
The direct C–H alkenylation of quinoline N-oxides as a suitable strategy for the synthesis of promising antiparasitic drugs
Vladimir V.
Kouznetsov
 *,
Leonor Y.
Vargas Méndez
,
Carlos E.
Puerto Galvis
and
Marlyn C.
Ortiz Villamizar
*,
Leonor Y.
Vargas Méndez
,
Carlos E.
Puerto Galvis
and
Marlyn C.
Ortiz Villamizar
Laboratorio de Química Orgánica y Biomolecular, CMN, Universidad Industrial de Santander, Parque Tecnológico Guatiguará, Km 2 Vía Refugio, Piedecuesta 681011, Colombia. E-mail: kouznet@uis.edu.co; vkuznechnik@gmail.com; Tel: +57 7 634 4000, ext. 3593
First published on 21st November 2019
Abstract
Functionalized quinolines are an important group of heterocyclic molecules with diverse, different, and interesting biological and pharmacological activities. Traditionally, several methods have been developed for the construction of this N-heterocyclic ring, but nowadays the functionalization of the quinoline scaffold has gained great interest and it is highly demand for the synthesis of quinoline derivatives. In this context, quinoline N-oxides have attracted considerable attention as a starting material for different transformations in organic chemistry, such as oxidations, deoxygenations, nucleophilic reactions, cycloadditions, aminations, etc. However, with the current need to extend the quinoline framework through the formation of new C–C bonds, in this review we will survey the recent developments in the direct alkenylation of quinoline N-oxides via the C(sp2)–H bond activation process, discussing metal-free and transition-metal catalysed protocols, and the regioselectivity during the synthesis of promising antiparasitic drugs.
Introduction
Among diverse classes of heterocyclic molecules, quinoline-containing compounds stand out as important compounds displaying a wide spectrum of interesting pharmacological activities and unique physicochemical properties.1 The quinoline ring is the key structural unit for numerous natural products and privileged skeletons in medicinal chemistry for the discovery of new drug leads.2 Inspired by quinine, an alkaloid isolated from the bark of the Cinchona tree (1820), and chimanine B, found in leaves and trunk bark of Galipea longiflora (1990), a plethora of synthetic quinolines have been isolated, identified and synthetized at large scale to study their antiparasitic properties like antileishmanial 1, antifungal 2 and 3, antiviral 4, antibacterial 5, and antitumor 6–8 activities (Fig. 1).3,4 | ||
| Fig. 1 Selected functionalized bioactive compounds and clinical candidates bearing the styryl quinoline ring. | ||
Parasitic diseases, or parasitosis, are a group of infectious diseases caused or transmitted by parasites such as: protozoa, helminths or ectoparasites.5 These infections are considered one of the major problems in public health since in 2013 diseases like malaria, leishmaniasis, trypanosomiasis, toxoplasmosis, giardiasis and amoebiasis caused more than one million deaths, and for 2017 an estimated 3.5 billion people were infected with intestinal parasites globally.6
Within the different approaches to control and treat parasitic infections,7 chemotherapy still remains as the preferred strategy, since no antiparasitic vaccines have been developed so far. However, the development of drug resistance to current derivatives suggests that new antiparasitic agents are needed with some urgency.8
For over a century, quinine 9 was used to treat malaria, playing an important role in the development of antiparasitic drugs.9 Its relatively low efficacy and tolerability inspired the synthesis of other potent antimalarial quinoline drugs like pamaquine 10 and chloroquine 11.10 Having a more short history, chimanine B 12, a simple 2-alkenylquinoline alkaloid, is a no less important model in drug research against protozoa.3 To date, several diverse quinoline compounds have been designed, prepared and evaluated as antiparasitic agents, e.g.13–15, determining that the pharmacophoric unit of those molecules is the quinoline ring, and that a robust substitution pattern of this core almost always results in active agents (Fig. 2).11 The considerable success of quinolines as parasitic drugs relies on the basis that these compounds disrupt a wide spectrum of biological targets common to the life cycle of most of the known parasites, such as hem detoxification, folate metabolism, fatty acid synthesis, DNA binding, protein farnesylation, and mevalonate-independent isoprenoid biosynthesis.12
 | ||
| Fig. 2 Antiparasitic compounds bearing the quinoline ring. | ||
Therefore, the challenge for the coming decades moved to organic chemists, by improving and developing efficient methodologies to construct quinoline molecules in a cleaner and safe way, without forgetting the inclusion of specific functionalization of the quinoline scaffold.
Synthesis of functionalized quinolines: conventional protocols versus C–H functionalization
Traditionally, the most useful methodologies for the synthesis of quinoline and its derivatives are frequently based on diverse well-known reactions such as Skraup, Doebner–von Miller, Friedländer, Pfitzinger, Conrad–Limpach, and Combes syntheses, as well as Gould–Jacobs, Meth–Cohn and Povarov reactions. All these are based on cyclocondensation or cyclization processes between aryl amines and carbonyl compounds (ketones and aldehydes) and are well documented in excellent reviews (Scheme 1).13–21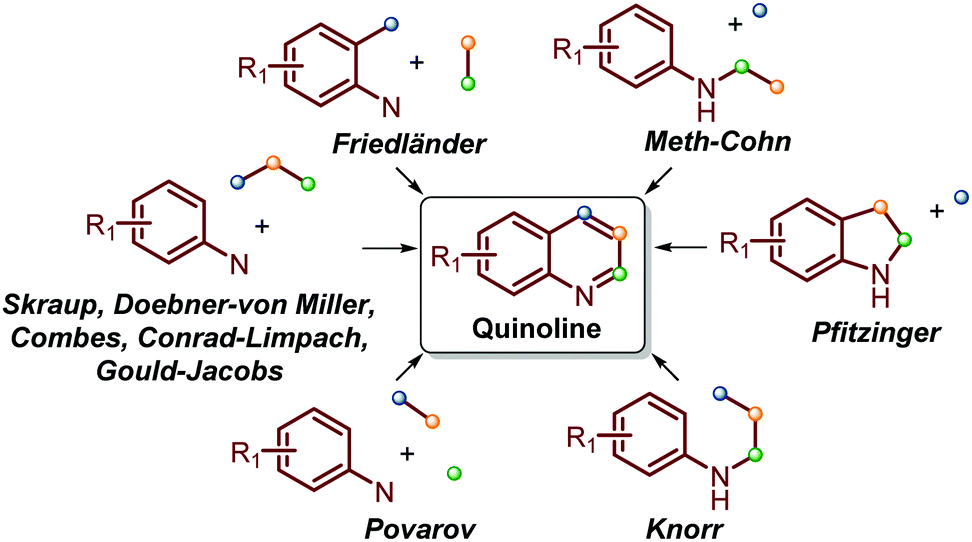 | ||
| Scheme 1 Traditional quinoline synthesis based on cyclocondensation/cyclization processes between aryl amines and carbonyl compounds. | ||
In general, these methods and their modifications or extensions under metal-free, transition-metal and acid-catalysis offer high efficacy, rapidity and good to moderate molecular diversification, but poor functional group specificity and regioselectivity.22 Additionally, many of these procedures suffer from various problems such as low yields, harsh reaction conditions, difficulties in work-up, expensive reagents, long reaction times and the use of toxic solvents.23 Therefore, new protocols for the efficient synthesis of structurally and functionally diverse quinolines are in high demand, and, in this sense, the current efforts of synthetic chemists are focused on the direct functionalization of the quinoline scaffold more than the construction of this core from cyclocondensation or cyclization processes.
Being commercially available or easily prepared at multi-gram scale in the laboratory, simple quinoline N-oxides have increasingly emerged as an ideal substrate to modify this N-heterocycle due to the ability of the N-oxide moiety to work as a directing group to perform and control the regioselectivity of the C–H functionalization.24–26 Under this approach, reactions such as acylation,27 sulfonylation,28–30 alkylation,31 acetoxylation,32 phosphorylation,33,34 arylation,35 amination36–38 and phenoxylation or alkoxylation39 have been performed through the C(sp2)–H functionalization of quinoline N-oxides using transition-metal complexes as catalysts. However, in pursuit of increased efficiency and environmental sustainability of this process, during recent years some interesting and elegant metal-free approaches have been developed but the challenge in this field is still open (Scheme 2).
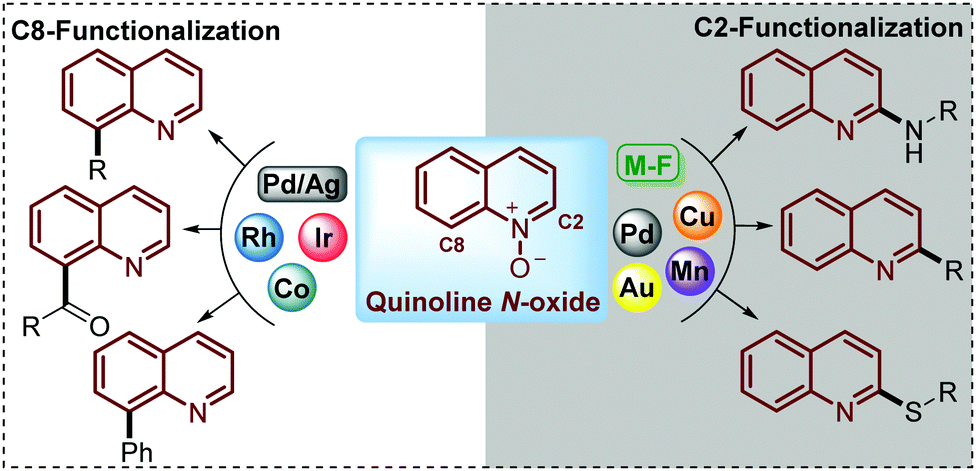 | ||
| Scheme 2 C2–H versus C8–H functionalization of quinoline N-oxides directed by the reaction conditions (catalyst, oxidant) with different substrates. | ||
To date, two main aspects of the C(sp2)–H functionalization of quinoline N-oxides have caught the attention of organic and medicinal chemists and constituted the major advantages of this approach. First, the incorporation of diverse functional groups at the quinoline ring has great pharmacological value since coupling can be performed with a vast number of commercially available and versatile substrates (C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C
C, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, alkyl halides, amines, tosyl chlorides, ketones, aldehydes, and nitriles, among others).40 Second, the regioselectivity of this coupling process can be controlled and mediated through specialized studies in organic synthesis, where the coupling at C-2 can be promoted either by metal-free conditions (M-F) or using metal complexes such as Pd, Au, Cu or Mn, while the coupling at C-8 has been only achieved with transition metal complexes such as Co, Rh and Ir catalysts (Scheme 2).41
C, alkyl halides, amines, tosyl chlorides, ketones, aldehydes, and nitriles, among others).40 Second, the regioselectivity of this coupling process can be controlled and mediated through specialized studies in organic synthesis, where the coupling at C-2 can be promoted either by metal-free conditions (M-F) or using metal complexes such as Pd, Au, Cu or Mn, while the coupling at C-8 has been only achieved with transition metal complexes such as Co, Rh and Ir catalysts (Scheme 2).41
Clearly, the wide robustness of the C–H functionalization of quinoline N-oxides will allow the formation of novel quinolines which cannot be prepared using the conventional methods depicted in Scheme 1 by incorporating pharmacologically relevant functional groups. One of those fragments is the CC double bond directly attached to the quinoline scaffold, which affords an enormous library of bioactive styryl (alkenyl) quinolines such as the ones shown in Fig. 1 and 2.
Thereby, among the current need for developing new strategies for the efficient and facile access of promising antiparasitic agents,42 this review was inspired by the tremendous work of pioneer research groups that have used the C–H functionalization of quinoline N-oxides to obtain styryl (alkenyl) quinolines under mild reaction conditions. Our discussion has been primarily organized based on the different C–H alkenylation reactions catalyzed by metal complexes and those performed under metal-free reaction conditions, highlighting in subsections the regioselectivity of these approaches.
Metal catalysed direct C–H alkenylation of quinoline N-oxides
 | ||
| Scheme 3 Pd-Catalysed C-2 alkenylation of quinoline N-oxides without external oxidant. | ||
A plausible mechanism of reaction was disclosed for this transformation by Liu's group, who studied the reaction between quinoline N-oxides 16 and different styrenes 19 (Scheme 4).45 Although PdCl2 was found to be a better catalyst for the alkenylation of isoquinoline N-oxides, the authors proposed that a C-2 palladated species 21 is formed via electrophilic addition oriented by the N-oxide moiety. Then, the coordination of 21 with styrene 19 followed by the insertion of the CC bond gave intermediate 22, which suffers β-H elimination, releasing the palladium hydride (HPdCl) and intermediate 23. Finally, the oxidation of HPdCl completed the catalytic cycle and the desired product was obtained (Scheme 4).
 | ||
| Scheme 4 Plausible reaction mechanism for the Pd-catalysed C-2 alkenylation of quinoline N-oxides. | ||
Fluorinated quinolines are important intermediates in pharmaceutical and materials sciences.46 With this aim, Lehecq et al. reported the C–H fluoroalkenylation of quinoline N-oxides (Scheme 5).47 Although the authors achieved the synthesis of the desired compounds 25 using a copper-based catalytic system from quinoline N-oxides 16 and gem-bromofluoroalkenes 24, the obtained yields did not exceed 37% (Scheme 5). After having observed that the conversion was always higher than the product yield, a palladium/copper cooperative catalytic system was applied to improve both the reaction rate and yield. In this context, the use of Pd(OAc)2/CuBr as a palladium and copper source was efficient to catalyse the coupling process using BINAP as a bidentate ligand and tBuOLi as a base, obtaining the desired products after 15 min and with moderate yields (Scheme 5). To explain the cooperative catalysis, it was proposed that copper plays a key role during the transmetalation step with palladium (21 to 22 in Scheme 4), while the bidentate ligand increases the reaction rate by facilitating this step and the reductive elimination step.
 | ||
| Scheme 5 Palladium/copper-catalysed fluoroalkenylation of quinoline N-oxides. | ||
In order to explore other coupling agents, allyl acetates 26 were suitable substrates for the direct C–H alkenylation of quinoline N-oxides 16 under palladium catalysis.48 This process was promoted by Pd(OAc)2, tri-tert-butylphosphonium salt, as the ligand precursor, and KF to furnished the corresponding C-2 alkenyl quinoline 27 after 16 h (Scheme 6). It was determined that the products were formed through an allylation/isomerization cascade process, in which the deprotonation/palladation was the key C–H activation step, while the isomerization goes through an oxidative hydropalladation, followed by the subsequent reductive dehydropalladation (Scheme 6).
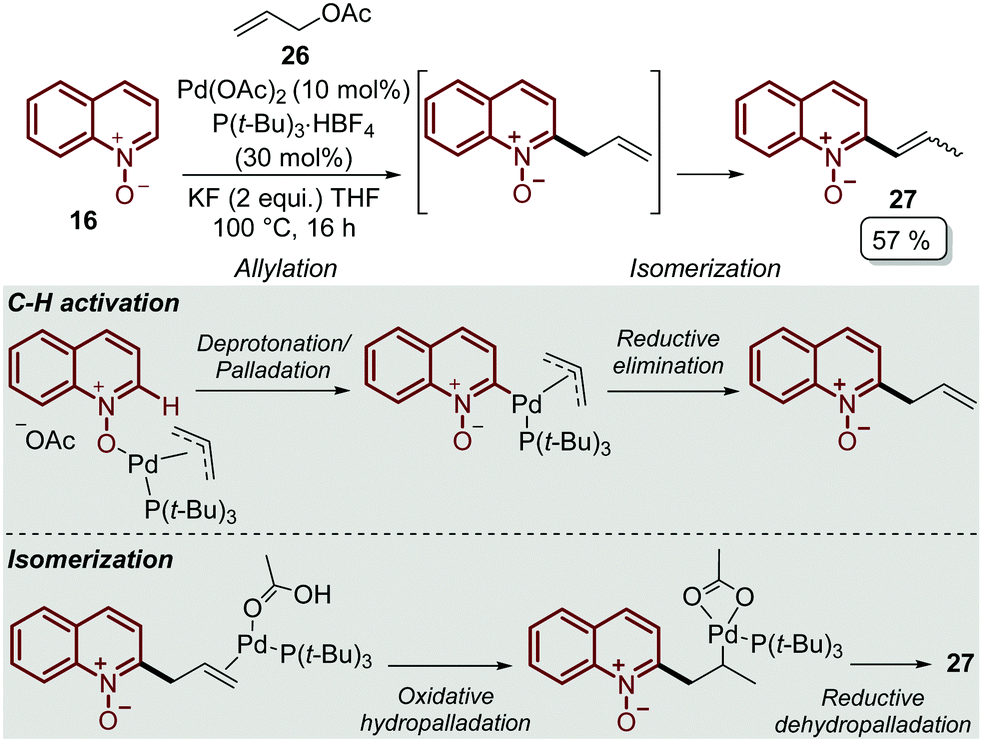 | ||
| Scheme 6 Pd-Catalysed C-2 alkenylation of quinoline N-oxides via the allylation/isomerization cascade process. | ||
Finally, this C-2 alkenylation was studied to examine the role of other palladium catalysts, such as PdCl2, Pd(CF3COO)2, Pd(CH3CN)2Cl2 and Pd(PPh3)2Cl2, but they were less efficient than Pd(OAc)2 for this transformation. On the other hand, it is clear that this reaction requires an oxidant, and among the most common (Ag2CO3, Ag2O, Ag2SO4, AgNO3, AgOAc, Cu(OAc)2, TBHP, and K2S2O8), silver carbonate has proven to be the most effective one. Although, the major contribution from Zhai and co-workers49 was the establishment of two more reaction variables: a weak base, like potassium acetate (KOAc), and an additive, pivalic acid (PivOH), to increase the reaction yields and to perform the C-2 alkenylation of quinoline N-oxides 16 with ethyl acrylate 17 (Scheme 7).
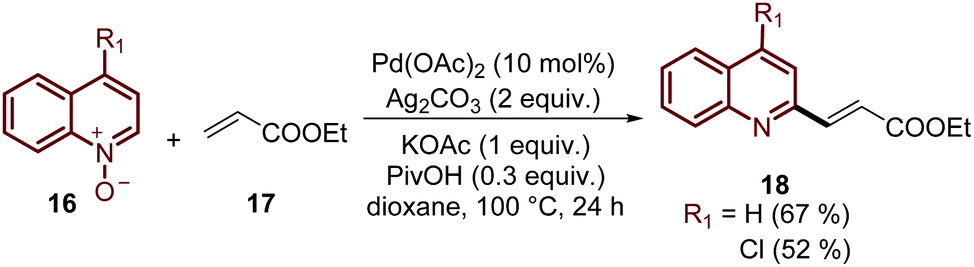 | ||
| Scheme 7 The use of an oxidant, base and additive in the Pd-catalysed C-2 alkenylation of quinoline N-oxides. | ||
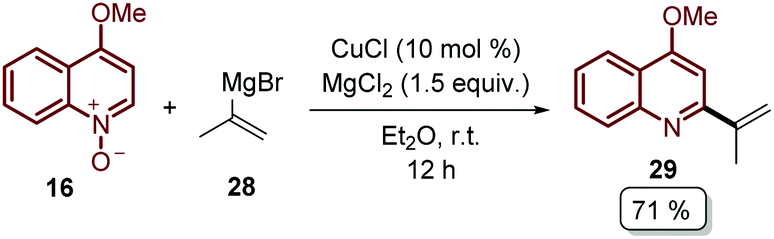 | ||
| Scheme 8 Cu-Catalysed C-2 coupling between quinoline N-oxides and Grignard reagents. | ||
It was not until 2014 that Shibata and Matsuo described the regioselective C-8 alkenylation of quinoline N-oxides 16 with alkynes 30, demonstrating for the first time that the N-oxide moiety acts as a directing group by forming a stable 5-membered metallacycle intermediate 32, which through a subsequent reductive elimination process afforded the alkenylated desired product 31 as a mixture of E- and Z-isomers. Another drawback of this initial approach was that the process required complicated and demanding reaction conditions: like [Rh(cod)2]OTf as a catalyst, xylylBINAP as a ligand, chlorobenzene as a solvent and a high reaction temperature, above 130 °C (Scheme 9).53
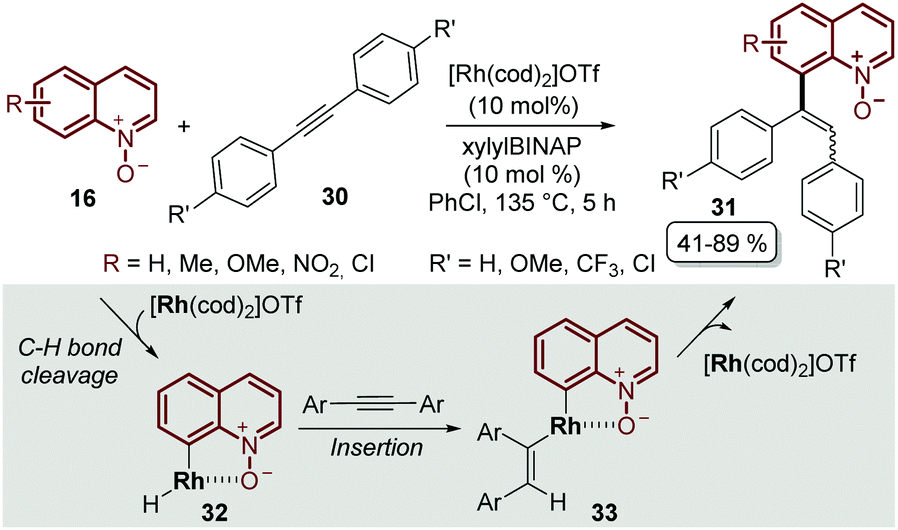 | ||
| Scheme 9 Cationic Rh(I) catalyst promoted direct C-8 alkenylation of quinoline N-oxides. | ||
Besides the problem of the diastereoselectivity when alkynes 30 are used as substrates during the C-8 alkenylation catalysed by rhodium complexes, an additional step is required to reduce the N-oxide group and afford the corresponding quinoline. In order to overcome these issues, Sharma and co-workers developed the direct C-8 functionalization of quinoline N-oxides with different alkenes, followed by the removal of the N-oxide moiety, in a single-step and in an orthogonal approach similar to the Pd-catalysed alkenylation discussed above.54 The combination of [Cp*RhCl2]2 and AgSbF6 was established as a catalytic system, while Cu(OAc)2·H2O was required as an oxidant and acetic acid was used as an additive, in order to promote the functionalization at the C-8 position along with the simultaneous removal of the oxygen atom in this reaction. Two main aspects of this study deserve to be highlighted: in the first place, these standard reaction conditions were robust enough to couple different quinoline N-oxides 16 with acrylates 17, styrenes 19 and unactivated olefins 34 to furnish a library of diverse C-8 olefinated quinolines 35–37 (Scheme 10). In addition, the authors studied the reaction mechanism, preparing, isolating and characterizing for the first time the five-membered rhodacycle 38 as the intermediate, which proves the behavior of the N-oxide moiety as a directing group (Scheme 10).
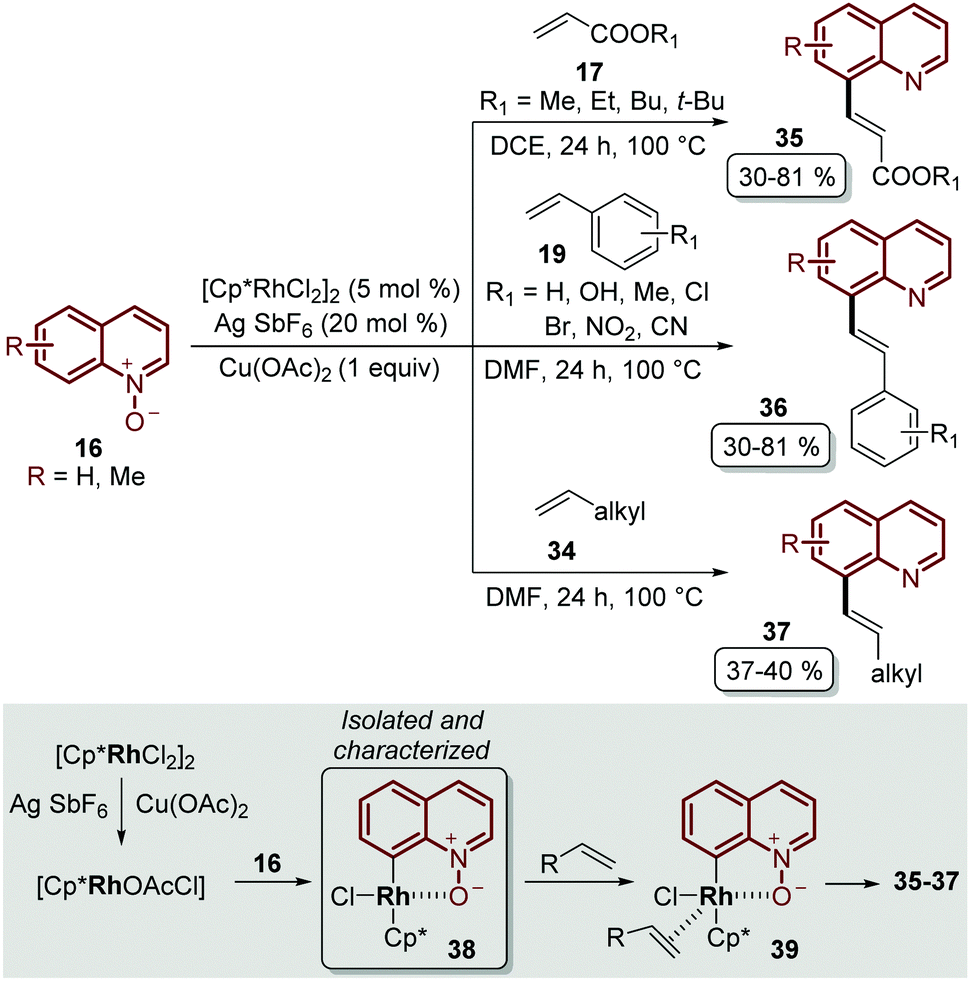 | ||
| Scheme 10 RhIII-Catalysed coupling of quinoline N-oxides with different olefins. C-8 functionalization and the identification of the rhodacycle intermediate 38. | ||
Recently, Sharma's group focused their efforts on improving their protocol by introducing molecular oxygen as an economic and clean oxidant for the coupling of quinoline N-oxides 16 with acrylates 17, styrenes 19 and unactivated olefins 34 (Scheme 11).55 Under these reaction conditions, which used a lower temperature, a library of diverse C-8 olefinated quinoline N-oxides 38–40 was obtained in good yields but with the drawback that the N-oxide moiety needed to be removed in an additional step.
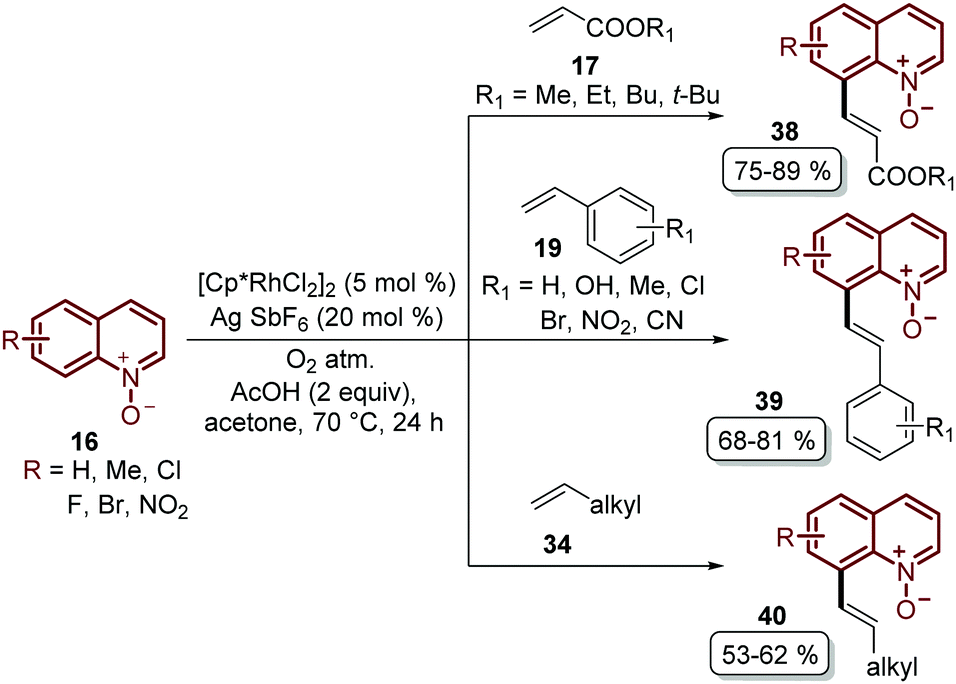 | ||
| Scheme 11 C-8 Functionalization of quinoline N-oxides and activated and unactivated olefins using the rhodium/O2 catalytic system. | ||
Metal-free C–H alkenylation of quinoline N-oxides
The transition-metal approaches described above for the C–H alkenylation of quinoline N-oxides still face challenges to some extent. First, some of the metal catalysts are expensive and require oxidants and additives that usually are even more expensive and sometimes difficult to prepare. Second, in most of the metal-catalysed protocols the desired products are obtained as quinoline N-oxide derivatives, so additional synthetic procedures are needed to remove the N-oxide moiety, which does not indeed meet the requirements of green chemistry.56 As was expected, organic chemists started to extend and develop alternative pathways to functionalized quinoline N-oxides under transition metal-free conditions to overcome these issues,57 and C–H alkenylation was not the exception.The first approach was reported by Bering and Antonchick in 2015,58 by coupling quinoline N-oxides 16 with alkenyl boronic acids 41 in DMSO at 110 °C to furnish a series of 2-styryl quinolines 20 under external oxidant- and metal-free conditions (Scheme 12). This reaction resembles the Petasis reaction,59 in which the quinoline N-oxide 16 contains a formal iminium-ion and a coordination site to complex with alkenyl boronic acids 41 to give intermediate 42, which would lead to olefin migration through the nucleophilic attack on the C2-position of the quinoline ring 43, followed by the elimination of boric acid with the N-oxide moiety (Scheme 12).
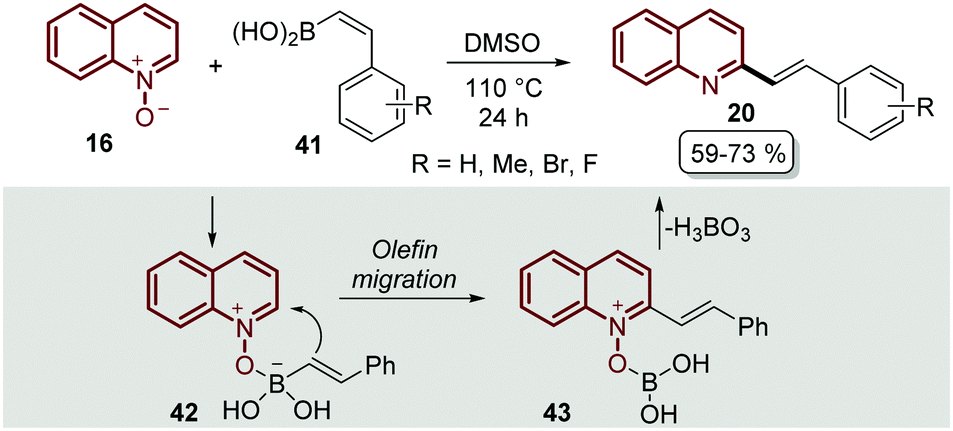 | ||
| Scheme 12 Oxidant- and metal-free C-2 functionalization of quinoline N-oxides with alkenyl boronic acids. | ||
To complement this approach, which was not applied to other classes of heteroalkenes and not required the prefunctionalization of this partner, as boronic species such as 44, a simple and efficient protocol was developed in which p-toluenesulfonic acid (PTSA) was employed as a catalyst for the coupling between quinoline N-oxides 16 and different styrenes 19, resulting in the corresponding 2-styryl quinolines 20 through an initial 1,3-dipolar cycloaddition followed by an acid-catalysed elimination, which in some cases resulted in quantitative yield (Scheme 13).60 Actually, following this protocol the authors established a two-step synthetic route for access, under smooth reaction conditions, to the alkaloid (−)-cuspareine 44, an antimalarial agent isolated from the bark of Galipea officinalis Hancock (Scheme 13).61
 | ||
| Scheme 13 PTSA-Catalysed C2-alkenylation of quinoline N-oxides with styrenes and heteroalkenes and its application in the total synthesis of (−)-cuspareine 44. | ||
Almost at the same time, but in independent work, Wang and co-workers reported the C-2 alkenylation of quinoline N-oxides 16 using a Brønsted acid as a catalyst.62 In their contribution, the easily available acetic acid (AcOH) proved to be the most effective additive to promote this transformation, which was carried out in DMSO as a solvent at 140 °C for 40 h (Scheme 14). Although the product yields turned out to be lower than the PTSA-catalysed approach (Scheme 13), under these reaction conditions quinoline N-oxides 16 were coupled with both acrylates 17 and styrenes 19, and the reaction was operationally simple and easy to scale up to gram scale.
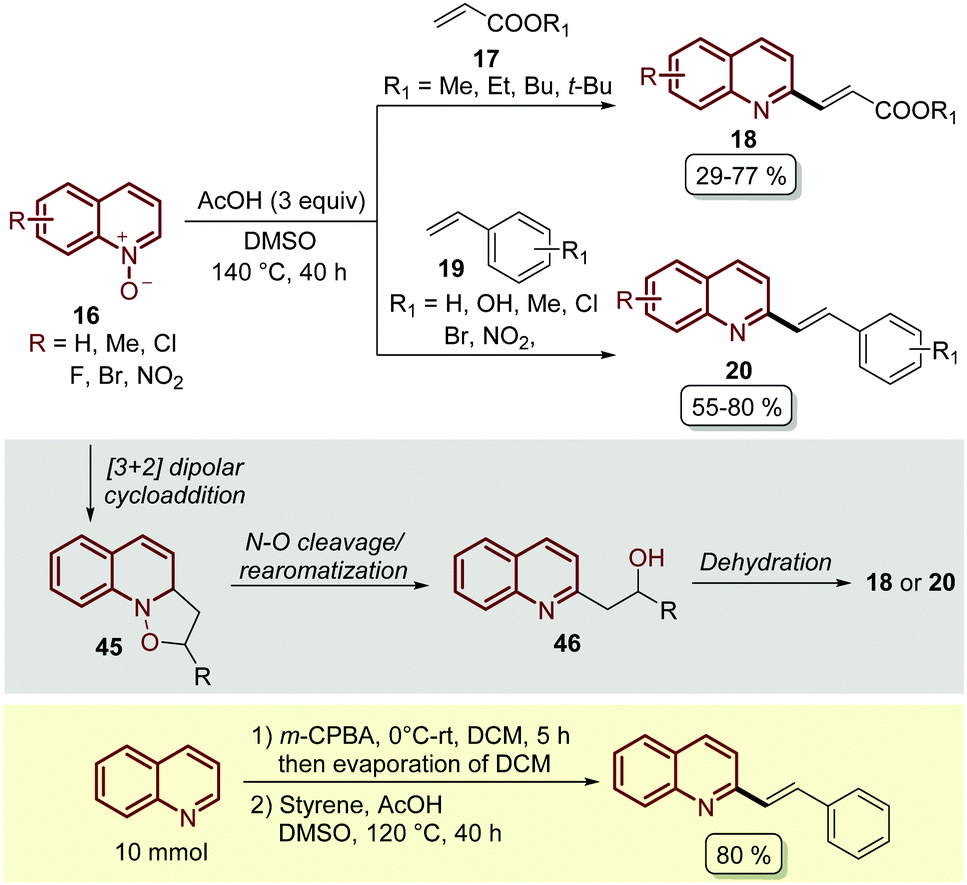 | ||
| Scheme 14 AcOH-Catalysed C2-alkenylation of quinoline N-oxides with olefins, possible reaction mechanism and one-pot oxidation/olefination of quinoline at the gram scale. | ||
In general, these Brønsted acid-catalysed reactions involve the same reaction mechanism, which first starts with the 1,3-dipolar cycloaddition between quinoline N-oxide and the corresponding olefin to form the five-membered isoxazolidine as intermediate 45. Then, the cleavage of the N–O bond and the rearomatization of the quinoline ring generate alcohol 46, which undergoes an acid catalysed elimination (dehydratation) to give the corresponding product (Scheme 14).
Wang and co-workers also demonstrated another interesting protocol in which quinoline, as a free-base, was in situ oxidized by meta-chloroperoxybenzoic acid (m-CPBA) and then subjected to olefination under the standardized reaction conditions, obtaining the desired product in 80% yield at a 10 mmol scale (Scheme 14).
Finally, we are pleased to close our review with the recent work reported by Wu's group, the same pioneers who described the first Pd-catalysed alkenylation of quinoline N-oxides 16 with different acrylates 17 (Scheme 3). During the last ten years, they studied this reaction and based on all the previous work published so far they introduced in 2019 the regioselective alkenylation of quinoline N-oxides 16 with styrenes 19 catalysed by iodine under metal- and external oxidant-free conditions (Scheme 15).63 With this approach, a broad range of 2-styryl quinolines 20 were prepared in moderate to excellent yields and the authors proposed a reaction mechanism, which is an ongoing study due to the novelty of their findings (Scheme 15).
 | ||
| Scheme 15 Iodine-catalysed C2-alkenylation of quinoline N-oxides with styrenes. | ||
Conclusion
Quinoline N-oxides have emerged as a new class of substrates suitable for the functionalization of the quinoline scaffold, and in this context the C–H functionalization of quinoline N-oxides via C–H bond activation is a dynamic field that can be developed as a synthetic strategy for the efficient, rapid and regioselective assembly of new bioactive quinoline based-molecules that are particularly significant for medicinal chemistry and the pharmaceutical industry, focused on antiparasitic agents.We reviewed and covered the metal and metal-free protocols described so far for the direct C–H alkenylation of quinoline N-oxides since the first report of this reaction (2009). Today, there is a wide broad spectrum of possibilities to perform the C–H alkenylation of quinoline N-oxides, which can be applied according to the resources and capacity of each laboratory: (i) using palladium complexes to couple simple alkenes at C-2, or employing ligands and additives to insert unreactive alkenes; (ii) making this process highly regioselective by using a rhodium catalyst and introducing the alkenyl fragment at C-8 and (iii) simply performing this process under accessible and rapid reaction conditions for biological purposes with metal-free conditions.
Nevertheless, some aspects of this reaction need further studies: (i) the use of other olefins as substrates, different from styrenes and acrylates; (ii) other cheap, abundant and less toxic metal complexes as catalysts; (iii) C-8 functionalization under metal-free reaction conditions and (iv) improving the reaction efficiency by using non-conventional reaction conditions like microwaves, ultrasound, ball-milling or flow chemistry.
Without any doubt, we believe that the development of green, cheap and robust catalytic systems for more efficient and selective functionalization of quinoline N-oxides will appear in the near future and certainly would enrich not only organic chemistry but also medicinal chemistry, especially antiparasitic drug research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by COLCIENCIAS (project No. RC-007-2017, Cod. 110274558597); MCOV thanks the National Program 727 for her doctoral fellowship and CPEG thanks the Fondo Nacional de Financiamiento para la Ciencia y la Tecnología y la Innovación “Francisco José de Caldas”, Conv. 811 for his postdoctoral fellowship.References
- P. Y. Chung, Z. X. Bian, H. Y. Pun, D. Chan, A. S. C. Chan, C. H. Chui, T. O. Tang and K. H. Lam, Future Med. Chem., 2015, 7, 947 CrossRef CAS PubMed
.
-
(a) R. Musiol, Expert Opin. Drug Discovery, 2017, 12, 583 CrossRef CAS
-
V. V. Kouznetsov, C. M. Gómez Meléndez, J. L. Valencia Peña and L. Y. Vargas-Méndez, in Discovery and Development of Therapeutics from Natural Products Against Neglected Tropical Diseases, ed. G. Brahmachari, Elsevier, 2019, pp. 87–164 Search PubMed
-
(a) R. S. Keri and S. A. Patil, Biomed. Pharmacother., 2014, 68, 1161 CrossRef CAS PubMed
- S. Momčilović, C. Cantacessi, V. Arsić-Arsenijević, D. Otranto and S. Tasić-Otašević, Clin. Microbiol. Infect., 2019, 25, 290 CrossRef PubMed
-
(a) P. R. Torgerson, B. Devleesschauwer, N. Praet, N. Speybroeck, A. L. Willingham, F. Kasuga, M. B. Rokni, X. N. Zhou, E. M. Fèvre, B. Sripa, N. Gargouri, T. Fürst, C. M. Budke, H. Carabin, M. Kirk, F. J. Angulo, A. Havelaar and N. de Silva, PLoS Med., 2015, 12, e1001920 CrossRef PubMed
-
(a) D. Horn and M. T. Duraisingh, Annu. Rev. Pharmacol. Toxicol., 2014, 54, 71 CrossRef CAS PubMed
- E. A. Ashley and A. Pyae Phyo, Drugs, 2018, 78, 861 CrossRef CAS PubMed
- X. F. Shang, S. L. Morris-Natschke, Y. Q. Liu, X. Guo, X. S. Xu, M. Goto, J. C. Li, G. Z. Yang and K. H. Lee, Med. Res. Rev., 2018, 38, 775 CrossRef CAS
- J. Wiesner, R. Ortmann, H. Jomaa and M. Schlitzer, Angew. Chem., Int. Ed., 2003, 42, 5274 CrossRef CAS
-
(a) S. Sandekerckhove and M. D’hooghe, Bioorg. Med. Chem., 2015, 23, 5098 CrossRef
-
(a) S. Bawa, S. Kumar, S. Drabu and R. Kumar, J. Pharm. BioAllied Sci., 2010, 2, 64 CrossRef CAS PubMed
- V. V. Kouznetsov, L. Y. Vargas-Méndez and C. M. Gómez-Meléndez, Curr. Org. Chem., 2005, 9, 141 CrossRef CAS
- S. Madapa, Z. Tusi and S. Batra, Curr. Org. Chem., 2008, 12, 1116 CrossRef CAS
- A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1 CrossRef PubMed
- W. Cieslik, M. Serda, A. Kurczyk and R. Musiol, Curr. Org. Chem., 2013, 17, 491 CrossRef CAS
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463 RSC
- J. B. Bharate, R. A. Vishwakarma and S. B. Bharate, RSC Adv., 2015, 5, 42020 RSC
- G. A. Ramann and B. J. Cowen, Molecules, 2016, 21, 986 CrossRef
- L. M. Nainwal, S. Tasneem, W. Akhtar, G. Verma, M. F. Khan, S. Parvez, M. Shaquiquzzaman, M. Akhter and M. M. Alam, Eur. J. Med. Chem., 2019, 164, 121 CrossRef CAS
- R. Sharma, P. Kour and A. Kumar, J. Chem. Sci., 2018, 130, 73 CrossRef
- S. V. Ley and I. R. Baxendale, Nat. Rev. Drug Discovery, 2002, 1, 573 CrossRef CAS PubMed
- C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed
- Y. Wang and L. Zhang, Synthesis, 2015, 289 Search PubMed
-
(a) A. M. Mfuh and O. V. Larionov, Curr. Med. Chem., 2015, 22, 2819 CrossRef CAS PubMed
- W. Z. Bi, C. Qu, X. L. Chen, S. K. Wei, L. B. Qu, S. Y. Liu, K. Sun and Y. F. Zhao, Tetrahedron, 2018, 74, 1908 CrossRef CAS
- G.-H. Li, D.-Q. Dong, X.-Y. Yu and Z.-L. Wang, New J. Chem., 2019, 43, 1667 RSC
- L.-Y. Xie, T.-G. Fang, J.-X. Tan, B. Zhang, Z. Cao, L.-H. Yang and W.-M. He, Green Chem., 2019, 21, 3858 RSC
- L.-Y. Xie, Y.-J. Li, J. Qu, Y. Duan, J. Hu, K.-J. Liu, Z. Cao and W.-M. He, Green Chem., 2017, 19, 5642 RSC
- K. Sun, X.-L. Chen, X. Li, L.-B. Qu, W.-Z. Bi, X. Chen, H.-L. Ma, S.-T. Zhang, B.-W. Han, Y.-F. Zhao and C.-J. Li, Chem. Commun., 2015, 51, 12111 RSC
-
(a) S. Han, P. Chakrasali, J. Park, H. Oh, S. Kim, K. Kim, A. K. Pandey, S. H. Han, S. B. Han and I. S. Kim, Angew. Chem., Int. Ed., 2018, 57, 12737 CrossRef CAS
- X. Chen, C. Zhu, X. Cui and Y. Wu, Chem. Commun., 2013, 49, 6900 RSC
- S.-J. Lee, H.-S. Kim, H.-W. Yang, B.-W. Yoo and C. M. Yoon, Bull. Korean Chem. Soc., 2014, 35, 2155 CrossRef CAS
- Y. Yang, C. Qu, X. Chen, K. Sun, L. Qu, W. Bi, H. Hu, R. Li, C. Jing, D. Wei, S. Wei, Y. Sun, H. Liu and Y. Zhao, Org. Lett., 2017, 19, 5864 CrossRef CAS PubMed
- R. Kumar, R. Kumar, A. K. Dhiman and U. Sharma, Asian J. Org. Chem., 2017, 6, 1043 CrossRef CAS
- L.-Y. Xie, S. Peng, L.-L. Jiang, X. Peng, W. Xia, X. Yu, X.-X. Wang, Z. Cao and W.-M. He, Org. Chem. Front., 2019, 6, 167 RSC
- L.-Y. Xie, S. Peng, L.-H. Lu, J. Hu, W.-H. Bao, F. Zeng, Z. Tang, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 7989 CrossRef CAS
- L.-Y. Xie, S. Peng, L.-L. Jiang, X. Peng, W. Xia, X. Yu, X.-X. Wang, Z. Cao and W.-M. He, Org. Chem. Front., 2019, 6, 167 RSC
- W.-Z. Bi, C. Qu, X.-L. Chen, L.-B. Qu, Z.-D. Liu, K. Sun, X. Li and Y.-F. Zhao, Eur. J. Org. Chem., 2017, 5125 CrossRef CAS
- C. You, C. Pi, Y. Wu and X. Cui, Adv. Synth. Catal., 2018, 360, 4068 CrossRef CAS
- D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507 RSC
-
(a) V. S. Gopinath, M. Rao, R. Shivahare, P. Vishwakarma, S. Ghose, A. Pradhan, R. Hindupur, K. D. Sarma, S. Gupta, S. K. Puri, D. Launay and D. Martin, Bioorg. Med. Chem. Lett., 2014, 24, 2046 CrossRef CAS PubMed
-
(a) L. C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS PubMed
- J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef CAS PubMed
- T. Guo, Y. Liu, Y.-H. Zhao, P.-K. Zhang, S.-L. Han and H.-M. Liu, Tetrahedron Lett., 2016, 57, 3920 CrossRef CAS
-
(a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed
- A. Lehecq, C. Hoarau, K. Rousée, C. Schneider, X. Pannecoucke, V. Levacher, J.-P. Bouillon and S. Couve-Bonnaire, Eur. J. Org. Chem., 2017, 3049 CrossRef CAS
- F. Roudesly, L. F. Veiros, J. Oble and G. Poli, Org. Lett., 2018, 20, 2346 CrossRef CAS PubMed
- K. Zhai, M. Lai, Z. Wu, M. Zhao, Y. Jing and P. Liu, Catal. Commun., 2018, 116, 20 CrossRef CAS
- O. V. Larionov, D. Stephens, A. Mfuh and G. Chavez, Org. Lett., 2014, 16, 864 CrossRef CAS PubMed
- L. C. Campeau, M. Bertrand-Laperle, J. P. Peclerc, E. Villemure, S. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3276 CrossRef CAS PubMed
- D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Atesin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167 CrossRef CAS PubMed
- T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 7, 1516 CrossRef
- R. Sharma, R. Kumar, I. Kumar and U. Sharma, Eur. J. Org. Chem., 2015, 7519 CrossRef CAS
- R. Sharma, R. Kumar and U. Sharma, J. Org. Chem., 2019, 84, 2786 CrossRef CAS
- C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS
-
(a) C. Sen and S. C. Ghosh, Adv. Synth. Catal., 2017, 360, 905–910 CrossRef
- L. Bering and A. P. Antonchick, Org. Lett., 2015, 17, 3134–3137 CrossRef CAS PubMed
-
(a) N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583 CrossRef CAS
- G. E. M. Crisenza, E. M. Dauncey and J. F. Bower, Org. Biomol. Chem., 2016, 14, 5820 RSC
- S. Chacko and R. Ramapanicker, J. Heterocycl. Chem., 2015, 52, 1902 CrossRef CAS
- H. Xia, Y. Liu, P. Zhao, S. Gou and J. Wang, Org. Lett., 2016, 18, 1796 CrossRef CAS PubMed
- Z. Zhang, C. Pi, H. Tong, X. Cui and Y. Wu, Org. Lett., 2017, 19, 440 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |