
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring supported lipid bilayers with n-type organic electrochemical transistors†
Malak
Kawan
a,
Tania C.
Hidalgo
 a,
Weiyuan
Du
b,
Anna-Maria
Pappa
c,
Róisín M.
Owens
c,
Iain
McCulloch
bd and
Sahika
Inal
*a
a,
Weiyuan
Du
b,
Anna-Maria
Pappa
c,
Róisín M.
Owens
c,
Iain
McCulloch
bd and
Sahika
Inal
*a
aKing Abdullah University of Science and Technology (KAUST), Division of Biological and Environmental Sciences and Engineering, Organic Bioelectronics Laboratory, Saudi Arabia. E-mail: sahika.inal@kaust.edu.sa
bKAUST, KAUST Solar Center, Division of Physical Sciences and Engineering, Saudi Arabia
cUniversity of Cambridge, Department of Chemical Engineering and Biotechnology, UK
dImperial College London, Department of Chemistry and Centre for Plastic Electronics, UK
First published on 11th June 2020
Abstract
Supported lipid bilayers (SLBs) have emerged as powerful model systems to study various membrane-governed cellular events. Conducting polymers are excellent materials to establish electrical communication with SLBs. However, forming SLBs that are defect-free on the existing library of electronic polymer films, which have not been designed to interface lipids, remains a challenge. Moreover, the existing polymers are predominantly p-type conductors, hindering the development of devices that can be superior to current technologies. In this work, we synthesized an n-type semiconducting polymer based on a naphthalene 1,4,5,8 tetracarboxylic diimide bithiophene (NDI-T2) backbone functionalized with bio-inspired, lysine-based side chains (L2). The lysine chains, that are oriented on the surface of the film, facilitated the assembly of the zwitterionic lipid vesicles into an SLB. The n-type polymer also proved to be an ideal channel material for the state-of-the-art bioelectronic transducer, i.e., organic electrochemical transistor (OECT). We used the n-type, accumulation mode OECTs to assess the quality of the SLB as well as to monitor the activity of a pore forming protein integrated into the SLB. Our work marks the first demonstration of a bio-functionalized n-type polymer, specifically designed for interfacing the lipid membrane, alongside the high operational stability in biologically relevant electrolytes and sufficient performance in microscale transistors for biosensing applications.
New conceptsWe report the very first ion channel sensor based on an n-type, accumulation mode, microscale organic electrochemical transistor (OECT), which interfaces a supported lipid bilayer (SLB) embedded with a pore-forming protein. Fluidity, mobility, and resistivity are key characteristics of native membranes in our cells where cation channels regulate numerous molecular mechanisms (e.g., the excitation of neurons). Interfacing with biomimetic membranes via electronic devices promises to expand our understanding of the function of these membranes in disease states. The challenge is to form SLBs on semi/conducting films while sustaining high sensitivity and operational stability. The novelty of this work lies in the uniquely engineered n-type semiconducting polymer functionalized with bioinspired side chains to create a desirable surface for the SLB. The “accumulation mode” operation of this n-type device is ideal for biosensing – the transistor switches ON as cations penetrate through the channel, indicating protein activity and SLB integrity. This is the first-time use of an n-type polymer to monitor biological activity, as well as the first demonstration of the air/water stability of an n-type OECT biosensor. The side-chain engineering of semiconducting polymers prompts the development of next-generation bioelectronic hybrids where electron-transporting and cation-sensitive polymers establish bilateral electronic communication with living systems. |
Introduction
Cell membranes regulate the selective transport of ions and biomolecules in and out of the cell, a function crucial for the healthy operation of a whole cascade of following cellular events. Acting as a shield between intra- and extra-cellular environments, the membrane also maintains the cell's integrity by providing a confined interface for cell-surface recognition, signaling, and transport. Monitoring the membrane in action allows us to understand, and maybe control, how it distinguishes the differences between various types of biosignals and their interactions with pathogens.1 These studies are key to unravel processes occurring at the molecular level and their contribution to disease state.Our knowledge of the structural and physical properties of the cell membrane has expanded tremendously since the first deposition of artificial model membranes on solid surfaces.2,3 Supported lipid bilayers (SLBs) are two dimensional assemblies that have emerged as a popular membrane model.4,5 The SLBs are formed typically via adsorption of lipid vesicles (sometimes coupled with membrane proteins) on solid substrates and their subsequent fusion into a bilayer. Their planar geometry enables characterization by various surface-sensitive techniques including fluorescence microscopy, atomic force microscopy (AFM),6 and quartz crystal microbalance with dissipation monitoring (QCM-D).6,7 The ideal substrates for SLB formation have hydrophilic surfaces, such as those of silicon dioxide,6 mica,7 and glass,8 as they allow for adsorption of vesicles; while interactions between the lipid vesicles and the surface play a role in their fusion. Owing to their insulating nature, silica-based substrates provide limited means (compatible only with optical microscopy methods) to monitor the SLB. Secondly, frictional coupling between such rigid surfaces and SLB-incorporated proteins increases the risk of protein denaturation due to a mechanical mismatch at the protein integrated SLB/substrate interface.9,10 This becomes a concern when studying membrane proteins such as ion channels because denaturation disrupts their function.10,11 Ion channels are of utmost importance regulating pathways of many cellular functions,12 ranging from growth to survival,13 and alterations in signaling cascades indicate disease state. Without ion channel proteins (or pore forming species such as toxins) embedded into the membrane, the transport across the bilayer is limited, giving rise to a highly resistive bilayer. Hence, when electronic substrates are used to interface SLBs, they offer a label-free platform to study ion channel activity as they are sensitive to ionic fluxes passing across the membrane.14 Furthermore, if these electronic substrates are made of soft materials, they can even act as “a cushion” and support the conformation of proteins integrated into the SLBs in contrast to lower-moduli counterparts.
(Semi)conducting polymers can fulfill the requirements of an electronic substrate ideal for interfacing SLBs due to a unique combination of properties they exhibit. When diligently designed, conjugated polymers can combine tissue-compatible softness, biocompatibility, and transparency, with mixed electronic/ionic conductivity and mechanical integrity in aqueous solutions.15,16 Integrated into micron scale channels of a high-performance electronic transducer, the organic electrochemical transistor (OECT), they form the basis of powerful biosensors.17 For instance, OECT based electrical impedance sensing platforms have monitored motion, morphology and integrity of barrier forming tissues cultured on top the transistor channel.18,19 Similarly, SLBs assembled on OECT channels have been used to monitor the effect of antibiotic on the bacterial membrane integrity,20 and effect of pore forming toxins on the archaeal membrane integrity.21 The SLBs were predominantly formed on OECT channels comprising the p-type (hole conducting) poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS) films, made from a commercially available dispersion. The SLBs on PEDOT:PSS however contained patches of bilayers with a number of vesicles remaining intact.21,22 This leads to poor modulation of OECT properties. The inherent complexity of the PEDOT:PSS dispersion formulations and the substantial swelling of the polymer film in water23 are expected to increase the surface heterogeneity and roughness, which might be responsible for the difficulties in obtaining a defect-free SLB. Moreover, the negatively charged PEDOT:PSS surface is not compatible with the fusion of all types of vesicles such as those that form negatively charged bacterial membranes. Recent work has shown another method for lipid bilayer formation on PEDOT:PSS, namely, solvent-assisted lipid bilayer formation, to bypass the stringent requirements of vesicle compositions and surface properties.21,24 This method is however only compatible with polymer films that are not soluble in alcohols as the film has to be exposed to lipid micelles which are suspended in an isopropanol/water mixture. This mismatch hinders the application of the method with different types of polymers that should be cast from non-orthogonal solvents. Overall, for some applications in organic bioelectronics, the limitations of PEDOT:PSS have been eclipsed by its set of advantages while new materials and device operations can provide favorable features for biological interfacing and biosensing.
A particular polymer class that is interesting to study lipid bilayers is the n-type (electron transporting) polymers as they specifically respond to cation fluxes. In contrast to PEDOT:PSS, these materials are insulators in the absence of a gate voltage, and they become conducting if cations are introduced into their bulk. This type of operation, known as “the accumulation mode”, is advantageous for ion channel monitoring, as the OECT current increases only when the electrolyte cations penetrate the channel.25 The SLB integrated channel blocks the vertical ion flow and hence keeps the device in OFF state (i.e., no current in the channel) at a given frequency of recording. When an ion channel is integrated to the SLB and it is open, it allows cations to flow towards the channel, leading to an abrupt increase in the OECT current, such large changes in drain current cannot be seen in depletion mode PEDOT:PSS based OECTs. Moreover, the accumulation mode OECTs have the advantage of requiring low power inputs compared to depletion mode devices (e.g., those comprising PEDOT:PSS) which are always ON. While the n-type, accumulation mode OECT is an excellent platform to monitor ion channel activity, the existing (and unfortunately limited) library of n-type polymers does not allow for SLB formation as they either lack functional groups that promote fusion or are not compatible with OECT operation.26 Developing bio-functional n-type semiconducting polymers that show operational stability at the solid/liquid interface will open up avenues in other applications as well, such as energy storage and conversion, neuromorphic computing, and thermoelectrics, to name a few.
In this work, we design an n-type polymer with a dual role; supporting a lipid bilayer and achieving high performance in OECT channels. The polymer contains lysine-based side chains tethered to a naphthalene 1,4,5,8 tetracarboxylic diimide bithiophene backbone, which enable interactions with zwitterionic vesicles while providing sufficient hydrophilicity to achieve high volumetric capacitance. The charged and hydrophilic surface of the polymer film enables vesicle adsorption, rupture, and fusion. We investigate the assembly of SLBs on the surface of these polymer films using fluorescence recovery after photobleaching (FRAP) and quartz crystal microbalance with dissipation monitoring (QCM-D). Taking advantage of the mixed conductivity of the polymer, we build micrometer scale n-type OECT channels, through which we monitor the integrity of the SLB. Incorporating gramicidin (GA) channels into this SLB and modulating its conductance via Ca2+ ions, we demonstrate the functionality and robustness of our microfabricated sensors. While highlighting this very first demonstration of an n-type, accumulation mode OECT for monitoring SLB integrity, we envision that such electron transporting and cation-sensitive polymers will provide a new avenue for interfacing lipid bilayers and various proteins that are integrated therein.
Results and discussion
An n-type semiconducting polymer for interfacing lipid bilayers
The conjugated polymer developed in this work comprises a backbone with electron-deficient naphthalene-1,4,5,8-tetracarboxylic diimide (NDI) and electron-rich bithiophene (T2) units (Fig. 1a). Previous work with the same conjugated backbone showed that functionalization with polar, ethylene glycol side chains results in a polymer film capable of water uptake and ion transport.25,27 These polymers showed stable electrochemical charging in aqueous electrolytes for about 2 hours,27 promising for biological applications at the interface with aqueous systems. As an alternative to ethylene glycol side chains, we attached lysine-based charged side chains to the backbone (see ESI† for the synthetic route). Lysine functionalization aimed to render the surface biologically relevant for zwitterionic lipid membrane components. We also expected the lysine chains to increase the hydrophilicity of the NDI-T2 backbone, facilitating the penetration of hydrated ions in the bulk of the film and their transport therein.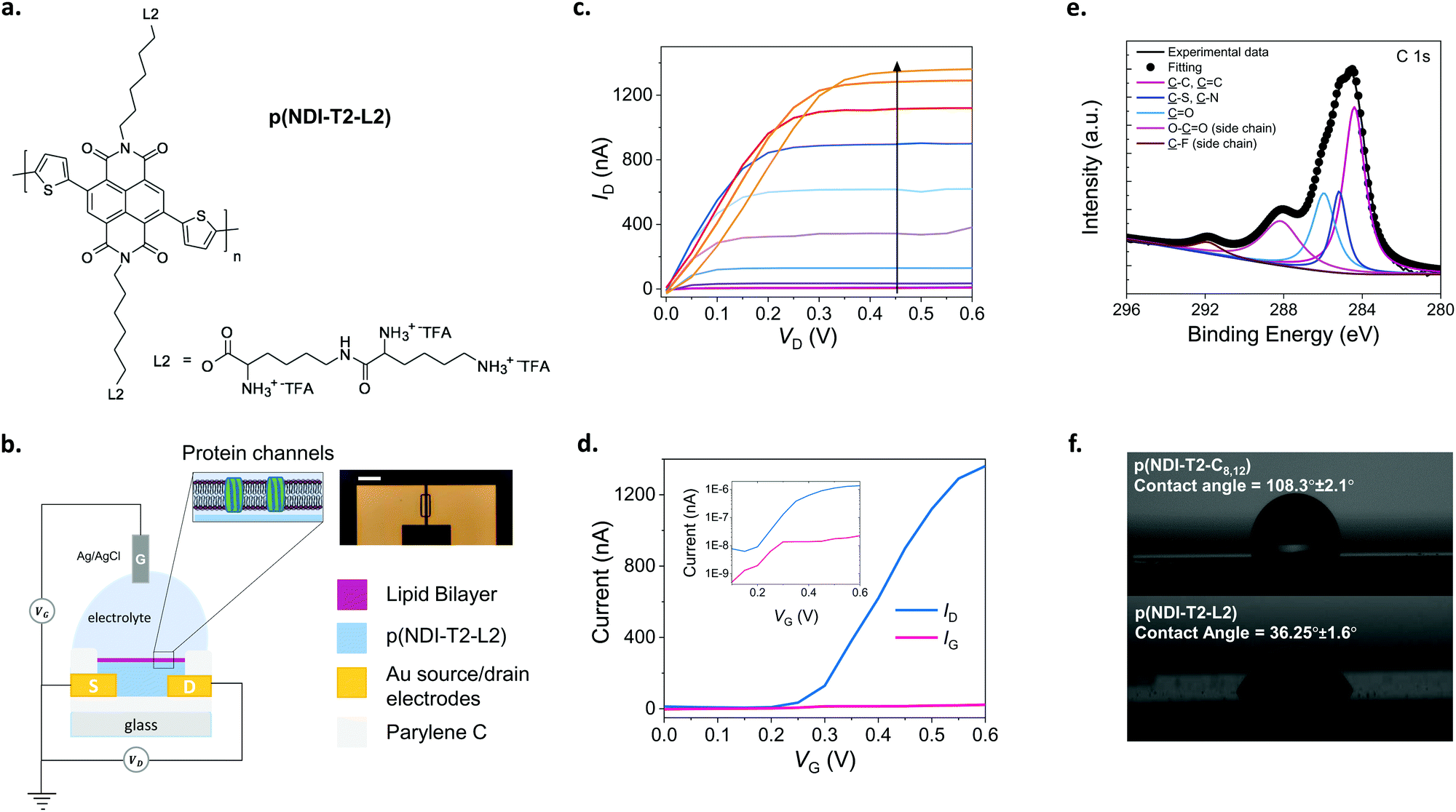 | ||
| Fig. 1 The n-type, lysinated semiconducting polymer and the accumulation mode OECT. (a) The chemical structure of the n-type semiconducting polymer, p(NDI-T2-L2). L2 corresponds to the lysine-based side chains with TFA being the trifluoroacetic acid. (b) Schematic of the OECT configuration alongside a microscope image of a channel (scale bar: 100 μm). The channel dimensions are 10 μm (length) × 100 μm (width), and the film thickness is ∼100 nm which was measured by a mechanical surface profilometer. The SLB (purple) with integrated gramicidin pores (green) is shown on the n-type channel (blue) suspended in PBS where Ag/AgCl was used as the gate electrode. (c) Output characteristics of a best performing p(NDI-T2-L2) OECT recorded in PBS after being treated with Ca2+. Gate voltage increases from 0 to 0.6 V with a step of 0.05 V, following the direction of the arrow. (d) Transfer characteristics of a best performing p(NDI-T2-L2) OECT measured in PBS at VD = 0.6 V along with the corresponding gate current. Curves are shown both on a linear scale, as well as on a logarithmic scale. (e) XPS spectrum of a p(NDI-T2-L2) film. (f) Water contact angle images for the p(NDI-T2-C8,12) and p(NDI-T2-L2) cast from chloroform and ethanol on glass substrates, respectively. | ||
We determined the ionization potential of our lysinated n-type polymer, named hereafter as p(NDI-T2-L2), using photoelectron spectroscopy in air (PESA). The p(NDI-T2-L2) exhibited a slightly higher ionization potential (5.9 eV) than the branched alkyl analogue, p(NDI-T2-C8,12) (5.7 eV). This result in contrast to the effect of ethylene glycol side chain functionalization on NDI-T2 based copolymers, which lowered the IP by interacting with the polymer backbone and increasing the electron density.25 The energetic difference between the work function of Au electrode and transport level of the n-type mixed conductors can be typically large, which leads to contact resistance in OECTs, inhibiting charge carrier injection/extraction. We have however reported for a similar n-type OECT that while contact resistance can be influential for low VG regime, at higher VG regime ion doping compensates this effect and hence n-type OECT performance is not governed by the energetic mismatches at the Au/polymer interface.28 The performance of such devices can however be further improved by ensuring wetting of the polymer on the electrode overlaps. The cyclic voltammetry (CV) curves of the polymer film recorded in phosphate buffered saline (PBS) show that that the reduction onset is around −0.2 V vs. Ag/AgCl, indicating the high electron affinity of the polymer (Fig. S1a, ESI†). The curve shows a reversible redox couple located at ca. −0.2/−0.3 V with reduction currents increasing drastically from −0.2 V onwards. The almost identical shape of 3 consecutive cycles suggests exceptional stability of the polymer film in the aqueous buffer (Fig. S1a, ESI†). The linear increase of the peak reduction current with scan rates suggests a diffusion-controlled charging for the film (Fig. S1b, ESI†). The magnitude of electrochemical impedance of the film recorded in PBS decreases drastically as the film is subject to doping potentials (<−0.2 V vs. Ag/AgCl) (Fig. S1c, ESI†). From the impedance spectrum recorded at −0.6 V vs. Ag/AgCl, we extract the volumetric capacitance of the film to be ca. 95 F cm−3 at 1 Hz (Fig. S1d, ESI†). The large change in the capacitance of the film induced by doping potentials in the aqueous medium suggests that this polymer will be ideal for use in an OECT. Therefore, we next fabricated OECTs with micrometer-scale channels patterned on Parylene C (PaC) coated glass substrates. PaC coating enhanced the adhesion of the polymer film to the substrate and ensured its integrity in the channel during the course of our experiments. We used an Ag/AgCl pellet as the gate electrode which was immersed in PBS in contact with the channel, as illustrated in Fig. 1b, on top of which the SLB was formed. The miniaturized dimensions of the OECT channel (a width of 100 μm and a length of 10 μm) was chosen to maximize the probability of a defect-free bilayer formed on top of channel while accommodating multiple channels on the chip covered by the same lipid bilayer solution. Fig. 1c shows the output characteristics of the best performing p(NDI-T2-L2) OECT. As the gate voltage (VG) exceeds 0.15 V, the film becomes conducting with the maximum source–drain current (ID) values reaching 1.3 ± 0.064 μA measured at a VG of 0.6 V and a source–drain voltage (VD) of 0.6 V. Fig. 1d shows the transfer characteristics of the same device along with the corresponding gate current. The low threshold voltage of the OECT (Vth = 0.22 V) is in agreement with the onset of the reduction current observed in the CV curve. The typical transconductance (gm = ∂ID/∂VG) of the micrometer scale devices is ca. 840 nS at VG = ≈0.45 V (see Fig. S2 for transconductance versus VG plots of various devices, ESI†). The device shows an ON/OFF ratio of ca. 220 (VD = VG = 0.6 V) and with the OFF currents matching the gate currents which are on the order of 1–22 nA (Fig. 1d). Fig. S2 (ESI†) shows data from other channels, following a similar trend. Future work aims to lower the leakage current further for these devices. Moreover, the devices showed minimal hysteresis with almost identical behavior, as observed from backward and forward voltage scans at different sweep rates (Fig. S3, ESI†). We next evaluated the aqueous stability of the devices by recording their steady-state and transient characteristics. The ON currents of devices measured after one week of first-time use were similar to those of fresh devices (Fig. S4a and b, ESI†). Note that within that week, the devices have been stored in air and used multiple times. Fig. S4c and d (ESI†) show that the current of the doped channel does not change over 100 cycles of continuous gate and drain sweeps. The devices also show no negligible change in the drain current as they were addressed with continuous square shaped gate bias pulses (VG = 0.5 V, 5 s) for one hour (Fig. S4e, ESI†). Table S1 (ESI†) summarizes the performance of p(NDI-T2-L2) OECTs used in this work. These results evidence the exceptional stability of the n-type polymer film in the OECT channel alongside the high gain and low threshold voltage of the devices, promising for electrical monitoring of the integrity of the SLB which will be assembled on top of the channel.
The lipid–surface interactions and the resulting fusion are governed by a complex interplay of various factors such as surface hydrophilicity, charge density, roughness as well as the rigidity of lipids, their composition, polarity and size.3,29–31 Although the exact mechanism is not completely understood, the transformation from a vesicle to a planar bilayer involves, firstly, the adsorption of small unilamellar vesicles on the substrate.3 After vesicle crowding and reaching a critical concentration on the surface, the vesicles deform, rupture, and spread, finally transforming into an SLB. Since conjugated polymer films typically have hydrophobic and uncharged surfaces, they may not be regarded as ideal candidates for vesicle fusion, where the vesicles are likely to remain intact.22 We, therefore, first investigated the elemental composition of the p(NDI-T2-L2) film surface using X-ray photoelectron spectroscopy (XPS). XPS analysis suggests that lysine groups are located at the uppermost surface of the film, alongside the backbone (Fig. 1e). We recorded XPS signals from organic fluorine and carboxylic acids, two molecular features that are only present in the side chains (Fig. S5, ESI†). We also found that the film exhibited a relatively hydrophilic surface, confirmed by a water contact angle measured to be 36.25° ± 1.6° (Fig. 1f, bottom). The effect of lysine groups on the surface polarity becomes more obvious when this value is compared to the contact angle measured for the alkylated analogue of NDI-T2 (p(NDI-T2-C8,12)), which is 108.3° ± 2.1° (Fig. 1f, top). We also found the zeta potential of the p(NDI-T2-C8,12) to be −59.07 ± 2.94 while the value becomes more positive for p(NDI-T2-L2) (−22.5 ± 0.55), attributed to the positively charged lysine groups accumulated on the surface. From the atomic force microscopy (AFM) image of the film in air (Fig. S6a, ESI†), we calculated a root mean square (RMS) roughness of 4.9 nm in a scan area of 5 μm by 5 μm, suggesting that the film surface may be compatible for the fusion of vesicles which were reported to prefer smooth surfaces.30–32 In PBS, this polymer swells only up to 8% of its initial thickness (Fig. S6b, determined by quartz crystal microbalance with dissipation monitoring studies, ESI†), a value significantly lower than that of PEDOT:PSS – which is about 80%.23 While water uptake enhances the softness of the film, we hypothesize that excess swelling could lead to a heterogenous surface, hindering SLB formation. Swelling is also suggested to be responsible for mechanical and electrochemical instability of conjugated polymer films subject to bulk charging.33 The minimized swelling of our polymer alongside its high OECT performance is an advantage for SLB integration on OECT channels made thereof. Taken together, p(NDI-T2-L2) is a promising mixed conductor to interface lipids and use in OECT channels thanks to the lysine modification at the side chains.
SLB formation on p(NDI-T2-L2) films
We used the 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) vesicles to assembly a SLB on top of the p(NDI-T2-L2) film (Fig. 2a). These zwitterionic lipids have a positively and a negatively charged moiety within their head groups (phosphocholine). DOPC is an unsaturated lipid with two fatty acid chains with a single double-bond in each chain, a common lipid used to form biologically relevant SLBs.2,34 We used two well-established methods to investigate whether a lipid bilayer is formed on the p(NDI-T2-L2) film and evaluate its quality by comparing our findings to those of the literature. Firstly, we used fluorescence recovery after photobleaching (FRAP) to determine the fluidity and diffusion of the lipid bilayer formed on the polymer surface. We mixed a small quantity of fluorophore-labeled vesicles (BODIPY-DHPE) with DOPC and fused them on the p(NDI-T2-L2) coated glass slides. A defined area was then photobleached and the recovery of the fluorescence intensity at that area was monitored in real-time. From the changes in intensity over time, the diffusion coefficient (D) of the lipids within the SLB and their mobile fraction (MF) were estimated. Fig. 2b shows FRAP images of DOPC lipids on a p(NDI-T2-L2) film where we observe the recovery of fluorescence signal at the bleached plot within 2 minutes, suggesting rupture of vesicles and their fusion into a SLB. The obtained values of D and MF (D = 2.1 ± 0.23 μm2 s−1, MF = 0.86 ± 0.05) and the fit for the data (Fig. S7, ESI†) are in agreement with values reported for SLBs formed on glass substrates (D = 1.5 ± 0.40 μm2 s−1, MF = 0.91 ± 0.01, Fig. S8a, ESI†), which are the ideal surfaces for SLB formation.35,36 These results suggest that the vesicle constituents of the SLB formed on top of the film have sufficient lateral diffusion. | ||
| Fig. 2 Characterizing lipid bilayer formation on p(NDI-T2-L2) films via FRAP and QCM-D. (a) Chemical structure of DOPC vesicles used to interface the polymer surface and a schematic depicting assembly of lipid vesicles into an SLB on the p(NDI-T2-L2) surface. (b) FRAP images taken before the beam and after photobleaching at t = 0 and t = 2 min (from left to right). (c) Frequency (hollow circles) and energy dissipation (filled circles) shift responses for DOPC lipid vesicles on p(NDI-T2-L2) coated sensor surface. Step 1: vesicle addition, Step 2: PBS rinse. The measurement values show the final frequency and energy dissipation shifts of the third harmonic. | ||
We next performed quartz crystal microbalance with dissipation monitoring (QCM-D) studies to follow vesicle fusion on pNDI-T2-L2 films coated on QCM-D crystals. This is a surface-sensitive technique that is used to monitor, in real-time, interactions of vesicles with the film surface and SLB formation through changes in the resonance frequency (Δf) and the dissipation of energy (ΔD) of the crystal. While Δf signals are associated with changes in mass uptake/release, ΔD signals are related to the viscoelastic properties of the layer. As the p(NDI-T2-L2) film is exposed to DOPC vesicles (step 1), we observe a monotonic increase in dissipation and a decrease in frequency, indicating that the DOPC vesicles are absorbing on the film surface (Fig. 2c). At t = 36 min, there is an abrupt decrease in ΔD while the decrease in Δf slows down. We attribute this behavior to the water loss from the adsorbed vesicles as they assembly into a bilayer resulting in simultaneous vesicle adsorption.37,38 As we replace the vesicle solution with PBS (step 2 in Fig. 2c), both signals stabilize with a final mass accumulating on the sample calculated to be 776 ng cm−2 (Δfmin = −39 Hz, calculated from Sauerbrey model, see experimental section for details) while the ΔD decreasing to ΔDfin = 1.1 × 10−6. The ΔD value decreased, indicating a strongly coupled membrane on the film surface and the change of the signal over time is comparable to the ideal case,39,40 which is an SLB on glass (ΔDfinal = 0.4 × 10−6) (Fig. S8b, ESI†). It is important to note that the presence of the thin n-type film on the sensor may lead to higher dissipation values. The incomplete increase in Δf, however, may suggest the presence of unfused vesicles on the surface alongside supported lipid bilayer patches.38 Note, however, that the vesicle solution will interface an area of 0.5 cm2 on our chip, covering 6 identical OECT channels underneath. Given the miniaturized dimensions of the OECT channel (100 × 10 μm2), the probability of a defect-free bilayer formed on top of channel is maximized compared to the case of these larger films used in FRAP and QCM-D experiments. Moreover, as we will see later (Fig. 3g), the FRAP monitored directly on top of the channel happens within 1 min after photobleaching, evidencing the bilayer formed therein.
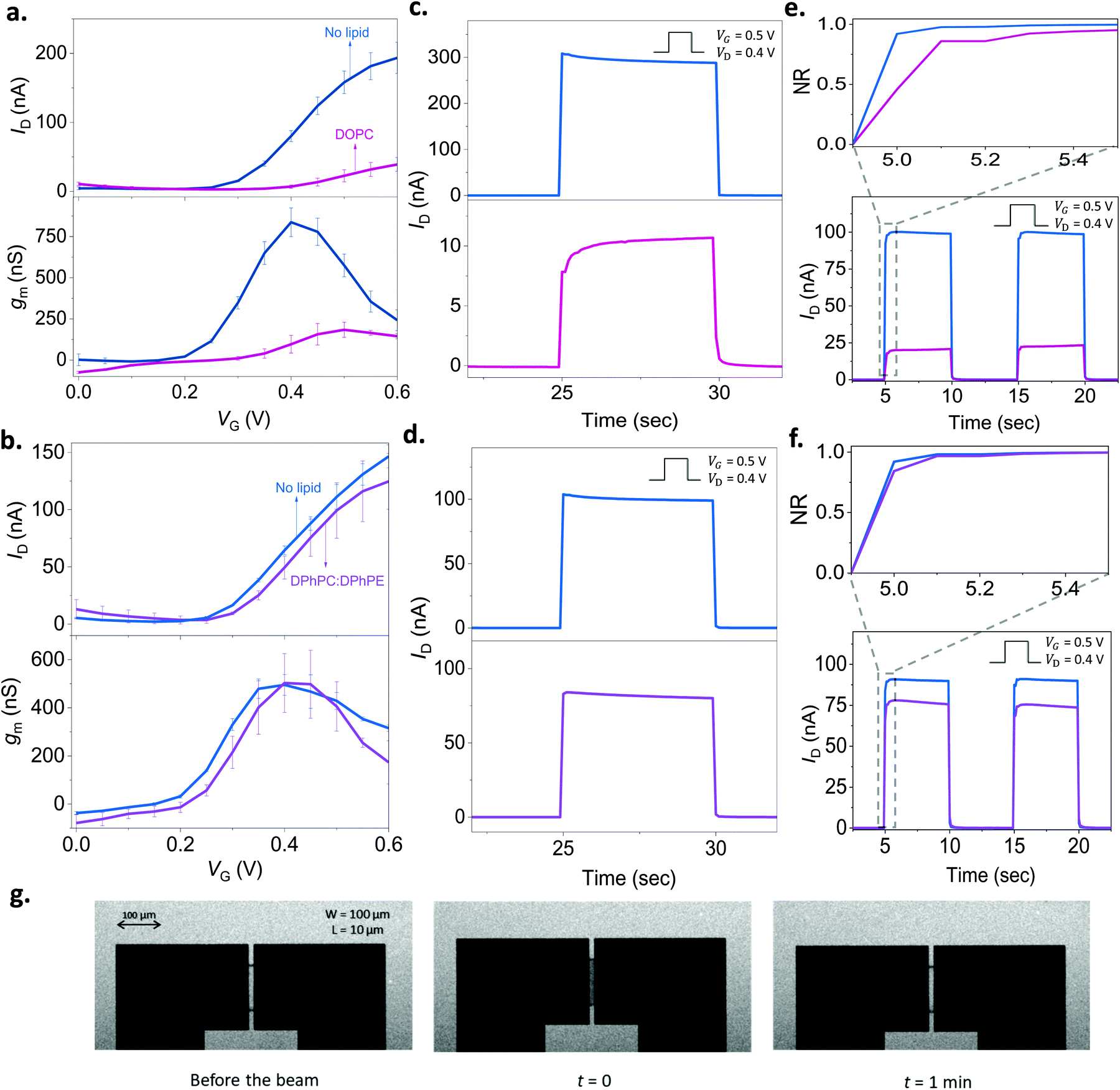 | ||
| Fig. 3 Monitoring the lipid bilayer with p(NDI-T2-L2) based OECTs. The gate voltage dependence of the drain current (ID, top) and the transconductance (gm, bottom) before (blue) and after the addition of lipid vesicles on the channel: (a) DOPC (pink), (b) DPhPC:DPhPE (purple). The gm is reported for VD = 0.6 V. The transient response of the channel before (blue) and after the addition of lipid vesicles: (c) DOPC (pink), (d) DPhPC:DPhPE (purple). (e) and (f) show the response after subtracting the capacitive current component alongside the normalized transient profile of the channel (x-axis: time, y-axis: NR) (inset) for DOPC and DPhPC:DPhPE, respectively. For the transients, VD was 0.4 V, and VG was 0.5 V applied for 5 seconds with a 5 second duty cycle. The error bars in all the data presented are derived from measurements from at least 3 different devices or more for each condition. g. FRAP images taken in the OECT channel showing the formation of DOPC based SLB. Note the transparency of the n-type film allowing to take these images. | ||
n-Type OECTs for monitoring SLB integrity
As we found out that a DOPC based SLB can be formed on the p(NDI-T2-L2) film, we next assembled the lipid bilayer on the micrometer scale p(NDI-T2-L2) channel of an OECT (see the schematic in Fig. 1b). Fig. 3a shows the current vs. gate voltage (transfer curve) and the gain (transconductance, gm) vs. gate voltage plots of these devices before and after the assembly of the SLB, evidencing distinct differences between the two conditions. Firstly, the p(NDI-T2-L2) OECT operates in accumulation mode in the aqueous electrolyte (PBS) gated with an Ag/AgCl pellet (recall Fig. 1c and d). Upon application of a positive gate voltage (VG), cations penetrate the film and compensate for the injected electrons, the net result being an increase in polymer conductance, thus the OECT drain current (ID). ID increases with an increase in VG, consistent with the accumulation mode operation. If the SLB formed on top of the p(NDI-T2-L2) is defect-free and resistive enough to block the vertical cation transport, the drain current would remain at low values, even when the gate bias is high. The formation of the SLB, indeed, causes a substantial reduction in ID (e.g., for VG = 0.4 V and VD = 0.6 V, ID = 80 ± 8.0 nA for the lipid-free vs. ID = 7.0 ± 2.4 nA for the SLB assembled channel) and prevents the device from reaching a high transconductance (gm = 837 ± 87 nS vs. gm = 96 ± 54 nS at VG = 0.4 V before and after DOPC fusion, respectively). These changes in the steady-state properties of the device are accompanied with a shift in the threshold voltage towards more positive values (Vth ≈ 0.22 V and 0.30 V before and after the lipid assembly, respectively). The sealing properties of the DOPC SLB become more evident when we compare the characteristics of OECT channels incubated with non-fusing vesicles. For this negative control experiment, we chose a mixture of two types of lipids, namely, 1,2-diphytanoyl-sn-glycero-3-phosphocholine and 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (DPhPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DPhPE; 7:3). These vesicles remained intact on the polymer film, confirmed by FRAP and QCM-D measurements (Fig. S9, ESI†). Fig. 3b shows that there are no notable differences in the output characteristics of the OECT upon adsorption of DPhPC:DPhPE vesicles on the channel. For example, at VG = 0.6 V and VD = 0.6 V, the maximum ID is measured to be 146 ± 10 nA before the addition of lipids and 124 ± 24 nA after their adsorption, suggesting that ions are able to penetrate the polymer when its surface is covered with (non-fusing) vesicles.
:DPhPE; 7:3). These vesicles remained intact on the polymer film, confirmed by FRAP and QCM-D measurements (Fig. S9, ESI†). Fig. 3b shows that there are no notable differences in the output characteristics of the OECT upon adsorption of DPhPC:DPhPE vesicles on the channel. For example, at VG = 0.6 V and VD = 0.6 V, the maximum ID is measured to be 146 ± 10 nA before the addition of lipids and 124 ± 24 nA after their adsorption, suggesting that ions are able to penetrate the polymer when its surface is covered with (non-fusing) vesicles.
It is not only the steady-state current, but the transient response is also sensitive to the presence of lipids. To determine the switch-ON/OFF time of the devices, we applied 5-second-long square-shaped pulses at the gate electrode with a duty cycle of 5 seconds and magnitude of 0.5 V. We monitored the changes in drain current during these pulses in real-time while the channel was biased at 0.5 V or 0 V. The latter condition (VD = 0 V) was performed to account for stray capacitance currents in the measured response. In Fig. 3c, we notice that the transient response profile of the OECT changes drastically once the DOPC assembles into a SLB on the channel. For the DPhPC:DPhPE vesicles, however, there is no significant change in the shape of the transients (Fig. 3d). Bernards and Malliaras have proposed a model for transient current response of OECTs describing two qualitatively different behavior: monotonic decay vs. spike-and-recovery.41 When the electronic transport of the organic film is faster than the ionic transport, the temporal response of ID for a given VG is monotonic. A spike-and-recovery, on the other hand, indicates that ionic charging is the speed limiting step, yielding a spike in the drain current which stabilizes exponentially.41 In our response time measurements, we observed spike and recovery behavior for the pristine (lipid-free) devices as well as for those covered with DPhPC:DPhPE vesicles. DOPC exposed channels, on the other hand, exhibit a monotonic decay (see the response of other channels in Fig. S10a–c, ESI†). When we subtracted the capacitive current component (measured at VD = 0 V) from the current transient recorded at VD = 0.4 V, we clearly observe that the channel switches ON more slowly following the addition of DOPC, a result of the barrier that the SLB imposes (Fig. S10c, ESI†). Overall, both the steady state and transient response of the n-type OECTs can be used to assess the integrity of the SLBs. A further demonstration of the SLB formation on the channel comes from the FRAP images that we took from these very same channels (Fig. 3g). The images show the recovery of the bleached spot on the micrometer scale channel 1 min after the exposure of the beam (Movie S1, ESI†). Thanks to the transparency of the polymer film, we could simultaneously optically monitor the SLB on the OECT channel, highlighting the multi-modality of our platform.
n-Type OECTs for monitoring the activity of a transmembrane channel
To assess the ability of our platform in monitoring channel activity, we incorporated a small peptide antibiotic, gramicidin A (GA), into the DOPC bilayer. GA has been used extensively to study interactions of peptides and lipids, providing mechanistic insight of membrane protein function.42 The ability of GA to form cation-selective channels for monovalent ions alongside its effect on macroscopic lipid organization have rendered this protein a gold standard for studies of protein–lipid model systems.43,44 In phospholipid membranes, conformations of GA may be distinguished by evaluating its ability to form cation permeable transmembrane channels.44 We assembled SLBs on OECT channels from a mixture of DOPC vesicles with various concentrations of GA. Fig. 4a shows the transfer characteristics of selected channels. The presence of GA in the bilayer has a clear impact on the channel current: the higher the concentration of GA in the bilayer, the higher the ON currents, reflecting the permeability of the n-type channel to cations. Fig. 4b demonstrates the effect of GA content of the SLB on OECT output current. We calculated the normalized difference between the maximum ON current of a pristine device (lipid free) and that of a GA-incorporated SLB incorporated device and plotted these “permeability” values as a function of the GA content of the SLB. We find that the SLB formed from a mixture of DOPC vesicles with 0.1 mg mL−1 of GA yields the highest permeability to cations (0.15, lowest bilayer sealing). We also report 0.025 mg mL−1 of GA content having the lowest permeability to ions (0.71, highest bilayer sealing) as opposed to 0.01 mg mL−1.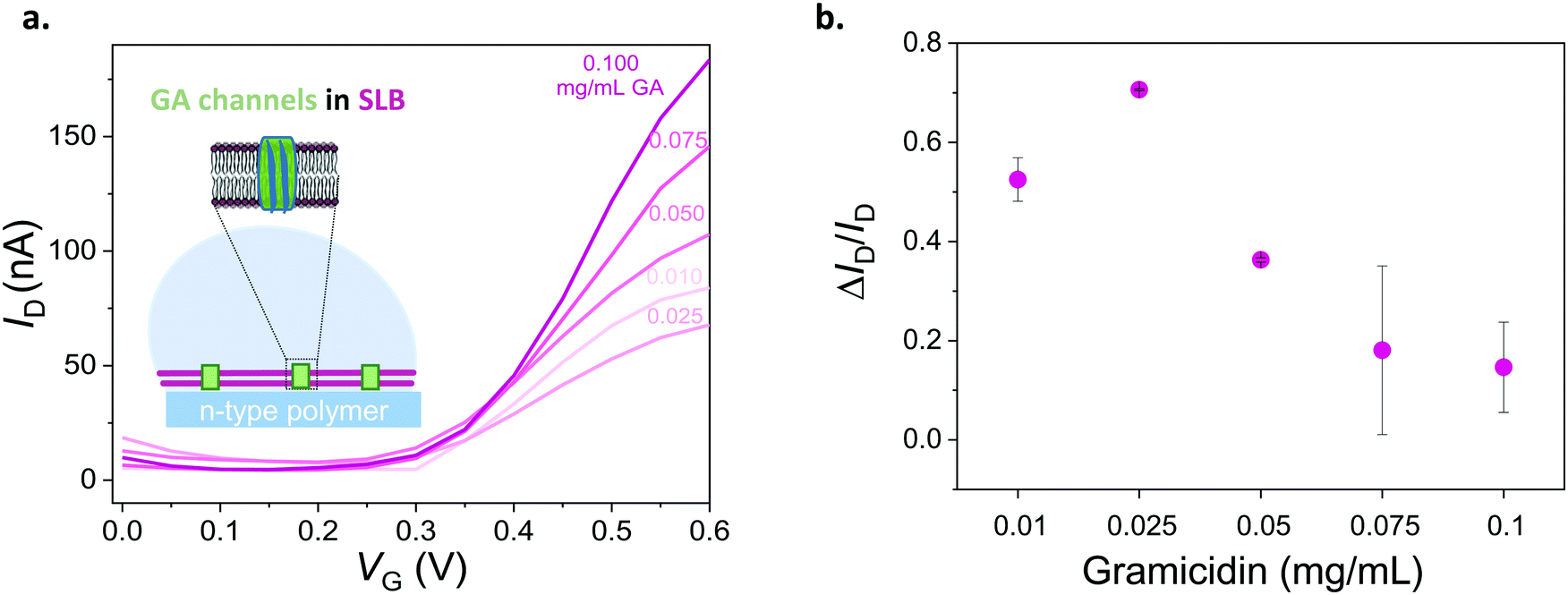 | ||
| Fig. 4 Electrical monitoring of a channel protein that is integrated into a lipid bilayer. (a) Transfer curves of devices with DOPC bilayers containing different concentrations of gramicidin (from 0.1 mg mL−1 to 0.01 mg mL−1). VD = 0.6 V. (b) Normalized responses of OECTs depicting the sealing properties of the DOPC lipid bilayer with respect to the GA concentration therein. Normalized response (ΔID/ID) is calculated as (ID − ISLB)/ID, where the maximum current of the GA-containing SLB integrated OECT is ISLB and that of the pristine device is ID. The error bars are the standard deviation of the measured average of at least 3 devices for the same condition (for 0.025 and 0.05 mg mL−1 of GA, error bars are from 2 devices). | ||
As we confirmed that GA integrated SLBs have permeability to cations, the next question concerns the conformation of GA in the SLB. GA is known to adopt two forms, channel (pore) forming or SLB integrated.45 While a pore forming confirmation would enable the cation permeability that we observed above, GA may, as well, simply be generating defects in the bilayer, making it leaky and, therefore, leading to similar changes in OECT characteristics. Besides providing selective transport to monovalent cations, GA pores are known to be blocked by Ca2+.46,47 We hypothesized that if the peptide forms pores in the membrane, the GA-integrated bilayer permeability should be compromised in the presence of Ca2+. We thus recorded the current response of the GA-incorporated SLB channels in the presence of increasing concentrations of CaCl2 in the medium. These experiments would also benchmark the sensitivity of our device to changes in the permeability of the bilayer, hence, the Ca2+ triggered activity of the channel.
First, we show that the presence of variable concentrations of CaCl2 has no effect on the integrity of the DOPC bilayer if the bilayer does not contain GA in the first place (Fig. 5a, see also Fig. S8 for results from other channels, ESI†). Then, we record the transfer characteristics of OECTs comprising the GA (0.1 mg mL−1) integrated bilayer in the presence of the same Ca2+ concentrations in the measurement solution. Fig. 5b demonstrates that an increase in Ca2+ concentration limits the currents that can otherwise be generated by the n-type channel. For instance, at a gate voltage of 0.4 V, we measure a drain current of 174 ± 14.2 nA when the measurement solution contained no CaCl2. In the presence of 0.94 M of CaCl2 in the solution, the current is, however, only 104 ± 5.9 nA (see Fig. S12, for data from other channels, ESI†). High concentrations of Ca2+ in the solution cause a ca. 50% reduction in the maximum current that can be otherwise drawn from the SLB integrated channels. We suggest that the divalent cation blocks the GA pores and dopant cations can no longer drift towards the polymer channel through these closed pores. The pores are closed by Ca2+ and we can see the effect in the OECT current. We note that with 0.56 M of Ca2+ in the solution, the channel shows slightly lower ID than with 0.94 M. Although the blocking mechanism is not fully understood, we suspect that at this high concentration of Ca2+, the GA pores are already fully blocked and the difference between 0.56 M and 0.94 M becomes negligible. An additional control experiment showed us that the n-type channel (in the absence of SLB) has ON currents increasing with Ca2+ concentration (Fig. S11; upper panel, ESI†). Interestingly, once measured at electrolytes with high CaCl2 concentrations, the channel becomes permanently doped, while its electrochemical charging ability remains unaffected and the device in fact achieves higher ON currents and transconductance values in PBS (Fig. S11; lower panel, ESI†). Cycling the devices in CaCl2 bearing solutions can thus be a strategy to improve the OECT ON currents and transconductance. Investigation of the origin of this increase and its optimization for this particular n-type polymer as well as applicability to other n-types polymers are beyond the scope of this current work. Overall, with these control experiments, we also rule out the possibility of a Ca2+ induced decrease in the SLB-free channel current and verify that the decrease in the current of GA integrated SLB devices is due to the Ca2+ ions blocking the GA pores.
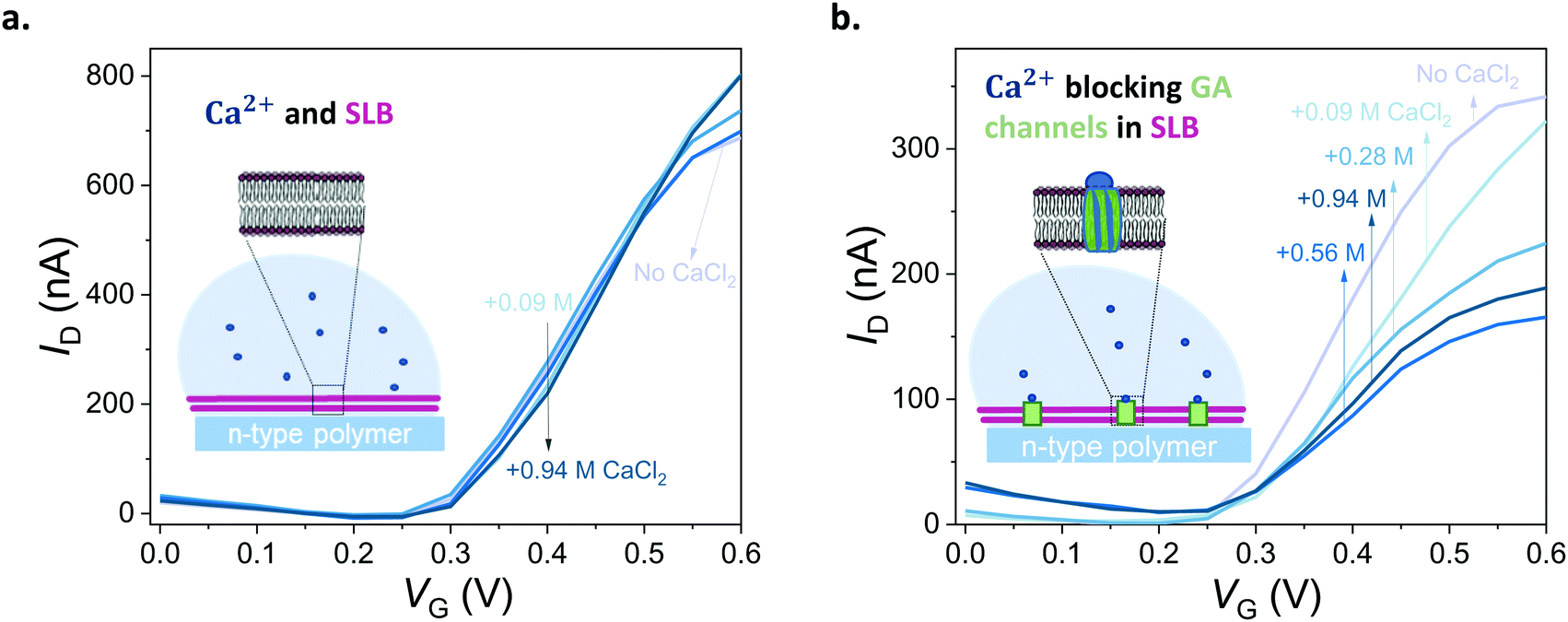 | ||
| Fig. 5 Blocking the ion channels of the lipid bilayer with Ca2+. (a) Transfer curves of a DOPC SLB integrated OECT in the presence of different concentrations of CaCl2 (0.09–0.94 M, lightest to darkest blue, respectively). (b) Transfer curves of an OECT channel comprising a DOPC bilayer with inbuilt GA (0.1 mg mL−1) in the presence of different concentrations of CaCl2 (0.09–0.94 M, lightest to darkest blue, respectively). VD = 0.6 V. CaCl2 in deionized water was titrated into PBS to reach the indicated concentrations. “No CaCl2” curve indicates Ca2+ free PBS. | ||
Conclusion
In this work, we designed an n-type polymer functionalized with lysine side chains, firstly, to create desirable surface properties for vesicle adsorption and fusion into an SLB and, secondly, to enable bulk cation charging at low voltages, hence high performance in OECT channels. We demonstrated, for the first time, SLB formation on an n-type polymer and accumulation mode OECT channels made thereof. The SLB formation and quality were evaluated using FRAP and QCM-D measurements, indicating a fluid and mobile bilayer. The micrometer scale, n-type OECT channels offered a sensitive platform to assess the quality of the SLB while its transparency allowed for monitoring the lipid bilayer optically. The OECT current and transient characteristics were sensitive to the presence and function of a pore forming protein integrated into the SLB and exhibited high operational stability. Showing the first example of a bio-functionalized n-type polymer and an n-type OECT interfacing a biomimetic platform, we demonstrated the advantages of developing new and improved chemistries for customized electronics for the biological interface. Future work will focus on enhancing the biomimicry of this platform and integrating catalytic and ligand-gated proteins into the SLB, paving the way for next-generation biosensors and bioelectronic hybrids.Experimental
Materials
The n-type polymer p(NDI-T2-L2) was synthetized according to the synthetic route described in the ESI.† All lipids were purchased from Avanti Polar Lipids (Alabaster, USA). Chloroform, phosphate-buffered-saline, calcium chloride, and gramicidin A were purchased from Sigma-Aldrich while 99% ethanol was purchased from VWR.Vesicle preparation
Lipids were dissolved in chloroform to reach the desired concentration. They were dried using Nitrogen flow for 5–10 min to remove chloroform then placed under vacuum overnight. They were re-suspended in PBS to reach 2 mg mL−1 concentration and sonicated in a sonication bath for 20–30 min. Multilamellar lipid vesicles were extruded through a polycarbonate membrane (pore size: 50 nm, Whatman Nucleopore) using a mini extruder (21 cycles) to make unilamellar vesicles. The average size of the vesicles was measured using dynamic light scattering (Malvern, Zetasizer Nano S). Lipid vesicles containing gramicidin were mixed together in chloroform, using a stock solution of gramicidin A (10 mg mL−1) to form a dilution series of desired concentrations, and then dried with a nitrogen gun, followed by the same extrusion process.Film preparation
Microscope cover glasses (24 × 32 mm; No. 1.5; VWR) and QCM-D crystals were cleaned in a NH4OH:H2O2:H2O (1:1:5) solution for 15 min at 80 °C. p(NDI-T2-L2) was dissolved in 99% ethanol at a concentration of 10 mg mL−1. It was spin coated at 1000 rpm for 30 sec with 1500 rpm of acceleration on any given surface and left to dry overnight at room temperature. Polydimethylsiloxane (PDMS) wells were assembled on the films to confine the lipid bilayer solution.
Contact angle measurements
The contact angle of the films was determined using a goniometer (KRUSS, DSA100, Germany). A drop of water (5 μL) was placed on top of the film and the mean value was obtained from 2 samples.Photo electron spectroscopy in air (PESA)
Measurements were performed with a Riken Keiki AC-2 PESA spectrometer with a power setting of 5 nW and a power number of 0.3. Samples were prepared on glass substrates by the spin-coating procedure as described above.Surface zeta potential
Zeta potential of the films was measured using Zetasizer Nano ZS (Malvern Instruments Ltd, UK). 7 mm × 4 mm glass coverslips were coated with the polymer. A zeta potential cell (ZEN1020) was used where polystyrene beads were the tracer particles. The Zeta potential was measured using the Zetasizer software based on the electrophoretic mobility of the tracer particles from the sample and the distance was measured in 4 different positions. The measurements were performed for 3 times and in 1× PBS (pH 7.4) as the electrolyte.X-ray photoelectron spectroscopy (XPS)
XPS was carried out using a KRATOS Analytical AMICUS instrument equipped with an achromatic Al Kα X-ray source (1468.6 eV). The source was operated at a voltage of 10kV and a current of 10mA generating a power of 100 Watts. The high-resolution XPS spectra were acquired using a step of 0.1 eV. The pressure in the analysis chamber was in the range of × 10−7 Pa during the course of the measurements.
Atomic force microscopy (AFM)
Atomic force microscopy images were obtained with a Veeco Dimension 3100 Scanning Probe System in non-contact mode using FESPA-V2 probes supplied by Bruker (Nominal Resonant Frequency: 75 kHz, Spring constant: 2.8 N m−1). Gwyddion software was used to analyze the data.Fluorescence recovery after photobleaching (FRAP)
FRAP was performed using an inverted Zeiss LSM 880 confocal microscope with Airyscan (Carl Zeiss Germany). Lipids were labeled with a fluorescent lipid (0.5% BODIPY DHPE, Thermo Fisher Scientific) and visualized using a 488 nm laser. Lipid vesicles were adsorbed on the pNDI-T2-L2 film and incubated overnight. A small scratch across the lipid bilayer was made using a needle to facilitate focusing at the bilayer plane using the microscope objective. The bilayer was then washed with PBS to remove unbound vesicles. A defined area was photobleached using the 488 nm-laser to evaluate membrane fluidity where fluorescence intensity recovery and mobile fraction was measured. Data were analyzed in the FRAP module in ZEN 2009 software, which calculated the mobile fraction percentage and fit the data after the subtraction of background and normalization. Diffusion coefficient (D) was calculated using the following equation: D = w2/4t1/2 where w is the radius of photobleached spot and t1/2 is time to reach half of the maximum fluorescence recovery intensity.22Quartz crystal microbalance with dissipation monitoring (QCM-D)
QCM-D measurements were carried out using Qsense Analyzer system (Biolin Scientific AB, Sweden). P(NDI-T2-L2) films were coated on SiO2 based QCM-D sensors as described above. The sensors were annealed at for 10 min at 100 °C to avoid film delamination. The experiments were performed at room temperature with a flow rate of 0.1 mL min−1 using a peristaltic pump. PBS was injected first and stabilized (baseline), then lipid vesicles were added (0.5 mg mL−1). The changes in dissipation (ΔD) and frequency (Δf) over time were analyzed as lipids adsorbed on the film surface. For the swelling experiment, the SiO2 sensor was ran in air and in PBS, then, the same sensor was coated with p(NDI-T2-L2) and measured in air and PBS again. The data were stitched using the QSoft software and the water-swollen mass (m) was calculated using the Sauerbrey equation with a selected overtone (n) (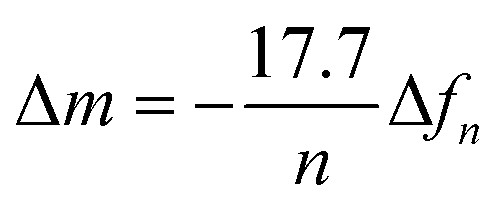 ).23 To calculate the final mass for the lipid bilayer, the difference in mass between PBS stabilized data and after the lipid bilayer formation on p(NDI-T2-L2) were calculated, which is related to Δf.
).23 To calculate the final mass for the lipid bilayer, the difference in mass between PBS stabilized data and after the lipid bilayer formation on p(NDI-T2-L2) were calculated, which is related to Δf.
OECT fabrication
All devices were fabricated using standard photolithographic procedures. Glass substrates were cleaned by immersion in hot piranha solution, rinsing with water, and dried under nitrogen. A final cleaning step was performed using oxygen plasma. A thin layer of Parylene C (SCS Labcoater 2) was vaporised on the substrates and adhered with 3-(trimethoxysilyl)propyl methacrylate (A174). Transistor geometry was transferred to the substrates using a bilayer resists structure (LOR 5B, Microchem; S1813, Shipley), and defined by sputtering 10 nm of Cr as an adhesion layer, followed by 100 nm of Au (Equipment Support Company Ltd ESCRD4). Two parylene C layers were later deposited: one to electrically insulate interconnects, which was also adhered by A174, and another to use a sacrificial layer for polymer patterning. Openings for polymer deposition were dry etched by O2 plasma (Oxford Instruments Plasmalab 100 – ICP 380). Each OECT chip contained 6 identical channels with a width of 100 μm and a length of 10 μm.OECT characterization
We characterized OECTs using Keithley 2612A with a customized LabVIEW software. Ag/AgCl electrode was used as the gate and PBS as the electrolyte. The gate voltage varied from 0 to 0.6 V, just as the drain voltage VD, with a step of 0.05 V. PDMS wells were placed on top of the OECT to contain the solutions. OECT measurements were performed in 1X PBS (i.e., 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, pH 7.4). Measurements in PBS were used as “No lipid” condition in all experiments unless stated otherwise. Lipid:PBS (5:3) mixture was then pipetted on the same devices and left overnight to form a SLB and measured the next day. Gramicidin (GA)-containing lipid vesicles were deposited and incubated as above to form a GA-integrated SLB. After forming the lipid bilayer with GA pores, different concentrations of CaCl2 were added sequentially to the devices followed by a 10–20 min incubation before characterization (total volume of the measurement solution = 80 μL). 0.25, 0.5, 0.75, and 1 M of CaCl2 were added consecutively to the devices resulting in final concentrations of 0.09, 0.28, 0.56, 0.94 M in solution, respectively, while the total volume solution remained constant with each CaCl2 titrations. We calculated the difference in the maximum ON current that was measured with a GA-incorporated SLB assembled device and its pristine condition (no lipid) and plotted these “permeability” values as a function of the GA content in the SLB.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. K. thanks Daniel team (Cornell) for FRAP related tips, and Dr Yi Zhang, Dr Shofarul Wustoni, Victor Druet, and Jokubas Surgailis (KAUST, Organic Bioelectronic Laboratory) for lipid work training, XPS analysis, AFM imaging, and QCM-D swelling analysis, respectively. A. M. P. acknowledges funding from the Oppenheimer Junior Research Fellowship and the Maudslay-Butler Research Fellowship at Pembroke College, Cambridge. This work is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. OSR-2018-CRG7-3709.References
- Y.-H. M. Chan and S. G. Boxer, Curr. Opin. Chem. Biol., 2007, 11, 581–587 CrossRef CAS PubMed.
- L. K. Tamm and H. M. McConnell, Biophys. J., 1985, 47, 105 CrossRef CAS PubMed.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips and M. B. Carter, Nat. Mater., 2011, 10, 389 CrossRef CAS PubMed.
- X. Zhou, J. M. Moran-Mirabal, H. G. Craighead and P. L. McEUEN, Nat. Nanotechnol., 2007, 2, 185 CrossRef CAS PubMed.
- R. Tero, M. Takizawa, Y.-J. Li, M. Yamazaki and T. Urisu, Langmuir, 2004, 20, 7526–7531 CrossRef CAS PubMed.
- R. P. Richter and A. R. Brisson, Biophys. J., 2005, 88, 3422–3433 CrossRef CAS PubMed.
- K. L. Weirich, J. N. Israelachvili and D. K. Fygenson, Biophys. J., 2010, 98, 85–92 CrossRef CAS PubMed.
- S. J. Johnson, T. M. Bayerl, D. C. McDermott, G. W. Adam, A. R. Rennie, R. K. Thomas and E. Sackmann, Biophys. J., 1991, 59, 289–294 CrossRef CAS PubMed.
- E. Sackmann, Science, 1996, 271, 43–48 CrossRef CAS PubMed.
- E. Sackmann and M. Tanaka, Trends Biotechnol., 2000, 18, 58–64 CrossRef CAS PubMed.
- K. Murata, K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J. B. Heymann, A. Engel and Y. Fujiyoshi, Nature, 2000, 407, 599–605 CrossRef CAS PubMed.
- D. E. Clapham, L. W. Runnels and C. Strübing, Nat. Rev. Neurosci., 2001, 2, 387–396 CrossRef CAS PubMed.
- W. Zhou, Y. Y. Wang, T.-S. Lim, T. Pham, D. Jain and P. J. Burke, Sci. Rep., 2015, 5, 9208 CrossRef CAS PubMed.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2013, 26, 679–685 CrossRef.
- S. Inal, J. Rivnay, G. G. Malliaras and I. McCulloch, Acc. Chem. Res., 2018, 51(6), 1368 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- J. Rivnay, M. Ramuz, P. Leleux, A. Hama, M. Huerta and R. M. Owens, Appl. Phys. Lett., 2015, 106, 8_1 CrossRef.
- L. H. Jimison, S. A. Tria, D. Khodagholy, M. Gurfinkel, E. Lanzarini, A. Hama, G. G. Malliaras and R. M. Owens, Adv. Mater., 2012, 24, 5919–5923 CrossRef CAS PubMed.
- C. Pitsalidis, A. M. Pappa, M. Porel, C. M. Artim, G. C. Faria, D. D. Duong, C. A. Alabi, S. Daniel, A. Salleo and R. M. Owens, Adv. Mater., 2018, 30, 1803130 CrossRef PubMed.
- Y. Zhang, S. Inal, C. Y. Hsia, M. Ferro, M. Ferro, S. Daniel and R. M. Owens, Adv. Funct. Mater., 2016, 26, 7304–7313 CrossRef CAS.
- Y. Zhang, S. Wustoni, A. Savva, A. Giovannitti, I. McCulloch and S. Inal, J. Mater. Chem. C, 2018, 6, 5218–5227 RSC.
- A. Savva, S. Wustoni and S. Inal, J. Mater. Chem. C, 2018, 6, 12023–12030 RSC.
- H. Su, H.-Y. Liu, A.-M. Pappa, T. C. Hidalgo, P. Cavassin, S. Inal, R. M. Owens and S. Daniel, ACS Appl. Mater. Interfaces, 2019, 11, 43799–43810 CrossRef CAS PubMed.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia and M. s. Zetek, Chem. Mater., 2018, 30, 2945–2953 CrossRef CAS PubMed.
- Y. Zhang, A. Savva, S. Wustoni, A. Hama, I. P. Maria, A. Giovannitti, I. McCulloch and S. Inal, ACS Appl. Bio Mater., 2018, 1, 1348–1354 CrossRef CAS.
- A. Giovannitti, C. B. Nielsen, D.-T. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras and J. Rivnay, Nat. Commun., 2016, 7, 13066 CrossRef CAS PubMed.
- A. F. Paterson, H. Faber, A. Savva, G. Nikiforidis, M. Gedda, T. C. Hidalgo, X. Chen, I. McCulloch, T. D. Anthopoulos and S. Inal, Adv. Mater., 2019, 31, 1902291 CrossRef PubMed.
- T. Cha, A. Guo and X.-Y. Zhu, Biophys. J., 2006, 90, 1270–1274 CrossRef CAS PubMed.
- R. Tero, Materials, 2012, 5, 2658–2680 CrossRef CAS.
- A. A. Duarte, S. L. Filipe, L. M. Abegão, P. J. Gomes, P. A. Ribeiro and M. Raposo, Microsc. Microanal., 2013, 19, 867–875 CrossRef CAS PubMed.
- J. A. Goodchild, D. L. Walsh and S. D. Connell, Langmuir, 2019, 35(47), 15352–15363 CrossRef CAS PubMed.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- H. Watson, Essays Biochem., 2015, 59, 43–69 CrossRef PubMed.
- C. Hamai, T. Yang, S. Kataoka, P. S. Cremer and S. M. Musser, Biophys. J., 2006, 90, 1241–1248 CrossRef CAS PubMed.
- C. Keller, K. Glasmästar, V. Zhdanov and B. Kasemo, Phys. Rev. Lett., 2000, 84, 5443 CrossRef CAS PubMed.
- R. Richter, A. Mukhopadhyay and A. Brisson, Biophys. J., 2003, 85, 3035–3047 CrossRef CAS PubMed.
- C. Merz, W. Knoll, M. Textor and E. Reimhult, Biointerphases, 2008, 3, FA41–FA50 CrossRef CAS PubMed.
- D. Meléndrez, T. Jowitt, M. Iliut, A. Verre, S. Goodwin and A. Vijayaraghavan, Nanoscale, 2018, 10, 2555–2567 RSC.
- T. J. Zwang, W. R. Fletcher, T. J. Lane and M. S. Johal, Langmuir, 2010, 26, 4598–4601 CrossRef CAS PubMed.
- D. A. Bernards and G. G. Malliaras, Adv. Funct. Mater., 2007, 17, 3538–3544 CrossRef CAS.
- E. Diamanti, E. Gutiérrez-Pineda, N. Politakos, P. Andreozzi, M. J. Rodriguez-Presa, W. Knoll, O. Azzaroni, C. A. Gervasi and S. E. Moya, Soft Matter, 2017, 13, 8922–8929 RSC.
- D. A. Bernards, G. G. Malliaras, G. E. S. Toombes and S. M. Gruner, Appl. Phys. Lett., 2006, 89, 053505 CrossRef.
- J. M. Muller, G. van Ginkel and E. E. van Faassen, Biochemistry, 1995, 34, 3092–3101 CrossRef CAS PubMed.
- J. A. Killian and D. W. Urry, Biochemistry, 1988, 27, 7295–7301 CrossRef CAS PubMed.
- D. A. Kelkar and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2011–2025 CrossRef CAS PubMed.
- F. Heitz and C. Gavach, Biophys. Chem., 1983, 18, 153–163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mh00548g |
| This journal is © The Royal Society of Chemistry 2020 |