
The oxygen vacancy in Li-ion battery cathode materials
Zhen-Kun
Tang
 a,
Yu-Feng
Xue
b,
Gilberto
Teobaldi
cde and
Li-Min
Liu
*b
a,
Yu-Feng
Xue
b,
Gilberto
Teobaldi
cde and
Li-Min
Liu
*b
aCollege of Physics and Electronics Engineering, Hengyang Normal University, Hengyang 421002, China
bSchool of Physics, Beihang University, Beijing, 100191, China. E-mail: liminliu@buaa.edu.cn
cScientific Computing Department, Science and Technology Facilities Council UKRI, Didcot OX11 0QX, UK
dStephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, L69 3BX Liverpool, UK
eSchool of Chemistry, University of Southampton, Highfield, SO17 1BJ Southampton, UK
First published on 24th August 2020
Abstract
The substantial capacity gap between available anode and cathode materials for commercial Li-ion batteries (LiBs) remains, as of today, an unsolved problem. Oxygen vacancies (OVs) can promote Li-ion diffusion, reduce the charge transfer resistance, and improve the capacity and rate performance of LiBs. However, OVs can also lead to accelerated degradation of the cathode material structure, and from there, of the battery performance. Understanding the role of OVs for the performance of layered lithium transition metal oxides holds great promise and potential for the development of next generation cathode materials. This review summarises some of the most recent and exciting progress made on the understanding and control of OVs in cathode materials for Li-ion battery, focusing primarily on Li-rich layered oxides. Recent successes and residual unsolved challenges are presented and discussed to stimulate further interest and research in harnessing OVs towards next generation oxide-based cathode materials.
1. Introduction
The growing concerns on energy availability, distribution and the environmental costs of energy production have turned development of clean and renewable solutions into a modern international priority. For practical applications to be widely accessible, renewable sources such as solar, wind and hydraulic energy need to be converted into secondary forms such as electricity.1–3 In order to solve the mismatch between generation of renewable energy and its demand and use further in time and space, the development of scalable and efficient energy storage technologies is essential. Among various energy storage technologies, electrochemical energy storage, that is, the use of batteries, has attracted growing attention as this technology is in principle capable to fulfil several of the requirements demanded by modern society for both mobile and stationary applications.4 In the past 20 years, the Li-ion battery has firmly established itself in the market of mobile terminal equipment, computer, mobile phones, electric vehicle, etc., thanks to its relatively high energy density, excellent charging and discharging performance and commercially suitable operational lifetimes.Being rechargeable, Li-ion batteries (LiBs) are a secondary battery technology. Their functioning depends on the movement of Li-ions between cathode and anode.5,6 Research in lithium batteries can be traced back to 1912. Gilbert N. Lewis was the first to propose and study the lithium metal battery. In 1958, Harris proposed to use organic electrolyte as the electrolyte in LiBs, opening for an era of intense study and rapid developments. In the 1980s, the development of transition metal oxides anode materials proposed by Goodenough for Li-ion battery,7,8 together with the emergence of practical graphite anodes,9 opened up the commercial era for LiBs. The capacity of available cathode materials in LiBs is generally lower than that of commercial anode materials.10 Whereas the capacity of a graphite anode is close to 400 mA h g−1, that of LiCoO2, a commercially used cathode material, is only about 140 mA h g−1, and that of LiFePO4 is about 160 mA h g−1. Thus, the specific capacity of commercially available LiBs is ultimately limited by the performance of available (stable) cathode materials. The good intercalation behaviour of the conventional cathode, layered LiCoO2, was first reported in the early 1980s,7,8 leading to successful commercialization of LiBs in 1991. Since then, LiMn2O4, LiFePO4, LiNixCoyMnzO2/LiNixCoyAlzO2,11 LiNixMn2−xO4,12 and Li-rich layered oxides such as Li[LixMnyNizCo1−x−y−z]O213 and Li2Ru1−xMxO3 (M = Mn, Sn, Ti)14,15 have also been used as cathode materials in commercial LiBs. Among these cathode materials, Li-rich layered oxide materials have attracted great attention due to their specific discharge capacity, which can reach 270 mA h g−1, holding great promise for the development of next generation cathodes for commercial Li-ion technologies.
By altering the physical and chemical properties of the hosting material, structural defects in battery electrodes can greatly affect the electro-chemical performance and lifetime of (Li-ion) batteries. Rational and controlled used of defects can enable tuning of key physical properties of materials such as, for instance, doping in semiconductors to enhance the electronic conductivity. Oxygen vacancies (OVs), as one of the most common and important defects in transition metal oxide materials, can effectively regulate the electronic structure, electrical conductivity and surface structure of transition metal oxides.16 At present, the main methods to obtain OVs in transition metal oxides are high temperature treatment,17–19 heat treatment in oxygen-deficient environments (H2, Ar, etc.),20–23 ion-doping,24,25 plasma jet irradiation,26–31 reducing flame roasting,32,33 supercritical fluid processing,34 gas-solid interface reaction35 and mechanical milling.36
Appropriately understood and suitably controlled creation of OVs in transition metal oxides cathode materials is an effective route to improving the capacity and charging-rate performance in LiBs by promoting Li-ion diffusion and electron transport. However, loss of oxygen from the pristine material can also cause phase transformations and cracks, which leads to performance degradation of the materials and, ultimately, failure of the Li-ion battery. Understanding the creation and diffusion of OVs in layered lithium metal oxides, and their effects on the physical and chemical properties of the hosting material is key to improve the capacity of LiBs and, in principle, highly valuable for development of new electrochemical applications (e.g. photo-electro-catalysis) of oxide materials too. To this end, here we review the latest research and progress in controlled creation of OVs and in understanding of their diffusion and their cumulative effects on the cathode performance. We also analyse and discuss emerging vacancy-mediated strategies to managing performance degradation in cathode materials for LiBs.
2. Controlled creation of OVs in cathode materials
OVs are the product of the removal of one oxygen atom (oxygen ion) from the lattice of metal oxides or other oxygen-containing compounds. Such removal results in oxygen loss and formation of a vacancy site. In short, OVs refer to the structural defect left by one oxygen ion escaping from its lattice. Because of its relatively low energy and ease of formation, the OV is the most widely intrinsic defect in transition metal oxides. OVs can affect profoundly the properties of semiconducting materials. The introduction of OVs has significant effects on the physical and chemical properties of materials, including the electronic structure, geometric structure, optical absorption and surface adsorption properties of the system. Firstly, OVs can effectively regulate the electronic structure of the material surface, altering the electronic or ionic conductance of the material. Second, the introduction of surface OVs leads to relaxation in the atomic layer closest to the surface, although the degree of relaxation is typically short-ranged. Thirdly, OVs introduce impurity energy levels in the material, which reduces the band gap width and further improves the optical absorption characteristics. Finally, the introduction of OVs on the surface of the material will also affect surface adsorption. Compared with the defect-free analogue, the introduction of OVs on the surface can make it is easier to adsorb molecules such as O2 and CO.37,38In the process of charging and discharging of LiBs, Li+ cations are reversibly intercalated and de-intercalated into and from the (two) battery electrodes. During battery charging, Li+ is de-intercalated from the cathode, and intercalated into the anode leaving it in a lithium rich state, following diffusion across the electrolyte. During discharge, the direction of Li+ movement is reversed, generating usable power, in the form of an electrical current in the external circuit.
Fig. 1 reports a schematic of the main components of a conventional rechargeable Li-ion battery. The battery cell comprises a positive electrode (cathode); a non-aqueous liquid electrolyte, typically LiPF6 salt in an organic solvent;39–41 and a negative electrode (anode), ordinarily based on graphite. During charging, application of a voltage difference between the electrodes forces Li+ ions to be extracted from the LiCoO2 lattice. Li+ ions diffuse through the electrolyte, to be eventually intercalated between the graphite sheets in the anode. During discharge, Li+ ions return to the cathode via the electronically insulating electrolyte, with electrons moving, as electrical current, along the external circuit, and providing extractable power from the device. The electrode half-reactions during discharge in a battery having graphite (C6) and LiCoO2 as anode and cathode, respectively, can be written as:
| Anode: LixC6 = xLi+(sol) + 6C + xe− | (1) |
| Cathode: Li1−xCoO2 + xLi+(sol) + xe− = LiCoO2 | (2) |
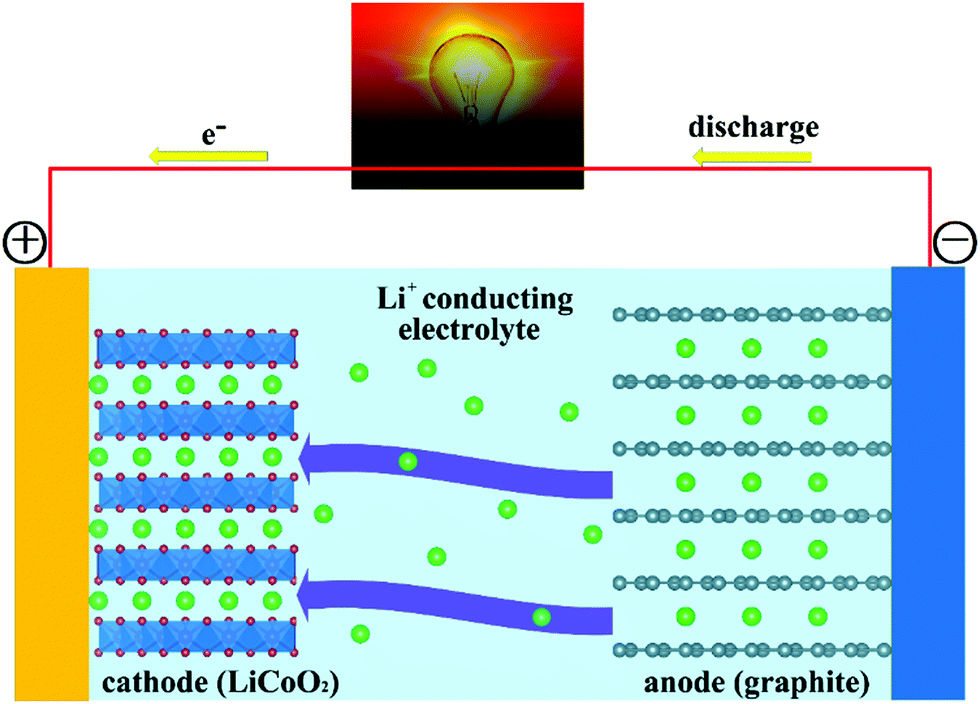 | ||
| Fig. 1 Schematic diagram of a rechargeable Li-ion cell. During charging, Li+ is de-intercalated from the cathode to be intercalated into the anode, following diffusion through the electrolyte. During discharge the direction of Li+ movement is reversed, generating usable power as electrical current in the external circuit. | ||
For commercial lithium batteries, thermochemical stability and high energy density (as achievable via high voltage, high capacity materials) are two important considerations. The main candidate cathode materials that are being investigated to achieve these aims include layered oxides (α-LiCoO2), Li-rich layered oxides [such as Li1.2Mn0.6Ni0.2O2 and Li(Li0.2Mn0.54Ni0.13Co0.13)O2], spinel oxides (LiMn2O4) and polyanionic materials (such as LiFePO4, Li2FeSiO4, and LiFeSO4F). Fig. 2 reports the structure of layered α-LiCoO2, Li-rich Li1.2Mn0.6Ni0.2O2, cubic LiMn2O4 spinel, and olivine-structured LiFePO4 cathode materials for LiBs.
 | ||
| Fig. 2 Atomic structures of four typical cathode materials for Li-ion battery. (a) layered α-LiCoO2, (b) layered Li-rich Li1.2Mn0.6Ni0.2O2, (c) cubic LiMn2O4 spinel, (d) olivine-structured LiFePO4. | ||
As one of the most important intrinsic defects in transition metal oxides, OVs can introduce localised states into the electronic structure of the material, which in turn can change the photoelectric properties of the cathode material. Table 1 list several available methods to introduce OVs in oxide cathode materials. For instance, Levasseur et al. synthesized oxygen-deficient LiCoO2−δ sample by annealing the stoichiometric LiCoO2 sample under a reduced O2 pressure.42 Lim et al. prepared Li2MnO3−δ by a carbo-thermal reduction and heat treatment at 850 °C.43 Armstrong et al. obtained the oxygen-deficient Li[Ni0.2Li0.2Mn0.6]O2 cathode materials by annealing after pre-heating the mixed precursors at 900 °C for 6 h in air.44 Xia et al. synthesized several series of Li1±xMn2−yO4±δ and Li1.05AlyMn1.95−yO4±δ samples with different degrees of oxygen defects by controlling synthesis protocols and temperature.45
| Cathode materials | Methods | Annealing temperature [°C] | Assistant/reaction environment | Ref. |
|---|---|---|---|---|
| LiCoO2−δ | High temperature treatment | 900 | Low O2 pressure | 42 |
| Li2MnO3−δ | High temperature treatment | 850 | 43 | |
| Li[Ni0.2Li0.2Mn0.6]O2 | High temperature treatment | 900 | Air | 44 |
| Li1+xAlyMn2−x−yO4±δ | High temperature treatment | 800–1000 | Air | 45 |
| Li[Ni0.5−xMn1.5+x]O4 | High temperature treatment | 600–900 | Air | 46 |
| LiNi0.5Mn1.5O4 | High temperature treatment | 700–1000 | O2 or air | 47 |
| LiNixMn2−xO4 | High temperature treatment | 600–800 | 48 | |
| LiNi0.5Mn1.5O4−δ | High temperature treatment | 700 | 49 | |
| Li1.2Mn0.54Ni0.13Co0.13O2 | High temperature treatment | 800–1000 | 50 | |
| Li1.2Mn0.54Ni0.13Co0.13O2 | High temperature treatment | 500–800 | Air | 51 |
| Li1.2Mn0.54Ni0.13Co0.13O2 | High temperature treatment | 450–900 | Air | 52 |
| Li1+yMn2−yO4−δ | Oxygen-deficient heat treatment | 700–800 | Air or N2 | 53 |
| LiTi2(PO4)3 | Oxygen-deficient heat treatment | 900–1000 | Air or N2 | 57 |
| Li1.12Mn0.55Ni0.145Co0.1O2 | Oxygen-deficient heat treatment | 250 | H2 | 54 |
| Li1+yMn2−yO4−δ | Oxygen-deficient heat treatment | 660 | Ar | 55 |
| LiV3O8 | Oxygen-deficient heat treatment | 40–400 | Ar | 56 |
| LiMn2O4 | Oxygen-deficient heat treatment | 800 | Quenched in liquid N2 | 58 |
| 0.5Li2MnO3·0.5LiNi0.44Co0.25Mn0.31O2 | Chemical assisted heat treatment | 445–750 | HNO3 | 59 |
| Li2MnO3 | Chemical assisted heat treatment | 350 | Stearic acid/Ar | 60 and 61 |
| Li2MnO3−x | Chemical assisted heat treatment | 255–265 | CaH2 or LiH | 62 |
| Li2MnO3−δ | Chemical assisted heat treatment | 380 | NaBH4 | 63 |
| Li(Li0.2Mn0.54Ni0.13Co0.13)O2 | Chemical assisted heat treatment | 130–350 | Carbon black (Super P) | 64 |
| Li1.143Mn0.544Ni0.136Co0.136O2 | Chemical assisted heat treatment | 300–600 | Na2S2O8 | 65 |
| Li[Li0.144Ni0.136Co0.136Mn0.544]O2 | Gas–solid interface reaction | 500–850 | Carbon dioxide/air | 35 |
| LiMgyCo1−yO2 | Ion-doping | 800 | 24 | |
| LiNi0.5Co0.2Mn0.3O2–SnO2 | Plasma bombarding and mechanical milling | 31 | ||
| Li1.2Mn0.56Ni0.16Co0.08O2 | High pressure, supercritical CO2 treatment | 35–120 | (Supercritical) CO2 | 66 |
Similar high temperature treatment is effective to introduce OVs also in Li[Ni,Mn]O4,46–49 and layered Li1.2Mn0.54Ni0.13Co0.13O250–52 cathode materials. Gu et al. observed a clear dependence between the amount of OVs in the sample and the time-length of the heat treatments.49 OVs up to with δ = 0.271 can be introduced in LiNi0.5Mn1.5O4−δ synthesized at 700 °C for 10 h. The value of δ, i.e. the content of OVs, was found to be tuneable up to 0.313 and 0.337 for samples synthesized at 700 °C for 20 h and 48 h, respectively. Not unexpectedly, different synthesis methods can lead to different concentrations of OVs.52 The Li1.2Mn0.54Ni0.13Co0.13O2 electrode synthesized by solvo-thermal approaches are reported to have many more OVs compared with the material synthesized by hydrothermal methods. Xia et al. found that annealing in oxygen-deficient environments results in higher concentration of OVs in Li1+yMn2−yO4−δ cathode materials.53 In addition, heat treatment in H2,54 Ar,55,56 N253,57 or sudden quenching in liquid N258 are also found to be effective methods to induce large amounts of OVs in layered-oxide cathode materials.
Chemical assisted heat-treatments are also often used to generate OVs in Li2MnO3 and layered Li[Ni,Co,Mn]O2 cathode materials. For example, oxygen-deficient cathode materials can be obtained by heating Li2MnO3 or Li[Ni,Co,Mn]O2 cathode materials with HNO3,59 stearic acid,60,61 CaH2/LiH,62 NaBH4,63 and carbon black (Super P),64 and Na2S2O8.65 Cheng et al. found that OVs can form in the process of Mg-doping of LiCoO2 to maintain the system in an overall electroneutral state according to an OV/Mg2+ charge compensation mechanism.24 Qiu et al. succeeded in creating OVs on the particle surface through the gas-solid interface reaction in lithium enriched layered oxides and carbon dioxide.35 Gas-solid interface reaction can achieve precise control of oxygen activity through uniformly creating OVs without affecting the structural integrity of Li-rich layered cathode materials. Chen et al. reported that OVs are effectively distributed on the surface of LiNi0.5Co0.2Mn0.3O2–SnO2 cathode materials under the interaction of plasma bombarding and mechanical milling.31 Ji et al. demonstrated that OVs can successfully form in Li1.2Mn0.56Ni0.16Co0.08O2 cathode materials under high-pressure, supercritical CO2 treatment.66Fig. 3 provides an overview of some typical oxygen-deficient cathode materials for LiBs together with their preparation protocol.
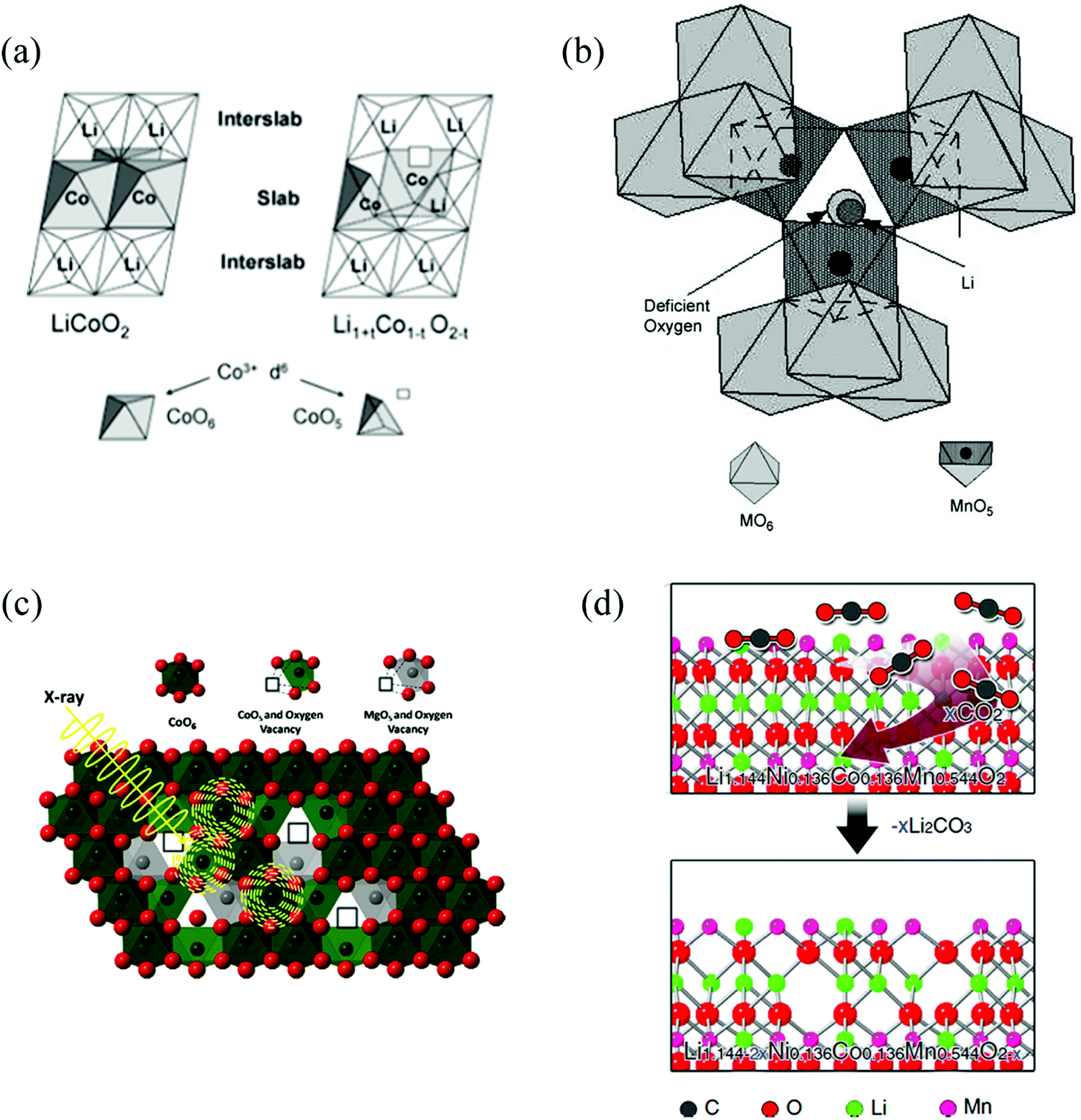 | ||
| Fig. 3 Selection of OV structures in different cathode oxide materials for LiBs. (a) LiCoO2−δ.42 Reprinted with permission from ref. 42. Copyright (2003) American Chemical Society. (b) Li1±xMn2−yO4±δ.45 Reprinted with permission from ref. 45, (c) LiMgyCo1−yO2.24 Reprinted with permission from ref. 24, (d) Li[Li0.144Ni0.136Co0.136Mn0.544]O2.35 | ||
3. Diffusion of OVs
Reversible creation and filling of Li vacancies in the cathode is one of the underpinning chemical processes in LiBs. By strongly affecting Li+ (de-)intercalation, the presence and local coordination of oxygen in the lattice is an extremely important factor for the performance and stability of cathode materials.16,67,68 It is fair to say that diffusion of OVs in lithium battery materials can be as important as the diffusion of Li-ions and deserves the utmost attention and precise understanding. Unfortunately, the diffusion of OVs and related processes are difficult to be observed directly and characterised experimentally. As a result, there are relatively few studies focussing on this aspect for Li-ion cathode materials.Fang et al.69 and Kubicek et al.70 have reported mechanisms for oxygen diffusion based on the presence of OVs in transition metal oxide Li-ion battery materials. Lee et al. studied OV-diffusion in LixMnO3 (0 < x < 2) cathode materials.71 They found that in the presence of large amounts (x < 1) of Li vacancies in LixMnO3 (x < 1), the oxygen migration barriers are too high for favourable diffusion of oxygen in the bulk material. However, as oxygen release from the particle surface is deemed likely, the ensuing local under-coordination and strain may facilitate surface oxygen diffusion. Another Lee et al. studied OV-diffusion and condensation in three types of cathode materials: LiCoO2, LiMn2O4, and Li2RuO3.72Fig. 4 summarises the main results from molecular dynamics simulation of LiMn2O4. Diffusion of OVs along a specific crystal direction causes continuous flow of OVs and transition metals, leading to eventual accumulation of OVs on a specific crystal surface, lattice mismatch and appearance of micro cracks in the single crystal cathode material.
 | ||
| Fig. 4 Migration model systems (Path A and Path B) and diffusion energy barriers of OVs in the orthogonal supercell of LiMn2O4 without and with the (110) propagation line of Mn-ions in DFT calculations. Li, Mn, diffusing Mn, oxygen and OVs are coloured violet, dark-gray, magenta, red and blue, respectively.72 Reprinted with permission from ref. 72. | ||
Yan et al. studied the oxygen “nanovoid” formation and migration in Li1.2Mn0.6Ni0.2O2 cathodes.73Fig. 5 shows the gradual propagation of the nanovoid-populated zone from the particle surface towards its interior during battery cycling. The formation of the oxygen nanovoid and its propagation from the surface towards the centre of the particle indicates accumulation of OVs from the particle surface into the bulk lattice during cycling. First principles calculations show that oxidising the oxide ions lowers both the formation energy and the diffusion barrier of OVs in the Li1.2Mn0.6Ni0.2O2 cathodes, which in turn promotes accumulation of OVs. Although OVs can be resolved in real time by state of the art in situ ACTEM combined with the HAADF technology, their images are still relatively fuzzy due to current limitations in the available resolution, see Fig. 5(a)–(f). Furthermore, the diffusion of OVs is a three-dimensional dynamic process, and surface OVs may diffuse into the bulk (or vice versa). This further increases the difficulty of direct experimental characterization, as surface sensitive technique cannot easily probe the bulk regions of materials. Fortunately, Density Functional Theory (DFT) can provide complementary or missing information about OVs diffusion at the atomic scale. Fig. 5(g)–(j) show the valence changes of both the Ni and O atoms during the charging process of a Li1.2Mn0.6Ni0.2O2 cathode. The calculated Ni electronic spin density confirms that the Ni atoms have different valence states as a function of the number of OVs in the sample. Additionally, the OV formation energy is calculated to sharply decrease once the anion redox reaction is triggered, indicating that hole-trapping at an oxygen site can promote formation of one OV.
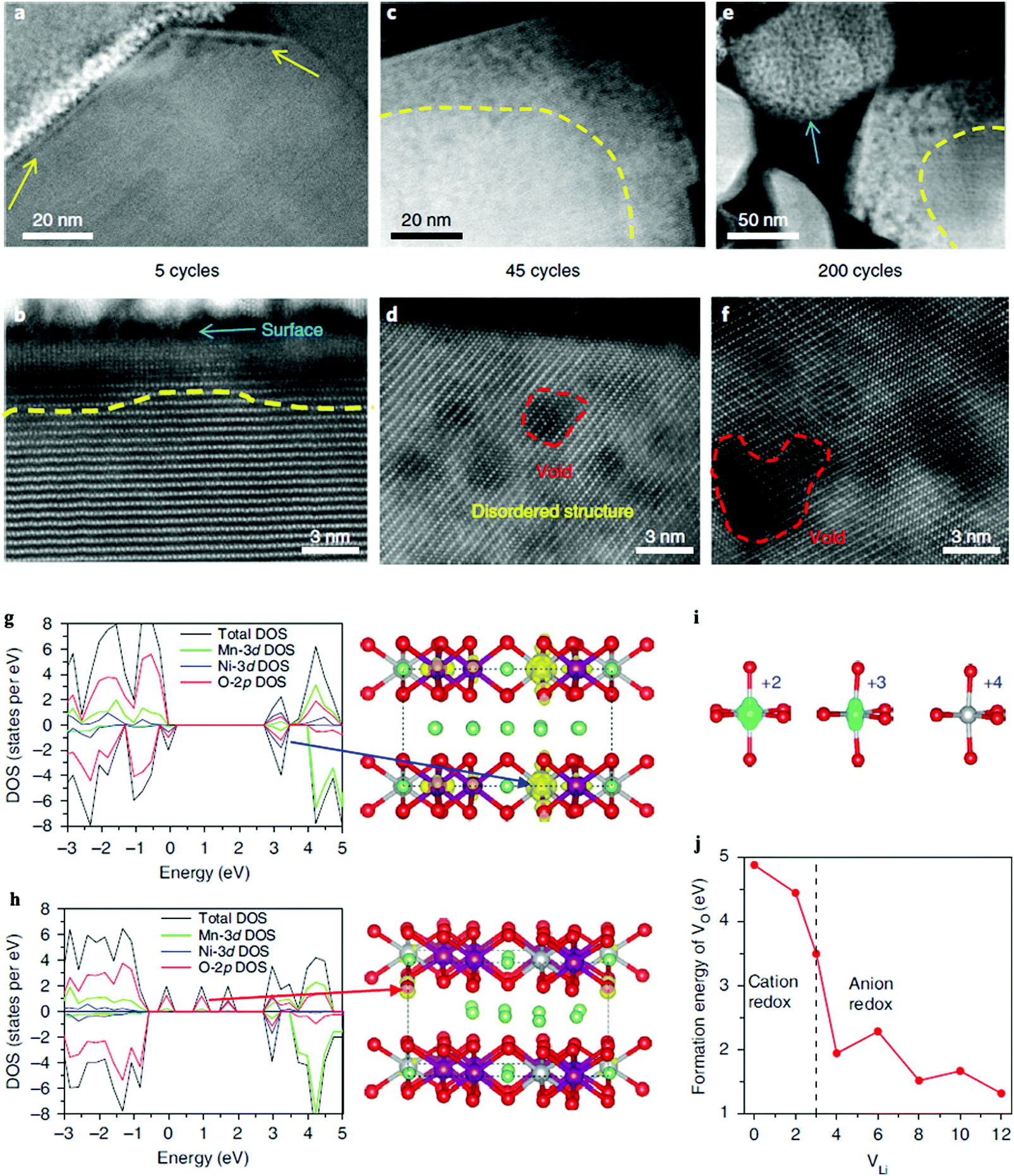 | ||
| Fig. 5 Spatial and temporal evolution of structural degradation from the surface into the bulk for a Li1.2Mn0.6Ni0.2O2 cathode. STEM-HAADF images show the gradual propagation of the (oxygen deficient) nanovoid-region from the surface to the interior of the particle during cycling. (a and b) Formation of the nanovoid region at the particle surface after five cycles is indicated by the yellow arrows in (a). The nanovoid shows up as a region of dark contrast. (c and d) After 45 cycles, the nanovoid region has grown. The red dashed line in (d) marks the nanovoid. (e and f) After 200 cycles, further growth of the nanovoid zone occurs. Note that the small particle, as indicated by the blue arrow in (e), is eventually fully populated by nanovoids. The dashed yellow lines in (b, c and e) mark the boundary between the nanovoid zone near the surface and the nanovoid-free region in the bulk. (g–j) Calculated electronic structure and formation energy of one OV in the charging process. (g) The calculated Density of States (DOS) after removing two Li ions, which leads to the oxidation of the Ni ions, as indicated by the blue arrow. (h) DOS after removing four Li ions, which leads to the oxidation of the oxide ions, as indicated by the red arrow. (i) Corresponding electronic spin density and valence states of the Ni atoms derived from the DOS of the charging process. (j) The OV formation energy as a function of the number of Li atoms removed from the supercell. The dashed line in (j) indicates the transition point of the valence change from Ni to O during removal of the Li ions. Purple, silver, green and red balls represent Mn, Ni, Li and O atoms, respectively.73 | ||
4. OV-induced effects in cathode materials
OVs can have profound influence on the physical and chemical properties of cathode materials, and greatly affect various performance indicators of LiBs. OVs can be used to very effectively tune the electronic structure, charge capacity, electrical conductivity, cation diffusion, surface structure and stability of cathode materials, thus strongly contributing to the lifetime and performance of LiBs. In the following, we provide an overview of OV-induced effects separating the discussion between Li-ion diffusion enhancement, performance degradation, and other materials physics and chemistry effects.a. OV-enhanced Li-ion diffusion and electrochemical performance
As early as 2002, Lu et al. found that the capacity of Li[NixLi1/3−2x/3Mn2/3−x/3]O2 cathode materials is closely related to the presence of OVs.74 In 2006, Armstrong et al. observed that oxygen loss causes an increase in capacity for Li[Ni0.2Li0.2Mn0.6]O2 cathode materials.44 Due to OV mediated charge compensation mechanisms, higher capacity and better cyclability can be obtained by introduction of magnesium (Mg2+) ions into the structure of LiCoO2.24 The introduction of vacancy-related hole states has been observed to be highly effective in improving electronic and Li-ion conductivity in LixCoO2.75 OVs can improve not only the capacity of cathode materials for LiBs; they can also enhance Li-ion diffusion and consequently reduce charge transfer resistance. Table 1 compactly presents a compilation of available results on the effects of OVs for electrochemical properties such as discharge capacity, Li+ diffusion coefficients, and charge transfer resistance of several oxygen-deficient cathode materials, namely Li2MnO3,60,61,63 LiV3O8,56 LiTi2(PO4)3,57 and Li[LiNiCoMn]O2.35,51,52,54,66Tan et al. have reported that OVs can increase the capacity of Li2MnO3−δ with respect to that pristine Li2MnO3 by nearly twenty times.63 After 100 charge–discharge cycles at 20 mA g−1, the residual capacity of Li2MnO3−δ is 135.1 mA h g−1, to be compared with 7.2 mA h g−1 for pristine Li2MnO3. Sun et al. also found that the occurrence of OVs in Li2MnO3 cathode materials leads to increased capacity and improved cycling performance.60,61 Besides beneficial effects on the cathode capacity, OVs in Li2MnO3 can also facilitate Li+ diffusion, increasing the electronic conductivity and improving stability during electrochemical cycling by suppressing charge-accumulation related degradation pathways (see below). In Li2MnO3 cathode materials, OVs lead to low valence Mn3+ ions due to charge compensation mechanisms. As demonstrated by experimental results on Li2MnO3/LiMn0.5Ni0.5O276 and Li2MnO3/LiMnO277 composite materials, partial substitution of Mn3+ with Mn4+ ions in Li2MnO3 can greatly improve the electrical conductivity and electrochemical performance of the cathode.
Similar electrochemical enhancement also occurs in oxygen-deficient LiV3O8 cathode materials, that display over 1 order of magnitude increase in electrical conductivity, and nearly 2 order of magnitude increased Li-ion diffusion with respect to the vacancy-free counterpart.56 Oxygen-deficient LiTi2(PO4)3 displays a discharge capacity of 93 mA h g−1, compared to 81 mA h g−1 for vacancy-free LiTi2(PO4)3 at 1C charge/discharge rates (140 mA g−1).57 In addition, the presence of OVs in this material was also found to increase the electronic conductivity by nearly two orders of magnitude with respect to the pristine vacancy-free substrate. First-principles simulations suggest that OVs can be formed in LiTi2(PO4)3 under oxygen-poor conditions, leading to formation of Ti3+ states and ensuing improvement of the overall electronic mobility for the material.57
Turning to Li-rich layered Li[Li,Ni,Co,Mn]O2 cathode materials, Abouimrane et al. found that heat-treated Li1.12Mn0.55Ni0.145Co0.1O2 shows better coulombic charge–discharge efficiency and electrochemical cyclability than the pristine material.54 Qiu et al. found that the capacity of Li [Li0.144Ni0.136Co0.136Mn0.544]O2 with OVs has no obvious attenuation after 100 cycles.35 Higher initial coulombic efficiency and improved rate capability are found also for oxygen-deficient Li1.2Mn0.56Ni0.16Co0.08O2,66 and Li1.2Mn0.54Ni0.13Co0.13O2.51,52 The occurrence of OVs, an expanded unit cell volume, and a larger amount of trivalent manganese (Mn3+) altogether lead to local environments with enhanced Li-ion conductivity, and better rate performance. Improved rate capability and enhanced cyclic stability also occurs when the cathode material is surrounded by nanostructured SnO231,78 and CeO279 with high concentration of OVs. Fig. 6 reports a compilation of cathode materials, whose stability and performance are improved by the engineered presence of OVs.
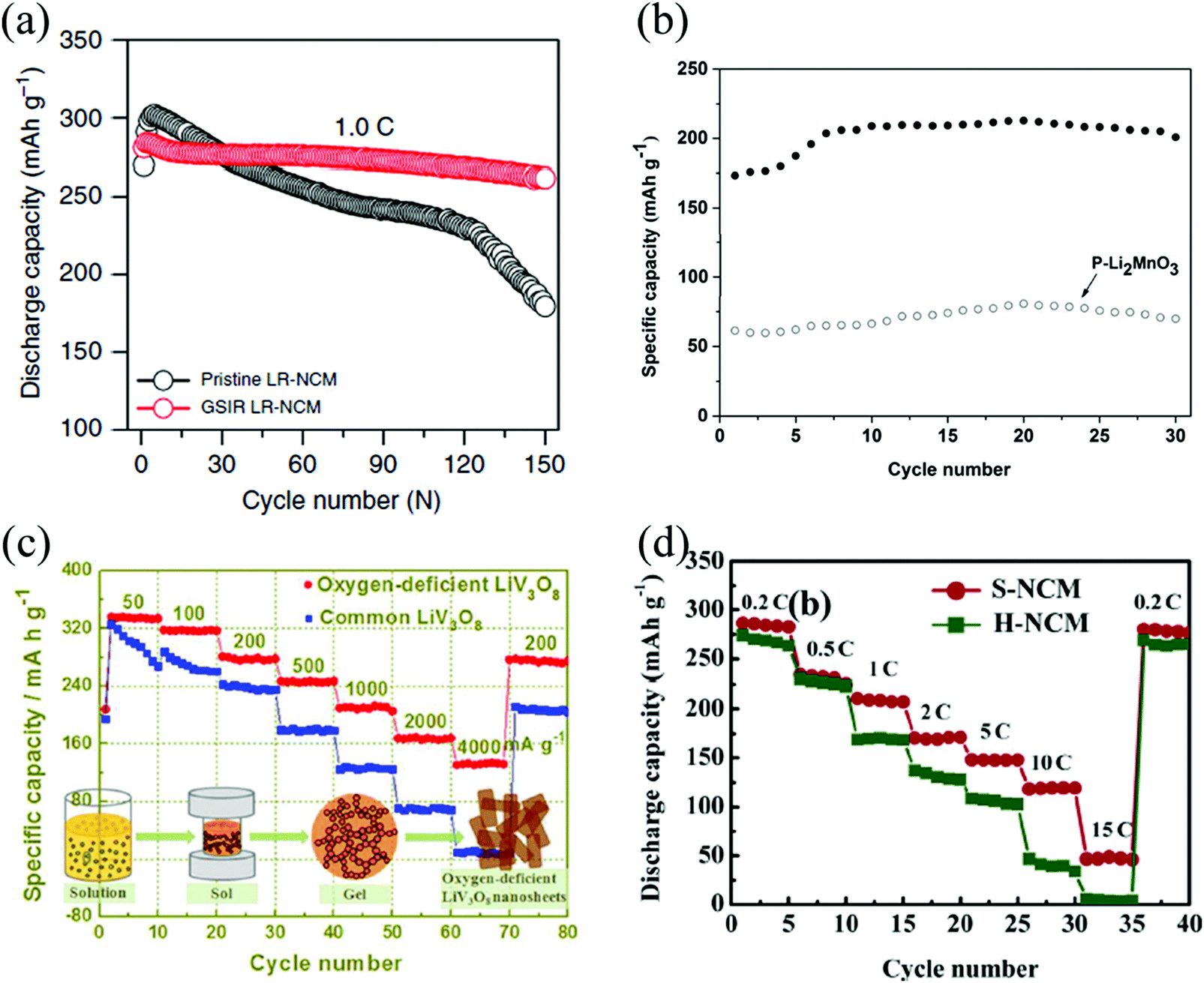 | ||
| Fig. 6 Cycling performance of the pristine and oxygen-deficient, (a) Li[Li0.144Ni0.136Co0.136Mn0.544]O2,35 (b) Li2MnO3.60 Reprinted with permission from ref. 60. Copyright (2017) American Chemical Society; discharge-rate capacity of the pristine and oxygen-deficient (c) LiV3O8.56 Reprinted with permission from ref. 56. Copyright (2017) American Chemical Society, (d) Li1.2Mn0.54Ni0.13Co0.13O2.52 Reprinted with permission from ref. 52. | ||
As shown in Table 2, OVs generally lead to lower charge transfer resistance or higher electrical conductivity for oxygen-deficient cathode materials. However, the charge transfer resistance does not increase linearly with the amount of OVs. For example, the charge transfer resistance for LiNi0.5Mn1.5O4−δ is 267.5 Ω, 271.3 Ω, and 405 Ω for samples prepared after 10 h (δ = 0.271), 20 h (δ = 0.313), and 48 h (δ = 0.337) of thermal treatment, respectively. It is obvious that the LiNi0.5Mn1.5O4−δ sample with the lowest concentration of OVs (δ = 0.271) exhibits the lowest charge transfer resistance.49 Similar electrochemical performance improvements are reported also for oxygen-deficient black SnO2−x,80 TiO2,81 MoO3−x,82 Li3VO4,18,20 Li4Ti5O12,19,30 TiP2O7,83 and TiNb2O734 anode materials as well as other electrode materials for Na-ion33,84,85 and Zn-ion86 batteries.
| Material | Capacity (mA h g−1)/cycles/current density (mA g−1) | Li+ diffusion coefficients (cm2 s−1) | Charge transfer resistance (Ω)/electrical conductivity (S cm−1) | Ref. | |||
|---|---|---|---|---|---|---|---|
| Pristine | OV | Pristine | OV | Pristine | OV | ||
| Li2MnO3 | 69.8/30/20 | 200.5/30/20 | 6.10 × 10−16 | 1.32 × 10−15 | 271.0 (Ω) | 226.5 (Ω) | 60 |
| Li2MnO3 | 7.2/100/20 | 135.1/100/20 | 161.0 (Ω) | 96.5 (Ω) | 63 | ||
| Li2MnO3 | 45.6/50/200 | 128.9/50/200 | 1.55 × 10−17 | 1.04 × 10−16 | 267.98 (Ω) | 132.2 (Ω) | 61 |
| LiV3O8 | 148/200/1000 | 197/200/1000 | 1.22 × 10−13 | 1.66 × 10−11 | 312.8 (Ω) | 145.2 (Ω) | 56 |
| LiTi2(PO4)3 | 81/N/140 | 93/N/140 | 4.4 × 10−8 (S cm−1) | 1.7 × 10−6 (S cm−1) | 57 | ||
| Li1.12Mn0.55Ni0.145Co0.1O2 | 171/20/320 | 196/20/320 | 3.2 × 10−7 (S cm−1) | 4.7 × 10−7 (S cm−1) | 54 | ||
| Li[Li0.144Ni0.136Co0.136Mn0.544]O2 | 179/100/250 | 262/100/250 | 35 | ||||
| Li1.2Mn0.56Ni0.16Co0.08O2 | 132.62/100/250 | 173.76/100/250 | 496.3 (Ω) | 354.7 (Ω) | 66 | ||
| Li1.2Mn0.54Ni0.13Co0.13O2 | 179/200/1250 | 225/200/1250 | 3.62 × 10−18 | 4.58 × 10−16 | 51 | ||
| Li1.2Mn0.54Ni0.13Co0.13O2 | 108.5/150/50 | 235/150/50 | 2.48 × 10−16 | 1.32 × 10−15 | 345.8 (Ω) | 243.3 (Ω) | 52 |
First principles density functional theory calculations show that the systems’ electronic band gaps are significantly reduced by the introduction of band-gap states associated with OVs. The electronic conductivity and discharge voltages of oxygen-deficient LiTiPO5 and LiTi2(PO4)3 may be increased by large concentration of OVs.87 Several mechanisms have been proposed to explain how OVs affects the electrochemical properties of cathode materials. The basic mechanisms for enhancement of Li-ion diffusion, and improvement of the electrochemical performance, include activation of Li-trapped states by OVs,35 enhancement of Li+ diffusion as a result of less sterically hindered diffusion channels owing to the presence of OVs,18,82,88 build-up of local electric fields originating from the unbalanced charge distribution,17,21 and reversible changes in the valence states for oxygen ions in oxidation–reduction process.14,15
In addition, due to the structural and electronic changes they introduce in the host, cation vacancies can also provide enhancement of performance in cathode materials.89 Compared with anion vacancies, metal cation vacancies are relatively more difficult to form.90 However, controlled formation of cation vacancies in cathode materials has been observed to improve the electrochemical performance of LiBs91 and zinc ion batteries (ZiBs).92
b. OV-related performance degradation in LiBs
Creation of OVs at suitable concentrations is an effective approach to increase the capacity of layered lithium transition metal oxides. However, OVs or more specifically the associated oxygen loss can also lead to phase transformations resulting in dead regions in the materials due to the introduced changes in electronic structure and promotion of further defect formation.93 The formation of large amounts of OVs inevitably leads to major Jahn–Teller triggered distortions of the host lattice. For tetragonal systems, the lattice constant c/a-ratio is a very sensitive parameter to reflect the state-of-charge, whose maximum value defines a stability limit for the layered oxides as cathode materials. The vacancy-induced stark volume reductions and changes in the c/a-ratio result in huge mechanical stress, which can cause cracks in the electrode particles. Hao et al. reported rapid capacity fade in LiMn2O4 cathodes caused by OVs.58 OVs result in lattice strain and large volume changes, which is the main cause of structural failure in LiMn2O4. The cracks caused by structural collapse along the (111) planes lead to mechanical collapse of the material during electrochemical cycling, leading to substantially increased charge-transfer resistance within the cell.Conversely, near stoichiometric oxygen content gives rise to more resilient structural integrity and stable electrochemical cycling.58 Zheng et al. found that a significant decrease of the valence of Mn ions can be detected in the cycled Li[Li0.2Ni0.2Mn0.6]O2 particles, which is below what expected for a detrimental Jahn–Teller distortion (+3.5) due to the formation of a large amount of OVs. The voltage fade can be correlated with Li ion insertion into the octahedral sites in the defect spinel-like structure and the disordered rock-salt structure.94 By experimental observation and finite element modelling, Mu et al. confirmed that crack formation can be attributed to the formation of OVs, oxygen release, and phase transformations. They observed that mechanical breakdown of charged Li1−xNi0.4Mn0.4Co0.2O2 materials proceeds via a two-step pathway involving both inter-granular and intra-granular crack formation. Owing to oxygen release, sporadic phase transformations from the layered structure to the spinel and/or rocksalt structures introduce local stress, which in turn initiates microcracks along grain boundaries and ultimately leads to the detachment of primary particles, i.e., to inter-granular crack formation. Furthermore, intra-granular cracks (pores and exfoliations) can also form, likely due to the accumulation of OVs and continuous phase transformations at the surfaces of the primary particles.95 Zhang et al. found that the oxygen loss and performance degradation of the layered cathode is a two-stage process with distinct release rates. Via in situ transmission electron microscopy observations of LiNi0.80Co0.15Al0.05O2, it was found that initial rapid oxygen loss generates high concentrations of OVs, which in turn results in the formation of an amorphous, vacancy-containing rock-salt layer on the particle surface. In the second stage, the slower oxygen loss rate allows recrystallization of this defective phase via coalescing of atomic OVs, which results in the formation of a cavity-containing surface layer with a crystalline rock-salt structure over the layered phase in the bulk.96
Recently, Lee et al. studied systematically the capacity attenuation of three types of cathode materials: spinel oxide (LiMn2O4, LMO), layered oxide (LiCoO2, LCO and Ni-rich materials), and Li-excess layered oxide (Li2RuO3 (LRO)). Fig. 7 shows the characterisation of microcracking for representative cathode materials after electrochemical cycling. The authors found that the capacity attenuation mechanism of cathode materials generally originates from the dynamics of OVs. All types of cathode materials have directional microcracking (planar defects) along the (1 1 1) cubic orientation of the oxygen sublattice. During electrochemical cycling, the occurrence of OVs, which are naturally present in transition metal oxides, causes continuous diffusion of OVs and transition metal ions toward the thermodynamically preferred state resulting in vacancy condensation and microcracking in specific orientations. Such cracks appear parallel to each other as seen by scanning electron microscopy. The arrangement of various lattice distortions, especially the stacking fault of oxygen-layers, provides direct evidence of OV accumulation.72 Yan et al. found that OVs created on the surface of Li1.2Mn0.6Ni0.2O2 migrate into the bulk during cycling, leading to the progressive surface-to-bulk degradation of the material.73 Thus, suppressing vacancy generation and blocking the OV-diffusion are the two critical ways to control the degradation of cathode caused by OVs. These approaches have been verified by introducing a vacancy band adjacent to the particle surface to effectively mitigate oxygen evolution,35 and by doping the surface to strongly bind oxygen, enhancing capacity retention upon cycling.97
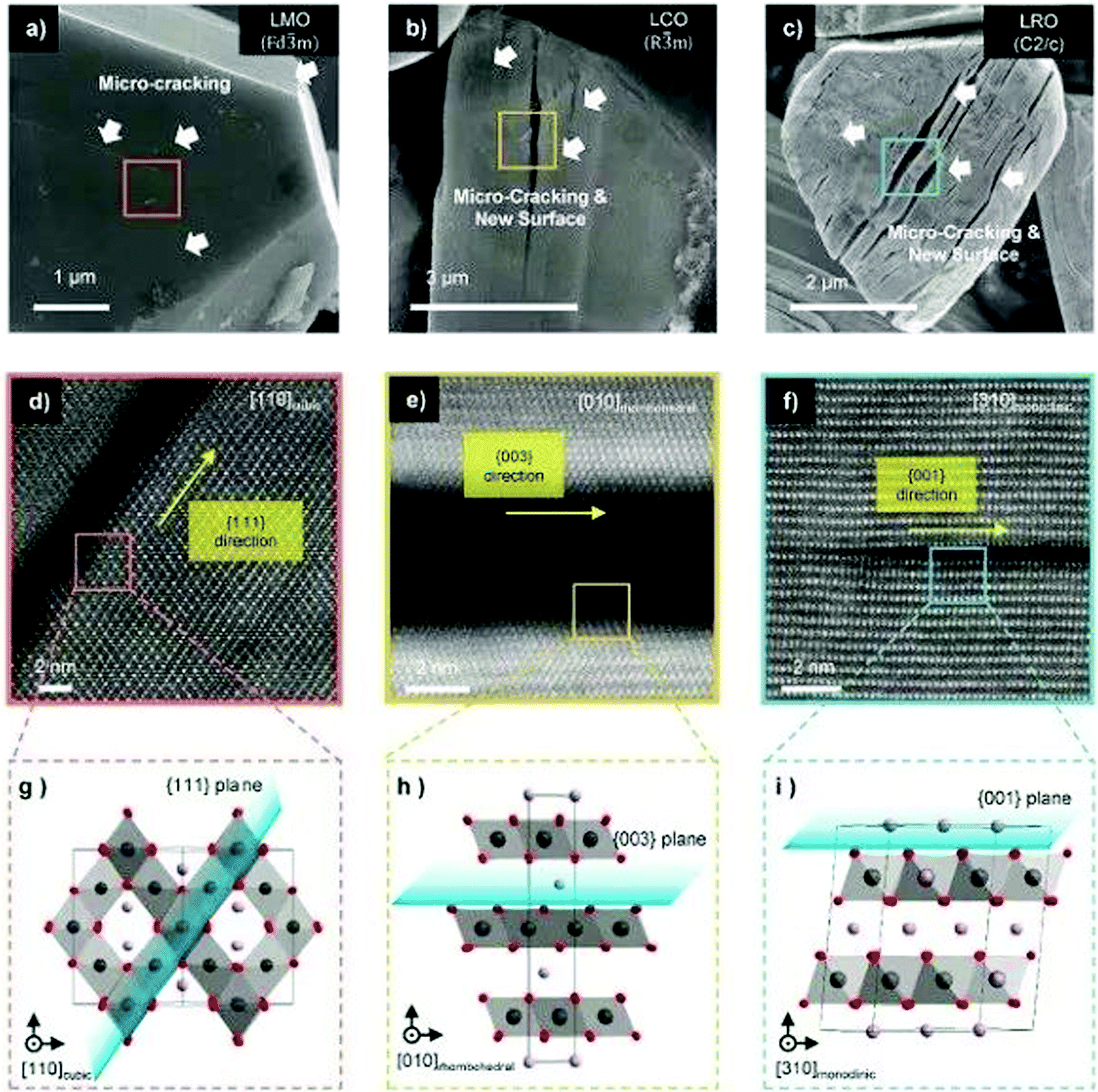 | ||
| Fig. 7 Characterization of microcracking for representative cathode materials after electrochemical cycling. (a–c) SEM image of specific orientation of microcrack formation on the single crystal of (a), LiMn2O4, (b) LiCoO2 and (c) Li2RuO3, respectively. White arrows indicate microcracks, which can generate new surfaces that continuously form new solid electrolyte interphases. (d–f) HAADF-STEM image of magnified micro-cracking regions. (g–i) Magnified atomic structure of the region highlighted in (d–f), showing the unit cell structure and orientation of the microcrack. The plane of the micro-crack is marked by the blue sheet. Li, Mn, and oxygen are coloured violet, dark-gray, and red, respectively.72 Reprinted with permission from ref. 72. | ||
c. Other physical and chemical effects of OVs in LiBs
It is established that occurrence of OVs leads to expansion of cell volume and structural distortion. In addition, OVs can also lead to some other chemical and physical phenomena in cathode materials, such as transition metal ions disorder,98–100 phase transition,45–47,53,101 changes in the diffusion of transition metal ions in the material,43,102etc.Sushko et al. found that neutral OVs in LiNi0.5Mn1.5O4−x promote substitutional Ni/Mn disorder and segregation of Ni-rich and Ni-poor regions. The former trap OVs, while the latter trap electrons associated with these vacancies. This leads to the creation of deep and shallow Mn3+ states, affecting the stability of lattice Li ions.98 Huang et al. found that OVs can result in Ni/Mn disorder in LiNi0.5Mn1.5O4−x cathode materials. In oxygen deficient LiNi0.5Mn1.5O4−x, the OVs tend to diminish the valence discrepancy between the Ni aggregated and the ordered phases, making the former energetically competitive and consequently resulting in disordered Ni/Mn distributions.99 Wang et al. showed that oxygen deficiency assists the migration of Ni and Mn ions from octahedral sites to tetrahedral ones in LiNi0.5Mn1.5O4 cathodes, and that, in parallel, migration of Ni and Mn ions favours formation of OVs.100
It has also been found that the order–disorder transitions for transition metal ions can initiate phase transition in cathode materials. Song et al. showed that the solubility limit for OVs in the disordered spinel phase Li[Ni0.5−xMn1.5+x]O4 is small at 600 °C. Above 700 °C, a reversible transition from spinel phase to rock-salt phase is initiated to accommodate oxygen loss. The transition from spinel to rock-salt is reversible, via the reinsertion of oxygen upon slow cooling to 700 °C, and the oxygen regained is not lost upon further cooling to room temperature.46 The crystal structures of spinel phase and rock-salt phase Li[Ni0.5−xMn1.5+x]O4 are schematically illustrated in Fig. 8. Kim found that Ni and Mn in LiNi0.5Mn1.5O4 have an ordered arrangement and change structure from face centred spinel (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) to primitive simple cubic (P4332) by additional heating at 700 °C.101 Liu et al. revealed that the occurrence and degree of Ni/Mn disordering are closely related with the formation of OVs and presence of Mn3+ ions. Slow cooling rate and post-annealing can result in low degrees of Ni/Mn disorder and OVs. The transition from disordered structure to ordered structure is reversible and depends on the presence of OVs. The two structures can be mutually interconverted by tuning synthesis temperature, cooling rate and post-annealing processing.47 The crystal structures of Ni/Mn ordered phase (P4332) and disordered phase (Fdm) LiNi0.5Mn1.5O4. Phase transitions caused by OVs can occur also in the cathode material with single-type transition metal ion. Xia et al. showed unambiguously two-phase coexistence for undoped spinel LiMn2O4 with oxygen deficiency.53 They also found that phase transition evolution in spinel Li1+xMn2−yO4±δ is greatly suppressed by Al doping.45
m) to primitive simple cubic (P4332) by additional heating at 700 °C.101 Liu et al. revealed that the occurrence and degree of Ni/Mn disordering are closely related with the formation of OVs and presence of Mn3+ ions. Slow cooling rate and post-annealing can result in low degrees of Ni/Mn disorder and OVs. The transition from disordered structure to ordered structure is reversible and depends on the presence of OVs. The two structures can be mutually interconverted by tuning synthesis temperature, cooling rate and post-annealing processing.47 The crystal structures of Ni/Mn ordered phase (P4332) and disordered phase (Fdm) LiNi0.5Mn1.5O4. Phase transitions caused by OVs can occur also in the cathode material with single-type transition metal ion. Xia et al. showed unambiguously two-phase coexistence for undoped spinel LiMn2O4 with oxygen deficiency.53 They also found that phase transition evolution in spinel Li1+xMn2−yO4±δ is greatly suppressed by Al doping.45
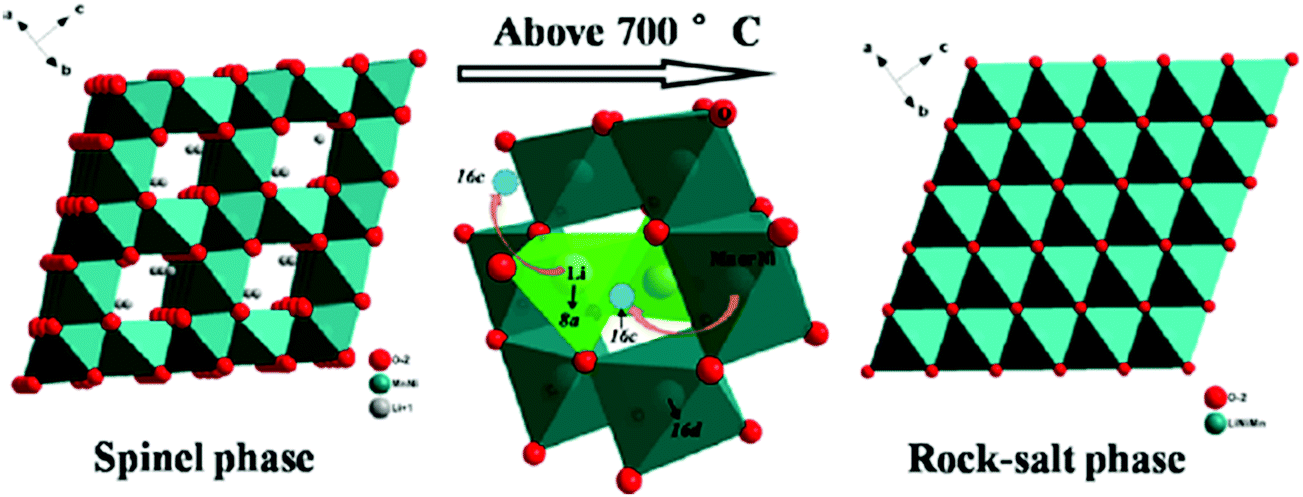 | ||
| Fig. 8 Phase transition from spinel to rock-salt in Li[Ni0.5−xMn1.5+x]O4.46 Reprinted with permission from ref. 46. Copyright (2012) American Chemical Society. | ||
The occurrence of OVs can also affect cation-diffusion in cathode materials by altering the steric and electrostatic hindrance in the corresponding diffusion channels and paths. Qian et al. demonstrated that the OVs assist the migration of transition-metal ions through a novel mechanism in layered Li20/28Ni1/4Mn7/12O2.102 The role of OVs for the Ni diffusion barriers is illustrated in Fig. 9(a). After the first charging, when most of the transition metal ions are fully oxidized, oxygen ions start to participate in the oxidation process (losing electrons) and OVs start to form with a formation energy of about 0.5–0.6 eV. Due to the slow oxygen migration, OVs mostly form in the surfaces and sub-surfaces region within the topmost 5–6 atomic layers. A significant fraction of the transition metal ions in these regions is therefore subjected to five (or even less) fold O-coordination due to the presence of either OVs or the broken metal-oxygen bonds on the particle's surface. The transition metal ions in those under-coordinated defect polyhedral sites become less stable and spontaneously migrate to the fully-coordinated polyhedral sites in the nearby Li layer.
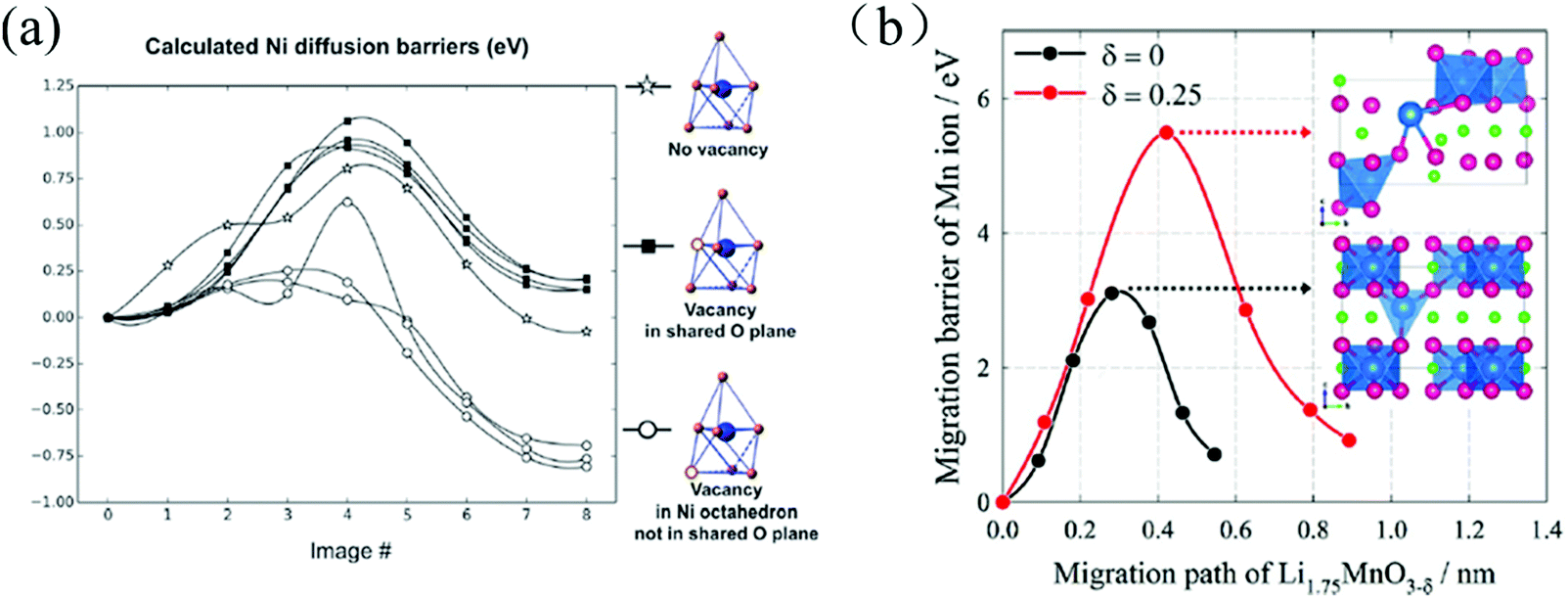 | ||
| Fig. 9 (a) Calculated Ni diffusion barriers with OVs in different positions of the Li20/28Ni1/4Mn7/12O2, lattice (vacancies in the tetrahedron but not in the shared plane are unstable).102 (b) Mn-Ion diffusion barriers as a function of the migration paths in Li1.75MnO3 (black) and Li1.75MnO3−δ (red); inset: the atomic structures of the diffusion transition state.43 Reprinted with permission from ref. 43. | ||
Lim et al. studied the effects of OVs on the diffusion barriers of Mn ions in the oxygen-deficient Li2MnO3−x cathode material. They found that OVs suppress oxidation of oxygen by lowering the redox potential for Mn reduction, and increasing the diffusion barriers for Mn ions (ΔEbarrier = 1.0–2.5 eV) as a result of a higher energy transition state for the diffusion event, see Fig. 9(b).43
5. Conclusions and outlook
In conclusion, OVs are closely related to the electron and ion transport properties of cathode materials for LiBs. The introduction of OVs is an effective strategy to improve the electrochemical performance of cathode materials for LiBs. OVs can increase the capacity and rate performance, accelerate Li-ion transport, hinder unwanted phase transformations, and reduce the energy barrier for the diffusion of transition metal cations. However, stabilizing OVs remains an only partially solved challenge, especially under oxidizing (oxygen rich) conditions under which the OVs are very reactive and their presence difficult to enforce or maintain. To the best of the authors’ knowledge, limited stabilization strategies are currently available, including oxygen deficient atmosphere, heteroatom doping, interface engineering, and specific crystalline plane. Given their clear potential for development of improved cathode materials, we hope this review will stimulate further interest and experimental as well as simulation work towards control of OVs in oxide based cathode materials.Based on the state of the art in the field as reviewed here, the most urgent avenues of development we envision are at the level of improving existing methods to controllably introduce and characterise OVs towards definition of novel and scalable compromises between enhancement of electrochemical performance and materials degradation. Important ongoing work is happening along these lines and it is reasonable to expect the rate of progress to further increase in the short future. Specifically, controlling the concentration of OVs and hindering their diffusion are effective strategies to restrain phase transformation and cracks in cathode materials for LiBs. In addition, the concentration and distribution of OVs can be controlled by different synthesis method. Heat treatment and ion doping can produce both surface and bulk oxygen vacancies. The corresponding concentration of oxygen vacancies can be controlled by varying the time extension of the heat treatment or doping-reaction. Plasma bombarding and mechanical milling produce OVs only on the surface or in the subsurface of metal oxides. However, these methods have the advantages of mild conditions, simple operation, fast and efficient, controllable defect content and easy large-scale production.
Many qualitative analysis methods can be used to characterize and analyse OVs, such as Raman spectroscopy, Electron Paramagnetic Resonance (EPR), X-ray Photoelectron Spectroscopy (XPS), X-Ray Absorption Fine Structure (XAFS), Photoacoustic Spectroscopy (PAS) and so on. The rapid development of electron microscopy has led to direct imaging of the atomic structure of samples. Scanning tunnelling electron microscopy (STEM) and high-resolution transmission electron microscopy (HRTEM) are widely used to characterize OVs. STEM is generally used to observe surface OVs and their interactions with external molecules, while HRTEM is used to observe surface and bulk OVs. Although the existence of OV defects can be seen by HRTEM, modern state of the art HRTEM imaging of OVs remain fuzzy due to the limitations in the achievable image resolution. The most advanced spherical aberration corrected Transmission Electron Microscope (ACTEM) has been shown to be capable of defects resolution, enabling quantitative study of their local environments and concentrations. Combined with High Angle Annular Dark Field (HAADF) images, it can also resolve three-dimensional reconstructions of materials, providing three-dimensional information about the distribution of defects and related lattice distortions. Progress in in situ TEM technologies has enabled direct, real time observation of processes in materials. In situ TEM can accordingly help scientists to better understand the role of defects in electrode materials for the functioning of lithium-ion batteries.
In spite of the impressive advances in modern experimental characterization approaches, challenges remain towards exhaustive insights into and understanding of OVs. Density Functional Theory (DFT) simulations can determine the structure and basic properties of materials, and achieve atomic level accuracy. DFT is the most effective method to study the geometric structure, vacancy defects and electrochemical characteristics of cathode materials for lithium-ion batteries. In addition, DFT has also been widely used to explore lithium insertion and de-intercalation mechanisms, and to model diffusion energy barriers, structural stability, lithium intercalation capacity mechanisms and similar key aspects of materials for LiBs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science Challenge Project (TZ2018004), the National Natural Science Foundation of China (No. 51602092, 11974037, 51861130360), the Science and Technology Innovation Project Foundation of Hunan Province (No. 2018RS3103), the Hunan Provincial Natural Science Foundation of China (No. 2020JJ4147), the Research Foundation of Education Bureau of Hunan Province, China (No. 19B076), and the Royal Society Newton Advanced Fellowships (No. NAF\R1\180242).References
- I. Dincer, Renewable Sustainable Energy Rev., 2000, 4, 157–175 CrossRef.
- H. Lund, Energy, 2007, 32, 912–919 CrossRef.
- N. L. Panwar, S. C. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS.
- M. G. S. R. Thomas, P. G. Bruce and J. B. Goodenough, Solid State Ionics, 1985, 17, 13–19 CrossRef CAS.
- D. Aurbach, M. D. Levi, E. Levi and A. Schechter, J. Phys. Chem. B, 1997, 101, 2195–2206 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS.
- D. D. MacNeil, Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1332 CrossRef CAS.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 142–147 CrossRef CAS.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS.
- M. Sathiya, K. Ramesha, G. Rousse, D. Foix, D. Gonbeau, A. S. Prakash, M. L. Doublet, K. Hemalatha and J. M. Tarascon, Chem. Mater., 2013, 25, 1121–1131 CrossRef CAS.
- Y. Wang, X. Xiao, Q. Li and H. Pang, Small, 2018, 14, 1802193 CrossRef.
- C. Hou, Y. Hou, Y. Fan, Y. Zhai, Y. Wang, Z. Sun, R. Fan, F. Dang and J. Wang, J. Mater. Chem. A, 2018, 6, 6967–6976 RSC.
- L. Chen, X. Jiang, N. Wang, J. Yue, Y. Qian and J. Yang, Adv. Sci., 2015, 2, 1500090 CrossRef.
- H. Xu, J. Chen, Y. Li, X. Guo, Y. Shen, D. Wang, Y. Zhang and Z. Wang, Sci. Rep., 2017, 7, 2960 CrossRef.
- K. Wang, C. Zhang, H. Fu, C. Liu, Z. Li, W. Ma, X. Lu and G. Cao, Chem. – Eur. J., 2017, 23, 5368–5374 CrossRef CAS.
- F. Cheng, T. Zhang, Y. Zhang, J. Du, X. Han and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 2474–2477 CrossRef CAS.
- Z. Sadighi, J. Huang, L. Qin, S. Yao, J. Cui and J.-K. Kim, J. Power Sources, 2017, 365, 134–147 CrossRef CAS.
- K. Liang, H. He, Y. Ren, J. Luan, H. Wang, Y. Ren and X. Huang, Ionics, 2020, 26, 1739–1747 CrossRef CAS.
- J. H. Cheng, C. J. Pan, C. Nithya, R. Thirunakaran, S. Gopukumar, C. H. Chen, J. F. Lee, J. M. Chen, A. Sivashanmugam and B. J. Hwang, J. Power Sources, 2014, 252, 292–297 CrossRef CAS.
- A. Roldán, M. Boronat, A. Corma and F. Illas, J. Phys. Chem. C, 2010, 114, 6511–6517 CrossRef.
- J.-Y. Shin, J. H. Joo, D. Samuelis and J. Maier, Chem. Mater., 2012, 24, 543–551 CrossRef CAS.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS.
- Q. Gan, H. He, K. Zhao, Z. He, S. Liu and S. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 7031–7042 CrossRef CAS.
- C.-K. Lan, S.-I. Chuang, Q. Bao, Y.-T. Liao and J.-G. Duh, J. Power Sources, 2015, 275, 660–667 CrossRef CAS.
- J. Zhu, J. Chen, H. Xu, S. Sun, Y. Xu, M. Zhou, X. Gao and Z. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 17384–17392 CrossRef CAS.
- Z. Chen, Y. Liu, Z. Lu, R. Hu, J. Cui, H. Xu, Y. Ouyang, Y. Zhang and M. Zhu, J. Alloys Compd., 2019, 803, 71–79 CrossRef CAS.
- D. Zhou, X. Xiong, Z. Cai, N. Han, Y. Jia, Q. Xie, X. Duan, T. Xie, X. Zheng, X. Sun and X. Duan, Small Methods, 2018, 2, 1800083 CrossRef.
- Z. Li, Y. Dong, J. Feng, T. Xu, H. Ren, C. Gao, Y. Li, M. Cheng, W. Wu and M. Wu, ACS Nano, 2019, 13, 9227–9236 CrossRef CAS.
- Y. Zhang, M. Zhang, Y. Liu, H. Zhu, L. Wang, Y. Liu, M. Xue, B. Li and X. Tao, Electrochim. Acta, 2020, 330, 135299 CrossRef CAS.
- B. Qiu, M. Zhang, L. Wu, J. Wang, Y. Xia, D. Qian, H. Liu, S. Hy, Y. Chen, K. An, Y. Zhu, Z. Liu and Y. S. Meng, Nat. Commun., 2016, 7, 12108 CrossRef CAS.
- G. Ou, Y. Xu, B. Wen, R. Lin, B. Ge, Y. Tang, Y. Liang, C. Yang, K. Huang, D. Zu, R. Yu, W. Chen, J. Li, H. Wu, L.-M. Liu and Y. Li, Nat. Commun., 2018, 9, 1302 CrossRef.
- W. Göpel, G. Rocker and R. Feierabend, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 3427–3438 CrossRef.
- X. Pan, M.-Q. Yang, X. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- L. O. Valøen and J. N. Reimers, J. Electrochem. Soc., 2005, 152, A882–A891 CrossRef.
- H. Yang, G. V. Zhuang and P. N. Ross, J. Power Sources, 2006, 161, 573–579 CrossRef CAS.
- Z.-K. Tang, J. S. Tse and L.-M. Liu, J. Phys. Chem. Lett., 2016, 7, 4795–4801 CrossRef CAS.
- S. Levasseur, M. Ménétrier, Y. Shao-Horn, L. Gautier, A. Audemer, G. Demazeau, A. Largeteau and C. Delmas, Chem. Mater., 2003, 15, 348–354 CrossRef CAS.
- J.-M. Lim, D. Kim, Y.-G. Lim, M.-S. Park, Y.-J. Kim, M. Cho and K. Cho, ChemElectroChem, 2016, 3, 943–949 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS.
- Y. Xia, H. Wang, Q. Zhang, H. Nakamura, H. Noguchi and M. Yoshio, J. Power Sources, 2007, 166, 485–491 CrossRef CAS.
- J. Song, D. W. Shin, Y. Lu, C. D. Amos, A. Manthiram and J. B. Goodenough, Chem. Mater., 2012, 24, 3101–3109 CrossRef CAS.
- H. Liu, X. Zhang, X. He, A. Senyshyn, A. Wilken, D. Zhou, O. Fromm, P. Niehoff, B. Yan, J. Li, M. Muehlbauer, J. Wang, G. Schumacher, E. Paillard, M. Winter and J. Li, J. Electrochem. Soc., 2018, 165, A1886–A1896 CrossRef CAS.
- D. Lu, J. Li, J. He, R. Zhao and Y. Cai, J. Phys. Chem. C, 2019, 123, 8522–8530 CrossRef CAS.
- Y. J. Gu, Y. Li, Y. B. Chen, H. Q. Liu, H. H. Zhou, H. F. Wang, Y. Q. Han and J. Zhang, Int. J. Electrochem. Sci., 2017, 12, 9523–9532 CrossRef CAS.
- H. Koga, L. Croguennec, M. Ménétrier, P. Mannessiez, F. Weill and C. Delmas, J. Power Sources, 2013, 236, 250–258 CrossRef CAS.
- Y. X. Cai, L. Ku, L. S. Wang, Y. T. Ma, H. F. Zheng, W. J. Xu, J. T. Han, B. H. Qu, Y. Z. Chen, Q. S. Xie and D. L. Peng, Sci. China Mater., 2019, 62, 1374–1384 CrossRef CAS.
- B. Chen, B. Zhao, J. Zhou, Z. Fang, Y. Huang, X. Zhu and Y. Sun, J. Mater. Sci. Technol., 2019, 35, 994–1002 CrossRef.
- Y. Xia, T. Sakai, T. Fujieda, X. Q. Yang, X. Sun, Z. F. Ma, J. McBreen and M. Yoshio, J. Electrochem. Soc., 2001, 148, A723 CrossRef CAS.
- A. Abouimrane, O. C. Compton, H. Deng, I. Belharouak, D. A. Dikin, S. T. Nguyen and K. Amine, Electrochem. Solid-State Lett., 2011, 14, A126 CrossRef CAS.
- X. Q. Yang, X. Sun, M. Balasubramanian, J. McBreen, Y. Xia, T. Sakai and M. Yoshio, Electrochem. Solid-State Lett., 2001, 4, A117 CrossRef CAS.
- H. Song, M. Luo and A. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 2875–2882 CrossRef CAS.
- J.-Y. Luo, L.-J. Chen, Y.-J. Zhao, P. He and Y.-Y. Xia, J. Power Sources, 2009, 194, 1075–1080 CrossRef CAS.
- X. Hao, X. Lin, W. Lu and B. M. Bartlett, ACS Appl. Mater. Interfaces, 2014, 6, 10849–10857 CrossRef CAS.
- S.-H. Kang, C. S. Johnson, J. T. Vaughey, K. Amine and M. M. Thackeray, J. Electrochem. Soc., 2006, 153, A1186–A1192 CrossRef CAS.
- Y. Sun, H. Cong, L. Zan and Y. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 38545–38555 CrossRef CAS.
- Y. Sun, L. Zan and Y. Zhang, Appl. Surf. Sci., 2019, 483, 270–277 CrossRef CAS.
- K. Kubota, T. Kaneko, M. Hirayama, M. Yonemura, Y. Imanari, K. Nakane and R. Kanno, J. Power Sources, 2012, 216, 249–255 CrossRef CAS.
- X. Tan, R. Liu, C. Xie and Q. Shen, J. Power Sources, 2018, 374, 134–141 CrossRef CAS.
- B. Song, H. Liu, Z. Liu, P. Xiao, M. O. Lai and L. Lu, Sci. Rep., 2013, 3, 3094 CrossRef.
- S. Han, B. Qiu, Z. Wei, Y. Xia and Z. Liu, J. Power Sources, 2014, 268, 683–691 CrossRef CAS.
- Y. Ji, R. Li, D. Mu, S. Sun, C. Dai and F. Ding, J. Electrochem. Soc., 2018, 165, A2880–A2888 CrossRef CAS.
- M. Oishi, C. Yogi, I. Watanabe, T. Ohta, Y. Orikasa, Y. Uchimoto and Z. Ogumi, J. Power Sources, 2015, 276, 89–94 CrossRef CAS.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák, G. Van Tendeloo, R. Dominko and J.-M. Tarascon, Science, 2015, 350, 1516–1521 CrossRef CAS.
- Q. F. Fang, X. P. Wang, Z. S. Li, G. G. Zhang and Z. G. Yi, Mater. Sci. Eng., A, 2004, 370, 365–369 CrossRef.
- M. Kubicek, A. Wachter-Welzl, D. Rettenwander, R. Wagner, S. Berendts, R. Uecker, G. Amthauer, H. Hutter and J. Fleig, Chem. Mater., 2017, 29, 7189–7196 CrossRef CAS.
- E. Lee and K. A. Persson, Adv. Energy Mater., 2014, 4, 1400498 CrossRef.
- S. Lee, W. Jin, S. H. Kim, S. H. Joo, G. Nam, P. Oh, Y.-K. Kim, S. K. Kwak and J. Cho, Angew. Chem., Int. Ed., 2019, 58, 10478–10485 CrossRef CAS.
- P. Yan, J. Zheng, Z.-K. Tang, A. Devaraj, G. Chen, K. Amine, J.-G. Zhang, L.-M. Liu and C. Wang, Nat. Nanotechnol., 2019, 14, 602–608 CrossRef CAS.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815 CrossRef CAS.
- T. Mizokawa, Y. Wakisaka, T. Sudayama, C. Iwai, K. Miyoshi, J. Takeuchi, H. Wadati, D. G. Hawthorn, T. Z. Regier and G. A. Sawatzky, Phys. Rev. Lett., 2013, 111, 056404 CrossRef CAS.
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085–1091 CrossRef CAS.
- Q. Zhang, T. Peng, D. Zhan and X. Hu, J. Power Sources, 2014, 250, 40–49 CrossRef CAS.
- C. Chen, T. Geng, C. Du, P. Zuo, X. Cheng, Y. Ma and G. Yin, J. Power Sources, 2016, 331, 91–99 CrossRef CAS.
- K. Yang, Y. Liu, B. Niu, Z. Yang and J. Li, Ionics, 2019, 25, 2027–2034 CrossRef CAS.
- W. Dong, J. Xu, C. Wang, Y. Lu, X. Liu, X. Wang, X. Yuan, Z. Wang, T. Lin, M. Sui, I.-W. Chen and F. Huang, Adv. Mater., 2017, 29, 1700136 CrossRef.
- X. Deng, Z. Wei, C. Cui, Q. Liu, C. Wang and J. Ma, J. Mater. Chem. A, 2018, 6, 4013–4022 RSC.
- H.-S. Kim, J. B. Cook, H. Lin, J. S. Ko, S. H. Tolbert, V. Ozolins and B. Dunn, Nat. Mater., 2017, 16, 454–460 CrossRef CAS.
- D. Bin, Y. Wen, Y. Yuan, Y. Liu, Y. Wang, C. Wang and Y. Xia, Electrochim. Acta, 2019, 320, 134555 CrossRef CAS.
- Y. Xu, M. Zhou, X. Wang, C. Wang, L. Liang, F. Grote, M. Wu, Y. Mi and Y. Lei, Angew. Chem., Int. Ed., 2015, 54, 8768–8771 CrossRef CAS.
- Y. Xu, M. Zhou, C. Zhang, C. Wang, L. Liang, Y. Fang, M. Wu, L. Cheng and Y. Lei, Nano Energy, 2017, 38, 304–312 CrossRef CAS.
- M. Liao, J. Wang, L. Ye, H. Sun, Y. Wen, C. Wang, X. Sun, B. Wang and H. Peng, Angew. Chem., Int. Ed., 2020, 59, 2273–2278 CrossRef CAS.
- L.-J. Chen, Y.-J. Zhao, J.-Y. Luo and Y.-Y. Xia, Phys. Lett. A, 2011, 375, 934–938 CrossRef CAS.
- T. Nakamura, H. Gao, K. Ohta, Y. Kimura, Y. Tamenori, K. Nitta, T. Ina, M. Oishi and K. Amezawa, J. Mater. Chem. A, 2019, 7, 5009–5019 RSC.
- M. Kim, D. Kim, Y. Wen, M. Kim, H. M. Jang, H. Li, L. Gu and B. Kang, Joule, 2019, 3, 1064–1079 CrossRef CAS.
- M. Martos, J. Morales and L. Sánchez, Electrochim. Acta, 2000, 46, 83–89 CrossRef CAS.
- F. Lu, Q. Chen, S. Geng, M. Allix, H. Wu, Q. Huang and X. Kuang, J. Mater. Chem. A, 2018, 6, 24232–24244 RSC.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS.
- R. Hausbrand, G. Cherkashinin, H. Ehrenberg, M. Gröting, K. Albe, C. Hess and W. Jaegermann, Mater. Sci. Eng., B, 2015, 192, 3–25 CrossRef CAS.
- J. Zheng, P. Xu, M. Gu, J. Xiao, N. D. Browning, P. Yan, C. Wang and J.-G. Zhang, Chem. Mater., 2015, 27, 1381–1390 CrossRef CAS.
- L. Mu, R. Lin, R. Xu, L. Han, S. Xia, D. Sokaras, J. D. Steiner, T.-C. Weng, D. Nordlund, M. M. Doeff, Y. Liu, K. Zhao, H. L. Xin and F. Lin, Nano Lett., 2018, 18, 3241–3249 CrossRef CAS.
- H. Zhang, B. M. May, F. Omenya, M. S. Whittingham, J. Cabana and G. Zhou, Chem. Mater., 2019, 31, 7790–7798 CrossRef CAS.
- S. Liu, Z. Liu, X. Shen, W. Li, Y. Gao, M. N. Banis, M. Li, K. Chen, L. Zhu, R. Yu, Z. Wang, X. Sun, G. Lu, Q. Kong, X. Bai and L. Chen, Adv. Energy Mater., 2018, 8, 1802105 CrossRef.
- P. V. Sushko, K. M. Rosso, J.-G. Zhang, J. Liu and M. L. Sushko, Adv. Funct. Mater., 2013, 23, 5530–5535 CrossRef CAS.
- Y. Chen, Y. Sun and X. Huang, Comput. Mater. Sci., 2016, 115, 109–116 CrossRef CAS.
- Z. Wang, Q. Su, H. Deng and Y. Fu, ChemElectroChem, 2015, 2, 1182–1186 CrossRef CAS.
- J. H. Kim, S. T. Myung, C. S. Yoon, S. G. Kang and Y. K. Sun, Chem. Mater., 2004, 16, 906–914 CrossRef CAS.
- D. Qian, B. Xu, M. Chi and Y. S. Meng, Phys. Chem. Chem. Phys., 2014, 16, 14665–14668 RSC.
| This journal is © The Royal Society of Chemistry 2020 |