400 s−1 under ambient conditions.22 Although there are some ambiguities, the core structure of the oxygen-evolving complex (OEC) is universally acknowledged to be a cubic Mn3CaO4 cluster linked to additional dangling Mn via μ-O atoms.23 Ligands including Cl− ions, water molecules and amino acid residues surround the Mn4CaO5 cluster to give the final crystal structure of the OEC, as shown in Fig. 3a. Structural mimicry of natural OECs is a feasible way to design artificial OECs. Limited by the accessible characterization and synthetic approach, it was not until 2015 that Zhang et al. reported a synthetic Mn4Ca-cluster similar to the native OEC in PSII with the two magnetic resonance signals assignable to the redox and structural isomerism in NP to produce O2 in the presence of H2O (Fig. 3b).24 However, as a complicated reaction involving multiple electrons and protons, catalytic water oxidation did not occur by using the artificial OEC metal complex. Challenges, such as the high overpotential and the vulnerability of WOCs under harsh oxidation conditions do remain.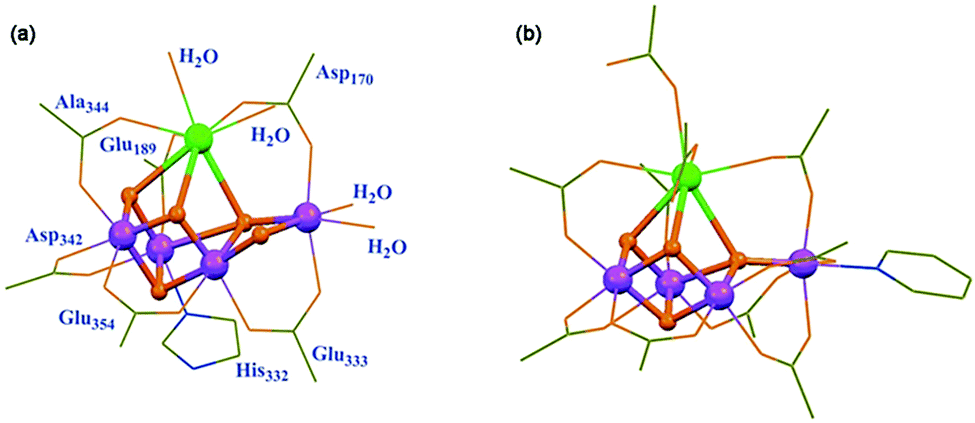 | ||
| Fig. 3 Crystal structures of the (a) native OEC and the (b) synthetic Mn4Ca complex. Reprinted with permission from ref. 24. Copyright 2015 Science. | ||
Designing metal complex catalysts to mimic PSII functionally started early in the 1980s. Meyer et al. reported the molecular catalyst [(bpy)2Ru(H2O)(μ-O)(H2O)Ru(bpy)2]4+ (bpy = 2,2′-bipyridine) for water oxidation, known as “Blue Dimer” (complex 1 in Fig. 5 and Table 1).25 From then on, Ru-based molecular catalysts have been regarded as potential candidates for natural PSII-OEC and have become the most efficient water-oxidizing molecular catalysts, which have been highlighted in many comprehensive reviews.26–28 In spite of multicore-Mn active sites in NP, theoretical and experimental results show that a single metal centre is active enough for water oxidation since Thummel et al. reported the first water oxidation catalyst based on a mononuclear molecular ruthenium complex.29 These findings expand the scope of WOCs based on metal complexes and lay a foundation for studying the critical mechanisms of O-O bond formation by transition metal complexes: water nucleophilic attack (WNA) and interaction of two M–O units (I2M) (Fig. 4).30,31
| WOCs | Photosensitizers/SEA/solvents | Light source | TON | TOF [s−1] |
|---|---|---|---|---|
| Complex 1 | — | — | 13.2 | 0.0042 |
| Complex 2 | — | — | 2000 | 41 |
| Complex 3 | [Ru(bpy)3]Cl2/Na2S2O8/pH 7.2 CH3CN/phosphate buffer solution | 150 W Xe lamp (380 nm) | >1255 | >13.1 |
| Complex 4 | [Ru(bpy)2(deeb)](PF6)2/Na2S2O8/pH 7.5 phosphate buffer solution | Halogen lamp (>400 nm) | 200 | — |
| Complex 5 | [Ru(bpy)2(dcbpy)]Cl2/Na2S2O8/pH 7.5 phosphate buffer solution | 500 W Xe lamp (>400 nm) | 51 | 0.28 |
| Complex 6 | [Ru(bpy)2(py-bpy)](PF6)2/50 mM Na2S2O8/phosphate buffer solution | Xe lamp (>400 nm) | — | 0.08 |
| Complex 7 | [Ru(bpy)3]Cl2/Na2S2O8/pH 9.0 sodium borate buffer solution | LED lamp (>420 nm) | 1610 | 11.1 |
| Complex 8 | [Ru(bpy)3]Cl2/Na2S2O8/pH 8.7 borate buffer solution | 3 W blue LED | 220 | 0.76 |
| Complex 9 | [Ru(bpy)3](ClO4)2/5 mM Na2S2O8/pH 8.5 borate buffer solution | Xe lamp (>400 nm) | 11.61 ± 0.23 | 0.158 |
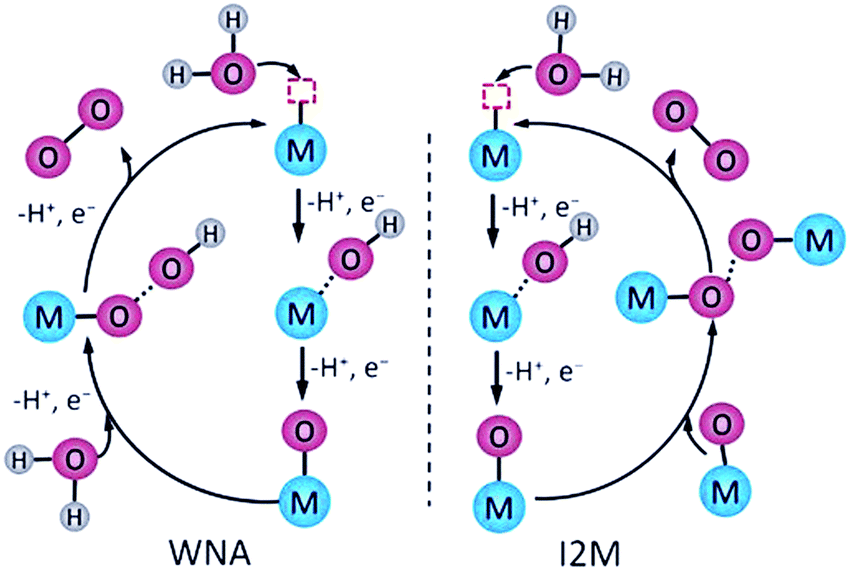 | ||
| Fig. 4 Schematics of the WNA and the I2M pathways to form O–O bonds catalysed by molecular catalysts. Reprinted with permission from ref. 30. Copyright 2020 Wiley-VCH. | ||
In the WNA mechanism, the O–O bond formation occurs by the nucleophilic attack of one H2O molecule to the high-valent MO intermediate. Alternatively, the I2M pathway forms an O–O bond via the interaction between two MO entities to an M–O–O–M species, which can release O2 upon further oxidization. Most of the water oxidation catalysts reported so far form O–O bonds through WNA, while the catalysts with a Ru-bda (bda = 2,2′-bipyridine-6,6′-dicarboxylic acid) framework, such as the complex 2 shown in Fig. 5, form O–O bonds mostly through I2M. As proposed by Sun et al., an uncommon seven-coordinated Ru(IV) dimer complex with [HOHOH]− bridging ligand was isolated as a critical intermediate for catalytic water oxidation via I2M.32 An exceptional case was proposed by Wurthner and coworkers in 2016, a metallosupramolecular macrocycle complex 3 that gathers three Ru(bda) centres was formed via the axial ligand connection, which allows water molecules to form O–O bonds through the WNA pathway (Fig. 5).33
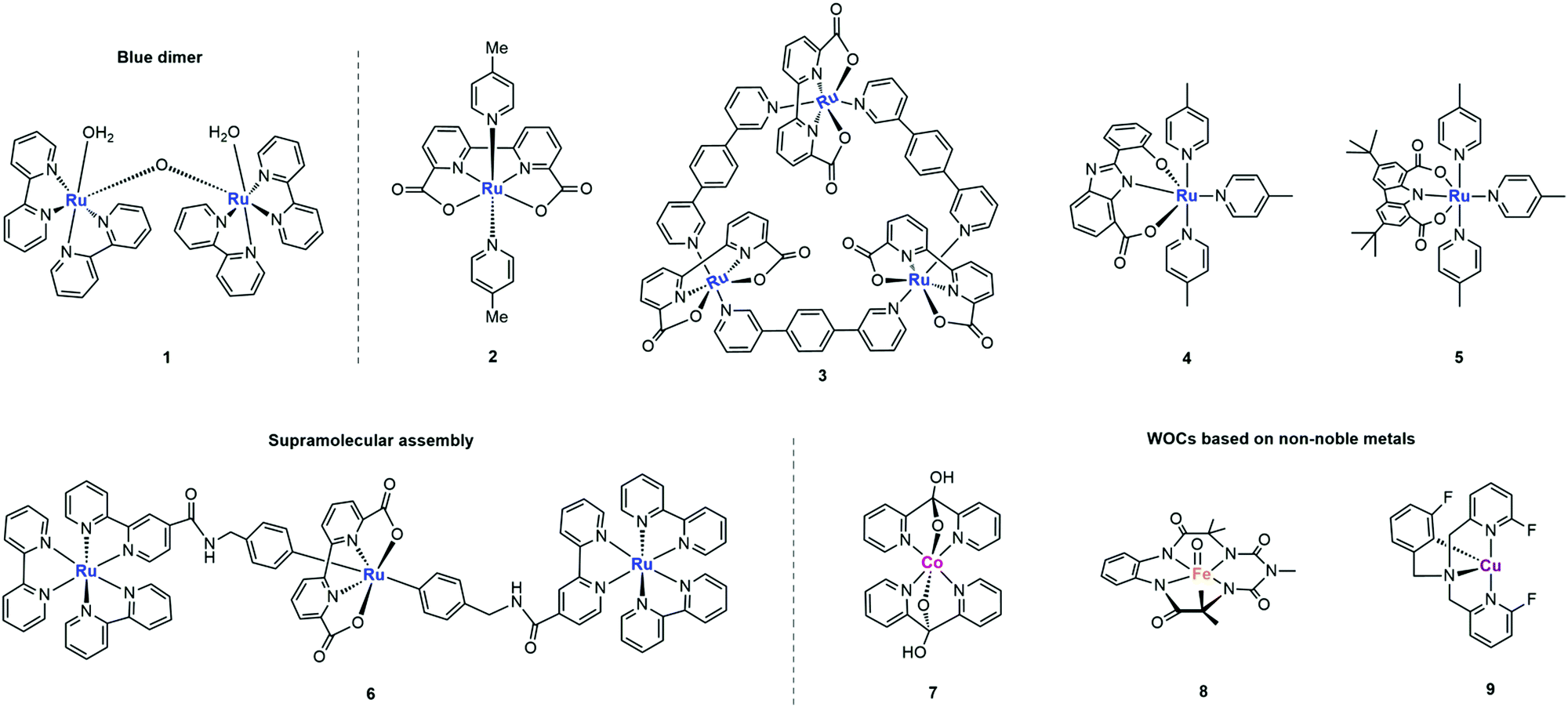 | ||
| Fig. 5 Representative structures of WOCs based on Ru and non-noble metals. | ||
The in-depth understanding of catalytic mechanisms has further promoted the application of molecular catalysts for water oxidation in the fields of thermocatalysis, electrocatalysis and photocatalysis.34,35 Early research studies emphasized the vital role of Proton Coupled Electron Transfer (PCET), which provides effective ways to reduce the redox potential, forms the Ru-oxo species through a narrow potential gap and stabilizes the high-valent metal–oxo intermediate.36 Accordingly, imidazole and carboxylate were introduced as redox and proton transfer mediators into the ligands of WOC by Akermark and coworkers.37 As shown in Fig. 5, the corresponding ruthenium complex 4 showed decreased redox potential as well as favourable coupled proton–electron transfer, which were able to catalyse water oxidation under neutral conditions with [Ru(bpy)3]3+ as the photosensitizer.
In the photocatalytic water-oxidation process, one of the main obstacles is the mismatch redox potentials between the catalyst and the photosensitizer. Both the push–pull effect (effect from electron donating and electron withdrawing groups) and the space effect (the space blocking effect caused by the proximity of some atoms or groups in a molecule) of ligands could affect the redox potential, activity and stability of the critical high valence intermediate RuVO. A charge-neutral mononuclear ruthenium complex RuL(pic)3 was synthesized with a carbazoledicarboxylate ligand, as shown in Fig. 5 (complex 5).38 Due to the rigid planar structure and strong electron-donating feature of the ligand, the Ru centre can be well stabilized at high redox states for efficient photochemical water oxidation. Alternatively, researchers aim to mimic the natural water-oxidizing environment to improve the catalytic activity. Accordingly, König et al. loaded the hydrophilic derivative of complex 2 and the photosensitizer onto the surface of the vesicle membrane to mimic the biological photosynthesis where both chromophores and catalytic units are bound to membranes. Compared to the homogeneous systems with dissociative components, this two-dimensional assembly greatly increased the local concentration of the redox counterparts and shortened the average distance between them for electron transfer.39
In addition to constructing intermolecular systems with separated catalysts and photosensitizers, Ru based catalysts can also form supramolecular assembly with a photosensitizer to achieve efficient intramolecular charge transfer. As shown in Fig. 5, Sun et al. proposed the first case of an active photochemical water oxidation assembly by coupling two ruthenium diamine chromophores with one [RuL(pic)2] catalyst (complex 6).40 The photocatalytic activity of this assembly was enhanced compared with that of the conventional multicomponent system.
On the other hand, the Mn4CaO5 cluster developed in nature as WOC suggests that the use of Mn or other non-noble metals is likely to achieve efficient light-driven water oxidation.41–46 However, few reports have been proposed on homogeneous Mn-based molecular catalysts for water oxidation, and most of them are utilized in electrocatalytic systems.47 The only case of a molecular Mn-based catalyst for photochemical O2 evolution was reported in 2016 by Streb et al.48 Lately, a molecular manganese vanadium oxide cluster consisting of a [Mn4O4]6+ cubane core, a tripodal [V4O16]6− polyoxovanadate and three acetate ligands served as a critical model compound to investigate the structure and mechanism of OEC in PSII, as the formation of a [MnIII2MnIV2O4]6+ intermediate is similar to the S1 state in the natural Kok cycle. A TOF (turnover frequency, referring to the number of H2 molecules produced per unit time at each catalytic active site (or catalyst)) of around 1.75 s−1 was observed under visible light irradiation with [Ru(bpy)3]2+ as the photosensitizer and persulphate (S2O82−) as the oxidant.
The complex with a Co4O4 cubane structure also achieved efficient photochemical O2 evolution, which was first reported by Patzke et al. as [CoII4(hmp)4(μ-OAc)2(μ2-OAc)2(H2O)2].49 The highly flexible architecture of monodentate acetate and aqua ligands makes it the closest mimic of PSII-OEC among the highly sought after cubane WOCs to date, providing mechanistic modelling to explore the water oxidation pathways of PSII. In addition, some N-coordinated cobalt complexes, such as cobalt porphyrins, cobalt salophen, and cobalt polypyridine complexes, are also reported as homogeneous catalysts for photocatalytic water oxidation (complex 7).50,51
At present, the reports on iron based molecular catalysts are very limited. On one hand, it is difficult to design and synthesize metallic iron catalysts with both suitable redox potential as well as relatively low overpotential. On the other hand, the stability of metallic iron catalysts is generally not satisfactory; especially under alkaline conditions, as they decompose into nanoparticles.52 In 2014, the first homogeneous photocatalytic water oxidation system based on an iron complex was reported (Fe-TAML, FeIII complex with biuret-modified tetra-amidomacrocyclic ligands, complex 8).53 For the first time, the generation of a high valence FeV(O) intermediate species during photocatalysis was detected as the active intermediate for water oxidation. No nanoparticles were observed after the measurement, indicating that the system was carried out through homogeneous catalysis.
Due to its inexpensiveness and easy accessibility to various oxidation states, Cu is also considered to be an ideal catalytic centre for water oxidation. However, there are few reports about Cu-based photochemical water oxidation compared with its wide application in the field of electrocatalysis. It was not until 2016 that the first homogeneous photocatalytic water oxidation catalyst based on a Cu complex was reported.54 With [Ru(bpy)3]2+ as the photosensitizer and Na2S2O8 as the electron acceptor, the water-soluble copper–polypyridine complex, [Cu(F3TPA)(ClO4)2], could catalyse water oxidation with a TON (turnover number, the number of H2 molecules produced at each catalytic active site (or catalyst)) of 11.61 ± 0.23 (complex 9). The substitution of fluorine (F) on pyridine ligands increases the stability of the metal centre. One of the keys to further develop Cu-based water oxidation catalysts is to reduce their overpotentials.55 Accordingly, a Cu(II) coordination polymer, [Cu(tza)2]n (Htza, tetrazole–1-acetic acid) with negatively charged carboxylate ligands, was synthesized to reduce the overpotential. Moreover, the nitrogen catenation ligand with both electronically rich and conjugated electron-withdrawing properties could maintain the catalytic activity of the system to the greatest extent (TOF 1.68 s−1 at pH 9.0).
As the key part in NP, water oxidation not only produces the most critical resource of O2 for mankind, but also provides necessary electrons and protons for the subsequent reduction reactions. In artificial photosynthetic systems, solar-powered water oxidation into O2 is also indispensable to realize the overall water splitting, which is the ultimate goal of mimicking natural photosynthesis. Over the years, many breakthroughs have been made in the design and synthesis of novel OECs that mimic PSII-OEC in structure and function. However, water oxidation usually requires a high overpotential and a high thermodynamic demand (ΔG ≈ 237 kJ mol−1), which create troublesome challenges in this field. How to stabilize the high-valent intermediates and how to reveal the mechanism of O–O bond formation are the key points of current research on molecular catalysts for photocatalytic water oxidation.
Molecular hydrogen evolution catalysts (HECs)
NP uses hydrogenase as the hydrogen-evolution catalyst (HEC) to reduce the protons released from the oxygen-evolving centre, thus completing the whole catalytic cycle. With remarkable catalytic activity (producing 6000–9000 molecules of H2 per second per site) and only ubiquitous elements like Fe, S and C, [FeFe]-hydrogenase ([FeFe]-H2ases) has great potential as a catalyst in the field of photocatalytic H2 production.56 However, limited by its vulnerability to O2 as well as the intricate purification technology, it is very difficult to directly apply hydrogenase extracted from microorganisms to artificial photosynthetic systems. Fortunately, with the in-depth research on the structure and mechanism of these important biological enzymes, it has been a hot research topic in recent years to mimic the active centre of hydrogenase in both structure and function for solar H2 evolution.57Fig. 6a shows the active site of [FeFe]-H2ase featuring a butterfly binuclear Fe–Fe cluster ([2Fe2S]) coordinated with CN− and CO ligands, an azadithiolate (adt2−) double bridge ligand as well as a cysteine-linked [4Fe4S] cluster.58 One of the Fe ions within [2Fe2S] is in an ideal six-coordinated octahedral configuration, whereas the other one shows only five-coordination with an open coordination site that is believed to be the active site for proton binding and H2 release. Thus, the [2Fe2S] cluster as the catalytic centre can capture and reduce protons to produce H2, and the [4Fe4S] cluster acts as a channel for electron transfer from and to the active site.
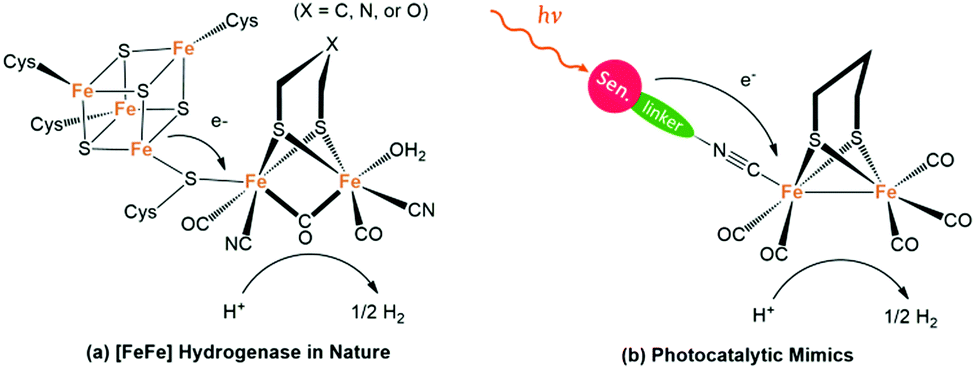 | ||
| Fig. 6 (a) Active site of natural [FeFe]-H2ase and (b) the photocatalytic mimics. | ||
It is worth mentioning that, the unique adt2− cofactor acts as a proximal proton acceptor site and plays a critical role in the mechanism of H2 evolution, which also provides inspiration for the design of complex catalysts.
At the early stage, scientists have focused on the structure modification of the [2Fe2S] subunit to tune their properties, such as the redox potentials and water solubility. Accordingly, the first reduction potentials of [2Fe2S] complexes can be tuned in the range of −1.18 to −1.67 V for the all-CO complexes with different dithiolate bridges in CH3CN, which enabled efficient electron transfer from the excited photosensitizer to the [2Fe2S] mimics.59 Since the pioneering work by Sun and Akermark in which they constructed intramolecular dyads consisting of photosensitizer and mimics, more and more attention has been paid to the interaction between the photosensitizers and the catalysts.60 On one hand, a series of molecular dyads and triads that are composed of a molecular photosensitizer and a [2Fe2S] catalytic centre have been constructed via covalent or coordinative linking, for desired photoinduced electron transfer (Fig. 6b). For instance, molecular triads with ferrocene as the electron donor, [FeFe]-H2ase mimics as the catalyst and a Ru-complex or zinc porphyrin as the photosensitizer (PS) were synthesized with enhanced photocatalytic H2 evolution.61 On the other hand, [FeFe]-H2ase mimics have been incorporated effectively with semiconducting PS. Accordingly, under the same conditions, the system with QDs as the PS exhibited a much higher catalytic activity than that of the system with [Ru(bpy)3]2+, due to the long-lived charge-separation state of the former.62 Since the first attempt by Wu et al. in 2011 with CdTe QDs and the first water-soluble [FeFe]-H2ases mimic, using semiconducting QDs as the PS to combine with a [FeFe]-H2ase mimic for H2 production has developed rapidly, and the catalytic efficiency has increased from null to TONs of hundreds of thousands.63 Strikingly, the synthetic [2Fe2S] catalytic centre, even the most popular one that could decompose generally within one-hour of irradiation, can serve as an effective catalyst for H2 production.
In NP, the active sites of [FeFe]-H2ase are surrounded by protein matrices, which protect the vulnerable cores and serve as a channel to input substrates and output products. Thus, in recent years, scientists have focused on not only the structural modification of [FeFe]-H2ase mimics, but also their surrounding biological environment. Specifically, the self-assembled system that comprises chitosan, [Fe2(CO)6(μ-adt)CH2C6H5], CdTe QDs and H2A was capable of producing H2 with a TON of up to (5.28 ± 0.17) × 104 with respect to the catalyst, nearly 4000-fold compared with that of the same system in the absence of chitosan (Fig. 7).64 Also, the catalytic stability was enhanced from 8 to 60 h in this self-assembled system. In other photocatalytic system, micelles, dendrimers and peptide scaffolds have also been used to mimic the natural protein environment for improved photocatalytic H2 evolution.65
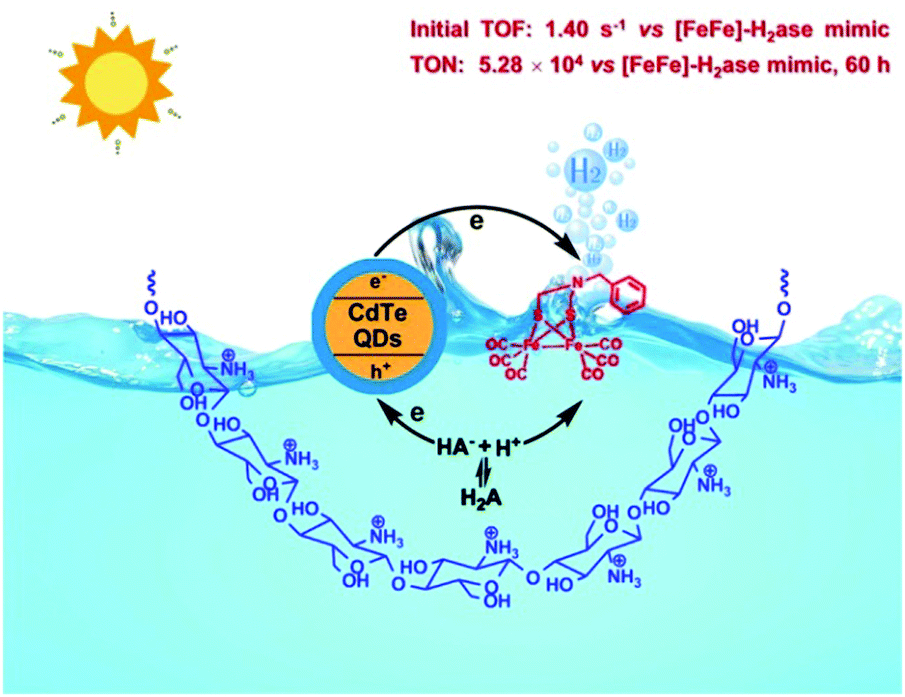 | ||
| Fig. 7 A schematic describing the H2 photogeneration of a chitosan-confined mimic of the diiron subsite of [FeFe]-H2ase in the presence of CdTe quantum dots and H2A. Reprinted with permission from ref. 64. Copyright 2013 Springer Nature. | ||
Compared with [FeFe]-H2ase, the catalytic H2 production rate of natural [NiFe]-H2ase is far behind, which is known to prefer H2 oxidation (>1000 s−1).66 The greater structural complexity of heterobimetallic centres of [NiFe]-H2ase mimics lagged far behind that of [FeFe]-H2ase mimics. Very recently, a [NiFe]-H2ase mimic, (dppe)Ni(μ-pdt)(μ-Cl)Ru(CO)2Cl, was synthesized based on the similar valence shell structures of RuII and FeII.67 Experimental and theoretical analyses revealed that mimic 1 in Fig. 8 worked as the pre-catalyst and H2 was produced only after being reduced to an unprecedented dimer intermediate 2 with an extraordinary TOF of 0.54 s−1, which offers inspiration for further design of heterobinuclear [NiFe]-H2ase mimics for advanced H2 evolution.
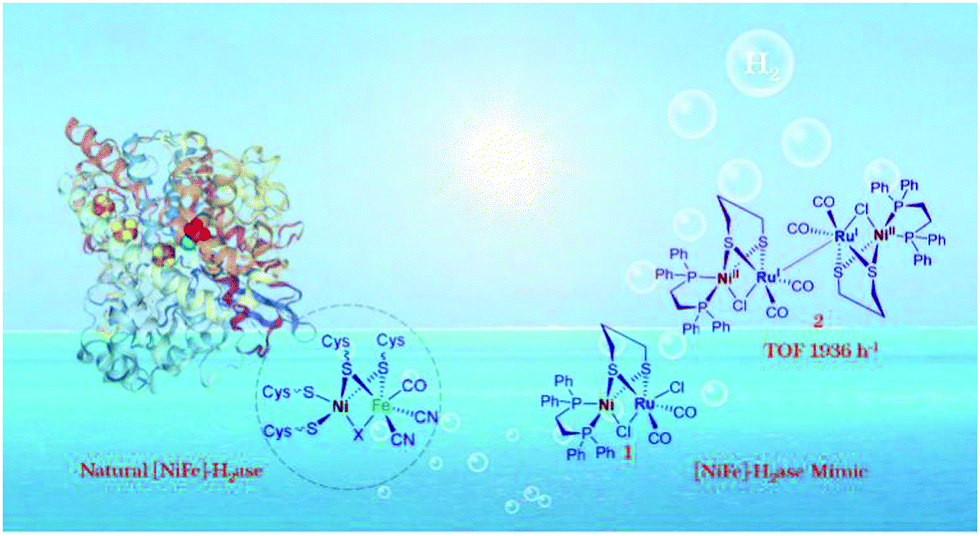 | ||
| Fig. 8 The structure and active site of natural [NiFe]-H2ase, the general structure of [NiFe]-H2ase mimics and the real dimer catalyst for exceptional H2 photogeneration. Reprinted with permission from ref. 67. Copyright 2020 Wiley-VCH. | ||
Fig. 9 shows the general catalytic mechanism for proton reduction at the metallic centre Mn+. Reduction of the active metal centre Mn+ followed by protonation generates the key metal-hydride intermediate (H–Mn+), which can react in two different ways.68 On one hand, the H–Mn+ reacts with a second metal-hydride to afford M(n−1)+ and generate H2via reductive elimination (homolytic pathway). On the other hand, H–Mn+ is further reduced and protonated to release H2, which is defined as the heterolytic pathway. The active site of natural [FeFe]-H2ase, for example, is believed to produce H2 by heterolysis, according to the H/D-isotope effect.69 Natural photosynthetic organisms evolve [FeFe]-H2ase with pendant amines in the second coordination sphere as a proton relay to promote the formation of H–Mn+ intermediate via PCET processes. Thus, an efficient metal complex catalyst should have unsaturated first coordination spheres as coordination sites for substrate binding. And exploring HECs that are conducive for the formation and stabilization of pivotal metal-hydride intermediates is widely regarded as an essential precondition for proton reduction.
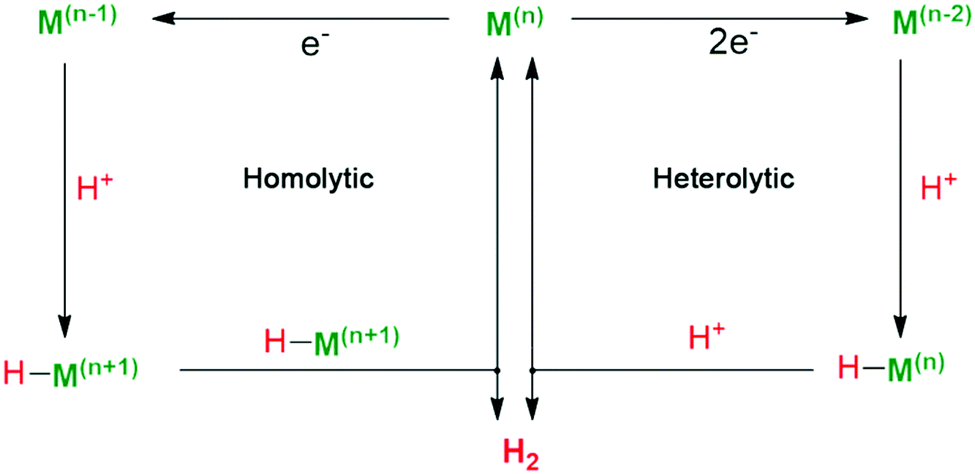 | ||
| Fig. 9 Proposed mechanistic pathways for H2 evolution by a metallic catalytic center. | ||
The photocatalytic activity of natural [FeFe]- and [NiFe]-H2ase and their mimics shows the potential of using non-noble metal catalysts to produce H2 in artificial photosynthesis. In this regard, complexes of cobalt and nickel have been found to be critical candidates.70 A so-called “DuBois-type” complex 10 with a bis(diphosphine) ligation was firstly described by DuBois and coworkers as [Ni(P2PhN2Ph)2](BF4)2 (Fig. 10).71 With the similarly positioned pendant amine, the catalytic rate of DuBois-type Ni complexes increased by three orders of magnitude compared with that of the corresponding carbon-based analogues. In 2011, Holland and coworkers reported the first case of photocatalytic H2 evolution based on this kind of nickel diphosphine catalyst.72 With Eosin Y (EY) as the light absorber and H2A as the SED, up to 2700 TON in 150 h was obtained (based on the catalyst) under visible light irradiation in water/acetonitrile mixed solvent at pH 2.25. Further functionalization with appropriate substitutes can not only improve the reactivity by regulating the electronic structure, but also promote the interaction between the catalyst and other components (complex 11).73
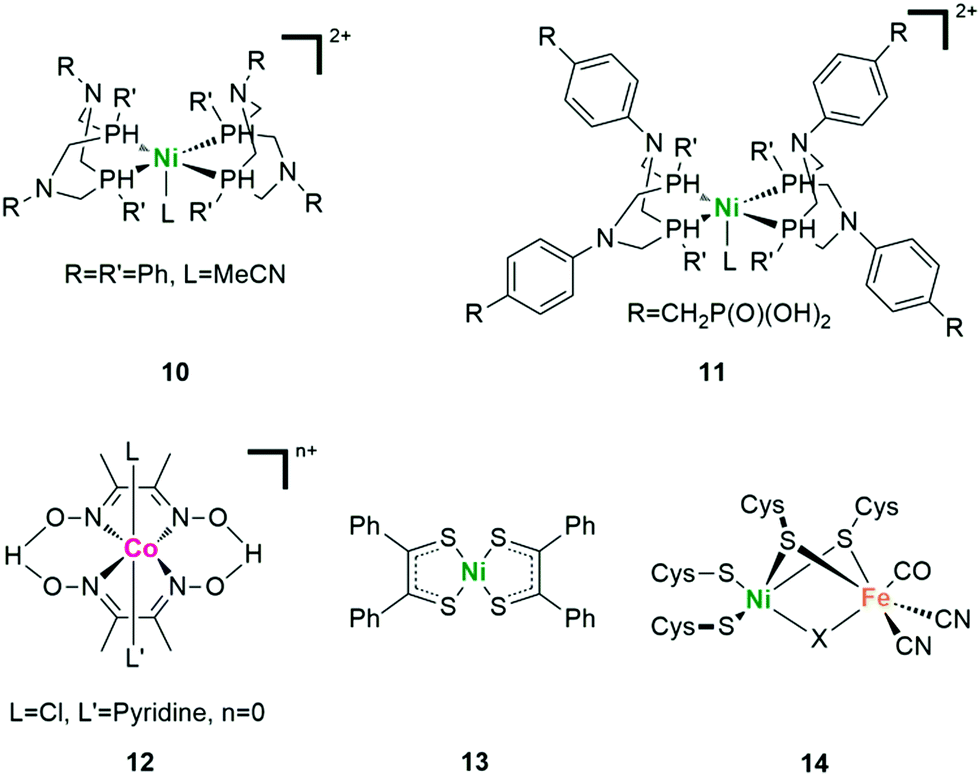 | ||
| Fig. 10 Representative structures of Ni-based, Co-based HECs and the active centre of [NiFe]-H2ase. | ||
The cobaltoximes that formed from the coordination of two glyoximato equatorial ligands with a cobalt ion are some of the most well-developed catalysts (Fig. 10 complex 12). Since firstly reported by Lehn and co-workers as [Co(dmgH)2] (dmgH2 = dimethylglyoxime) in 1983, a large number of diimine/dioxime cobalt HECs have been described.74 Most of them showed high activity in organic media while low stability in water, especially under acidic reductive conditions. Mechanism studies have revealed that the cobalt catalyst underwent glyoximate ligand exchange during photocatalysis.75 Such an exchange was not expected due to the bidentate nature of these ligands as well as the H-bonding between the glyoximate ligands that holds the two monoanionic chelates in the equatorial positions of a tetragonally distorted octahedral complex. To solve this problem, the addition of extra dmgH2 ligands (dmgH = dimethylglyoxime) or triphenylphosphine (PPh3) into the system as well as the replacement of the H bridges with BFx bridges in cobaloxime complexes was used to significantly improve the stability of the photosystems.76,77
Different substitutes of axial pyridine ligation in cobaloxime complexes can not only tune their catalytic performance, but also provide more possibilities for the construction of hybrid systems with semiconducting photosensitizers. Chen et al. fabricated CdSe/ZnS core/shell QDs functionalized with a cobaloxime catalyst via a phosphonate linkage.78 Transient spectra show that the electron transfer from the light-excited QDs to cobaloxime occurs within an average time scale of 105 ps, followed by a much slower carrier recombination process that takes more than 3 ns. This long-lived charge separation state enables the hybrid system to produce more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 turnovers of H2 per QD in 10 h when using TEOA as SED.
000 turnovers of H2 per QD in 10 h when using TEOA as SED.
HECs based on redox non-innocent ligands like sulfur donors are also very popular for photocatalytic H2 evolution.79 As shown in Fig. 10, Kisch and coworkers reported the first photocatalytic H2 evolution system with nickel dithiolene complex 13.80 This kind of complex has a bioinspired aspect, since the nickel ion has mixed N/S ligation as found in [NiFe]-H2ase (Fig. 10 complex 14). The redox processes may occur on both the ligands as well as the metal centre, making such complexes potential reservoirs of charge in the multielectron processes. Additionally, ligand sites corresponding to S and/or N donors are available for protonation, thus allowing these systems to assemble two protons and two electrons to make H2 from stepwise electron-transfer and protonation. Examples of such cobalt- and nickel-based dithiolene catalysts were both systematically studied in photocatalytic systems with excellent H2 evolution efficiency.81,82
Moreover, cobalt complexes stabilised by other nitrogen donor ligands have also shown remarkable photocatalytic performance in aqueous systems.83 For instance, polypyridyl scaffolds as ligands not only provide desired stability to the low-valent Co metal centre throughout the catalytic cycle, but also minimize structural reorganization within the molecular architecture. In 1981, Sauvage et al. reported the first photosystem based on a [Co(bpy)n]2+ (n = undefined) complex for H2 evolution, where a Co(I) bipyridine intermediate was detected.84 Nowadays, polypyridyl–cobalt complexes are a diverse family featuring pyridine- and bpy-derived tetra- and penta-dentate ligands.85,86 Castellano et al. evaluated ten distinct Co2+-based polypyridyl catalysts for light induced H2 evolution using high-throughput screening with [Ru(bpy)3]2+ as the PS and H2A as the SED.87 They proposed that the complexes with tetradentate ligands yield more active photocatalytic compositions than their counterparts bearing pentadentate ligands with high catalytic stability. Moreover, molecules with cis-open coordination sites seem to be impressively more active than those with trans-open sites, providing guidelines for the development of molecular compositions. Very recently, three water-soluble polymer catalysts with appending Co porphyrins and different side-chain groups were synthesized to mimic the natural enzyme reaction centre and to explore the effect of residual groups on the catalytic performance.88 Attributed to the strong interaction between the positively charged polymer chain and the negative CdSe QDs after protonation, the highest photocatalytic activity of 2.7 × 104 TON came from the catalyst with amino side-chain groups.
As mentioned above, the activity, stability, and even molecular solubility of the pre-synthesized metal complexes can be easily tuned through rational structural design for artificial H2 production. Here, it is worth noting that the metal catalysts formed in situ also show great potential for their convenient preparation and satisfying catalytic activity. One typical system was proposed by Eisenburg and coworkers, who proposed that the DHLA dissociated from CdSe QDs may coordinate with Ni2+ to form a soluble nickel(II)-DHLA species to serve as the real catalytic center.89 Simultaneously, Wu and coworkers constructed an artificial photosynthetic system by directly adding Co2+ ions into MPA-functionalized CdTe QD solutions.90 Upon irradiation, the dangling bonds (S2− from photogenerated hole oxidation of MPA) on the surface of QDs interact with Co2+ ions for H2 generation with remarkable activity. This methodology can be adapted to other earth-abundant metal centres, including Ni2+, Cu2+, Fe2+ and Mn2+, where nickel ions showed the highest catalytic activity of a TON up to 137500.91 Subsequent characterization studies provided direct evidence of photoinduced electron transfer (PET) from QDs to Ni2+ active sites.92
Furthermore, the metal complex as the catalysts for proton reduction and water oxidation can be not only used in a homogeneous system, but also integrated onto the electrode surface of PEC cells. For example, by using a low-cost cobalt porphyrin complex (CoTCPP) as the catalyst on the surface of a three-dimensional BiVO4 film, a 2-fold enhancement of photocurrent density was observed.93 Another typical work was proposed by Reisner et al. in 2016 as shown in Fig. 11. A phosphonic acid-modified nickel(II) bisdiphosphine catalyst (NiP) and an Fe(II) catalyst based on a phosphonic acid-modified tris(2-picolyl)amine (TPA) ligand (FeP) were loaded on the surface of a TiO2 photocathode and WO3 photoanode as the reduction and oxidation catalysts, respectively.94 Water splitting with an approximate 2:1 ratio of H2:O2 observed shows the possibility of using a metal complex as the catalyst to realize the overall water splitting in PEC cells. It should be mentioned that the current state of O2 evolution complexes in photocatalytic systems lags far behind that of H2 evolution catalysts. The mismatch of catalytic performance limits the further integration and development of solar-driven overall water splitting. Presently, there is an urgent need to know how to improve the binding stability, increase the loading amount of catalysts, and promote the interaction between the light absorber and the catalyst at the electrode interface.
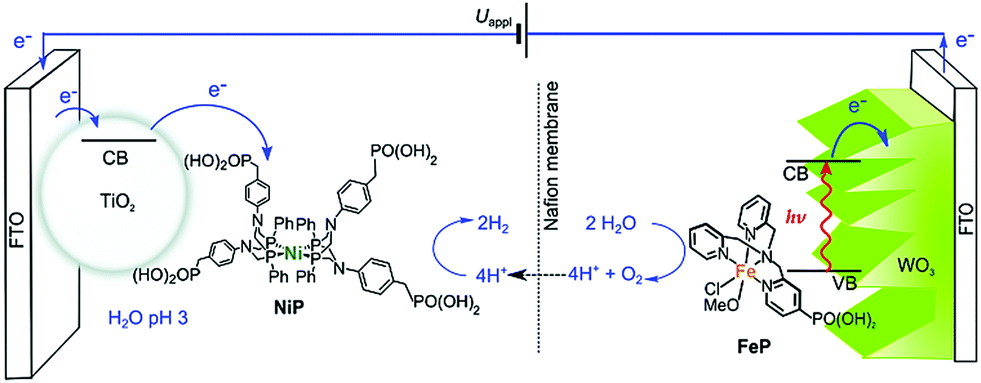 | ||
| Fig. 11 Scheme of PEC water splitting with the TiO2|NiP hydrogen evolution cathode wired to the WO3|FeP oxygen evolution photoanode. Reprinted with permission from ref. 94. Copyright 2016 Royal Society of Chemistry. | ||
Molecular CO2 reduction catalysts
Unlike anaerobic bacteria that use hydrogenases to reduce protons for H2 evolution, most green plants utilize the protons and electrons released by PSII to fix CO2 in the atmosphere, thus providing energy for life on Earth and maintaining the balance of carbon and oxygen in the atmosphere. Mimicking the natural processes to convert the greenhouse gas CO2 into valuable chemicals and fuels using solar energy is of great significance to ease the growing energy crisis and excessive CO2 emission.95 Unfortunately, the multielectron reduction of CO2 by artificial photochemical transformation often shows poor selectivity with a variety of products, including carbon monoxide, formic acid, formaldehyde, methanol, etc.96 In particular, reduction in aqueous solution involves a serious competition process of H2 production. Therefore, appropriate catalyst design is often unavoidable to minimize the energy barriers of the overall process and control the selectivity of the products.97Although the inherent thermodynamic stability of CO2 makes its reductive activation challenging, a variety of enzymes can interact with CO2. For instance, [NiFe]-CODHs (CODHs-carbon monoxide dehydrogenases) are metalloenzymes present in many anaerobic microorganisms that catalyse the reversible conversion of CO2 to carbon monoxide (CO).98 The active centre in [NiFe]-CODHs is a [Ni–4Fe–5S] cluster, as shown in Fig. 12a. Crystal analysis after incubation with NaHCO3 resulted in a structure consistent with the binding of CO2. Notably, a two-atom bridge with Ni–C and Fe–O bonds could be detected (Fig. 12b). Both CO2-derived O atoms are closely associated with the two nearby protein residues. Plausible mechanisms for CO2 conversion include the formation of a {NiIIFeII(OH)} species and the reduction to afford a Ni0FeII intermediate.99,100 In this mechanism, a metal-built frustrated Lewis pair instead of the CO2 radical anion, is considered to be responsible for the selective transformation of CO2 to exclusively afford CO, with a TOF of 0.194 s−1.101
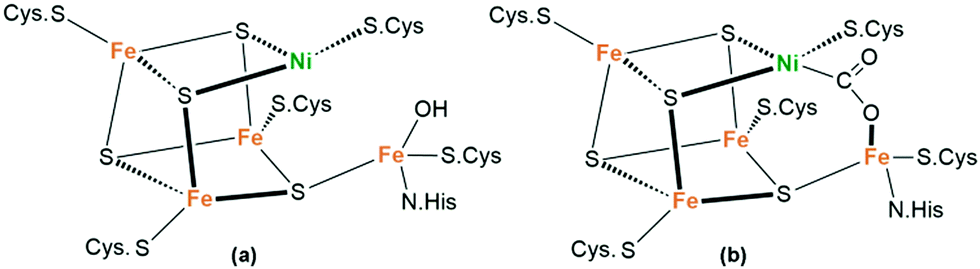 | ||
| Fig. 12 Structure of the [NiFe]–CODH active site with (a) a reduced nickel center in the absence of a substrate and (b) bound CO2 after incubation with NaHCO3. | ||
In 2010, a CO2-reducing enzyme CODH I from the anaerobic microbe Carboxydothermus hydrogenoformans (Ch) was firstly introduced to an artificial CO2 photoreduction system by Armstrong and coworkers.102 In the presence of [Ru(bpy)3]2+ modified TiO2 nanoparticles, the sensitized hybrid enzyme–nanoparticle system could catalyse a controlled, two-electron reduction giving CO as a clean product, which shows the feasibility of constructing artificial photosystems using biological enzymes. However, the sensitivity of natural enzymes requires all the preparation stages of their system in an anaerobic glove box. If artificial catalysts can be used to simulate the function and activity of these enzymes, it is undoubtedly an attractive research direction. In this respect, transition metal complexes have the advantages of their easily tuned redox potential, trapping of CO2via coordination to the metal centre, and valence jump of the metal oxidation state in response to the multielectron reduction processes.
Fig. 13 shows the competing pathways for CO and HCOOH formation from CO2 at a metallic centre Mn+,103 in which the metal-hydride intermediate influenced the final product distribution of CO2 reduction. In the first pathway, CO2 gets inserted into the metal-hydride via the electrostatic attractions between the polarized O–C and M–H bonds, setting the electrophilic C atom near the nucleophilic hydride moiety. Alternatively, electron-rich metals with free binding sites tend to activate CO2 in the form of monodentate C-coordination, leading to a metallo–carboxylate species with O-atoms sterically available for protonation. Generally, the insertion method often affords HCOOH as a product, whereas the coordination method tends to yield CO.
 | ||
| Fig. 13 The competing pathways for CO and HCOOH formation from CO2. | ||
In the 1980s, Lehn et al. used Re(I) complexes [fac-Re(bpy)(CO)3L (bpy = 2,2′-bipyridine; L = Cl− or Br−)] to achieve the first selective CO2 reduction to CO under UV irradiation.104 Since then, metal complexes with single-metal or bi-metallic centres involving Re, Ru, Ni, Co, Mn, and Fe have been widely examined as catalysts for CO2 photoreduction.105,106 Like the photoinduced electron transfer processes that exist in NP, the metal complex catalyst can be covalently bonded to photosensitizer units to form supramolecular photocatalysts for efficient intramolecular electron transfer. To this end, supramolecular photocatalysts such as Ru–Re, Ru–Ni binuclear or even multinuclear complexes have been constructed.107 Detailed investigations showed that the type and the length of the bridging ligands, the redox potential of both the catalytic and photosensitive units, as well as the types of substituents can be reasonably controlled.108 According to Ishitani and coworkers, covalently bridged Ru(II) multinuclear complexes composed of both photosensitizers and catalysts were able to photocatalyze the reduction of CO2 to formic acid (HCOOH) with NADH model compounds as the SED in a mixed solution of dimethylformamide (DMF) and TEOA.109 The ratio between the photosensitizer units and catalyst units strongly affected the photocatalytic performance. For a trinuclear supramolecular composed of two photosensitizer units and one catalyst unit, a TOF of up to 11.6 min−1 and corresponding 671 TON can be measured. Noncovalent interactions, such as hydrogen bonding, have also been found to be able to construct supramolecular systems based on metal complex catalysts.110
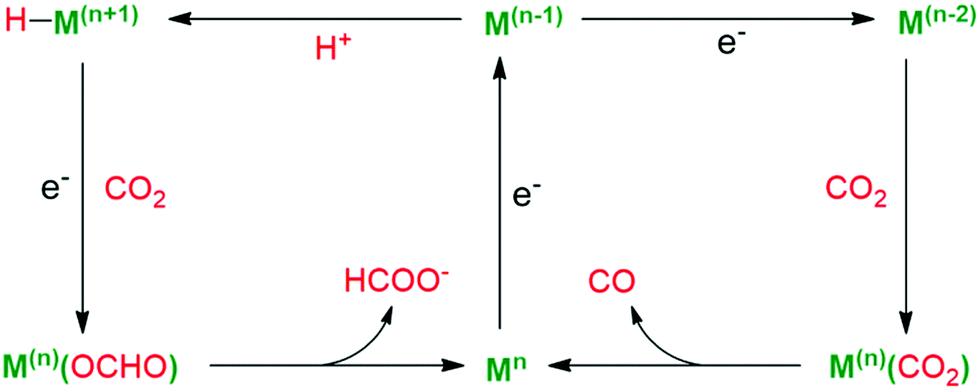
Multicomponent systems with separated photosensitizers and catalysts have been reported based on well-designed metal complex catalysts. The unique [Ni–4Fe–5S] cluster of [NiFe]-CODHs provides a critical design of homogeneous catalysts. Considering the evidence proving the importance of sulfur ligation for CO2 reduction in CODHs, a Ni complex bearing an S2N2-type tetradentate ligand was synthesized and investigated as a catalyst for CO2 photoreduction (complex 15 in Fig. 14) by Kojima et al.111 With [Ru(bpy)3]2+ as the photosensitizer and BIH as the SED, a QY of up to 1.42% was obtained. Inspired by the natural RuBisCo using Mg ions as a Lewis-acidic active site for efficient CO2 fixation, two noncoordinating pyridine pendants were further introduced into the above catalyst, which act as “artificial arms” to capture Mg2+ ions in the vicinity of the Ni center (complex 16).112 With [Ru(bpy)3]2+ as the PS and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as the SED, a higher quantum yield (QY) for CO production could be obtained, as compared to that of their counterparts without Lewis-acid capturing sites. Mass spectrometry and 1H NMR measurements confirmed the generation of Mg2+-bound Ni complex 17. DFT calculation shows that the cooperatives between Ni and Mg centres for the stabilization of a Ni–CO2 intermediate is the key to increase the catalytic efficiency of the system.
 | ||
| Fig. 14 Ni complexes for CO2 photoreduction bearing an S2N2-type tetradentate ligand as the photocatalyst and a Ni(II) complex bearing an S2N2-tetradentate ligand with two noncoordinating pyridine pendants as the photocatalyst. | ||
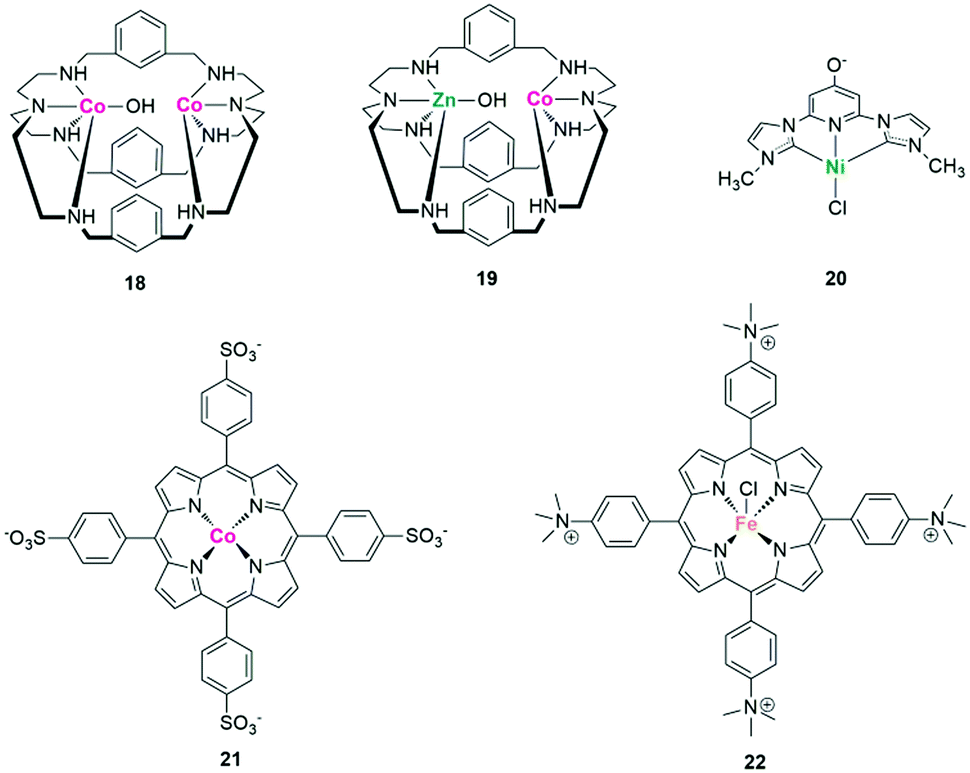
Through rational molecular design of the metal centres and ligands, the catalytic activity, selectivity and even the reaction mechanism can be tuned and promoted. Lu et al. proposed that by replacing one CoII in the dinuclear homometallic [CoCo(OH)L1](ClO4)3 complex 18 (CoCo) with a ZnII unit, the resulting dinuclear heterometallic CoZn catalyst 19 showed a much improved activity for photochemical CO2 reduction (Fig. 15).113 The enhanced performance was attributed to the strengthened dinuclear metal synergistic catalysis. That is, the CoII acts as the catalytic centre and the ZnII serves as the assistant catalytic site to strongly bind with OH−, thus greatly promoting the C–OH cleavage of the OC–OH intermediate.
 | ||
| Fig. 15 Representative structures of several CO2 photoreduction complex catalysts. | ||
In addition to considering the influence of metal centres, the regulation of ligands greatly influences the photocatalytic performance. Ishida et al. found that the amide groups at the 5,5′-positions of 2,2′-bipyridyl ligands (L) affect the reduction potential of the trans-(Cl)-[Ru(L)(CO)2Cl2]-type complexes.114 The C-connected amide group is twisted with respect to the bipyridyl plane, whereas the N-connected counterpart is within the plane. DFT calculations show that the slight twist of the former raises its LUMO level, causing a negative shift of the first reduction potential (Ep) of the Ru-based catalyst. Correspondingly, the logarithm of the TOF value increases with the negative shift of Ep until it reaches the reduction potential of the [Ru(bpy)3]2+ photosensitizer. Even a remote substituent within the complex catalyst also significantly controls visible-light-driven CO2 reduction. Papish et al. found that for a nickel(II) pincer complex 20 with C–N–C ligands, the introduction of the π electron donor group (O−) on the para-position of pyridine resulted in greatly enhanced CO2 photoreduction performance (Fig. 15).115 Protonation, however, turns off its catalytic activity, highlighting the importance of selecting rational remote-substituents.
The improvement of product selectivity and inhibition of H2 production are some of the most challenging issues in the field of CO2 photoreduction, especially in pure water. A water-soluble cobalt porphyrin complex 21 ([{meso-tetra(4-sulfonatophenyl)porphyrinato}-cobalt(III)], CoTPPS) was synthesized by Sakai et al. as a catalyst for the photoreduction of CO2 in fully aqueous media, which shows a selectivity to CO up to 91% (Fig. 15).116 Preparation of syngas, a mixture of CO and H2 for methanol production, is another way to solve the competitive process of proton reduction. By anchoring both a CO-producing ReI catalyst and a hydrogen-evolution CoIII catalyst onto TiO2 particles, a ternary hybrid catalyst was established for photocatalytic syngas production.117 Due to the controllable loading mode and amounts of molecular catalyst, the ratio of H2 to CO in the formed syngas can be widely tuned from 1:2 to 15:1 by changing the ratio of the two catalysts.
Considering the product distribution, it has attracted much attention to control the types of products through molecular structure regulation. According to Robert and coworkers, an iron tetraphenylporphyrin (TPP) complex 22 functionalized with tetramethylammonium groups was a highly efficient catalyst to realize the eight-electron reduction of CO2 to CH4 with an Ir-dye as the PS and TEA as the SED (>420 nm, Fig. 15).118 CH4 was achieved via a successive two-step reduction, in which CO2 was firstly reduced to CO and then to CH4. Mechanism studies show that the initial product CO was connected to FeII and further reduced to CH4via a postulated FeI–formyl (FeI–CHO) intermediate, which can be greatly stabilized through the space interactions between the positive charged tetramethylammonium groups and the partial negative charges on the CHO species bound to the metal. The non-substituted tetraphenyl Fe porphyrin, however, only gives CO and H2 under the same irradiation conditions, possibly due to its much more negative redox potentials and the absence of phenyl ring substitution to stabilize the intermediates involved in hydrocarbon production.
Transition metal complexes are also good candidates to couple with a variety of materials, e.g., semiconductors, carbon materials, polymers and mesoporous organosilicas.119 By anchoring the catalyst, [Ru(dcbpy)(bpy)(CO)2]2+ or [Ru-(dcbpy)2(CO)2]2+ to an N-doped Ta2O5 semiconductor, Motohiro et al. fabricated a hybrid CO2 photoreduction system in a CO2-saturated MeCN/TEOA (5:1) solution.120 The selectivity for visible-light-induced reduction of CO2 to HCOOH was more than 75% and the QY was 1.9% at 405 nm. They proposed that the linkage between the complex and the semiconductor is essential for efficient electron transfer from the excited state of the semiconductor to the metal complex. Very recently, Robert and coworkers constructed a hybrid CO2 photoreduction system with only earth-abundant elements via covalent grafting a Co-quaterpyridine complex to the surface of g-C3N4 through an amide linkage, as shown in Fig. 16a.121 Visible light irradiation of the molecule–material hybrid in the presence of BIH furnished CO with impressive selectivity (98%) and long-term durability, since no degradation was observed after 4 days of visible light illumination.
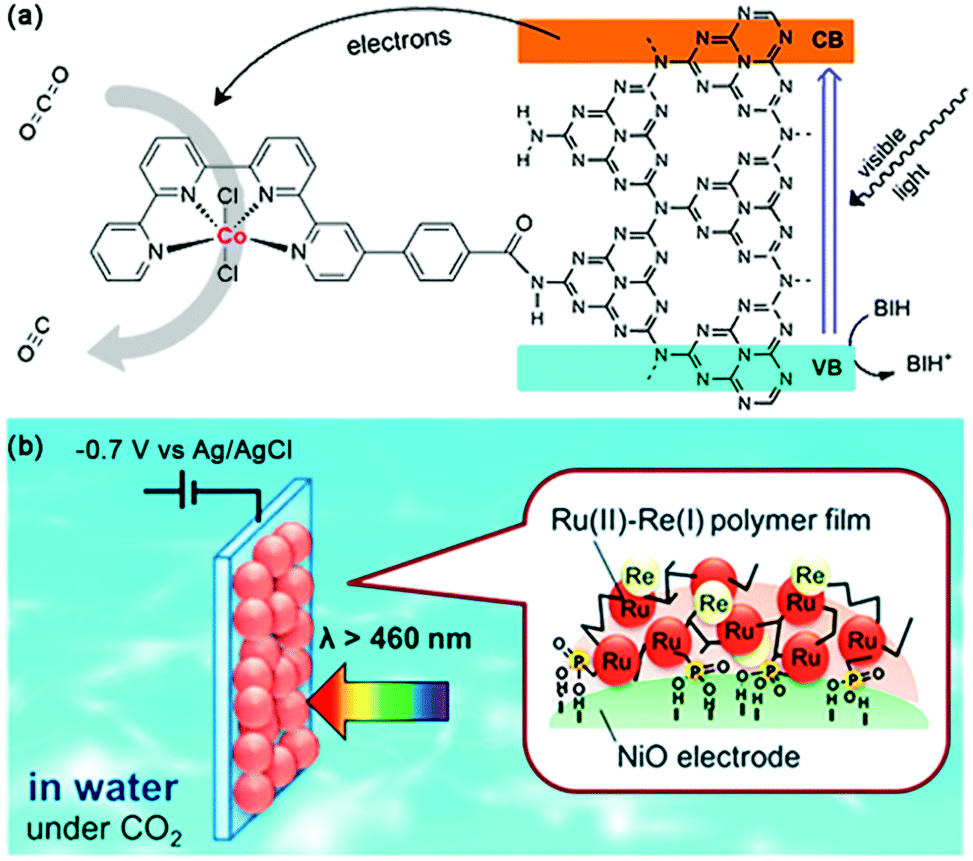 | ||
| Fig. 16 (a) Visible-light-driven CO2 reduction by a cobalt complex covalently linked to mesoporous carbon nitride. Reprinted with permission from ref. 121. Copyright 2020 American Chemical Society. (b) PolyRuRe/NiO photocathode for CO2 photoreduction. Reprinted with permission from ref. 124. Copyright 2019 American Chemical Society. | ||
By introducing molecular catalysts to the electrode surface, CO2 reduction can also be coupled with water oxidation, just like photosynthesis in nature.122 Different methods can realize the loading of these reduction catalysts to the photocathode surface. At present, the well-developed method is chemical bonding. Grätzel et al. introduced a phosphonated Re(bpy)(CO)3Cl catalyst to the surface of a light-harvesting p-type Cu2O electrode via an interfaced nanostructured TiO2 scaffold for CO2-to-CO conversion.123 Compared to the corresponding planar devices, a 40-fold enhancement of the catalytic photocurrent could be observed based on the mesoporous-TiO2-modified Cu2O photocathode, representing the first demonstration of utilizing phosphonate linking groups to immobilize molecular catalysts onto the surface of the photoelectrode. Recently, Ishitani et al. developed a novel immobilization method based on electrochemical polymerization for a polyRuRe/NiO photocathode bearing both a Ru(II)–Re(I) supramolecular photocatalyst and a Ru(II) tris-diimine photosensitizer (Fig. 16b).124 This strategy was considered to increase the amount of metal complex adsorbed on the electrode surface while also suppressing desorption. CO generation was enhanced by a factor of 2.5 under visible light irradiation than that of the electrode bearing only methyl phosphonic acid anchors and an improved FE (from 57% to 85%) was also detected at −0.7 V vs. Ag/AgCl.
Recently, a tandem PEC cell was reported by Reisner and coworkers who can realize bias-free solar syngas production.125 Commercially available cobalt(II) meso-tetrakis(4-methoxyphenyl) porphyrin (CoMTPP) was firstly immobilized via π–π stacking interactions onto carbon nanotube (CNT) sheets. Then the composite was integrated onto the surface of a perovskite-based photocathode. By using an n-type BiVO4 film as the photoanode, the perovskite–BiVO4 PEC tandem device could realize bias-free syngas production coupled to water oxidation for three days under 1 sun irradiation in an aqueous CO2 saturated solution. The corresponding conversion efficiencies for solar-to-H2 and solar-to-CO are 0.06 and 0.02%, respectively. Similar to the PEC water splitting systems, the PEC CO2 conversion is also limited by the sluggish kinetics of water oxidation. Developing catalysts with high selectivity, high efficiency and high stability requires a deeper understanding of the catalytic mechanisms. At present, most reported complex catalysts can only show good catalytic performance in CO2 saturated systems. How to achieve efficient photoconversion of CO2 in a low concentration or even an atmospheric atmosphere may be one of the directions that need to be paid attention to in the future.
Molecular organic transformation catalysts
As mentioned before, while photosynthesis in nature oxidizes water to produce O2, the released protons and electrons can be used for reductive H2 evolution and overall water-splitting. Artificial photosynthesis reproduces the natural chemical transformation in a redox-neutral way (reactions with no external reducing or oxidizing agent). Water oxidation occurring at the photoanode can be coupled with proton reduction or CO2 reduction at the photocathode. Taking one step further, artificial photosynthesis may use photogenerated carriers to catalyse redox-neutral chemical reactions in a one-spot reaction (two or more steps taken in one reactor for the synthesis). This strategy can overcome the consumption of sacrificial reagents in traditional transition-metal catalysis. One typical example is proposed, as shown in Fig. 17, to generate a persistent ketyl radical and a transient α-carbonyl radical in situ from aromatic β-ketoesters using a fac-Ir(ppy)3 photocatalyst under visible-light irradiation.126 The persistent ketyl radicals can undergo pinacol coupling, while the transient acarbonyl radical can be intercepted by an alkyne to afford a series of highly substituted 1-naphthols in good to excellent yields. Remarkably, this photocatalytic method avoids the normal requirement for stoichiometric external oxidant and reductant. The easy-to-get photocatalyst, broad substrate scope, mild reaction conditions and high functional-group tolerance make this reaction a useful synthetic tool.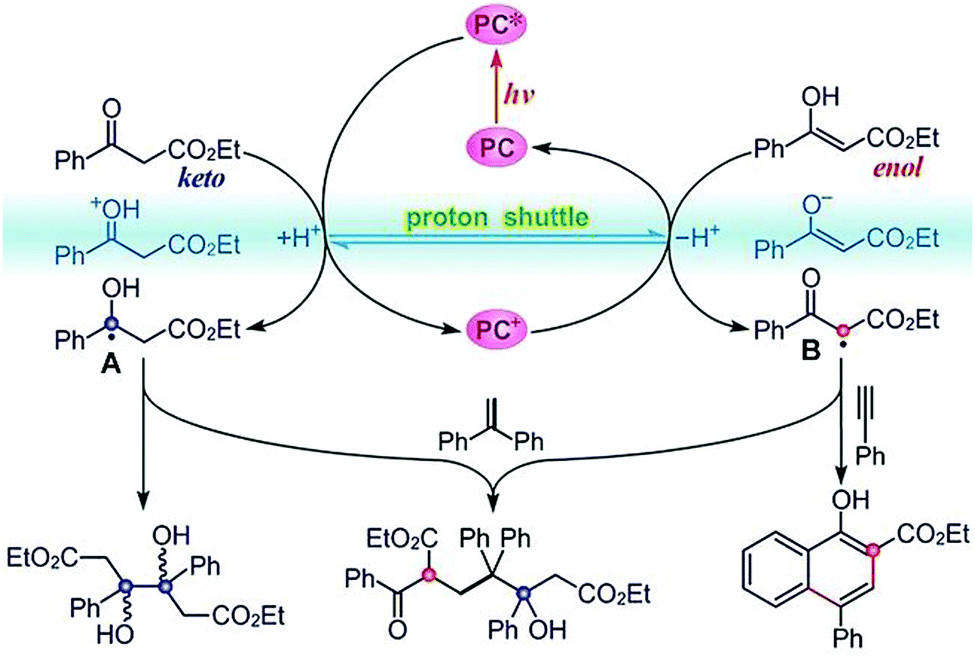 | ||
| Fig. 17 Photoredox catalysis of aromatic β-ketoester for the in situ production of transient and persistent radicals for organic transformation. Reprinted with permission from ref. 126. Copyright 2020 Wiley-VCH. | ||
The redox-neutral reaction design provides enlightenment for the construction of chemical bonds, especially the activation of inert bonds. As an important bond-forming reaction, the oxidative cross-coupling reactions have been recently considered as a powerful and straightforward tool to realize the direct C–H functionalization for accessing carbon–carbon (C–C) or carbon–heteroatom (C–X) bonds.127,128 However, the catalytic conversion often requires the addition of stoichiometric sacrificial oxidants to activate the inert C–H or X–H bond and remove H atoms, which is not suitable for the substrates sensitive under oxidative conditions and does not meet the requirements of atom economy (converting as much of the reactants as possible into the final products) and sustainable chemistry. In this regard, a new reaction called cross-coupling with H2 evolution (CCHE) was proposed.129,130 A typical CCHE system combines a photocatalyst with a proton reduction cocatalyst to create a dual catalyst system. After irradiation, the former initiate electron transfer to generate the radical cation or radical anion of the substrate, while the latter captures electrons from the substrates or reaction intermediates to reduce the protons eliminated from the reactive scaffolds (C–H/C–H or C–H/X–H bonds) into H2. This bond formation strategy not only avoids the use of sacrificial oxidants but also releases H2 gas as the sole byproduct, which can be further used as an efficient energy source.
The pioneer work in this field was reported by Wu and coworkers in 2013 by using EY and a graphene-supported RuO2 (G-RuO2) nanocomposite as the PS and catalyst, respectively.131 Catalysed activation and functionalization of the C(sp3)-H bonds adjacent to amino groups was achieved via CCHE reaction with various tetrahydroisoquinoline derivatives and indole derivatives under visible light irradiation. Later on, the first cobalt catalysed CCHE reaction, where a Co(dmgH)2Cl2 molecule was used to capture the electron and proton for cross-coupling reactions between a variety of isoquinoline and indole substrates, and H2 was the only by-product (Fig. 18a).132 Mechanistic insights revealed that, an efficient CCHE reaction relies on the successive electron transfer from the isoquinoline substrate to the excited [eosin Y]* to generate a [eosin Y]˙− radical anion, which then delivers the electron to Co(dmgH)2Cl2 for proton reduction to H2. Since then, photocatalytic CCHE reactions have been widely reported to functionalize less reactive C(sp3)–H and C(sp2)–H bonds for C–C bond construction. Because of its powerful ability for both proton reduction and hydrogen atom transfer, the family of cobaloxime complexes has become widely used as cocatalysts in this field.133
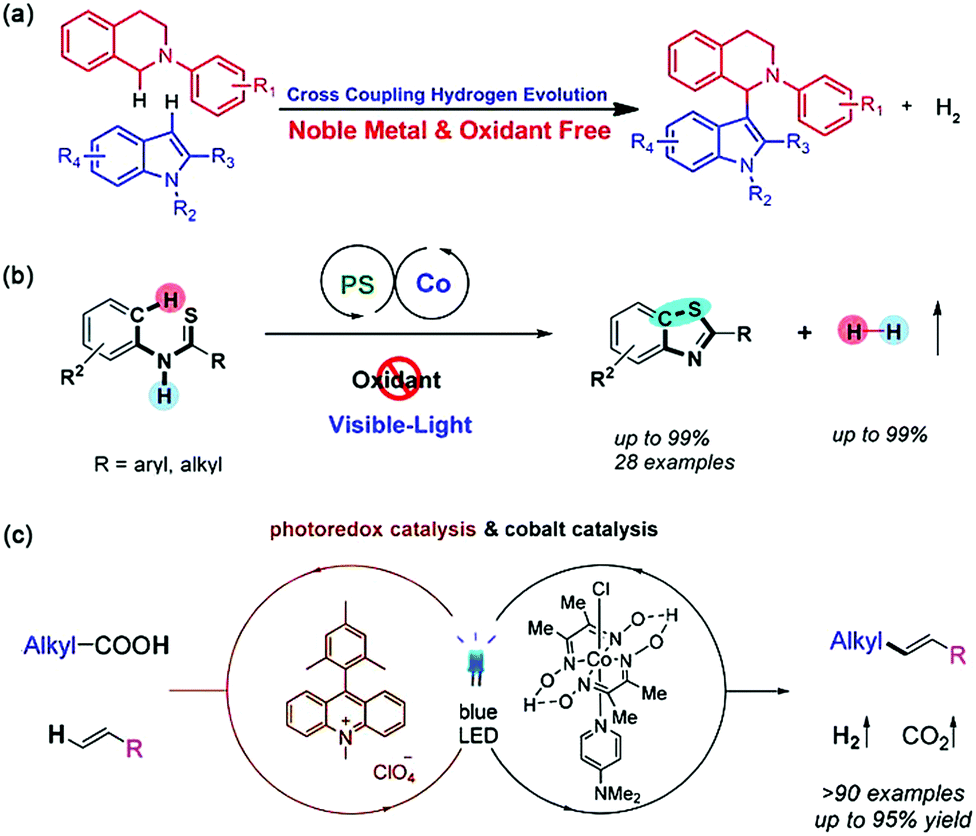 | ||
| Fig. 18 (a) The first cobalt complex catalyzed CCHE reaction between isoquinoline and indole substrates. Reprinted with permission from ref. 132. Copyright 2014 Wiley-VCH. (b) Oxidant-free aromatic C–H thiolation to construct C–S bonds. Reprinted with permission from ref. 136. Copyright 2016 American Chemical Society. (c) Schematic diagram for the decarboxylative Heck-type coupling. Reprinted with permission from ref. 140. Copyright 2018 American Chemical Society. | ||
Two ideal reactions were reported using this strategy: one-step amination of benzene to aniline and one-step hydroxylation of benzene to phenol with the combination of QuH+ (1-methylquinolinum ion) or QuCN+ (3-cyano-1-methylquinolinum ion) as the photocatalyst and a cobalt complex as the H2 evolution catalyst.134 Aniline was synthesized from the reaction of benzene with ammonia while phenol from the reaction of benzene with water. An equivalent amount of H2 was the sole byproduct in these reactions. Both of them are proposed to proceed via nucleophilic addition to the arene radical cation followed by the cobaloxime-facilitated removal of one electron and two protons. With excellent yields and selectivity, this strategy provides a blueprint for aromatic C–H functionalization under unusually mild conditions with no sacrificial oxidants. Kinetic study reveals that the arene radical cation species serve the key role for the C–H/N–H cross-coupling. With Ru(bpy)3(PF6)2 as the photocatalyst and Co(dmgH)2Cl2 as the cocatalyst, a variety of N-arylazoles were successfully synthesized with H2 evolution, whereas C(sp3)–H bonds were unaffected.135 Selective C(sp2)–H bond amination of arenes was also achieved by utilizing heterocyclic azoles as nitrogen sources.
Intramolecular or intermolecular C–C/C–X bond construction for building valuable five/six-membered heterocyclic compounds can be achieved by photocatalytic CCHE reactions. In 2016, Wu and coworkers reported an intramolecular oxidative cyclization between aromatic C–H bonds and enamine C–H bonds of N-aryl enamines into indoles with good to excellent yields under visible light irradiation (Fig. 18b).136 Mechanistic investigations reveal that the system proceeds via visible-light-catalysed oxidation of the enamines, followed by intramolecular radical addition to yield the indoles smoothly. Another intramolecular photocatalytic CCHE reaction was reported by Lei and Wu, which can realize the C–H functionalization/C–S formation to form benzothiazoles with no external oxidant.137 With both the Ru(bpy)32+ photocatalyst and cobaltoxime cocatalyst, N-phenylthioamides were transformed to 2-aryl- or 2-alkylbenzothiazoles with H2 as the only side product. The unexpected oxidation byproduct amides, which are often generated in oxidative cyclization of thiobenzanilides, can be completely avoided. Using a photo/cobaloxime dual catalytic system, the intermolecular photocatalyzed dehydrogenative [4+2] annulation between styrene derivatives with electron-rich dienophiles can also be achieved to form poly-substituted aromatics.138 The in situ generated alkene radical cation species were proposed as the key intermediate for the annulation reaction with good regioselectivity.
Due to the unique features of cobaloxime complexes to form alkyl cobalt derivatives as well as the tendency to generate alkyl radicals, diverse types of reactions can be designed using these metal complexes.139 Accordingly, Wu et al. proposed a room temperature Heck-type coupling using inactivated aliphatic acids and terminal alkenes as feedstocks (Fig. 18c).140 The scope of this reaction was quite broad in that more than 90 olefins were synthesized in good to excellent yields. A radical-based mechanism was proposed. That is, in the presence of a base, the excited photosensitizer ([Mes-Acr-Me+]*) converted the substrates of carboxylic acids to alkyl radicals. Then, a radical addition to the alkenes occurred and subsequently a β-H abstraction by a planar Co(II) intermediate formed from the cobaloxime catalyst resulted in the formation of Heck-type products.
Furthermore, a photoinduced asymmetric cross-dehydrogenative coupling reaction between tertiary amines and simple ketones was achieved using a synergistic tri-catalytic hydrogen-transfer strategy.141 In combination with a Ru(bpy)3(PF6)2 photocatalyst and a Co(dmgH)2pyCl catalyst, a chiral primary amine catalyst was used to react with ketones for chiral enamine production. With nitrobenzene derivatives as the hydrogen acceptor, the synergistic effect between the photocatalyst and cobaltoxime catalyst was able to transform tertiary amines into iminium ions. Cross-coupling products with high chemo-, region- and enantio-selectivity were obtained through the reaction between the in situ generated iminiums and enamines.
Generally speaking, introducing phosphine groups into molecules can significantly improve their bioavailability as well as the intrinsic optical and electronic features. Recently, Wu and coworkers developed a straightforward and atom economic strategy to synthesize alkenylphosphine oxides via a free radical pathway (Fig. 19).142 Different from the well-established photocatalytic systems, this reaction uses cobaloxime as the only photocatalyst to initiate the photocatalytic reaction of H-phosphine oxide [P(O)–H] with alkene and alkyne to selectively produce alkenylphosphine oxides in good to excellent yields. No external photosensitizer and sacrificial oxidant were needed. Mechanistic studies showed that, the excited cobaloxime complex could activate [P(O)–H] into its reactive radical species phosphinoyl radical [P(O)·], which was subsequently utilized to functionalize alkenes and alkynes. Meanwhile, the electron and proton eliminated from the P(O)–H functionality could be captured to form intermediates such as CoIII–H or CoII for a hydrogen transfer reaction. The formation of both the phosphinoyl radical and cobalt intermediates could be experimentally detected, which reveals the value-added potential of cobaloxime in photocatalysis.
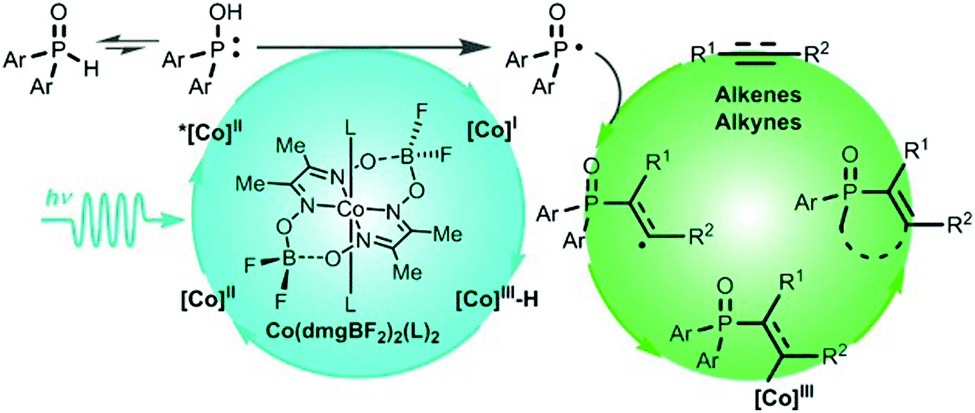 | ||
| Fig. 19 Plausible mechanism for the reaction of H-phosphine oxide and terminal alkene. Reprinted with permission from ref. 142. Copyright 2019 American Chemical Society. | ||
Conclusions
Inspired by NP, artificial photosynthetic systems based on oxidation or reduction half reactions have been widely considered by researchers for efficient solar-to-chemical energy conversion. Once optimized, the oxidative processes, such as water oxidation, can be coupled to reductive reactions like H2 evolution or CO2 fixation, enabling a more practical small molecule transformation. More importantly, the mimicry of natural processes provides researchers with important insights into organic synthesis, particularly the activation of inert bonds. In this respect, catalysts play a crucial role as NP produces various enzymes as catalytic active sites to achieve molecular conversion.Lessons learned from NP have stimulated researchers to design abundant metal complexes as promising catalysts for energy-related photocatalytic small-molecule transformation. At present, metal complexes can be facilely tuned on the molecular scale via metal centers, ligands, functional substitutes, binding sites, and co-ordination number, thus regulating their photophysical, redox, and catalytic properties. The metal centres not only provide suitable binding sites for the reactive small molecules, but also show specific selectivity to certain substrates, which is important for artificial photosynthesis. Compared with these well-developed non-noble metal complexes used in photocatalytic H2 production, Ru, Re and other precious metals are still outstanding in the field of CO2 photoreduction and water photooxidation. From an economic and sustainable viewpoint, high-performance catalysts based on non-noble metals should be further developed, although the troublesome challenges are that a higher overpotential and stronger basic conditions are often required, under which conditions some of these low-cost catalysts decompose into metal oxides to serve as the true catalysts. In addition to the metal center, the flexible ligand-tunability provides more possibilities for the application of metal complexes in the field of small molecule transformation. How to design and synthesize ligands with higher performance under the guidance of a catalytic mechanism is a prerequisite in catalyst design.
In NP, the secondary coordination spheres are derived to protect the catalytic centers as well as to input substrates and output products. Artificial photosystems have been focused on exploring the influence of secondary coordination effects on the catalytic activity of systems based on metal complexes. The next step should pay more attention to the interactions between the catalysts and other components like photosensitizers, sacrificial reagents and other additives, similar to the well-organized integration of light absorber, charge transfer chains and reaction units in natural photosynthesis. Furthermore, the strategy of building a heterostructure through integrating with inorganic materials such as semiconductors should be considered for their merits such as light absorption, charge separation and stability.
In addition to developing more efficient complex catalysts through structural and functional optimization, theoretical exploration and in situ characterization of catalytic mechanisms, carrier dynamics, and inactivation processes are also required to deepen the mechanistic understanding of the catalytic process. On one hand, these theoretical analyses can support the experimental results, and more importantly, they may guide the design and synthesis of molecules as well as the system construction.
From the perspective of small molecule transformation, the artificial photosynthesis offers a shortcut to take advantage of the efficient principles of the primary photosynthetic reaction. The greater diversity in molecular design of metal complexes and the smaller components than their natural counterparts in artificial systems have higher potential for small molecule activation. Photochemical methods, coupled with time-resolved spectroscopy and theoretical analysis, provide unique opportunities to identify and monitor intermediates in the reaction cycle. The kinetic measurements can shed light on rate-determining processes and the mechanism as a whole. Once optimized, the catalytic conversions of oxidation and reduction can be coupled to get rid of the dependence on sacrificial reagents. At present, the idea has been realized by constructing photoelectrochemical cells and CCHE reactions. However, the mismatch between the catalytic performance of the oxidative and reductive reactions are still far from meeting the requirements of practical applications. Challenges and opportunities require and stimulate further exploration in bioinspired metal complexes to achieve more efficient, stable, low-cost and environmentally friendly artificial photosynthetic systems for a bright future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Ministry of Science and Technology of China (2017YFA0206903), the National Natural Science Foundation of China (22088102 and 21861132004), the Strategic Priority Research Program of the Chinese Academy of Science (XDB17000000), the Key Research Program of Frontier Sciences of the Chinese Academy of Science (QYZDY-SSW-JSC029), and the K. C. Wong Education Foundation.Notes and references
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434–447 CrossRef CAS.
- H. Dau, E. Fujita and L. Sun, ChemSusChem, 2017, 10, 4228–4235 CrossRef CAS.
- D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS.
- X.-L. Ma, J.-C. Liu, H. Xiao and J. Li, J. Am. Chem. Soc., 2018, 140, 46–49 CrossRef CAS.
- N. Nelson, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 856–863 CrossRef CAS.
- G. W. Brudvig, Philos. Trans. R. Soc., B, 2008, 363, 1211–1219 CrossRef CAS.
- F. Gloaguen and T. B. Rauchfuss, Chem. Soc. Rev., 2009, 38, 100–108 RSC.
- F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, Chem. – Eur. J., 2018, 24, 1471–1493 CrossRef.
- T. Morikawa, S. Sato, K. Sekizawa, T. Arai and T. M. Suzuki, ChemSusChem, 2019, 12, 1807–1824 CrossRef CAS.
- M. Wang, K. Han, S. Zhang and L. Sun, Coord. Chem. Rev., 2015, 287, 1–14 CrossRef CAS.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- J. Cogdell Richard, T. Gardiner Alastair and C. Leroy, Philos. Trans. R. Soc., A, 2012, 370, 3819–3826 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–966 RSC.
- S. Kaufhold, L. Petermann, R. Staehle and S. Rau, Coord. Chem. Rev., 2015, 304–305, 73–87 CrossRef CAS.
- A. M. Kluwer, R. Kapre, F. Hartl, M. Lutz, A. L. Spek, A. M. Brouwer, P. W. N. M. van Leeuwen and J. N. H. Reek, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10460–10465 CrossRef CAS.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS.
- Y.-J. Yuan, Z.-T. Yu, D.-Q. Chen and Z.-G. Zou, Chem. Soc. Rev., 2017, 46, 603–631 RSC.
- M. Ahmed and I. Dincer, Int. J. Hydrogen Energy, 2019, 44, 2474–2507 CrossRef CAS.
- H. Zhang and A. Lei, Asian J. Org. Chem., 2018, 7, 1164–1177 CrossRef CAS.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473(7345), 55–60 CrossRef CAS.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS.
- Q.-Q. Yang, X. Jiang, B. Yang, Y. Wang, C.-H. Tung and L.-Z. Wu, iScience, 2020, 23, 100969 CrossRef CAS.
- L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS.
- R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS.
- S. Romain, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef CAS.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS.
- M. Schulze, V. Kunz, P. D. Frischmann and F. Würthner, Nat. Chem., 2016, 8, 576–583 CrossRef CAS.
- M. A. Asraf, H. A. Younus, M. Yusubov and F. Verpoort, Catal. Sci. Technol., 2015, 5, 4901–4925 RSC.
- S. Fukuzumi and D. Hong, Eur. J. Inorg. Chem., 2014, 645–659 CrossRef CAS.
- M. D. Symes, Y. Surendranath, D. A. Lutterman and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 5174–5177 CrossRef CAS.
- M. D. Kärkäs, T. Åkermark, E. V. Johnston, S. R. Karim, T. M. Laine, B.-L. Lee, T. Åkermark, T. Privalov and B. Åkermark, Angew. Chem., Int. Ed., 2012, 51, 11589–11593 CrossRef.
- Z. Lu, Y. Gao, H. Chen, Z. Liu and L. Sun, Dalton Trans., 2017, 46, 1304–1310 RSC.
- M. Hansen, F. Li, L. Sun and B. König, Chem. Sci., 2014, 5, 2683–2687 RSC.
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417–2420 CrossRef CAS.
- J. Lin, X. Meng, M. Zheng, B. Ma and Y. Ding, Appl. Catal., B, 2019, 241, 351–358 CrossRef CAS.
- J. Lin, B. Ma, M. Chen and Y. Ding, Chin. J. Catal., 2018, 39, 463–471 CrossRef CAS.
- J. Lin, X. Liang, X. Cao, N. Wei and Y. Ding, Chem. Commun., 2018, 54, 12515–12518 RSC.
- Y. Liu, R. Xiang, X. Du, Y. Ding and B. Ma, Chem. Commun., 2014, 50, 12779–12782 RSC.
- F. Song, R. Moré, M. Schilling, G. Smolentsev, N. Azzaroli, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2017, 139, 14198–14208 CrossRef CAS.
- F. Song, K. Al-Ameed, M. Schilling, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2019, 141, 8846–8857 CrossRef CAS.
- H. J. Hou, Materials, 2011, 4, 1693–1704 CrossRef CAS.
- B. Schwarz, J. Forster, M. K. Goetz, D. Yücel, C. Berger, T. Jacob and C. Streb, Angew. Chem., Int. Ed., 2016, 55, 6329–6333 CrossRef CAS.
- F. Evangelisti, R. Güttinger, R. Moré, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2013, 135, 18734–18737 CrossRef CAS.
- Y. Zhao, J. Lin, Y. Liu, B. Ma, Y. Ding and M. Chen, Chem. Commun., 2015, 51, 17309–17312 RSC.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- G. Chen, L. Chen, S.-M. Ng, W.-L. Man and T.-C. Lau, Angew. Chem., Int. Ed., 2013, 52, 1789–1791 CrossRef CAS.
- C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS.
- R.-J. Xiang, H.-Y. Wang, Z.-J. Xin, C.-B. Li, Y.-X. Lu, X.-W. Gao, H.-M. Sun and R. Cao, Chem. – Eur. J., 2016, 22, 1602–1607 CrossRef CAS.
- X. Jiang, B. Yang, Q.-Q. Yang, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2018, 54, 4794–4797 RSC.
- M. Frey, ChemBioChem, 2002, 3, 153–160 CrossRef CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS.
- E. J. Lyon, I. P. Georgakaki, J. H. Reibenspies and M. Y. Darensbourg, Angew. Chem., Int. Ed., 1999, 38, 3178–3180 CrossRef CAS.
- A. P. S. Samuel, D. T. Co, C. L. Stern and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 8813–8815 CrossRef CAS.
- Y. Na, M. Wang, J. Pan, P. Zhang, B. Åkermark and L. Sun, Inorg. Chem., 2008, 47, 2805–2810 CrossRef CAS.
- H.-Y. Wang, G. Si, W.-N. Cao, W.-G. Wang, Z.-J. Li, F. Wang, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2011, 47, 8406–8408 RSC.
- J.-X. Jian, C. Ye, X.-Z. Wang, M. Wen, Z.-J. Li, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2016, 9, 2083–2089 RSC.
- K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS.
- J.-X. Jian, Q. Liu, Z.-J. Li, F. Wang, X.-B. Li, C.-B. Li, B. Liu, Q.-Y. Meng, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Nat. Commun., 2013, 4, 2695 CrossRef.
- X. Li, M. Wang, D. Zheng, K. Han, J. Dong and L. Sun, Energy Environ. Sci., 2012, 5, 8220–8224 RSC.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS.
- L.-Z. Wu, X.-Z. Wang, S.-L. Meng, H.-Y. Xiao, K. Feng, Y. Wang, J.-X. Jian, X.-B. Li and C.-H. Tung, Angew. Chem., Int. Ed., 2020, 132, 18558–18562 CrossRef.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC.
- J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2009, 253, 1476–1494 CrossRef CAS.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS.
- M. P. McLaughlin, T. M. McCormick, R. Eisenberg and P. L. Holland, Chem. Commun., 2011, 47, 7989–7991 RSC.
- M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Nouv. J. Chim., 1983, 7, 271–277 CAS.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS.
- P. Zhang, M. Wang, J. Dong, X. Li, F. Wang, L. Wu and L. Sun, J. Phys. Chem. C, 2010, 114, 15868–15874 CrossRef CAS.
- P. Zhang, P.-A. Jacques, M. Chavarot-Kerlidou, M. Wang, L. Sun, M. Fontecave and V. Artero, Inorg. Chem., 2012, 51, 2115–2120 CrossRef CAS.
- J. Huang, K. L. Mulfort, P. Du and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 16472–16475 CrossRef CAS.
- C. F. Wise, D. Liu, K. J. Mayer, P. M. Crossland, C. L. Hartley and W. R. McNamara, Dalton Trans., 2015, 44, 14265–14271 RSC.
- R. Henning, W. Schlamann and H. Kisch, Angew. Chem., Int. Ed. Engl., 1980, 19, 645–646 CrossRef.
- Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667–1670 CrossRef CAS.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS.
- D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027–2036 CrossRef CAS.
- M. Kirch, J. M. Lehn and J. P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- L. Tong, R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2014, 136, 4881–4884 CrossRef CAS.
- M. Nippe, R. S. Khnayzer, J. A. Panetier, D. Z. Zee, B. S. Olaiya, M. Head-Gordon, C. J. Chang, F. N. Castellano and J. R. Long, Chem. Sci., 2013, 4, 3934–3945 RSC.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477–1488 RSC.
- L. Xie, J. Tian, Y. Ouyang, X. Guo, W. Zhang, U.-P. Apfel, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 15844–15848 CrossRef CAS.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS.
- Z.-J. Li, X.-B. Li, J.-J. Wang, S. Yu, C.-B. Li, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2013, 6, 465–469 RSC.
- Z.-J. Li, J.-J. Wang, X.-B. Li, X.-B. Fan, Q.-Y. Meng, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2013, 25, 6613–6618 CrossRef CAS.
- Z.-J. Li, X.-B. Fan, X.-B. Li, J.-X. Li, C. Ye, J.-J. Wang, S. Yu, C.-B. Li, Y.-J. Gao, Q.-Y. Meng, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2014, 136, 8261–8268 CrossRef CAS.
- B. Liu, J. Li, H.-L. Wu, W.-Q. Liu, X. Jiang, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 18577–18583 CrossRef CAS.
- T. E. Rosser, M. A. Gross, Y.-H. Lai and E. Reisner, Chem. Sci., 2016, 7, 4024–4035 RSC.
- E. J. Maginn, J. Phys. Chem. L., 2010, 1, 3478–3479 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS.
- C. L. Drennan, J. Heo, M. D. Sintchak, E. Schreiter and P. W. Ludden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11973–11978 CrossRef CAS.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281–1285 CrossRef CAS.
- C. Darnault, A. Volbeda, E. J. Kim, P. Legrand, X. Vernède, P. A. Lindahl and J. C. Fontecilla-Camps, Nat. Struct. Mol. Biol., 2003, 10, 271–279 CrossRef CAS.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS.
- K. A. Grice, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS.
- S. Lian, M. S. Kodaimati, D. S. Dolzhnikov, R. Calzada and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 8931–8938 CrossRef CAS.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., A, 2015, 25, 106–137 CrossRef CAS.
- M. Khalil, J. Gunlazuardi, T. A. Ivandini and A. Umar, Renewable Sustainable Energy Rev., 2019, 113, 109246 CrossRef CAS.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS.
- E. Fujita, C. Creutz, N. Sutin and B. S. Brunschwig, Inorg. Chem., 1993, 32, 2657–2662 CrossRef CAS.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2017, 139, 6538–6541 CrossRef CAS.
- D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS.
- T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS.
- Y. Kuramochi, K. Fukaya, M. Yoshida and H. Ishida, Chem. – Eur. J., 2015, 21, 10049–10060 CrossRef CAS.
- D. B. Burks, S. Davis, R. W. Lamb, X. Liu, R. R. Rodrigues, N. P. Liyanage, Y. Sun, C. E. Webster, J. H. Delcamp and E. T. Papish, Chem. Commun., 2018, 54, 3819–3822 RSC.
- A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 4867–4874 CrossRef CAS.
- J.-S. Lee, D.-I. Won, W.-J. Jung, H.-J. Son, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2017, 56, 976–980 CrossRef CAS.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS.
- D.-I. Won, J.-S. Lee, J.-M. Ji, W.-J. Jung, H.-J. Son, C. Pac and S. O. Kang, J. Am. Chem. Soc., 2015, 137, 13679–13690 CrossRef CAS.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T.-C. Lau, J. Bonin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef CAS.
- X. Chang, T. Wang, P. Yang, G. Zhang and J. Gong, Adv. Mater., 2019, 31, 1804710 CrossRef.
- M. Schreier, J. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Grätzel, J. Am. Chem. Soc., 2016, 138, 1938–1946 CrossRef CAS.
- R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS.
- X.-L. Yang, J.-D. Guo, H. Xiao, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 5365–5370 CrossRef CAS.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS.
- Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682–2685 RSC.
- B. Chen, L.-Z. Wu and C.-H. Tung., Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS.
- Q.-Y. Meng, J.-J. Zhong, Q. Liu, X.-W. Gao, H.-H. Zhang, T. Lei, Z.-J. Li, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2013, 135, 19052–19055 CrossRef CAS.
- J.-J. Zhong, C.-J. Wu, Q.-Y. Meng, X.-W. Gao, T. Lei, C.-H. Tung and L.-Z. Wu, Adv. Synth. Catal., 2014, 356, 2846–2852 CrossRef CAS.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS.
- Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-Y. Meng, L.-Z. Wu and C.-H. Tung, J. Am. Chem. Soc., 2016, 138, 10080–10083 CrossRef CAS.
- L. Niu, H. Yi, S. Wang, T. Liu, J. Liu and A. Lei, Nat. Commun., 2017, 8, 14226 CrossRef.
- C.-J. Wu, Q.-Y. Meng, T. Lei, J.-J. Zhong, W.-Q. Liu, L.-M. Zhao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Catal., 2016, 6, 4635–4639 CrossRef CAS.
- G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS.
- G. Zhang, Y. Lin, X. Luo, X. Hu, C. Chen and A. Lei, Nat. Commun., 2018, 9, 1225 CrossRef.
- X. Sun, J. Chen and T. Ritter, Nat. Chem., 2018, 10, 1229–1233 CrossRef CAS.
- H. Cao, H. Jiang, H. Feng, J. M. C. Kwan, X. Liu and J. Wu, J. Am. Chem. Soc., 2018, 140, 16360–16367 CrossRef CAS.
- Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef CAS.
- W.-Q. Liu, T. Lei, S. Zhou, X.-L. Yang, J. Li, B. Chen, J. Sivaguru, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 13941–13947 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |