
Pseudo-enantiomeric carbohydrate-based N-heterocyclic carbenes as promising chiral ligands for enantiotopic discrimination†
Alexander S.
Henderson
,
John F.
Bower
* and
M. Carmen
Galan
 *
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: john.bower@bris.ac.uk; m.c.galan@bris.ac.uk
First published on 8th April 2020
Abstract
The practical synthesis of carbohydrate-based NHC–Rh complexes bearing C1 or C3 sterically differentiated positions, accessed by glycosylation or SNAr strategies, is reported. These catalysts exhibit pseudo-enantiomeric behaviour in the hydrosilylation of acetophenone. We show that steric bulk at C1 gives preference for (S)-phenyl-1-ethanol, while bulk at C3 leads to the (R)-enantiomer. These results represent the first example of pseudo-enantiomeric carbohydrate-based NHC ligands leading to enantiotopic discrimination.
The search for stable and effective N-heterocyclic carbene (NHC) transition metal (TM) complexes is of great interest for catalytic applications.1 NHCs with tailored electronic and steric properties have become indispensable in many applications of transition metal and main group chemistry.2 For instance, the success of NHC ligands in non-stereoselective reactions, such as cross-coupling and olefin metathesis,3 has prompted the incorporation of homochiral units into NHC scaffolds and this offers exciting new opportunities in reaction development.4 Typical strategies to monodentate NHC ligands suitable for asymmetric TM-catalysis have employed commercially available enantioenriched amines,5 featured the resolution of racemic material6 and involved chiral pool substrates, such as amino acid and natural product derivatives.7 Enantiopure NHCs encompassing a diverse array of ligand architectures have been evaluated in a plethora of asymmetric reactions and, although highly effective examples exist, the search for increasingly more efficient systems is paramount.4–7 However, a limiting factor and general problem in the synthesis and assessment of novel NHC ligands is the ability to obtain readily modifiable building blocks, such as enantioenriched amines.8
The abundance of inherently chiral, biologically derived saccharide material provides useful starting compounds with a range of stereochemistries for ligand syntheses. Indeed, the application of carbohydrates as the enantioinduction component in stereoselective catalysis is frequently encountered within the literature and a multitude of efficient ligands have been reported.9 Towards this end, our laboratory and others have described the use of carbohydrate-based NHCs for asymmetric catalysis in an effort to solve the issues highlighted above.10 Our initial report disclosed the expedient assembly of C2-symmetric NHC ligands from per-alkylated β-glucosamines.10a Upon successful ligation to [Rh(cod)Cl]2, the carbohydrate-based NHC complexes (e.g.1, Scheme 1A, eqn (1)) were evaluated in the asymmetric hydrosilylation of ketones (e.g.2, Scheme 1A, eqn (2)).2,11 Ultimately, catalyst 1, featuring permethylated β-glucoside residues attached to the NHC moiety, was most efficient and provided phenyl-1-ethanol (3) in modest er (81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 R:S).10a
:19 R:S).10a
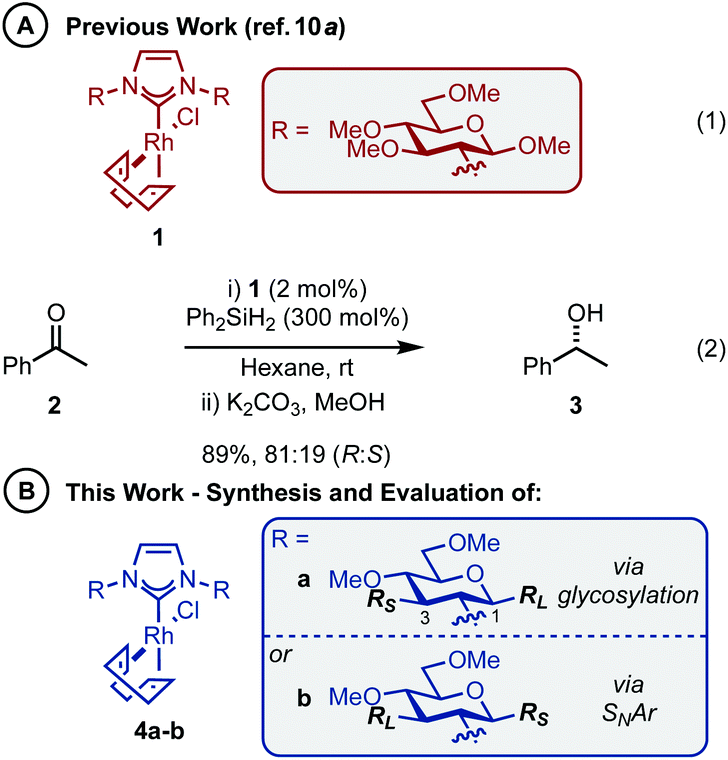 | ||
| Scheme 1 Carbohydrate-based NHC complexes. RL = large group; RS = small group. | ||
One of the advantages of carbohydrate chiral synthons is the ability to chemoselectively incorporate functional groups at each of the different OH groups. We thus sought to achieve greater asymmetric induction from 1 by sterically differentiating the C1 and C3 positions which flank the connecting position (C2) between the carbohydrate and the imidazolylidene scaffold (4a–b, Scheme 1B). Practically, this can be realised in two different ways: (1) by employing a carbohydrate unit with a large group (RL) at C1 and a small group (RS) at C3, as depicted in 4a, or (2) vice versa as in 4b. We surmised that an aryl group would serve as an effective RL candidate as this motif is readily available. From a synthetic viewpoint, installation of an aryl residue (e.g. phenol or thiophenol) at C1 by stereoselective glycosylation is well known.12 Accessing a related C3–OH arylated carbohydrate derivative is conceptually more challenging, however, this could be achieved using SNAr-based methdology recently developed in our laboratory.13 Therefore, our aims were to synthesize sterically differentiated carbohydrate amines, access the corresponding C2-symmetric imidazolium salts, ligate to a Rh source and subsequently evaluate the complexes in the hydrosilylation of acetophenone (2).
Thioglycoside 5,14 synthesized in two steps from D-glucosamine HCl in 70% yield (see ESI† for details), was chosen as it was a commonly used building block in carbohydrate synthesis and thus easily accessible. 5 was globally deprotected by stirring with NaOMe in methanol to remove the ester groups, this was followed by addition of aq. NaOH at reflux to give amine intermediate 615 (Scheme 2A). Cu-catalyzed azide formation from 6 with TfN3 gave triol 7 in 78% yield over the three steps. Methylation of the remaining hydroxyl positions using MeI and NaH and subsequent azide hydrogenation (H2, Pd/C) gave C1 sterically differentiated amine 8a in an 81% yield. It is pertinent to note that a methyl ether (OMe) was selected as the RS group based on our previous observations.10a Elaboration of 8a into imidazolium salt 9a was achieved in 41% yield by condensation with aq. glyoxal and then cyclization with MOMCl (Scheme 2B).
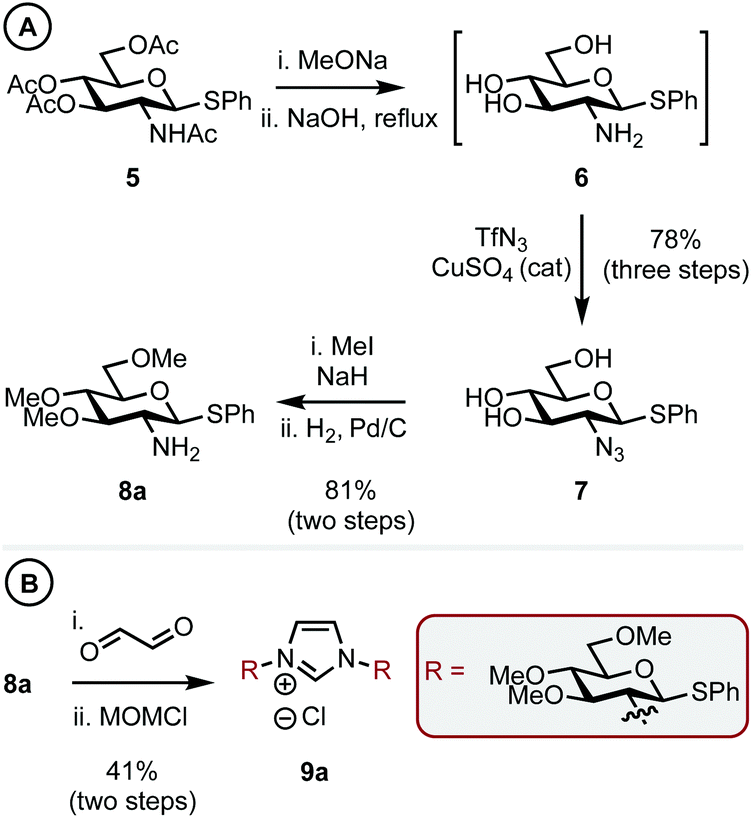 | ||
| Scheme 2 Synthesis of a carbohydrate-based imidazolium salt with steric bulk at C1. | ||
With a C1 sterically differentiated carbohydrate-based NHC·HCl salt in hand, our attention turned to the C3 analogue (Scheme 3A and B).16 The synthesis of carbohydrate-aryl ether 10 was accomplished by an SNAr reaction between the appropriate fluoro-arene and the corresponding OH-bearing carbohydrate.13 Next, under sonication, TsOH acid catalyzed deprotection of the benzylidene acetal revealed the O4 and O6 hydroxyls, which were methylated as before to give 11 in 95% overall yield. Heterogeneous hydrogenation gave amine 8b and the corresponding imidazolium salt (9b) was accessed in 44% yield utilizing the same procedure outlined before.
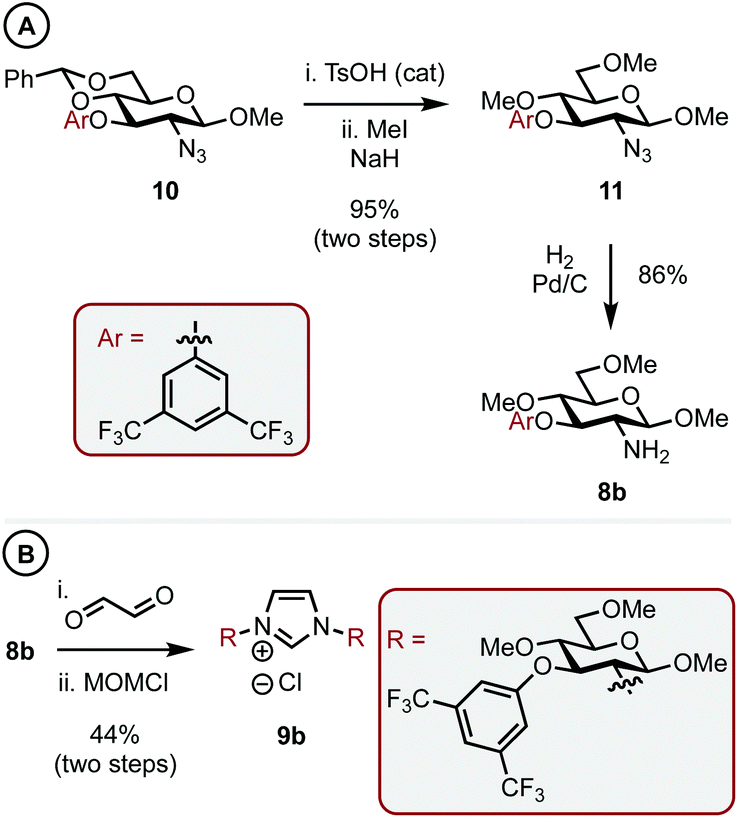 | ||
| Scheme 3 Synthesis of a carbohydrate-based imidazolium salt with steric bulk at C3. | ||
Imidazolium salt 9a was deprotonated by tBuONa at rt and ligated to [Rh(cod)Cl]2 which afforded complex 12a in usable yield (Scheme 4, (1)). NHC·HCl 9b required the action of tBuOK at 100 °C to effect ligation and this allowed isolation of 12b in 72% yield (Scheme 4, (2)). Unfortunately, neither complex was crystalline but HRMS confirmed product formation (see the Experimental section) and the carbenic 13C NMR resonance was visible for 12b (δC: 189.2 ppm). The 1H NMR data of 12a–b is similar to other β-glucoside-based NHC–Rh compounds and the spectra also featured significant peak broadening, which further supports metal ligation.10a In 12b the backbone imidazolylidene protons were not obscured and appeared at δH: 6.95 and 6.80 ppm respectively, indicating that the complex does not possess C2-symmetry and, therefore, it is likely the pyranoside rings are each in unique environments. From this information we infer that our NHC ligands are dynamic in solution. Indeed, Grubbs and co-workers have observed rotation around CCarbohydrate–NImidazolylidene bonds in metathesis catalysts featuring carbohydrate-based NHC ligands.10c
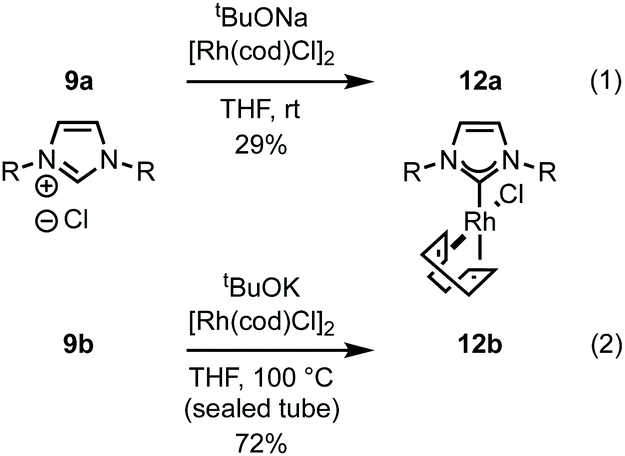 | ||
| Scheme 4 Ligation of 9a–b to [Rh(cod)Cl]2. | ||
Next, complexes 12a–b were evaluated in the hydrosilylation of acetophenone as model system and both were found to be catalytically competent (Table 1). Interestingly, although the enantiomer ratios (er) of the product were modest, 12a resulted in the isolation of 3 with a distinguishable preference for the S-enantiomer (30:70 R:S, entry 1), whereas 12b gave preference for the R-enantiomer (75:25 R:S, entry 2). Carbohydrates largely exist in nature in the D-configuration and this causes complications when employing them as ligands because sufficient quantities of the antipode are not normally available. Therefore, an asymmetric process must often rely on pseudo-enantiomeric ligands to give access to each enantiomer of the product.17 While pseudo-enantiomerism has been documented in carbohydrate-based ligands, such as phosphinites18 and phosphites,19 and carbohydrate-based organocatalysts,20 to the best of our knowledge this is the first example concerning a carbohydrate-based NHC ligand.
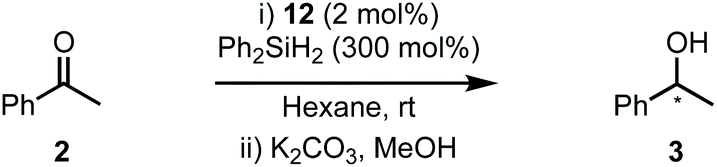
Encouraged by the results, we envisaged optimizing the asymmetric induction potential of the carbohydrate-based NHC ligands by altering the size and substitution pattern of the C3 aryl group.21 Commercially available fluoro-aryls bearing a range of structural motifs (ortho-, meta- and para-substitution patterns; biaryls and bicyclics) were introduced using SNAr, expediently delivering a small library of carbohydrate-aryl ethers (13a–e) as previously described.13 The ethers were then transformed into the required amines (15a–e) by benzylidene acetal cleavage, methylation of the OHs (14a–e) and reduction of the azide functionality as before (Scheme 5).
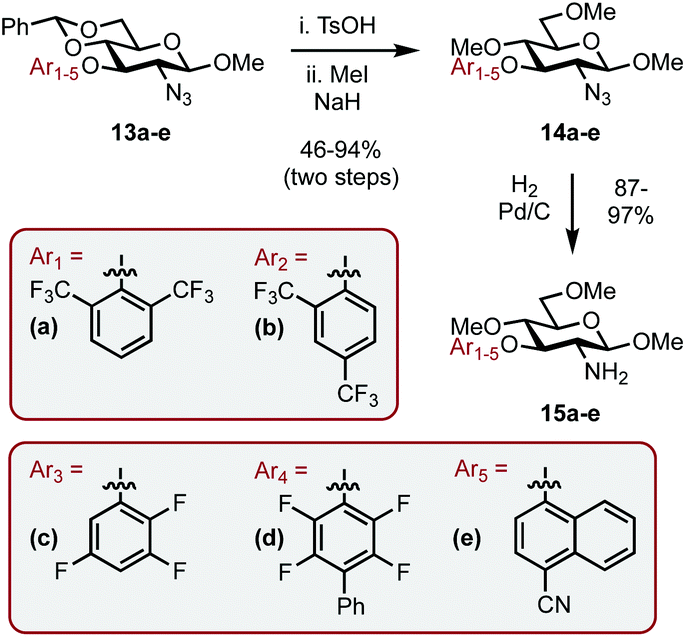 | ||
| Scheme 5 Synthesis of SNAr modified amines 15a–e. | ||
The SNAr modified amines were swiftly elaborated into the corresponding imidazolium salts (16a–e) by treatment with aq. glyoxal and then MOMCl (Scheme 6). Deprotonation with tBuOK and ligation to Rh was performed in THF at reflux to give complexes 17a–e in 40–99% yields. While the yields for both of these steps were predominantly good, the example featuring Ar1 (bearing ortho–ortho aryl substitution, e.g.13a–17a) was consistently low yielding despite our best efforts and this was attributed to the level of steric bulk present in the system.
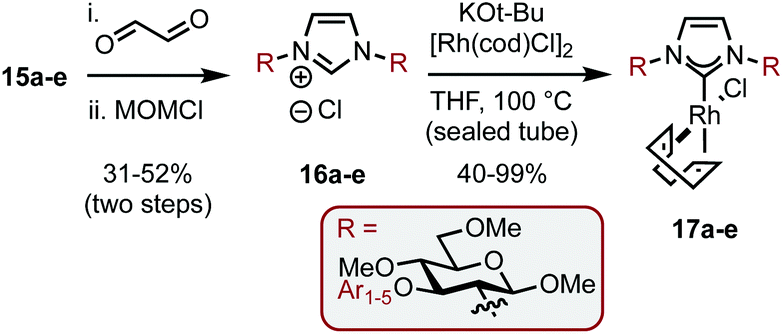 | ||
| Scheme 6 Synthesis of complexes 17a–e. | ||
Complexes 17a–e were then evaluated in the hydrosilylation of 2 and the results are displayed in Table 2. Catalysts 17a–b, featuring an ortho-CF3 substituted aryl group (Ar1–2), gave negligible stereoinduction in 3 (entries 1 and 2). However, 17c (entry 3) resulted in the isolation of 3 in high yield and with the greatest er to date (86:14). Both 17d and 17e (entries 4 and 5) afforded 3 with detectable er's but were ultimately less efficient systems.
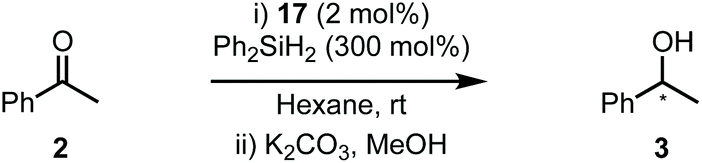
Conclusions
In summary, a series of pseudo-enantiomeric carbohydrate-based NHC-ligands featuring sterically differentiated groups at positions C1 or C3 of the carbohydrate were synthesized by simple glycosylation or SNAr strategies and evaluated in the Rh-catalyzed hydrosilylation of acetophenone. We show that ligands bearing a bulky group at C1 lead to a preference for (S)-phenyl-1-ethanol, while the presence of large groups at C3 afforded preferentially the (R)-enantiomer. Our results suggest this effect is achieved by the steric differentiation of the carbohydrate's C1 and C3 positions which flank the attachment to the imidazolylidene scaffold. This “pseudoenantiomeric” design feature is shown in Fig. 1.22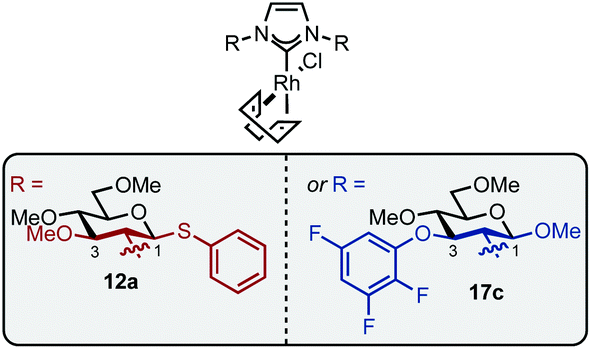 | ||
| Fig. 1 Pseudo-stereochemical ligands 12a and 17c. | ||
While our efforts to achieve a high level of asymmetric induction in the benchmark hydrosilylation reaction11,12 have been less successful, we have surpassed our previous result9 and shown, to the best of our knowledge for the first time, that the judicial functionalization of carbohydrate synthons can lead to pseudo-enantiomeric NHC-ligands for asymmetric catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
ASH thanks EPSRC Bristol Chemical Synthesis CDT EP/G036764/1, JFB thanks Royal Society for a University Research Fellowship and MCG thanks the ERC (COG: 648239) and EPSRC CAF EP/L001926/1 and EPSRC EP/J002542/1 for funding.Notes and references
- (a) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS; (b) N-Heterocyclic Carbenes, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2014 Search PubMed.
- (a) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994.3 CrossRef PubMed; (b) S. Kumar, J. Heterocycl. Chem., 2019, 56, 1168 CrossRef CAS.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed.
- (a) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (b) F. Wang, L. J. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef CAS; (c) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed.
- Selected examples: (a) D. Enders, H. Gielen, G. Raabe, J. Runsink and J. H. Teles, Chem. Ber., 1996, 129, 1483 CrossRef CAS; (b) W. A. Herrmann, L. J. Goossen, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1996, 35, 2805 CrossRef CAS; (c) T. J. Seiders, D. W. Ward and R. H. Grubbs, Org. Lett., 2001, 3, 3225 CrossRef CAS PubMed.
- Selected examples: (a) E. P. Kündig, T. M. Seidel, Y. X. Jia and G. Bernardinelli, Angew. Chem., Int. Ed., 2007, 46, 8484 CrossRef PubMed; (b) C. T. Check, K. P. Jang, C. B. Schwamb, A. S. Wong, M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2015, 54, 4264 CrossRef CAS PubMed.
- Selected examples: (a) F. Glorius, G. Altenhoff, R. Goddard and C. Lehmann, Chem. Commun., 2002, 2704 RSC; (b) S. Würtz, C. Lohre, R. Fröhlich, K. Bergander and F. Glorius, J. Am. Chem. Soc., 2009, 131, 8344 CrossRef PubMed; (c) P. J. Czerwinski and M. Michalak, Synthesis, 2019, 51, 1689 CrossRef CAS.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed.
- Selected reviews: (a) M. Diéguez, O. Pàmies and C. Claver, Chem. Rev., 2004, 104, 3189 CrossRef PubMed; (b) Carbohydrates: Tools for Stereoselective Synthesis, ed. M. M. K. Boysen, Wiley-VCH, Weinheim, 2013 Search PubMed; (c) A. S. Henderson, J. F. Bower and M. C. Galan, Org. Biomol. Chem., 2016, 14, 4008 RSC; (d) R. Pretorius, J. Olguin and M. Albrecht, Inorg. Chem., 2017, 56, 12410 CrossRef CAS PubMed; (e) M. E. Cucciolito, A. D'Amora, G. De Feo, G. Ferraro, A. Giorgio, G. Petruk, D. M. Monti, A. Merlino and F. Ruffo, Inorg. Chem., 2018, 57, 3133 CrossRef CAS PubMed; (f) J. C. Shi, N. Lei, Q. S. Tong, Y. R. Peng, J. F. Wei and L. Jia, Eur. J. Inorg. Chem., 2007, 2221 CrossRef CAS; (g) J. P. Byrne, P. Musembi and M. Albrecht, Dalton Trans., 2019, 48, 11838 RSC; (h) B. K. Keitz and R. H. Grubbs, Organometallics, 2010, 29, 403 CrossRef CAS PubMed; (i) W. H. Zhao, V. Ferro and M. V. Baker, Coord. Chem. Rev., 2017, 339, 1 CrossRef CAS; (j) Z. G. Zhou, Y. Y. Yuan, G. H. Xu, Z. W. Chen and M. Li, Prog. Chem., 2019, 31, 351 Search PubMed; (k) T. Shibata, H. Hashimoto, I. Kinoshita, S. Yano and T. Nishioka, Dalton Trans., 2011, 40, 4826 RSC.
- (a) A. S. Henderson, J. F. Bower and M. C. Galan, Org. Biomol. Chem., 2014, 12, 9180 RSC; (b) F. Tewes, A. Schlecker, K. Harms and F. Glorius, J. Organomet. Chem., 2007, 692, 4593 CrossRef CAS; (c) B. K. Keitz and R. H. Grubbs, Organometallics, 2009, 29, 403 CrossRef PubMed; (d) M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J.-P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213 CrossRef CAS PubMed.
- For selected examples on asymmetric Rh-catalysed hydrosilylation of ketones see: (a) K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613 CrossRef CAS; (b) W.-L. Duan, M. Shi and G.-B. Rong, Chem. Commun., 2003, 2916 RSC; (c) L. H. Gade, V. César and S. Bellemin-Laponnaz, Angew. Chem., Int. Ed., 2004, 43, 1014 CrossRef CAS PubMed; (d) N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Angew. Chem., Int. Ed., 2009, 48, 1609 CrossRef CAS PubMed; (e) J. Lee, H. Hahm, J. Kwak and M. Kim, Adv. Synth. Catal., 2019, 361, 1479 CrossRef CAS.
- Selected reviews: (a) P. J. Garegg, in Adv. Carbohydr. Chem. Biochem, ed. H. Derek, Academic Press, 1997, vol. 52, p. 179 Search PubMed; (b) M. Jacobsson, J. Malmberg and U. Ellervik, Carbohydr. Res., 2006, 341, 1266 CrossRef CAS PubMed.
- A. S. Henderson, S. Medina, J. F. Bower and M. C. Galan, Org. Lett., 2015, 17, 4846 CrossRef CAS PubMed.
- T. Nishiyama, Y. Kusumoto, K. Okumura, K. Hara, S. Kusaba, K. Hirata, Y. Kamiya, M. Isobe, K. Nakano, H. Kotsuki and Y. Ichikawa, Chemistry, 2010, 16, 600 CrossRef CAS PubMed.
- T. Buskas, P. J. Garegg, P. Konradsson and J. L. Maloisel, Tetrahedron: Asymmetry, 1994, 5, 2187 CrossRef CAS.
- The use of an identical aryl group for the C1 and C3 sterically differentiated NHC ligands would have been ideal, however, for proof of principle aryls compatible with the respective glycosylation and SNAr methodologies were initially used.
- For a review see: V. Benessere, M. Lega and F. Ruffo, J. Organomet. Chem., 2014, 771, 105 CrossRef CAS.
- T. V. RajanBabu, T. A. Ayers, G. A. Halliday, K. K. You and J. C. Calabrese, J. Org. Chem., 1997, 62, 6012 CrossRef CAS.
- M. T. Reetz and T. Neugebauer, Angew. Chem., Int. Ed., 1999, 38, 179 CrossRef CAS.
- L. Fang, L. Yan, F. Haeffner and J. P. Morken, J. Am. Chem. Soc., 2016, 138, 2508 CrossRef CAS PubMed.
- It is pertinent to note that given the pseudo-enantiomeric relationship between the C1 and C3 positions, a promising aryl moiety at C3 could also be mirrored at C1 by glycosylation with the appropriate phenol or thiol.
- Sulfur-containing molecules can act as ligands outright, however, we have no evidence for a Rh-S interaction in complex 12a. For a review on sulfur-containing ligands in asymmetric catalysis see: H. Pellissier, Tetrahedron, 2007, 63, 1297 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, and HPLC traces for novel compounds. See DOI: 10.1039/d0ob00155d |
| This journal is © The Royal Society of Chemistry 2020 |