
Carbon-based materials for photo- and electrocatalytic synthesis of hydrogen peroxide
Xiaoyi
Hu
,
Xiangkang
Zeng
,
Yue
Liu
,
Jun
Lu
and
Xiwang
Zhang
 *
*
Department of Chemical Engineering, Monash University, Clayton, VIC 3168, Australia. E-mail: xiwang.zhang@monash.edu
First published on 14th July 2020
Abstract
The high demand for hydrogen peroxide (H2O2) has been dominantly supplied by the anthraquinone process for various applications globally, including chemical synthesis and wastewater treatment. However, the centralized manufacturing and intensive energy input and waste output are significant challenges associated with this process. Accordingly, the on-site production of H2O2via electro- and photocatalytic water oxidation and oxygen reduction partially is greener and easier to handle and has recently emerged with extensive research aiming to seek active, selective and stable catalysts. Herein, we review the current status and future perspectives in this field focused on carbon-based catalysts and their hybrids, since they are relatively inexpensive, bio-friendly and flexible for structural modulation. We present state-of-the-art progress, typical strategies for catalyst engineering towards selective and active H2O2 production, discussion on electro- and photochemical mechanisms and H2O2 formation through both reductive and oxidative reaction pathways, and conclude with the key challenges to be overcome. We expect promising developments would be inspired in the near future towards practical decentralized H2O2 production and its direct use.
 Xiaoyi Hu | Xiaoyi Hu is currently a Ph.D. candidate in the Department of Chemical Engineering at Monash University under the supervision of Prof. Xiwang Zhang. She received her M.S. Degree in Materials Science from Xiamen University, China, in 2014. Her current research focuses on the synthesis of carbon-based catalyst materials for solar-driven oxidation and water treatment. |
 Xiwang Zhang | Xiwang Zhang is a Professor in the Department of Chemical Engineering at Monash University, and the Director of ARC Research Hub for Energy efficient Separation. His research interests focus on membrane and advanced oxidation technologies. Prof. Zhang was the recipient of the prestigious Australian Research Fellowship and Larkins Fellowship. Prof. Zhang has authored more than 160 peer-reviewed journal papers, including Nature Materials, Energy & Environmental Science, Science Advances, Nature Communications, Advanced Materials, J. Am. Chem. Soc, Advanced functional materials, Angew. Chem. Int. Ed., and ACS Catalysis. |
1. Introduction
Along with the rapid advances of society, the unsustainability of fossil fuels as the main energy source and the resulting environmental disruption arouse pressing needs for alternative energy sources and green solutions. Hydrogen peroxide (H2O2) is an important green chemical that has illustrated great application potentials in many fields1,2 as a clean oxidant in the paper industry,3–5 wastewater treatment,6,7 disinfection8–11 and chemical synthesis,12–15 and as a promising energy carrier in fuel cells.16–18 The advantages of having the highest concentration of active oxygen (47.1 wt%) and producing the cleanest by-product (H2O) make H2O2 particularly appealing amongst various oxidants.19 Currently, over 95% of H2O2 is produced industrially via the mature anthraquinone (AQ) process, consisting of a four-step cycle19 (Fig. 1a): (i) hydrogenation of AQ, (ii) oxidation of hydrogenated AQ to regenerate it and yield H2O2, (iii) extraction-purification-concentration of H2O2 and (iv) reclaiming the working solutions. However, although the H2O2 yield is high in this large-scale synthetic process, it involves intensive energy input and chemical waste output. Furthermore, side reactions (Fig. 1a-v) produce organic by-products and consume AQ when the aromatic ring is hydrogenated, and consequently AQ cannot be recovered fully during this cycling process. Moreover, there are safety issues associated with the storage and transportation of unstable H2O2 right from the centralized factory to the end users.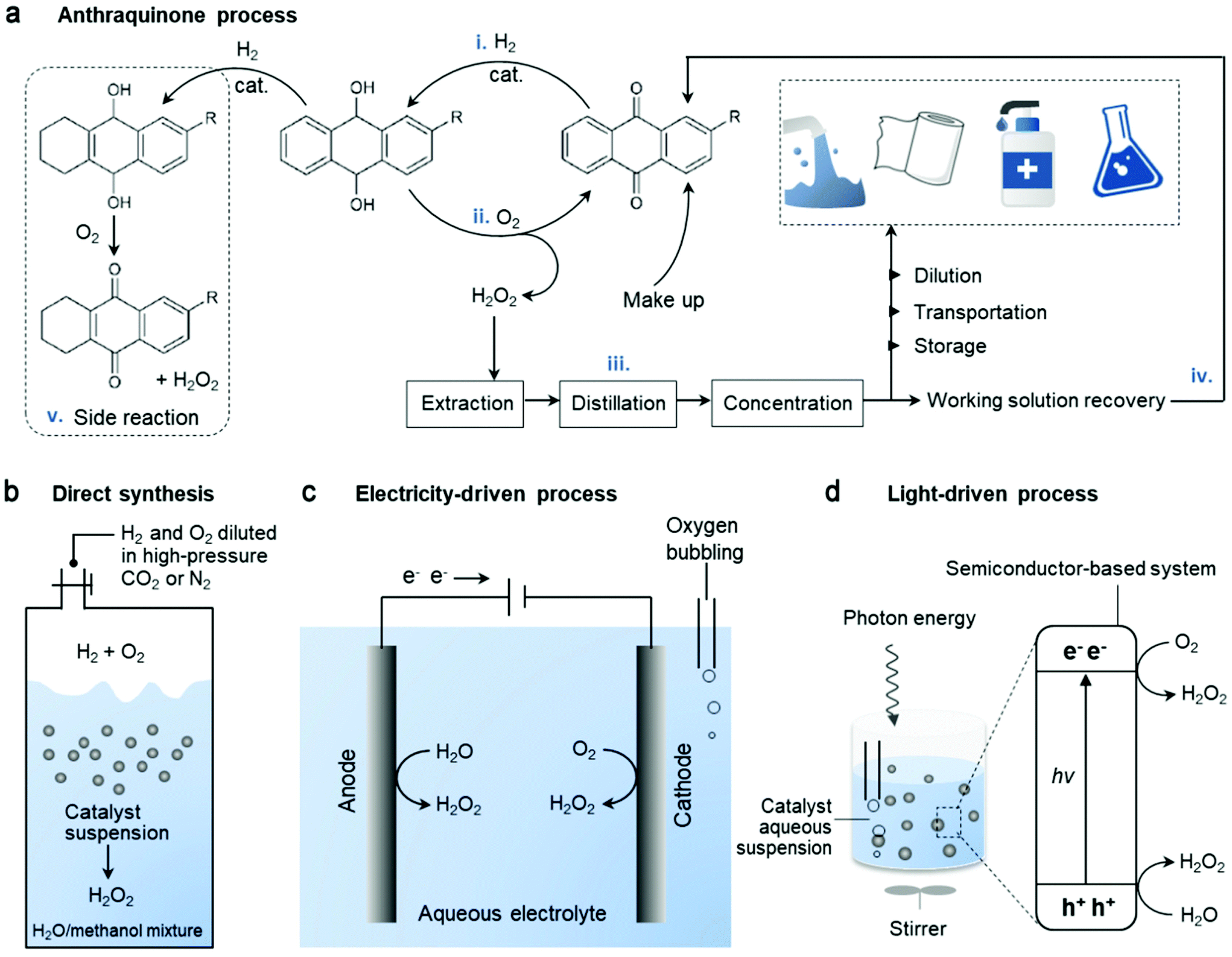 | ||
| Fig. 1 Simplified schematic of the different catalytic technologies employed in the production of H2O2. (a) Industrialized anthraquinone process using H2 and O2, following a four-step cycle (i–iv), ending with various applications. Side reaction (v) consumes AQ. (b) Direct synthesis from H2 and O2 with inert gas carrier and mixed solvent. Adapted with permission.20 Copyright 2019 American Association for the Advancement of Science. Decentralized (c) electrocatalysis and (d) photocatalysis from water oxidation or oxygen reduction mainly via a two-electron pathway. | ||
Considering that in many applications, only H2O2 diluents are required, it is unnecessary to endure the risks and energy cost associated with H2O2 concentration and its transportation, when it can be produced locally for on-site point of use.21,22 The major applications including pulp bleaching and chemical synthesis require H2O2 concentration of less than 9 wt%. Moreover, concentrations as low as 3 wt% and 0.1 wt% are enough for many medical uses and the remediation of chemical and microbial contaminations, respectively.23 Thus, the subsequent dilution of the high concentration H2O2 product (up to 70 wt%) obtained in the AQ process is required, resulting in an energy wastage. Therefore, the direct synthesis of H2O2 from H2 and O2 appear to be an alternative and the toxic by-products can be minimized without AQ as the reaction carrier (Fig. 1b).24,25 Unfortunately, besides the expected reaction of H2 + O2 → H2O2, a series of undesirable reactions such as 2H2 + O2 → 2H2O also occur, which are even more thermodynamically favored. However, the major challenge involves handling of the explosive gaseous mixture of H2/O2, which requires some inert carrier gases (such as nitrogen, carbon dioxide or argon) to reduce the danger.20 Consequently, the selectivity and productivity are partially sacrificed. To date, the expensive noble metal catalysts have been exclusively used for higher selectivity and activity in this reaction. Hence, alternative routes for the localized synthesis of H2O2, which are cost-effective, mild, efficient, and environmentally benign, are still in demand.
The recently emerging H2O2 production from water and oxygen driven by electricity (Fig. 1c) or light (Fig. 1d) has attracted significant attention.22,26–30 Compared to the aforementioned two technologies, it is much greener and easier to handle with lower operating risks. Generally, three key factors are considered and tuned to optimize the performance of catalysts including: (i) catalyst material engineering, (ii) reaction setup, and (iii) reaction conditions (solution composition, pH, promoters, and others). In particular, the catalyst system as a critical element towards the selectivity of H2O2 formation has been the main focus. A wide variety of materials such as metal/metal alloys, metal oxides, carbon nanomaterials, graphene, graphitic carbon nitrides and polymer semiconductors and their hybrids have been demonstrated to be effective as electrocatalysts31–34 and photocatalysts,35–38 among which carbon-based materials have attracted significant interest. Much effort has been made in the design of carbon-incorporated catalyst systems for the selective and active production of H2O2 since they are relatively inexpensive, bio-friendly and flexible for structure modulation. Hence, this review presents the current status and future perspectives, focusing on a wide range of carbon-based catalysts. It includes the state-of-the-art progress, typical strategies for catalyst engineering towards H2O2 production, discussion of the electro- and photochemical mechanism and H2O2 formation through both reductive and oxidative reaction pathways. Finally, we also present the key challenges to be overcome towards practical decentralized H2O2 production and its direct use.
2. Basics in catalytic synthesis of hydrogen peroxide from water and oxygen
2.1. Oxygen reduction and water oxidation reactions
Theoretically, H2O2 can be formed from H2O and O2via electrocatalysis or photocatalysis (in the presence of photoabsorbers).28,39,40 It involves the oxygen reduction reaction (ORR) and water oxidation reaction (WOR) mainly through a two-electron (2e−) pathway, which competes with the four-electron transfer towards H2O and O2 (possible redox pathways are summarized in Fig. 2).28 To date, much progress has been reported for the 2e− ORR, while the 2e− WOR has been rarely explored until very recently. However, the possible electron transfer pathways are much more complex in the water/oxygen system. The electron-reduction of oxygen can undergo 1e− (eqn (1)), 2e− (eqn (2)), and 4e− (eqn (3)) processes, yielding the superoxide anion O2˙− (˙OOH is the protonated form of O2˙−), H2O2 and H2O, respectively, as follows:28| O2 + e− → O2˙− E° = −0.33 V vs. RHE | (1) |
| O2 + 2e− + 2H+ → H2O2 E° = 0.68 V vs. RHE | (2) |
| O2 + 4e− + 4H+ → 2H2O E° = 1.23 V vs. RHE | (3) |
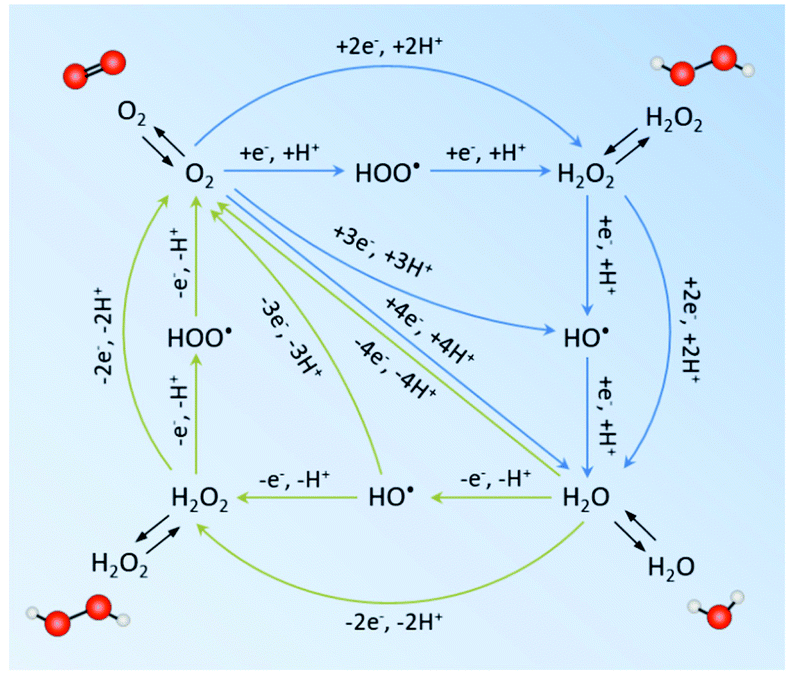 | ||
| Fig. 2 Possible reduction pathways from O2 to H2O (blue) and oxidation pathways from H2O to O2 (green). The black arrows denote the adsorption and desorption/diffusion of species from the bulk solution and the catalyst. Oxygen and hydrogen atoms are represented as red and white balls, respectively. Stoichiometric H2O is omitted for clarity.28 | ||
The direct 2e− process and a sequential two-step 1e− process are both considered feasible to produce H2O2, depending on how strong the intermediate OOH* (where * denotes a site on the catalyst surface) adsorbs on the catalyst surface. For example, in the case of weak OOH* adsorption, O2˙− dissolves in the bulk solution and may undergo further reduction upon re-interaction with the active sites on the catalyst surface. On the other hand, the WOR can proceed via 1e− (eqn (4)), 2e− (eqn (5)), and 4e− (eqn (6)) transfer to form hydroxyl radicals ˙OH, H2O2 and O2 as follows:39
| H2O → ˙OH + e− + H+ E° = 2.73 V vs. RHE | (4) |
| 2H2O → H2O2 + 2e− + 2H+ E° = 1.76 V vs. RHE | (5) |
| 2H2O → O2 + 4e− + 4H+ E° = 1.23 V vs. RHE | (6) |
Similarly, the adsorption energies of the intermediates for the 1e− and 2e− WOR (i.e. OH* and O*) are key factors that influence the following steps to be the dissociation of OH* to form ˙OH, or further oxidation to H2O2 or H2O. More complexity arises from the undesirable decomposition of H2O2 through spontaneous disproportionation (2H2O2 → 2H2O + O2) or homolysis (H2O2 → 2˙OH). Accordingly, in an electrochemical or photochemical unit, H2O2 can be formed via either reaction leading to H2O2 or indirect pathways.
2.2. Selectivity and activity towards H2O2
Importantly, the screening of efficient and selective catalyst materials is a priority in the electrocatalytic and photocatalytic production of H2O2. The trends in electrochemical ORR and WOR to H2O2 have been theoretically predicted by density functional theory (DFT) calculations on different classes of materials including pure metals and metal alloys (Fig. 3a),33 metal oxides (Fig. 3b),41 and carbon-based materials,28 with experimental supporting evidence for some. Here, we introduce briefly the fundamental theoretical basis, which is suggested to be applicable to other materials. The oxidation pathway (Fig. 3b) is taken as an example,41 and the reduction pathway follows a similar rule. The theoretical limiting potential (UL, the lowest potential for all reaction steps being downhill in free energy) is plotted as a function of Gibbs free energy of binding the one-electron oxidation intermediate OH* (ΔGOH*), presenting a volcano relationship. Specifically, the overpotential (denoted as the difference between UL and equilibrium potential 1.76 V) follows the same correlation as ΔGOH*, thus the catalytic activity. The lowest overpotential (zero) at the peak of the volcano suggests a highest activity of an ideal catalyst with the ΔGOH* value located at ∼1.76 eV. Otherwise, it will need certain overpotential to overcome either the uphill OH* formation (on the right hand of the peak) or the 1e− reduction of OH* to H2O2 (on the left hand of the peak). | ||
| Fig. 3 (a) Calculated oxygen reduction volcano plot for the two-electron (blue) and four-electron (red) reduction of O2, with the limiting potential plotted as a function of ΔGHO* (lower horizontal axis) and ΔGHOO* (upper horizontal axis). The equilibrium potentials for the two-electron and four-electron pathway are shown as the dotted line and dashed line, respectively. The range of interesting OH* free energy for high selectivity and activity is highlighted with the greyscale gradient at its edges, recognizing limitations to the accuracy of DFT. Reproduced with permission.33 Copyright 2013 Springer Nature. (b) Activity volcano plots based on calculated limiting potentials as a function of calculated ΔGOH* for the two-electron oxidation of H2O to H2O2 (black) and the four-electron oxidation to O2 (blue). The corresponding equilibrium potentials for each reaction are shown in dashed lines. Reproduced with permission.41 Copyright 2017 Springer Nature. | ||
Regarding the selectivity towards H2O2, there is a suitable range for ΔGOH*, and thus the OH* binding is not too weak or too strong. Specifically, catalysts with weaker OH* are less likely to undergo 4e− oxidation; however, if ΔGOH* is higher than the free energy for ˙OH formation (2.4 eV), ˙OH will dissolve before two-electron reduction occurs. On the other hand, O* binding should be weak enough to form H2O2 (with free energy of 3.5 eV) rather than undergoing further oxidation, hence ΔGO* ≥ 3.5 eV. Considering the general scaling relation between ΔGO* and ΔGOH* (ΔGO* = 2ΔGOH* + 0.28), the down limit is then set to be 1.6 eV. With the range of 1.6 eV ≤ ΔGOH* ≤ 2.4 eV, one can expect catalysts with high selectivity and activity towards H2O2.
Therefore, it is important to characterize the selectivity of catalysts experimentally. Currently, rotating disk electrode (RDE) analysis can be used to determine the preferred electron transfer number for electrochemical ORR.22 A rotating ring-disk electrode (RRDE) in a three-electrode cell can further provide quantitative measurement of the produced H2O2 from oxygen reduction. Selectivity can be calculated in two ways, i.e. faradaic efficiency and the fraction of oxygen used to produce H2O2. However, some researchers have suggested that these analyses may have some inherent limitations, and instead, used a modified hermetically sealed electrochemical H-cell to quantitatively measure the oxygen consumption and H2O2 production (described as the total electrons consumed per oxygen molecule (e−/O2)).34 For the water oxidation reaction, selectivity can be evaluated via the faradaic efficiency, which is defined as the ratio of charge converted to H2O2 to the total number of charge transferred. The above theory is generally used as reference for photochemical processes; however, the light response and separation of photogenerated electrons on the conduction band (CB) and holes (h+) on valence band (VB) are other important factors.
3. Typical carbon-based catalysts
3.1. Polymeric carbon nitride and its hybrids
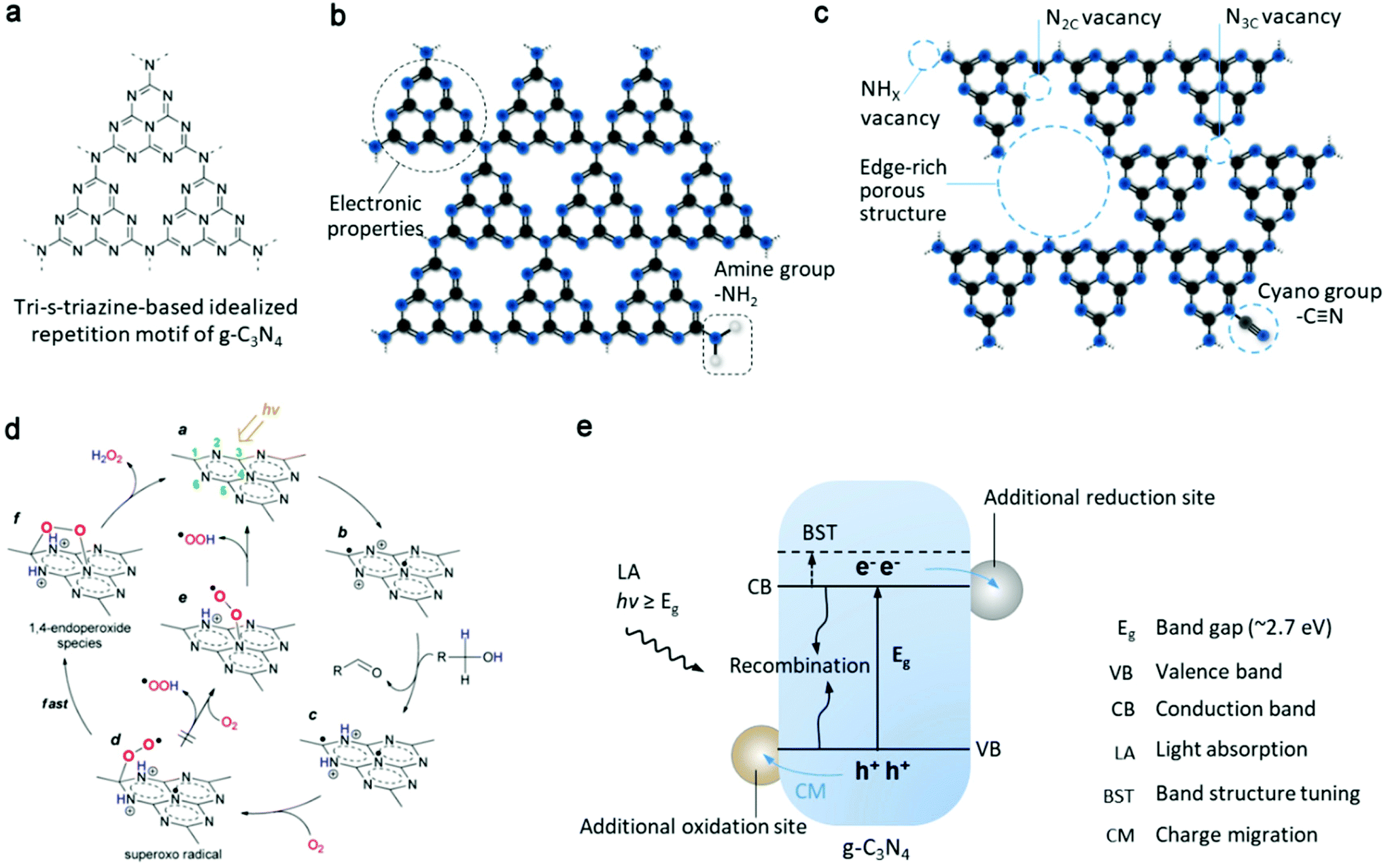 | ||
| Fig. 4 (a) Tri-s-triazine structure on g-C3N4 sheet. (b) Intrinsic C–N aromatic rings on the g-C3N4 sheet define its electronic properties, rich with amine groups at the edge of g-C3N4. (c) Different nitrogen vacancies, concept of porous structure and cyano functional group as several possible active sites. (d) Proposed mechanism for selective H2O2 formation on the surface of photoactivated g-C3N4. Reproduced with permission.38 Copyright 2014 American Chemical Society. (e) Photoinduced process of g-C3N4 semiconductor, including light absorption, generation and migration of charge carriers for surface redox reaction or recombination. Band structure tuning, additional reduction and oxidation sites (e.g. by modification with organic molecules, cocatalysts or other semiconductors) are some of the general approaches for the optimization of the photoactivity of g-C3N4. | ||
The pioneering work38 used g-C3N4 for photosynthesis of H2O2 and realized visible-light excitation (>420 nm) and higher selectivity (90%) compared to that of the previously reported TiO2 system. According to the subsequent work by the same group, the origin of this performance is the intermediate 1,4-endoperoxide species (Fig. 4d) on the surface of g-C3N4, which selectively promote the two-electron ORR route (pathway is shown in Fig. 5a), whereas the peroxo species coordinated with Ti4+ on the surface of TiO2 only enhance the selectivity up to 32.8%. Also, the decomposition decreased in the absence of ultraviolet range irradiation. However, their findings greatly depended on the oxidation half-reaction of alcohols (such as benzyl alcohol or ethanol) on the photogenerated h+ side, which provides protons and electrons for the reduction of O2 to H2O2. Subsequently, they further demonstrated water oxidation to directly generate O2 and H+ (eqn (6)) by modifying g-C3N4 with aromatic diamide49 and graphene50 for band structure tuning (mainly for positively shifted VB position) and electron trapping, respectively (pathway is similar to that in Fig. 5c). This solar-to-chemical reaction involving only water, O2 and metal-free catalysts is particularly promising for the cheap and sustainable on-site production of H2O2, paving the way for further development of g-C3N4-based catalysts towards higher activity and selectivity.
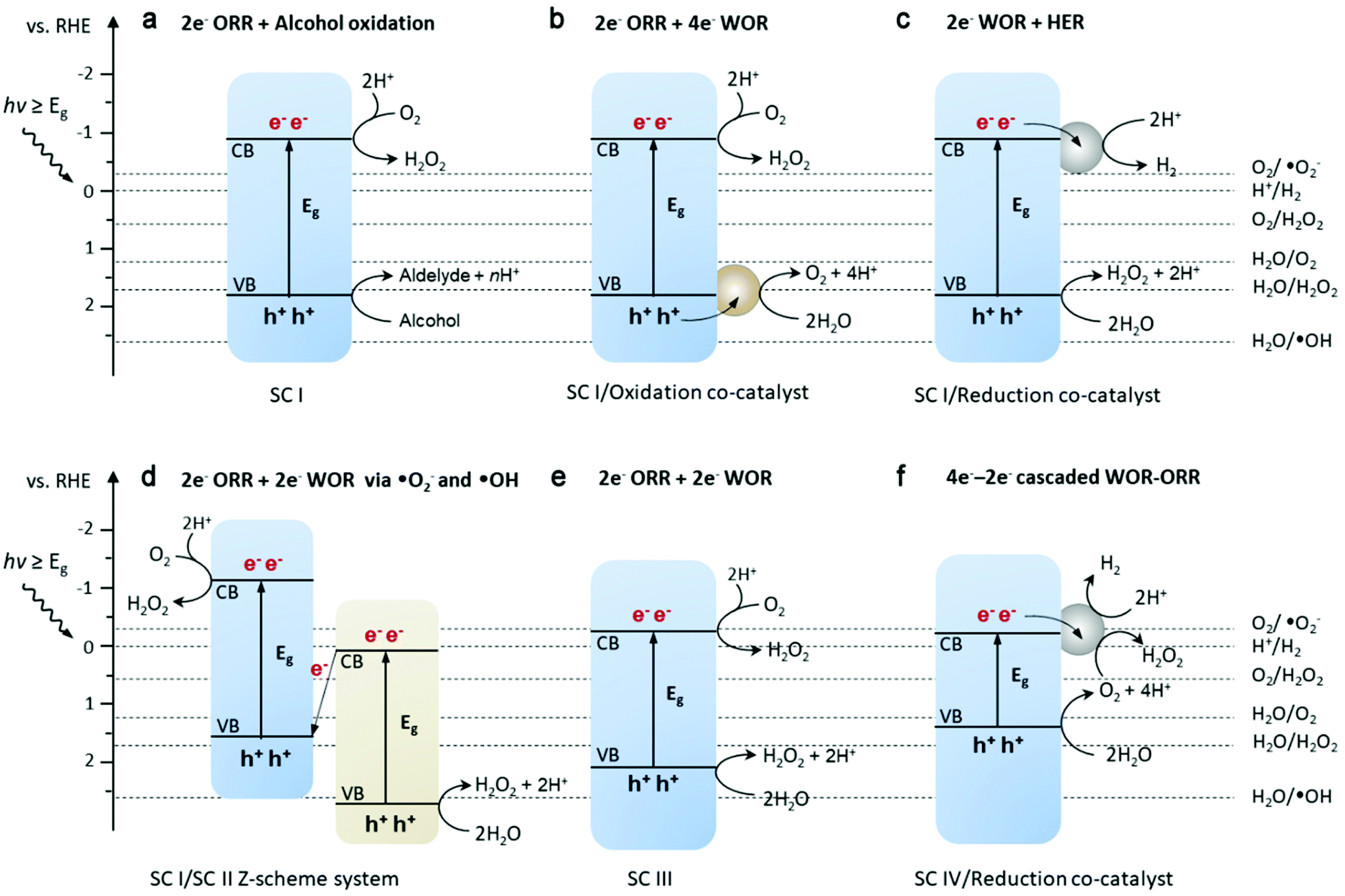 | ||
| Fig. 5 Different redox pathways for photochemical H2O2 formation (a–f) and selected redox reaction potentials regarding the oxygen reduction reaction (ORR) and water oxidation reaction (WOR). SC and HER are abbreviations of semiconductor and hydrogen evolution reaction, respectively. SCs I–IV are different semiconductors in previously reported specific systems for the photocatalytic synthesis of H2O2. | ||
Due to carbon vacancies and amino group termination, g-C3N4 demonstrated a great improvement (by a factor of 14) in non-sacrificial oxygen reduction to H2O2, and altered the pathway from a sequential 1e− ORR to a direct 2e− ORR.55 The reduced symmetry of g-C3N4 and the electron delocalization were explained as critical points for these results. Ye's group investigated how two types of nitrogen vacancies (NHX and N2C, Fig. 4c) in polymeric carbon nitride (PCN) affected the reductive formation of H2O2.57 According to DFT calculation, they found a redistributed local energy level and changed Fermi level. When combining experiments with controlled vacancies, they explained that NHX assisted charge separation, whereas N2C activated oxygen more selectively in the 2e− pathway. Regarding the precise control of defects, the fabrication procedure should be considered seriously.
The functional groups on the carbon nitride surface can directly act as catalytic active sites by regulating the adsorption of reactants and energy profile of reactions. The above mentioned KOH post-treatment of g-C3N4 and calcination resulted in the formation of NHX and N2C vacancies, but in another work by Tian and coauthors,58 the one pot calcination of KOH and g-C3N4 precursor (urea) basically introduced cyano groups (Fig. 4c) on the N position shared by tri-s-triazine units, resulting in a different modulation of properties and catalytic optimization. The concurrent breakage of pyridine nitride and creation of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds were observed after annealing the g-C3N4 matrix under a reductive atmosphere. The resultant variation in electronic structure including deviation in electrons from melem units and new intermediate energy level led to a better photochemical performance for the generation of H2O2.52 By grafting hydroxyl groups on the surface of PCN, an ultrahigh apparent quantum yield (AQY, 52.8%) was reported for the photocatalytic synthesis of H2O2 in ethanol solution.59 The hydroxyl groups helped lower thermodynamic threshold for the conversion of O2 into O2˙− (eqn (1)) and its oxidation to ˙OH could selectively oxidize ethanol to provide protons on the VB side for the subsequent reaction from O2˙− to H2O2 with electron/proton transfer. Nonetheless, the activity was reduced during the recycling test because of the oxidation of the hydroxyl groups.
N bonds were observed after annealing the g-C3N4 matrix under a reductive atmosphere. The resultant variation in electronic structure including deviation in electrons from melem units and new intermediate energy level led to a better photochemical performance for the generation of H2O2.52 By grafting hydroxyl groups on the surface of PCN, an ultrahigh apparent quantum yield (AQY, 52.8%) was reported for the photocatalytic synthesis of H2O2 in ethanol solution.59 The hydroxyl groups helped lower thermodynamic threshold for the conversion of O2 into O2˙− (eqn (1)) and its oxidation to ˙OH could selectively oxidize ethanol to provide protons on the VB side for the subsequent reaction from O2˙− to H2O2 with electron/proton transfer. Nonetheless, the activity was reduced during the recycling test because of the oxidation of the hydroxyl groups.
Modification with oxygen functional groups is one common approach in developing effective carbon-based catalysts.54 In oxygen-rich g-C3N4 catalysts, the C–O–C carbon–oxygen group could greatly lower the energy of protonation of the g-C3N4 structure compared to –NH2 and –OH groups, thus improving the ORR activity with preference for two-electron transfer, and an AQY of 10.2% was obtained at 420 nm. Recently, a simple co-polymerization of melem and barbituric acid introduced C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in the carbon nitride matrix, which resulted in a positive shift in the top of VB, allowing the oxidation of H2O to O2 and inducing a sequential 1e− ORR with expended visible light absorption.53 Notably, oxidized carbon was also reported to exhibit high 2e− ORR selectivity and activity using other carbon-based catalysts, for example, carbon nanotubes (CNT)32 and graphene.34 Thus, this approach and the disclosed mechanisms are highly recommended to be the focus of further studies.
O groups in the carbon nitride matrix, which resulted in a positive shift in the top of VB, allowing the oxidation of H2O to O2 and inducing a sequential 1e− ORR with expended visible light absorption.53 Notably, oxidized carbon was also reported to exhibit high 2e− ORR selectivity and activity using other carbon-based catalysts, for example, carbon nanotubes (CNT)32 and graphene.34 Thus, this approach and the disclosed mechanisms are highly recommended to be the focus of further studies.
Doping of heteroatoms may alter the reaction pathway to H2O2. A two-in-one strategy60 incorporating a porous structure and phosphorous doping enabled H2O2 production under visible light illumination via both indirect 2e− WOR on the VB side and 2e− ORR on the CB side. The production rate (1968 μmol g−1 h−1) recorded was among the highest for pure water photocatalytic systems driven by visible light. The authors deduced that H2O2 was formed by the combination of two ˙OH from one-electron water oxidation (eqn (4)) on the h+ side and a sequential two-step single electron activation of O2 on the e− side. In contrast, in another similar system (P doped porous g-C3N4 nanosheets),61 the direct 2e− ORR was dominant due to the down-shifted bottom of the CB. Generally, the oxidation potential of the ˙OH/H2O reaction is more positive than the top of the VB of pristine g-C3N4, and thus the detection of ˙OH failed for many g-C3N4-based catalysts (not referring to hybrid catalysts). Nevertheless, the two-channel photocatalytic synthesis of H2O2 is extremely attractive for practical solar-to-chemical application in the future, although more detailed mechanism studies need to be further conducted for a clear reaction pathway to H2O2. In addition, halogen doping was also found to be effective for the photocatalytic synthesis of H2O2 over g-C3N4 nanorods.62 Besides, the incorporation of multiple heteroelements into PCN with other possible configurations was demonstrated by Choi's group by simple calcination of melamine in the presence of potassium salts.63,64 The AQYs were boosted by 17–25 times in comparison to that of the unmodified carbon nitride (under monochromatic irradiation of 420 and 320 nm). The incorporation of heteroatoms (K, P, and O) was characterized by various spectroscopic methods, though it was difficulty to give the exact structure and synthetic mechanism of the modified carbon nitride, for which the building blocks are usually different from ideal theoretical models. However, due to the earth-abundant precursors and easy synthesis, this optimization route could be possibly scaled up.
To reveal the relation between the porosity and activity, Shiraishi's group65 fabricated a group of g-C3N4 catalysts via the silicon-templated thermal polymerization of cyanamide. It was found that a moderate surface area showed optimized activity for H2O2 production. However, a further increase in surface area had a negative effect on the selectivity of g-C3N4 towards the 2e− ORR since the increase in the number of intrinsic amine groups in mesoporous g-C3N4 made it easier to drive the 4e− ORR, where the authors proposed that melem units are the active sites for selective 2e− ORR. Hence, excess defects in porous g-C3N4 may reduce the H2O2 selectivity by decreasing the amount of inherent active sites on g-C3N4, and accordingly a larger surface area, which is believed to decrease the charge/mass transfer and accelerate the reaction kinetics, becomes less important. Subsequently, Wang's group66 fabricated carbon nitride aerogels via a sol–gel process by simply leaving a water suspension of carbon nitride nanoparticles to stand, which had highly exposed –NHX and hydroxyl groups to promote physical interaction forces, and thus form a hydrogel. The resulting aerogels exhibited an increased production of H2O2 from water and oxygen. Both the hierarchical porous structure and functional groups including –CN and hydrogen-containing groups synergistically contributed to the enhancement.
The strong anchoring of metal compounds on g-C3N4 is attributed to the delocalization of long electrons induced by element-doping (such as P) in the g-C3N4 matrix. Xue et al.71 found that substitutional P facilitated the binding of CoxNiyP clusters onto g-C3N4 (CoxNiyP-PCN) by P+-Pδ−-Coδ+/Niδ+ centers. Photogenerated electrons were much favorable to be transferred to these charged centers, and subsequently the CoxNiyP clusters, leaving holes on the g-C3N4 substrate. The calculated H binding energy on CoNiP clusters, ΔGH*, was close to zero (the top of hydrogen evolution reaction volcano plots). Hence, this catalyst system resulted in a unique water splitting route to H2 on the CoNiP clusters and H2O2 on g-C3N4via a direct 2e− WOR pathway (Fig. 5c). The hybridization method is also a crucial point to be considered since robust interfacial contact and uniform dispersion are widely verified to be significant, for example when loading metallic nanostructures onto g-C3N4. Accordingly, Cai et al.72 modified ultrathin g-C3N4 with the redox of dopamine via a one-pot polymerization to obtain a homogeneous decoration of silver (Ag@U-g-C3N4-NS). The activity for H2O2 production was improved since the Ag nanoparticles preferred photoformation of H2O2 over its decomposition. Meanwhile, Ag@U-g-C3N4-NS presented outstanding performance in the degradation of common organic pollutants, demonstrating both the application of environmental remediation and green chemical synthesis. To maximize the interfacial contact area, the 2D/2D geometry is thought helpful for a tight interface and promoted mass/charge interaction between two components.73 Besides metal cocatalysts (which are often nanoparticles), metallic sulfides (such as MoS2)74 can serve as e− acceptors and additional active sites. In our previous work,75 the in situ growth of metallic MoS2 on g-C3N4 nanosheets showed an even loading of MoS2 nanolayers, while only random agglomeration could be formed via the ex situ mixing. Consequently, impressively enhanced activity for the photocatalytic synthesis of H2O2 was achieved via increased interfacial charge transfer, leading to a better performance in bacterial inactivation by the photogenerated germicide H2O2.
Moreover, flexible modification by various organic molecules50,76,77 has been another strategy of great interest for the photocatalytic synthesis of H2O2 using g-C3N4. For example, Yang and coauthors68 assembled perylene imide (PI) on rich inherent amine edges of g-C3N4 nanosheets to form a Z-scheme heterojunction. The photoexcited h+ left on the VB of PI could oxidize water to ˙OH and the following combination of two ˙OH enabled the formation H2O2. Together with the 2e− ORR pathway, this Z-scheme system resulted in two-channel photocatalytic H2O2 production (Fig. 5d), indicating the importance in band alignment design for semiconductor-based photocatalyst hybrids. Recently, the g-C3N4 photocatalyst was modified with cationic polyethylenimine (PEI) molecules by simply mixing in aqueous suspension due to their opposite surface charge.78 The variation in local electronic environment was demonstrated by both DFT calculation and experimental characterization. The possible route for H2O2 formation was a sequential two-step 1e− ORR based on the O2-dependent performance and detection of O2˙−. The intermolecular electronic interaction in PEI/g-C3N4 together with intrinsically protonated PEI serving as a H+ relay promoted both the activity and selectivity for the photocatalytic synthesis of H2O2. As mentioned above, AQ can selectively serve as an oxygen reduction site, and cobalt species can act as a water oxidation site for O2 evolution. In a recent report, loading both of them as reductive and oxidative sites on C3N4 resulted in inhibited charge recombination for an enhanced H2O2 production rate and selectivity. A center/edge approach was proposed to separately load single-atom Co and AQ molecules based on the rational design of the fabrication procedure, such as homogeneous dispersion of Co precursor and rich amino groups at the edge of C3N4 to bind with carboxylic groups in the AQ precursor, instead of unregulated loading and distribution of two cocatalysts.79
In the electrocatalytic production of H2O2, an example of a g-C3N4-based gas diffusion electrode (GDE) was reported, where by covalently modifying with AQ, the generation rate of H2O2 increased by 20 times and 4 times, respectively, compared to that of the bare g-C3N4 GDE and AQ-g-C3N4 conventional immersed electrode.80 g-C3N4 was proven to be an amiable substrate for chemically bonding AQ (20 wt%) as an electrocatalytic redox reaction center for H2O2 formation from 2e−-ORR, although the overpotential (900 mV) was relatively large for an optimal performance due to the weak conductivity of g-C3N4 and the maximum faradaic efficiency was only 42.2% (which could be caused by hydrogen evolution reaction (HER) on the g-C3N4 surface covered with AQ). Furthermore, combining g-C3N4 with a conventional electrode (activated carbon (AC) fiber) could drive the photoelectrochemical (PEC) production of H2O2 to directly degrade phenol.81
Reduced graphene oxide (rGO) is another well-known electron mediator for photoexcited charge separation on semiconductors.50,82,83 Electronic interactions were found in some multiple-component photocatalyst systems based on g-C3N4, where rGO accepted photoexcited e− from g-C3N4 and promoted the reductive production of H2O2 (inherent oxygen functional groups on rGO possibly being the active sites).
3.2. Graphene and its hybrids
Specifically, thermally reduced graphene oxide was studied as an electrocathode to catalyze oxygen reduction to H2O2.34 The overpotential was as low as 10 mV and completely selective 2e− ORR was realized, exceeding that of the state-of-the-art catalysts reported at that time (Fig. 6b). According to the FTIR spectra (Fig. 6a), the peak assigned to the ring ether groups at the sheet edge of 600 °C mrGO (mild reduction of graphene oxide) was much more obvious than that of the other samples. Combined with results of a series of spectroscopic analyses and in situ Raman spectroscopy (which has not been exploited before), the sp2-hybridized carbon near-ring ether defects were identified as the active sites, while for F-mrGO (few-layered mrGO) the epoxy groups on the basal planes were the active sites (Fig. 6c). The likely overoxidation during the fabrication of graphene (typically by Hummers’ method) induces abundant in-plane carbon lattice defects, which may improve the catalytic activity when the density of defects is appropriate or negatively influence the performance when the defects are excessive (one possible reason is the lower electrical conductivity).87 This was explored by Han and coauthors using graphene/GO precursors with different defect densities in the electrochemical reaction for 2e− ORR to H2O2.88 After ammonium hydroxide treatment of the precursors to induce N doping, the highest selectivity was found with the lowest defect density precursor, and the activity was optimized at 0.2 V (vs. RHE). This indicates that the content of in-plane carbon lattice defects should be controlled when designing carbon-based electrocatalysts for the production of H2O2.
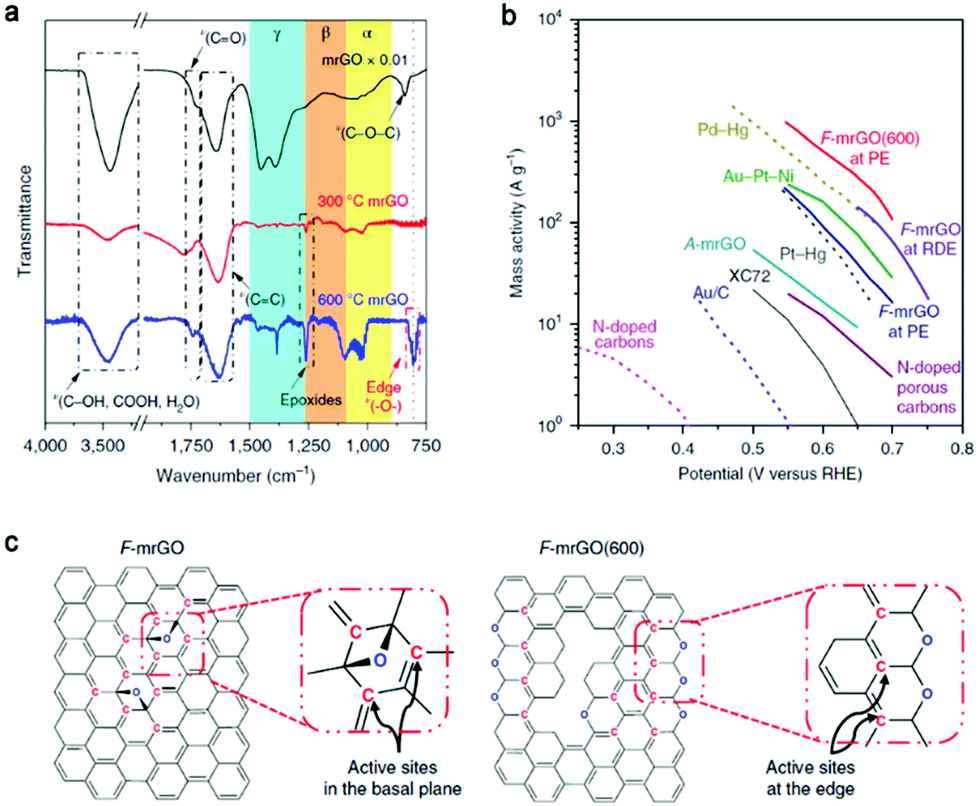 | ||
| Fig. 6 (a) FTIR spectra of various mrGO powder samples. (b) Mass activity of different electrocatalysts for H2O2 production. The data presented as dashed lines was measured in acidic conditions (0.1 M HClO4) and that as solid lines was measured in basic conditions (0.1 M KOH). (c) Idealized schemes of proposed low-overpotential active sites on F-mrGO and F-mrGO (600). Reproduced with permission.34 Copyright 2018 Springer Nature. | ||
Hydrogen peroxide is commonly used in advanced oxidation processes to degrade organic pollutants. Su and coauthors modified a graphite felt cathode with regulable N-doped graphene.89 According to their investigation, graphite N was proven to favor the 2e− reduction of O2, while pyridinic N catalyzed H2O2 to produce ˙OH, which was the dominant oxidant for the following phenol degradation. Compared to conventional electro-Fenton system, the dependence on solution pH was greatly minimized in this electrochemical process.
Notably, bare graphene oxide, possessing a semiconducting band gap, was demonstrated to directly catalyze H2O2 production driven by simulated sunlight in the absence of an organic electron donor.90 A higher production rate was achieved at a higher pH; however, more severe photocorrosion occurred because of the possible reduction of graphene oxide.
Besides the commonly used graphene sheet, graphene quantum dots (GQD) with dual dopants (nitrogen and sulfur) were anchored on the surface of TiO2 and the photocatalytic generation of H2O2 was superior to bare TiO2 and TiO2 modified with undoped GQD and N-doped GQD. According to the theoretical analyses, the authors suggested that N- and S-codoped GQD could offer active sites for the formation of *OOH and proton relays, thus promoting proton-coupled electron transfer for O2 reduction to H2O2.92 However, dual dopants are not always beneficial for the catalytic formation of H2O2. As is known, the 2e− ORR was an undesirable reaction and needed to be suppressed for the selective ORR through a 4e− pathway. The additional doping of boron or phosphorous on a N-doped graphene electrocatalyst facilitated the 4e− ORR by minimizing H2O2 production because it enhanced the asymmetry of the spin density or electron transfer on the basal plane of the N-doped graphene and reduced the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of graphene.98 In the case of the electrochemical formation of H2O2 from O2 by graphene-based hybrids, Nb2O5/rGO sheets improved the H2O2 production in acidic media in contrast to the pristine rGO cathode, although the enhancement was not significant.99 In an electro-Fenton system, when a traditional graphite-based gas diffusion cathode was integrated with rGO, it presented a better performance for the removal of organic dye (98% removal rate over 60 min) and a lower energy consumption.100
3.3. Nanostructured carbons beyond graphene
Carbon nanostructures, including nanodots, nanohorns, nanotubes, nanosized architectures and porous carbon with flexible structure engineering (Fig. 7) have been widely utilized in catalyzing the synthesis of H2O2.7,32,101–108 They act as active catalysts, substrates (mostly to support metals or metal alloys nanoparticles due to their large surface area and high conductivity),27 and photogenerated charge carrier mediators. They can be optimized in terms of heteroatom doping,109–114 surface oxidization,32,115 porous structure tuning,108,116 and defect design117 with in-depth studies aiming to reveal their structure-to-activity mechanisms. Some of the experimental results showed their comparable and even better performances relative to metal catalysts.32,108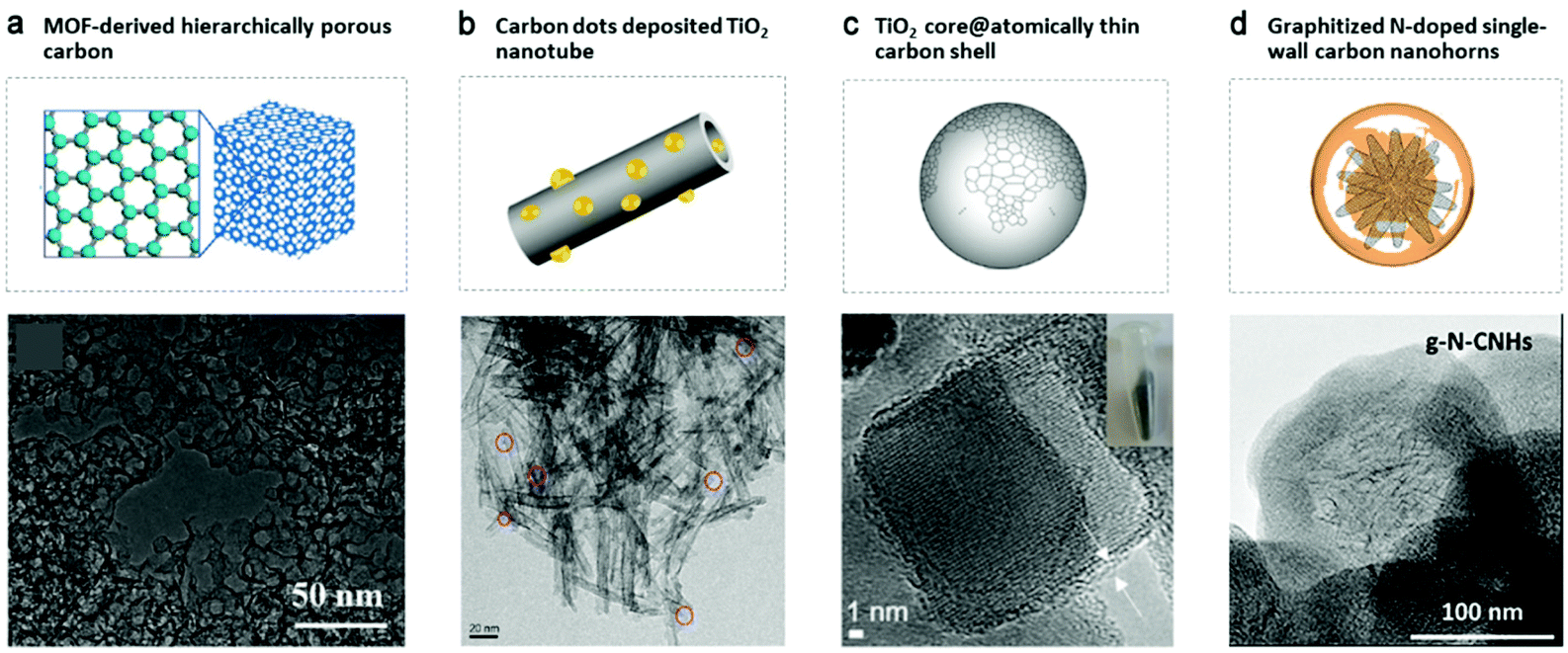 | ||
| Fig. 7 Nanostructured carbon catalysts. (a) Metal–organic framework (MOF)-derived hierarchically porous carbon (fabricated via the hydrothermal growth of MOF following pyrolysis at 1100 °C under an H2 atmosphere). Reproduced with permission.108 Copyright 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) Hybrids of TiO2 nanotubes with carbon dots, obtained from P25 TiO2 by NaOH, HNO3 and citric acid treatment. Reproduced with permission.103 Copyright 2019 Elsevier. (c) TiO2 naoparticle@C core–shell structure (through pyrolytic decarboxylation of the adsorbed aromatic compounds). Reproduced with permission.105 Copyright 2019 American Chemical Society. (d) Graphitized N-doped single-wall carbon nanohorns (g-N-CNHs), prepared via a multi-step protocol: oxidization of CNHs, combination with dopamine hydrochloride and polymerization, and thermal treatment at 700 °C under an argon atmosphere. Reproduced with permission.102 Copyright 2018 Elsevier. | ||
 | ||
| Fig. 8 Plots of H2O2 current (a) and selectivity (b) at 0.6 V as a function of oxygen content for O-CNTs with various oxidation times, demonstrating that both the activity and selectivity correlate linearly with the oxygen content. (c) H2O2 current (0.6 V) comparison of SP, O-SP, AB and O-AB, suggesting that the oxidation process is generally applicable for carbon materials. (d) Different oxygen functional group type configurations. The carbon atoms denoted by a blue circle are the active sites under investigation (M = H and Na). (e) Calculated two-electron (solid black) ORR-related volcano plot for the electro-reduction of oxygen to H2O2 displayed with the limiting potential plotted as a function of ΔGOOH*. The equilibrium potential for the two-electron ORR is shown as the dashed black line. Reproduced with permission.32 Copyright 2018 Springer Nature. | ||
The assembly of different low-cost carbon materials can potentially enhance their electrochemical performance. For example, Khataee et al. systematically compared the cathodic generation of H2O2 on a bare graphite electrode and modified ones (with immobilized AC and CNT).125 Both AC and CNT were found to be beneficial in the electrogeneration of H2O2 with an improvement of 3- and 7-fold, respectively, compared to bare graphite. A suitable applied current, air flow rate, and acidic electrolyte formed optimized operating conditions. Amorphous carbon, as another important carbon electrode characterized by mixed sp2/sp3 carbon bonds, was examined in terms of correlation between sp2 carbon content and capability of producing H2O2 with varying percentages of sp2 C bond over a wide range by tuning the fabrication parameters.126 The maximum H2O2 production was afforded by the highest content of sp2 C bond. A very impressive overall performance for the electroreduction of O2 to H2O2 was achieved.102 A metal-free catalyst system was prepared, which possessed a unique carbon nanohorn core, suitable porosity, and a profitable distribution of two types of doped N sites (Fig. 7d). The catalyst system worked well under acidic, physiological and alkaline conditions (pH = 1, 7.4 and 13, respectively) with a faradaic efficiency up to 98% and relatively low overpotential.
3.4. Emerging organic catalysts
Using organic materials with abundant feedstocks as catalysts can offer sustainable and affordable regulation of reactions in terms of environment remediation and energy generation, especially when solar light can solely drive the catalytic reaction in the case of organic semiconductors.127,128 They often show flexibility in tuning their chemical and electronic structure. Some exciting achievements have been presented recently.The stability of organic catalysts themselves against photocorrosion is a precondition. Głowacki's group129,130 developed several highly stable organic systems for the photo(electro)catalytic reduction of oxygen to H2O2 in a wide range of pH (roughly 1–12), i.e. H-bonded pigments (Fig. 9a) and biscoumarin-containing acenes, with no detectable self-degradation. Notably, these organic catalysts were able to produce H2O2 in the absence of sacrificial alcohol and the catalyst dosage-normalized H2O2 yield was as high as 4060 mg g−1 h−1 (Fig. 9b), exceeding the pioneering results on g-C3N4 (0.7 mg g−1 h−1). However, the concentration accumulated was only 3 mM. The mechanism was proposed to be similar to that of the quinone/hydroquinone system, where the organic molecules act as reaction carriers, following the steps of: (i) photoexcitation of molecules from the ground state, (ii) protonation and photoreduction of the excited basic carbonyl groups resulting in a reduce enal state, (iii) nucleophilic attack on dissolved O2 and formation of a tetrahedral intermediate, and (iv) dissociation of the intermediate releasing HO2−, which abstracts a proton from another enol unit, thus regenerating the ground-state molecule and yielding one H2O2 molecule.
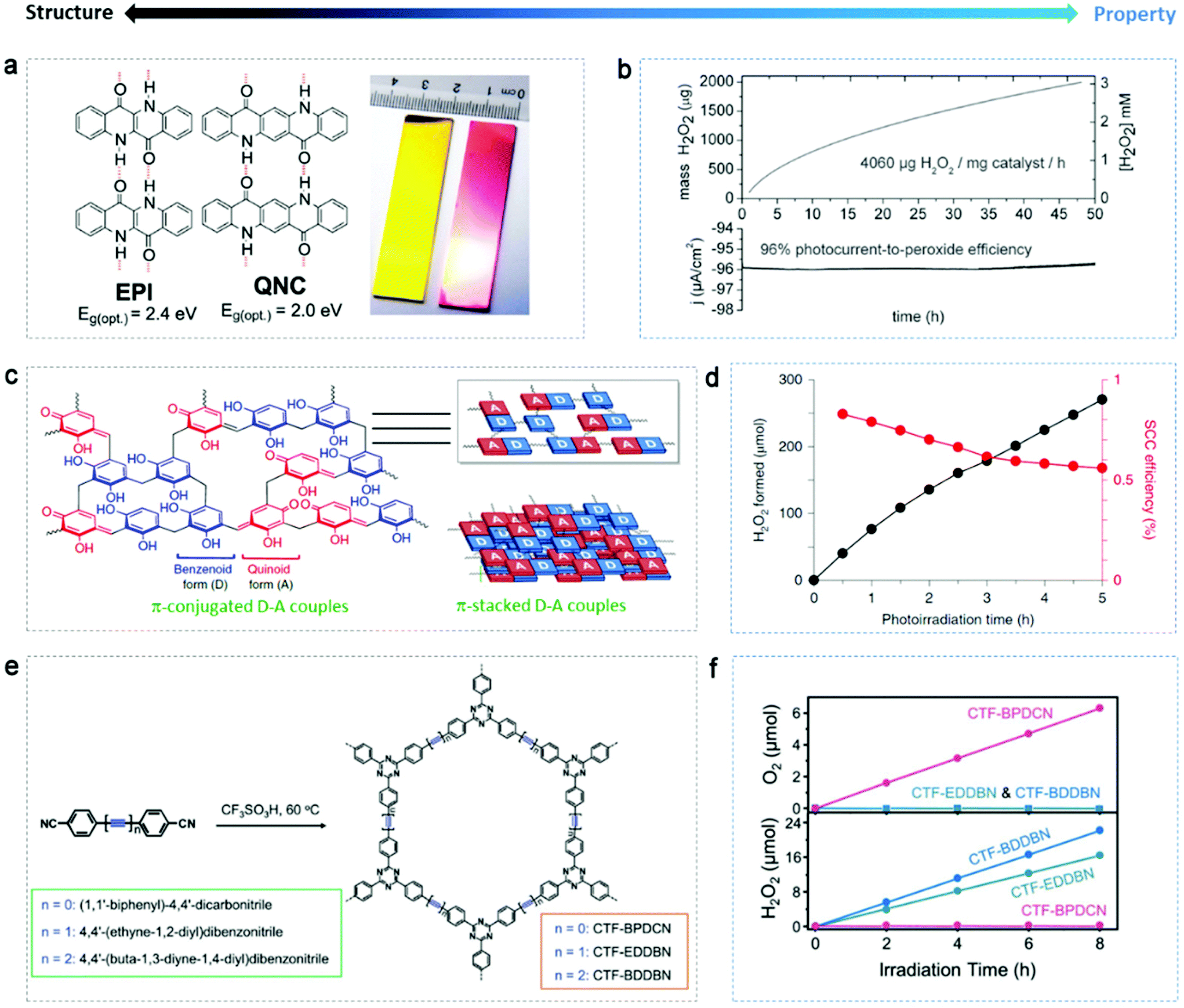 | ||
| Fig. 9 Organic semiconductors for photo(electro)catalytic H2O2 production. (a) Hydrogen-bonded pigment semiconductor epindolidione (EPI) and quinacridone (QNC) and a digital picture of EPI (yellow) and QNC (red) photoelectrodes. (b) Photocathodic H2O2 evolution in a two-compartment cell with EPI on a Cr/Au working electrode. Reproduced with permission.129 Copyright 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) Fundamental structure and π-conjugated and π-stacked D–A structure of RF resins. (d) Changes in the amounts of H2O2 generated on RF523 and SCC efficiency. Adapted with permission.37 Copyright 2019 Springer Nature. (e) Scheme of the synthesis of covalent triazine frameworks (CTF) from their corresponding precursors. (f) Time-dependent formation of H2O2 or O2 by WOR with different CTF. Reproduced with permission.131 Copyright 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Very recently, Shiraishi's group37 presented crosslinked polymer semiconductors, resorcinol-formaldehyde (RF) resins (Fig. 9c), increasing the efficiency of artificial photosynthesis (not only H2O2 production) on powder catalysts towards an unprecedentedly high level (above 0.5% solar-to-chemical conversion efficiency). The increase in temperature during hydrothermal reaction for the fabrication of the RF resins had a positive influence on their catalytic activity, with the resin obtained at 523 K generating the maximum H2O2 of 62 μmol in 24 h. A relatively stable and high SCC was reached when increasing the catalyst dosage and photoreaction temperature (Fig. 9d), even though self-oxidation of the resins was observed in the initial stage, which is quite common with organic catalysts. The authors conducted a detailed investigation on the relation between resin structure and optical/electronic properties. Briefly, the methylene-crosslinked π-conjugated quinoid (electron acceptor)-benzenoid (electron donor) resorcinol chains formed a low HOMO–LUMO gap and the graphitic π-stacked chains led to a hybridized HOMO–LUMO level and low bandgap (2.0 eV, active with 700 nm photons). The suitable band structure of the RF resins enabled the photoreduction of O2 to H2O2 in pure water (2H2O + O2 → H2O2). Hence, this inexpensive and efficient metal-free semiconductor with one-pot synthesis and wide light-response is particularly promising for the photosynthesis of H2O2.
In comparison to the dominant 2e− ORR (or a sequential two-step 1e− ORR), water oxidation towards H2O2 has not yet been well investigated and optimized on carbon-based catalysts; however, it has a higher atom utilization efficiency. Accordingly, Xu's group131 managed reaction the pathway for the photosynthesis of H2O2 based on the control of the chemical structure of covalent triazine frameworks (CTF) with the modification of acetylene (–CC–) and diacetylene (–CC–CC–) moieties (Fig. 9e). The functionalized CTF generated H2O2 by both oxygen reduction and water oxidation via a two-electron process (pathway is shown in Fig. 5e), while only the former occurred on the C–C triple-bond-free CTF (Fig. 9f). ΔGOH* was used as a descriptor to analyze whether or not the formation of OH* intermediate is thermodynamically favored compared to the formation of ˙OH radical (ΔG˙OH). When acetylene or diacetylene was the active site, ΔGOH* was much lower, which explained the novel 2e− WOR on the CC functionalized CTF, where charge separation was also encouraged.
Nevertheless, photocatalysis by organic materials is still in its infancy, and thus attempted trials would be encouraged even with unsatisfactory experimental findings at present. A mass-produced biopolymer, lignin, was examined for the photochemical generation of H2O2.132 Lignin underwent autooxidation, i.e. degradation, simultaneously with reductive H2O2 formation. The oxidation of oxalate, the added electron donor, competed with that of lignin and the redox reaction in total was increased simultaneously, reaching a maximum turnover number (TON) of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 254 μmol g−1. Although, lignin failed to be a valid photocatalyst, the green H2O2 production upon the degradation of lignin or the byproducts of photooxidation can be potentially of interest in some applications.
254 μmol g−1. Although, lignin failed to be a valid photocatalyst, the green H2O2 production upon the degradation of lignin or the byproducts of photooxidation can be potentially of interest in some applications.
The control of both the composition and microstructure of catalysts at the molecular level is essential for disclosing the mechanism of reactions. Thus, for the study of the electrochemical production of H2O2 and to gain theoretical insight, Briega-Martos et al.133 selected an aza-fused π-conjugated microporous polymer (Aza-CMP, Fig. 10a), which was assembled from molecular building blocks, with rich pyridinic N as the single dopant. Aza-CMP presented a high H2O2 selectivity under a tiny overpotential, while post-treatment with Co(II) catalyzed the further reduction of H2O2 to H2O (Fig. 10b). The DFT results indicated that the hydrogenation of pyridinic N could destabilize neighboring carbon atoms slightly, which became the adsorption sites for O2 to form superoxide species (from the partial Mulliken charges in Fig. 10d) in a monodentate chemisorbed state (Fig. 10c). The chemisorbed state resulted in the less stable adsorption of oxygen molecules compared to physisorbed state, which restrained the breaking of the O–O bond, leading to the partial reduction of O2 to H2O2. Although the activity of this system was not high, it provided insight into the ORR mechanism on polymer catalysts.
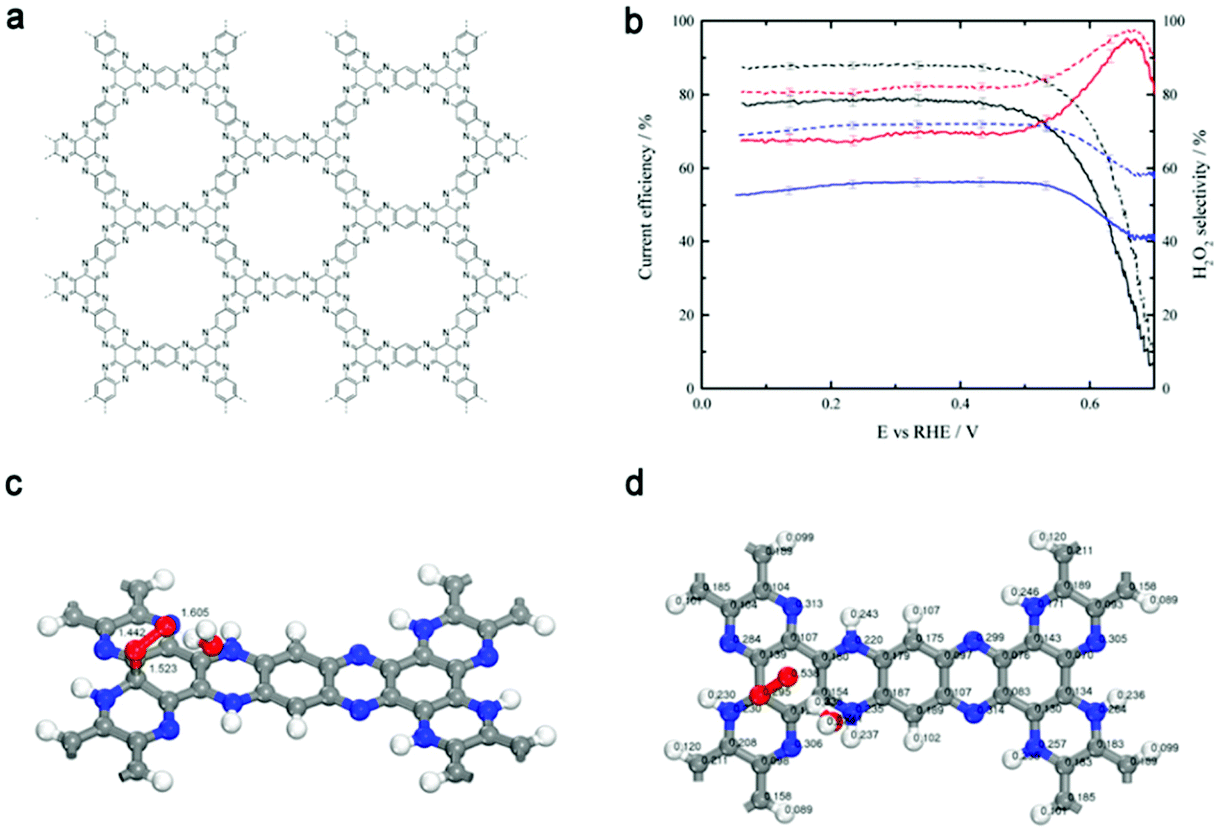 | ||
| Fig. 10 (a) Structure of the aza-fused π-conjugated microporous polymer (Aza-CMP). (b) Current efficiency (solid lines) and H2O2 selectivity (dashed lines) for the ORR reaction on the Aza-CMP (red line), Aza-CMP@Co (blue line), and glassy-carbon (black line) electrodes in O2-saturated 0.1 M NaOH solution. (c and d) Monodentate chemisorbed state of molecular oxygen on the carbon atom. (c) Adsorbent-adsorbate-solvent geometry (lengths given in Å). (d) Mulliken partial charges (e). Explicit water molecule in addition to a continuum model was used as a solvation effect treatment. Reproduced with permission.133 Copyright 2017 American Chemical Society. | ||
Despite the intriguing advantages of the light-driven production of H2O2, its efficiency is still far from satisfactory since the separation and migration of photogenerated charge carriers lack a driving force. PEC cells can induce spatial charge separation. Thus, a metal-free polyterthiophene (pTTh) coating was used as photocathode for oxygen reduction to H2O2, and a record high H2O2 concentration of 110 mM was accumulated in alkaline electrolyte (pH ∼ 13).134 When combined with the NiFeOx/BiVO4 photoanode as an O2 evolution catalyst, the authors constructed a bias-free system (Fig. 11a), which retained outstanding activity and stability. The 2e− and 4e− pathways were accompanied with the key steps of C-OOH and CO-OH cleavage, respectively (Fig. 11b). According to the calculation results, C-OOH cleavage to release peroxide ion HO2− (S3 → S0) was favored by a relatively lower energy and this reaction rate was ∼218 times faster than the rate for hydroxide ion formation (S3 → S4) via CO-OH cleavage. The energy profile was affected by pH, with a lower pH value to facilitate the 4e− process, making the pH a sensitive regulator.
 | ||
| Fig. 11 (a) Illustration of the unbiased H2O2 production device, with pTTh as the photocathode and NiFeOx/BiVO4 as the photoanode in 0.1 M KOH and 1 M borate buffer electrolyte, respectively. (b) Proposed reaction cycles of H2O2 production with the energetically most feasible active sites. S0 is the bare surface and Sx (x = 1, 2, 3, 4, and 5) are the structures of the intermediate states involved in the ORR. Red, gray, yellow and black balls represent oxygen, hydrogen, sulphur and carbon, respectively. Reproduced with permission.134 Copyright 2020 Royal Society of Chemistry. | ||
4. Highlighted carbon-based materials for 2e− or 4e− ORR
To maximize the energy capacity, the 4e− ORR is a desirable reaction in fuel cells and metal-batteries.135,136 Extensive studies have been carried out to develop carbon-based catalysts, which are abundant in nature, to replace platinum catalysts for the 4e− reduction. However, the exact origin and structure of the ORR active sites remain unclear. Here, we present some important experimental and theoretical highlights on carbon platforms. For the 4e− ORR, carbon nanostructures doped with nitrogen, such as N-doped CNT and graphene, have shown similar activity and superior stability and durability to commercial Pt/C catalysts, especially in alkaline electrolyte.137–139 Among the configurations of nitrogen species, pyridinic and pyrrolic nitrogen atoms are mostly likely to facilitate the 4e− ORR process, while their counterparts without N doping preferably form H2O2 first (substantially go reduction or oxidation, but usually the direct 4e− route affords higher complete ORR activity and thus is more desirable). The electron-accepting nitrogen induces charge delocalization, which can make adjacent carbon atoms positively charged, to affect the adsorption of O2 and breakage of O–O bond and to function as oxygen reduction active sites. This hypothesis was further evidenced in a recent report using graphite model catalysts with N-doping more precisely controlled.120 The role of pyridinic N was emphasized again. Sometimes, metal and nitrogen coordinations are thought to be the exact reduction sites. However, each conclusion is largely dependent on synthesis of the catalyst materials. High temperature annealing of carbon materials under N-rich atmosphere (e.g. ammonia) or using N-containing precursors often results in active N-sites, while N doping realized by hydrothermal reaction (150 °C, with NH4OH) does not demonstrate a similar trend (reduced electron number was calculated to be ∼2.7).140 Instead, it was found to facilitate the anchoring of the active metal sites. Moreover, even with a similar doping method, active nitrogen could be doped as quaternary type rather than pyridinic or pyrrolic N.135 Other factors such as geometry (alignment or roughness of carbons) are less related to the electrochemical mechanism for the ORR, but relevant to the electrokinetics. For the 2e− ORR, N-doping was also repeatedly reported beneficial to form H2O2 selectively (particularly for oxygen electroreduction), mostly using mesoporous carbon materials.101,112,114 However, clear mechanisms have not been reported to date. We speculate that the high exposure of surface/edges in these porous structures may affect the doping position of nitrogen and alter the way that the N sites regulate the ORR mechanism. In addition, doping with phosphorus and halogen atoms result in an enhancement in solar-driven H2O2 production.On the other hand, many cases related the oxygen-containing functional groups on carbon catalysts (e.g. g-C3N4, rGO, CNT, AC and BDD) with active and selective 2e− oxygen reduction, as discussed in the above.7,32,34,54,115 The oxidization methods vary. In oxidized carbon nitride, according to the theoretical energy evolution for each reaction step, C–O–C was much more favorable in energy than that of other functional groups. In oxidized graphene, the configurations of oxygen groups have an obvious influence on activity towards H2O2 (as a function of ΔGOOH*, Fig. 8d and e, scaling linearly with ΔGOH*, i.e. ΔGOOH* = ΔGOH* + 3.2 ± 0.2 eV). The characterization evidence provided by Kim et al. to support the oxygen defect and property relationship was relatively solid. Just as they suggested, the etheric group should be eliminated when designing 4e− ORR carbon catalysts. In other words, etheric group can be intentionally introduced for further developing high quality 2e− ORR catalysts.
In theoretical studies, both activity and selectivity could be examined using ΔGOH* as a descriptor (Fig. 3). The catalysts with ΔGOH* close to the top of the 2e− ORR volcano plots can potentially catalyze the reaction selectively and actively, which is also applicable as well for water oxidation towards H2O2. Tremendous efforts have led us to a deeper understanding of the ORR mechanism; however, new insights are still needed with characteristic evidence of catalysts and direct monitoring of the reaction intermediates.
5. Conclusions and perspectives
The last few years have witnessed fast development in the electro- and photocatalytic synthesis of H2O2 from water and oxygen in the aspects of engineering catalyst materials, optimizing operating conditions and designing reaction setups. Some inexpensive carbon-based catalysts show comparable (or even exceeding) electrochemical activity and selectivity to their noble metal counterparts. For example, the superior performance of oxidized carbon under alkaline conditions may allow the direct use of bulk solution in applications such as paper bleaching and the treatment of acidic wastewater.32,34,141 The performance of carbon-based photocatalysts has also improved significantly in pure water systems, where electron/proton donors are no long necessary. Considering these advances, we may expect a massive reduction in capital input when these technologies are put into large-scale use with efficient, selective and cheap carbon-based catalysts, simultaneously being sustainable. However, some challenges are concluded here as suggestions for problems further works may deal with firstly.Common problems for both electro- and photocatalytic systems are impurity of the bulk solution (other than water) and the self-degradation of catalysts. Generally, H2O2 solutions are desirable with moderate pH, and thus additional electron or hole scavengers and strong acidic or alkaline electrolytes should be avoided for most end users. During the redox reaction to form H2O2, catalysts themselves are likely to be oxidized or reduced when charge carriers accumulate on their surface for both anodic/cathodic electrodes and photoexcited semiconductors. Therefore, the intrinsic properties of materials against certain overpotentials or photocorrosion are important to be considered, and this importance becomes more significant in long-term operation. For example, WOR cocatalysts for H2O to O2 may relieve the photooxidation of carbonaceous 2e− ORR photocatalysts by transferring h+ to the active oxidation sites on the cocatalyst.
Another big challenge is to increase the H2O2 yield, which has been limited to several millimoles per litre per hour for most reported systems despite the modification of catalysts (with a very few exceptional yields of 89–116 mM h−1 in electrochemical synthesis).32,108 As is known, some materials originally reported as photocatalysts have been later employed as electrocatalysts (such as g-C3N4),45 and vice versa (such as WO3 and BiVO4).28 The major difference between them is that photoabsorbers are requisite in a light-driven process. Thus, expended light response to the visible range should be valued to mitigate H2O2 decomposition in the ultraviolet range. Higher conductivity is another critical factor to increase the charge transfer in both processes while in photocatalysis the separation of photoexcited e−/h+ pairs must be intentionally guided to withstand their fast recombination. However, there are several reports showing exciting performances by carbon-based materials. Porous carbon achieved H2O2 yield of 222.6 mM from electrosynthesis,108 while only <30 mM could be used in sewage treatment and disinfection. A PEC system with a polymer photocathode produced 110 mM H2O2, which could be directly used in certain applications (for example, pulp and paper bleaching or H2O2 fuel cell for the generation of electricity).134 Although other photocatalytic systems yielded less concentrated H2O2, they were capable of in situ fading dyes and killing bacteria.75,100 Instead of predicted practical applications, a remarkable case was demonstrated that 50 ppm red basic Fuchsin dye in flowing water could be instantly degraded by continuously produced H2O2 solution (24 μmol min−1) from water oxidation.142 To fulfill wider and higher demands, an accumulation of >570 mM H2O2 (∼2 wt%)143 is desired by upgrading the state-of-the-art generation rate by several folds in the following development.
All the above properties (optical, electronic and physicochemical) are related to the structure of materials, which also define the origin of selectivity and activity towards H2O2 formation. Nevertheless, some of the mechanical explanations remain rather speculative, especially when catalyst systems are prepared elaborately.144,145 More direct experimental and simulated evidence is fundamentally needed based on the precisely controlled catalyst structures/components for in-depth understanding of their structure-to-property correlation, which is essential for the rational design of future catalysts. For instance, the bottom-up wet chemical approach could be employed for the more controllable synthesis of catalyst materials.146 To gain insight, a combination of theory, computational studies, and sophisticated in situ/in operando characterization techniques will be beneficial.147 Accordingly, researchers on heterogeneous catalysis can learn from each other to inspire ideas.
Besides the catalyst, the reaction can be boosted by designing the system setup. Since the reduction of oxygen to H2O2 involves the interaction among the catalyst, O2 and liquid source of H+ (photon incident also matters in photocatalysis), the reaction is supposed to be faster if one can facilitate their interaction.149 Recently, an air–liquid–solid triphase photocatalytic architecture was reported to verify this concept (Fig. 12a).148 O2 from air was able to directly diffuse from the backside of the superhydrophobic (aerophilic) carbon fiber substrate to the front deposited Au-TiO2 photocatalysts in a triphase system and the production rate was 44 times that of its liquid–solid diphase counterpart. Notably, the diffusion coefficient of O2 in air (2.0 × 10−1 cm2 s−1) is about four orders of magnitude higher than that in H2O (2.1 × 10−5 cm2 s−1). However, the H2O2 concentration increased slowly since decomposition was found to be dominant over generation. Thus, a subsequent separation step may solve this issue.150 An interfacial engineering method was also demonstrated for anodic H2O2 generation on carbon electrodes (glassy carbon, Fig. 12b, and porous carbon fiber paper).142 Hydrophobic polymers deposited on carbon electrodes locally confined O2 (from 4e− WOR), thus shifting ΔGOH* to the peak of the volcano, and consequently improving the activity and selectivity of the 2e− WOR to H2O2. Finally, authors combined this anode to oxidized CNT cathode in a flow cell for the 2e− WOR/2e− ORR strategy to directly fade organic contamination continuously. In the future, our reasonable yet ambitious anticipation is that electro- and photochemical H2O2 synthesis systems will be moving towards more active, selective and stable carbon-based catalysts, with affordable raw materials and feasible devices to open up opportunities in wide practical applications.
 | ||
| Fig. 12 (a) Schematic illustration of the triphase photocatalytic O2 to H2O2 reaction system with Au-decorated TiO2 photocatalysts immobilized on the porous superhydrophobic carbon fibers. Adapted with permission.148 Copyright 2019 Elsevier. (b) Schematic showing the assumed possible reaction pathway of H2O to H2O2 tuning by local confined oxygen on glassy carbon after hydrophobic treatment. Reproduced with permission.142 Copyright 2020 Springer Nature. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the Australian Research Council (DP180102062). X. Hu acknowledges the scholarships from Monash Graduate Scholarship and Monash International Postgraduate Research Scholarship.References
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS PubMed
.
-
C. W. Jones, Applications of hydrogen peroxide and derivatives, Royal Society of Chemistry, 2007 Search PubMed
- R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2006, 45, 206–222 CrossRef CAS
- R. A. Rozendal, E. Leone, J. Keller and K. Rabaey, Electrochem. Commun., 2009, 11, 1752–1755 CrossRef CAS
- M. Melchionna, P. Fornasiero and M. Prato, Adv. Mater., 2019, 31, 1802920 CrossRef PubMed
- K. Kosaka, H. Yamada, K. Shishida, S. Echigo, R. A. Minear, H. Tsuno and S. Matsui, Water Res., 2001, 35, 3587–3594 CrossRef CAS PubMed
- I. Yamanaka and T. Murayama, Angew. Chem., Int. Ed., 2008, 47, 1900–1902 CrossRef CAS PubMed
- P. Drogui, S. Elmaleh, M. Rumeau, C. Bernard and A. Rambaud, J. Appl. Electrochem., 2001, 31, 877–882 CrossRef CAS
- R. Pedahzur, H. I. Shuval and S. Ulitzur, Water Sci. Technol., 1997, 35, 87–93 CrossRef CAS
- C. A. Martínez-Huitle and S. Ferro, Chem. Soc. Rev., 2006, 35, 1324–1340 RSC
- M. Ksibi, Chem. Eng. J., 2006, 119, 161–165 CrossRef CAS
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed
-
G. Strukul, Catalytic oxidations with hydrogen peroxide as oxidant, Springer Science & Business Media, Netherlands, 2013 Search PubMed
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang and C. Mesters, Science, 2020, 367, 193–197 CAS
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310–8311 CrossRef CAS PubMed
- A. Sanli and A. Aytaç, Int. J. Hydrogen Energy, 2011, 36, 869–875 CrossRef CAS
- S. A. Mousavi Shaegh, N.-T. Nguyen, S. M. Mousavi Ehteshami and S. H. Chan, Energy Environ. Sci., 2012, 5, 8225–8228 RSC
- D. N. Prater and J. J. Rusek, Appl. Energy, 2003, 74, 135–140 CrossRef CAS
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Catal. Today, 2015, 248, 3–9 CrossRef CAS
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS
- M. N. Young, M. J. Links, S. C. Popat, B. E. Rittmann and C. I. Torres, ChemSusChem, 2016, 9, 3345–3352 CrossRef CAS PubMed
- M. G. Clerici and P. Ingallina, Catal. Today, 1998, 41, 351–364 CrossRef CAS
- J. García-Serna, T. Moreno, P. Biasi, M. J. Cocero, J.-P. Mikkola and T. O. Salmi, Green Chem., 2014, 16, 2320–2343 RSC
- J. S. Hargreaves, Y.-M. Chung, W.-S. Ahn, T. Hisatomi, K. Domen, M. C. Kung and H. H. Kung, Appl. Catal., A, 2020, 117419 CrossRef CAS
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 1801909 CrossRef
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. P. de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS
- S. Fukuzumi, Y. M. Lee and W. Nam, Chem. – Eur. J., 2018, 24, 5016–5031 CrossRef CAS PubMed
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201911609
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 CrossRef
- M. Teranishi, S.-I. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850–7851 CrossRef CAS PubMed
- Z. Haider, H.-I. Cho, G.-H. Moon and H.-I. Kim, Catal. Today, 2019, 335, 55–64 CrossRef CAS
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS PubMed
- B. O. Burek, D. W. Bahnemann and J. Z. Bloh, ACS Catal., 2019, 9, 25–37 CrossRef CAS
- X. Shi, S. Siahrostami, G.-L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- D. Deng, K. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218 CrossRef CAS PubMed
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281–7285 CrossRef CAS PubMed
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS PubMed
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed
- Z. Zhu, H. Pan, M. Murugananthan, J. Gong and Y. Zhang, Appl. Catal., B, 2018, 232, 19–25 CrossRef CAS
- Z. Teng, W. Cai, S. Liu, C. Wang, Q. Zhang, S. Chenliang and T. Ohno, Appl. Catal., B, 2020, 271, 118917 CrossRef CAS
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC
- S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26–35 CrossRef CAS
- R. Wang, X. Zhang, F. Li, D. Cao, M. Pu, D. Han, J. Yang and X. Xiang, J. Energy Chem., 2018, 27, 343–350 CrossRef
- Y. Xie, Y. Li, Z. Huang, J. Zhang, X. Jia, X.-S. Wang and J. Ye, Appl. Catal., B, 2020, 265, 118581 CrossRef
- J. Tian, D. Wang, S. Li, Y. Pei, M. Qiao, Z. H. Li, J. Zhang and B. Zong, ACS Sustainable Chem. Eng., 2020, 8, 594–603 CrossRef CAS
- W. Hou, Y. Li, S. Ouyang, H. Chen, J. Ye, X. Han and Y. Deng, Chem. Commun., 2019, 55, 13279–13282 RSC
- J. Cao, H. Wang, Y. Zhao, Y. Liu, Q. Wu, H. Huang, M. Shao, Y. Liu and Z. Kang, J. Mater. Chem. A, 2020, 8, 3701–3707 RSC
- L. Zhou, J. Feng, B. Qiu, Y. Zhou, J. Lei, M. Xing, L. Wang, Y. Zhou, Y. Liu and J. Zhang, Appl. Catal., B, 2020, 267, 118396 CrossRef
- C. Zhang, J. Bai, L. Ma, Y. Lv, F. Wang, X. Zhang, X. Yuan and S. Hu, Diamond Relat. Mater., 2018, 87, 215–222 CrossRef CAS
- G.-H. Moon, M. Fujitsuka, S. Kim, T. Majima, X. Wang and W. Choi, ACS Catal., 2017, 7, 2886–2895 CrossRef CAS
- S. Kim, G.-H. Moon, H. Kim, Y. Mun, P. Zhang, J. Lee and W. Choi, J. Catal., 2018, 357, 51–58 CrossRef
- Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2015, 5, 3058–3066 CrossRef CAS
- H. Ou, P. Yang, L. Lin, M. Anpo and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 10905–10910 CrossRef CAS PubMed
- Y. Yang, Z. Zeng, G. Zeng, D. Huang, R. Xiao, C. Zhang, C. Zhou, W. Xiong, W. Wang, M. Cheng, W. Xue, H. Guo, X. Tang and D. He, Appl. Catal., B, 2019, 258, 117956 CrossRef CAS
- L. Yang, G. Dong, D. L. Jacobs, Y. Wang, L. Zang and C. Wang, J. Catal., 2017, 352, 274–281 CrossRef CAS
- Y. Zheng, Z. Yu, H. Ou, A. M. Asiri, Y. Chen and X. Wang, Adv. Funct. Mater., 2018, 28, 1705407 CrossRef
- Y. Liu, X. Zeng, X. Hu, J. Hu and X. Zhang, J. Chem. Technol. Biotechnol., 2019, 94, 22–37 CrossRef CAS
- F. Xue, Y. Si, M. Wang, M. Liu and L. Guo, Nano Energy, 2019, 62, 823–831 CrossRef CAS
- J. Cai, J. Huang, S. Wang, J. Iocozzia, Z. Sun, J. Sun, Y. Yang, Y. Lai and Z. Lin, Adv. Mater., 2019, 31, 1806314 CrossRef PubMed
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed
- X. Hu, X. Zeng, Y. Liu, J. Lu, S. Yuan, Y. Yin, J. Hu, D. T. McCarthy and X. Zhang, Appl. Catal., B, 2020, 268, 118466 CrossRef
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, X. Li, B. Li, W. Dong and Y. Zhou, Appl. Catal., B, 2020, 272, 118970 CrossRef CAS
- X. Zeng, Y. Liu, Y. Kang, Q. Li, Y. Xia, Y. Zhu, H. Hou, M. H. Uddin, T. R. Gengenbach, D. Xia, C. Sun, D. T. McCarthy, A. Deletic, J. Yu and X. Zhang, ACS Catal., 2020, 10, 3697–3706 CrossRef CAS
- C. Chu, Q. Zhu, Z. Pan, S. Gupta, D. Huang, Y. Du, S. Weon, Y. Wu, C. Muhich, E. Stavitski, K. Domen and J.-H. Kim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6376 CrossRef CAS PubMed
- Q. Zhu, Z. Pan, S. Hu and J.-H. Kim, ACS Appl. Energy Mater., 2019, 2, 7972–7979 CrossRef CAS
- F. Yu, Y. Wang, H. Ma and G. Dong, Int. J. Hydrogen Energy, 2018, 43, 19500–19509 CrossRef CAS
- J. Xiong, X. Li, J. Huang, X. Gao, Z. Chen, J. Liu, H. Li, B. Kang, W. Yao and Y. Zhu, Appl. Catal., B, 2020, 266, 118602 CrossRef CAS
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, ChemCatChem, 2018, 10, 2070–2077 CrossRef CAS
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954–17961 CrossRef CAS PubMed
- Ç. Ö. Girit, J. C. Meyer, R. Erni, M. D. Rossell, C. Kisielowski, L. Yang, C.-H. Park, M. F. Crommie, M. L. Cohen, S. G. Louie and A. Zettl, Science, 2009, 323, 1705–1708 CrossRef PubMed
- J.-H. Zhong, J. Zhang, X. Jin, J.-Y. Liu, Q. Li, M.-H. Li, W. Cai, D.-Y. Wu, D. Zhan and B. Ren, J. Am. Chem. Soc., 2014, 136, 16609–16617 CrossRef CAS PubMed
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed
- L. Han, Y. Sun, S. Li, C. Cheng, C. E. Halbig, P. Feicht, J. L. Hübner, P. Strasser and S. Eigler, ACS Catal., 2019, 9, 1283–1288 CrossRef CAS
- P. Su, M. Zhou, X. Lu, W. Yang, G. Ren and J. Cai, Appl. Catal., B, 2019, 245, 583–595 CrossRef CAS
- W.-C. Hou and Y.-S. Wang, ACS Sustainable Chem. Eng., 2017, 5, 2994–3001 CrossRef CAS
- L. Zheng, J. Zhang, Y. H. Hu and M. Long, J. Phys. Chem. C, 2019, 123, 13693–13701 CrossRef CAS
- L. Zheng, H. Su, J. Zhang, L. S. Walekar, H. Vafaei Molamahmood, B. Zhou, M. Long and Y. H. Hu, Appl. Catal., B, 2018, 239, 475–484 CrossRef CAS
- G.-H. Moon, W. Kim, A. D. Bokare, N.-E. Sung and W. Choi, Energy Environ. Sci., 2014, 7, 4023–4028 RSC
- J. Xu, Z. Chen, H. Zhang, G. Lin, H. Lin, X. Wang and J. Long, Sci. Bull., 2017, 62, 610–618 CrossRef CAS
- X. Zeng, Z. Wang, G. Wang, T. R. Gengenbach, D. T. McCarthy, A. Deletic, J. Yu and X. Zhang, Appl. Catal., B, 2017, 218, 163–173 CrossRef CAS
- X. Zeng, Z. Wang, N. Meng, D. T. McCarthy, A. Deletic, J.-H. Pan and X. Zhang, Appl. Catal., B, 2017, 202, 33–41 CrossRef CAS
- H.-I. Kim, O. S. Kwon, S. Kim, W. Choi and J.-H. Kim, Energy Environ. Sci., 2016, 9, 1063–1073 RSC
- C. H. Choi, M. W. Chung, H. C. Kwon, S. H. Park and S. I. Woo, J. Mater. Chem. A, 2013, 1, 3694–3699 RSC
- J. F. Carneiro, M. J. Paulo, M. Siaj, A. C. Tavares and M. R. V. Lanza, J. Catal., 2015, 332, 51–61 CrossRef CAS
- Z. Zhang, H. Meng, Y. Wang, L. Shi, X. Wang and S. Chai, Electrochim. Acta, 2018, 260, 112–120 CrossRef CAS
- T.-P. Fellinger, F. Hasché, P. Strasser and M. Antonietti, J. Am. Chem. Soc., 2012, 134, 4072–4075 CrossRef CAS PubMed
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, Chem, 2018, 4, 106–123 CAS
- R. Ma, L. Wang, H. Wang, Z. Liu, M. Xing, L. Zhu, X. Meng and F.-S. Xiao, Appl. Catal., B, 2019, 244, 594–603 CrossRef CAS
- C. Zhu, M. Zhu, Y. Sun, Y. Zhou, J. Gao, H. Huang, Y. Liu and Z. Kang, ACS Appl. Energy Mater., 2019, 2, 8737–8746 CrossRef CAS
- T. Lee, H. T. Bui, J. Yoo, M. Ra, S. H. Han, W. Kim and W. Kwon, ACS Appl. Mater. Interfaces, 2019, 11, 41196–41203 CrossRef CAS
- X. Zhang, J. Fu, Y. Zhang and L. Lei, Sep. Purif. Technol., 2008, 64, 116–123 CrossRef CAS
- H. Zhao, Y. Chen, Q. Peng, Q. Wang and G. Zhao, Appl. Catal., B, 2017, 203, 127–137 CrossRef CAS
- Y. Liu, X. Quan, X. Fan, H. Wang and S. Chen, Angew. Chem., Int. Ed., 2015, 54, 6837–6841 CrossRef CAS PubMed
- J. Zhang, G. Zhang, S. Jin, Y. Zhou, Q. Ji, H. Lan, H. Liu and J. Qu, Carbon, 2020, 163, 154–161 CrossRef CAS
- S. Mavrikis, M. Göltz, S. Rosiwal, L. Wang and C. Ponce de León, ACS Appl. Energy Mater., 2020, 3, 3169–3173 CrossRef CAS
- J. Park, Y. Nabae, T. Hayakawa and M.-A. Kakimoto, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS
- X. Sheng, N. Daems, B. Geboes, M. Kurttepeli, S. Bals, T. Breugelmans, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2015, 176–177, 212–224 CrossRef CAS
- K. Zhao, Y. Su, X. Quan, Y. Liu, S. Chen and H. Yu, J. Catal., 2018, 357, 118–126 CrossRef
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS
- J. O. Thostenson, E. Ngaboyamahina, K. L. Sellgren, B. T. Hawkins, J. R. Piascik, E. J. D. Klem, C. B. Parker, M. A. Deshusses, B. R. Stoner and J. T. Glass, ACS Appl. Mater. Interfaces, 2017, 9, 16610–16619 CrossRef CAS PubMed
- Y.-H. Lee, F. Li, K.-H. Chang, C.-C. Hu and T. Ohsaka, Appl. Catal., B, 2012, 126, 208–214 CrossRef CAS
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, ACS Sustainable Chem. Eng., 2018, 6, 311–317 CrossRef CAS
- C. J. Shearer, A. Cherevan and D. Eder, Adv. Mater., 2014, 26, 2295–2318 CrossRef CAS PubMed
- M. Melchionna, P. Fornasiero and M. Prato, Adv. Mater., 2019, 31, 1802920 CrossRef PubMed
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361 CrossRef CAS PubMed
- R. A. Sidik, A. B. Anderson, N. P. Subramanian, S. P. Kumaraguru and B. N. Popov, J. Phys. Chem. B, 2006, 110, 1787–1793 CrossRef CAS PubMed
- Y. Zhu, S. Qiu, F. Deng, F. Ma and Y. Zheng, Sci. Total Environ., 2020, 722, 137853 CrossRef CAS PubMed
- X. Lu, D. Wang, K.-H. Wu, X. Guo and W. Qi, J. Colloid Interface Sci., 2020, 573, 376–383 CrossRef CAS PubMed
- Y. Liu, Y. Zhao, Y. Sun, J. Cao, H. Wang, X. Wang, H. Huang, M. Shao, Y. Liu and Z. Kang, Appl. Catal., B, 2020, 270, 118875 CrossRef CAS
- A. R. Khataee, M. Safarpour, M. Zarei and S. Aber, J. Electroanal. Chem., 2011, 659, 63–68 CrossRef CAS
- R. Pandiyan, N. Delegan, A. Dirany, P. Drogui and M. A. El Khakani, Carbon, 2015, 94, 988–995 CrossRef CAS
- L. Wang, Y. Zhang, L. Chen, H. Xu and Y. Xiong, Adv. Mater., 2018, 30, 1801955 CrossRef PubMed
- J. Sun and Y. Wu, Angew. Chem., Int. Ed., 2020, 59, 10904–10908 CrossRef CAS PubMed
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef
- M. K. Węcławski, M. Jakešová, M. Charyton, N. Demitri, B. Koszarna, K. Oppelt, S. Sariciftci, D. T. Gryko and E. D. Głowacki, J. Mater. Chem. A, 2017, 5, 20780–20788 RSC
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef CAS PubMed
- E. Miglbauer, M. Gryszel and E. D. Głowacki, Green Chem., 2020, 22, 673–677 RSC
- V. Briega-Martos, A. Ferre-Vilaplana, A. de la Peña, J. L. Segura, F. Zamora, J. M. Feliu and E. Herrero, ACS Catal., 2017, 7, 1015–1024 CrossRef CAS
- W. Fan, B. Zhang, X. Wang, W. Ma, D. Li, Z. Wang, M. Dupuis, J. Shi, S. Liao and C. Li, Energy Environ. Sci., 2020, 13, 238–245 RSC
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 RSC
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed
- Z. Chen, S. Chen, S. Siahrostami, P. Chakthranont, C. Hahn, D. Nordlund, S. Dimosthenis, J. K. Nørskov, Z. Bao and T. F. Jaramillo, React. Chem. Eng., 2017, 2, 239–245 RSC
- C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen, X. Sun, S. Siahrostami, K. Chan and H. Wang, Nat. Catal., 2020, 3, 125–134 CrossRef CAS
- M. F. Easton, A. G. Mitchell and W. F. K. Wynne-Jones, Trans. Faraday Soc., 1952, 48, 796–801 RSC
- S. Sorcar, Y. Hwang, C. A. Grimes and S.-I. In, Mater. Today, 2017, 20, 507–515 CrossRef CAS
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 78 CrossRef
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- Z. Liu, X. Sheng, D. Wang and X. Feng, iScience, 2019, 17, 67–73 CrossRef CAS PubMed
- Q. Zhang, M. Zhou, G. Ren, Y. Li, Y. Li and X. Du, Nat. Commun., 2020, 11, 1731 CrossRef CAS PubMed
- Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 5402–5406 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |