
Remote-controlled multi-enzyme system for enhanced tumor therapy via dark/light relay catalysis†
Ying
Chen‡
,
Zi-Hao
Li‡
,
Jing-Jing
Hu
,
Si-Yuan
Peng
,
Lei
Rong
*,
Yunxia
Sun
* and
Xian-Zheng
Zhang
 *
*
Key Laboratory of Biomedical Polymers of Ministry of Education & Department of Chemistry, Wuhan University, Wuhan 430072, P. R. China. E-mail: rong-lei@outlook.com; yx-sun@whu.edu.cn; xz-zhang@whu.edu.cn
First published on 11th October 2019
Abstract
Nanozymes have been widely used in biomedicine, especially in tumor therapy. However, inadequate H2O2 supply in the tumor microenvironment (TME) and uncontrolled catalytic activity of nanozymes in vivo restrict their practical application. Here, a dark/light relay strategy is proposed to realize adequate H2O2 supply in the dark reaction stage and selectively perform desired catalytic activities for toxic reactive oxygen species (ROS) generation to kill tumors by remote light control. A tumor membrane camouflaged and glucose oxidase (GOx) loaded hollow mesoporous Prussian blue (mGPB) nanosystem is designed to target tumor tissues for homologous aggregation of membranes. The cascaded catalysis of superoxide dismutase (SOD) and catalase (CAT)-like activities inherited from hollow mesoporous Prussian blue (HMPB) efficiently catalyze endogenous O2−˙ to O2, which contributes to the oxidative decomposition of glucose to produce H2O2 by loaded GOx. Moreover, mGPB nanoparticles are found to utilize H2O2 to produce ˙OH and 1O2 under NIR irradiation via other light-dependent dual-catalytic properties, acting as peroxidase (POD) and oxidase (OXD). By dark/light relay catalysis, we successfully overcome the limited H2O2 supply in TME and achieve precise ROS generation, displaying prominent tumor suppression in mouse xenograft models.
New conceptsDue to inadequate H2O2 supply in the tumor microenvironment (TME), catalytic therapy of ˙OH generation struggles to achieve satisfactory therapeutic effects. In this work, we report a dark/light relay strategy to realize an adequate H2O2 self-supply in the dark reaction stage and selectively perform desired catalytic activities for toxic reactive oxygen species (ROS) generation to kill tumors by remote light control. We designed a tumor membrane camouflaged and glucose oxidase (GOx) loaded hollow mesoporous Prussian blue (mGPB) nanozyme to target tumor cells. In the dark stage, mGPB nanoparticles efficiently accelerate the GOx-mediated oxidative decomposition of glucose to produce H2O2 by in situ O2 regeneration. Under NIR irradiation, mGPB nanoparticles are found to utilize H2O2 to produce ˙OH and 1O2 for tumor therapy. This dark/light relay strategy successfully overcomes the limited H2O2 supply in TME and achieves precise ROS generation, displaying great potential in tumor catalytic therapy. |
Nanozymes are nanomaterials with intrinsic enzyme-like activities and have been widely used in biomedicine since they were reported in 2007,1 such as antibacterial materials, biosensors and immunoassays.2–4 In particular, a growing number of research reports indicate that nanozymes exhibit great potential in reactive oxygen species (ROS)-based tumor therapy.5–7 Tumor cells with increased intrinsic ROS stress compared to normal cells are more vulnerable to attack by exogenous ROS.8 The peroxidase (POD)-like activity and oxidase (OXD)-like activity of nanozymes, as the main enzyme-like activities for ROS generation in vivo, could catalyze H2O2 or O2 to produce toxic free radicals, respectively, and are recognized as potential modalities to kill tumors.9,10 In contrast to photodynamic therapy (PDT), another widely applied ROS-based anti-tumor modality, the nanozyme system possesses higher stability for the higher resistance of nanozymes to light and complex physiological environments than photosensitizers.11 Additionally, high enzymatic activity further guarantees the excellent performance of nanozymes in physiological environments and the low cost of nanozymes makes it possible to scale up.
For the abnormal metabolism of tumor cells, endogenous H2O2 is generally recognized to be overproduced in the TME compared with normal tissue and has been used to generate toxic free radicals via POD-like catalysis.12 However, the amount of intratumoral H2O2 (50–100 μM) is still insufficient for adequate toxic free radicals generation to destroy tumor cells.13–15 To achieve satisfactory tumor therapy, considerable efforts have been devoted to intratumoral H2O2 supply.14,16–18 As a H2O2-producing strategy, the glucose oxidase (GOx)-mediated catalysis of glucose to generate H2O2 has attracted extensive interest since it was applied in tumor therapy for its excellent catalytic efficiency and adequate glucose supply in vivo.6,19–23 Considering the contradiction between the aerobic property of GOx and hypoxia in TME, the local regeneration of O2 becomes extremely important to maximize the efficacy of GOx.
To increase O2 in TME, endogenous O2 production and exogenous O2 delivery have developed into two main methods.24,25 Very recently, nanozymes (such as CeO2, Prussian blue, etc.) with superoxide dismutase (SOD) and catalase (CAT)-like activities, acting as endogenous O2 producers, have been combined with chemotherapeutic agents or photosensitizers to enhance the efficacy of chemotherapy or photodynamic therapy.26–28 However, many nanozymes usually possess multiple enzyme activities and exhibit different enzyme-like activities to different environments (pH, temperature, light, etc.).11,29,30 For instance, Fe2O3 nanozymes exhibit POD-like activity for ˙OH generation in acidic pH while showing CAT-like activity for O2 generation in neutral pH. The activity of the former is favored under the demand of ˙OH while the latter is preferred under the demand of O2. Therefore, when applying nanozymes to in vivo diagnosis and therapy, selectively performing the desired catalytic activity is another critical issue for the off-target activity of nanozymes as it would seriously impair their desired activity.
Inspired by the perfect combo of dark reaction and photoreaction in photosynthesis,31 a dark/light relay strategy is proposed to realize adequate substrate supply in the dark reaction stage and selectively perform desired catalytic activities for enhanced tumor therapy by remote light control (Scheme 1). GOx, which could catalyze the oxidation of glucose to generate H2O2 in the presence of O2, was loaded into HMPB to gain HMPB@GOx (GPB) nanoparticles. Without near-infrared (NIR) 660 nm irradiation, GPB showed SOD and CAT-like activities as the same with HMPB. By cascaded catalysis of the SOD and CAT-like activities of GPB, the endogenous O2−˙ was transformed to O2. The generation of O2 further accelerated the catalytic decomposition of glucose by GOx in GPB to realize the supply of H2O2 for POD-like catalysis. When exposed to irradiation, the enzyme-like activities of HMPB in GPB nanoparticles were found to transform from anti-oxidant to pro-oxidant for significantly enhanced POD-like activity. In the meantime, the OXD-like activity of HMPB was identified to exist under irradiation for 1O2 generation. Then, the HMPB@GOx nanoparticles were further coated with a murine mammary carcinoma (4T1) cell membrane to obtain HMPB@GOx@Membrane (mGPB). Due to the immune escape and homotypic targeting ability of the homogenous tumor cell membrane,32,33 mGPB would accumulate in 4T1 tumor tissues for accurate catalytic therapy. Combining a sufficient H2O2 supply and selective ROS (˙OH, 1O2) generation by remote control, our strategy showed great anti-tumor potential in 4T1 tumor xenograft mice models.
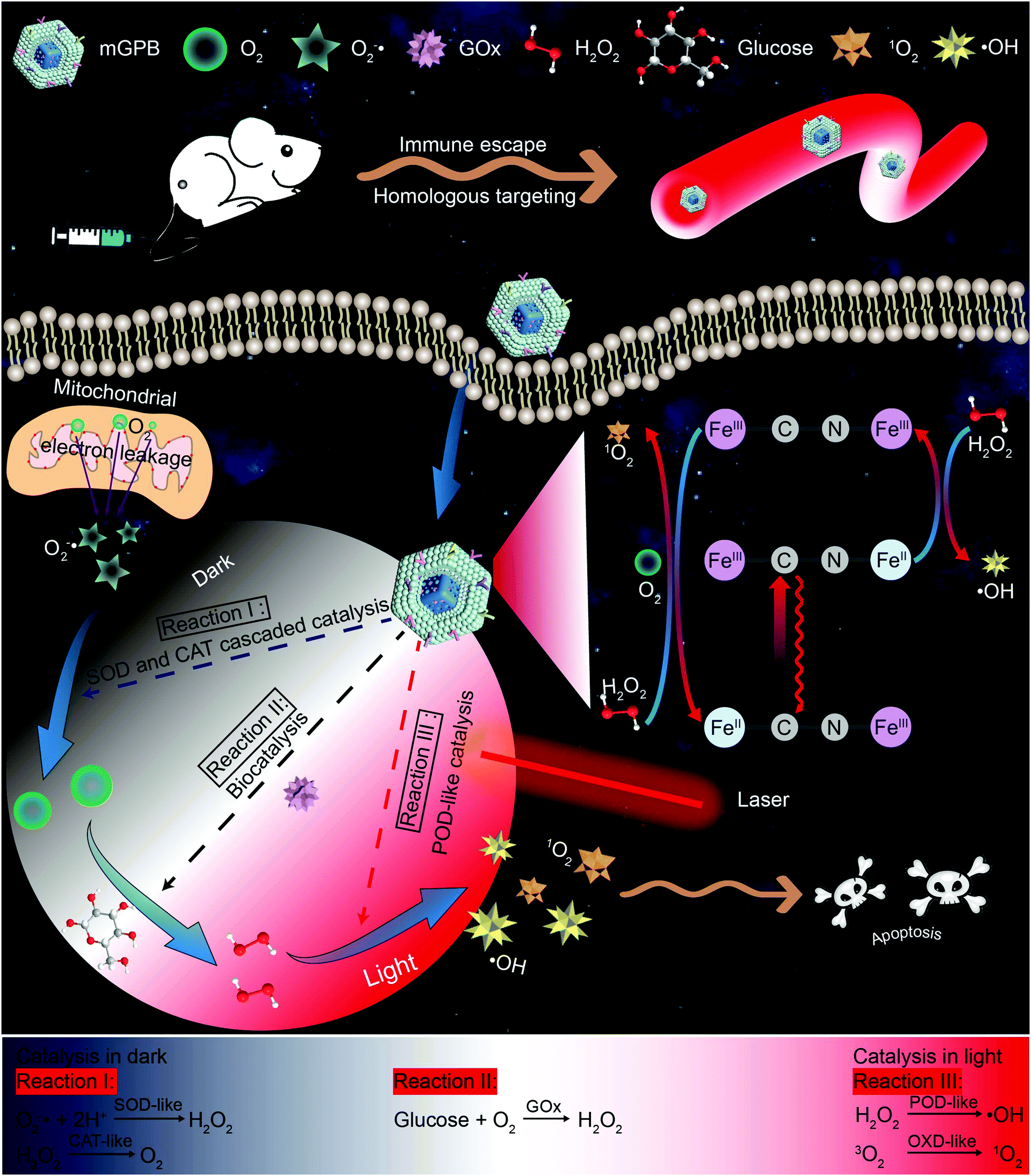 | ||
| Scheme 1 Schematic illustration of enhanced tumor therapy via dark/light relay catalysis. Membrane camouflaged and GOx loaded hollow mesoporous Prussian blue (mGPB) accelerates the catalysis of GOx to produce adequate H2O2 by in situ O2 generation in the dark. With adequate H2O2 supply, mGPB exhibits excellent POD-like activities to produce ROS for tumor therapy in the presence of NIR 660 nm irradiation. | ||
Firstly, Prussian blue nanocubes (PB) were successfully synthesized from K3[Fe(CN)6] and polyvinyl pyrrolidone (PVP) according to a reported method,34 as shown in transmission electron microscopy (TEM) images (Fig. S1, ESI†). After acid etching, hollow mesoporous Prussian blue nanocubes (HMPB) were obtained (Fig. S2, ESI†). TEM images (Fig. 1A) showed HMPB contained a hollow cavity and mesoporous shell. Nitrogen adsorption–desorption isotherms also confirmed that the morphology of HMPB had changed to a hollow mesoporous structure (Fig. S3A, ESI†) with a pore size of around 8.73 nm (BJH method, Fig. S3B, ESI†), which could be utilized to encapsulate biomacromolecules efficiently. Then, GOx with a size of 5.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm × 6.0 nm × 7.7nm was loaded into HMPB to obtain HMPB@GOx (GPB) (Fig. 1B).7 After loading, the hydrodynamic size of the nanoparticles (162.2 nm) increased to ∼182.0 nm (Fig. 1C), and the zeta potential changed from +42 to +27 mV (Fig. 1D). EDX spectrum and element mapping further verified the successful loading of GOx for the corresponding sulfur and phosphorus elements (Fig. 1E and F). Additionally, powder X-ray diffraction (PXRD) analysis (Fig. 1G) and the Fourier transform diffraction pattern of GPB (Fig. S4, ESI†) demonstrated the pure face-centered-cubic phase. The UV-vis absorbance spectra showed a typical peak at 720 nm (Fig. 1H), which could be attributed to the metal-to-metal charge transfer (MMCT) between FeII and FeIII.35 Thermogravimetric analyzer (TGA) tests revealed that the GOx was nearly 9.75% in the GPB (Fig. 1I), which was consistent with the result measured with the BCA Protein Assay Kit (Fig. S5, ESI†).
nm × 6.0 nm × 7.7nm was loaded into HMPB to obtain HMPB@GOx (GPB) (Fig. 1B).7 After loading, the hydrodynamic size of the nanoparticles (162.2 nm) increased to ∼182.0 nm (Fig. 1C), and the zeta potential changed from +42 to +27 mV (Fig. 1D). EDX spectrum and element mapping further verified the successful loading of GOx for the corresponding sulfur and phosphorus elements (Fig. 1E and F). Additionally, powder X-ray diffraction (PXRD) analysis (Fig. 1G) and the Fourier transform diffraction pattern of GPB (Fig. S4, ESI†) demonstrated the pure face-centered-cubic phase. The UV-vis absorbance spectra showed a typical peak at 720 nm (Fig. 1H), which could be attributed to the metal-to-metal charge transfer (MMCT) between FeII and FeIII.35 Thermogravimetric analyzer (TGA) tests revealed that the GOx was nearly 9.75% in the GPB (Fig. 1I), which was consistent with the result measured with the BCA Protein Assay Kit (Fig. S5, ESI†).
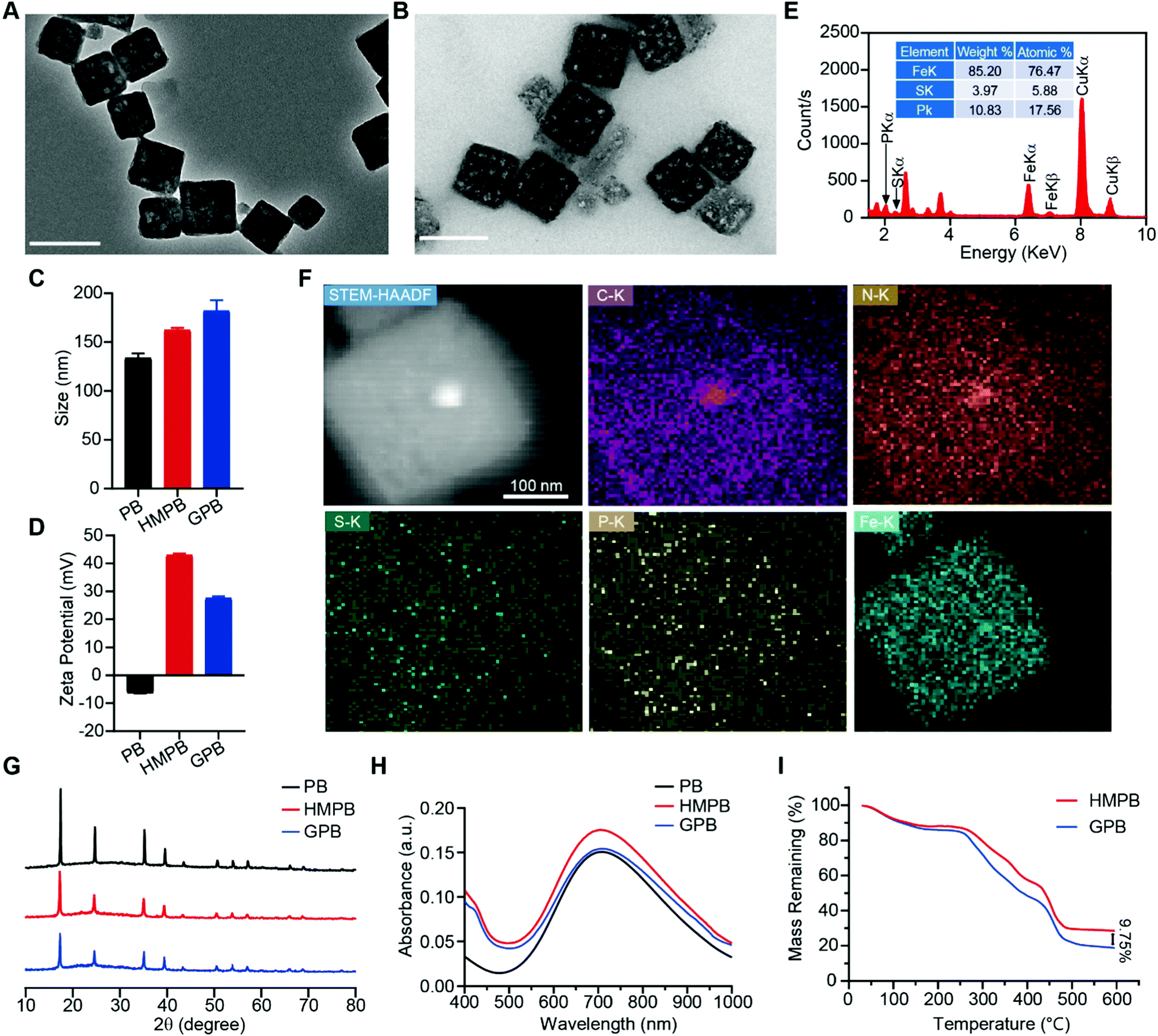 | ||
| Fig. 1 (A) TEM images of HMPB. Scale bar = 200 nm. (B) TEM images of GPB. Scale bar = 200 nm. (C) Hydrodynamic size distribution of PB, HMPB and GPB. (D) Zeta potential of PB, HMPB and GPB. (E) EDX spectrum of GPB. (F) STEM-HAADF image and the corresponding element mapping images of GPB. (G) PXRD pattern of PB, HMPB and GPB. (H) UV-vis absorption spectra of PB, HMPB and GPB. (I) TG weight loss profiles of HMPB and GPB. | ||
After verifying the successful synthesis of the GPB nanoparticles, their catalytic properties were detected. The catalytic capability of GOx in GPB, which catalyzed the glucose oxidation to produce H2O2, was verified by a Hydrogen Peroxide Assay Kit (Fig. 2A). The pH drop during the GOx catalytic reaction was also observed as the reaction progressed (Fig. 2B). Considering the O2 requirements during the GOx catalytic process (Fig. 2C), O2 regeneration was recognized to facilitate the catalysis of GOx.20 To produce O2in situ, the SOD-like activity of GPB to reduce the superoxide radical (O2−˙) was then investigated by the WST-1 method. As shown in Fig. 2D, the SOD-like activity of GPB showed concentration-dependence and light irradiation had a negligible influence on the activity. In addition, the CAT-like activity of GPB to decompose H2O2 into O2 and H2O was verified by the real-time production of O2 and light irradiation showed a slight increase in the enzyme-like activity (Fig. 2E). After realizing a sufficient H2O2 supply via the SOD-like/CAT-like/GOx catalysis of GPB, the pro-oxidative catalysis of GPB to H2O2 was investigated with NIR 660 nm irradiation. Rhodamine B (RhB) dye was chosen to evaluate whether highly active free radicals existed (Fig. 2F).36 In the group of GPB and H2O2 with NIR irradiation, RhB was obviously degraded (Fig. 2G and H), implying the generation of highly active free radicals under light irradiation. When H2O2 was replaced by glucose, RhB could also be degraded significantly for the catalysis of GPB on glucose to produce H2O2. As the control, no obvious fluorescence change of RhB occurred for the group without GPB or without light irradiation (Fig. S6A and B, ESI†). The main catalysis of GPB was shown in Fig. 2I, and the catalysis in the dark accelerated the supply of substrates for the catalysis in light. Considering that temperature could affect the enzyme-like activity of the nanozymes, the photothermal effect of GPB under NIR 660 nm (150 mW cm−2) was investigated. However, GPB nanoparticles showed weak photothermal effects when their concentration was below 20 ppm (Fig. S7, ESI†).
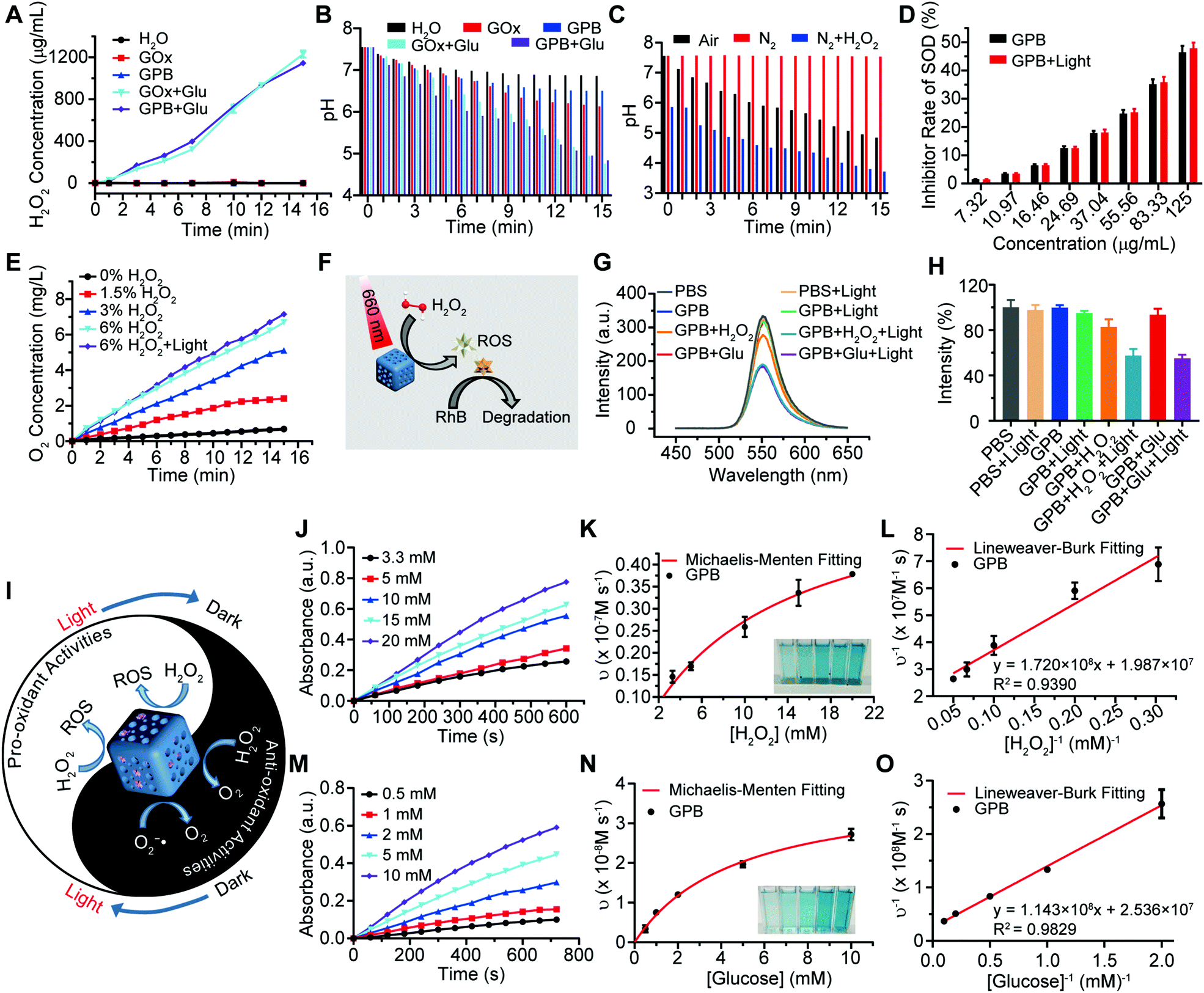 | ||
| Fig. 2 (A) The H2O2 generation of GOx and GPB solution in the absence and presence of glucose. (B) The pH value changes of GOx and GPB solution in the absence and presence of glucose. (C) The pH value changes of GPB solution with glucose under different O2 content. (D) SOD-like activity of GPB measured by WST reaction. (E) Generation of O2 in the presence of H2O2 during the CAT-like catalysis. (F) Schematic illustration of catalytic reactions of GPB under NIR irradiation. (G) Fluorescence spectra of RhB degradation. (H) Relative quantification of RhB degradation. (I) Schematic illustration of anti- and pro-oxidative enzyme-like catalytic reactions of GPB related to NIR irradiation. (J) Time-dependent absorbance of GPB with the addition of different concentrations of H2O2 under NIR 660 nm irradiation. (K) Michaelis–Menten kinetics of GPB with H2O2 added (n = 3). (L) Lineweaver–Burk plotting of GPB with H2O2 added (n = 3). (M) Time-dependent absorbance of GPB with the addition of different concentrations of glucose under NIR 660 nm irradiation. (N) Michaelis–Menten kinetics of GPB with glucose added (n = 3). (O) Lineweaver–Burk plotting of GPB with glucose added (n = 3). | ||
Further, the catalytic kinetics of GPB were determined by 3,3′,5,5′-tetramethyl-benzidine (TMB) for oxidized TMB (oxTMB) with absorption at 652 nm.7,9 As shown in Fig. 2J–O, with NIR irradiation, the catalytic reactions between GPB and H2O2 or glucose showed the typical Michaelis–Menten kinetics, respectively. Moreover, the Michaelis constant (KM) and maximal reaction velocity (Vmax) were calculated to be 8.66 mM and 5.03 × 10−8 m s−1 for H2O2 while 4.51 mM and 3.94 × 10−8 m s−1 for glucose according to the Lineweaver–Burk equation. The KM value for glucose declared that the catalytic activity would reach half of the maximum catalytic activities of GPB under 4.51 mM glucose, indicating that the efficient free radical generation in vivo for the glucose level of cancer cells was far below 8 mM.7,37 When all the GPB nanoparticles were bonded to the glucose substrate, the catalytic reaction proceeded at a velocity of up to 3.94 × 10−8 m s−1, showing excellent free radical generation for tumor catalytic therapy.
To illuminate the catalytic mechanism of GPB under NIR irradiation, the possible intermediates were detected. Terephthalic acid (PTA), a highly selective fluorescent probe for hydroxyl radicals (˙OH), was used as an indicator of ˙OH.10 As shown in Fig. 3A and B, no obvious fluorescence signal at 430 nm was detected in the GPB and H2O2 group without NIR irradiation. Upon irradiation, fluorescence intensity increased significantly, indicating the production of ˙OH, which could be contributed to the POD-like activity of GPB. In addition to ˙OH, the production of singlet oxygen (1O2) was also detected by using the fluorescent probe Singlet Oxygen Sensor Green (SOSG) (Fig. 3D), consistent with previous reports.38 The generation of 1O2 under NIR irradiation could contribute to the OXD-like activity of GPB, which was further confirmed by replacing the catalytic substrate from H2O2 to O2 (Fig. S8, ESI†). Additionally, the electron spin resonance (ESR) spectrum, as a more direct method to recognize radicals, was used to further identify active free radicals.39 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and 2,2,6,6-tetramethylpiperide (TEMP), as trapping probes, were employed to demonstrate the existence of ˙OH and 1O2, respectively. The characteristic spectra of the DMPO/˙OH adduct (1:2:2:1, quartet signal) and the TEMP/1O2 (TEMPO) adduct (1:1:1, triplet signal) were observed after NIR irradiation (Fig. 3C and E). The 1:1:1 signal peak appearing in Fig. 3C might be due to the ring-opening product of DMPO by 1O2 attacked.40 Besides, the changes of GPB during the catalytic reactions under NIR irradiation were investigated. The UV-vis spectra were determined to monitor the reaction between GPB and H2O2 (Fig. 3F). When exposed to irradiation, a weak absorption peak of 415 nm, consistent with the characteristic absorption of FeIIIFeIII(CN)6,41 appeared compared with the group without irradiation. However, the weak absorption peak disappeared immediately following addition of reduction reagent dithiothreitol (DTT). This observation indicates that the oxidative product FeIIIFeIII(CN)6 may exist as an intermediate during the reaction.
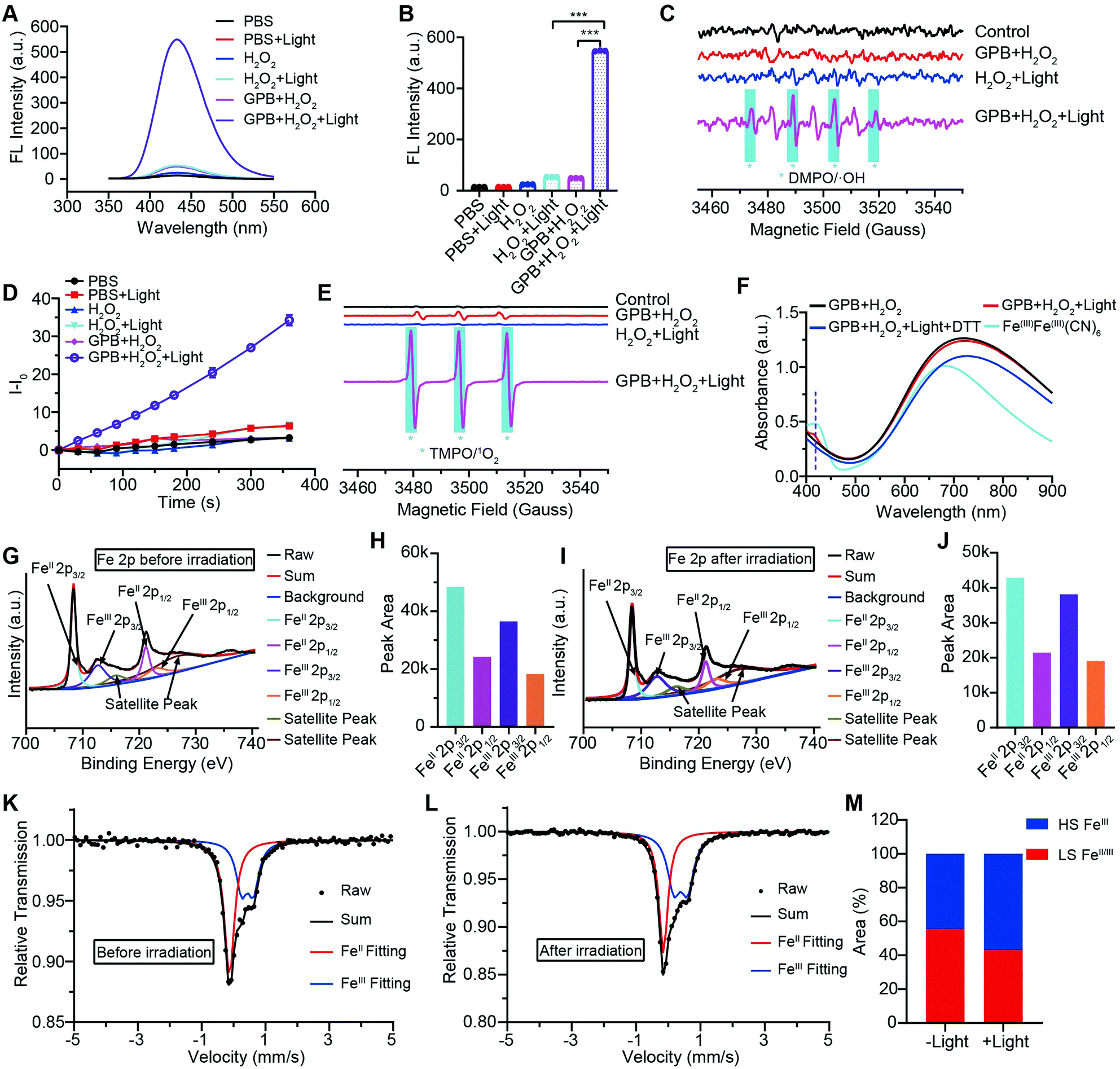 | ||
| Fig. 3 (A) Fluorescence spectra of PTA to demonstrate the generation of ˙OH. (B) Fluorescence intensity of PTA at 430 nm. (C) ESR spectra of the DMPO/˙OH adducts. (D) SOSG fluorescence changes to demonstrate the generation of 1O2. (E) ESR spectra of TEMP/1O2 adducts. (F) UV-vis spectra of GPB with different treatment. (G) High-resolution Fe 2p spectra of GPB without NIR irradiation. (H) Peak area of Fe 2p spectra without NIR irradiation. (I) High-resolution Fe 2p spectra of GPB with NIR irradiation. (J) Peak area of Fe 2p spectra with NIR irradiation. (K) Room temperature 57Fe Mössbauer spectra of GPB with irradiation and (L) without irradiation. (M) Relative quantitative analysis of 57Fe Mössbauer spectra. | ||
XRD patterns were then measured to illustrate possible structural changes. All the XRD samples (Fig. S9, ESI†) showed almost the same diffraction peaks at 2θ = 17.4°, 24.7°, 35.2°, 39.5°, 43.5°, 50.6°, 53.9°, and 57.1°, which were indexed to (100), (110), (200), (210), (211), (220), (300) and (310) facets of PB with the face-centered-cubic phase (JCPDS no. 01-0239), indicating no significant change in the crystal structure of GPB under irradiation. Apart from the crystal structure, the valence state changes of the Fe element were detected by X-ray photoelectron spectroscopy (XPS) and 57Fe Mössbauer measurements. The surface element compositions (C/O/N/Fe) of GPB, revealed by XPS, were changed from 49.64/11.8/30.95/7.61 to 57.87/12.76/24.4/4.97 under NIR irradiation (Fig. S10 and Table S1, ESI†). The increase in the O/Fe ratio indicated the valence change of Fe. The high resolution XPS of Fe 2p further illustrated the valence change (Fig. 3G and I). The Fe 2p spectra showed six typical peaks, where the peaks appearing at about 708.5 eV and 712.7 eV correspond to FeII 2p3/2 and FeIII 2p3/2 and the peaks of about 721.2 eV and 723.0 eV were indexed to FeII 2p1/2 and FeIII 2p1/2. Besides, the other two peaks at about 716.0 eV and 726.8 eV were considered to be the satellite peaks.42 The fitting results of Fe 2p are shown in Fig. 3H and J. Based on the peak area analysis of Fe 2p, the peak area ratio of FeIII (FeIII 2p3/2 and FeIII 2p1/2) to FeII (FeII 2p3/2 and FeII 2p1/2) was changed from 0.75/1 to 0.89/1 under NIR irradiation, indicating the oxidation of FeII after NIR treatment. 57Fe Mössbauer spectroscopy (Fig. 3K–M) was further used to investigate electron-transfer in GPB during NIR treatment. The specific fitting results were shown in Table S2, ESI.† In the 57Fe Mössbauer spectra, the singlet peak corresponding to FeII and the doublet peak considered to be FeIII were observed. After NIR treatment, the FeIII/FeII ratio changed to 55.4/44.6 from 43.1/56.9, indicating that the significant active free radical generation under NIR treatment might originate from the valence change of Fe after NIR irradiation. Based on these results, the possible mechanism for the catalytic reaction of GPB to produce ROS under NIR irradiation in the presence of H2O2 was speculated to be reactions (1)–(4).
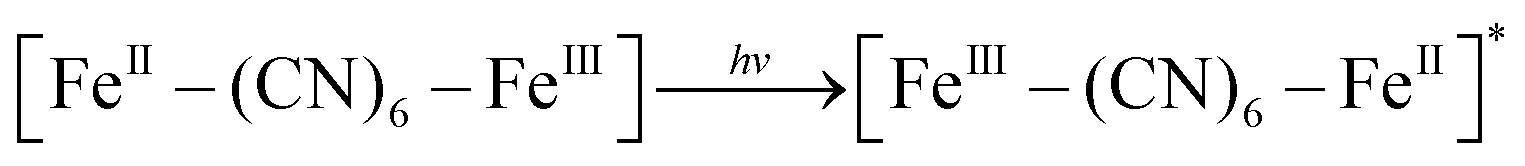 | (1) |
| [FeIII–(CN)6–FeII]* + H2O2 → [FeIII–(CN)6–FeIII] + ˙OH + OH− | (2) |
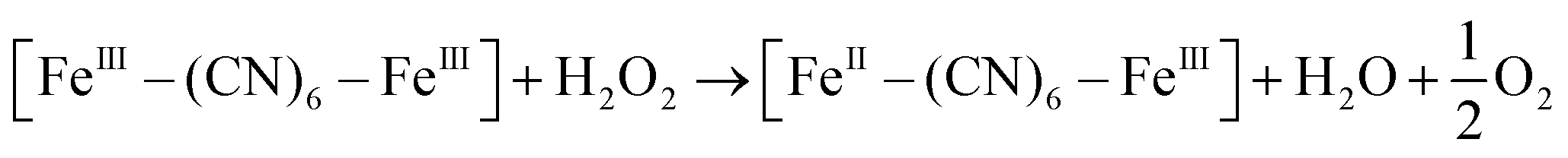 | (3) |
| [FeIII–(CN)6–FeIII] + 3O2 → [FeII–(CN)6–FeIII] + 1O2 | (4) |
Encouraged by the excellent ability of GPB to regulate ROS, its therapy effects were assessed in 4T1 tumor cells. To enhance the stability of GPB at physiological conditions and the auto-aggregation to 4T1, a 4T1 tumor cell membrane was used to coat GPB to obtain GPB@Membrane (mGPB) by an extrusion method (Fig. 4A). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 4B) and Fourier transform infrared spectroscopy (FTIR) (Fig. S11, ESI†) verified the successful coating of the 4T1 membrane. The hydrodynamic size and zeta-potential of the nanoparticles changed to ∼220 nm and −11.2 mV (Fig. 4C) respectively. UV-vis (Fig. S12, ESI†) and XRD patterns (Fig. S13, ESI†) also demonstrated the successful formation of mGPB. In particular, the hydrodynamic size and the polydispersity index (PDI) of mGPB were almost constant over 7 days bestowing it with good stability in storage conditions (Fig. 4D). Next, the immune evasive function and targeting ability to its source 4T1 of mGPB were evaluated by co-incubating Cy5-labeled mGPB with RAW 264.7 murine macrophages and 4T1, respectively. As expected, evident red fluorescence of Cy5 was observed in 4T1 cells compared with COS7, HeLa, and CT26 cells (Fig. 4E) and negligible macrophage engulfment was found in RAW 264.7 cells after treatment with mGPB nanoparticles rather than those treated with GPB nanoparticles (Fig. 4F). To identify the kind of intracellular ROS, fluorescence probe aminophenyl fluorescein (APF) was chosen to detect hydroxyl radical production of mGPB in 4T1 cells for selective reaction with ˙OH to form fluorescein.43 As illustrated in Fig. 4G, HMPB coated by a 4T1 membrane (mPB) was chosen as the control. The evident fluorescence was observed in 4T1 cells after NIR irradiation treatment in the group of mGPB and mPB for significantly enhanced POD-like activities of Prussian blue. Benefiting from the sufficient H2O2 supply generated by GOx, the group of mGPB showed the strongest fluorescence. In addition, broad ROS probe dichlorofluorescein diacetate (DCFH-DA) and specific ROS scavengers DMSO (˙OH) and NaN3 (1O2) were used to further verify the kind of ROS. As illustrated in Fig. 4H and Fig. S14 (ESI†), mPB and mGPB with NIR irradiation were 8.6-fold and 1.6-fold stronger, respectively, than those without NIR irradiation as measured by quantitative flow cytometry analysis. After adding NaN3 (4%), no significant fluorescence changes were observed, while the fluorescence intensity of DCFH-DA decreased dramatically after treatment with DMSO (87%) or broad spectrum antioxidant ascorbic acid (91%) (Fig. 4I and Fig. S15, ESI†), implying that the pro-oxidative activities of mGPB under NIR irradiation primarily stemmed from ˙OH. Heartened by the encouraging outcomes of ROS generation, the toxicity of the mGPB nanoparticles was examined by MTT in vitro. In Fig. 4J, the negligible toxicity of mPB nanoparticles in 4T1 cells indicated their good biocompatibility. mGPB showed the lowest IC50 value as well as the highest inhibition effect on cell growth after 15 min light exposure, indicating that mGPB had the optimal therapeutic efficacy for enhanced POD and OXD-like catalysis under NIR irradiation. The similar results were verified by the fluorescence live/dead cell staining in Fig. S16 (ESI†). In addition, a large number of apoptotic cells (∼46.5%) was observed by Annexin V-FITC (AV) apoptosis assay after treatment with mGPB and light irradiation for catalytic therapy (Fig. S17, ESI†).
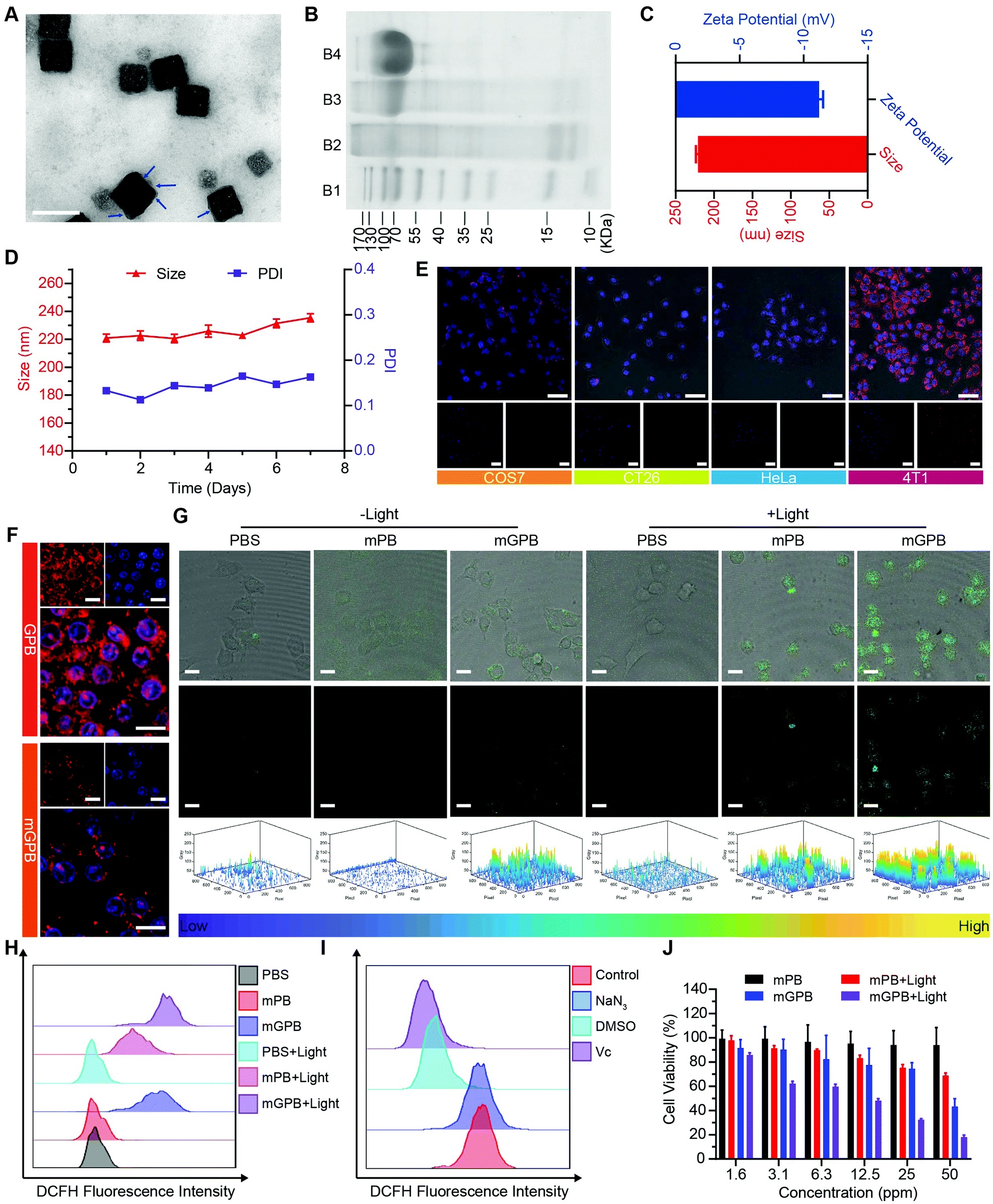 | ||
| Fig. 4 (A) TEM images of mGPB. Scale bar = 200 nm. (B) SDS-PAGE protein analysis. (B1: Maker; B2: 4T1 cell membrane; B3: mGPB; B4: GOx). (C) The hydrodynamic size and zeta-potential of mGPB. (D) Hydrodynamic size distribution and PDI changes of mGPB over 7 days. (E) CLSM images of COS7 cells, HeLa cells, CT26 cells and 4T1 cells after treatment with Cy5-labeled mGPB stained with nuclear dye. Scale bar = 50 μm. (F) CLSM images of RAW 264.7 cells after treatment with Cy5-labeled GPB or mGPB stained with nuclear dye. Scale bar = 20 μm. (G) CLSM image of 4T1 cells with different conditions stained with APF. Scale bar = 50 μm. (H) Flow cytometry analysis of 4T1 cells after stain with DCFH-DA. (I) Flow cytometry analysis of 4T1 cells after treated with different ROS inhibitors. (J) Cell viability of 4T1 cells treated with different concentration of mPB and mGPB under 660 nm laser irradiation. | ||
To verify the targeting ability of mGPB nanoparticles in vivo, a 4T1 tumor-bearing mice model was built to examine tumor fluorescence imaging and bio-distribution by an IVIS imaging system. After intravenous injection of DiR-labeled nanoparticles to 4T1 tumor bearing mice, fluorescence intensity accumulated in tumor was observed at different times in the tumor site. As shown in Fig. 5A, obvious fluorescence of DiR-mGPB was observed at 12 h. The fluorescence intensity increased gradually and reached a maximum value at 60 h. Moreover, that fluorescence intensity was retained within 96 h, indicating the long retention of mGPB nanoparticles for long-term treatment. However, no strong fluorescence of DiR-GPB was found in the tumor issue within 96 h (Fig. 5B). Semiquantitative analysis exhibited 2 times higher fluorescence intensity in the tumor issue of DiR-mGPB group than that of DiR-GPB group after 12 h (Fig. 5C). In addition, the main organs and tumor sacrificed at 96 h showed a similar trend (Fig. 5D and E). This remarkable difference might be attributed to the immune escape and homologous targeting of mGPB after coating with the 4T1 membrane. Based on the achievement of efficient tumor accumulation of mGPB nanoparticles, the in vivo anti-tumor study of mGPB nanoparticles was carried out in 4T1 tumor-bearing mice. During treatment of 14 days, the therapeutic effect was evaluated by monitoring the changes of the relative tumor volume every day (Fig. 5F). Obviously, no significant therapeutic effect was also observed after treatment with the PBS group with or without irradiation. Meanwhile, the GPB group with light irradiation for 6 min had a low therapeutic effect. However, after treatment with mGPB nanoparticles and light irradiation, the tumor growth was significantly inhibited. As illustrated in Fig. 5G, changes of body weights were monitored every day and no notable difference was observed, indicating the negligible systemic toxicity of mGPB. After treatment for 14 d, all mice were sacrificed and the tumors were collected. The weight of each group was shown in Fig. 5H, and the photographs of tumors in different groups were taken in Fig. 5I. The results further confirmed the highly effective anticancer effect of the mGPB nanoparticles in vivo. Besides, hematoxylin and eosin (H&E) staining (Fig. 5J) of tumor was obtained, the maximum amount of tumor cells were dead in the mGPB nanoparticles with the light irradiation group. The proliferation activity of tumor tissues under different treatments was characterized by Ki67 staining (Fig. 5K). Compared with other groups of cells, the cancer cells treated with mGPB nanoparticles in the presence of NIR irradiation almost lost their ability to proliferate, and it was very important for the long-term inhibition of tumors. In addition, the therapeutic effect was further confirmed by terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling (TUNEL) staining (Fig. S18, ESI†). These results indicated that mGPB nanoparticles had a satisfactory tumor suppressive effect. Additionally, several physiological and biochemical indexes were measured to assess the in vivo systemic toxicity. The concentrations of alanine aminotransferase (ALT), aspartate transaminase (AST), total protein (TP), albumin (ALB) and bilirubin (TBIL) had no significant difference indicating the low systemic toxicity of mGPB nanoparticles to the liver (Fig. S19A, ESI†). The concentrations of blood urea nitrogen (BUN), blood glucose (GLU) and creatinine (CRE) also had no significant difference after various treatments, indicating the low systemic toxicity of mGPB nanoparticles to the kidney (Fig. S19B, ESI†). The results of the routine blood examination further indicated the low systemic toxicity of the mGPB nanoparticles (Table S3, ESI†). In addition, no obvious physiological morphology changes were found through H&E staining against the major organs (heart, liver, spleen, lung, and kidney) in different groups (Fig. S20, ESI†), implying that the nanoparticles had good biocompatibility in vivo.
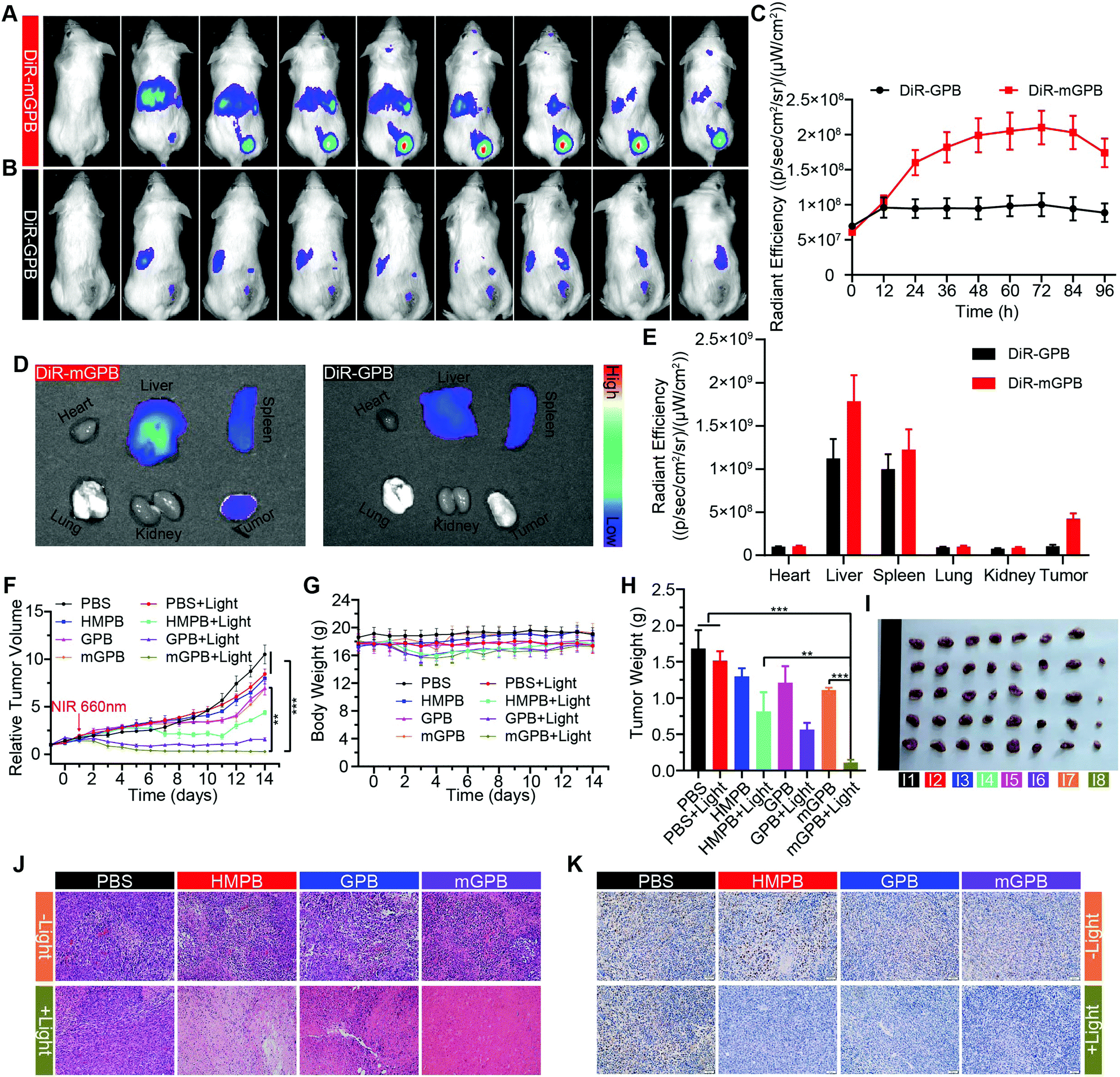 | ||
| Fig. 5 (A) In vivo fluorescence imaging of 4T1 tumor-bearing mice after intravenously injection of DiR-mGPB and (B) DiR-GPB. (C) Corresponding fluorescence intensity of tumor issue after treated with DiR-mGPB and DiR-GPB (n = 3). (D) Ex vivo fluorescence imaging of tumor and major organs from mice sacrificed at 96 h post injection. (E) Corresponding fluorescence intensity of (D). (F) The relative tumor volume change of 4T1 tumor-bearing mice after various treatments during the 14 days’ therapy. (G) The body weight change of 4T1 tumor-bearing mice. (H) The average tumor weight of the mice at the 14th day. (I) Photographs of 4T1 tumor tissues after various treatments. (I1: PBS; I2: PBS + Light; I3: HMPB; I4: HMPB + Light; I5: GPB; I6: GPB + Light; I7: mGPB; I8: mGPB + Light). (J) H&E staining of tumor tissues. (K) Ki67 staining of tumor tissues after different treatments. Results are presented as means ± standard deviation (SD) (n = 5) (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
Conclusions
In summary, a multi-enzyme system (mGPB) with self-sufficient H2O2 supply and photoselective multienzyme-like activities was developed for enhanced tumor catalytic therapy. Once in the tumor cells for homologous targeting ability of homogenous tumor cell membrane, mGPB could effectively utilize the cellular metabolic waste (O2−˙, H2O2) to produce oxygen by cascaded SOD/CAT-like catalysis and then further exhaust glucose to generate plenty of H2O2 with O2 supply via GOx in the absence of NIR irradiation. The H2O2, derived from the catalysis in dark, serves as substrates for the POD-like catalysis of mGPB under NIR irradiation, leading to sufficient production of highly toxic ˙OH to attack tumors. Moreover, the possible catalytic mechanism for photoselective POD-like catalysis of mGPB was revealed by high resolution XPS and 57Fe Mössbauer spectroscopy. NIR irradiation efficiently promoted the metal-to-metal charge transfer of FeII and FeIIIvia a cyanide bridge and then an electron was transferred to H2O2 from FeII to generate ˙OH. By dark/light relay, the multienzyme-like activities of mGPB were efficiently combined to realize the supply of H2O2 and selective ROS (˙OH, 1O2) generation. In vivo experiments further demonstrate the high-efficiency anti-tumor effect of mGPB. Rationally coordinating the multienzyme-like activities of the nanozyme related to different conditions (pH, temperature, light, etc.) could not only break the limitations of complex tumor microenvironments (hypoxia, insufficient H2O2, low pH, etc.) but also could expand a horizon for selective catalysis in vivo, which has great potential in precision tumor catalytic therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51703168, 5187316, 51833007 and 51690152).Notes and references
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS.
- B. Xu, H. Wang, W. Wang, L. Gao, S. Li, X. Pan, H. Wang, H. Yang, X. Meng, Q. Wu, L. Zheng, S. Chen, X. Shi, K. Fan, X. Yan and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 4911–4916 CrossRef CAS.
- H. Sun, Y. Zhou, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2018, 57, 9224–9237 CrossRef CAS.
- Q. Wang, H. Wei, Z. Zhang, E. Wang and S. Dong, Trends Anal. Chem., 2018, 105, 218–224 CrossRef CAS.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579 CrossRef CAS.
- K. Fan, J. Xi, L. Fan, P. Wang, C. Zhu, Y. Tang, X. Xu, M. Liang, B. Jiang, X. Yan and L. Gao, Nat. Commun., 2018, 9, 1440 CrossRef.
- Z. Wang, Y. Zhang, E. Ju, Z. Liu, F. Cao, Z. Chen, J. Ren and X. Qu, Nat. Commun., 2018, 9, 3334 CrossRef.
- J. Zhang, S. Wu, X. Lu, P. Wu and J. Liu, Nano Lett., 2019, 19, 3214–3220 CrossRef CAS.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., Int. Ed., 2016, 55, 2101–2106 CrossRef CAS.
- Q. Chen, C. Liang, X. Sun, J. Chen, Z. Yang, H. Zhao, L. Feng and Z. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5343–5348 CrossRef CAS PubMed.
- J.-X. Fan, M.-Y. Peng, H. Wang, H.-R. Zheng, Z.-L. Liu, C.-X. Li, X.-N. Wang, X.-H. Liu, S.-X. Cheng and X.-Z. Zhang, Adv. Mater., 2019, 31, 1808278 CrossRef.
- Z. Tang, H. Zhang, Y. Liu, D. Ni, H. Zhang, J. Zhang, Z. Yao, M. He, J. Shi and W. Bu, Adv. Mater., 2017, 29, 1701683 CrossRef.
- L. Zhang, S.-S. Wan, C.-X. Li, L. Xu, H. Cheng and X.-Z. Zhang, Nano Lett., 2018, 18, 7609–7618 CrossRef CAS.
- Z. Yang, Y. Dai, C. Yin, Q. Fan, W. Zhang, J. Song, G. Yu, W. Tang, W. Fan, B. C. Yung, J. Li, X. Li, X. Li, Y. Tang, W. Huang, J. Song and X. Chen, Adv. Mater., 2018, 30, 1707509 CrossRef.
- L.-S. Lin, T. Huang, J. Song, X.-Y. Ou, Z. Wang, H. Deng, R. Tian, Y. Liu, J.-F. Wang, Y. Liu, G. Yu, Z. Zhou, S. Wang, G. Niu, H.-H. Yang and X. Chen, J. Am. Chem. Soc., 2019, 141, 9937–9945 CrossRef CAS.
- L.-H. Fu, C. Qi, J. Lin and P. Huang, Chem. Soc. Rev., 2018, 47, 6454–6472 RSC.
- L. Feng, R. Xie, C. Wang, S. Gai, F. He, D. Yang, P. Yang and J. Lin, ACS Nano, 2018, 12, 11000–11012 CrossRef CAS PubMed.
- W. Fan, N. Lu, P. Huang, Y. Liu, Z. Yang, S. Wang, G. Yu, Y. Liu, J. Hu, Q. He, J. Qu, T. Wang and X. Chen, Angew. Chem., Int. Ed., 2017, 56, 1229–1233 CrossRef CAS.
- Y. Zhang, Y. Yang, S. Jiang, F. Li, J. Lin, T. Wang and P. Huang, Mater. Horiz., 2019, 6, 169–175 RSC.
- L.-H. Fu, C. Qi, Y.-R. Hu, J. Lin and P. Huang, Adv. Mater., 2019, 31, 1808325 CrossRef.
- Y. Cheng, H. Cheng, C. Jiang, X. Qiu, K. Wang, W. Huan, A. Yuan, J. Wu and Y. Hu, Nat. Commun., 2015, 6, 8785 CrossRef CAS.
- Y. Zhang, F. Wang, C. Liu, Z. Wang, L. Kang, Y. Huang, K. Dong, J. Ren and X. Qu, ACS Nano, 2018, 12, 651–661 CrossRef CAS.
- C. Xu, Y. Lin, J. Wang, L. Wu, W. Wei, J. Ren and X. Qu, Adv. Healthcare Mater., 2013, 2, 1591–1599 CrossRef CAS.
- W. Zhang, S. Hu, J. J. Yin, W. He, W. Lu, M. Ma, N. Gu and Y. Zhang, J. Am. Chem. Soc., 2016, 138, 5860–5865 CrossRef CAS.
- Z. L. Yang, W. Tian, Q. Wang, Y. Zhao, Y. L. Zhang, Y. Tian, Y. X. Tang, S. J. Wang, Y. Liu, Q. Q. Ni, G. M. Lu, Z. G. Teng and L. J. Zhang, Adv. Sci., 2018, 5, 1700847 CrossRef PubMed.
- A. Asati, C. Kaittanis, S. Santra and J. M. Perez, Anal. Chem., 2011, 83, 2547–2553 CrossRef CAS PubMed.
- Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685–3689 CrossRef CAS PubMed.
- D. W. Lawlor and G. Cornic, Plant, Cell Environ., 2002, 25, 275–294 CrossRef CAS PubMed.
- W. Zou, Nat. Rev. Cancer, 2005, 5, 263 CrossRef CAS.
- S. Y. Li, H. Cheng, B. R. Xie, W. X. Qiu, J. Y. Zeng, C. X. Li, S. S. Wan, L. Zhang, W. L. Liu and X. Z. Zhang, ACS Nano, 2017, 11, 7006–7018 CrossRef CAS.
- W. Chen, K. Zeng, H. Liu, J. Ouyang, L. Wang, Y. Liu, H. Wang, L. Deng and Y.-N. Liu, Adv. Funct. Mater., 2017, 27, 1605795 CrossRef.
- T. Uemura, M. Ohba and S. Kitagawa, Inorg. Chem., 2004, 43, 7339–7345 CrossRef CAS PubMed.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- R. A. Nascimento, R. E. Ozel, W. H. Mak, M. Mulato, B. Singaram and N. Pourmand, Nano Lett., 2016, 16, 1194–1200 CrossRef CAS.
- B. Zhou, B.-P. Jiang, W. Sun, F.-M. Wei, Y. He, H. Liang and X.-C. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 18036–18049 CrossRef CAS.
- L. Gao, R. Liu, F. Gao, Y. Wang, X. Jiang and X. Gao, ACS Nano, 2014, 8, 7260–7271 CrossRef CAS PubMed.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang, J. Gao, J. Wang, Y. Huang, T. Zhang and B. Liu, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS.
- M. Pyrasch and B. Tieke, Langmuir, 2001, 17, 7706–7709 CrossRef CAS.
- N. Wang, W. Ma, Y. Du, Z. Ren, B. Han, L. Zhang, B. Sun, P. Xu and X. Han, ACS Appl. Mater. Interfaces, 2019, 11, 1174–1184 CrossRef CAS.
- K. Ni, G. Lan, C. Chan, B. Quigley, K. Lu, T. Aung, N. Guo, P. La Riviere, R. R. Weichselbaum and W. Lin, Nat. Commun., 2018, 9, 2351 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00583h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |