
Calcium cyclic carboxylates as structural models for calcium carbonate scale inhibitors†
Yuexian
Hong
a,
Dmitry S.
Yufit
a,
Nathalie
Letzelter
b and
Jonathan W.
Steed
 *a
*a
aDepartment of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk
bProcter & Gamble Newcastle Innovation Centre, Longbenton, Whitley Road, Newcastle upon Tyne, NE12 9BZ, UK
First published on 16th March 2020
Abstract
Cyclic oligocarboxylic acids are the most commonly explored phosphate-free inhibitors for calcium carbonate scale deposition. The structural chemistry of calcium complexes of candidate inhibitors has the potential to give insight into inhibitor mode of action and design. We report a series of calcium compounds of cyclic oligocarboxylic acids of (1α,3α,5α)-1,3,5,-cyclohexanetricarboxylic acid (CHTCA); cyclohexane-1,2,4,5-tetracarboxylic acid (CHTTCA); 1,2,3,4,5,6-cyclohexanehexacarboxylic acid (CHHCA); 1,1-cyclohexandiacetic acid (CHDAA) and cis,cis,cis,cis-1,2,3,4-cyclopentanetetracarboxylic acid (CPTCA) to understand the relationship between ligand stereochemistry and calcium ion coordination mode.
Introduction
Calcium carbonate precipitation has been studied for more than a century because of its importance in paleoclimate reconstructions,1 ocean acidification,2–4 and biomineralization.5 Calcium carbonate is also a model system for studying nucleation and crystallization mechanisms from ion solutions.6–8 With the discovery of a new hemihydrate phase of calcium carbonate,9 the study on the crystallization as well as inhibition processes has become particularly topical. Despite the tremendous amount of work on calcium carbonate crystallisation, the deposition of calcium carbonate in water-cycling industrial and domestic systems remains a problem.10,11 The use of antiscalants, especially small molecules, still seems the easiest way to achieve scale control.12 While phosphate-containing inhibitors such as the sodium salt of hydroxyethyl diphosphonic acid (HEDP, Fig. 1) can inhibit all forms of calcium carbonate in a variety of conditions, environmental concerns have given rise to increasing interest in phosphate-free inhibitors.13 Phosphorus-free oligocarboxylic acids represent potentially environmentally friendly alternatives because of the highly oxophilic nature of Ca2+ both observed in biominerals14 and inside organisms.15 In previous work, we have showed that the cyclic oligocarboxylic acids cis,cis,cis,cis-1,2,3,4-cyclopentanetetracarboxylic acid (CPTCA) and 1,2,3,4,5,6-cyclohexanehexacarboxylic acid (CHHCA, Fig. 1) are highly substrate and polymorph specific inhibitors of CaCO3 under domestic dishwasher conditions, with CPTCA inhibiting aragonite growth on polymethylmethacrylate (PMMA) while CHHCA inhibits calcite growth on glass. Use of both compounds together provides a broadly effective scale inhibition system.16 Testing of a wide range of oligocarboxylic acids reveals that subtle changes in structure can lead to substantial variation in inhibition performance. As a result, the relationship between structure, calcium binding mode and inhibitor performance remains an open question.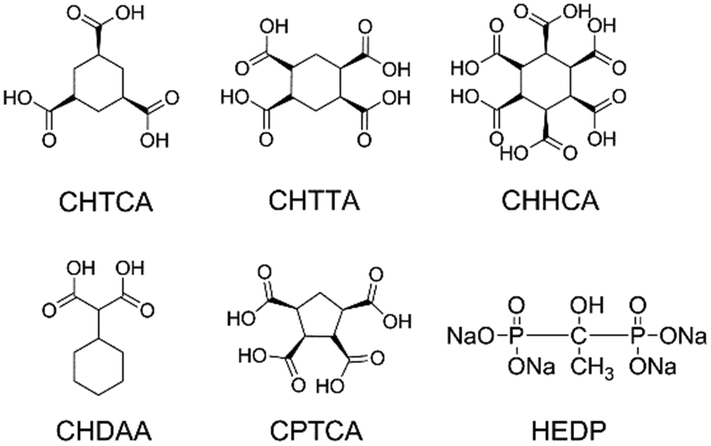 | ||
| Fig. 1 Structures of key cyclic polycarboxylic acids and the common phosphorus based inhibitor, HEDP. Compounds shown are: (1α,3α,5α)-1,3,5,-cyclohexanetricarboxylic acid (CHTCA); cyclohexane-1,2,4,5-tetracarboxylic acid (CHTTCA); 1,2,3,4,5,6-cyclohexanehexacarboxylic acid (CHHCA); 1,1-cyclohexandiacetic acid (CHDAA); cis,cis,cis,cis-1,2,3,4-cyclopentanetetracarboxylic acid (CPTCA); tetra sodium salt of hydroxyethene 1,1-diphosphonic acid (HEDP). | ||
In previous work, we examined the structures of two calcium complexes of the selective inhibitors CPTCA and CHHCA in an attempt to address the question of their inhibition specificity in contrast to the broad spectrum inhibition performance of HEDP.16 In the present work, we undertake a much more extensive study of the ligand conformation and calcium binding properties of a range of cyclic oligocarboxylic acids and attempt to elucidate the factors that contribute to the CaCO3 scale inhibition efficiency of CPTCA and CHHCA in particular. We report the synthesis and X-ray crystal structures of a range of newly synthesised calcium salts of cyclic oligo-carboxylates (Fig. 1), in conjunction with a discussion on electrostatic and stereochemical factors of these potential inhibitor structures. In all cases, single crystal structures could only be obtained at acidic pH, in contrast to the basic conditions prevailing in a dishwasher under which inhibition efficiency has been assessed.3 However, the stereochemistry of ligand itself as well as its bonding mode with the calcium cation is still likely to be informative. The understanding of structures of the known inhibitors can provide useful guidelines for future inhibitor design.
Results
Calcium complexes of 1,2,3,4-cyclopentanetetracarboxylic acid
Reaction of Ca(OH)2 or CaO with the successful inhibitor cis,cis,cis,cis-1,2,3,4-cyclopentanetetracarboxylic acid (cis-CPTCA) in deionised water (DI water), in ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by solvent evaporation results in the crystallisation of [Ca(μ-C9H8O8)(H2O)4]n (1), reported previously.16 Interestingly, reactions of CaO with 1,2,3,4-cyclopentanetetracarboxylic acid (CPTCA, a mix of trans- and cis-isomers), in 1:1 ratio, in deionised (DI) water, afforded two new crystalline products that were characterised by X-ray crystallography, [Ca(H2O)8][Ca(C9H8O8)2(H2O)4] (3), and [Ca(C9H9O8)2(H2O)4] (4). After four months, this mixture (Fig. S1a†) transformed into a mixture of 1 and 4 (ESI† Fig. S1b). Compound 4 is obtained as a mixture with unreacted CPTCA (Fig. S3†). This residual CPTCA is likely to be unreacted trans-CPTCA following incorporation of the cis-form into the product. A further new complex of formula [Ca(H2O)8][Ca(C9H8O8)2(H2O)4]·2H2O (2) was obtained upon evaporation at room temperature upon reacting CPTCA with CaO in a 2:1 metal:ligand ratio in DI water. Compound 2 transforms into compound 1 (Fig. S2†) during the drying process and subsequent repeated crystallisation failed to isolate it. Although the new complexes were obtained from a mixture of trans- and cis-CPTCA, all of the calcium complexes isolated are those of the all-cis-CPTCA. While yields were not measured in order to avoid the risk of deterioration of the sample by dehydration upon removal from the mother liquor, generally products were abundant and the correspondence of the crystals studied by X-ray diffraction with the bulk material was confirmed by comparison of calculated and experimental XRPD data, except where discussed. The ligand in its dianionic form is only found in compound 1, while the ligands in the other three crystal forms are all singly deprotonated (Fig. 2).
:1, followed by solvent evaporation results in the crystallisation of [Ca(μ-C9H8O8)(H2O)4]n (1), reported previously.16 Interestingly, reactions of CaO with 1,2,3,4-cyclopentanetetracarboxylic acid (CPTCA, a mix of trans- and cis-isomers), in 1:1 ratio, in deionised (DI) water, afforded two new crystalline products that were characterised by X-ray crystallography, [Ca(H2O)8][Ca(C9H8O8)2(H2O)4] (3), and [Ca(C9H9O8)2(H2O)4] (4). After four months, this mixture (Fig. S1a†) transformed into a mixture of 1 and 4 (ESI† Fig. S1b). Compound 4 is obtained as a mixture with unreacted CPTCA (Fig. S3†). This residual CPTCA is likely to be unreacted trans-CPTCA following incorporation of the cis-form into the product. A further new complex of formula [Ca(H2O)8][Ca(C9H8O8)2(H2O)4]·2H2O (2) was obtained upon evaporation at room temperature upon reacting CPTCA with CaO in a 2:1 metal:ligand ratio in DI water. Compound 2 transforms into compound 1 (Fig. S2†) during the drying process and subsequent repeated crystallisation failed to isolate it. Although the new complexes were obtained from a mixture of trans- and cis-CPTCA, all of the calcium complexes isolated are those of the all-cis-CPTCA. While yields were not measured in order to avoid the risk of deterioration of the sample by dehydration upon removal from the mother liquor, generally products were abundant and the correspondence of the crystals studied by X-ray diffraction with the bulk material was confirmed by comparison of calculated and experimental XRPD data, except where discussed. The ligand in its dianionic form is only found in compound 1, while the ligands in the other three crystal forms are all singly deprotonated (Fig. 2).
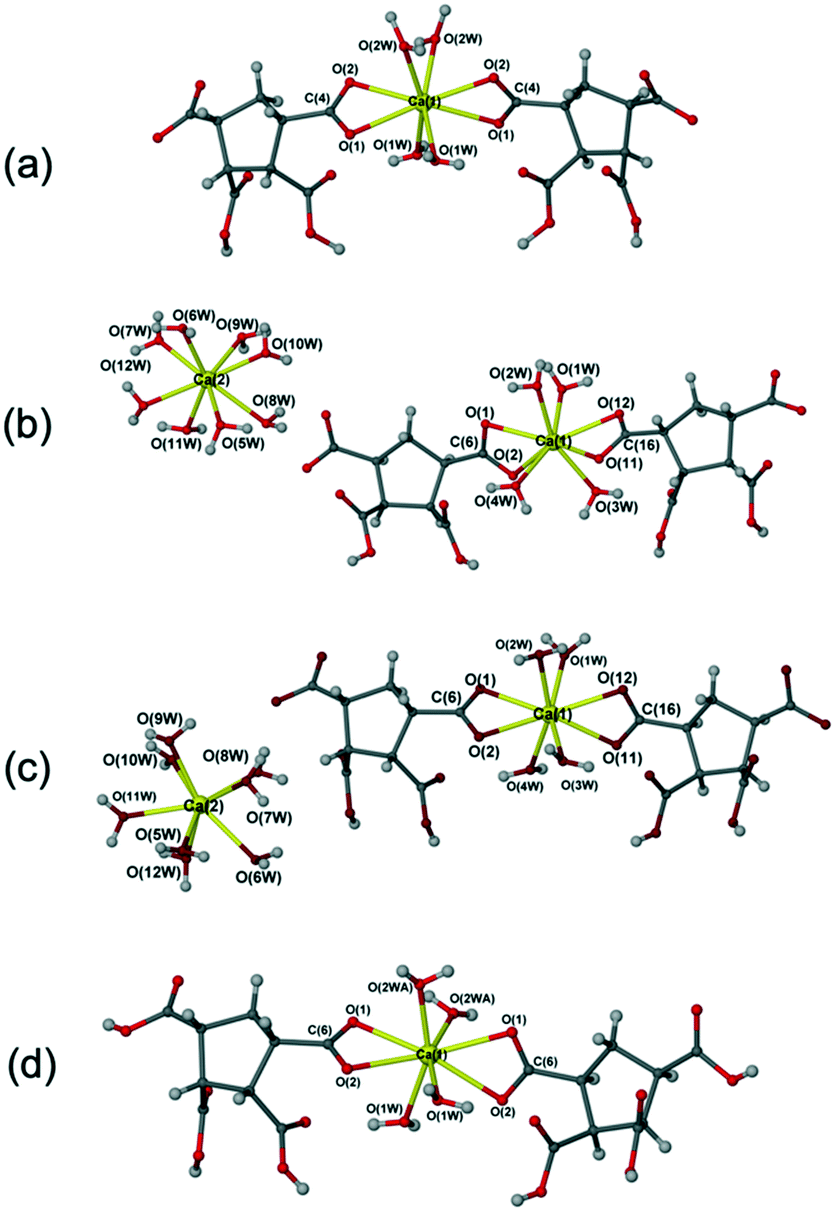 | ||
| Fig. 2 Calcium complexes of CPTCA. (a) Structure of [Ca(μ-C9H8O8)(H2O)4]n (1);16 (b) structure of [Ca(H2O)8] [Ca(C9H8O8)2(H2O)4]·2H2O (2); (c) structure of [Ca(H2O)8] [Ca(C9H8O8)2(H2O)4] (3); (d) structure of [Ca(C9H9O8)2(H2O)4]n (4). | ||
Compound 2 is a higher hydrate of compound 3 with two additional lattice water molecules. The product obtained was dependent primarily on the ratio of metal salt and ligand added. The new complexes 2 and 3 were side products of crystallisation with CPTCA (a mix of trans- and cis-) in a metal:ligand ratio of 2:1 and 1:1, respectively. Attempts to prepare larger quantities of 2 and 3 were unsuccessful, possibly because they are less stable phases and transform into 1 or 4 on drying. Therefore, only the X-ray structures of 2 and 3 are reported without further characterisation. The new complex 4 was fully characterised by single crystal X-ray diffraction, elemental analysis, X-ray powder diffraction, IR spectra and TGA analysis (see the Experimental section and Fig. S3–S5†).
Calcium complexes of 1α,3α,5α-cyclohexanetricarboxylic acid
Layering solutions of 1α,3α,5α-cyclohexanetricarboxylic acid (CHTCA) on top of an aqueous solution of Ca(OH)2 and allowing evaporation at room temperature resulted in two new complexes [Ca3(C9H9O6)2(H2O)4]·3H2O (5) and [Ca(C9H10O6)(H2O)2]·H2O (6) which were characterised by X-ray crystallography. The new materials were obtained with metal:ligand ratio of 1:2, and 1:1, respectively. Complex 5 was obtained in pure form from methanol (Fig. S6†) or mixed with a small amount of 6 from ethanol (Fig. S9†). Pure 6 was obtained using acetone (Fig. S10†), THF, or ethyl acetate as the solvent for the ligand solution. However, the XRPD pattern collected at room temperature for 6 is different to the simulated XRPD pattern derived from the 120 K single crystal X-ray data (Fig. S10†). To explore this discrepancy, a variable-temperature study was carried out. This showed the transformation of 6 into a new hemihydrate phase [Ca(C9H10O6)(H2O)2]·0.5H2O (7) at 290 K, from which the simulated XRPD pattern corresponds well with the experimental diffractogram collected at room temperature (Fig. S10†). The hemihydrate 7 in space group C2/c has half a free water molecule less per formula unit than compound 6 (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). Compounds 6 and 7 contain an infinite array of CHTCA groups, Ca2+ ions and water molecules held together by hydrogen bonding and calcium coordination of the oxygen atoms from both CPTCA and water molecules, forming a 3D Ca–O–Ca framework. The calcium coordination environment in 6 and 7 is different to one another. The calcium ions in 7 are all seven coordinate binding to five oxygen atoms from four individual CHTCA ligands, while in 6 with one calcium ion is seven coordinate and the other eight coordinate (Fig. 3c and d).
). Compounds 6 and 7 contain an infinite array of CHTCA groups, Ca2+ ions and water molecules held together by hydrogen bonding and calcium coordination of the oxygen atoms from both CPTCA and water molecules, forming a 3D Ca–O–Ca framework. The calcium coordination environment in 6 and 7 is different to one another. The calcium ions in 7 are all seven coordinate binding to five oxygen atoms from four individual CHTCA ligands, while in 6 with one calcium ion is seven coordinate and the other eight coordinate (Fig. 3c and d).
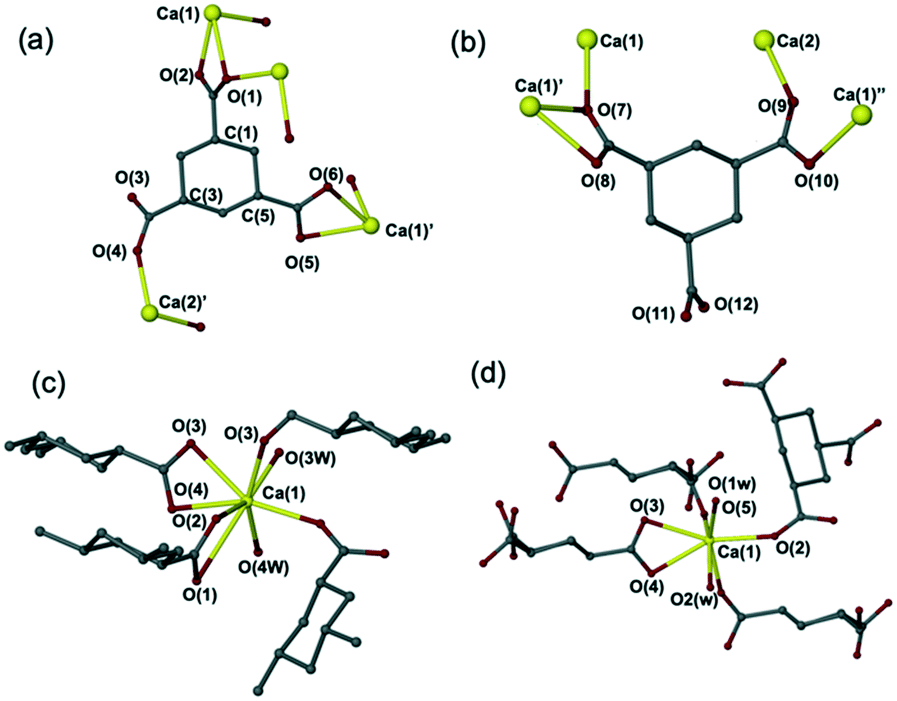 | ||
| Fig. 3 (a) The binding of ligand CHTCA to the calcium ions in [Ca3(C9H9O6)2(H2O)4]·3H2O (5), and (b) [Ca(C9H10O6)(H2O)2]·H2O (6). Selected interatomic distances in 5 (Å): Ca(1)–Ca(2) = 3.836, Ca(1)–Ca(1)′ = 9.492, Ca(1)′–Ca(2)′ = 8.044. Selected bond lengths in 6 (Å): Ca(1)–Ca(2) = 5.095, Ca(1)′–Ca(1)′′ = 9.377, Ca(1)–Ca(1)′ = 4.001, Ca(1)′′–Ca(2) = 5.192. (c) The calcium coordination environment in [Ca(C9H10O6)(H2O)2]·H2O (6); (d) the calcium coordination environment in [Ca(C9H10O6)(H2O)2]·0.5H2O (7). | ||
The Experimental section shows the full characterization data for the new complexes 5 and 6 by single crystal X-ray diffraction, elemental analysis, X-ray powder diffraction, IR spectroscopy, and TGA. The crystal data of 7 from the variable-temperature study are also provided. The XRPD, IR and TGA data for complexes 5 and 6 are listed in the Supporting Information in Fig. S6–S9 and S10–S12,† respectively.
Calcium complexes of 1,2,4,5-cyclohexanetetracarboxylic acid
Reactions of Ca(OH)2 with a commercial mixture of cis- and trans-1,2,4,5-cyclohexane tetracarboxylic acid (CHTTCA) in DI water followed by solvent evaporation produced a mixture of two new complexes [Ca2(C10H8O8)(H2O)7]·2H2O (8) and [Ca(C10H10O8)(H2O)6]·H2O (9). Complex 8 was obtained as the dominant phase while 9 was a minor phase (Fig. S13†). A pure sample of 8 can be obtained by using a solvent layering method in ethanol with metal:ligand ratio of 1:2 (Fig. S14†) or 1:1. The full characterisation for the new complexes 8 and 9 by single crystal X-ray diffraction (Fig. 4), elemental analysis, X-ray powder diffraction, IR spectroscopy, and TGA analysis are described in the Experimental section. The XRPD, IR and TGA data for complexes 8 and 9 can also be found in ESI† in Fig. S14–S16 and S17 and S18,† respectively. A further new compound 10 was also obtained using methanol as the solvent with metal:ligand ratio of 1:1.5. X-ray crystallography shows that compound 10 has formula [Ca(C10H12O8)2(H2O)4]n but the structure determination is not presented in detail because of poor crystal quality. However, the calculated XRPD pattern derived from the approximate model corresponds well with the experimental XRPD pattern suggesting that the broad details of the structure are correct and the material is a single phase. The preliminary crystal structure along with elemental analysis, XRPD pattern, IR spectroscopy and TGA-MS can be found in Fig. S19–S22† and in the Experimental section.
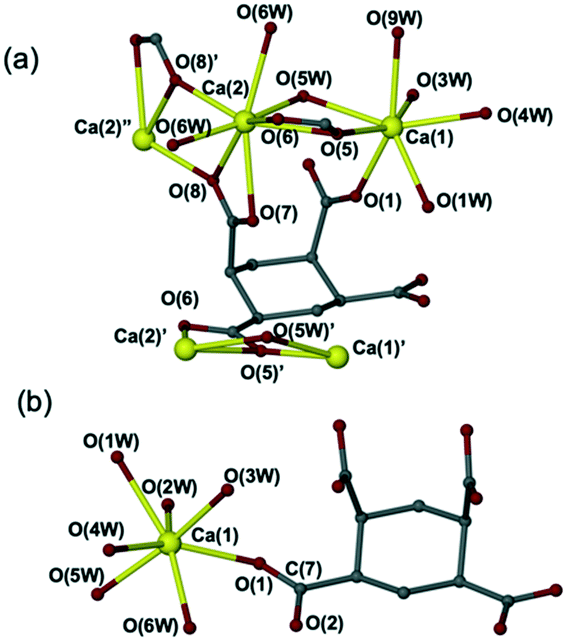 | ||
| Fig. 4 Crystal structures of (a) [Ca2(C10H8O8)(H2O)7]·2H2O (8) and (b) [Ca(C10H10O8)(H2O)6]·H2O (9). | ||
The X-ray crystallographic analysis revealed that only calcium complexes of the all-cis isomer of CHTTCA preferentially crystallised. This phenomenon was also observed in the crystallisation of the CPTCA derivatives. While this may represent a higher calcium affinity of the all-cis isomer it could also arise from a lower solubility of the all-cis complexes.17 It seems that the calcium ions are capable of isolating the all-cis-isomer of CHTTCA and leaving the other isomer in solution when using a commercially available mixture.
Calcium complexes of cyclohexane-1,2,3,4,5,6-hexacarboxylic acid
Layering a solution of cyclohexane-1,2,3,4,5,6-hexacarboxylic acid (CHHCA) in butan-1-ol onto an aqueous solution of Ca(OH)2 in a ratio of 1:1 followed by evaporation at room temperature afforded [Ca(C12H11O6)2(H2O)4]·1.25H2O (11) (published previously16). Crystals of the hydrated free acid C12H12O12·H2O (12), were also obtained from methanol with metal:ligand ratio of 1:1. This represents a third crystal form of CHHCA in addition to the known tri- and tetrahydrates.18,19 The CHHCA conformation is similar in all three hydrates.
Calcium complexes of 1,1-cyclohexandiacetic acid
The reaction of calcium hydroxide with 1,1-cyclohexandiacetic acid (CHDCA) in a 1:2 ratio in DI water followed by solvent evaporation gave [Ca(C10H15O4)2(H2O)2]n (13) at room temperature (Fig. 5). The new complex 13 was fully characterised by single crystal X-ray diffraction, elemental analysis, X-ray powder diffraction, IR spectroscopy, and TGA analysis (see Experimental section and Fig. S23–S25†).
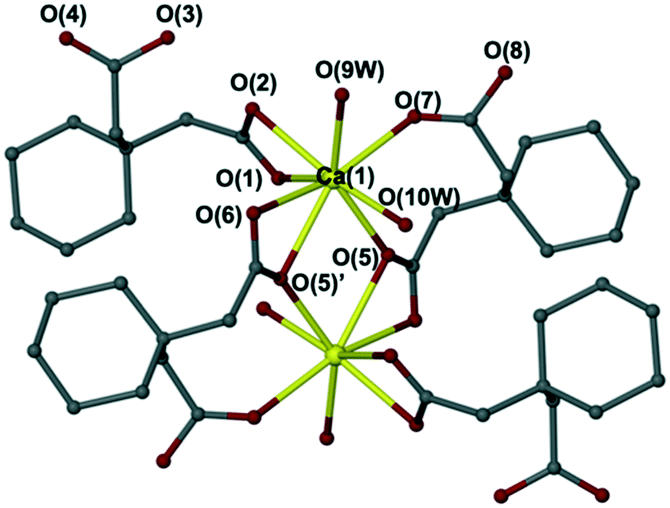 | ||
| Fig. 5 The crystal structure of [Ca(C10H15O4)2(H2O)2]n (13), shows the eightfold coordination. | ||
Discussion
The binding mode in the calcium-cyclic poly(carboxylic acid)s
In the cyclic polycarboxylate coordination compounds studied, two different carboxylate-binding modes are observed. The carboxylate groups can bind the calcium cation either in a monodentate fashion (via one of the carboxylate O atoms) or in a bidentate four-membered ring chelate mode (via both carboxylate oxygen atoms). No obvious relationship is observed in the binding mode and position of carboxylate on the ligand backbone. While a bidentate carboxylate mode is likely to be inherently more stable than its monodentate counterpart, both modes are equally observed in all ligands. The final coordination mode is likely to represent a balance between the coordination ability of the ligand, chelate ring strain, crystal packing effects and specific hydrogen bonding interactions. The Ca–Ocarboxylate distance in monodentate binding ranges from 2.31 to 2.43 Å, while the Ca–Ocarboxylate distance in bidentate mode is longer; 2.43 to 2.60 Å. The former is comparable to the distance of Ca–Ophosphate in HEDP, while the latter is close to Ca–OOH in HEDP. Dudev20 has shown that the energy difference between these two modes is relatively small. As can be seen from Table 1, the Ca–O bond lengths for both HEDP and polycarboxylic acids are similar to those in CO32− salts of Ca2+. Therefore, the greater effectiveness of phosphate-based inhibitors over carboxylate-containing inhibitors is likely to be rooted in the high polarity of the whole phosphate functional group as well as a better electrostatic balancing capability towards the Ca2+ cation given the dianionic charge of the –PO32− group. In contrast, it takes two COO− groups to give charge balance rather than one PO32−, and this fact may determine the way a carboxylic-based inhibitor approaches the growing crystal or nucleus surface. Moreover, COO− groups will need to adopt a well-defined, selective structure in order to achieve the critical charge density requirements.| Compound | Ca–O distance range (Å) |
|---|---|
| Calcite | 2.35 |
| Aragonite | 2.42–2.65 |
| Vaterite | 2.37–2.39 |
| HEDP-Ca | 2.35 (Ca–Ophosphate), 2.42 (Ca–OOH) |
| Cyclic oligo (carboxylic acid)s | 2.31–2.41 (monodentate); 2.41–2.55 (bidentate) |
| BHCA | 2.35 (monodentate); 2.51–2.64 (bidentate) |
Relationship between the stereochemistry of cyclic poly carboxylic acids and electrostatic interaction with calcium
The COOH groups attached to the alicyclic ring can be either axial or equatorial radiating away from the centre of the ring (Fig. 6). The spatial orientation of the COOH functionalities attached to the 5-membered ring follows a similar distribution pattern as those observed in the chair conformation of the 6-membered ring.21 Around the ring, any two adjacent COOH groups follow a pattern of alternate equatorial/axial distribution. The coordination to calcium does not affect the functional group orientation which depends on the isomeric form of the ligand. Each ligand exhibits a unique characteristic spatial orientation and hence characteristic distribution of metal-binding carboxylate groups. The relative rigidity of cyclic carboxylates compared to their acyclic analogues may explain why subtle changes in molecular structure leads to significant variation in CaCO3 crystallization inhibition ability as the spatial charge density varies considerably with different inter-functional group distances and orientations. It seems that in order to be an effective inhibitor towards a specific mineral a ligand must adopt specific structural disposition. A rigid, cyclic backbond is likely to contribute to this required degree of organisation of the distances between and spatial orientation of the coordinating groups.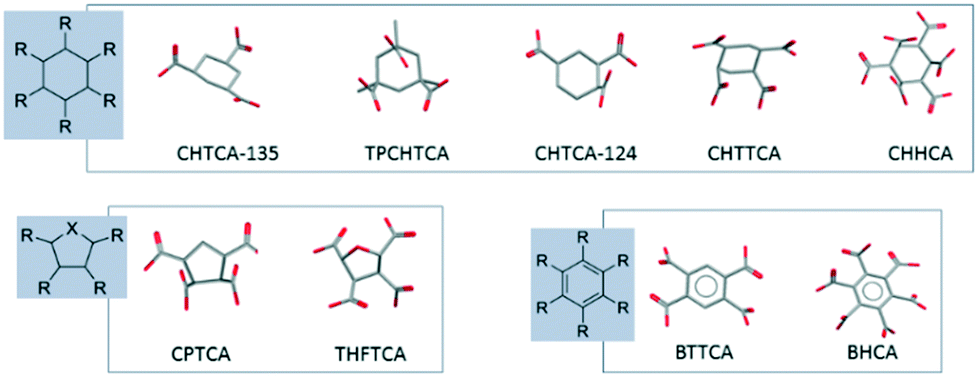 | ||
| Fig. 6 The orientation of the COOH along the cyclic 6-membered ring, 5-membered ring and benzene ring. TPCHTCA (DEVCOW);24 THFTCA (ZEZLEV);25 BTTCA (PYMELL12);26 BHCA (MELLIT).27 | ||
Two studies on crystallization inhibition showed that only a certain type of molecule with a specific binding motif gives rise to significant inhibition effects towards a specific mineral.22,23 For example, the inhibition study of a series of linear α,ω-dicarboxylates toward calcite (110) showed that good inhibition efficiency is only obtained when two carboxylate groups are connected by one or two carbon atoms. The distance between two carboxylate carbon atoms ranges from 2.5 to 3.2 Å, and this could be the distance range required to achieve the best balancing charge density for calcite.
Oligo(carboxylic acid)s with a bulkier backbone need to be more structurally preorganised so as to exhibit effective interactions to calcium ions during nucleation compared to the versatile HEDP. Since, not all oligo(carboxylic acid)s with four COOH groups are effective inhibitors, this difference in performance must lie in structural and stereochemical factors of the ligand, which in return has a marked effect on the primary electrostatic interactions with the metal cations on the growing mineral surface. Mann23 has observed that malonate (n = 1, C–C = 2.5–2.6 Å) in a cis-conformation has the best charge density for electrostatic interaction among a series of α,ω-dicarboxylates (CH2)n(CO2H)2 (Ca/malonate = 3). When n = 0, it leads to the crystallization of calcium oxalate while when n > 2 this leads to a separation in spatial charge density, hence reduced inhibition efficiency.
Conclusions
The metal affinity of an inhibitor as well as its disruptive ability towards periodic packing in nucleating clusters is of equal importance when assessing inhibition efficiency. In this work, only the affinity of the inhibitor toward minerals is discussed, the disrupting ability requires further exploration. Even if electrostatic interactions are the primary interactions, electrostatic factors are correlated with the structural and stereochemical factors, therefore, only when the inhibitor structure simultaneously satisfies the electrostatic, stereochemical and structural factors, can the inhibitor achieve a satisfactory performance.22 Inhibitor performance depends on the structure of the whole molecule. A subtle difference in molecular structure has a marked effect in the electrostatic interactions, hence the disrupting/weakening effectivity in the bond formation.15,23Due to the difficulty in obtaining the exact structures for the given conditions, the analysis of crystal structure itself cannot provide a definite understanding on ‘interaction–activity’ relationship. However, it is especially useful to understand the structural and stereochemical information of the inhibitor ligands and their static state interaction with the lattice ions. A systematic understanding of the relationship between the local spatial charge density and its inhibition ability would also be useful.
Experimental
Materials
All other solvents and reagents were obtained from standard commercial sources and used without further purification.Synthesis of coordination complexes
379 reflections collected, 6836 unique (Rint = 0.0950). Final GOF = 1.044, R1 = 0.0885, wR2 = 0.2058, R indices based on 6836 reflections with I ≧ 2σ(I) (refinement on F2), 477 parameters, 2 restraints.
102 reflections collected, 7638 unique (Rint = 0.0719). Final GOF = 1.042, R1 = 0.0663, wR2 = 0.1565, R indices based on 7638 reflections with I ≧ 2σ(I) (refinement on F2), 513 parameters, 28 restraints.
Analysis calc. for [Ca(C9H9O8)2(H2O)4]: elemental analysis is consistent with the loss of one water molecule. Calc. for [Ca(C9H9O8)2(H2O)3]: C, 36.95%; H, 4.10%. Found: C, 36.99%, H, 4.25%. Calc. for [Ca(C9H9O8)2(H2O)4]: C, 35.85%; H, 4.32%. Water loss in TGA calculated for 4: 11.9% for four water molecules, found: 11.4%. IR (cm−1): 3017 (w), 2971 (w), 2941 (w), 1738 (s), 1521 (m), 1446 (m), 1406 (m), 1366 (s), 1314 (w), 1287 (w), 1256 (w), 1228 (m), 1217 (s), 1206 (s), 1145 (w), 1115 (w), 1032 (w), 1007 (w), 940 (w), 876 (w), 833 (w), 811 (w), 735 (w), 707 (w), 670 (m), 618 (m). Crystal data: M = 602.47, colourless plates, orthorhombic, Pccn, a = 36.2540(18) Å, b = 5.5867(3) Å, c = 11.6210(6) Å, V = 2353.7(2) Å3, Z = 4, Dc = 1.700 g cm−3, μ = 0.368 mm−1, F000 = 1256.0, T = 120 K, 24873 reflections collected, 2837 unique (Rint = 0.0950). Final GOF = 1.106, R1 = 0.0816, wR2 = 0.1971, R indices based on 2837 reflections with I ≧ 2σ(I) (refinement on F2), 212 parameters, 0 restraints.
502 reflections collected, 3110 unique (Rint = 0.0977). Final GOF = 0.974, R1 = 0.0516, wR2 = 0.1424, R indices based on 3110 reflections with I ≧ 2σ(I) (refinement on F2), 243 parameters, 10 restraints.
, a = 8.5782(9) Å, b = 12.5674(14) Å, c = 12.8876(14) Å, α = 64.439(3)°, β = 89.726(3)°, γ = 89.647(3)°, V = 1253.3(2) Å3, Z = 4, Dc = 1.634 g cm−3, μ = 0.541 mm−1, F000 = 648.0, T = 120 K, 13401 reflections collected, 6328 unique (Rint = 0.0872). Final GOF = 1.021, R1 = 0.0835, wR2 = 0.2567, R indices based on 6328 reflections with I ≧ 2σ(I) (refinement on F2), 355 parameters, 0 restraints. Elemental analysis is consistent with the observed conversion to the partially dehydrated form 7. Calc. for 6: C, 35.03%; H, 5.19%. Found: C, 36.69%; H, 5.29%. Calc. for 7: [Ca(C9H10O6)(H2O)2]·0.5H2O: 36.21%; H, 5.03%. Water loss in TGA calculated is 17.52% for three water molecules, found: 17.21%. IR (cm−1): 3517 (m, b), 3205 (m, b), 2940 (m), 2867 (m, b), 2634 (w, b), 2381 (w), 2347 (w), 2279 (w), 2162 (w), 2051 (w), 1980 (w), 1701 (m), 1624 (w), 1580 (s), 1548 (s), 1448 (m), 1402 (s), 1356 (m), 1334 (m), 1307 (m), 1286 (w), 1272 (w), 1242 (m), 1185 (m), 1137 (w), 1105 (w), 1034 (w), 970 (w), 917 (w), 894 (w), 845 (w), 807 (w), 773 (w), 720 (w), 672 (w), 664 (w). Crystal data for 7: M = 298.28, colourless plate, monoclinic C2/c, a = 21.4940(8) Å, b = 13.6060(5) Å, c = 8.6138(14) Å, β = 90.270(3)°, V = 2519.07(15) Å3, Z = 8, Dc = 1.573 g cm−3, μ = 0.553 mm−1, F000 = 1248.0, T = 290.0 K, 18483 reflections collected, 3348 unique (Rint = 0.0594). Final GOF = 1.048, R1 = 0.0462, wR2 = 0.1245, R indices based on 3384 reflections with I ≧ 2σ(I) (refinement on F2), 181 parameters, 0 restraints.
906 reflections collected, 5002 unique (Rint = 0.0595). Final GOF = 1.090, R1 = 0.0541, wR2 = 0.1453, R indices based on 5002 reflections with I ≧ 2σ(I) (refinement on F2), 358 parameters, 23 restraints.
700 reflections collected, 4669 unique (Rint = 0.0458). Final GOF = 1.113, R1 = 0.0353, wR2 = 0.0801, R indices based on 4669 reflections with I ≧ 2σ(I) (refinement on F2), 331 parameters, 92 restraints.
548 reflections collected, 10883 unique (Rint = 0.1089, Rsigma = 0.0910). Final GOF = 1.061, R1 = 0.0915, wR2 = 0.2595, R indices based on 10883 reflections with I ≧ 2σ(I) (refinement on F2), 358 parameters, 12 restraints.
993 reflections collected, 3225 unique (Rint = 0.1135). Final GOF = 1.049, R1 = 0.0516, wR2 = 0.1448, R indices based on 3225 reflections with I ≧ 2σ(I) (refinement on F2), 282 parameters, 0 restraints.
, a = 7.07770(10) Å, b = 11.1674(2) Å, c = 15.7081(3) Å, α = 106.469(3)°, β = 96.639(3)°, γ = 101.751(3)°, V = 1145.63(3) Å3, Z = 2, Dc = 1.376 g cm−3, μ = 0.326 mm−1, F000 = 508.0, T = 120 K, 25088 reflections collected, 6654 unique (Rint = 0.0314, Rsigma = 0.0292). Final GOF = 1.012, R1 = 0.0295, wR2 = 0.0748, R indices based on 6654 reflections with I ≧ 2σ(I) (refinement on F2), 416 parameters, 6 restraints.
Physical measurements
Microanalysis on compounds 2 and 9 was performed by London Metropolitan University (London) using a Thermo Scientific (Carlo Erba) Flash 2000 Elemental Analyser. Other samples were run using an Exeter Analytical E-440 Elemental Analyser (Durham). Thermogravimetric analysis (TGA) were carried out using a Perkin Elmer Pyris 1 TGA coupled with a 500 amu Hiden mass spectrometer (MS), which allows the analysis of the volatiles generated by the thermal degradation of materials. The volatiles monitored by the MS are CO, CO2, and H2O. Thermal analysis is carried out from 20 °C to 500 °C at a rate of 10 °C min−1. The TGA figures were plot using Pyris manager. Data was collected by attenuated total reflectance infrared spectrometer (ATR-IR). ATR-IR spectra were recorded on a Perkin-Elmer Spectrum 100 using a diamond compression cell. The IR spectra were collected by pressing the probe against the diamond and scanning the 4000–400 cm−1 region at a resolution of 4 cm−1. Typically, 32 or 64 scans were conducted for each spectrum. The resulting IR spectra were analysed using KnowItAll.28 Peak intensity is described as strong (s), medium (m), weak (w), or broad (b). X-ray powder diffraction patterns were collected on a Bruker D8 Advance powder diffractometer using a nickel-filtered copper X-ray radiation (λ = 1.5406 Å). Powder diffractometer operates at 40 mA and 45 kV. The 2θ range for crystalline material from 5° to 50° is recorded at a scan rate of 0.02° per step. The outcome data was plot in Plots2 software.29 The lack of CHN, TGA, IR and PXRD data for some samples is because continuing attempts to prepare suitable bulk quantities for analysis have been unsuccessful.Single-crystal structure determination
The data collection temperature (see individual descriptions) was maintained by open flow N2 Oxford Cryostream (Oxford Cryosystems) devices. All data were collected using MoKα radiation (λ = 0.71073 Å). The data for compounds 12 and 13 were collected on a Bruker D8 Venture (Photon100 CMOS detector, IμS-microfocus source, focusing mirrors) 3-circle diffractometer. Data for 2 and 9 were collected on an Agilent XCalibur (Sapphire-3 CCD detector, graphite monochromator) 4-circle kappa diffractometer. Data for the remaining compounds were collected on a Bruker SMART CCD 6000 3-circle diffractometer (fine-focus sealed tube, graphite monochromator, Monocap optics). Data sets were corrected for Lorentz and polarization effects and the effects of absorption. All structures were solved by a direct method and refined by full-matrix least squares on F2 for all data using Olex-2 (ref. 30) and SHELXTL.31 All non-disordered, non-hydrogen atoms were refined in an anisotropic approximation. In most structures, the hydrogen atoms were placed in calculated positions and refined in riding mode. The structures were visualised using X-Seed.32 Molecular graphics were produced using POV-Ray.33Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Durham-Procter & Gamble CEMENT partnership for financial support (to Y. H.). We acknowledge Dr. Chunhai Wang for assistance with PXRD analysis.Notes and references
- J. W. Beck, R. L. Edwards, E. Ito, F. W. Taylor, J. Recy, F. Rougerie, P. Joannot and C. Henin, Science, 1992, 257, 644–647 CrossRef CAS PubMed.
- F. J. Millero, Chem. Rev., 2007, 107, 308–341 CrossRef CAS PubMed.
- S. Goffredo, F. Prada, E. Caroselli, B. Capaccioni, F. Zaccanti, L. Pasquini, P. Fantazzini, S. Fermani, M. Reggi, O. Levy, K. E. Fabricius, Z. Dubinsky and G. Falini, Nat. Clim. Change, 2014, 4, 593 CrossRef CAS PubMed.
- M. Cusack and A. Freer, Chem. Rev., 2008, 108, 4433–4454 CrossRef CAS PubMed.
- S. Mann, Biomineralization: principles and concepts in bioinorganic materials chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- J. J. De Yoreo, P. U. Gilbert, N. A. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky and J. F. Banfield, Science, 2015, 349, 6760 CrossRef PubMed.
- H. Cöelfen and M. Antonietti, Mesocrystals and nonclassical crystallization, John Wiley & Sons, Chichester, 2008 Search PubMed.
- J. B. Mullin, Crystallization, Butterworth-Heineman, Oxford, 4th edn, 2001 Search PubMed.
- Z. Y. Zou, W. Habraken, G. Matveeva, A. C. S. Jensen, L. Bertinetti, M. A. Hood, C. Y. Sun, P. Gilbert, I. Polishchuk, B. Pokroy, J. Mahanid, Y. Politi, S. Weiner, P. Werner, S. Bette, R. Dinnebier, U. Kolb, E. Zolotoyabko and P. Fratzl, Science, 2019, 363, 396–400 CrossRef CAS PubMed.
- Z. Amjad, Advances in crystal growth inhibition technologies, Springer, Boston, MA, 2002 Search PubMed.
- J. Chung, I. Granja, M. G. Taylor, G. Mpourmpakis, J. R. Asplin and J. D. Rimer, Nature, 2016, 1–5 CAS.
- J. Ihli, Y.-Y. Kim, E. H. Noel and F. C. Meldrum, Adv. Funct. Mater., 2013, 23, 1575–1585 CrossRef CAS.
- M. M. Reddy and A. R. Hoch, J. Colloid Interface Sci., 2001, 235, 365–370 CrossRef CAS PubMed.
- N. A. Sommerdijk and G. D. With, Chem. Rev., 2008, 108, 4499–4550 CrossRef CAS PubMed.
- J. Chung, I. Granja, M. G. Taylor, G. Mpourmpakis, J. R. Asplin and J. D. Rimer, Nature, 2016, 536, 446–450 CrossRef CAS PubMed.
- Y. Hong, N. Letzelter, J. S. O. Evans, D. S. Yufit and J. W. Steed, Cryst. Growth Des., 2018, 18, 1526–1538 CrossRef CAS.
- T. Anthonsen, B. Larsen and O. Smidsrød, Acta Chem. Scand., 1972, 26, 2988–2989 CrossRef CAS.
- P. Thuery, CCDC 877683: Experimental Crystal Structure Determination, 2004, DOI:10.5517/ccyg9c1.
- S. Bruckner, L. Malpezzi Giunchi, G. Di Silvestro and M. Grassi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 586–590 CrossRef.
- T. Dudev and C. Lim, J. Phys. Chem. B, 2004, 108, 4546–4557 CrossRef CAS.
- H. D. Orloff, Chem. Rev., 1954, 54, 347–447 CrossRef CAS.
- S. N. Black, L. A. Bromley, D. Cottier, R. J. Davey, B. Dobbs and J. E. Rout, J. Chem. Soc., Faraday Trans., 1991, 87, 3409–3414 RSC.
- S. Mann, J. M. Didymus, N. P. Sanderson and B. R. Heywood, J. Chem. Soc., Faraday Trans., 1990, 86, 1873–1880 RSC.
- J. Rebek, L. Marshall, R. Wolak, K. Parris, M. Killoran, B. Askew, D. Nemeth and N. Islam, J. Am. Chem. Soc., 1985, 7476–7481 CrossRef CAS.
- J. F. Feil-Jenkins, K. L. Nash and R. D. Rogers, Inorg. Chim. Acta, 1995, 236, 67–74 CrossRef CAS.
- Q. B. Bo, S. Y. Zhao, Z. W. Zhang, Y. L. Sheng, Z. X. Sun, G. X. Sun, C. L. Chen and Y. X. Li, CCDC 608247: Experimental Crystal Structure Determination, 2014, DOI:10.5517/ccndxwt.
- S. Darlow, Acta Crystallogr., 1961, 14, 159–166 CrossRef CAS.
- KnowItAll academic edition, Bio-Rad Laboratories, 2005.
- Plot2: a scientific 2D plotting program for OS X, Version 2.6.5 (9969), 2018.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- L. J. Barbour, XSeed: A Program for the manipulation and display of crystallographic models.
- POV-Ray, Persistence of Vision Raytracer, 2012.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information in CIF format has been deposited with the Cambridge Crystallographic Data Centre, deposition numbers CCDC 1984518–1984528. XRPD data, IR spectra and TGA scans are available as supplementary information. For ESI and crystallographic data in CIF or other electronic format see DOI:10.1039/d0ce00243g |
| This journal is © The Royal Society of Chemistry 2020 |