
Advanced functional polymer materials
Kaojin
Wang
a,
Kamran
Amin
b,
Zesheng
An
 c,
Zhengxu
Cai
d,
Hong
Chen
e,
Hongzheng
Chen
f,
Yuping
Dong
d,
Xiao
Feng
g,
Weiqiang
Fu
d,
Jiabao
Gu
a,
Yanchun
Han
h,
Doudou
Hu
i,
Rongrong
Hu
a,
Die
Huang
a,
Fei
Huang
j,
Feihe
Huang
k,
Yuzhang
Huang
a,
Jian
Jin
l,
Xin
Jin
m,
Qianqian
Li
n,
Tengfei
Li
o,
Zhen
Li
n,
Zhibo
Li
p,
Jiangang
Liu
h,
Jing
Liu
i,
Shiyong
Liu
q,
Huisheng
Peng
r,
Anjun
Qin
a,
Xin
Qing
s,
Youqing
Shen
i,
Jianbing
Shi
d,
Xuemei
Sun
r,
Bin
Tong
d,
Bo
Wang
g,
Hu
Wang
k,
Lixiang
Wang
h,
Shu
Wang
t,
Zhixiang
Wei
b,
Tao
Xie
u,
Chunye
Xu
v,
Huaping
Xu
w,
Zhi-Kang
Xu
x,
Bai
Yang
c,
Yanlei
Yu
s,
Xuan
Zeng
y,
Xiaowei
Zhan
o,
Guangzhao
Zhang
z,
Jie
Zhang
a,
Ming Qiu
Zhang
aa,
Xian-Zheng
Zhang
y,
Xiao
Zhang
s,
Yi
Zhang
ab,
Yuanyuan
Zhang
g,
Changsheng
Zhao
ac,
Weifeng
Zhao
ac,
Yongfeng
Zhou
m,
Zhuxian
Zhou
i,
Jintao
Zhu
ad,
Xinyuan
Zhu
m and
Ben Zhong
Tang
*aae
c,
Zhengxu
Cai
d,
Hong
Chen
e,
Hongzheng
Chen
f,
Yuping
Dong
d,
Xiao
Feng
g,
Weiqiang
Fu
d,
Jiabao
Gu
a,
Yanchun
Han
h,
Doudou
Hu
i,
Rongrong
Hu
a,
Die
Huang
a,
Fei
Huang
j,
Feihe
Huang
k,
Yuzhang
Huang
a,
Jian
Jin
l,
Xin
Jin
m,
Qianqian
Li
n,
Tengfei
Li
o,
Zhen
Li
n,
Zhibo
Li
p,
Jiangang
Liu
h,
Jing
Liu
i,
Shiyong
Liu
q,
Huisheng
Peng
r,
Anjun
Qin
a,
Xin
Qing
s,
Youqing
Shen
i,
Jianbing
Shi
d,
Xuemei
Sun
r,
Bin
Tong
d,
Bo
Wang
g,
Hu
Wang
k,
Lixiang
Wang
h,
Shu
Wang
t,
Zhixiang
Wei
b,
Tao
Xie
u,
Chunye
Xu
v,
Huaping
Xu
w,
Zhi-Kang
Xu
x,
Bai
Yang
c,
Yanlei
Yu
s,
Xuan
Zeng
y,
Xiaowei
Zhan
o,
Guangzhao
Zhang
z,
Jie
Zhang
a,
Ming Qiu
Zhang
aa,
Xian-Zheng
Zhang
y,
Xiao
Zhang
s,
Yi
Zhang
ab,
Yuanyuan
Zhang
g,
Changsheng
Zhao
ac,
Weifeng
Zhao
ac,
Yongfeng
Zhou
m,
Zhuxian
Zhou
i,
Jintao
Zhu
ad,
Xinyuan
Zhu
m and
Ben Zhong
Tang
*aae
aState Key Laboratory of Luminescent Materials and Devices, Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, Center for Aggregation-Induced Emission, South China University of Technology, Guangzhou 510640, China
bCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China
cState Key Laboratory of Supramolecular Structure, and Materials, College of Chemistry, Jilin University, Changchun 130012, China
dBeijing Key Laboratory of Construction Tailorable Advanced Functional Materials and Green Applications, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China
eCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, China
fMOE Key Laboratory of Macromolecular Synthesis and Functionalization, State Key Laboratory of Silicon Materials, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
gSchool of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China
hState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun 130022, China
iCenter for Bionanoengineering and College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
jState Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
kState Key Laboratory of Chemical Engineering, Center for Chemistry of High-Performance & Novel Materials, Department of Chemistry, Zhejiang University, Hangzhou 310027, China
lCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China
mSchool of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China
nDepartment of Chemistry, Wuhan University, Wuhan 430072, China
oDepartment of Materials Science and Engineering, College of Engineering, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Peking University, Beijing 100871, China
pCollege of Polymer Science and Engineering, Qingdao University of Science and Engineering, Qingdao 266042, China
qCAS Key Laboratory of Soft Matter Chemistry, Hefei National Laboratory for Physical Sciences at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, China
rState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, and Laboratory of Advanced Materials, Fudan University, Shanghai 200438, China
sDepartment of Materials Science & State Key Laboratory of Molecular Engineering of Polymers, Fudan University, Shanghai 200433, China
tInstitute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
uState Key Laboratory of Chemical Engineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
vHefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, China
wDepartment of Chemistry, Tsinghua University, Beijing 100084, China
xMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
yKey Laboratory of Biomedical Polymers of Ministry of Education & Department of Chemistry, Wuhan University, Wuhan 430072, China
zDepartment of Polymer Science and Engineering, South China University of Technology, Guangzhou 510640, China
aaKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, GD HPPC Lab, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China
abPCFM Lab, GD HPPC Lab, Guangdong Engineering Technology Research Center for High-performance Organic and Polymer Photoelectric Functional Films, State Key Laboratory of Optoelectronic Materials and Technologies, School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou 510275, China
acCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China
adSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
aeDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong 999077, China. E-mail: tangbenz@ust.hk
First published on 31st March 2020
Abstract
The research on advanced functional polymers is being driven by the fast-growing demand for new functional materials that can be used in revolutionary technologies. Polymers can be endowed with functions by using certain special preparation methods or by introducing functional groups or fillers into materials. These functions are either intrinsically possessed by materials or actuated by external stimuli. In this review, we present an overview of the recent developments made in the research hotspots of functional polymers, encompassing polymerization methodologies, luminescent polymers, photovoltaic polymers, other electronic and optical polymers (including low-k polyimides and second-order nonlinear optical polymers), biorelated polymers (particularly those for biomedical applications), supramolecular polymers, stimuli-responsive polymers, shape-memory polymers, separation polymer membranes, energy storage polymers, and covalent organic framework polymers. The concepts, design strategies at the molecular level, preparation methods, classifications, properties, potential applications, and recent progress made in such polymers are summarized. Challenges and future perspectives of each type of functional polymers are also addressed, including research efforts regarding the design and fabrication of functional polymers for serving the increasing demand for new materials.
1. Introduction
Conventional polymers refer to materials that are used in our daily lives, such as plastics, artificial fibers, rubbers, and paint coatings. With the development of modern society, however, special functions are required in certain fields, where conventional polymers cannot serve the full needs, thereby promoting the development of novel polymers with specific functions. Such polymers are called advanced functional polymers that have potential applications in many fields such as energy harvesting and storage, wireless communication industry, biomedicine, oil/water or gas separation, and intelligent and bionic industries.1,2 Functional polymer science, developed in the 1960s, is an interdisciplinary field encompassing polymer chemistry, polymer physics, materials science, biology, energy, nanoscience, and environment.3 Besides the mechanical properties of conventional polymeric materials, advanced functional polymeric materials can also possess chemical reactivity, photosensitivity, electrical conductivity, catalytic properties, biocompatibility, biological activity, pharmacological properties, selective separation, and energy conversion. They have become an important cornerstone of the modern industry and advanced technology. A considerable amount of attention has been paid toward the development of new functional polymers to meet the major demand necessary for the development of modern society.Advanced functional polymers follow their own characteristics and laws. The first is that the syntheses of monomers are usually complex and diverse, which involve organic chemistry and metal catalysis. Second, new polymerization reactions, methods, mechanisms, and catalysts are needed to be developed because of the introduction of functional monomers. Third, their functions are not only related to the chemical structures of the polymer chains but also closely related to their aggregate states. Therefore, it is necessary to combine the synthesis, processing, and morphology of polymers to obtain excellent functions. The cutting-edge development of other disciplines can be used to create new materials and improve the performance of the existing polymeric materials because the field of functional polymeric materials is an interdisciplinary subject.
Advanced functional polymers have drawn much attention from researchers in many fields. As a result, a number of review papers have been published with different emphases on this particular subject. Almost all of them involve specific aspects of advanced functional polymers, such as conductive polymers,4 polymer micelles or vesicles for drug and gene delivery,5,6 polymer gels,7,8 polymer acceptors,9–11 or/and donors12 for use in organic solar cells (OSCs), biomedical polymers,13,14 shape-memory polymers (SMPs),15–17 stimuli-responsive polymers,18,19 separation polymer membranes,20,21 luminescent polymers,22,23 covalent organic framework (COF) polymers,24 and supramolecular polymers (SPs).25 All the above reviews only focused on one specific aspect of functional polymers. To the best of our knowledge, no reviews have summarized and reported all the research hotspots of advanced functional polymers. For a better understanding of advanced functional polymers in a comprehensive manner, a review of functional polymeric materials is necessary. Owing to advanced functional polymers covering too many topics, we mainly present research directions studied by the authors’ own groups in this review. We start by discussing polymerization methodologies of advanced functional polymers and then focus on several research directions involving functional polymers, such as luminescent polymers, photovoltaic polymers, other electronic and optical polymers, biorelated polymers, SPs, stimuli-responsive polymers, SMPs, separation polymer membranes, energy storage polymers, and COF polymers.
2. Polymerization methodologies
The key element of a functional polymer is its preparation methodology, which can facilitate the creation of new functional polymers and improve their performance. Much effort has been devoted toward the development of new polymerizations, which are not only owing to research interests but also because of various potential applications of polymers. Theoretically, polymerizations can be classified into chain-growth and step-growth polymerizations. Chain-growth polymerization, i.e., addition polymerization, requires the initiation of a monomer to begin the chain-growth process, which involves the addition of monomers to growing free radicals or cationic or anionic chains. As the name implies, the chain of step-growth polymerization grows in a stepwise manner. Condensation polymerization and alkyne-based click polymerization usually exhibit the features of step-growth polymerization. Ring-opening polymerization (ROP) possesses many features of chain-growth polymerization, but it may also exhibit some features of step-growth polymerization. Free-radical polymerizations usually comprise four processes: initiation, propagation, chain transfer, and termination. If chain transfer and termination can be minimized during the process of polymerization, the resultant polymer may be controlled to a certain level. The corresponding polymerization is called controlled radical polymerization (CRP).In this section, the research content includes the preparation of new functional monomers and the development of new polymerization reactions. It is essential to develop a polymerization reaction featuring advantages such as simplicity, efficiency, mild conditions, good selectivity, atomic economy, and easily available raw materials for the preparation of polymeric materials with newer structures and functions. Due to space limitations, we will discuss several representative polymerizations for the preparation of functional polymers: (1) CRP (chain-growth polymerization) for the preparation of olefin functional polymers; (2) alkyne-based click polymerization (step-growth polymerization); (3) multicomponent polymerization (MCP) based on triple-bond building blocks used for the synthesis of alkyne functional polymers (step-growth polymerization); and (4) ROP (possessing the features of both chain- and step-growth polymerizations) for the preparation of degradable and renewable polymer materials by the use of new catalysts and renewable biomonomers.
2.1 CRP
CRP or reversible-deactivation radical polymerization (RDRP) has reshaped the field of polymer chemistry and materials science by allowing nonexperts to access well-defined polymers that can be prepared under mild conditions for a myriad of applications. CRP relies on either reversible termination or degenerative chain transfer to control the equilibrium between dormant and active radicals; polymerization control is realized by biasing the equilibrium toward the dormant species. As such, the concentration of active radicals is minimized, and side reactions such as bimolecular termination and irreversible chain transfer are diminished. Although CRP techniques are not living polymerizations, they do exhibit features analogous to living polymerization (e.g., living anionic polymerization), such as pseudo-first-order polymerization kinetics, predictable molecular weight with a narrow distribution, and high end-group fidelity. Despite having been extensively studied for more than 20 years, some exciting new trends have recently emerged, such as externally regulated CRP and enzyme-participated CRP. These advances provide new opportunities to control polymer architectures and functions, as well as the development of sustainable pathways for synthesizing materials under greener conditions.External stimuli, such as light, electric field, or ultrasound, can provide useful means to regulate the activation or alteration of the polymerization rate of CRP by switching the stimuli ON/OFF or changing the input intensity/magnitude. In 2011, Matyjaszewski et al. reported an electrochemically mediated atom transfer radical polymerization (eATRP) process, whereby applying suitable potential reduces the air-stable Cu(II)/ligand deactivator to the Cu(I)/ligand activator to induce polymerization, and the polymerization rate can be modulated by varying the applied potential to control the activator/deactivator ratio.27 Inspired by the visible-light photoredox catalysis for organic transformations, Fors and Hawker et al. developed visible-light-mediated ATRP using fac-[Ir(ppy)3] as the photoredox catalyst, which allowed the photomodulation of polymerization kinetics by switching the light ON/OFF.28 However, transition-metal photocatalysts are expensive and may potentially contaminate the obtained polymers: a similar problem was found in traditional ATRP when using a Cu/ligand catalyst. To address this challenge, the same team further developed a metal-free ATRP using a highly reducing organic photocatalyst, namely, PTH (Fig. 1).26 In contrast to fac-[Ir(ppy)3], PTH was compatible with a broader range of functional monomers, such as 2-(dimethylamino)ethyl methacrylate. The field of using organic photocatalysts for conducting metal-free or organic-compound-catalyzed ATRP (O-ATRP) was further developed by Miyake et al., who introduced diaryl dihydrophenazine as another class of strongly reducing photoredox catalysts.29 On the basis of computational studies, it was suggested that the ability to access photoexcited intramolecular charge transfer states is important in minimizing fluorescence and facilitating electron transfer from the photoexcited photocatalyst to the substrate, thereby improving the efficiency and control of ATRP.30 However, the light absorption wavelength of existing organic photoredox catalysts for ATRP is limited to ultraviolet or blue light; therefore, significant efforts have been devoted toward the design and synthesis of long-wavelength-absorbing photoredox catalysts, e.g., by extending the conjugation length.31,32 Photoredox-catalyzed reactions have also been coupled with another type of widely used CRP, namely, reversible addition–fragmentation chain transfer (RAFT) polymerization, which was pioneered by Boyer et al. in 2014 and was called the photoinduced electron/energy transfer RAFT (PET-RAFT) (Fig. 2).33–40 PET-RAFT is extremely versatile as evident by the variety of photocatalysts (metal complexes, organic molecules, biocatalysts, and nanoparticles (NPs)) and therefore a wide range of wavelengths covering the visible as well as near-infrared (NIR) spectra, a diverse families of monomers, and various polymerization media. In particular, some of these photocatalysts are capable of reducing triplet oxygen into its singlet state, thereby enabling polymerizations to be conducted with oxygen tolerance.33 This trait has been harnessed to facilitate flow polymerization and high-throughput synthesis at lower volumes.41
 | ||
| Fig. 1 Metal-free ATRP mediated by 10-phenothiazine (PTH) as a photoredox catalyst. Adapted from ref. 26 with permission. Copyright (2014) American Chemical Society. | ||
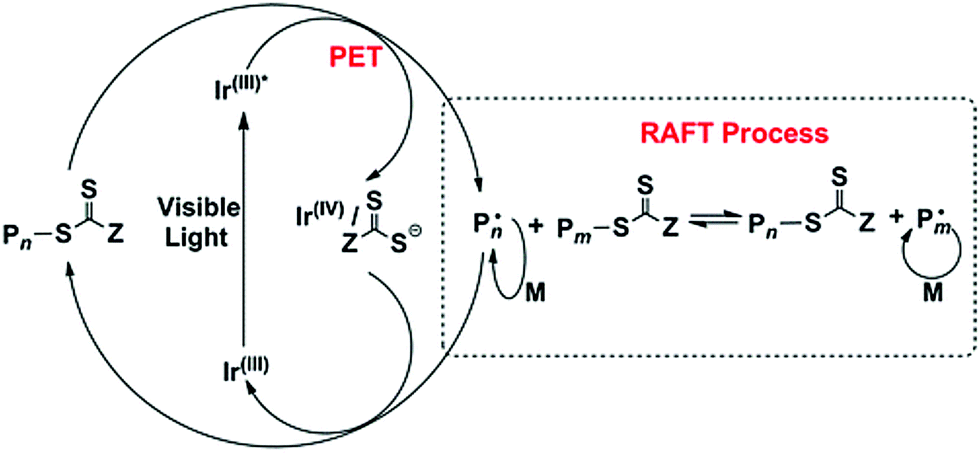 | ||
| Fig. 2 Mechanism of PET-RAFT using fac-[Ir(ppy)3] as a photoredox catalyst. Adapted from ref. 33 with permission. Copyright (2014) American Chemical Society. | ||
Despite being highly versatile, only a few photocatalysts have been employed for aqueous polymerization. A limitation of aqueous polymerization when using photoredox catalysis is that the catalysts typically exhibit large conjugation and therefore tend to aggregate in water, which, in turn, reduces their performance. To address this problem, An et al. introduced a host–guest strategy to improve water solubility and reduce the aggregation of photoredox catalysts. Enhanced polymerization rates were demonstrated with respect to cucurbit[7]uril (CB[7]) binding with the photocatalyst, Zn(II) meso-tetra(4-naphthalylmethylpyridyl) porphyrin.42 This host–guest strategy is simple and effective with minimal synthesis efforts and allows tuning the photochemical properties of supramolecular photocatalysts.
The rapid progress in visible-light-controlled CRP has offered exciting opportunities for the preparation of advanced materials under biologically friendly conditions. A notable example along this line was the work by Hawker and Soh et al.,43 who reported a cytocompatible strategy using Eosin Y as the photocatalyst for directly grafting high-density structurally defined functional polymers from living cell surfaces with high cell viability. This advancement was enabled by fast polymerization kinetics using a biocompatible polyethylene glycol (PEG)–acrylamide monomer and equimolar chain transfer agent and cocatalyst, which resulted in conversion of <30% within 5 min. In addition, ultrasound has been investigated as a mechanical stimulus to modulate the CRP, involving both ATRP and RAFT.44,45
Further, we have witnessed significant interest in biocatalyzed CRP in recent years, which promises greener pathways for precision polymer synthesis owing to the mildness and high efficiency of enzymatic catalysis.47,48 The enzymes that have been explored to control radical polymerizations are mainly oxidoreductases such as metalloproteins or flavoproteins. Metalloproteins containing a transition-metal complex as the cofactor have been examined as ATRP catalysts given their similar properties to traditional ATRP catalysts (e.g., Cu/ligand complex). However, metal complexes that are tightly bound to the protein matrix in metalloproteins can yield a catalyst with much lower toxicity and that is more robust toward functional monomers. For example, the polymerization of monomers with complexing capabilities toward transition metals is difficult to control by traditional ATRP. In a notable recent work, Bruns et al. reported the controlled polymerization of N-vinylimidazole under aqueous ATRP conditions catalyzed by laccase, a multi-copper-containing oxidoreductase.49 Laccase could be quantitatively separated from the polymerization solution via ultracentrifugation fitted with filter membranes having a suitable molecular weight cutoff. The ability to access low-dispersity functional poly(N-vinylimidazole) without metal contamination permits its applications as biomaterials and energy materials. Some peroxidase metalloproteins such as horseradish peroxidase (HRP) catalyze the oxidation of electron-rich compounds and generate radical intermediates, which can be harnessed to initiate RAFT polymerization or activate ATRP by reducing a Cu(II)/ligand deactivator into a Cu(I)/ligand activator. This strategy was first introduced by An et al. in RAFT polymerization initiated by HRP/hydrogen peroxide/acetylacetone—a three-component system.50 Predictable molecular weights ranging from 5 × 103 to 520 × 103 g mol−1, low narrow molecular weight distributions, and pseudo-first-order kinetics were observed for the polymerizations of various monomers in solution, dispersion, and biological media. Utilizing a biological Fenton reaction between hemoglobin and hydrogen peroxide to generate hydroxyl radicals, Qiao et al. reported a very interesting blood-catalyzed RAFT polymerization process, which may permit in vivo cell engineering using synthetic polymers.51 One of the grand challenges of CRP is its oxygen sensitivity. Recently, this challenge has been successfully tackled by the use of flavin-containing oxidases, which can effectively remove dissolved oxygen from the polymerization solution, allowing CRP to be conducted without prior degassing via traditional inert gas purging or freeze–pump–thaw operations. Since the first demonstration of oxygen-tolerant RAFT enabled by glucose oxidase (GOx) deoxygenation by Stevens et al.,52 applying oxidase to enable oxygen-tolerant RAFT and ATRP has attracted significant attention because this elegant strategy not only simplifies the synthesis operation but facilitates combinatorial synthesis methods to be developed, which is expected to play a key role in functional material discovery by high-throughput structure–property relationship screening.53–56 Remarkably, enzymatic cascade catalysis that involves oxidase deoxygenation and peroxidase radical generation has been demonstrated to be an elegant and powerful strategy for the efficient synthesis of well-defined polymers with oxygen tolerance. An et al. reported the use of GOx–HRP dual enzymes for controlled RAFT polymerization under ambient conditions, in which GOx deoxygenation provided hydrogen peroxide in situ for the HRP oxidation of acetylacetone to initiate well-controlled RAFT polymerization.50 Later on, inspired by the oxidase-peroxidase cascade catalysis found in root fungi, they reported a new cascade involving P2Ox and HRP for the efficient synthesis of decablock copolymers in open vessels and ultrahigh-molecular-weight polymers (up to 2.3 × 106 g mol−1) in close vessels without prior deoxygenation (Fig. 3).46 The GOx–HRP cascade was also employed by Matyjaszewski et al. for oxygen-tolerant ATRP, in which the radicals generated by the cascade catalysis was used to reduce Cu(II)/ligand to activate the polymerization (Fig. 4).57
 | ||
| Fig. 3 Pyranose oxidase (P2Ox)–HRP enzymatic cascade catalysis for the synthesis of multiblock and ultrahigh-molecular-weight (co)polymers with oxygen tolerance. Adapted from ref. 46 with permission. Copyright (2017) Wiley-VCH. | ||
 | ||
| Fig. 4 “Oxygen-fueled” ATRP. Adapted from ref. 57 with permission. Copyright (2018) Wiley-VCH. | ||
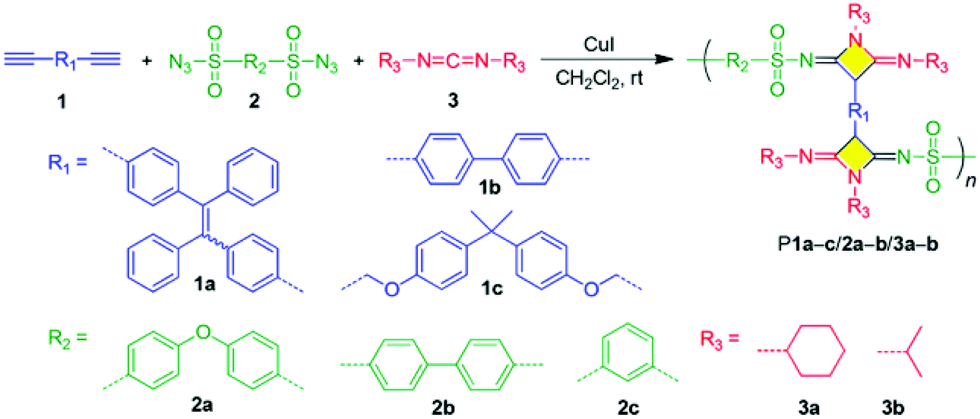
2.2 Alkyne-based click polymerization
Since it was coined by Sharpless et al. in 2001, click chemistry has inspired a lot of scientists in various fields.58 Attracted by the outstanding merits of click reactions, such as high efficiency, atom economy, mild reaction conditions, good functional tolerance, simple isolation procedure, and regio-/stereoselectivity, polymer chemists have made great effort to develop click reactions into versatile polymerization techniques, that is, click polymerizations.59,60 Until now, besides Cu(I)-catalyzed azide–alkyne click polymerization (CuAACP) evolved from a typical click reaction of Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC), numerous click polymerizations such as Diels–Alder click polymerization, thiol-based click polymerization, and amino-based click polymerization have also been established, which inherit the fascinating features of click reactions. In particular, alkyne-based click polymerizations combining the advantages of click reaction and the rich chemistry of alkynes have drawn considerable attention of polymer chemists.61 As shown in Fig. 5, diversified alkyne-based click polymerizations including azide–alkyne click polymerizations (AACPs), thiol–yne click polymerizations, and emerging amino–yne click polymerizations as well as hydroxyl–yne click polymerizations, have been exploited to fabricate functional polymers for specific applications.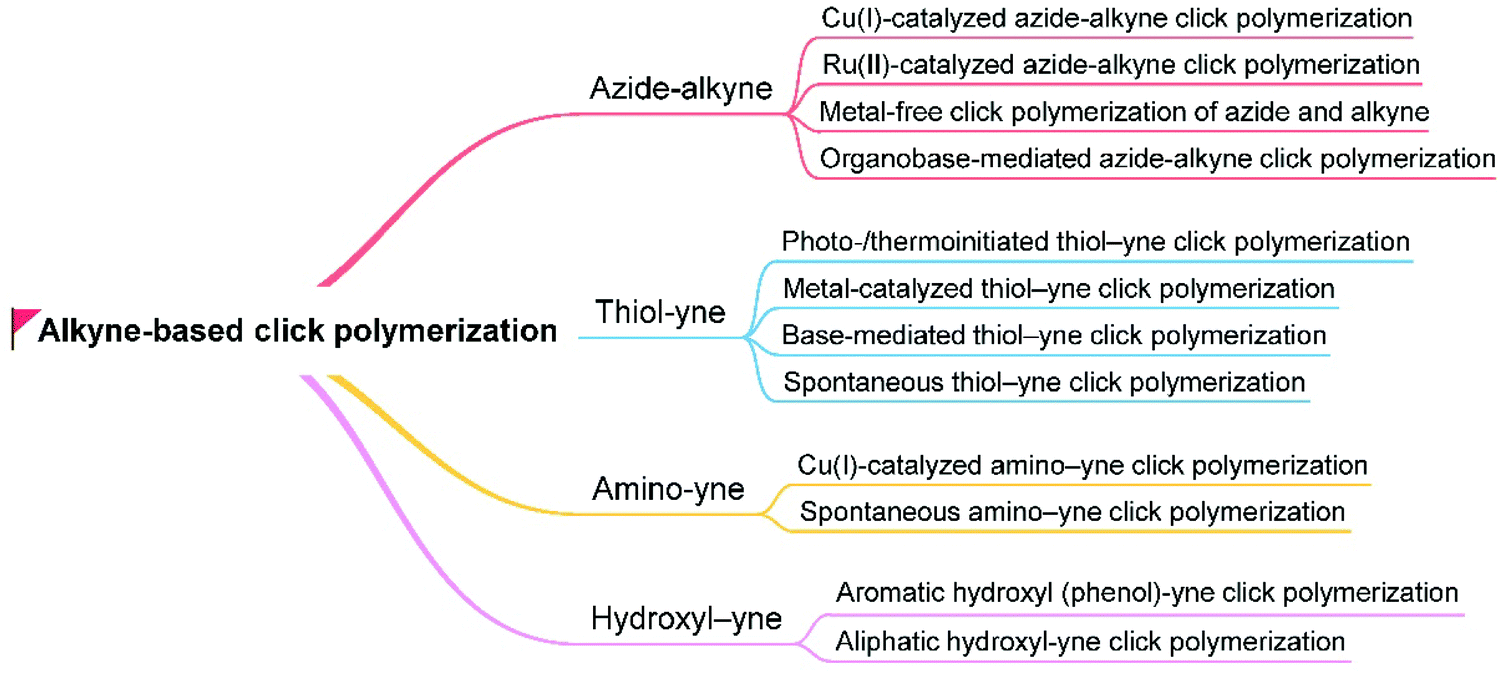 | ||
| Fig. 5 Alkyne-based click polymerization. | ||
Over the past few decades, CuAACPs—the most famous click polymerizations—have been widely applied in the construction of linear and hyperbranched polytriazoles (PTAs) with varied properties, such as amphipathy, self-assembly, self-healing, photoluminescence, aggregation-induced emission (AIE), chemosensing, ionic conductivity, light harvesting, optical nonlinearity, and catalytic activity (Fig. 6a).62 In recent years, considerable progress in the methodology has also been made in the development of CuAACPs. Generally, CuAACPs is a step-growth process. By taking advantage of the complexation between triazole rings and Cu(I) catalyst, Gao et al. successfully established a novel chain-growth CuAACP of multifunctional ABm (m ≥ 2) monomers and prepared a series of hyperbranched PTAs with high molecular weights (up to 430 × 103 g mol−1), low dispersities, and high degrees of branching (DB).63–66 Considering the fact that CuAACPs could only afford 1,4-regioregular PTAs, Qin and Tang et al. developed the ligand-controlled regiodivergent Ru(II)-catalyzed azide–alkyne click polymerization (RuAACP), which can selectively offer 1,4- or 1,5-regioregular PTAs (Fig. 6b).67,68 With this RuAACP in hand, they systematically investigated the structure–property relationship of regioregular PTAs. Although these transition-metal-catalyzed AACPs have become powerful tools for the preparation of functional PTAs with diverse structures, metal catalyst residues are difficult to be completely removed from PTAs owing to the strong coordination between the metallic species and nitrogen atoms in the triazole rings, which are detrimental to their applications in the areas of biomaterials and optoelectronic materials.69 The usage of supported Cu(I) catalysts for AACPs can reduce the metal residue contents in the resultant PTAs, which stimulates the development of the metal-free click polymerization (MFCP) of azides and alkynes without the risk of metal catalyst residues.70,71 With aroylacetylenes as activated alkyne monomers, Qin and Tang et al. successfully developed a thermal-initiated MFCP process for the preparation of 1,4-regioregular PTAs (Fig. 6c).72–75 To avoid the tedious and difficult synthesis of aroylacetylene monomers, readily available propiolate monomers with similar structures were then designed and applied in MFCP (Fig. 6d).76–78 In addition to these MFCPs of azides and activated alkynes, the MFCPs of alkynes and activated azide of 4,4′-diazidoperfluorobenzophenone were also established by them (Fig. 6e).79,80 They prepared several 1,4-regioregular PTAs with well-defined structures, which could be used in the areas of chemosensing, self-healing, photonic patterning, nonlinear optics, etc. Recently, Qin and Tang et al. reported the AACPs mediated by the organobases of tetramethylammonium hydroxide (NMe4OH) and phosphazene base (t-BuP4); consequently, 1,5-regioregular PTAs were successfully produced (Fig. 6f).81,82
 | ||
| Fig. 6 AACPs: (a) CuAACP, (b) RuAACP, and (c) MFCP of aroylacetylene and azide monomers; (d) MFCP of propiolate and azide monomers; (e) MFCP of 4,4′-diazidoperfluorobenzpphenone and alkyne monomers; and (f) organobase-mediated AACPs. | ||
Another emerging alkyne-based click polymerization reaction is thiol–yne click polymerization, which can proceed in the bis-addition or mono-addition manner to afford polythioethers or poly(vinyl sulfide)s (PVSs).83 In general, the mechanism of thiol–yne click polymerizations can be categorized into three types, namely, radical-initiated, transition-metal-catalyzed, and base-mediated processes. Inspired by the original work of Perrier et al. in 2009,84 photo-/thermo-initiated thiol–yne click polymerizations, typical radical-initiated thiol–yne click polymerizations, and those occurring through a bis-addition process, were widely investigated and applied in the preparation of hyperbranched polythioethers with high DB and various other properties, such as high refractivity, biodegradability, and liquid crystallinity (Fig. 7a). By using disubstituted aromatic alkynes as monomers, Voit et al. successfully exploited a novel thermo-initiated thiol–yne click polymerization reaction, which proceeded via a mono-addition process to produce PVSs (Fig. 7b).85 Diverse hyperbranched PVSs with high refractive indices were obtained, which have been applied in the fabrication of planar all-polymer photonic crystals and organic light-emitting diodes.86,87 Theoretically, the mono-addition of thiol to alkyne can occur in the form of Markovnikov or anti-Markovnikov addition, and the produced vinyl sulfides can be regio- and stereoisomers. To obtain PVSs with well-defined structures, Tang et al. developed a transition-metal-catalyzed thiol–yne click polymerization reaction with regiospecificity and stereoselectivity.88 By using the Rh(PPh3)3Cl catalyst, anti-Markovnikov additive PVSs with high stereoregularities (E-isomer contents up to 100%) could be obtained in high yields (Fig. 7c). Meanwhile, with propiolates as activated monomers and diphenylamine as a promoter, organobase-mediated thiol–yne click polymerization was also established for the construction of Z-stereoregular PVSs (Z-isomer contents up to 81.4%) (Fig. 7d).89 More recently, Qin and Tang et al. reported an efficient inorganic base-mediated thiol–yne click polymerization reaction in the presence of K3PO4, which afforded PVSs with Z-stereoregularities up to 100% (Fig. 7e).90 In particular, they also found that the click polymerization of thiols and aromatic alkynes could proceed in an anti-Markovnikov additive manner by simple mixing in tetrahydrofuran (THF) without the assistance of external stimuli and additional catalysts, indicating the spontaneity of this click polymerization reaction (Fig. 7f).91,92 A series of PVSs with linear and hyperbranched structures were prepared by this click polymerization reaction.
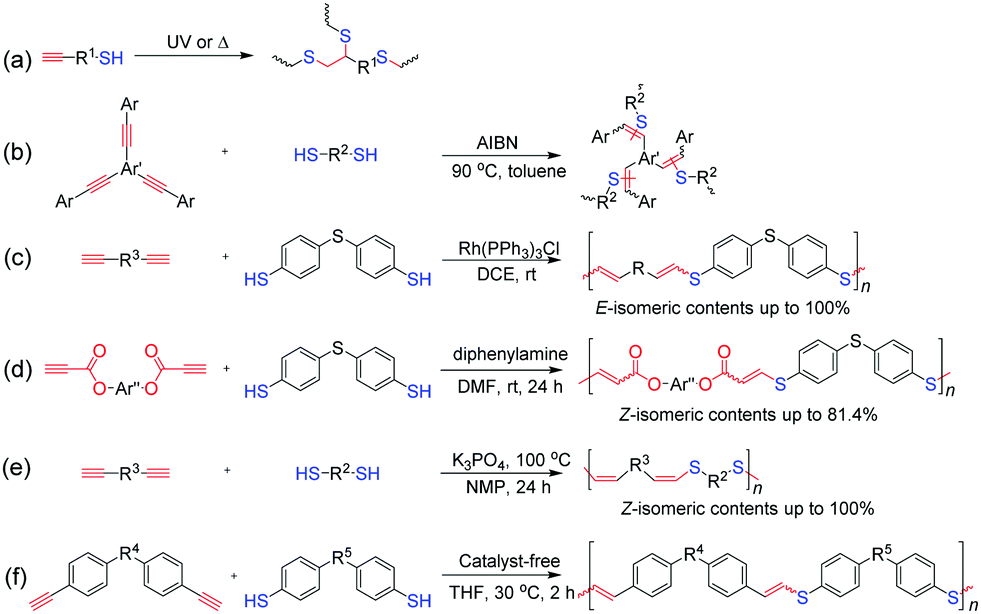 | ||
| Fig. 7 Thiol–yne click polymerizations: (a) photo-/thermo-initiated thiol–yne click polymerization, (b) thermo-initiated thiol–yne click polymerization based on disubstituted aromatic alkynes, (c) transition-metal-catalyzed thiol–yne click polymerization, (d) organobase-mediated thiol–yne click polymerization, (e) K3PO4-mediated thiol–yne click polymerization, and (f) spontaneous thiol–yne click polymerization. | ||
Besides the promising progress in AACPs and thiol–yne click polymerizations, new types of alkyne-based click polymerizations with great potential have also been developed in the past few years. In 2016, Qin and Tang et al. reported Cu(I)-catalyzed amino–yne click polymerization for the preparation of nitrogen-containing polymers (Fig. 8a).93 The polymerizations of diamines and diynes were performed in bulk under the catalyst of CuI in a regiospecific and stereoselective manner, producing Z-stereoregular polyenamines with high molecular weights (up to 13.5 × 103 g mol−1) in high yields. In the subsequent year, a simple and powerful spontaneous amino–yne click polymerization reaction was successfully developed for the first time by them (Fig. 8b). Poly(β-aminoacrylate)s with high molecular weights (Mw up to 64.4 × 103 g mol−1) and excellent yields could be produced by this polymerization reaction under mild conditions, and the resultant polymers possessed good solubility and high thermal stability. Furthermore, the polymers with tetraphenylethylene (TPE) moieties in the main chains were AIE-active and could be used in the sensing of explosives and specific lysosome labeling, indicating that this click polymerization could be widely applied in diverse areas.94
 | ||
| Fig. 8 Amino–yne click polymerizations: (a) Cu(I)-catalyzed amino–yne click polymerization, (b) propiolate-based spontaneous amino–yne click polymerization, and (c) ethynylsulfone-based spontaneous amino–yne click polymerization. | ||
Anti-Markovnikov additive poly(β-aminoacrylate)s with E-isomer contents of 100% were afforded in excellent yields after stirring the mixtures of dipropiolates and diamines in DCM for 3 h at room temperature. Taking advantage of its high efficiency and fantastic spontaneity, this amino–yne click polymerization reaction was successfully developed into a fancy intracellular polymerization reaction by the use of aroylacetylene instead of propiolate as the monomer.95 Interestingly, turn-on fluorescent imaging and in situ killing of cells could be realized by this in vivo amino–yne polymerization reaction. In spite of its several advantages, such propiolate-based spontaneous amino–yne click polymerization still has some shortcomings. For example, only aliphatic secondary diamines could polymerize with propiolate derivatives in a regio- and stereospecific manner. The polymerization lost its stereospecificity or even failed when aliphatic primary diamines or aromatic diamines were adopted to polymerize with propiolate derivatives, which limits its applications and promotes the development of new spontaneous amino–yne click polymerization. Considering the fact that the reactivity of ethynyl groups could be enhanced by increasing the electron-withdrawing ability of the adjacent groups, Qin and Tang et al. designed and synthesized highly active ethynylsulfone monomers in which the ethynyl groups were connected with stronger sulfonyl groups instead of ester groups.96 By the meticulous optimization of polymerization conditions, an ethynylsulfone-based spontaneous amino–yne click polymerization process was developed and regio-/stereoregular poly(β-aminovinylsulfone)s (PAVSs) could be obtained by polymerizing ethynylsulfones with various amines including aliphatic/aromatic primary/secondary ones (Fig. 8c). Because of the strong electron-withdrawing ability of sulfonyl groups, a dynamic property of these PAVSs could be observed, which endows them with degradability via an amine-exchange process.
To further expand the structures of polymers based on triple-bond building blocks, hydroxyl–yne click polymerizations have also been developed by Qin and Tang et al. in recent years.97 As shown in Fig. 9a, bis(aroylacetylene) and diphenol were polymerized in the presence of the Lewis base of 4-dimethylamino-pyridine (DMAP) in THF at room temperature, affording anti-Markovnikov additive poly(vinylene ether ketone)s (PVEKs) with high molecular weights (up to 35.2 × 103 g mol−1) and E-isomer contents as high as 100% at higher yields. Subsequently, regiospecific hydroxyl–yne click polymerization was exploited by the use of superbase t-BuP4 as the catalyst for the preparation of poly(vinyl ether)s (PVEs) (Fig. 9b).98 Both PVEKs and PVEs containing abundant acid-labile vinyl ether sequences in the polymer backbones exhibit degradability under strong acid conditions, suggesting the promising potential of hydroxyl–yne click polymerizations in the biomedical field.
 | ||
| Fig. 9 Hydroxyl–yne click polymerizations: (a) DMAP-catalyzed aromatic hydroxyl (phenol)–yne click polymerization and (b) superbase t-BuP4-catalyzed aliphatic hydroxyl–yne click polymerization. | ||
In short, the recent progress made in alkyne-based click polymerizations, including azide–alkyne, thiol–yne, amino–yne, and hydroxyl–yne click polymerizations and their subtypes, have been addressed. Moreover, the concept, mechanism, and design strategies of AIE-active polymers have also been introduced. All the polymerization methods and reactions summarized by us may provide researchers and engineers with additional approaches to fabricate desirable advanced functional polymers.
2.3 MCP based on triple-bond building blocks
MCP, a new strategy in polymer science, provides a series of advantages, such as easy preparation of monomers, mild reaction conditions, controllable polymer structures, and atom economy. It is well known that both isocyanide and alkyne are active chemical compounds with triple bonds. Isocyanide-based MCPs have been used to synthesize functional polyamides, namely, poly(ester amide)s, polyoxazolines, polyesters, and hyperbranched polymers (HBPs). Meanwhile, alkyne-based MCPs have also been recently reported by polymerizing diynes–primary amines–aldehydes and diynes–disulfonylazide–amino esters. Dialkyl acetylenedicarboxylate (DAAD) is a commercial chemical and has been widely used in cycloaddition reactions. Isocyanides can attack DAAD to afford zwitterionic species, which are readily trapped by carbon electrophiles and then converted into a pyrrole, furan, ketenimine, dihydropyridine, etc.Many limitations, such as narrow monomer scope, low conversion, and more possible side reactions, have hindered the development of MCP. The Cu(I)-catalyzed multicomponent reactions (MCRs) based on sulfonyl azide, alkyne, and third nucleophiles inherit the advantages of Cu(I)-catalyzed azide–alkyne cycloaddition and exhibit promising potential in polymer synthesis. Based on these reactions, Choi et al. were the first to develop the new click-reaction-based MCPs of diynes, sulfonyl azides, and diamines/diols.99,100 These polymerizations perfectly adapted to the “graft through” strategy: a series of graft and dendronized polymers with huge bulky side chains were afforded with excellent results by the use of monofunctional alkyne macromolecules.101 Tang et al. reported the facile and efficient click-polymerization-based MCP of diynes, disulfonyl azide, and salicylaldehyde, or o-hydroxylacetophenone catalyzed by Cu(I) and trimethylamine at room temperature (Fig. 10, Route 1). This polymerization approach inherits the remarkable merits of both MCRs and click reactions, such as simple operation, high reaction efficiency and isolation yields, mild reaction conditions, and common substrates. The resultant polymers with high molecular weights (Mw up to 64.6 × 103 g mol−1) possessed outstanding film-forming ability, high thermal stability, and good morphological stability. The obtained polymers with bright film emission and high photosensitivity could be facilely fabricated into well-resolved 2D and 3D patterns by treating their films with UV light.102
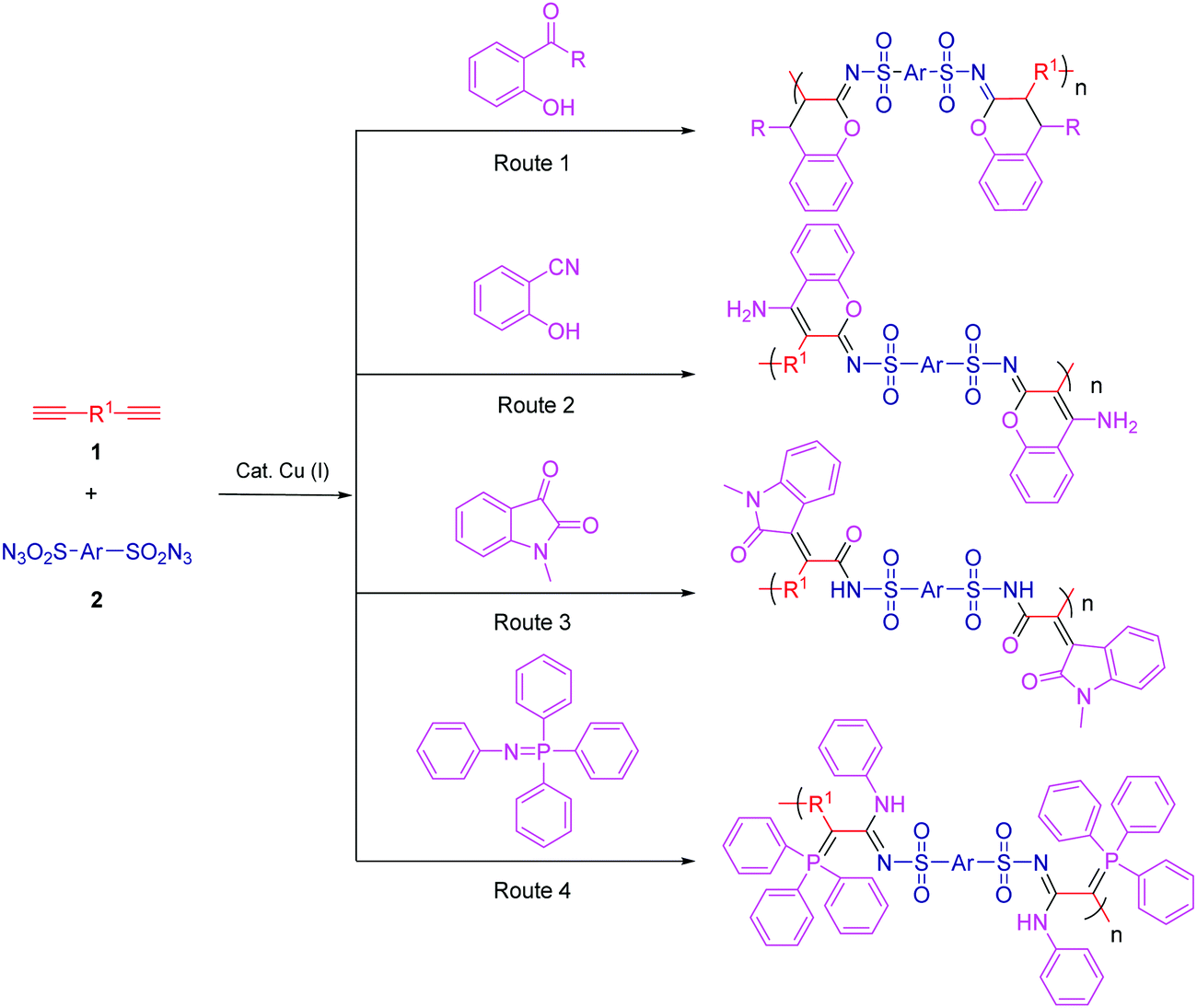 | ||
| Fig. 10 Copper(I)-catalyzed click-polymerization-based MCPs of diynes, disulfonyl azides, and third components. | ||
Based on the strategy of changing the third components, various click-reaction-based MCPs have been developed to fabricate functional polymers with complex structures. Hydroxybenzonitrile underwent a similar reaction mechanism to yield fused heterocycles-containing polymers (Fig. 10, Route 2).103 Because of the presence of multiple heteroatoms and amino groups in the polymer chains, the resultant polymers enjoyed antibacterial properties and potential sensitivity and selectivity of Ru3+ detection. In addition, N-protected isatins can act as a nucleophile for attacking the highly reactive intermediate to yield oxindole-containing poly(N-acylsulfonamide)s (Fig. 10, Route 3). These polymers enjoyed reversibly tunable hydrophilicity by undergoing structural changes when using LiOH and HCl.104 Heteroatoms can be conveniently introduced into the polymer chains via the third component. Iminophosphorane was utilized as the third monomer to develop a new MCP involving diynes and disulfonyl azides in order to fabricate N-, O-, S-, and P-containing heteroatom-rich polymers (Fig. 10, Route 4).105 The obtained polymers exhibited good solubility in polar solvents, good film-forming ability, and high refractivity. The possible interaction between the heteroatom and metal ion endowed the polymer with excellent selectivity in Pd2+ fluorescence detection. Moreover, this powerful MCP provides straightforward access to the synthesis of heteroatom-rich HBPs by the use of multifunctional alkynes and disulfonyl azides.106
When compared with five- or six-membered heterocycles, the in situ generation of small-ring heterocycles in polymer chains has encountered a formidable challenge due to difficulties during synthesis and inherent low stabilities. By replacing o-hydroxylacetophenone with carbodiimides, Tang et al. also developed a novel three-component polymerization route for fabricating functional polymers bearing heteroatom-rich multisubstituted azetidine rings (Fig. 11). This synthesis strategy exhibits advantages such as readily available monomers, simple operation, high efficiency, mild condition, environmental friendliness, and high atom economy. The strained rings in the polymer main chains enable them to efficiently undergo ring-opening post-modification under simple acidic conditions to transform into new polymers with amide and amidine units. It is noteworthy that the obtained azetidine-containing polymers showed obvious clusteroluminescence characteristic because the clustering of heteroatoms and conformational-rigidification-induced formation of efficient through-space conjugation played an important role in such an unconventional luminescence phenomenon.107
 | ||
| Fig. 11 Three-component polymerization using the small heterocycles route. Adapted from ref. 107 with permission. Copyright (2018) American Chemical Society. | ||
More recently, Tang et al. reported an efficient MCP strategy toward chalcogen-rich multifunctional poly(vinyl sulfone)s with regio-/stereoselectivity in high yields. This polymerization proceeded smoothly at room temperature under the protection of nitrogen and could yield functional poly(vinyl sulfone)s that could serve as effective fluorescent bioprobes for use in cellular imaging.108 Another significant work is the preparation of a series of multifunctional oxazine-containing polyheterocycles with AIE characteristics, which are generated in situ by metal-free one-pot A3 polymerizations of terminal diynes, dialdehydes, and ureas. These obtained polymers exhibited outstanding fluorescence response to protonation and deprotonation. Based on this, a fast-response reversible fluorescent sensor for ammonia can be used to detect biogenic amines and seafood spoilage.109
In 2014, Tang et al. explored the first multicomponent tandem polymerizations (MCTPs) for the synthesis of conjugated polymers (CPs), which could yield functional CP materials by the three-component reaction of alkyne, aroyl chloride, and ethyl 2-mercaptoacetate (Fig. 12, Route 1).110 After that, by combining the Sonogashira coupling reaction and other reactions, a series of one-pot, two-step, three-component coupling–addition tandem reactions and polymerizations were developed by them. For example, by utilizing the Sonogashira coupling reaction and the hydrothiolation reaction of electron-deficient alkynone intermediates, the multicomponent tandem reactions (MCTRs) and the corresponding MCTPs of alkynes, carbonyl chlorides, and aliphatic/aromatic thiols were investigated, which could afford sulfur-rich CPs with high molecular weights (Mw up to 59.1 × 103 g mol−1), high regioselectivity, and good stereoregularity in high yields (Fig. 12, Route 2).111 Through the one-pot sequential Sonogashira coupling of alkyne and carbonyl chloride, as well as the hydroamination of the internal alkyne, nitrogen-containing CPs with high regioselectivity and stereoselectivity could be obtained by the efficient MCTPs of alkynes, carbonyl chlorides, and primary amines (Fig. 12, Route 3).112 Heterocycle-containing conjugated polypyrazoles were also synthesized by the efficient MCTPs of alkyne, carbonyl chloride, and hydrazines/aromatic diynes through the combination of Sonogashira coupling–Michael addition–cyclocondensation reactions in a one-pot procedure (Fig. 12, Route 4).113 Furthermore, the one-pot MCTPs of alkynes, carbonyl chloride, and Fischer's base were developed to afford conjugated poly(diene-merocyanine)s with satisfactory molecular weights (Mw up to 10.9 × 103 g mol−1) and higher yields (up to 81%) (Fig. 12, Route 5).114 The MCTPs of N-(2-iodophenyl)-3-phenyl-N-tosylpropiolamide, aromatic terminal alkynes, and diamines were explored to afford poly(indolone)s with unique structures and high molecular weights (Mw up to 30.4 × 103 g mol−1) in high yields.115 In addition, by combining Glaser coupling–nucleophilic addition–heterocyclization–oxidation reactions, Hu and Tang et al. reported the four-step MCTPs of diyne, guanidine hydrochloride, DMSO, and O2, affording conjugated poly(pyrimidine)s with well-defined and tunable structures.116 These works have demonstrated the promising potential of MCTPs: various reactions with compatible conditions could be integrated into a single polymerization route and could afford polymers with diverse structures, which considerably extends the scope of such polymer structures.
 | ||
| Fig. 12 MCTPs of alkynes, carbonyl chlorides, and third components. | ||
Moreover, Hu and Tang et al. also developed the facile metal-free one-pot MCTPs of activated internal alkynes, aromatic diamines, and formaldehyde with high efficiency and enhanced convenience, which enabled the facile preparation of polyheterocycles with well-defined structures, regulated sequence, high molecular weights (Mw up to 69.8 × 103 g mol−1), and high yields (up to 99%) (Fig. 13).117 In these MCTPs, functional heterocycles were directly constructed from the polymerization of commercially available, inexpensive, simple monomers; further, the resultant polymer structures were inaccessible by other synthesis approaches. Moreover, disubstituted internal alkynes used as monomers could considerably enrich the monomer variety, product diversity, and functionalities.117 Based on this, they constructed four HBPs with different topological structures having unique advantages and strong designability of these MCTPs; this provides a synthesis platform for the preparation of hyperbranched polyheterocycles with diverse functionalities.118
 | ||
| Fig. 13 Metal-free MCTPs of alkynes, aromatic diamines, and formaldehyde. Adapted from ref. 117 with permission. Copyright (2017) American Chemical Society. | ||
Inspired by the small molecule reaction reported by other groups and experiences in the triple-bond chemistry field, Dong et al. developed four new methods for preparing heterocyclic-containing functional polymers based on the multicomponent cyclopolymerization (MCC) of isocyanides, DAAD, and third component in the presence of a metal-free catalyst, which have been committed toward the development of new functional materials based on triple-bond reactions.119,120 Indeed, a series of heterocyclic-containing functional polymers with good solubility and thermal stability were successfully prepared in high yields (Fig. 14). Therefore, MCC may be a powerful tool for fabricating new types of multifunctional polymers.
 | ||
| Fig. 14 MCCs based on isocyanides, DAAD, and third component. | ||
First, catalyst-free one-pot multicomponent spiropolymerization (MCS) was developed for spiropolymer synthesis by using diisocyanides and alkynes in the presence of CO2 as the monomer (Fig. 14, Route 1). The target spiropolymers contained 1,6-dioxospiro[4,4]nonane-3,8-diene as the spirostructural unit in the main chain with different types of spacer groups.121 As a kind of atom-economical polymerization process, this new MCS reaction can serve as the beginning of spiropolymer preparation and can have wide potential applications in materials science. This work inspired the use of dialdehydes instead of carbon dioxide to investigate the possibility of multicomponent cyclopolymerization (Fig. 14, Route 2). This approach afforded poly(amine-furan-arylene)s (PAFAs) with high molecular weights (Mw up to 76.4 × 103 g mol−1), comprising 3,4-substituted furan that was formed in situ during the polymerization process, with good thermal stability and film-processing properties. The single crystal of the model compound (MC) indicated that the carbonyl group on the furan ring and the adjacent amino group had an intramolecular hydrogen bond to enhance the conjugation of the main chain, which exhibited the characteristic of black materials that have high absorption property and high thermal stability.122 Recently, they extended this system to additional aromatic dialdehyde monomers, including those with other heterocyclic spacers (e.g., pyrimidine, quinoxaline, pyridine, and thiophene). A series of poly(furan-amine)s (PFAs) based on these aromatic heterocyclic dialdehydes were successfully prepared under catalyst-free polymerization conditions. Interestingly, these PFAs can be degraded into volatile products with low molecular weights under UV irradiation. Therefore, this MCC process can yield useful photodegradable materials, which may offer a new platform for the design of multifunctional PAFs.123
Second, a catalyst-free MCS route for the facile construction of heteroatom-containing spiropolymers has been developed by Dong et al. (Fig. 14, Route 3). Here, 4,7-bis[alkyl(aryl)imino]-2-phenyl-3-oxa-6-thia-1-azaspiro[4.4]nona-1,8-dienes as a structural unit was instantly formed by the conversion of benzoyl isothiocyanate into spirosegments. With respect to monomer versatility, procedure simplicity, and high atom economy of the spiropolymerization process, this catalyst-free catalyzed MCS process opens up newer opportunities for the preparation of advanced spiropolymer materials. They further replaced benzoyl isothiocyanate with 4,4′-bis(2-bromoacetyl)biphenyl and constructed polymers containing iminolactone arylene (PIAs) by using catalyst-free MCP (Fig. 14, Route 4). The results of this analysis indicated that PIAs exhibited good solubility, aggregation-enhanced emission (AEE) properties, and high refractive indices under visible light (400–800 nm). Moreover, in the presence of bromomethyl groups in the side chain, PIAs were endowed with better performance because of post-functionalization.124
The utilization of sulfur remains a challenge for the efficient and environmentally friendly transformation of byproducts into commercial offerings based on their abundant storage but limited consumption in the petroleum industry. Polysulfide anions with high nucleophile content generated from elemental sulfur and aliphatic amines can become involved in attacks on both alkynes and isocyanides to obtain sulfur-based functional groups. Hu and Tang et al. reported the efficient and metal-free MCPs of elemental sulfur, diynes, and aliphatic diamines in pyridine at 100 °C to obtain well-defined polythioamides with high molecular weights (Mw up to 127.9 × 103 g mol−1) (Fig. 15, Route 1).125 The resulting polythioamides exhibited unconventional luminescence behavior without any typical fluorophores, which can be attributed to the presence of “heterodox clusters” formed with numerous lone-pair-containing electron-rich heteroatoms. With the presence of sulfur in polymer chains, they possessed high refractivity index of 1.80–1.66 in a wide wavelength ranging from 400 to 1700 nm.
 | ||
| Fig. 15 MCPs based on elemental sulfur, aliphatic diamines, and diynes/isocyanide. Reproduced from ref. 125 and 126 with permission. Copyright (2015 and 2018) from American Chemical Society. | ||
Polythioureas have attracted considerable attention in many fields due to their special properties such as self-healing characteristic, excellent electrical performance, and metal removal ability. Inspired by the polymerization reaction discussed earlier, isocyanides were utilized to replace alkynes as monomers to develop another new element, namely, sulfur-based auto-catalyzed MCP comprising diisocyanides and aliphatic diamines for the synthesis of polythioureas (Fig. 15, Route 2).126 The analysis based on in situ IR technique indicated that the high reactivity of isocyanide endowed the polymerization with high efficiency and quick reaction at both ambient temperature and 100 °C, affording a series of high-molecular-weight polythioureas (Mw up to 242.5 × 103 g mol−1) with well-defined structures. Due to the high affinity and selectivity of thiourea to mercury(II), the obtained polythioureas exhibited high sensitivity and selectivity in mercury fluorescence detection and excellent removal efficiency (>99.99%) toward Hg2+ pollution, which implied that these sulfur-based materials have feasible potential applications in the recovery of heavy metals.
In summary, several types of MCPs based on triple-bond building blocks by Tang and Dong et al. are briefly introduced, including Cu(I)-catalyzed MCPs, MCTPs, MCCs, MCPs based on elemental sulfur, etc. All these MCPs may supply many useful tools for designing and fabricating advanced functional polymers.
2.4 ROP of biorenewable poly(γ-butyrolactone) (PγBL)
Preparation methods for olefin and alkyne polymers, which are nonrenewable and nondegradable, have been addressed earlier. In this subsection, the ROP of renewable monomers is addressed. Aliphatic polyesters have become a class of important biomaterials that have been widely used as drug delivery carriers, tissue engineering scaffolds, and food packaging materials because of their promising biocompatibility and biodegradability. In particular, the ROP of cyclic esters or lactones has been applied in the preparation of polyesters with well-defined structures on a large scale.127 Recently, PγBL has attracted considerable interest since γ-butyrolactone (γ-BL) can be obtained from succinic acid, which is the most promising biomass-derived compound to replace petroleum-derived chemicals.128,129 On the other hand, poly(4-hydroxybutyrate) (P4HB), chemically identical to PγBL, exhibits a suitable degradation rate in vivo and can avoid the undesirable accumulation of acidic degradation products as compared to other aliphatic polyesters. Remarkably, PγBL can be completely depolymerized back to its monomeric form of γ-BL by simple heating. Therefore, PγBL is a truly sustainable polymer, which is fully biorenewable and completely recyclable.130However, γ-BL has been ignored as a monomer in polymer science for a long time due to the presence of a five-membered ring with low strain energy and related unfavorable thermodynamics involved in the ROP process. In 2016, Hong and Chen et al. demonstrated a milestone work and successfully prepared high-molecular-weight PγBL via ROP at lower temperatures.130 PγBL with high molecular weight (up to 30.0 × 103 g mol−1) and high monomer conversion (up to 90%) could be fabricated for the first time by utilizing Ln[N(SiMe3)2]3 or yttrium-amide-catalyzed coordination ROP at −40 °C (Fig. 16a). The resulting PγBL possessed either linear or cyclic topology (Fig. 16b) depending on the catalyst/initiator nature and ratio. Subsequently, they demonstrated that the organic superbase of 1-tertbutyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)-phosphoranylidenamino]-2λ5,4λ5catenadi (t-BuP4, Fig. 16a) can also catalyze the ROP of γ-BL at lower temperatures.131 In this case, the superbase t-BuP4 can abstract the α-proton of the monomer to generate the reactive enolate species and therefore directly initiate the ROP of γ-BL. For example, conversion of 30.4% after 12 h could be achieved, and a PγBL sample with Mn of 26.4 kg mol−1 and Đ (Mw/Mn) of 1.79 was obtained when using t-BuP4 alone (1.0 mol% loading) in toluene at −40 °C. Moreover, a combination of alcohol with the initiator formed the ion pair [t-BuP4H+⋯OBn−] and led to an even more effective ROP system, as evidenced by the high monomer conversion (up to 90%) and high-molecular-weight PγBL (Mn up to 26.7 kg mol−1). Recently, Zhang et al. reported the dual organocatalyst t-BuP4/(thio)urea for the ROP of γ-BL with increased activity and enhanced selectivity in comparison to those obtained when using t-BuP4 alone.132 The turnover frequencies (TOFs) of these t-BuP4/(thio)urea pair-catalyzed processes were as high as 125 h−1, while the resultant PγBLs had molecular weights as high as 64.3 kg mol−1. Besides the homopolymerization of γ-BL, Hong and Chen et al. also reported the effective copolymerization of γ-BL with ε-CL or δ-VL using t-BuP4 as the catalyst.133 The copolymerization produced random copolyesters with high molecular weights (up to 135 kg mol−1) as well as an unprecedented level of γ-BL incorporation (up to 84.0 mol%). The successful synthesis of these copolyesters with high γ-BL incorporations of >50 mol% led to the discovery of the eutectic phase of the γ-BL/ε-CL copolymer with a eutectic temperature, Teu, of 11.0 °C and eutectic composition, Xeu, of 66.0 mol% γ-BL; therefore, the copolyester with this composition was a viscous liquid at room temperature, although the two constituent homopolyesters were semicrystalline solids.
 | ||
| Fig. 16 (a) Metal catalysts and organocatalysts for the ROP of γ-BL; (b) proposed mechanism for the ROP of γ-BL by La[N(SiMe3)2]3. Reproduced from ref. 130 and 131 with permission. Copyright (2016) from Nature Publishing Group. | ||
On the other hand, Li et al. reported that the cyclic trimeric phosphazene base (CTPB, Fig. 17a) was a highly efficient organocatalyst for the ROP of γ-BL and can be used to prepare well-defined PBL with high conversions (up to 98%) below −50 °C within 4 h.134 The resulting PγBLs have high molecular weights (up to 22.9 kg mol−1) and relatively narrow mass distributions (Đ = 1.27–1.50). In contrast to t-BuP4, CTPB alone could not activate the monomer and therefore showed no activity in the absence of alcohol, as indicated by the in situ NMR analyses. The structural analyses of a low-molecular-weight polymer obtained in the presence of BnOH by MALDI-TOF suggested that the linear polymers with BnO/H chain ends were the primary products. More recently, Li et al. successfully synthesized a series of PBL-b-poly(L-lactide) (PLLA) diblock copolyesters with different compositions via a one-pot method by the sequential ROP of γ-BL and L-LA in the presence of CTPB as the catalyst (Fig. 17c).135 Although the first ROP of γ-BL could not consume all the monomers, the unreacted γ-BL could not homopolymerize or copolymerize with L-LA after the addition of the L-LA/THF solution. The PBL-b-PLLA copolyesters showed improved thermal stability as compared to the PBL homopolymer according to the thermogravimetric analysis (TGA) results. For example, the TGA curve of PγBL53-b-PLLA97 displayed a one-step decomposition profile with Td,5% value of 326 °C and Td,max value of 368 °C, both of which are higher as compared to those of the PγBL homopolymer (Td,5% of 273 °C and Td,max of 334 °C). Differential scanning calorimetry (DSC) results revealed the microphase separation in the PγBL-b-PLLA block copolymer (BCP), in which two separate melting transitions were present. Amphiphilic copolymers with PγBL as the hydrophobic segment were also successfully prepared by the use of PEG as the macroinitiator and CTPB or sodium hydride as the catalyst (Fig. 17d).136 Both catalytic/initiating systems showed moderate control over the ROP of γ-BL and successfully produced PEG-b-PγBL diblock copolymers with varied molecular weights. A suitable activation temperature (−40 °C) was crucial to obtain moderate to high γ-BL conversions and satisfactory molecular weight of PEG-b-PγBL with CTPB as the catalyst. The self-assembly behavior of the obtained amphiphilic PEG-b-PγBL diblock copolymer was preliminarily investigated with dynamic light scattering (DLS) and transmission electron microscopy (TEM) modalities. The TEM results revealed that the amphiphilic PEG-b-PγBL copolymer could self-assemble into micelles and vesicles in water, making it to be a promising candidate as a drug delivery carrier. Given the advantages of PγBL mentioned above, amphiphilic PEG-b-PγBL is expected to exhibit superior performances as compared to other existing amphiphilic aliphatic polyesters in biomedical applications.
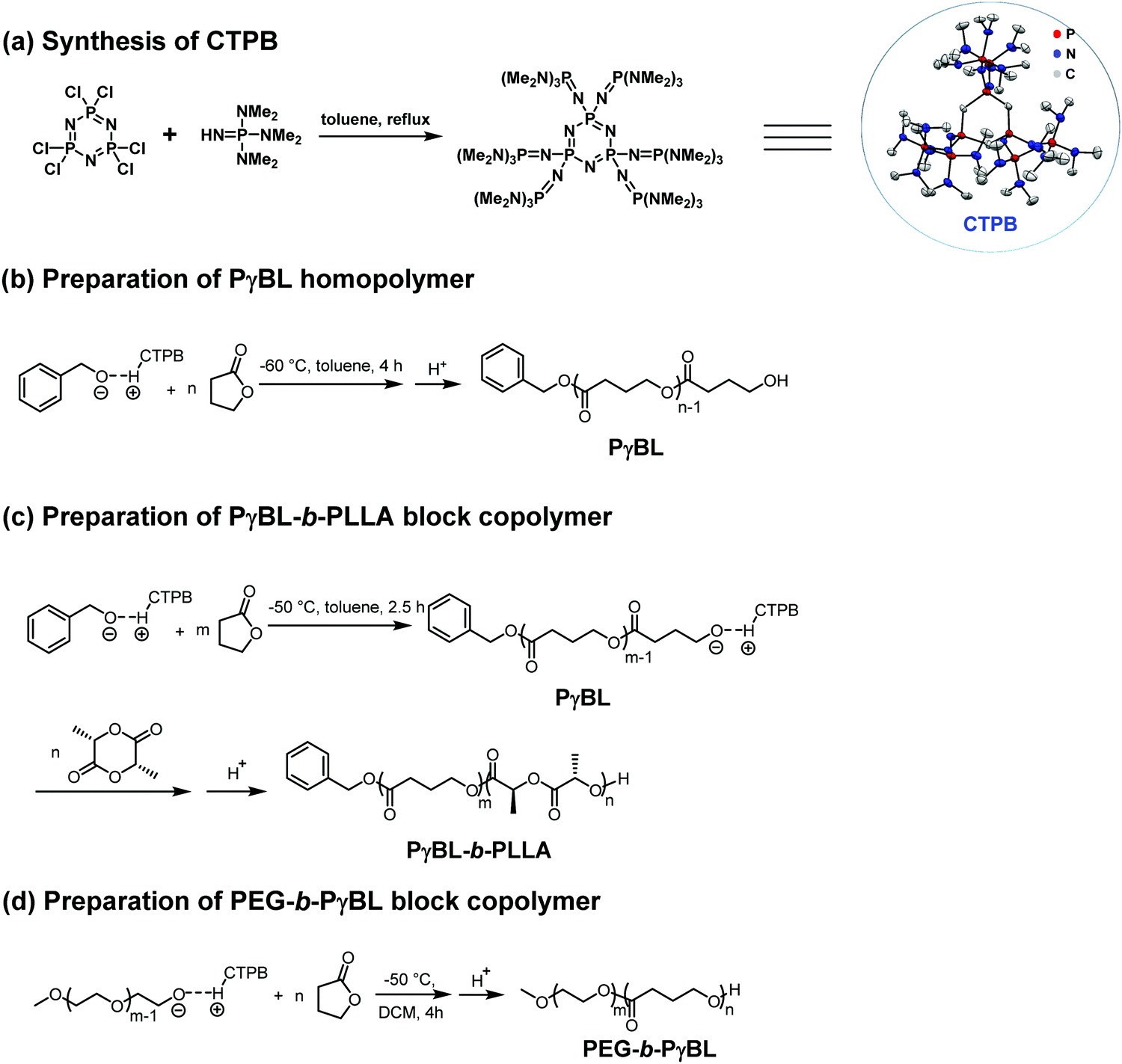 | ||
| Fig. 17 (a) Synthesis of CTPB and using it for the preparation of PγBL-containing (co)polymers (b–d). Reproduced from ref. 134–136 with permission. Copyright (2016, 2017, and 2018) from Wiley-VCH, Royal Society of Chemistry, and American Chemical Society. | ||
Although comprehensive studies have been conducted in the past few years, the development of new catalysts having both high activity and selectivity for the ROP of γ-BL remains a challenging subject. Dual catalytic systems would be a valuable alternative. The construction of PγBL-containing copolymers with complex but well-defined monomer sequences and varying topologies, e.g., graft or comb-like copolymers, should be paid more attention.
In this section, the developed preparation methods and polymerization reactions of functional polymer materials in the past few years are addressed, including CRP, alkyne-based click polymerization, MCP based on triple-bond building blocks, and ROP. Although new progresses in the preparation methods have been achieved in the past few years, particularly with regard to sequence-controlled polymerization and MCP, there are still a number of challenges in this field, such as building relationships between the polymer structures and functions, realizing controllable synthesis and function integration, and tailoring and modifying functional polymers. Therefore, more efforts should be directed toward the development of synthesis methodologies, allowing us to prepare more varied functional polymer materials, improve their performances, and reduce their costs of preparation.
3. Luminescent polymer materials
Luminescent materials are capable of converting other energy forms into light energy and subsequent light emission. They can be classified into three types: inorganics, organic molecules, and polymeric materials. Luminescent polymers can be classified into two subtypes based on the luminescent principle: fluorescent and phosphorescent polymers. According to the spin statistical distribution, only 25% of singlet (S) excitons and 75% of triplet (T) excitons are generated when a molecule in the ground state (S0) is excited by external light or electricity. For fluorescent polymers, only S excitons can be utilized, while T excitons can only be dissipated as thermal energy due to their spin-forbidden characteristic. Phosphorescent polymers can use the spin-forbidden triplet state under the action of the rotating orbital coupling of heavy metal atoms. Therefore, 100% internal quantum efficiency (IQE) can be achieved. Although many methods have been used to improve the device efficiency of fluorescent polymers, all of them are based on the use of T excitons. The most popular one is that the T excitons are converted into S excitons by an upconversion process, generating fluorescence (S1 → S0). Many theories have been developed in recent years, including thermally activated delayed fluorescence (TADF), hybridized local charge transfer (HLCT), and triplet–triplet annihilation (TTA). Theoretically, 100% IQE can be achieved when using the above methods. The actual device efficiency of fluorescent polymers obtained by the use of the above methods is usually lower than that of phosphorescent polymers. In this section, phosphorescent polymers such as carbonized polymer dots (CPDs) and electrophosphorescent polymers (PhPs) have been discussed. For fluorescent polymers, only AIE polymers are addressed; interested readers can refer to other reviews.137,1383.1 AIE polymers
AIE, a groundbreaking concept coined by Tang et al. in 2001, has attracted considerable attention because of its attractive applications in diverse areas.139 Researchers from all over the world have been working in this promising area, and significant progresses have been made. A large variety of AIE molecules with promising structural diversity have been developed, such as TPE, hexaphenylsilole (HPS), distyreneanthracene (DSA), and tetraphenylpyrazine (TPP) (Fig. 18).140–143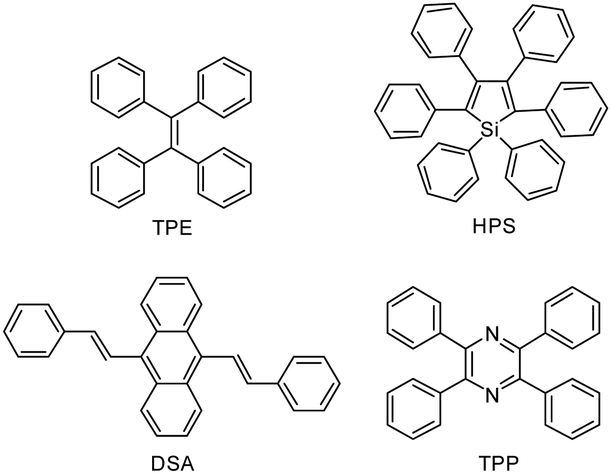 | ||
| Fig. 18 Chemical structures of typical AIE luminogens. | ||
When compared with AIE-active low-mass molecules, AIE polymers have been less explored.144 However, with the research of AIE polymers being constantly performed, an increased number of superior performances have been found, which, in turn, promote their development. According to the principles involved in the restriction of intramolecular motion as well as structural rigidification, the intramolecular motion of AIE units in polymer chains can be further restricted in comparison to their solid-state counterparts, leading to stronger emissions. Therefore, when luminogens with AIE characteristics (referred to as “AIEgens”) are involved in polymer chains, steric hindrance can be enhanced because of the long-chain segment of AIE polymers, hindering the free rotation of the luminogenic units. Evidently, intramolecular motion can be more easily restricted in polymers than that in low-mass molecules, resulting in a relatively more emissive solution state for polymers.145 Furthermore, AIE polymers possess many advantages over low-mass molecules, such as processability, ease of functionalization, structural diversity, and good thermal stability, facilitating their potential practical applications. Until now, a large number of AIE polymers have been reported, such as nonconjugated linear AIE polymers, conjugated linear AIE polymers, and nonlinear AIE polymers. In addition, the applications of AIE polymers in optoelectronic, fluorescent chemosensor, biological, and other fields have been summarized in some excellent reviews.146,147
Although various synthesis strategies have been used to fabricate AIE polymers, all of them have the same core principle, i.e., incorporating AIEgens into polymer structures, such as TPE, HPS, DSA, and TPP. Generally, two methods have been employed to incorporate AIEgens into polymers, i.e., post-functionalization and direct polymerization.147 The post-functionalization method can transform AIE-inactive polymers into AIE polymers via different polymer reactions. For example, AIEgens are usually linked to polymers as pendants or connectors via chemical reactions. Jin et al. converted the synthesized zwitterionic copolymer into an AIE polymer by grafting TPE molecules to the polymer side chains via pH-cleavable hydrazine bonds. The resultant AIE copolymer could self-assemble into spherical micelles and it could swell under acidic conditions after the cleavage of TPE, which may find applications in bioimaging and drug delivery.148 In addition, the direct polymerization method can also be used to fabricate AIE polymers via the polymerization of low-mass molecules. Tang et al. designed and prepared a series of AIE-active polymers by polymerizing aliphatic hydroxyl groups and AIEgen-containing diynes. The obtained PVEs exhibit typical AIE characteristics, i.e., stronger emissions in the aggregate state than that in the solution state, which can be used in the detection of explosives.98,149,150
Besides fluorescence, room-temperature phosphorescence (RTP) has also been reported in natural compounds and polymers, such as starch, cellulose, and chitosan. These natural macromolecules cannot emit in solutions, but they are capable of emitting bright phosphorescence in the solid states. Therefore, AIE polymers can emit not just fluorescence but also RTP, which is an important parameter for exploring the unknown territory of molecular luminescence.
3.2 CPDs
RTP has attracted increasing attention owing to its long-lived luminescence, large Stokes shift, and high signal-to-noise ratio, facilitating its applications in the fields of optoelectronics, photocatalytic reactions, molecular imaging, security aspects, chemical sensors, and so forth.151,152 Currently available RTP materials suffer from problems such as scarce resources, high cost, toxicity, limited species, and complex synthesis. Therefore, there is an urgent need to develop new classes of metal-free RTP materials.Carbon dots (CDs) have been recognized and proposed as luminescent nanomaterials since 2004. Until now, types of CDs have been continuously increasing, and they can be mainly divided as follows: graphene quantum dots, carbon quantum dots, and CPDs. Emerging CPDs can most likely resolve the abovementioned problems. In fact, CPDs are carbon-based nanodots with obviously polymeric, slightly graphitized, and inaccurate chemical structures. Their interior is a highly crosslinked network structure, and the outer part retains a large number of hydrophilic flexible polymer chains and functional groups (Fig. 19).
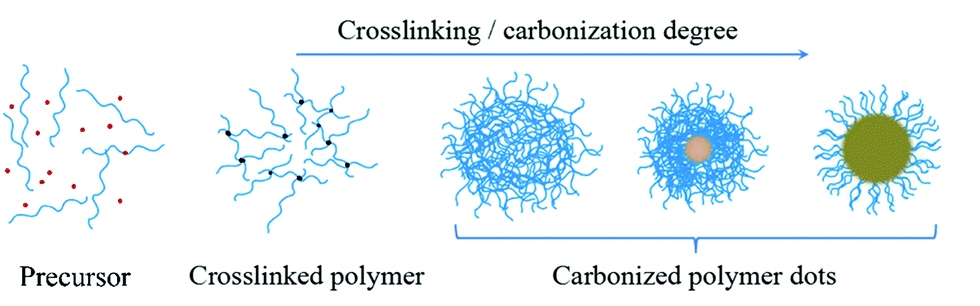 | ||
| Fig. 19 Schematic illustrations of the formation process and structure of CPDs. | ||
The formation of CPDs usually necessitates four processes, namely, dehydration, condensation, crosslinking, and carbonization, which enable them to inherit the molecular properties of their raw materials, as well as the properties of both polymers and quantum dots induced by the polymerization and carbonization reactions. More importantly, the abundant energy levels of CPDs increase the probability of intersystem crossing (ISC), and their covalently crosslinked framework structures considerably suppress nonradiative transitions, exactly following the generation principle of phosphorescence. Therefore, promising RTP can be probably achieved from CPDs by means of smarter design.
Yang et al. were the first to describe a feasible and facile route to prepare metal-free RTP materials by directly constructing CPDs via hydrothermal synthesis without the compositing of additional matrices.153 As a model system, CPDs synthesized from polyacrylic acid and ethylenediamine (EDA) exhibited unique RTP properties in air, as expected, with lifetime up to 658.11 ms. These CPDs synthesized with amide or imide structures, alongside the crosslink enhanced emission (CEE) effect, were confirmed to contribute toward the generation of RTP in CPDs by both contrast experiments and computational simulations (Fig. 20). First, chemical reactions easily occurred among the abundant functional groups. EDA serving as a crosslinker reacted with extensive dangling carboxyl groups on the polymer chains, forming luminescent centers and covalently crosslinked self-matrix frameworks. Second, effective crosslinking could shorten the distance between the functional groups. New distributions of energy levels can be formed by electron overlap from coupled luminescence units in nanoconfined space that can effectively reduce the energy gap (ΔEST) and facilitate the generation of intrinsic triplets. Third, the widespread existence of supramolecular interactions in the interior of CPDs can also decrease vibrations and rotations, thereby inhibiting any nonradiative relaxation. The reported guiding results demonstrate the potential of CPDs as a universal route for achieving effective metal-free RTP. Recently, they expanded the method from polycondensation to polyaddition, confirming the universality and feasibility of the hydrothermal crosslinking and carbonization processes for CPDs to achieve the RTP property.154
 | ||
| Fig. 20 Theoretical analysis of the CEE effect: (a) energy level diagrams of single luminescence units and (b) coupled luminescence units. (c) Schematic illustration of the CEE effect on RTP properties in CPDs. Reprinted from ref. 153 with permission. Copyright (2018) Wiley-VCH. | ||
Inspired by related reports, increasing number of researchers have noticed the advantages of CPDs on RTP performance and a range of significant studies have been rapidly performed. For example, Lin et al. successively achieved RTP from two similar CPD systems based on the aforementioned CEE effect. One involved the preparation of CPDs with RTP performance by the microwave irradiation of ethanolamine and phosphoric acid. Gram-scale CPDs and the longest RTP lifetime (1.46 s) could be achieved.155 Another study described the hydrothermal treatment of EDA and phosphoric acid for obtaining CPDs. The as-synthesized CPDs could realize the conversion from fluorescence (180 °C) to ultralong RTP (280 °C) by an external heating stimulus. They supposed that the relatively low temperature afforded intertwined CEE-type CPDs with abundant sub-fluorophores. After heating to a higher temperature, parts of the intertwined polymer chains in the CPDs could be further reacted via crosslinking and dehydration/carbonization to form compact cores, which behaved as a matrix to immobilize the sub-fluorophores, eventually inducing RTP performance. The system demonstrated the potential of CPDs as stimuli-responsive optical materials (Fig. 21a).156 Subsequently, Qu et al. utilized the difference in luminescence responses between several CPDs and polyvinyl alcohol composites annealed at different temperatures. They proposed the concept of thermal-treatment-controlled multilevel fluorescence/phosphorescence data encryption and further developed the application of CPDs in the stimuli-responsive field.157 Moreover, Feng et al. designed and synthesized novel fluorine and nitrogen codoped CPDs via one-step solvothermal treatment. The CPDs possessed good aqueous solubility, blue pH-stabilized fluorescence, and green pH-responsive RTP with outstanding reversibility. Based on these interesting properties, they demonstrated the data encoding/reading strategy using basic or acidic aqueous CPD solutions for advanced anticounterfeiting via time-resolved luminescence imaging techniques, as well as the steganography of complex patterns (Fig. 21b–e).158
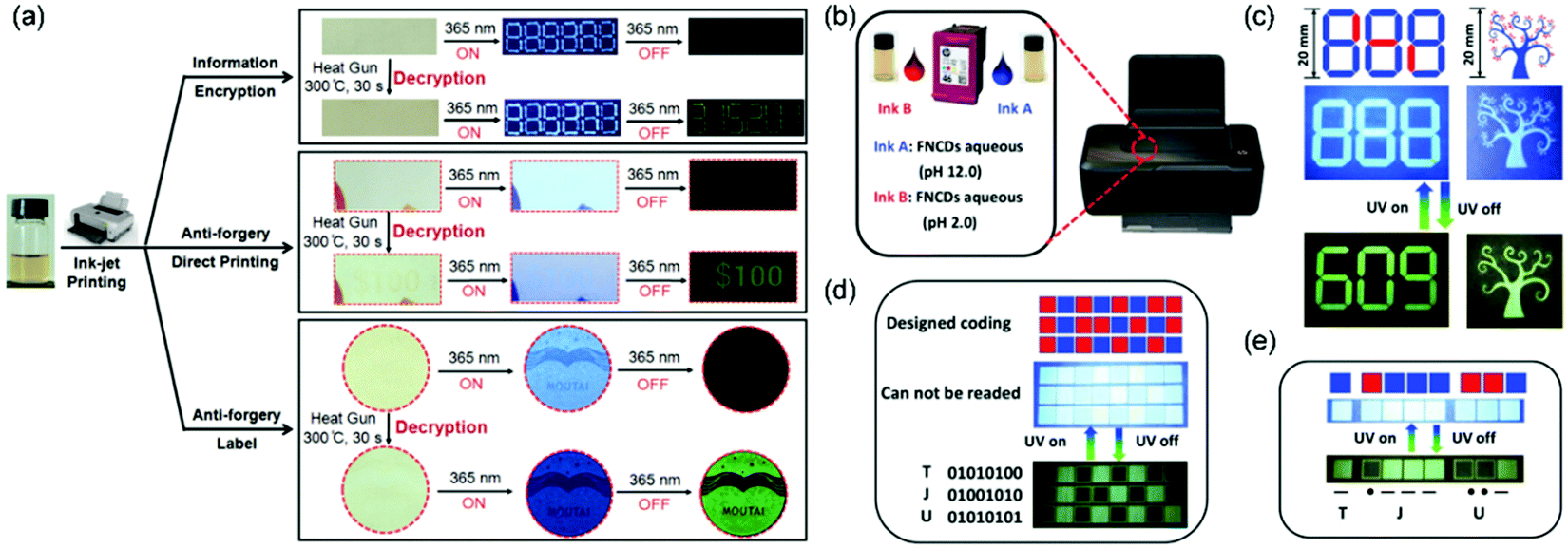 | ||
| Fig. 21 (a) Potential applications of F-CD-based ink in information encryption, direct printing on a coupon, and Moutai Liquor anticounterfeiting tags. (b) Schematic illustrations of a representative customized tricolor inkjet cartridge. (c) Demonstration of lifetime encoding for security applications. Digital photographs of (d) binary-coded and (e) Morse-coded two-color microarray data storage chips. Reprinted from ref. 156 and 158 with permission. Copyright (2018) Wiley-VCH. | ||
Given the wide range of chemical precursors and possible synthesis conditions, these approaches can lead to a wide variety of CPD materials with diverse engineered RTP properties. A careful selection of precursors (such as combinations of polyacid, polyamine, polyol, diacid, diamine, and other potential multifunctional compounds) and different synthesis routes (such as solvothermal and ultrasonic-/microwave-assisted hydrothermal/solvothermal treatment), as well as even some specific addition polymerization reactions, can provide a wide range of potential routes for the formation of CPD materials for use in particular applications. Further studies should focus on making RTP materials brighter, with different emission colors and improved quenching resistance, as well as simultaneously being observable in aqueous solutions. Moreover, the emergence of novel biosystems is also anticipated, which can further expand the RTP properties for specific biomaterials.
3.3 PhPs
PhPs, another important class of phosphorescent polymers, refer to instances in which phosphorescent emitters (e.g., Ir(III) and Pt(II) complexes) are incorporated into the polymer main chains or side chains via covalent bonds.159 Owing to the strong spin–orbital coupling of the phosphorescent emitters, PhPs can harvest both S and T excitons to achieve 100% IQE, which is four-fold higher than that of fluorescent polymers. To develop efficient PhPs, several requirements need to be addressed. First, the triplet energy level (ET) of polymer hosts should be higher than that of the phosphorescent emitters; otherwise, triplet back energy transfer from the phosphorescent emitters to the polymer hosts can occur, and the emission of phosphorescent emitters can get quenched. Second, the polymers should have suitable highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) energy levels to facilitate charge injection from the electrode to the emissive layer and then create charge transport to the recombination zone. Third, phosphorescent emitters should exhibit high photoluminescent quantum efficiency (PLQY) to convert all the generated excitons into photons in order to achieve efficient electroluminescence. Finally, PhPs should exhibit excellent solubility in common solvents to form high-quality films and good thermal and morphological stabilities for the fabrication of long-durable light-emitting devices. In the past few years, several efficient PhPs have been synthesized by incorporating phosphorescent emitters into conjugated or nonconjugated polymer hosts to form a host/dopant system. In this subsection, recent progresses made in the two abovementioned kinds of PhPs, i.e., full-color emission and white-light emission PhPs, is briefly addressed. | ||
| Fig. 22 Full-color-emitting PhPs. Reproduced from ref. 161–163 and 165 with permission. Copyright (2012, 2012, 2010, and 2011) from American Chemical Society, Royal Society of Chemistry, and Wiley-VCH. | ||
To design red-emitting PhPs (PhP-Rs) and green-emitting PhPs (PhP-Gs), CPs with lower ET values (2.10–2.60 eV) can be selected as the hosts. For instance, PhP-Rs (Fig. 22d) were reported by the use of polyfluorene (PF) with ET value of 2.10 eV as the host and Ir(ppq)2(acac) (ppq: 2,4-diphenylquinolyl-N-C2′) as the phosphorescent emitter. A single-layer device based on this polymer exhibited superior EL performance with the maximum LE of 5.0 cd A−1 and CIE coordinates of (0.63, 0.35). A further optimization of the device structure by inserting an alcohol-soluble electron injection layer could improve the LE to 8.3 cd A−1.163 In addition, it was found that the charge balance of the polymer host played a crucial role in determining the device efficiency of the PhP-Rs. By alteration of the polymer host from PF to poly(fluorene-alt-carbazole) (PFCz) and polycarbazole (PCz), the LE decreased from 5.0 to 0.3 cd A−1 and 0.04 cd A−1, respectively, which could be attributed to the imbalanced hole and electron flux in PFCz and PCz caused by their higher hole mobilities than electron mobilities.164 For the development of PhP-Gs, polycarbazoles are selected as the hosts owing to their higher ET values (2.40 eV). For example, three kinds of PhP-Gs have been developed by using PF, PFCz, and PCz with different ET values as the polymer hosts and 1,2-diphenyl-benzoimidazole-based Ir complexes as the green phosphorescent emitter (Fig. 22b, PhP-G). Since the ET level gradually increased from 2.20 to 2.40 and 2.60 eV from PF to PFCz and PCz, respectively, the triplet back energy transfer from the phosphorescent emitter to the polymer host could be effectively inhibited. As a result, the maximum LE of the PhPs with a double-layer device configuration was significantly improved from 0.3 (PF) to 33.9 cd A−1 (PCz).165
To improve device efficiency, all-phosphorescent SWPs were reported by simultaneously grafting blue- and yellow-emitting phosphors into a bipolar, high-ET poly(arylene ether) host. Different from all-fluorescent and fluorescent/phosphorescent hybrid ones, all-phosphorescent SWPs were supposed to fully utilize the S and T excitons by phosphorescent dopants. Based on this consideration, FIrpic and (fbi)2Ir(acac) were selected as blue- and yellow-emitting phosphors, respectively, for the design of all-phosphorescent SWPs. A series of all-phosphorescent SWPs (Fig. 23, WPB75Y7) were successfully prepared by tailoring the feed ratios of monomers. The obtained polymers showed balanced blue emissions from FIrpic and yellow emissions from (fbi)2Ir(acac) to yield white emission. Among the SWPs, polymers containing 7.5 mol% FIrpic and 0.7 mol% (fbi)2Ir(acac) exhibited the best device performance with the maximum LE of 18.4 cd A−1, maximum PE of 8.5 lm W−1, peak EQE of 7.1%, and CIE coordinates of (0.31, 0.43). Even at a brightness level of 1000 cd m−2, the LE was as high as 14.2 cd A−1, indicating slow efficiency roll-off. These LE and PE values were much higher than those of all-fluorescent and fluorescent/phosphorescent hybrid SWPs, shedding light on the significance of all-phosphorescent SWPs.167
 | ||
| Fig. 23 White-emitting PhPs. Reproduced from ref. 167 and 168 with permission. Copyright (2012 and 2018) from American Chemical Society and Royal Society of Chemistry. | ||
To realize high PE, new all-phosphorescent SWPs (Fig. 23, SWP1–FIrpic, SWP1–FIrOBu, SWP2–FIrOBu, and SWP3–FIrOBu) were synthesized by the use of a combination of high-HOMO-level blue-emitting phosphor and high-HOMO-level nonconjugated poly(arylene phosphine oxide) host. On one hand, by changing the common blue-emitting phosphor (FIrpic) to a homemade blue-emitting phosphor (FIrOBu) containing electron-donating alkoxy groups on ligands, the HOMO level could be elevated from −5.64 to −5.28 eV, which matches well with the HOMO level of the polymer host. Therefore, hole scattering between the blue-emitting phosphors and polymer hosts can be eliminated, resulting in a reduced turn-on voltage from 4.2 to 3.4 V. On the other hand, the HOMO level of the polymer host can also be tuned from −5.64 to −5.20 eV by the use of alkoxy-substituted carbazole dendrimers instead of using the tert-butyl-capped carbazole as the side chain, coming close to the work function of the anode. Consequently, the hole injection barrier at the polymer/anode interface was lowered, and the turn-on voltage was reduced to 2.8 V. As a result, a low turn-on voltage of 2.8 V, record EQE of 18.0%, and forward PE of 52.1 lm W−1 could be achieved for SWPs. Such remarkable progress makes SWPs outperform currently used polymer blends for solution-processed white organic light-emitting diodes (WOLEDs) and comparable with the low-mass molecules used for vacuum-deposited WOLEDs.168
Recent progresses made in AIE polymers, CPDs, and PhPs are introduced in this section. The preparation methods, design strategies, material systems, and device performances of these luminescent polymers are emphasized. Although significant advances have been made, many challenges require redressal for the development of phosphorescent polymers. For example, their material design, photophysical properties, and device processes need to be improved. The future directions for phosphorescent polymers can focus on the following aspects: long-life blue emissions, high-efficiency white lighting, and low-cost nonprecious metal-containing ones.
4. Photovoltaic polymer materials
One of the strategies to resolve energy shortage and environmental pollution is to develop solar energy conversion. Solar cells are classified into two types: inorganic solar cells and OSCs. Inorganic solar cells that are based on inorganic semiconducting materials have achieved great advances. For example, crystalline silicon solar cells have been commercialized for many years. When compared with inorganic solar cells, OSCs based on polymer materials have many advantages, such as low cost, lightweight, low toxicity (or nontoxic), flexibility, high absorption coefficient, semitransparency, and easy and large-area fabrication.169 Moreover, photovoltaic functional polymers have a wide range of properties, and their optical properties (absorption range and intensity), electronic properties (energy level and carrier mobility), and film morphology can be effectively controlled by chemical methods.Conventional OSC devices generally comprise the active layer, interfacial layers, and electrodes (Fig. 24). The active layer of bulk-heterojunction (BHJ) OSCs is generally composed of two kinds of semiconductor materials, namely, electron donor (D) and electron acceptor (A), which exhibit microphase separation between the donors and acceptors at the scale of 10–20 nm. Many strategies have been used to obtain OSCs with excellent power conversion efficiencies (PCEs), such as designing active layer materials (including donors and acceptors), controlling the morphology of the active layer, and optimizing the fabrication methods as well as structures of the OSC devices. Exciton diffusion, charge separation, and charge transport, which occur in the active layer, require appropriate nanoscale phase-separated interpenetrating network (IPN) structures. Therefore, controlling the morphology of the active layer is crucial. In addition, water/alcohol-soluble conjugated polymers (WSCPs) can be used as interfacial layer materials to optimize the processing methods of OSC devices. The development of electron donors has been more comprehensively investigated than electron acceptors for the past decade; interested readers should refer to other reviews in relation to electron donors.170,171
 | ||
| Fig. 24 Structure of a BHJ solar cell. | ||
Among acceptors, fullerene derivatives have dominated this field for two decades, and single-junction fullerene-based devices have exhibited PCEs up to 11%.172 However, shortcomings of fullerene derivatives, such as weak absorption in the visible region, difficult synthesis and purification, and low tunability of energy levels, hinder further improvements in device performance. In recent years, non-fullerene acceptors (NFAs) have attracted increasing attention, among which rylene diimides and fused-ring electron acceptors (FREAs) are two promising material categories.9,173 When compared with fullerenes and rylene diimides, FREAs exhibit several superiorities, such as stronger and red-shifted absorption in the visible and NIR regions and more tunable energy levels. Currently, OSCs based on FREAs have achieved PCEs of over 17%.174 In this section, NFAs, control morphology of the active layer, and solution processing using WSCPs have been addressed.
4.1 ITIC-based fused-ring electron acceptors
In 2015, Zhan et al. reported the star acceptor of ITIC based on a fused-ring core, indacenodithieno[3,2-b]thiophene (IDTT), with four 4-hexylphenyl side chains and two 2-(3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (IC) end groups (Fig. 25).175 As-cast devices based on the polymer donor PTB7-Th and ITIC exhibited PCEs up to 6.8%, which was the new record for fullerene-free OSCs at that time. Since then, the ITIC family has begun to replace the dominant position of fullerene acceptors, inaugurating a new era—the era of NFAs. In general, the FREA molecule consists of three parts: electron-donating aromatic fused-ring core, electron-withdrawing terminal groups, and side chains. FREAs exhibit good solubility in chloroform, chlorobenzene, and o-dichlorobenzene, good thermal stability with a high decomposition temperature of over 300 °C, and tunable electronic properties with different energy levels and variable absorption edges from 700 to 1100 nm. Meanwhile, their solubility, crystallinity, energy level, absorption, and charge mobility can be modulated by fused-ring core engineering, end-group engineering, and side-chain engineering. | ||
| Fig. 25 Fused-ring core engineering of FREAs. | ||
 | ||
| Fig. 26 End-group engineering of FREAs. | ||
 | ||
| Fig. 27 Side-chain engineering of FREAs. | ||
Since the discovery of ITIC, extensive efforts in new materials synthesis have been expended in the avenues of fused-ring core engineering, end-group engineering, and side-chain engineering.204,205 Besides, morphology control and device optimization also play vital roles in the improvement of device performance. Nowadays, the PCEs of FREA-based OSCs have exceeded 17%, and it is believed that PCEs in the range of 18–20% could be possible in a couple of years. However, several challenges still persist before OSCs can enter the markets: (1) novel FREAs with high electron mobility and low-cost preparation; (2) more in-depth mechanism study about the driving force and energy loss;206,207 (3) simpler and more stable device fabrication methods for commercialization.
4.2 Unfused-core electron acceptors (UCEAs)
Although the efficiency of OSCs based on FREAs is developing fast, the stability and cost of OSCs are the other two important factors to be considered in order to meet the requirements of future practical applications. Therefore, designing new materials that can effectively balance the parameters of efficiency, stability, and cost is urgently required. To realize the above considerations, Chen et al. proposed a strategy by utilizing noncovalent interactions to construct UCEAs with an acceptor–donor–core–donor–acceptor (A–D–C–D–A) structure (Fig. 28). Such a strategy can not only yield high-performance NFAs with adjustable optoelectronic properties but also reduce complexities encountered during synthesis. | ||
| Fig. 28 Diagram of UCEAs and chemical structures of representative molecules. | ||
Chen et al. first reported DF-PCIC UCEA (Fig. 28) that constructs an unfused donor core by connecting one 2,5-difluorobenzene unit with two cyclopentadithiophene (CPDT) units. Among the structural moieties in UCEA, noncovalent interactions, such as the H–F hydrogen bond, has been employed to reinforce the rigidity and planarity of the molecular structure.208 It is noteworthy that noncovalent interactions, such as O–S interaction and S–N conformational lock, have proven to be effective in the design of NFAs.209,210 By the condensation of the unfused core with the IC terminals, the obtained UCEA and DF-PCIC exhibited planar and stackable geometry with the A–D–C–D–A-type structures (Fig. 29), which not only facilitates intramolecular charge transfer (ICT) but also improves the intermolecular carrier transport. Moreover, DF-PCIC also exhibits similar absorption (up to around 800 nm) and energy levels to those of FREAs. When DF-PCIC was blended with the wide-bandgap polymer donor PBDB-T, solar cell devices exhibited good PCE of 10.14% with high FF of 0.72. Interestingly, they displayed good morphological stability upon high-temperature annealing (130, 150, and 180 °C, respectively). On the basis of the one-on-one comparison of DF-PCIC-, ITIC-, and PC71BM-based active layers, the one comprising DF-PCIC was found to be more stable than the others, which may be because the flexibility of the mediated structure built via noncovalent interactions prevents over-aggregation upon thermal treatment.
 | ||
| Fig. 29 Geometry, absorption, and energy levels of DF-PCIC and device performance of PBDB-T:DF-PCIC-based OSCs. | ||
Moreover, the molecular engineering of UCEAs has been performed, yielding enhanced ICT effect, absorption, electron mobility, and modulated energy levels of new acceptors. These efforts include the introduction of fluorine (HF-PCIC) or chlorine (HC-PCIC) atoms at the IC terminals, attachment of the methoxyl group on the benzene-based core (HFO-PCIC), extension of the conjugated length of IC terminals (DF-PCNC), replacement of benzene-based core with thiophene-based core (HF-TCIC), and so on.211–215 After selecting suitable polymer donors, such as PBDB-T or its fluorinated analog PBDB-TF, obvious enhancement in JSC can be observed (over 17 mA cm−2 for HF-PCIC-based OSCs; over 18 mA cm−2 for HC-PCIC- and DF-PCNC-based OSCs; over 20 mA cm−2 for HF-TCIC-based OSCs) for the relevant devices. Therefore, HF-PCIC-, HC-PCIC-, and DF-PCNC-based OSCs could yield PCEs over 11%. For PBDB-TF:HC-PCIC-based OSCs, the subsequent use of PC71BM as the third component to optimize the morphology and enhance the charge transfer can yield solar cells that exhibit large improvements in quantum efficiencies (from ∼70% to over 80%) with outstanding PCE of 12.36%.212 The above efforts from Chen et al. demonstrate that UCEAs possess good structural variability for tuning the material properties for obtaining high-performance OSCs.
New acceptors with fully unfused structures have also been recently developed by Chen et al., such as ICTP that is constructed from single aromatic units within 2 or 3 synthesis steps.216 With the assistance of noncovalent interactions, they identified that these potentially rotatable acceptor structures allow adapting stackable yet thermally stable conformation in a condensed solid, facilitating the new molecules to not only exhibit feasible solution processability but also excellent film characteristics. PCE of 4.43% was achieved for ICTP with the PBDB-T polymer. Further, PCE of over 10% can also be obtained for such a type of simple electron acceptor, revealing that there is still considerable space for achieving better device performance. Notably, such unfused acceptors can be synthesized with C–H activation coupling reaction and purified with recrystallization, largely reducing the synthesis complexity of electron acceptors.
To enable the efficient working of fullerene-free OSCs, allowing small driving forces is essential to achieve higher voltages and better performance for the OSCs. Very recently, Chen et al. performed a systematical study on the above issue with UCEAs and revealed profound understandings on the charge transfer process in non-fullerene blends. Due to the adjustable energy levels of UCEAs, they chose three UCEAs, namely, DF-PCIC, HC-PCIC, and FO-PCIC, developed by their group, as well as three polymer donors, namely, PTQ10, PBDB-TF, and PBDB-T; therefore, six blends with various HOMO offsets ranging from −0.05 to 0.21 eV were formulated (Fig. 30).217 Through the aid of bilayer hole-only devices and transient absorption spectra, they found that the occurrence of hole transfer is independent of the HOMO offsets, but the hole transfer rate was influenced by the HOMO offsets. The blend of PTQ10/DF-PCIC with slightly negative HOMO offset of −0.05 eV yielded a really slow hole transfer rate of ∼400 ps, while blends such as PTQ10/HC-PCIC and PBDB-TF/HC-PCIC with HOMO offsets of ≥0 eV could yield faster hole transfer rates of ≤4.6 ps. As a result, small HOMO offsets of 0 and 0.06 eV can enable achieving higher PCEs of 10.42% and 11.75% for the PTQ10:HC-PCIC blend and PBDB-TF:HC-PCIC blend, respectively. This work illustrates the fact that the modulation of HOMO offsets ranging between 0 and 0.1 eV has promising potential to realize breakthroughs in OSC efficiencies and energy loss.
 | ||
| Fig. 30 Diagram of hole transfer for different blends with various HOMO offsets and hole transfer rates. Adapted from ref. 217 with permission. Copyright (2019) American Chemical Society. | ||
In summary, from the abovementioned discussion, we can understand that UCEAs possess advantages with respect to the following aspects: (1) flexibility and diversity in molecular design, (2) reduction in synthesis complexity of new materials, and (3) efficient performance and good stability. UCEAs would also be good choices for the design of high-performance and stable NFAs.
4.3 Control morphology of active layer during solution processing
CPs and their corresponding blends have promising potential applications in organic optoelectronic devices, such as organic thin-film field-effect transistors (OTFTs), OSCs, and OLEDs, due to their advantages such as a combination of optoelectronic properties and solution processing, e.g., inkjet printing, roll-to-roll printing, etc. Part of the challenge of the application of CP semiconductors is the deeper understanding of the effect of the microstructure on the macroscopic electrical properties. Different device applications have different requirements for the polymer microstructures (Fig. 31). In this subsection, we focus on how to control the morphology of the polymers from the viewpoint of thermodynamics or kinetics during solution processing, and the relationship between morphology and device performance is also established. | ||
| Fig. 31 Requirements of the active layer for OTFTs, oligo(p-phenylene vinylene) (OPVs), and OLEDs. | ||
Charge transport in semiconducting polymers occurs within the polymer backbones and between different chains due to the electronic coupling between the rings, which is related to their microstructure. In order to obtain higher carrier mobility, the CP films should possess a percolated network with long-range order, high crystallinity, and appropriate crystal orientation. However, the entanglement of the CP caused by the flexible side chain and rigid backbone suppresses the intrachain mechanism. Furthermore, the fast crystallization kinetics during the film-forming process yields several grain boundaries among the crystals. Hence, the high-density nanowires of the CP are desirable to improve the charge transport. However, the entanglement of the CP chain severely impedes the nucleation to grow nanowires at a higher solution concentration. Han et al. controlled the equilibrium of entanglement, disentanglement, nucleation, and growth by tuning the interaction between the solvent and CP, temperature, ultrasonic oscillation, adding additives, blending with flexible polymer, etc.218–221 Once the disentangled polymer chains could nucleate to form nanowires, the equilibrium between entanglement and disentanglement is broken and the entangled polymer chains get further disentangled to self-assemble into nanowires in the solution or during the film-forming process. Furthermore, directional solvent evaporation and zone casting were employed to fabricate aligned fibers. Evidently, when the molecular crystallization rate matched the solvent evaporation rate, the polymers tended to form a highly oriented film and the order parameter reached as high as 0.97.
The morphology of the active layer of OSCs plays a crucial role in determining the PCE of the devices since it is closely related to the photoelectric conversion process. The ideal morphology of the active layer should comprise a pure donor and acceptor phase with a domain size of 10–20 nm and a large interfacial mixed phase, which guarantees efficient exciton separation, carrier transport, and carrier collection. However, due to the competition and coupling between crystallization and phase separation, the active layer usually possessed poor morphology, including the “sea-island” phase separation structure, low crystallinity, and inconsistent molecular orientation. Han et al. proposed that the application of the liquid–solid solution phase transformation resulted in phase separation, fractional crystallization, and confined crystallization; further, controlled film-forming kinetics could optimize the film morphology.221–223 They selected the P3HT:O-IDTBR blend as a model system, and they proposed to control the film-forming kinetics, i.e., the crystallization of P3HT occurred ahead of O-IDTBR, to get an IPN with higher crystallinity and optimized lateral and vertical phase separations. The optimized film morphology boosted the PCE from 4.45 to 7.18%, which is the highest performance in P3HT binary non-fullerene solar cells. In addition, the molecular orientation in polymer blends is decisive to the charge transfer efficiency of these π-conjugated photovoltaic devices. They used epitaxial crystallization, confined crystallization, and tuning the solution state or film-drying time to control the D/A molecular orientation.224,225 In P3HT/N2200 blends, molecular orientation and phase separation were controlled by chain segment and molecular movement. P3HT crystallization with face-on or edge-on orientation for different post-annealing processes is dependent on the P3HT chain segment and molecular movement confined by crystalline N2200 or broken out of N2200 crystalline confinement. For the conjugated PTB7-Th and N2200 polymers, the transformation of preferential molecular orientation from the face-on state to the edge-on state occurs when extending the film-drying time and changing the processing solvents. Therefore, the PCE of the devices was improved from 0.53 to 3.52%.
4.4 WSCPs
WSCPs are a type of CP that possess solubility and processability in water-/alcohol-like polar solvents. When compared with most traditional CPs with hydrophobic alkyl groups that impart solubility in nonpolar/low-polar solvents, WSCPs possess highly polar or ionic side chains that facilitate excellent water/alcohol solubility and processability.226–228 The water/alcohol solubility of WSCPs provides a promising opportunity to fabricate multilayer films using orthogonal solvents, which can be compatible with the multilayered fabrication of optoelectronic devices (Fig. 32a).229–231 Moreover, the conjugated backbones of WSCPs endow them with semiconductivity that can facilitate the charge transport characteristics for various applications.232–234 WSCPs have been widely used as electron/hole transport materials (ETMs/HTMs) in optoelectronic devices,226,227 including OLEDs, OSCs, perovskite solar cells (PVSCs), and organic field-effect transistors (OFETs). For example, in OSC devices, a thin layer of PFN or PFN-Br acting as the ETM can improve electron collection at the cathode.235 Evidently, WSCPs in OSCs can reduce the work function of the metal electrode and optimize the active layer morphology, block holes, and dope PCBM, all of which contribute toward the enhancement of device performance.236,237 However, the low electron mobilities of PFN and PFN–Br hinder their application as thick ETMs in large-area OSCs, while a thicker ETM is much more compatible with large-area OSCs processed by the roll-to-roll technique.238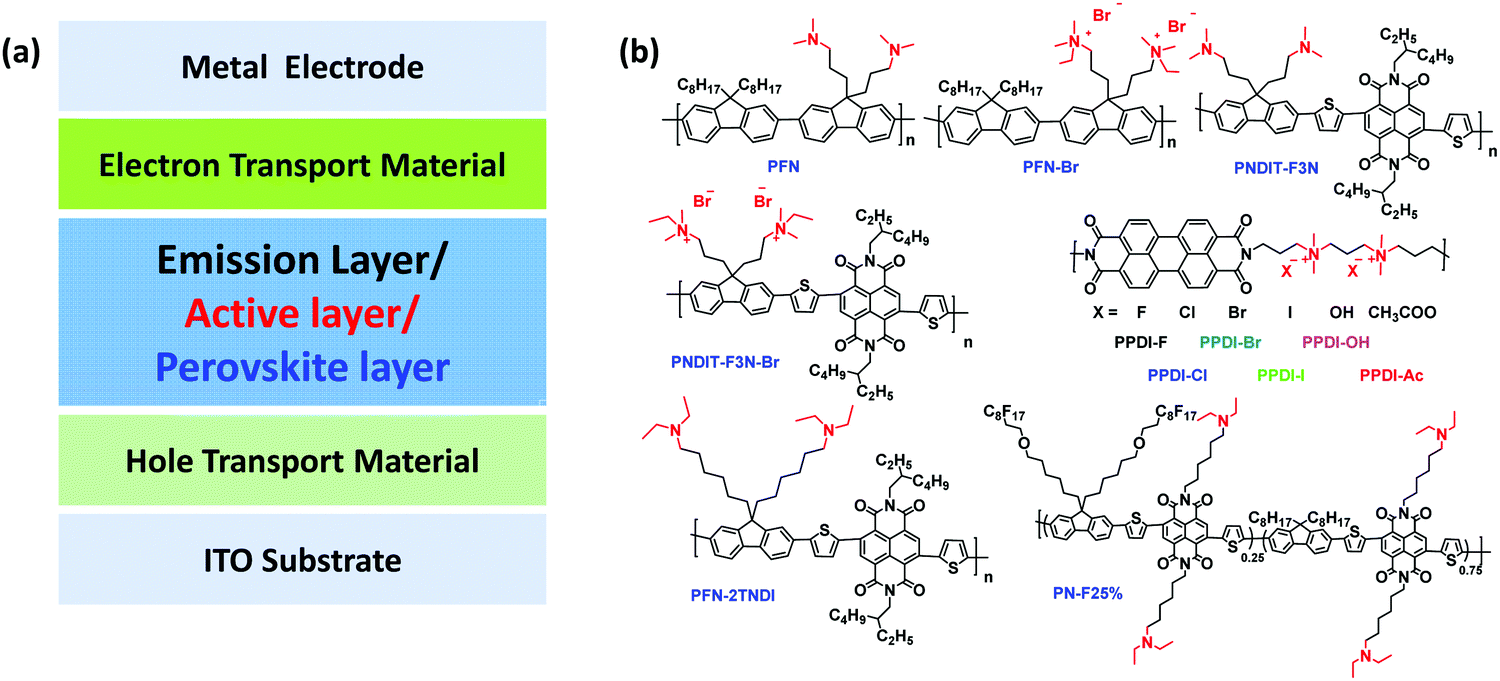 | ||
| Fig. 32 (a) Structure of optoelectronic devices; (b) representative WSCPs. | ||
To overcome this problem, a series of novel WSCPs with self-doping behaviors have been recently developed by rational design on the polar side chains and n-type conjugated backbones.239 Self-doping behaviors were observed in these n-type WSCPs due to the electron transfer from polar groups to n-type conjugated backbones. A special finding in these WSCPs was that their self-doping behaviors could be regulated by changing the polar group species. Wu et al. reported that neutral amino-group-functionalized WSCPs (PNDIT-F3N, Fig. 32b) employed a light-induced doping mechanism, while the ammonium-bromide-group-functionalized WSCPs (PNDIT-F3N-Br) could be self-doped without light (Fig. 33).239 Consequently, PNDIT-F3N exhibited a photoinduced conductivity-enhancing property, while the conductivity of PNDIT-F3N-Br showed no difference in dark and light (Fig. 33). Moreover, the self-doping behaviors and charge transport properties of WSCPs could be readily regulated by changing the counterion type and energy levels of the conjugated backbones in WSCPs.240–242 For example, Hu et al. prepared a series of perylene-diimide-based polyelectrolytes (PPDI-X, Fig. 32b) with different counterions (F−, Cl−, Br−, I−, CH3COO−, and OH−) and investigated their self-doping behaviors. Evidently, polyelectrolytes with F−, OH−, and CH3COO− as anions are strongly self-doped, and polyelectrolytes with Cl−, Br−, and I− possess weak self-doping properties.240
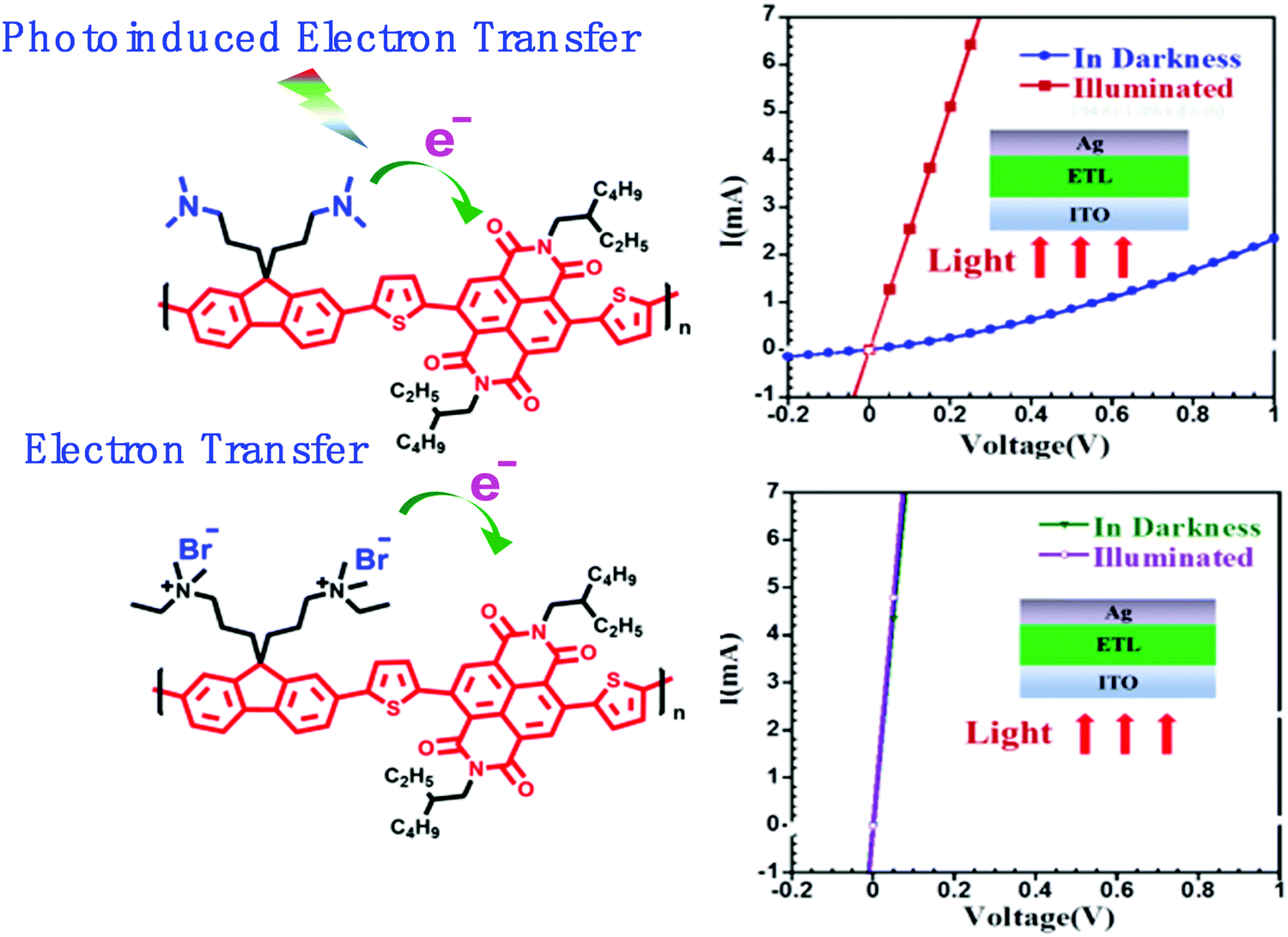 | ||
| Fig. 33 Different doping processes of PNDIT-F3N and PNDIT-F3N-Br. Adapted from ref. 239 with permission. Copyright (2016) American Chemical Society. | ||
These self-doped WSCPs showed great promise as thickness-insensitive ETMs to enable highly efficient OSCs.239,240 With PffBT4T-2OD/PC71BM as an active layer, high PCE of 10.11% for devices with 5 nm PNDIT-F3N-Br and prominent PCE of 8.04% for devices with 100 nm PNDIT-F3N-Br can be achieved.239 Moreover, these self-doped WSCPs also worked well in non-fullerene solar cells.243,244 Sun et al. reported that PNDIT-F3N largely improved electron collection and reduced charge recombination at the cathode of non-fullerene solar cells. One particular finding was that PNDIT-F3N in the interlayer also worked as the third component and induced charge transfer from the donor in the active layer to PNDIT-F3N, contributing to the photocurrent output of non-fullerene solar cells.243 Moreover, it was found that a thicker ETM could also slow down and block the diffusion of metal atoms (from the metal electrode), resulting in much-improved device stability.245
In addition to the successful application of self-doped WSCPs in single-junction OSCs, WSCPs also showed promising applicability as interconnection materials for tandem OSCs.246,247 Zhang et al. reported that an interconnection layer constructed by PNDIT-F3N-Br/Ag (2 nm)/poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) could efficiently protect the front cell from the erosion caused by the solutions used for processing the back cell, enabling the successful fabrication of tandem OSCs.246 More recent works have shown that all the solution-processable interconnection layers for tandem OSCs could be realized by combining a WSCP-based HTM and ETM, affording highly efficient electron–hole recombination as well as much improved photovoltaic performance.247
WSCPs were also discovered as efficient ETMs for PVSCs due to their excellent interface modification capability. Amino-functionalized WSCPs can improve interfacial contact between the perovskite layer and metal cathode, resulting in more efficient electron collection at the cathode.248,249 Sun et al. found that the amino groups in PFN-2TNDI (Fig. 32b) could passivate the interface traps of perovskite layer, reducing interface recombinations and improving the device performance of PVSCs.248 Tian et al. further introduced perfluorooctane side chains into a self-doped CP and obtained the hydrophobic polymer PN-F25% (Fig. 32b) with excellent water resistance capability. PN-F25% could act as a high-performance bifunctional ETM for PVSCs due to its amino and fluoro side chains, which not only enhanced electron collection but also significantly improved the device stability of PVSCs.249
In general, the design of electron acceptors in the active layer, including fused-ring and UCEAs, control morphology of the active layer using solution processing, and WSCPs are summarized. Recent progresses, molecular design strategies, and structure–property relationships of fused-ring and UCEAs are initially addressed briefly. The factors affecting the morphology of the active layer and the corresponding strategies are subsequently introduced. After that, the progress of WSCPs that can act as interfacial layers is presented. In the future, the development of high-performance electron donors and acceptors will still remain an important research direction. For instance, the high carrier mobility of electron acceptors can meet the needs of high-efficiency thick-film OPV devices. Acceptor materials that can be processed by environmentally friendly solvents can meet the requirements for the preparation of low or nontoxic OPV devices. Moreover, improvements in device stability and lifetime of OPV devices can be another important research direction.
5. Other electronical and optical polymer materials
Besides the aforementioned polymer materials, there are still other electronical and optical polymers that are very important for basic research and practical applications, such as conducting polymers and photosensitive polymers. These polymers can also be divided into numerous subtypes. For example, conducting polymers include two different subtypes of polymer materials. One is a polymer composite filled with conductive fillers such as carbon nanotubes (CNTs), graphene oxide, metal particles, etc. The other subtype is a polymer that can generate and propagate charge carriers along its backbones. In this section, we will introduce some other functional polymers that are also very important for applications in our daily life. Polyimide can be used to fabricate the flexible electrode of lithium-ion batteries (LIBs) with carbon materials;250 it can also be used in the wireless communications industry due to its low dielectric constant (k). Second-order nonlinear optical (NLO) polymers have been drawing considerable attention due to their potential applications in photonic devices. Therefore, we address these functional polymers with respect to their preparation methods, design strategies, and potential applications in this section.5.1 Intrinsic low-k polyimide materials
With the development of the wireless communication industry, the high speed and high-frequency transmission, particularly the fifth generation of wireless communications technology (5G technology), has been a hot research subject.251 Both signal transmission speed (V) and signal propagation loss rate (α) are closely related to k and dielectric loss of the substrate materials, which can effectively reduce the resistance–capacitance delay, line-to-line crosstalk noise, and power dissipation. As a result, the accuracy and integrity of signal transmission can be ensured, and the service life of electronic components can also be improved.252 Therefore, low-k and low-loss dielectric materials are highly desirable in the wireless communications industry.There are two ways to design low-k dielectric polymers. One is the introduction of nanosized air voids (k ∼ 1) into the bulk polymers. The dielectric constant of these porous polymers can be reduced to a certain level, but the overall properties of the materials can be considerably damaged, limiting their practical applications.253 The other is to design the dielectric properties of materials at the molecular level. For example, some chemical bonds with lower molar polarizability (such as the –C–F bond) or some rigid and shape-persistent components (such as triptycene moiety) can be introduced into the molecular structures of the polymer.254 When compared with porous polyimide materials, the intrinsic low-k polyimide materials have practical application value with regard to both preparation process and comprehensive performance. However, the k values of most of the developed low-k polyimide materials can only reach around 2.3. Therefore, the development of flexible high-performance intrinsic low-k or ultralow-k polymer materials remains a formidable challenge in the microelectronics industry.
Recently, a highly efficient design strategy to fabricate high-performance intrinsic low-k polyimides has been developed by Zhang and Xu et al. by the use of propeller-like structures (such as triphenylamine, triphenylmethane, and tristyrene units) in order to form a series of novel functional diamine monomers with rigid nonplanar large conjugated structures.255–261 By regulating the chemical structures and aggregation structures of the polymers at the molecular level, low- and even ultralow-k polyimide films with excellent comprehensive properties can be obtained (Fig. 34).260
 | ||
| Fig. 34 Relationship between k and Tg of the polyimides developed by Zhang's group and the reported intrinsic low-k polyimides. Reprinted from ref. 260 with permission. Copyright (2019) from Springer. | ||
Liu et al. designed and synthesized novel, simple, and efficient diamine (TriPEDA and TPEDA)-containing rigid nonplanar conjugated triphenylethylene and TPE moieties through the Wittig–Horner and Suzuki coupling reactions.255,256 A series of high-performance functional polyimides were consequently prepared by the dipolymerization of TriPEDA/TPEDA and four dianhydrides (Fig. 35). Because of the introduction of the aromatic rigid nonplanar triphenylethylene/TPE structure, the polyimides exhibited the following: low k (2.63–2.76); excellent mechanical properties with high tensile strength and high tensile modulus in the range of 115–130 MPa and 2.2–2.7 GPa, respectively; and high glass transition temperatures ranging between 359 and 443 °C. Moreover, these organo-soluble polyimides exhibited special fluorescent and resistive switching (ON/OFF) characteristics, as the maximum fluorescence emission of the four polyimides was observed at 425–505 nm in the NMP solution and 470–541 nm in the film states.
 | ||
| Fig. 35 Low-k polyimides containing triphenylethylene and TPE moieties. Adapted from ref. 255 and 256 with permission. Copyright (2012 and 2013) American Chemical Society and Wiley-VCH. | ||
Chen et al. prepared novel fluorinated aromatic polyimides by the conventional two-step imidization of 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA) and diamines (TriPMPDA and TriPMMDA)-bearing triphenyl methane moiety (Fig. 36).257 Both flexible and tough polyimide films exhibited intrinsic low k values of 2.56 and 2.33 at a frequency of 10 kHz, respectively, due to the introduction of bulky triphenyl methane side groups as well as tortuous backbone structures. In addition, they showed light color, high thermal stability, moderate mechanical property, and more importantly, excellent solubility in common organic solvents. Therefore, both these functional polyimides possessed attractive potential applications in the field of high-performance flexible polymer interlayer materials.
 | ||
| Fig. 36 Low-k polyimides containing triphenylmethane moieties. Adapted from ref. 257 with permission. Copyright (2016) Elsevier Ltd. | ||
Bei et al. successfully synthesized three polyimides (PPy6F, mBPPy6F, and mTPPy6F) containing the same rigid nonplanar conjugated diphenylpyridine structure in the main chains and a different number of benzene rings in the side chains (Fig. 37).258 The polyimides having one, two, and three benzene rings in the pendant group showed intrinsic k values of 2.81, 2.61, and 2.44, respectively. Owing to the rigid nonplanar conjugated diphenylpyridine structure in the main chain, PPy6F had a lower k value of 2.81 as compared to that of the traditional polyimide 6FDA-ODA (3.0). The k values of PPy6F, mBPPy6F, and mTPPy6F presented regular decreasing tendency. Based on the results of WAXD, morphology, and density studies, it was revealed that the free volume of the polyimides increased with an increase in the number of benzene rings in the pendant group. Meanwhile, mTPPy6F showed excellent comprehensive properties, such as Tg of 342 °C, decomposition temperature (Td,5%) of 551 °C, tensile strength of 105 MPa, low moisture absorption of 0.61%, and good solubility, making it a competitive material for potential applications in microelectronics industries.
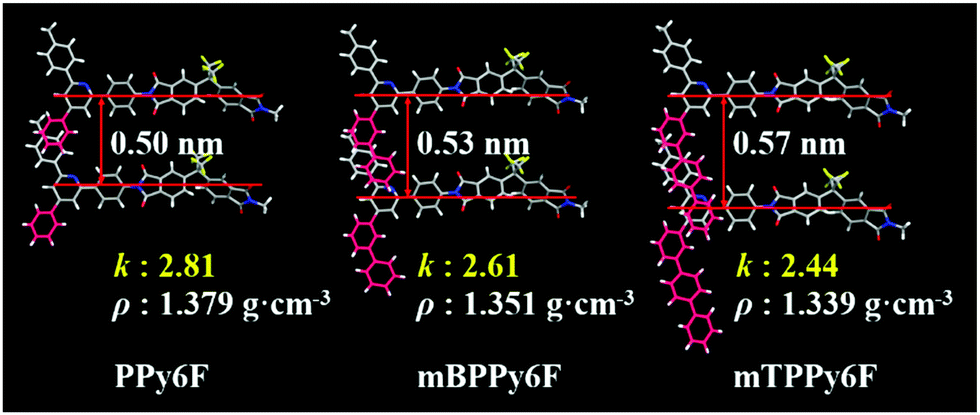 | ||
| Fig. 37 Intrinsic low-k polyimide containing the diphenylpyridine structure. Adapted from ref. 258 with permission. Copyright (2017) Royal Society of Chemistry. | ||
Furthermore, a series of high-performance multifunctional polyimides with intrinsic ultralow-k values (∼1.93) as well as exceptional thermostability and solubility (Tg as high as 494 °C, and, at the same time, worthwhile solubility in common organic solvents) were successfully designed and synthesized by introducing a typical aromatic rigid trifluoromethyl-containing moiety with special nonplanar and conjugated characteristics into the polymer backbone (Fig. 38).259,260 In addition, these polyimides showed light color (even colorlessness in one case) and transparency as well as electrical bistability characteristics (ON/OFF ratio as high as 107; working voltage as low as 1.5 V). The excellent thermal stability and solubility allowed them to undergo the high-temperature process (over 400 °C) in the preparation of photoelectric devices (such as PVD and PECVD) or the highly efficient, continuous roll-to-roll process. Ideally, the as-synthesized polymers are potential candidates for practical applications in the fields of ultra-large-scale integration (ULSI), high-performance polymer memory devices, flexible displays, thin-film photovoltaic industries, and wearable electronics.
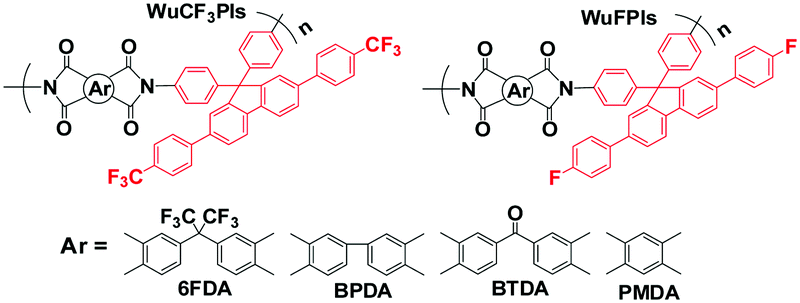 | ||
| Fig. 38 Multifunctional polyimides containing trifluoromethyl moieties. Adapted from ref. 259 and 260 with permission. Copyright (2017 and 2019) Royal Society of Chemistry and Springer. | ||
A bulk dielectric polymer film with an intrinsic ultralow-k value of 1.52 at 10 kHz was successfully synthesized based on the novel polyimide, FPTTPI (Fig. 39).261 It is well known that this k value is the highest among the intrinsic ultralow-k polymers in the literature. More importantly, such outstanding dielectric properties remained stable until up to 280 °C. The excellent ultralow dielectric properties could be mainly attributed to the large free volume (sub-nanoscale), which intrinsically existed in the amorphous region of polymeric materials. Meanwhile, FPTTPI also showed excellent thermal stability and mechanical properties, Tg of 280 °C, decomposition temperature of 530 °C, and residual of 63% at 800 °C under N2. It was soluble in common solvents, which makes it possible to be used in simple spin-on or efficient, low-cost, and continuous roll-to-roll processes.
 | ||
| Fig. 39 Dielectric properties of the FPTTPI film and a DuPont Kapton polyimide film. Reprinted from ref. 261 with permission. Copyright (2015) American Chemical Society. | ||
In general, polyimides have shown potential applications in the wireless communications industry due to their low-k values and low dielectric losses. Therefore, the design strategies and preparation methods for intrinsic low-k polyimides with good properties are discussed. Although a polyimide with an intrinsic ultralow-k value (as low as 1.52) has been obtained, it still remains a challenge to obtain a lower k value while retaining other worthwhile properties.
5.2 Second-order NLO polymers and dendrimers
Organic second-order NLO materials have attracted considerable attention owing to their potential applications in photonic devices, such as high-speed electrooptic modulators, optical switches, and frequency converters. Until now, many strategies and approaches have been proposed to meet the basic requirements of practical applications, including large optical nonlinearity, low optical loss, and excellent temporal stability of dipole orientation. A major problem encountered in organic NLO materials is to overcome the strong dipole–dipole interactions among the chromophores with donor–π–acceptor (D–π–A) structures. Therefore, it is crucial to efficiently translate the large β values of organic chromophores into high macroscopic NLO activities of polymers, which is mainly based on the orderly arrangement of the chromophore moieties with noncentrosymmetric forms. However, due to the strong electrostatic interaction of the chromophore moieties with electronic push–pull structures, the centrosymmetric arrangement is the natural state with nearly no macroscopic second-order NLO coefficient. Therefore, many approaches have been explored to reverse the orientation of such chromophores, with the aim to achieve favorable molecular arrangement, for instance, a poling process. Further, the optimized molecular shapes and electronic properties of chromophores play essential roles in molecular packing of thin films, which are mainly based on the site isolation principle, that is, the more spherical the molecular structure, the larger is the reduction in dipole–dipole interactions.Hence, systematical studies were conducted by Li et al. to optimize the molecular alignment of organic chromophores in both polymers and dendrimers. First, according to the site isolation principle, the larger the sizes of the side groups, the better was the isolation effect; this led to the noncentrosymmetric arrangement of the chromophores. On the other hand, this decreased the loading density of the chromophore moieties, which could weaken the NLO coefficient (d33). Therefore, there could exist a most suitable state in which the minimum size of the isolation group could prevent interactions between the adjacent chromophore moieties, and the maximum loading density could be guaranteed at the same time. To reach this state, Li et al. conducted systematic work and proposed the concept of a “suitable isolation group (SIG).” That is, for a given chromophore, there could be a SIG present to ensure the noncentrosymmetric arrangement of the chromophores, thereby achieving possibly improved macroscopic NLO properties in the organic materials. For example, various NLO effects could be obtained in linear polymers bearing the same nitroazobenzene chromophore modified by different isolation groups and different linkage modes. In most of the cases, it was clear that the NLO effect initially increased and then decreased with greater sizes of isolation groups: P2 exhibited the largest d33 value with the phenyl moiety as the SIG. However, for different linkage modes, SIG is considerably different for the different arrangements of chromophores as the natural state (Fig. 40).262 Since the isolation group was not the valid unit for the NLO effect, they subsequently proposed a more efficient strategy by the introduction of isolated chromophores bearing different polarities with the main ones. As shown in Fig. 40, P11 with alternative chromophore moieties (nitroazobenzene and sulfonylazobenzene ones) afforded a larger d33 value (116.8 pm V−1) as compared to those obtained from polymers with single chromophores (78.1 and 34.3 pm V−1), confirming that favorable arrangement can be achieved with tunable dipole–dipole interactions.263
 | ||
| Fig. 40 Structures of linear polymers P1–11 and schematic illustrations of isolation groups and isolation chromophores. Adapted from ref. 262 and 263 with permission. Copyright (2007 and 2013) American Chemical Society and Royal Society of Chemistry. | ||
As the most promising candidates for second-order NLO materials, dendrimers with a special topological structure were also employed with the optimized chromophores as repeat units (Fig. 41). Excitingly, because the SIG concept was utilized, the NLO effect could be dramatically enhanced with increasing dendritic generations because of the dendrimer (dendritic) effect.264 Moreover, this could be further enhanced by modifying the topological structures. As shown in Fig. 41, X-type dendrimers exhibited almost parallel alignment of the chromophore units with fixed orientation as the initial state, which could help in maintaining the noncentrosymmetric arrangement during the electronic poling process.265,266 Accordingly, the Janus dendrimer X-1 possessed a d33 value of 299 pm V−1, which is the highest value reported so far for polymers containing a simple azo-chromophore moiety, further confirming the advantage of Janus dendrimers and the key role of the molecular arrangement for the NLO property. With structures similar to those of dendrimers, HBPs can be prepared very easily, typically by one-pot syntheses, which are likely to have more industrial and commercial values.267 By introducing isolation chromophores derived from AB4-type monomers by means of click chemistry, the resultant HBPs demonstrated good NLO properties and optical transparency, making them promising candidates for practical applications.268
 | ||
| Fig. 41 Chemical structures of the high-generation dendrimer HGD1 and X1 and X2 with a Janus structure. | ||
Furthermore, when considering dendrimers with perfect conformations and linear and HBPs with easy synthesis strategies, their combined advantages can be exploited by the introduction of dendrimers into linear or HBPs with rational molecular designs. Main-chain dendronized polymers, namely, MDPG1 and MDPG2 (Fig. 42), which were prepared by the macromonomer approach, simultaneously exhibited the advantages of both main-chain polymers (which usually demonstrate good stability of the NLO effect) and dendronized polymers (which usually possess a large NLO coefficient).269 With the incorporation of low-generation dendrimers as the monomer in the HBP backbone, a novel kind of polymer, namely, dendronized hyperbranched polymers (DHPs) (Fig. 42), was proposed by Li et al. in 2013.270,271 This special system could maintain both high NLO efficiency and improved NLO thermal stability, partially combining the advantages of HBPs and dendrimers. For an A2-type monomer with a dendrimer as the side chain, after being polymerized with a B3-type monomer of the TPA–borate ester, polymer DHP1 exhibited a high d33 value of 133 pm V−1, and the decay temperature was as high as 110 °C, which were much better than those of the corresponding dendrimer (100 pm V−1, 30 °C).
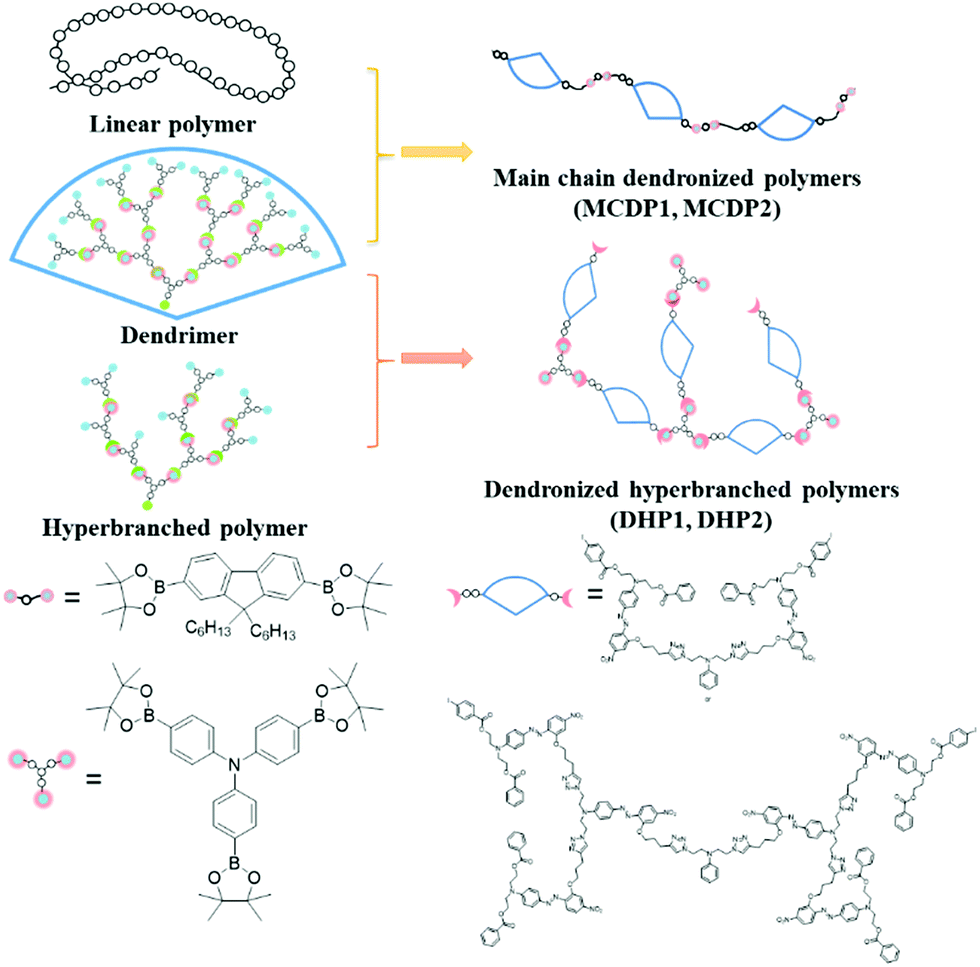 | ||
| Fig. 42 Chemical structures of the main-chain dendronized polymers and DHPs. | ||
In summary, based on some typical examples, recent progresses made in NLO dendrimers and polymers as developed by Li et al. are demonstrated, mainly focusing on molecular design ideas at the molecular level, as well as an attempt to determine the structure–property relationships to aid the rational design of better NLO materials for further investigations and possibly render designs that can be emulated by other functional polymers.
6. Biorelated polymer materials
Due to the aging of the global population, environmental problems caused by economic development have resulted in higher incidence of major diseases such as cancer, cardiovascular diseases, and diabetes; further, these patients tend to be younger. It is highly desirable to develop new technologies for diagnosing and treating diseases. Polymer materials have been playing an increasing important role in the diagnosis and treatment of major diseases such as cancer, cardiovascular and cerebrovascular diseases, and infectious diseases. A large number of functional polymers, such as micelles, gels, composites, fibers, particles, brush polymers, SMPs, etc., have been investigated for biomedical applications for many years. Their characteristics such as thermal and chemical stabilities, shape, dimensions, surface charge, surface chemistry, mechanical properties, and porosity may be tuned by using certain strategies to achieve the targeted functionalities that can meet the demands of specific biomedical applications. Therefore, recent progress in functional polymers and their corresponding strategies have become highly desirable. In this section, we only introduce polymers that are still in the state of basic research, including polymerization for protein modification, medical imaging materials, drug and gene delivery materials, biomedical surface–interface materials, integrated disease diagnosis and treatment materials, etc. Marine biofouling can accelerate biocorrosion and enhance hydrodynamic drag; therefore, it is highly desirable to develop new coatings for antibiofouling. Hence, we also address this type of functional polymers on the basis of preparation methods, design strategies, and applications in this section. Polymer gels and SMPs for biomedical applications will be addressed in the subsequent sections.6.1 In situ polymerization for protein modification
After thousands of years of evolution, the natural structures of proteins are considered to be in their most optimized form. When used in therapeutics, biosensors, or industrial catalysts, catalytical proteins play key roles in enhancing the quality of life. Most of these proteins, however, have currently been utilized in brand new areas, far away from their original ecologies, resulting in an inevitable need for structural modification. For example, cells make superoxide dismutase (SOD) to control intracellular superoxide from DNA mutation, while SOD has been used in sunblock to protect our skin from sunburn. A new application environment breeds additional modifications to the protein structure. Until now, various chemical strategies have been explored to facilitate protein modification for better functions, including liposome encapsulation, polymeric conjugation, supramolecular encapsulation, inorganic adsorption, and so on.272,273Polymeric materials are the most popular choices for protein modification for obtaining diverse functions: in situ polymerization of the protein structure is a feasible and efficient method. A generic in situ polymerization reaction for proteins comprises two steps: (1) amine-reactive acrylates are conjugated to the protein surface in order to introduce polymerizable groups; (2) initiating the in situ polymerization of hydrophilic monomers in aqueous media at room temperature yields a thin polymeric shell around each protein molecule. Such a polymeric shell could offer not only enhanced thermodynamic stability to the protein but also additional functions depending on the type of monomer.274
For example, protein therapeutics usually suffer from high immunogenicity, which might result in shorter circulation times and allergies; moreover, the in situ polymerization of a nonfouling polymeric shell, such as poly(2-methylacryloyloxyethyl phosphorylcholine) (pMPC) or poly(N-vinylpyrrolidone) (PVP) on the surface of protein therapeutics, can shield immunological recognition and clearance, yielding high biological stability for protein therapeutics (Fig. 43).275–278 A prolonged plasma circulation time can be observed (as high as 7 days) with low level of immunogenicity. This in situ polymerization technique can be universally applied to a library of proteins, with different sizes, surface charges, and structures.
 | ||
| Fig. 43 (a) In situ polymerization route of the MPC monomer for building nonfouling surfaces on proteins; (b) SPECT/CT images of a mouse after the intravenous administration of radioactive 125I-labeled pMPC-modified protein; a significant concentration of protein persists even after 120 h; (c) serum urate oxidase (UOx) activities after the repetitive administration of native UOx or UOx nanocapsules (n(UOx)), confirming the ability of the nanocapsules to evade an adaptive immune system. Reprinted from ref. 276 with permission. Copyright (2016) from Springer. | ||
Furthermore, the in situ copolymerization of two or three monomers can also ensure that the protein structure exhibits multiple functions. For example, the copolymerization of MPC and specific monomers can induce additional functions to protein therapeutics, such as targeted delivery (with folic-acid-modified monomers) as cancer therapy or blood–brain barrier (BBB) penetration (with TAT-modified monomer or mustard-modified monomers) as a part of Parkinson's therapy (Fig. 44).279–282 Based on a prolonged circulation time as well as low immunogenicity, all these therapies exhibited significant delivery efficiency. A proper mechanism might involve a longer circulation time that can statistically provide more possibilities; based on this, the specific interaction between the protein and targets becomes significant.
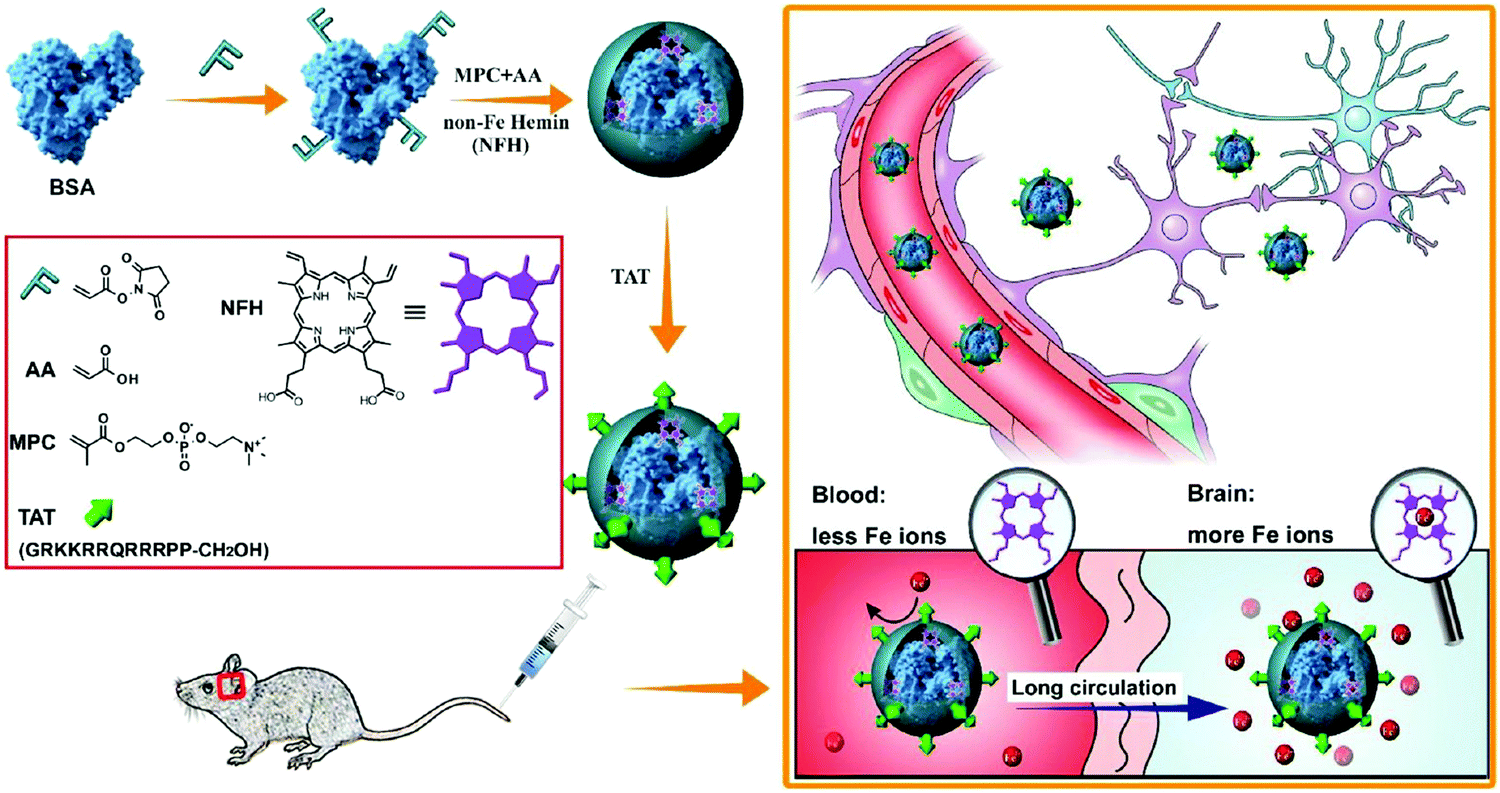 | ||
| Fig. 44 TAT-modified protein obtained by in situ copolymerization and its mechanism of BBB penetration to treat Parkinson's disease. Reprinted from ref. 280 with permission. Copyright (2017) American Chemical Society. | ||
As most functional proteins favor aqueous media, the hydrophobic modification of proteins is always cumbersome, which might afford an unstable dispersed system with protein denaturation. However, hydrophobic modification is desirable for certain applications, such as heterogeneous catalysis and electrochemistry applications. The in situ copolymerization of one hydrophobic monomer with one hydrophilic monomer can offer the opportunity to introduce hydrophobic segments into the protein structure, which might induce additional photoelectronic properties to the protein functions.
For example, in the electrochemistry applications of proteins, such as biosensors and biofuels, the protein converts matter signals (substrate concentrations) into electrical signals via substrate catalysis. Most proteins yield a fast electron turnover rate, resulting in high sensitivity or power output. However, the protein structures have super low conductivity, and the distance between the active center and protein surface is considerably higher than the electron tunneling distance obtained from the Marcus theory; therefore, the electron transfer rate is much lower in applications. Jin et al. designed a cofactor-like molecule, 2′-diallyamino-ethyl flavin (DAA-Flavin), derived from riboflavin and incorporated as a crosslinker into the polyacrylamide (PAAm) network around the GOx surface by in situ polymerization (Fig. 45).283 This incorporated DAA-Flavin could act as an alternative electron acceptor to interact with the active centers of GOx in the presence of the substrate, as well as initiate direct electron transfer to the protein surface. Based on these functions, DAA-Flavin-modified GOx can yield enhanced catalytic activity toward glucose as well as the direct sensing of glucose concentration via fluorescence signal changes.
 | ||
| Fig. 45 (a) Synthesis routes of DAA-Flavin and its redox mechanism; (b) in situ copolymerization of a hydrophobic monomer and a hydrophilic monomer. Reprinted from ref. 283 with permission. Copyright (2018) American Chemical Society. | ||
A variety of polymerization techniques, such as free radical polymerization, ATRP, and RAFT, can be applied in situ for protein modification under mild reaction conditions. Based on these mature polymerization processes, the modification of proteins has become controllable and variable. Because of the diverse choice of polymeric materials, in situ polymerization offers promising diversities in protein structures and functions. Last but not least, the scale-up manufacturing of such modified proteins has become bright and worthwhile.
6.2 CPs for biosensing, imaging, and therapy
CPs are organic macromolecules that are characterized by large π-electronic delocalized backbones, which are capable of effectively coordinating electronic coupling to enhance the light-harvesting and light-amplifying abilities, as discussed in Section 4. It is worth mentioning that the tuning of the compositions and structures of the polymer backbones and pendants allows control over their optical properties. As a result, CPs provide a versatile platform in resolving biomedical issues, such as designing optical biosensors as well as fluorescence imaging and therapeutics.284–287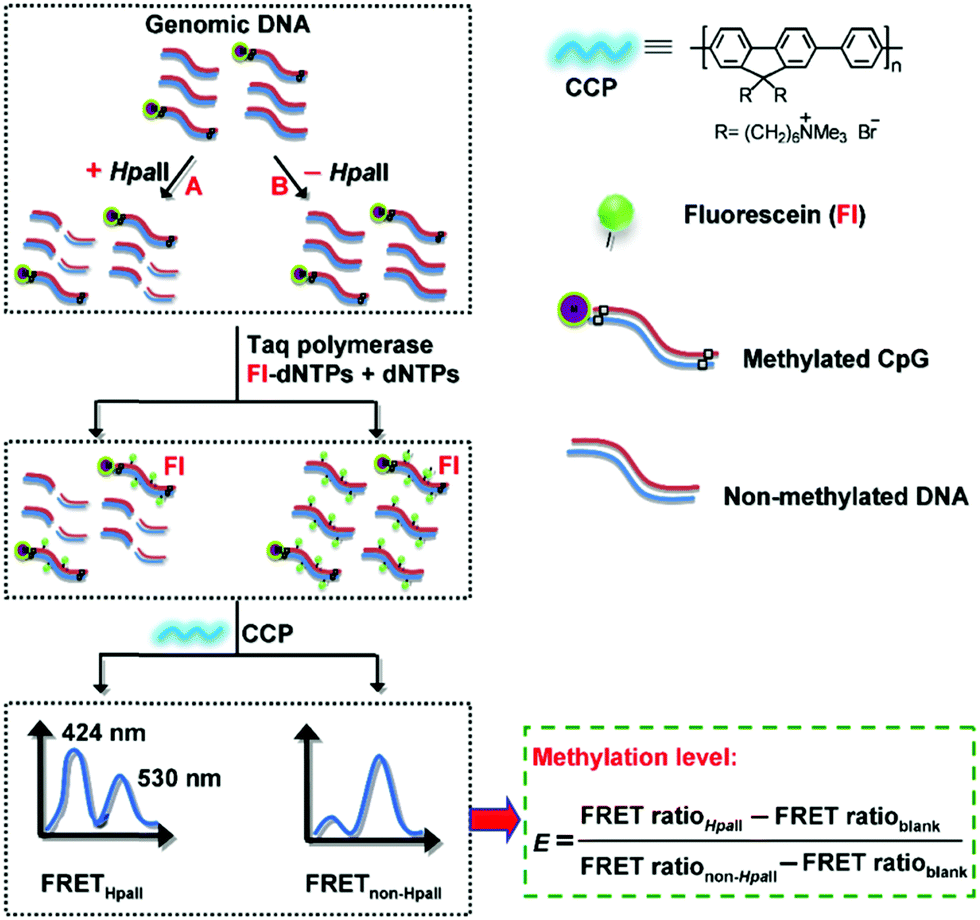 | ||
Fig. 46 Schematic illustration of the CCP-based FRET technique for DNA methylation detection. The methylation level (E) of DNA is calculated according to FRET ratios (I530![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm/I424nm) of HpaII-treated, untreated, and blank control samples. Reprinted from ref. 288 with permission. Copyright (2012) from Nature Publishing Group. nm/I424nm) of HpaII-treated, untreated, and blank control samples. Reprinted from ref. 288 with permission. Copyright (2012) from Nature Publishing Group. | ||
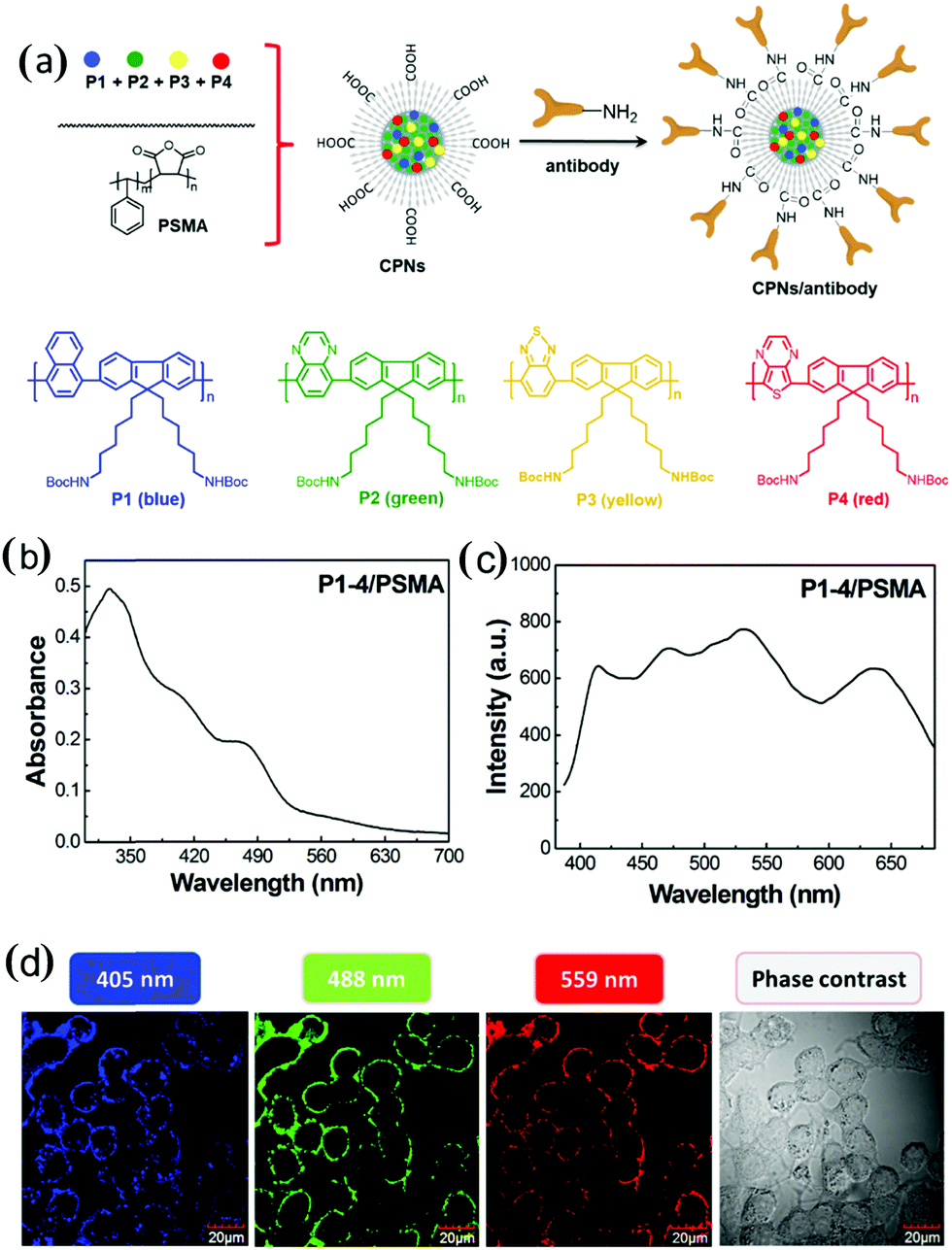 | ||
| Fig. 47 (a) Schematic illustration of the preparation of P5–8/PSMA CPNs and the conjugation of antibody to the CNPs for target imaging. (b) UV-vis absorption and (c) fluorescence emission spectra of CPNs in aqueous solutions (λex = 360 nm). (d) Multichannel fluorescence images of MCF-7 cells incubated with CPNs/anti-EpCAM. The excitation wavelengths are 405, 488, and 559 nm, respectively. Adapted from ref. 289 with permission. Copyright (2014) Wiley-VCH. | ||
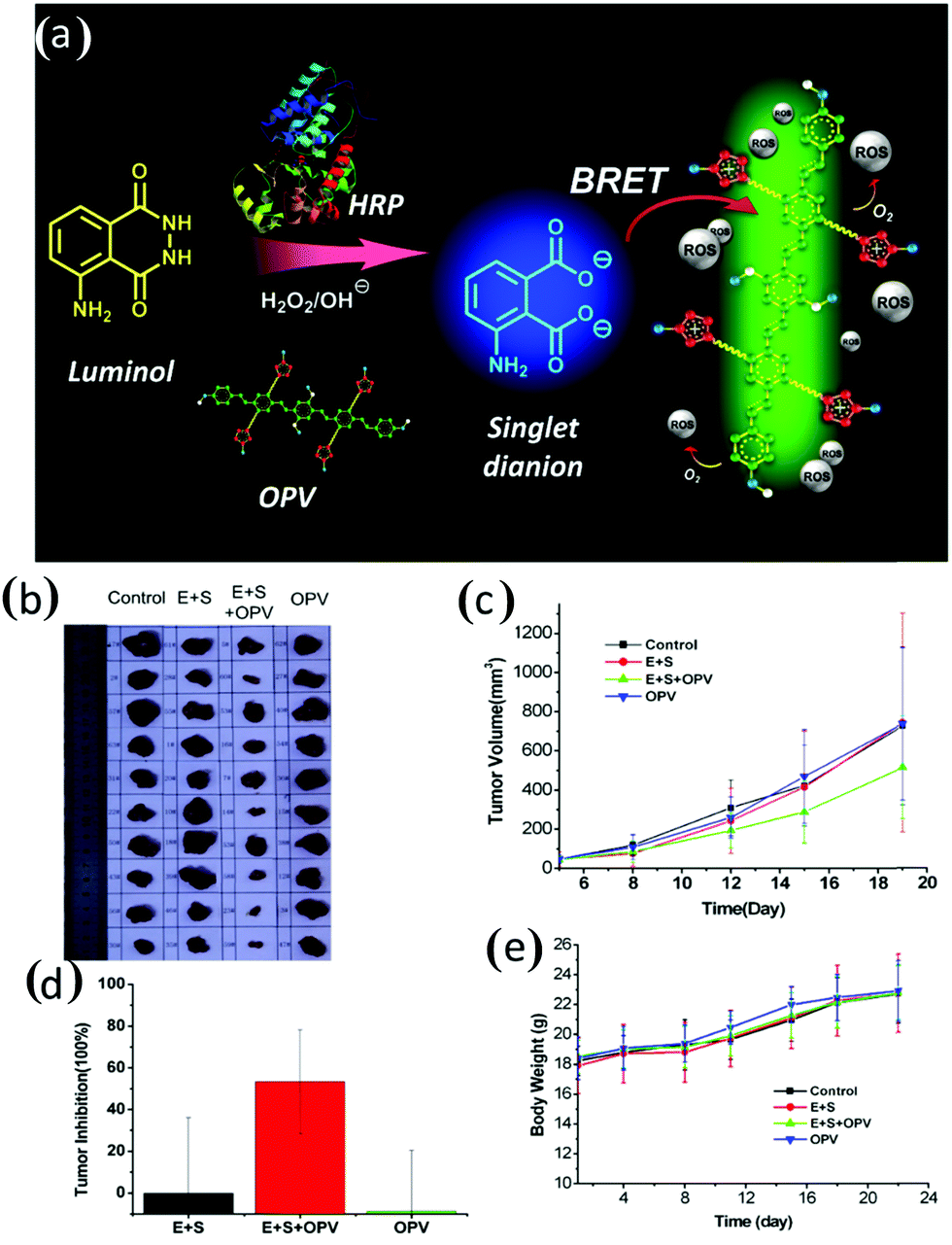 | ||
| Fig. 48 (a) Schematic illustration of the BRET system for PDT and the synthesis of cationic OPV as a photosensitizer. (b) Representative photographs of mice and tumors collected from the tumor-bearing mice before and after 18 days of treatments with the BRET system. (c) Tumor volume as a function of treatment time. (d) Tumor inhibition of mice after 18 days of treatment. (e) Body weight curves after various treatments. Adapted from ref. 291 with permission. Copyright (2012) American Chemical Society. | ||
Recently, Wang et al. constructed, for the first time, an electricity-driven luminous system based on electrochemiluminescence (ECL) for killing pathogenic bacteria, where ECL was used as excitation of the photosensitizer instead of a physical light source in order to produce ROS (Fig. 49).292 The mechanism for ECL therapeutics depends on the perfect spectral overlap and energy transfer from the ECL generated by luminol to the photosensitizer (cationic OPV), thereby sensitizing the surrounding oxygen molecule into ROS. Furthermore, stretchable and persistent hydrogel devices could be designed by integrating stretchable hydrogel, persistent ECL, and antibacterial function into hydrogel matrices. This novel strategy avoids the use of an external light source, making the process simple, convenient, and controllable. This affords new fields for ECL beyond sensors and also opens up another novel model for PDT.
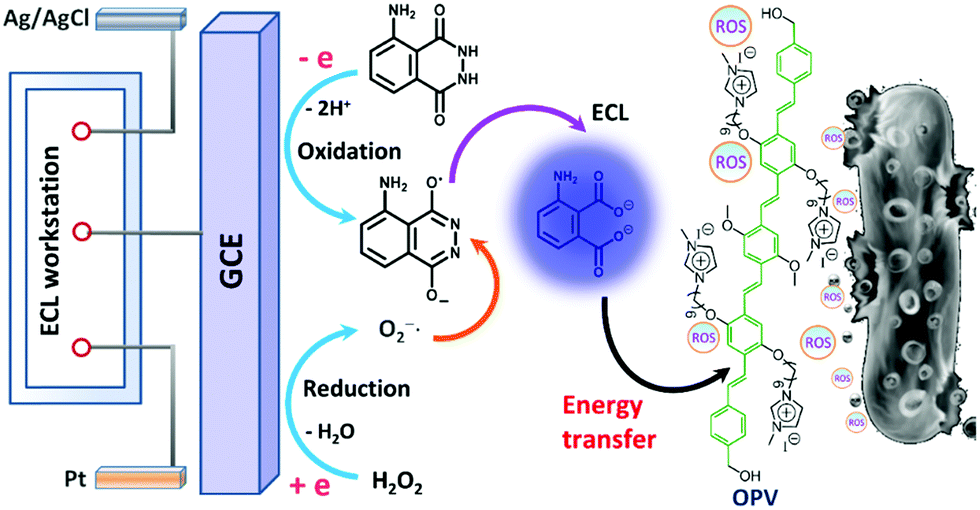 | ||
| Fig. 49 Schematic illustration of electricity-driven mechanism for ECL therapeutics to produce ROS and kill pathogenic bacteria. Ag/AgCl electrode: reference electrode; Pt electrode: counter electrode; glassy carbon electrode (GCE): working electrode. Reprinted from ref. 292 with permission. Copyright (2018) American Chemical Society. | ||
CPs are rapidly emerging as a new photonic platform that can not only be used to monitor and image biophysical markers and tumor metastasis both in vitro and in vivo but also deliver drugs and intelligently trigger photo-attacks. It is believed that the considerable progress made in CPs and their hybrid materials can result in tremendous advances in noninvasive diagnosis, synergistic therapy, and theranostics. There is still plenty of space for further improvement, and further development in this field can promise fascinating applications that can overthrow the currently used operation modes in the field of disease imaging and therapeutics.
6.3 Stimulus-responsive polymers for anticancer drug and gene delivery
It is difficult for cancer nanomedicine to deliver therapeutic agents from the injection site to the action site inside tumor cells due to the presence of multiple physiological barriers in the delivery steps. Earlier studies have shown that the optimal nanoproperties to achieve high delivery efficacy vary for the different delivery steps. Therefore, stimulus-responsive polymers have been developed to endow cancer nanomedicines with self-adaptive nanoproperties to maximize their efficiency at each step. In this subsection, the current designs of stimulus-responsive polymers for cancer nanomedicine to realize 3S transitions have been summarized.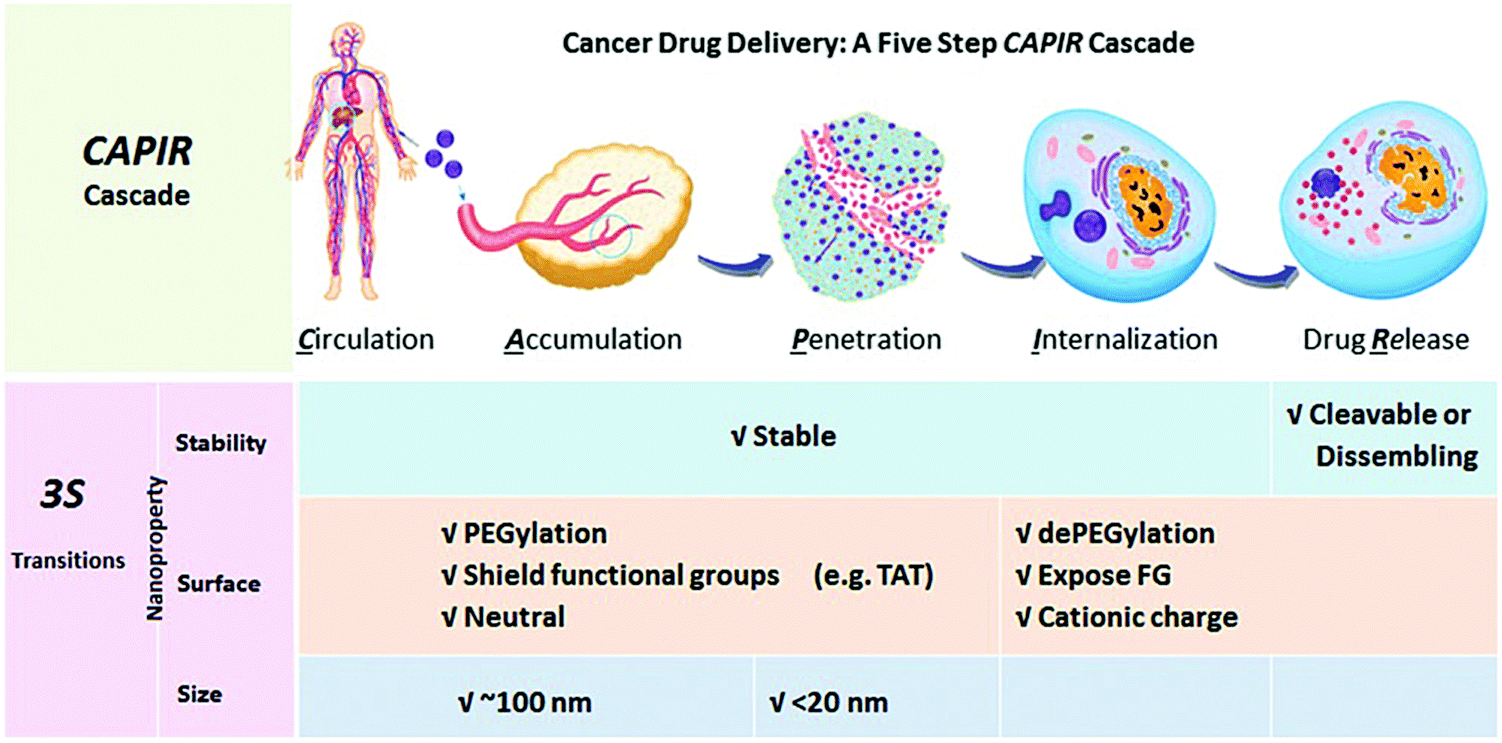 | ||
| Fig. 50 Cancer drug delivery: the CAPIR cascade and 3S nanoproperty transitions for nanomedicines to obtain high overall efficiency. Reprinted from ref. 295 with permission. Copyright (2017) from Wiley-VCH. | ||
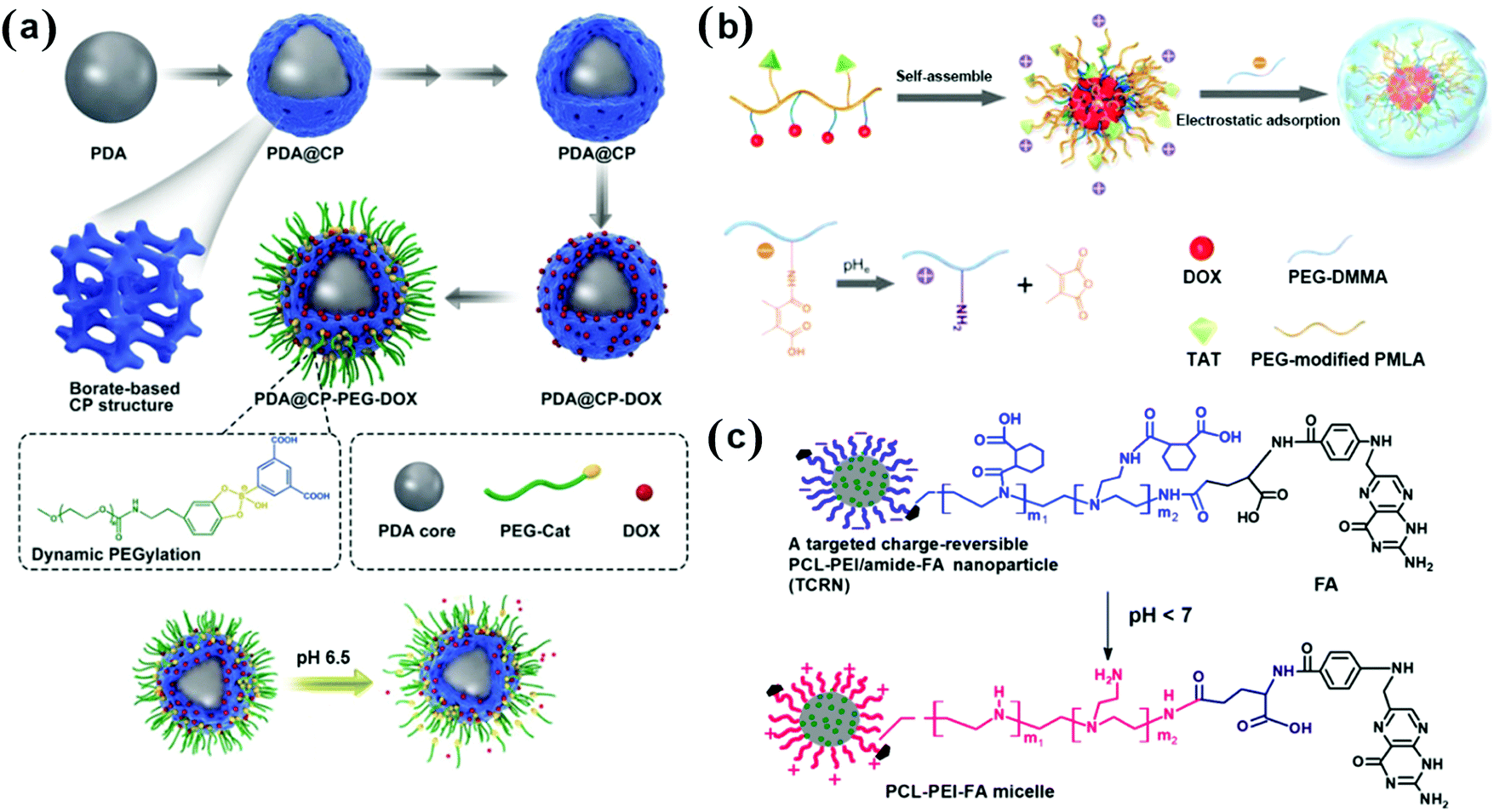 | ||
| Fig. 51 Representative stimulus-responsive polymers for surface nanoproperty transition. (a) Fabrication of dynamically PEGylated, borate-coordination-polymer-coated PDA nanoparticle. (b) Preparation of PEG–DMMA-coated nanocomplex. (c) Structure of the targeted charge-reversal nanoparticle. Reprinted from ref. 307, 310, and 322 with permission. Copyright (2018, 2017, and 2007) from Wiley-VCH and Ivyspring International. | ||
Two general approaches have been used to realize this transition. The first approach is to mask the cationic CPPs via electrostatic interactions by polyanions or polyanions-conjugated PEG. The surface polyanions were designed to shed off in response to pH or enzymes. For instance, 2,3-dimethylmaleic anhydride (DMMA)-amide functionalized PEG and poly(methacryloyl sulfadimethoxine) (PSD)-linked PEG were used to mask the CPPs on NPs.310,311 The DMMA amides underwent a negative-to-positive charge reversal in the acidic tumor microenvironment and therefore abolished their electrostatic interactions with CPPs (Fig. 51b). Hyaluronic acid (HA) was also used to complex with the CPP-functionalized NPs. In tumors, the surface HA degraded by the overexpressed hyaluronidases (HAase) and the exposed CPP for fast tumor cell internalization.312 Another approach is to directly connect the cationic CPPs with short zwitterions through cleavable linkers so that the polyanions can stick on the CPPs and mask the cationic charges. The linkers are chosen to be cleavable by pH, enzymes, or light.313–318 The breakage of linkers weakens the interaction and makes the polyanions fall off. The other strategy is dePEGylation. Apparently, removing the surface PEG chains would expose the target motifs such as folic acid and Arg-Gly-Asp (RGD) anchored with stable linkers.305,319,320
Acidity-triggered charge reversal is commonly used to realize this transition. Amine-containing polymers, such as polyethylenimine (PEI), poly(L-lysine) (PLL), poly(2-aminoethyl methacrylate hydrochloride) (PAMA), amino-functionalized polyphosphoester, and chitosan, were reacted with anhydrides to produce acid-labile β-carboxylic amides, which decomposed in weak acidity to regenerate the amines carrying cationic charges.321–330 As an example, Shen et al. were the first to demonstrate the charge-reversal concept using acid-labile β-carboxylic-amides-functionalized poly(ε-caprolactone) (PCL)–PEI nanoparticle (Fig. 51c). The resulting PCL–PEI/amide micelles underwent a surface charge transition as the pH decreased from 7.4 to 6.0. Subsequently, it was demonstrated that the pH sensitivity of the amides could be tuned by introducing substituents or unsaturated double bonds.322,331 Subsequently, Wang et al. designed a series of charge-reversal systems using more labile amides.303,328,329,332
As alternatives, the protonation/deprotonation of carboxyl or amino groups has been applied to charge-reversal nanomedicine design.333 Tuning the ratios of carboxyl to amino groups or using zwitterionic groups afforded materials that were capable of charge transition.334–340 The advantage of such charge transition is its instant response to pH variations. In a recent study,340 a gene delivery system was fabricated via complexing PEI, PEG–histidine (His)–glutamic acid (Glu) copolymer, and p53 plasmid. By adjusting the ratio of PEI to copolymer, the resulting DNA complex could undergo a negative-to-positive charge reversal as the pH decreased from 7.4 to 6.8. In addition, charge-reversal nanomedicines triggered by other stimuli, such as enzymes or gas molecules, were also reported.308,341
Dendrimers have mostly been used to develop size-transitional multistage delivery systems.346–351 As an example, Shen et al. engineered a lipid-coated dendrimer nanoassembly system to address the size dilemma.346 The nanoassembly was a 45 nm-diameter PEGylated liposome encapsulated with almost 27 dendrimers (5 nm in diameter). Once accumulated at the tumor site, the lipid membrane fused with the cancer cell membrane to induce the rupture of the nanoassembly. The released small dendrimers then penetrated into deeper areas of the tumor. Meanwhile, the 2-(N,N-diethylamino)ethyl termini on the dendrimers protonated in the acidic tumor microenvironment, triggering cellular uptake and release of the hydrophobic payloads (Fig. 52). Likewise, the dendrimer complex was also designed to disassemble in response to a photoacoustic shockwave.352 Recently, a micelle-in-liposome hybrid nanomedicine was designed to release micelles (∼42 nm) from the liposomes (∼94 nm) at the tumor microenvironment by responding to both MMP-9 and low pH.353 In addition, size-shrinkable nanomedicines were designed by the use of enzyme-degradable substrates such as MMP-2-cleavable peptides and gelatin.354–356 The degradation of polymer particles is another approach for size transition. For instance, a nanomedicine composed of ROS-responsive poly(thioketal phosphoester), chlorin e6, and doxorubicin (DOX) could shrink from 154 to 72 nm after the degradation of thioketal backbones by ROS produced under light irradiation.357
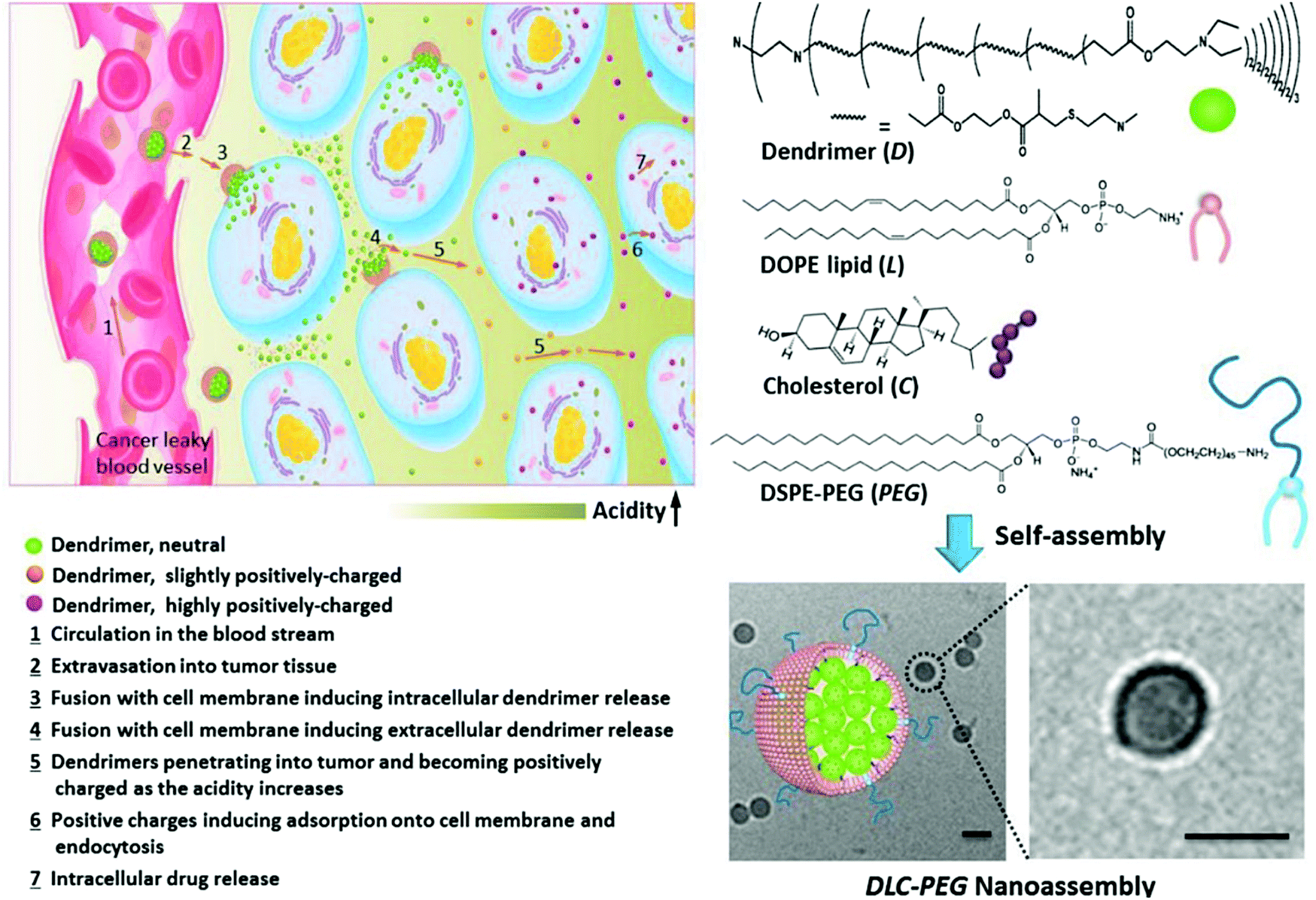 | ||
| Fig. 52 Schematic of the cluster-bomb-like nanoassembly. Reprinted from ref. 346 with permission from Wiley-VCH. | ||
For noncovalently encapsulated drugs, a transition can be realized by the swelling or dissociation of the hydrophobic cores in response to low pH, ROS, enzymes, or adenosine-5′-triphosphate (ATP).372–378 For instance, in the ATP-triggered stability transition, ATP-binding moieties (such as aptamers, chaperonin, phenylboronic acid, or metal ions) were integrated into the cores of the nanomedicines.379–383 After being internalized into the cancer cells, the concentrated ATP molecules were competitively bound to the motifs, causing the disassociation or conformational transition of nanomedicines followed by drug release. Recently, prodrugs, which are converted into active forms by responding to specific signals, loaded into polymeric NPs form a special type of nanomedicine. For instance, Shen et al. developed an enzyme and ROS-dependent cascade amplification strategy to specifically release the drug to cancer cells.384 β-Lapachone, a ROS generator, was coencapsulated with a ROS-sensitive DOX prodrug into the micelles. In cancer cells, the overexpressed NAD(P)H:quinone oxidoreductase-1 (NQO1) enzyme enabled β-lapachone to produce an elevated level of ROS, which decomposed the prodrug and released free DOX in the cascade amplification strategy. This was also demonstrated in other systems.385–388
Polymer gene delivery systems generally have low gene expression efficiency mainly because the tight packing via strong interactions between the cationic polymers and genes makes it difficult to release DNA in the cells for expression.293,298,389 Degradable cationic polymers, which can degrade into low-molecular-weight chains and therefore loose binding to the DNA, have been widely explored to enhance gene release.340,390–394 However, the resulting positively charged short chains may still interfere with gene transfection. To overcome this limitation, Shen et al. developed intracellular positive-to-negative charge-reversal polymers to abolish the binding to complete DNA release, thereby yielding enhanced gene expression.395–398 In one design, the cationic polymers were made to respond to intracellular ROS, which converted them into negatively or neutrally charged (Fig. 53).395,396,399 The polymers degraded quickly in half an hour in the presence of H2O2. In another design, the cationic polymers were made susceptible to esterase-catalyzed hydrolysis (Fig. 54). The hydrolysis of the phenolic acetate triggered charge reversal.397
 | ||
| Fig. 53 (a and b) A positive-to-negative convention mechanism of surface charge for gene carriers. (c) Schematic representation of cationic-lipid-coated ROS-responsive polyplexes for gene delivery. Reprinted from ref. 395, 396 and 399 with permission. Copyright (2017, 2016, and 2018) from Wiley-VCH and American Chemical Society. | ||
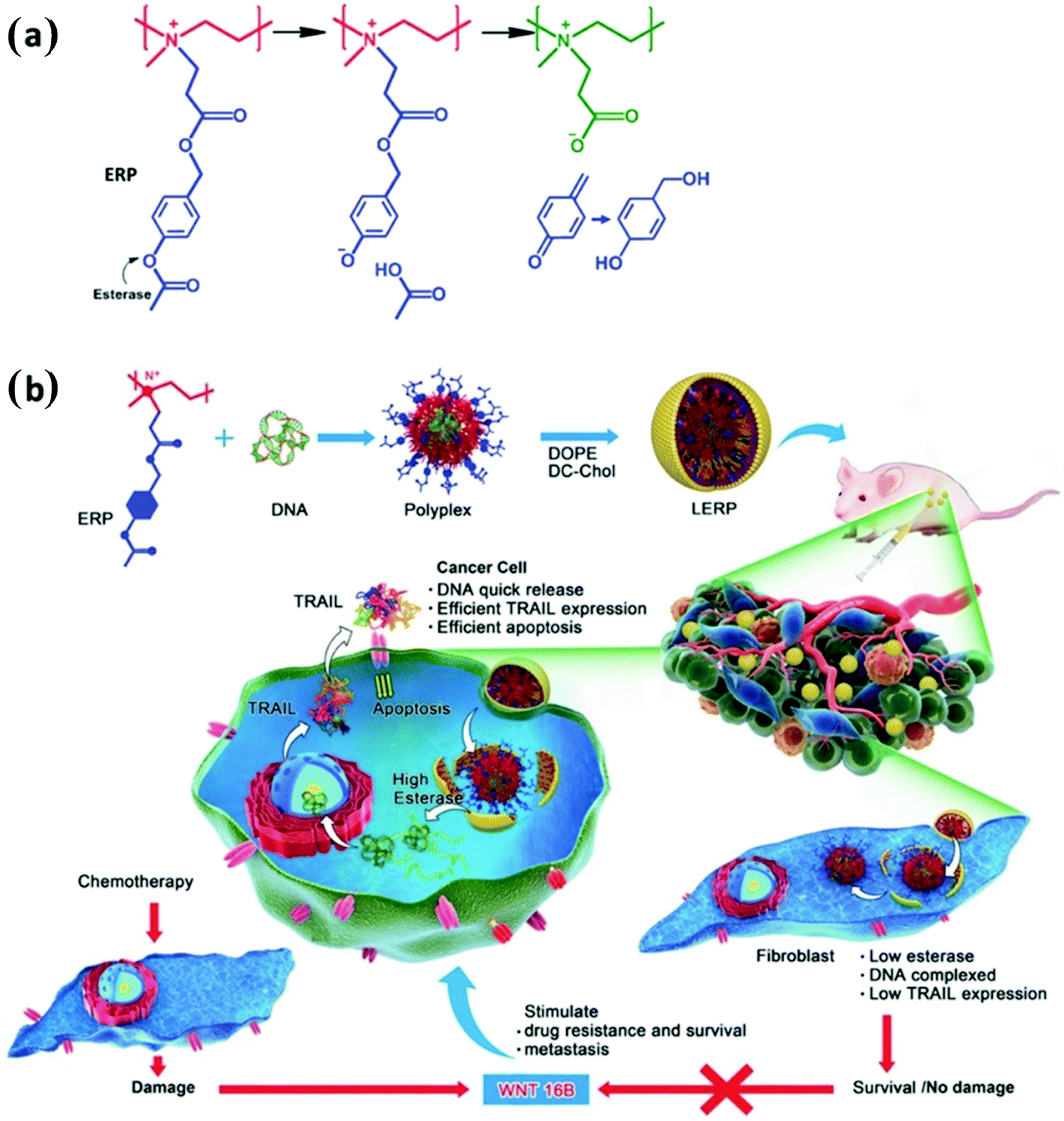 | ||
| Fig. 54 Esterase-responsive charge-reversal polymer. (a) Mechanism of esterase-induced charge reversal of the gene carrier. (b) Illustration of selective gene delivery to cancer cells. Reprinted from ref. 397 with permission. Copyright (2016) Wiley-VCH. | ||
Complicated physiological environments in the human body yield a diverse array of barriers that need to be efficiently overcome by cancer nanomedicines with multiple functions. The 3S nanoproperty transition concept provides a design principle for the integration of necessary functions into a single nanomedicine. Stimuli-responsive polymers are excellent materials to realize such 3S nanoproperty transitions. Further design of biocompatible, highly selective and sensitive, and simply structured polymers is needed for developing highly potent and translational nanomedicines.
6.4 Polymeric vectors for drug and gene delivery
Therapeutic agents, such as chemical compounds, genes, photosensitizers, and proteins, are fragile. They can undergo biodegradation in the organism's physiological environment before they can reach their targets. Therefore, powerful “bodyguard” vectors have seen a rising demand, which can protect their clients from both in vitro and in vivo dangers and facilitate their delivery to the target sites. In the past few decades, the field of drug and gene delivery has grown tremendously by the development of a wide range of advanced delivery systems. Such delivery systems offer novel technologies and promising strategies for precise diagnosis and therapy, including chemotherapy, radiotherapy, immunotherapy, PDT, photothermal therapy (PTT), and ultrasound therapy. Polymer-based delivery systems have been continuously receiving significant attention due to their biocompatibility, higher carrying capacity, structure controllability, low cost, and production ability in large amounts. A number of polymeric delivery systems have been designed for controlled drug and gene release applications, which are capable of responding to a range of external stimuli (e.g., light, heat, magnetic fields, and ultrasound) as well as internal stimuli (e.g., pH, ionic strength, metabolic enzymes, glutathione (GSH), ROS, and ATP).Each disease has its own characteristics as well as microenvironments, which play key roles in disease development and progression. For instance, the tumor microenvironment is a unique cellular environment in which the tumor exists, and it comprises the surrounding blood vessels, immune cells, fibroblasts, bone-marrow-derived inflammatory cells, lymphocytes, signaling molecules, and extracellular matrix (ECM). A tumor can change its microenvironment, which, in turn, can affect the tumor progression and metastasis as a result of its interactions with the host. Accordingly, the microenvironment of the disease provides various internal stimuli, which can be considered as appropriate triggers for smart delivery systems that may cause changes in the phase, size/shape, reactivity, permeability, and surface wettability. Moreover, after conjugating with the targeting ligands (e.g., monoclonal antibodies, peptides, or small molecules), such vectors can be used to target the microenvironment with high specificity and greater affinity. This subsection discusses the use of polymeric vectors to deliver therapeutic drug molecules and bioactive genes inside the organism. The recent contribution by Zhang et al. to this field is summarized, and promising applications in cancer immunotherapy, targeted drug delivery, and highly efficient gene transfection are also demonstrated.
Light-sensitive systems have been used in diverse applications for the development of novel intelligent materials and systems. Light or thermal stimuli can be used to tailor the physical and chemical properties of vectors, such as viscosity, refractive index, conductivity, pH, solubility, wettability, mechanical properties, and polymer morphology. Following this idea, Zhang et al. developed photoresponsive polymers possessing very attractive characteristics. Taking inspiration from nature, the biomimetic concept has been integrated into drug delivery systems. In a recent study by Zhang et al., spinach thylakoid fragments were recombined with lipid molecules to synthesize a hybrid proteoliposome, also called a highly efficient life-support intracellular opto-driven system (HELIOS), for the generation of ATP.400 With red-light irradiation, HELIOS could improve the intracellular ATP concentration to 1.38–2.45 times in various cell lines. Moreover, it was noticed that HELIOS-mediated ATP generation could comprehensively promote the cell functions such as protein synthesis and insulin secretion. At the organ and individual levels, it was proven that HELIOS rescued a mouse heart from myocardial infarction and sustained the life of fasting zebrafish models.
In addition, tumor hypoxia has attained the status of a core hallmark of cancer that globally affects the entire tumor phenotype. Reversing tumor hypoxia might offer alternative therapeutic opportunities for current anticancer therapies. To address this challenge, Zhang et al. further designed a photosynthetic leaf-inspired abiotic/biotic nanothylakoid (PLANT) system by fusing the thylakoid membrane with synthetic NPs for efficient O2 generation in vivo (Fig. 55).401 Under laser irradiation at 660 nm, the PLANT system exhibited intracellular O2 generation. In vivo, it was found that this PLANT system could not only normalize the entire metabolic network but also adjust the abnormal structure and function of the tumor vasculature. This PLANT system could significantly enhance the efficacy of phototherapy or antiangiogenesis therapy. This facile approach for normalizing the tumor microenvironment can have promising potential in tumor therapies.
 | ||
| Fig. 55 Schematic diagram of tumor normalization induced by the PLANT system. Reprinted from ref. 401 with permission. Copyright (2018) American Chemical Society. | ||
Tumor hypoxia can also considerably suppress the therapeutic efficacy of PDT, mainly because the generation of toxic ROS in PDT is highly oxygen-dependent. Different from ROS, the generation of oxygen-irrelevant free radicals is oxygen-independent. Zhang et al. synthesized a therapeutic strategy based on the light-induced generation of free radicals for cancer therapy.402 An initiator of 2,2-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (AIBI) was chosen as the radical source, which was loaded in the hollow interiors of gold nanoclusters (AuNCs), followed by the coating of thermal-responsive poly(N-isopropylacrylamide-co-acrylamide) as the gatekeeper to form the AIBI@AuNC copolymer. Under NIR irradiation, the plasmonic heating effect of AuNCs could induce the decomposition of the initiator to generate alkyl radicals (R˙), which could increase the oxidative stress (OS) and cause DNA damage in the cancer cells, finally leading to apoptotic cell death under different oxygen tensions. As a proof of concept, this research opens up a new field to use various free radicals for cancer therapy.
Recently, bacteria-mediated drug delivery has attracted considerable attention, particularly for tumor therapy. Beyond utilizing their tumor-targeting ability, chemical explorations have been made to directly conjugate drug-loaded nanomaterials and tumor-targeting bacteria. As a biological strategy, genetic engineering has also been used to program bacteria to become in situ anticancer agents. As a further extension, the same group designed a hybrid system of CCN@E. coli to achieve photocontrolled bacterial metabolite therapy (Fig. 56).403 By combining both selectivity of biosynthetic enzymes and robustness of photocatalytic nanomaterials, the bacterial NO generation could be controlled with light irradiation. In addition, self-powered nonpathogenic microorganisms were used to overcome the shortcomings of classical nanomaterials in tumor targeting and organ penetration. The results obtained from proteomics also suggested that the immune response may get involved in this therapy, and the PMT strategy may lead to useful actuation in cancer immunotherapy.
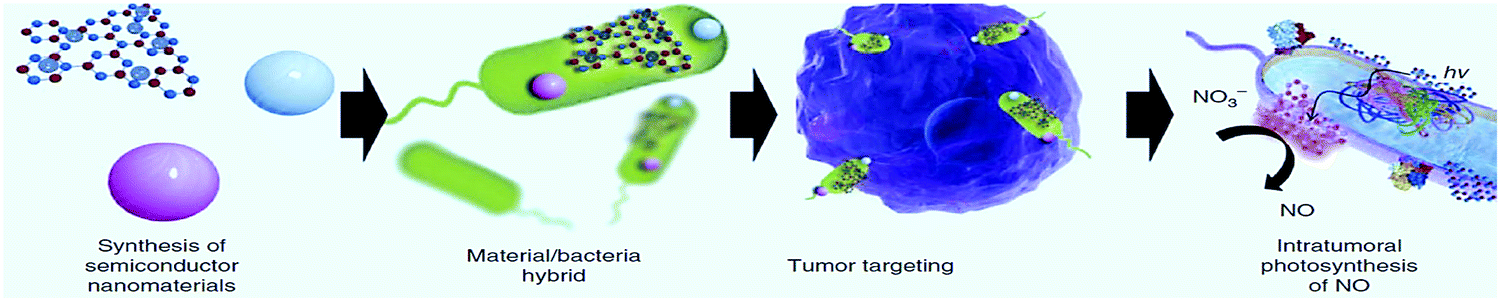 | ||
| Fig. 56 Schematic diagram of the preparation of a PMT system. Reprinted from ref. 403 with permission. Copyright (2018) Nature Publishing Group. | ||
Oral drug administration is widely adopted for diverse drugs and is convenient to use due to its capability to reach different parts of the body via the bloodstream. However, it is generally not feasible for biomacromolecular antitumor drugs, such as proteins and nucleic acids, due to the limited absorption through the gastrointestinal tract and poor tumor targeting. Zhang et al. recently engineered a noninvasive thermally sensitive programmable therapeutic system using E. coli MG1655 as a vehicle for tumor treatments via oral administration.404 Thermally sensitive programmable bacteria (TPB) are transformed with plasmids expressing the therapeutic protein TNF-α and then decorated with biomineralized gold nanoparticles (AuNPs) to afford TPB@Au. AuNPs and TNF-α plasmids efficaciously protected by TPB in the gut can be transported into internal microcirculation via the transcytosis of microfold cells (M cells). The antitumor vehicles accumulate at the tumor sites due to the anaerobic bacterial feature of homing onto the tumor microenvironments. In vitro and in vivo experiments verify the successful delivery of AuNPs and TNF-α plasmids by TPB. Importantly, under remote activation, the expression of TNF-α at the tumor sites can be precisely controlled by the heat generated from the photothermal AuNPs to induce therapeutic actions.
The ability of the engineered cells to communicate with other cells in vivo and interact with the microenvironment of the disease allows them to perform many complex tasks. Inspired by the biological nature of red blood cells (RBCs) as the primary oxygen supplier in mammals, they engineered an aggressive manmade RBC (AmmRBC) to combat the hypoxia-mediated resistance of the tumors toward PDT.405 In particular, the complex formed between hemoglobin and enzyme-mimicking PDA and the PDA-carrying photosensitizer were encapsulated inside the biovesicle that was engineered from recombined RBC membranes. Owing to the same origin of the outer membranes, AmmRBCs shared excellent biocompatibility with the parent RBCs. The introduced PDA played the role of antioxidative enzymes existing inside the RBCs to effectively prevent the oxygen-carrying hemoglobin from oxidation damage during circulation. This biomimetic engineering could facilitate accumulation in tumors, permit efficient in situ oxygen supply, and impose strong PDT efficacy toward the extremely hypoxic tumor with complete tumor elimination.
The development of multifunctional delivery systems that combine targeting, diagnostic, and therapeutic functions within single nanoscale agents is another design trend. Zhang et al. synthesized telluride molybdenum (MoTe2) nanosheets with wide NIR absorbance functionalized with PEG-cyclic arginine-glycineaspartic acid tripeptide (PEG-cRGD).406 After loading a chemotherapeutic drug (DOX), MoTe2–PEG-cRGD/DOX was used for combined PTT and chemotherapy. With high photothermal conversion efficiency, this system exhibited favorable cell-killing ability under NIR irradiation. Owing to the cRGD-mediated specific tumor targeting, it showed efficient accumulation in tumors to induce a strong tumor ablation effect. The in vitro and in vivo experimental results demonstrated that this theranostic nanoagent could be readily degradable in normal organs to enable rapid excretion and avoid long-term retention/toxicity, holding promising potential to effectively treat tumors.
Metastasis and recurrence are two unavoidable and intractable problems in cancer therapy. Immunotherapy might be an effective approach to resolve these problems, but the high heterogeneity of the tumor tissue, inefficient presentation of tumor antigen, and deficient targeting ability of therapy can usually blunt the efficacy of immunotherapy and hinder its clinical applications. In such a circumstance, Zhang et al. developed an approach based on combining photodynamic and immunological therapy.407 The chimeric peptide PpIX-1MT was synthesized, which integrates the photosensitizer PpIX with the immune checkpoint inhibitor 1MT via a caspase-responsive peptide sequence, Asp-Glu-Val-Asp (DEVD), to realize a cascaded synergistic effect. The functional peptide could form NPs in PBS and accumulate in the tumor areas via the EPR effect. Upon light irradiation at 630 nm, the PpIX-1MT NPs produced ROS, induced the apoptosis of cancer cells, and facilitated the expression of caspase-3 and production of tumor antigens; these triggered an intense immune response. The subsequently released 1MT upon caspase-3 cleavage could further strengthen the immune system and effectively facilitate the activation of CD8+ T cells. This cascaded synergistic effect could effectively inhibit both primary and lung metastasis tumors, which may provide a solution for clinically resolving tumor recurrence and metastasis.
Gene therapy has been receiving considerable attention as an efficient treatment of inherited genetic disorders and a wide range of severe diseases. Not only efficient but also rapid transfection is required since it can maximize the bioavailability of vector-carried genes prior to cellular excretion. However, the “rapid” goal has been less investigated so far in the field of vector-aided transfection research. To address this challenge, Zhang et al. synthesized the lysosome-targeting acidity-responsive nanoassembly as gene vectors, which proved the amazing potency to mediate the “superfast” transnuclear gene transport and gene transfection with high efficiency both in vitro and in vivo (Fig. 57).408 The nanoassembly was constructed on the pH-reversible covalent boronic acid–diol coupling between 1,3-diol-rich oligoethylenimine (OEI-EHDO) and phenylboronic-acid-modified cholesterol (Chol-PBA). The nanoassembly-mediated transfection at 8 h could afford an outcome that was comparable to that achieved at 48 h by the golden standard of PEI25k; high transfection efficiency could be retained during 48 h. The in vitro and in vivo results exhibited the low toxicity of this biodecomposable nanoassembly.
 | ||
| Fig. 57 Acidity-responsive gene delivery for “superfast” nuclear translocation. Reprinted from ref. 408 with permission. Copyright (2016) from Elsevier Ltd. | ||
With the prominent progress of polymeric delivery systems, novel biomaterials with high biocompatibility and versatile functions are urgently needed. Zhang et al. explored the hierarchical micro-/nanostructures of human hair that can be further broken into hierarchical microparticles (HMPs) and hierarchical nanoparticles (HNPs) with top-down procedures.409 When compared with commercialized carriers, such as liposomes or albumin NPs, the obtained particles exhibited high hemocompatibility and negligible immunogenicity. Furthermore, these materials also exhibited peculiar abilities with respect to light absorption and free radical scavenging. Evidently, HMP and HNP could prevent UV-induced damage in skin and relieve symptoms of cataract in vitro. Moreover, both HMP and HNP showed satisfactory photothermal conversion ability. By using microcomputed tomography and intravital fluorescence microscopy, it was found that warfarin-loaded HMP could prevent vein thrombosis in mice. In another aspect, HNP modified with tumor-targeted aptamers exhibited dramatic antineoplastic effect and suppressed 96.8% tumor growth in vivo. Therefore, the multifaceted materials described here might provide new vectors for addressing biomedical challenges.
Although considerable advances have been achieved in the field of drug and gene delivery, limitations in polymer-based delivery approaches still persist, which have to be overcome before they can be applied as standard practice in clinics. With regard to drug delivery systems, further developments are certainly needed in this direction, particularly with regard to interactions between the specific microenvironmental conditions of disease and functional design of biomaterials. Determining a clear and stringent understanding of the delivery mechanisms is also necessary to address in vivo safety concerns. With regard to gene delivery systems, although polymeric nonviral vectors are generally less efficient in delivering DNA and initiating gene expressions in vivo as compared to viral vectors, they have promising potential for producing large quantities under the required GMP conditions. There is certainly higher interest in developing new generations of nonviral vectors, which can not only find the target cells but also put the gene in the correct position on the host genome. Until now, the effective delivery of multiple CRISPR components in vivo into host cells still remains a formidable challenge. With the robust development of functional biomaterials, smart polymer-based carriers might become potent vectors for the delivery of CRISPR/Cas9 systems. These developments are very exciting for the drug and gene delivery fields and could have an enormous impact on pharmaceutical technology and clinical treatments. Numerous challenges in the delivery field need to be addressed, and considerable efforts should be made in this direction before translating them into clinics.
6.5 Polyprodrug amphiphiles for drug delivery and theranostics
The use of a polyprodrug strategy for drug delivery provides a plethora of advantages such as customized structural design, ultrahigh drug loading contents, avoidance of premature leakage, and bespoke multifunctional integration.410 Recently, Liu et al. proposed a new concept, namely, polyprodrug amphiphiles, to further enrich the connotation and broaden the vision of polyprodrug strategies. Polyprodrug amphiphiles are a type of specific amphiphilic BCPs, in which one of the blocks is composed of prodrug units. The drug molecules are generally covalently attached to the polymer backbones through stimuli-responsive linkages. The design of stimuli-responsive drug-based monomers may provide the following unique advantages: (1) increasing the water dispersibility and blood circulation stability of water-insoluble and chemically labile drugs; (2) avoiding premature drug release and reducing the systemic cytotoxicity of drugs; (3) optimizing pharmacokinetics and pharmacodynamic performance and elevating the therapeutic efficacy; and (4) integrating diagnostic/imaging functions to develop theranostic nanovectors. In a typical example, an anticancer drug, namely, camptothecin (CPT), was monomerized with reduction-sensitive disulfide linkage, which was subsequently polymerized into amphiphilic PEG-b-PCPTM via the RAFT polymerization technique by using a PEG-based macroRAFT agent.411 The CPT loading contents could be delicately tuned and over 50 wt% CPT contents could be facilely loaded. Remarkably, PEG-b-PCPTM could be self-assembled into four types of distinct morphologies, namely, spherical micelles, large compound vesicles (LCVs), smooth disks, and unprecedented staggered lamellae with spiked peripheries (Fig. 58). Further studies revealed that, in sharp contrast to the other three types of nanoassemblies, staggered lamellae possessed extended blood circulation time, unique endocytic pathways, and the fastest cellular internalization rate. Moreover, the four types of nanostructures exhibited drastic CPT release profiles, disassembly kinetics, and in vitro cytotoxicities. These observations highlighted the importance of self-assembled morphologies in drug delivery applications and demonstrated that morphology control should be an important consideration in the design of nanomedicines. In this context, Hubbell et al. functionalized ibuprofen (IBU)—a well-known antiinflammatory drug—with 2-hydroxyethyl acrylamide via the formation of an ester bond.412 The resultant monomer could be readily polymerized via RAFT polymerization to form amphiphilic PEG-b-PIBU copolymers by the use of a PEG-based macroRAFT agent. The self-assembly of PEG-b-PIBU copolymers led to the formation of both spherical and worm-like micelles, depending on the block ratios of PEG and PIBU. The self-assembly morphology played a critical role in the IBU release behavior and exerted an antiproliferative effect on human cervical carcinoma (HeLa) and murine melanoma (B16-F10) cells.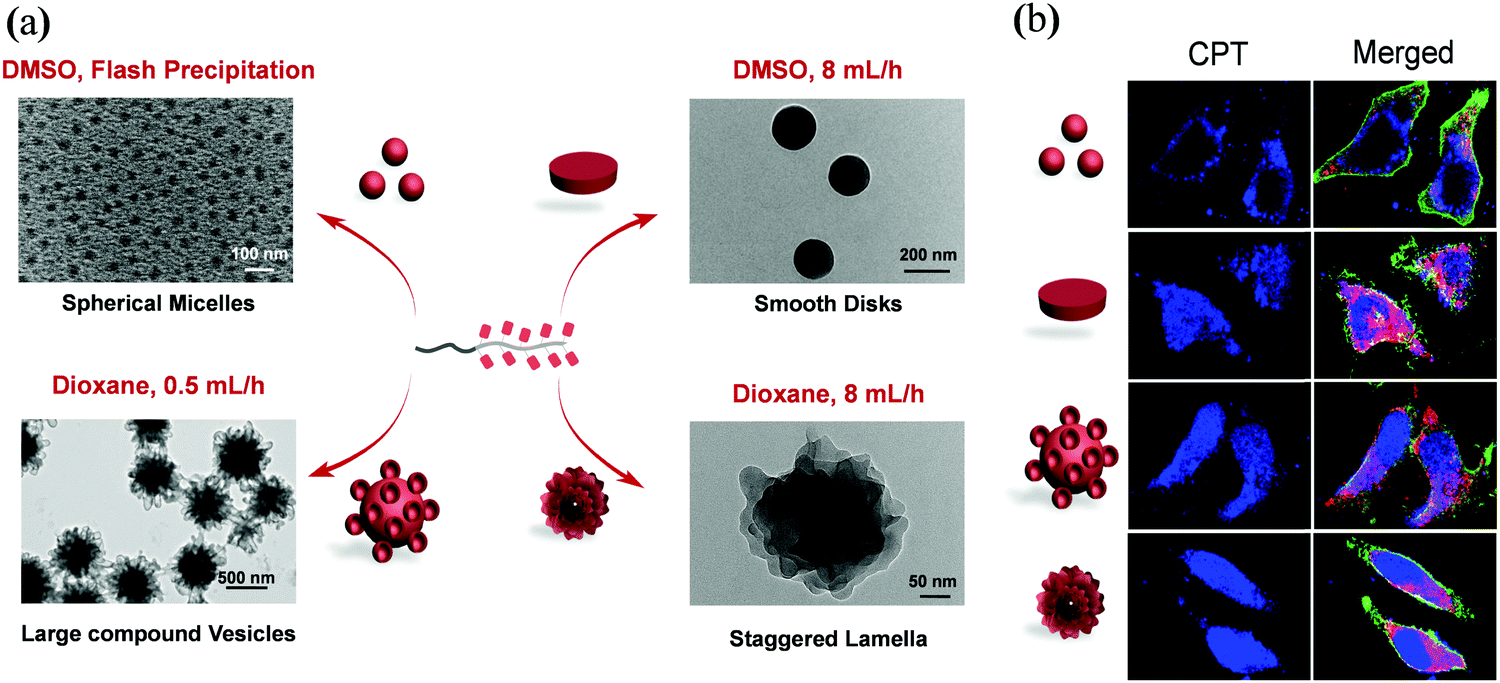 | ||
| Fig. 58 (a) Self-assembly of PEG-b-PCPTM polyprodrug amphiphiles with the formation of four types of nanostructures. (b) Distinct cellular internalization of these four types of nanostructures. Adapted from ref. 411 with permission. Copyright (2013) American Chemical Society. | ||
Indeed, the polyprodrug approach provides a general and robust procedure to fabricate highly efficient nanocarriers with extremely high drug loading contents.413–417 Moreover, the design of stimuli-responsive drug-based monomers renders it possible to fabricate polyprodrug amphiphiles with varying chain topologies and to integrate other functions. For example, besides linear polyprodrug amphiphiles, hyperbranched polyprodrug amphiphiles were synthesized by the polymerization of CPTM using a methacrylate-monomer-based RAFT agent. Moreover, a magnetic resonance imaging (MRI) contrast agent was initially embedded within the hydrophobic inner cores with suppressed relaxivity, but the MRI signals could be switched on when being exposed to cytosolic-abundant GSH, which also led to the release of the CPT drug by the cleavage of the disulfide linkage (Fig. 59). As such, both the release process of CPT drug and therapy outcomes could be monitored in situ and reported on the basis of enhanced MRI signals, which was fairly beneficial for the development of personalized nanomedicines. Notably, the self-assembled nanoassemblies of polyprodrug amphiphiles were subjected to extensive dilution and higher shear force in the bloodstream upon administration, disintegrating the assembled nanostructures and compromising the therapeutic efficacy. Hyperbranched polyprodrug amphiphiles can, however, exist in the form of unimolecular micelles even after extensive dilution, providing long-term stability when applied for drug delivery.
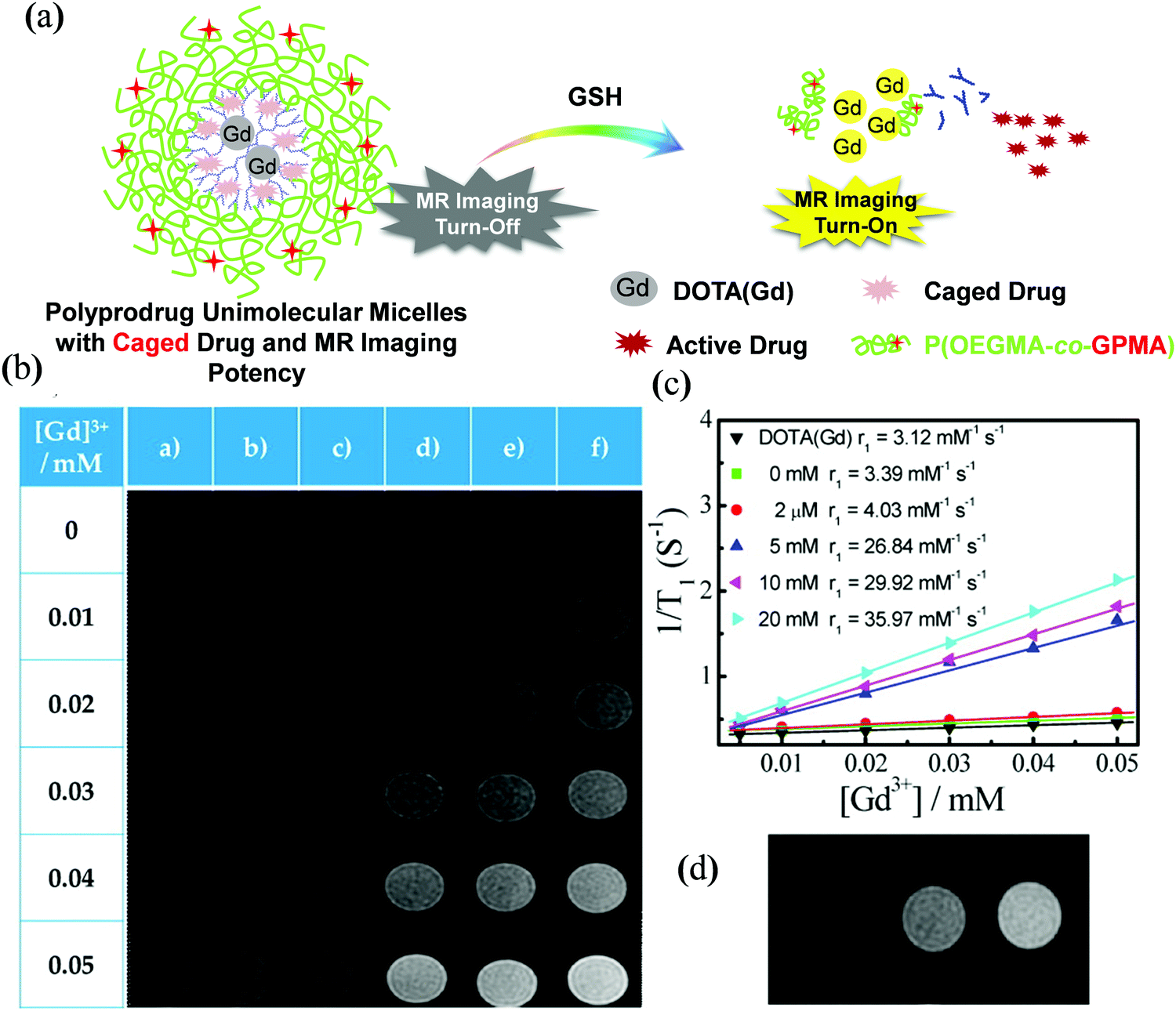 | ||
| Fig. 59 Hyperbranched polyprodrug amphiphiles with caged CPT prodrug and MRI contrast agents within the inner cores shielded by hydrophilic P(OEGMA-b-GPMA) coronas exhibit GSH-triggered intact CPT release and enhanced MRI relaxivity (OEGMA: oligo(ethylene glycol)methyl ether methacrylate; GPMA: N-(3-guanidinepropyl) methacrylamide). Adapted from ref. 413 with permission. Copyright (2015) American Chemical Society. | ||
The fabrication of polyprodrug amphiphiles represents an emerging but promising approach to design novel therapeutic platforms. The tunable chemical structures, ultrahigh drug loading contents, and versatile self-assembled morphologies can not only provide an opportunity to unravel the interrelation between the assembled nanostructures and therapeutic performance but also shed light on how to overcome the drawbacks encountered in conventional drug carriers. Recently, the direct linkage of hydrophobic and hydrophilic drugs without recourse to other components enabled prodrug amphiphiles to afford drug loading content of 100 wt%.418 This example suggests that the drug loading contents of polyprodrug amphiphiles could be remarkably elevated by the synergistic introduction of drugs with distinct water solubility. Future direction in this topic can include, but not be limited to, the expansion of drug categories, screening of stimuli-responsive linkages, and introduction of targeting motifs. Meanwhile, the detailed self-assembly mechanism of polyprodrug amphiphiles and how to precisely control the self-assembled morphologies should be conducted to achieve controlled self-assembly behavior. Moreover, considerable effort should be devoted toward the assessment of the in vivo behavior and therapeutic performance of polyprodrug amphiphiles and preclinical evaluations could be carried out at an appropriate time.
6.6 Interactions of biomaterial surfaces/interfaces
Biomaterials come into contact with biological environments (e.g., blood, body fluids, tissues) through their surfaces, and the immediately occurring biointerfacial interactions determine the ultimate biocompatibility and the realization of biofunctionality of such biomaterials.419–421 The communication between biological structures (e.g., cells) and physical objects (e.g., biomaterials) occurs at their interface (called the biointerface), and the surface of the given biomaterial is one of the most important elements to determine the outcomes of this communication. Biointerface, an interdisciplinary subject among the diverse fields of physics, chemistry, biology, materials science, and biotechnology, is becoming one of the most innovative and expanding areas of science and technology. In the recent decade, considerable efforts have been devoted toward the creation of synthetic biointerfaces either to mimic the biological microenvironment for the fundamental study of cell biology or to endow the required functions for specific applications.422,423 For the construction of functional biointerfaces, scientists and engineers have developed a series of surface modification techniques, namely, layer-by-layer (LbL) assembly, surface-initiated polymerization, plasma polymerization, dopamine- or metal–phenolic-networks-assisted deposition, and so on.424–428 The resultant biointerfaces have been used to investigate cellular behaviors such as adhesion, proliferation, migration, and differentiation in response to surface properties, to improve the blood compatibility and antithrombotic ability for blood-contacting materials and devices, to kill pathogenic bacteria and eliminate biofilm for implants, and to detect analytes and analyze specific biomolecular interactions for biosensors and biochips.429–435 In addition, the functionalization of the biointerfaces of nanomaterials is also a research hotspot for controlled and targeted drug delivery.436,437Among a series of biointerfacial interactions, protein adsorption has been well recognized as the first significant event, which plays an important role in events such as cell adhesion, platelet activation, and host responses.438–441 On the positive side, the adsorption of adhesion proteins on cell culture substrates is beneficial for the promotion of cell adhesion and proliferation, which is crucial for regenerative medicine and tissue engineering.442,443 The selective immobilization of bioactive proteins on the matrix is an important step for the fabrication of biosensors and microarrays.444,445 On the negative side, nonspecific protein adsorption on synthetic material surfaces can result in several undesired effects such as thrombosis, bacterial infection, and biofouling contamination, leading to the failure of implant materials or the reduction in the sensitivity of diagnostic devices.446,447 Therefore, understanding and further regulating such interactions between proteins and biomaterial surfaces have become an increasingly important goal for a range of scientific fields and applications.448–450
The acquisition of an accurate amount of adsorbed proteins on material surfaces is highly desired for the study of protein–surface interactions and is essential for the evaluation of antifouling properties. However, obtaining protein adsorption in the nanogram range or comparing subtle differences in protein adsorption between various surfaces remains challenging for currently available techniques. Although several methods have been used to determine the quantity of protein adsorbed, the results usually differ. Unfortunately, the reasons leading to such differences are always ignored, which are unfavorable for the rational determination of protein adsorption on a given surface. In this regard, Chen et al. compared the three most commonly used quantitative characterization methods, namely, radiolabeling, surface plasma resonance (SPR), and quartz crystal microbalances (QCM), to systematically discuss their applicability for the evaluation of protein adsorption using a series of model gold surfaces with gradient hydrophilicity.451 They found that when converting SPR signals into protein adsorption, the conversion coefficient was not a fixed value, but it varied with the amount of adsorption. The variation in protein adsorption with wettability measured by QCM was different than that measured by SPR or radiolabeling, possibly because QCM is used to measure both adsorbed proteins and their associated water molecules. In addition, due to the acoustic principle of QCM, it has performance limitations when working with surfaces modified with thick viscous layers. In order to explore this effect, a series of surfaces modified with self-assembled monolayers or hydrophilic polymer brushes with different thicknesses were prepared, and the protein adsorption on these surfaces measured by QCM and SPR were compared.452 The experimental results showed that for larger proteins that are expected to adsorb mainly “on top” of the polymer brushes, QCM could not detect a small amount of protein adsorption if the thickness of the polymer brushes exceeded a certain “critical value”; such a critical value increased with polymer viscosity. However, for smaller proteins that are expected to adsorb mainly at the chip substrate and within the polymer brushes, the amount of protein adsorption is detectable by QCM. In conclusion, these findings suggest that QCM data indicating very low protein adsorption on surfaces grafted with polymer brushes should take into account these considerations and should be generally treated with caution.
As mentioned above, the regulation of protein adsorption on biomaterial surfaces (selective binding of favorable proteins and repelling nonspecific proteins) will be beneficial for the promotion of favorable events and the prevention of undesirable events. Taking blood-contacting biomaterials and devices as examples, the initially adsorbed protein layer on the surfaces trigger adverse responses such as plasma coagulation and thrombosis generation. In order to resolve these serious problems, various approaches have been developed to improve the blood compatibility of surfaces by controlling the adsorption of plasma proteins. These modified surfaces can be generally divided into three types: (1) bioinert surfaces that prevent thrombus formation by resisting the adsorption of nonspecific proteins (particularly fibrinogen, which is abundant in plasma and plays a major role in blood coagulation); (2) anticoagulant surfaces that inhibit thrombus formation by suppressing thrombin activity (the key protein in the coagulation process); and (3) fibrinolytic surfaces that destroy the clot (thrombus) once formed by selectively binding essential proteins in the fibrinolytic system. Here, we focus on the recent progresses made in the development of fibrinolytic surfaces by Chen et al.; further, the first two strategies are not described in detail due to space limitations; interested readers should refer to other excellent reviews.431,453,454
Generally, fibrinolytic surfaces mimic the naturally occurring fibrinolytic processes in the body by the generation of a clot-lysing enzyme called plasmin to break down the nascent clot before it poses any danger.455 The two main proteins involved in this process were plasminogen (Plg)—a precursor of plasmin—and a plasminogen activator (e.g., tissue plasminogen activator, tPA). Therefore, the main strategy for building fibrinolytic surfaces is to endow the surfaces with the capability to selective bind Plg and/or tPA in significant quantities when they come into contact with blood. Inspired by natural binding events involved in protein recognition, the bioaffinity ligand for Plg and tPA—ε-lysine with free carboxyl and ε-amino groups—is screened out and used for the construction of fibrinolytic surfaces. In the initial works of Chen et al., they attached ε-lysine to surfaces using hydrophilic polymers (e.g., PEG and poly(2-hydroxy-ethyl methacrylate) (PHEMA)) as spacers; the resultant surfaces could both reduce nonspecific protein adsorption and platelet adhesion as well as capture Plg from plasma with a high degree of selectivity.456,457 When treated with tPA and incubated in plasma, these surfaces could lyse the clots formed on them or adjacent to them. This clot-lysing ability increased with a decrease in the length of the spacer and increase in the density of ε-lysine. To simplify the preparation process and avoid multistep surface modification procedures, they designed an ε-lysine-containing monomer, namely, lysyl methacrylate (LysMA), and used it for either graft copolymerization with HEMA from surfaces containing vinyl groups or copolymerization with HEMA for further blending with polyurethane (PU).458,459 The Plg-binding capacity of the copolymer films could be simply regulated by varying the compositions of LysMA and HEMA. To further increase the local density of ε-lysine, Chen et al. synthesized a multivalent ligand, namely, a β-cyclodextrin derivative bearing seven ε-lysine ligands (CD-lysine7); they demonstrated that an increase in the local density of ε-lysine resulted in higher Plg adsorption and higher Plg binding affinity, possibly due to the well-established “multivalent effect.”460 In particular, using CD-lysine7 and other bioactive ligands, Chen et al. developed a series of dual-functional blood-compatible surfaces via a sequential coimmobilization strategy.461 Surfaces were first modified with a copolymer of poly(2-hydroxy-ethyl methacrylate-co-1-adamantan-1-ylmethyl methacrylate) (P(HEMA-co-AdaMA)), which served as a linker/spacer for the subsequent sequential attachment of functional peptides (e.g., REDV (Arg-Glu-Asp-Val) peptide with the capability to promote adhesion and proliferation of endothelial cells) via covalent bonding and CD-lysine7 host–guest interactions, respectively.461 The resultant surfaces exhibited bioactivities toward both the ligands; more importantly, neither of the two functions of peptide and lysine gets compromised by the presence of the other, providing enhanced blood compatibility.
The abovementioned works mainly focused on the selective binding of Plg; evidently, Plg alone is insufficient to generate plasmin as it necessitates the activation of the bound plasminogen. However, the activators of Plg normally exist at extremely low concentrations in bodily fluids. Therefore, surfaces are expected to either release the preloaded tPA or capture tPA from plasma. In this regard, taking advantage of the fact that Plg has higher affinity than tPA toward surface-bound ε-lysine, Chen et al. developed a new tPA-releasing concept based on protein–protein displacement.462 This concept was realized by the use of a PU fibrous mat modified with ε-lysine for the incorporation of tPA via specific interaction with lysine residues. As shown in Fig. 60, in contact with the plasma, the preloaded tPA was released from the surface by the displacement of Plg; the amount of released tPA correlated with the amount of adsorbed Plg. This tPA-releasing system provides dual mechanisms for clot lysis, namely, by tPA release from the surface and by plasmin generation on the surface. In addition to using a shared ligand for Plg and tPA, Chen et al. also developed a dedicated affinity ligand specific to tPA, i.e., the ARMAPE (Ala-Arg-Met-Ala-Pro-Glu) peptide.463,464 This peptide is derived from plasminogen activator inhibitor-1 (PAI-1) and is involved in the initial binding of tPA to PAI-1. The binding of tPA through the ARMAPE peptide has a particular advantage in protecting tPA from inactivation by PAI-1 in vivo, while the full enzymatic activity of tPA is retained. Taking advantage of ε-lysine and ARMAPE in exhibiting high affinity separately toward Plg and t-PA, a dual-affinity surface formed by immobilizing the two ligands on PU was developed.465 Differing from the tPA-releasing materials, this dual-affinity surface could continuously enrich Plg from blood and generate plasmin in situ to lyse fibrin clots formed on the surface.
 | ||
| Fig. 60 Schematic illustration of the concept of tPA release based on tPA–Plg displacement. Adapted from ref. 462 with permission. Copyright (2016) Royal Society of Chemistry. | ||
Surface modification with bioactive agents capable of combating thrombosis is a widely used strategy for developing antithrombotic biomaterials. However, the exposure of blood to antithrombotic agents on the material surface may cause hemostatic disorders under normal conditions. An ideal blood-contacting material should initiate antithrombotic activity once the thrombus begins to form, while it remains “bioinert” in the normal blood environment. To this end, Chen et al. developed a thrombosis-responsive surface coating with the ability to lyse fibrin as it forms.466,467 Thrombin generation is a key event in blood coagulation/thrombosis and therefore has been used as a physiologic stimulus or trigger in the design of this coating. As shown in Fig. 61, the coating consisted of nanocapsules (NCs) in which the fibrinolysis activator t-PA was encapsulated in a thrombin-degradable hydrogel shell. This t-PA NC surface was stable in normal plasma along with the release of t-PA at a constant rate and efficiently lysing fibrin clots when thrombin was present. In addition, it could release sufficient t-PA for clot lysis in response to endogenous thrombin generated during the clotting process in whole blood.
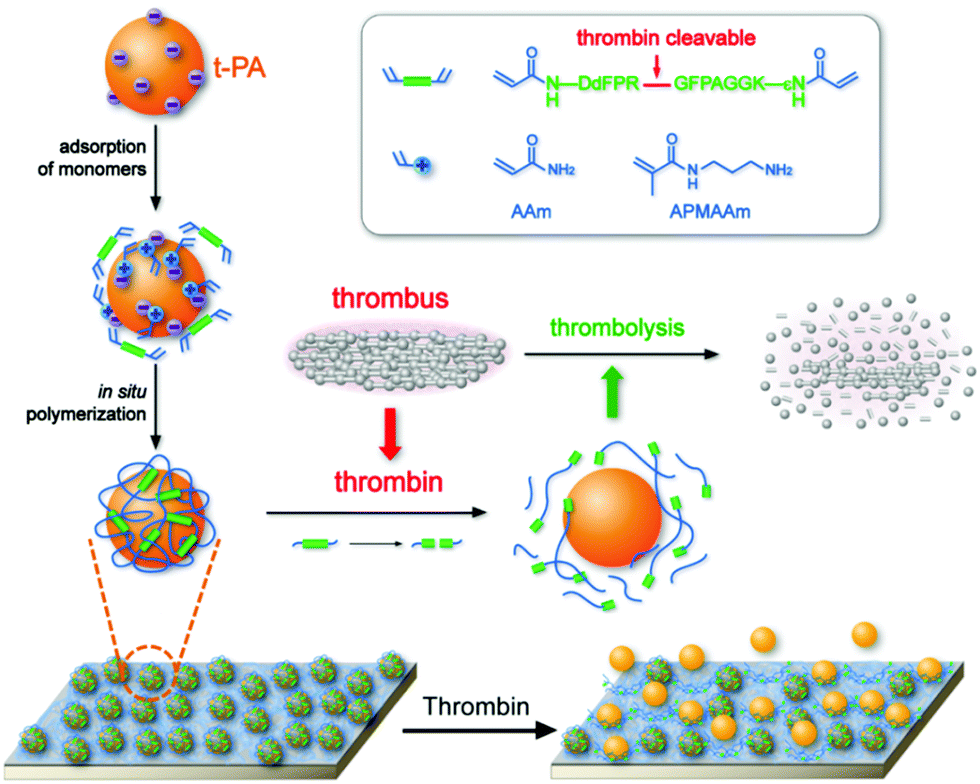 | ||
| Fig. 61 Schematic illustration of thrombosis-responsive thrombolytic coating based on the thrombin-degradable nanocapsules of t-PA. Adapted from ref. 466 with permission. Copyright (2017) from Wiley-VCH. | ||
6.7 Biodegradable polymers for marine antibiofouling
Marine biofouling formed by the settlement and accumulation of microorganisms, plants, and animals on surfaces immersed in seawater has profound effects on the maritime industry. It accelerates biocorrosion, increases hydrodynamic drag (and therefore fuel consumption), and transports invasive species. More than 4000 fouling species live in the sea, and they are fairly different from area to area; they also vary with temperature, salinity, pH, and other conditions. Therefore, marine biofouling is a worldwide problem. Since tributyltin-based coatings have been banned due to their negative effects on the marine environment, it has become imperative to develop eco-friendly antifouling systems.To combat marine biofouling, Zhang et al. proposed the concept of dynamic surface antifouling, where the dynamic surface involves a changeable surface that continuously renews itself in seawater and therefore prevents fouling organisms from landing and adhesion (Fig. 62).468,469 Dynamic surface antifouling was confirmed by observing the settlement of model microfouling organisms on the surfaces of degradable polymers via digital holographic microscopy (DHM), which can reveal the 3D real-time motion of a microorganism. The trajectories of bacteria in the near-surface range with respect to the mean square displacement (MSD) showed that when the polymer degradation rate is sufficiently high, a large portion of bacteria may exhibit superdiffusive motion rather than subdiffusive motion, which represents a transient state before irreversible adhesion. Atomic force microscopy (AFM) measurements demonstrated that adhesion forces between a bacteria-covered colloidal probe and the surface decrease as the degradation rate increases. Accordingly, polymer degradation alters bacterial motion such that they are not able to land on but have to escape from the dynamic surface.
 | ||
| Fig. 62 Schematic of dynamic surface antifouling. Reprinted from ref. 468 with permission. Copyright (2019) Royal Society of Chemistry. | ||
Based on this strategy, Zhang et al. developed coatings using PU with a biodegradable backbone: these coatings exhibited excellent antifouling performance in marine field tests.470 Furthermore, they synthesized PCL-based PU with triisopropylsilyl acrylate (TIPSA), where the main chains are degradable and the side chains are hydrolyzable in the marine environment. Therefore, the coating surface continuously renews even under the static condition.471 Marine field tests demonstrated that such a coating has excellent antifouling efficiency over 3 months in natural seawater. The renewal rate of such degradable PU could also be regulated by fouling-resistant side groups, such as N-(2,4,6-trichlorophenyl)maleimide or 2-(dimethylamino) ethyl methacrylate.472 The combination of a dynamic surface and fouling-resistant groups can significantly improve the antifouling efficiency against marine microorganisms (Fig. 63).
 | ||
| Fig. 63 Dynamic surface formed by PU with polyester as the soft segments and hydrolyzable side chains. Reprinted from ref. 468 with permission. Copyright (2019) Royal Society of Chemistry. | ||
To prolong the service life of a coating, a small amount of antifoulant can be added. The system consisting of degradable PU with biobased soft segments such as poly(1,6-hexamethylene adipate) and 4,5-dichloro-2-octyl-isothiazolone (DCOIT, a relatively eco-friendly organic antifoulant) exhibited stable and persistent release of the antifoulant, and the cumulative release linearly increased with time (correlation coefficient >0.99). Such a coating was free from biofouling after 6 months of immersion in the sea.473 DCOIT could be replaced with natural compounds such as butenolide derived from marine bacteria such as Streptomyces sp.474 In the presence of enzymes secreted by a microorganism, PU degraded faster and released more antifoulants, indicating that the environment-adaptive system exhibited higher antifouling ability under heavier fouling pressure.
A dynamic surface can also be constructed by poly(ester-co-acrylate)s synthesized by hybrid copolymerization or ROP. These reactions allow inserting ester bonds in the backbone of polyacrylates with intrinsically remarkable coating performance, generating degradable polyacrylate coatings. A copolymer of methyl methacrylate (MMA) and 2-methylene-1,3-dioxepane (MDO) can enzymatically and hydrolytically degrade because of esters in the main chains, and the degradation rate increased with the ester content.475 A polymer coating with a low amount of DCOIT exhibited excellent antifouling performance in seawater. Because of the diversity of vinyl monomers, the degradable polyacrylates could be imparted with various functionalities. For instance, the copolymer of MDO, MMA, and tributylsilyl methacrylate with degradable main chains and hydrolyzable side groups exhibits not only controllable surface renewal but also self-smoothing ability, reducing the hydrodynamic drag of ships.476,477 A coating consisting of such a polymer and DCOIT can effectively resist biofouling in the sea for a long time. Further, degradable polyacrylates with fouling-resistant moieties has excellent antifouling and mechanical performances. For example, copolymer carrying the tertiary carboxybetaine (TCB) ester can be hydrolyzed in seawater to generate zwitterions with resistance to nonspecific protein and bacteria (both marine bacteria and clinically related bacteria).478 Moreover, these zwitterions were renewed by main-chain degradation, which was favorable to maintain both mechanical robustness and fouling resistance.
In short, biodegradable polymers for marine antibiofouling based on the strategy of dynamic surface antifouling are summarized. Fouling-resistant side groups or antifoulants can be introduced into biodegradable polymers for prolonging the service life of coatings. Moreover, the idea of dynamic surface antifouling was tested to be valid. In the future, environment-adaptive dynamic surfaces that respond to fouling pressure, temperature, salinity, and navigation speed need to be designed, which can yield high efficiency and long service life.
In this section, in situ polymerization methods used for protein modification are initially summarized, which offer many potential applications, such as targeted delivery as cancer therapy or BBB penetration as Parkinson's therapy, biosensor, and biofuel. Then, CPs are briefly introduced for their DNA sensing, bioimaging, and therapeutics. After that, various functional polymers used for drug and gene delivery are discussed, namely, stimuli-responsive polymers, polymer vectors, and polyprodrug amphiphiles. Finally, surfaces/interfaces between biomaterials and bioenvironments are discussed, because they play a critical role in biomedical applications. Although significant advances in biomedical polymer materials have been achieved in recent years, there are still many challenges that are needed to be addressed. For example, it is still difficult to obtain imaging materials with precise imaging performances and good biosafety. The biosafety of polymer materials used in drug delivery is not easy to achieve because they possess the ideal delivery function. Researchers should put a considerable amount of effort to obtain materials with ideal specific functions and biosafety.
7. SP materials
Beyond polymers based on covalent bonds, SPs are produced by a combination of supramolecular chemistry and polymer science.479–484 They refer to aggregates formed by the noncovalent interactions of low-mass molecular/polymeric building blocks and exhibit polymer properties in solution or in bulk. Noncovalent interactions comprise hydrogen bonding, host–guest interactions, ionic bonding, π–π stacking, charge transfer interactions, metal–ligand coordination, hydrophobic interactions, etc. Besides conventional polymer properties, SPs exhibit thermal response because they comprise noncovalent bonds with lower bond energy. That is, SPs can be degraded into their building blocks when the temperature is increased, and SPs are reformed when the temperature is decreased; this is a better processing property than that observed in covalent polymers. Moreover, SPs can also show response to other external stimuli, such as light, electricity, and chemicals. The reversibility of noncovalent bonds endows SPs with unique properties, such as self-adaptation and self-healing, which are difficult to be achieved by covalent polymers. In addition, using easy-to-operate noncovalent bonds, various properties and functions can be facilely integrated into SPs and can be reversibly tailored, endowing SPs with potential applications in the robotic industry, biomedicine, and sensors, which has attracted wide attention in this field. In this section, subsequent discussions are divided into four research directions of SPs due to space limitations: responsive fluorescent SP gels, selenium-containing polymer materials, self-assembly of hyperbranched-based SPs, and BCPs.7.1 Responsive fluorescent SP gels
Responsive fluorescent SP gels, a kind of smart gel with a three-dimensional (3D) network structure containing fluorophores, are crosslinked by noncovalent interactions, such as multiple hydrogen bondings, electrostatic interactions, π–π interactions, metal–ligand coordination, van der Waals interactions, and host–guest interactions.485–490 In particular, these noncovalent connections affect the fluorescence properties of the chromophores because their state of aggregation and energy transfer can be regulated by the assembly–disassembly process. Considering these interesting properties, responsive fluorescent SP gels have facilitated the evolution of new materials with applications in fluorescent sensors, biological probes, and imaging agents.491–493For example, Huang et al. reported a novel yet facile approach to prepare white-light-emitting fluorescent polymeric materials resulting from the aggregation of a single fluorophore (Fig. 64).494 This aggregation resulted from the energetically favorable self-assembly of polymers via intermolecular quadruple hydrogen bonding. This advanced material was shown to be an ideal candidate for constructing intelligent information display/storage devices. A protected quick response (QR) code was fabricated based on this white-light-emitting fluorescent supramolecular gel. This code was shown to hide the information under natural light, yet it displayed it under UV light. Therefore, this code was protected and only readable under a specific condition. Furthermore, due to the dynamic nature of hydrogen bonds, the SP gel and the resultant QR code showed self-healing abilities, which is crucial for their applications. Information in an intentionally damaged QR code is incomplete and cannot be read under UV-light irradiation. However, the code can be mended based on the interfacial self-assembly of gels through multiple hydrogen bonding; subsequently, the protected information can be recovered. This novel QR code with improved information security and durability against mechanical damage can find applications in various fields.
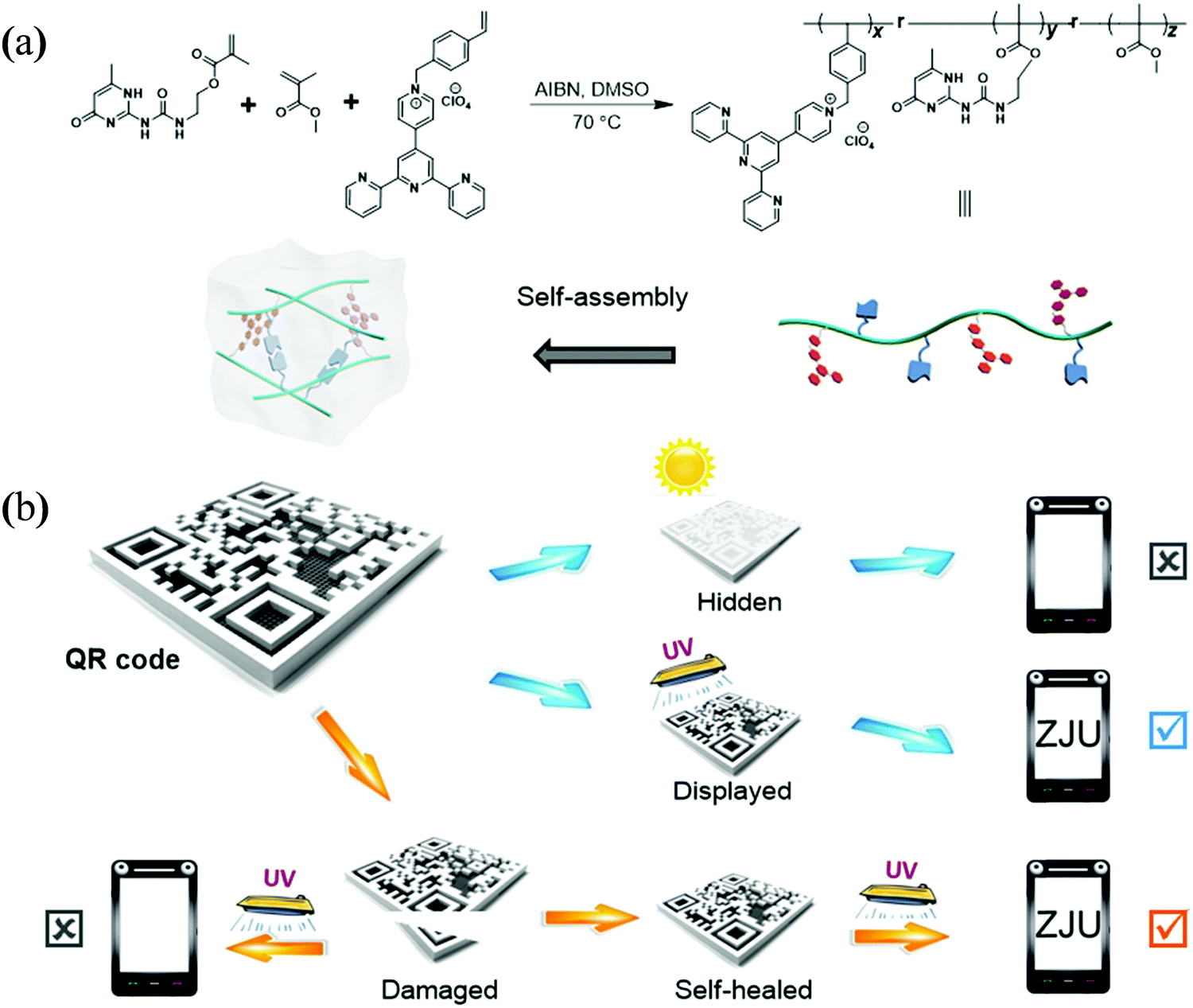 | ||
| Fig. 64 (a) Synthesis of the polymer and its self-assembly in solution to form a white-light-emitting fluorescent SP gel via intermolecular hydrogen bonding. (b) Schematic representation of data recording, data security protection, and data recovery of a quick response code constructed using this gel. Adapted from ref. 494 with permission. Copyright (2017) Royal Society of Chemistry. | ||
Then, Huang et al. constructed a novel bioresponsive supramolecular fluorescent hydrogel by electrostatic interactions between poly(sodium p-styrenesulfonate) and a TPE derivative containing two quaternary ammonium cations (Fig. 65).495 This self-assembly process led to the aggregation of TPE, which is a typical luminogen with AIE properties, thereby endowing this hydrogel with AIE properties. Moreover, this supramolecular fluorescent hydrogel was responsive to biomolecules. When ATP was added to this system, the network structure of the hydrogel was destroyed and the aggregation extent of TPE was decreased, which induced the transition from the gel state to the solution state and decreased the fluorescence intensity of the hydrogel at the same time. Subsequently, after ATPase was added to the solution, ATP was decomposed and the electrostatic interactions between the polymer and TPE were rebuilt, transforming solution into gel and the fluorescence intensity of the hydrogel was recovered. Therefore, the fluorescent supramolecular hydrogel was bioresponsive. Simultaneously, this fluorescent supramolecular hydrogel could cross the cell membrane and enter the cytoplasm. The hydrogel system could be potentially applied to characterize the biooxidation process in the cytoplasm. In addition, it can be further used to prepare supramolecular systems for other bioapplications, such as drug delivery, bioprobes, and biological detection.
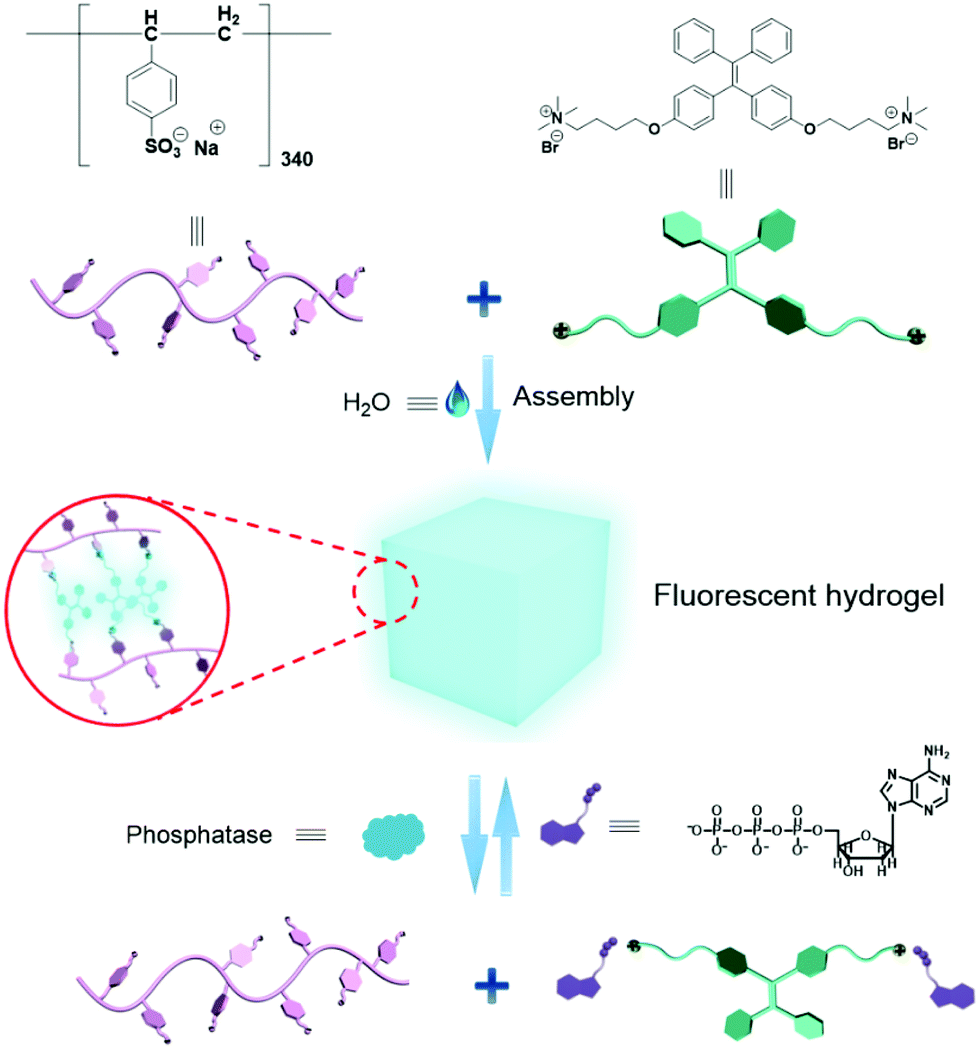 | ||
| Fig. 65 Chemical structures of poly(sodium p-styrenesulfonate) and TPE derivative, and the schematic representation of the formation and bioresponsive properties of the supramolecular fluorescent hydrogel. Reprinted from ref. 495 with permission. Copyright (2018) Royal Society of Chemistry. | ||
7.2 Selenium-containing dynamic covalent polymer materials
Dynamic covalent bond (DCB), which was first proposed by Jean-Marie Lehn in 1999,496 refers to a unique chemical bond that can cleave, reform, and exchange under certain stimuli.497,498 For the past decade, it has been extensively applied in research fields such as adaptive materials and supramolecular chemistry.499 In 2014, Xu et al. reported the discovery of diselenide covalent chemistry.500–502 Diselenide-containing molecules can undergo an exchange reaction simply under visible-light irradiation without additional additives via a radical mechanism.The unique visible-light stimuli response behavior of diselenide bonds was applied to produce different types of functional materials. Diselenide bonds containing PU were fabricated,503 which could self-heal under visible-light stimuli due to the dynamic exchange of diselenide bonds (Fig. 66a–c). Different contents of diselenide bonds could lead to different mechanical properties and self-healing properties. Moreover, employing a laser as the light source not only accelerated the healing process but also offered the potential for remote healing. Next, the crosslinker, glycerol, was added to the earlier recipe, and a thermoset diselenide bond containing PU was produced,504 which could serve as a shape-memory material. In particular, when the material was heated above 80 °C, it can be bent from its original shape (permanent shape) to another shape (temporary shape). This temporary shape could be memorized by cooling to room temperature. When heating back to 80 °C, the shape could recover from the temporary shape back to the permanent shape (Fig. 66d). It was found that a higher content of diselenide bonds would lead to better shape-memory behavior. Moreover, since the diselenide bonds are light-sensitive, stress relaxation could occur under visible-light stimuli. As a result, it is possible for the material to transfer from a temporary shape to a permanent shape via visible-light irradiation in shape-memory cycles. This is a special property as most of the thermoset shape-memory material only has one fixed permanent shape once formed. As an application demonstration, one can use a commercialized projector to pattern material with designed shapes (Fig. 66e). In a recent study, Xu et al. reviewed that Se–S bonds are also a dynamic covalent bond.505 The dynamic metathesis between diselenide and disulfide could be manipulated by light at different wavelengths. Based on this, a disulfide and a diselenide containing PU were prepared with light-triggered attachable–cleavable behavior. When they are attached together and irradiated by UV light, the Se–S exchange reaction can connect two materials into one piece and could be stretched up to 75% strain. However, for visible light at a wavelength above 410 nm, the welded material would be separated back into two pieces as the Se–S bonds were cleaved back into Se–Se and S–S bonds.
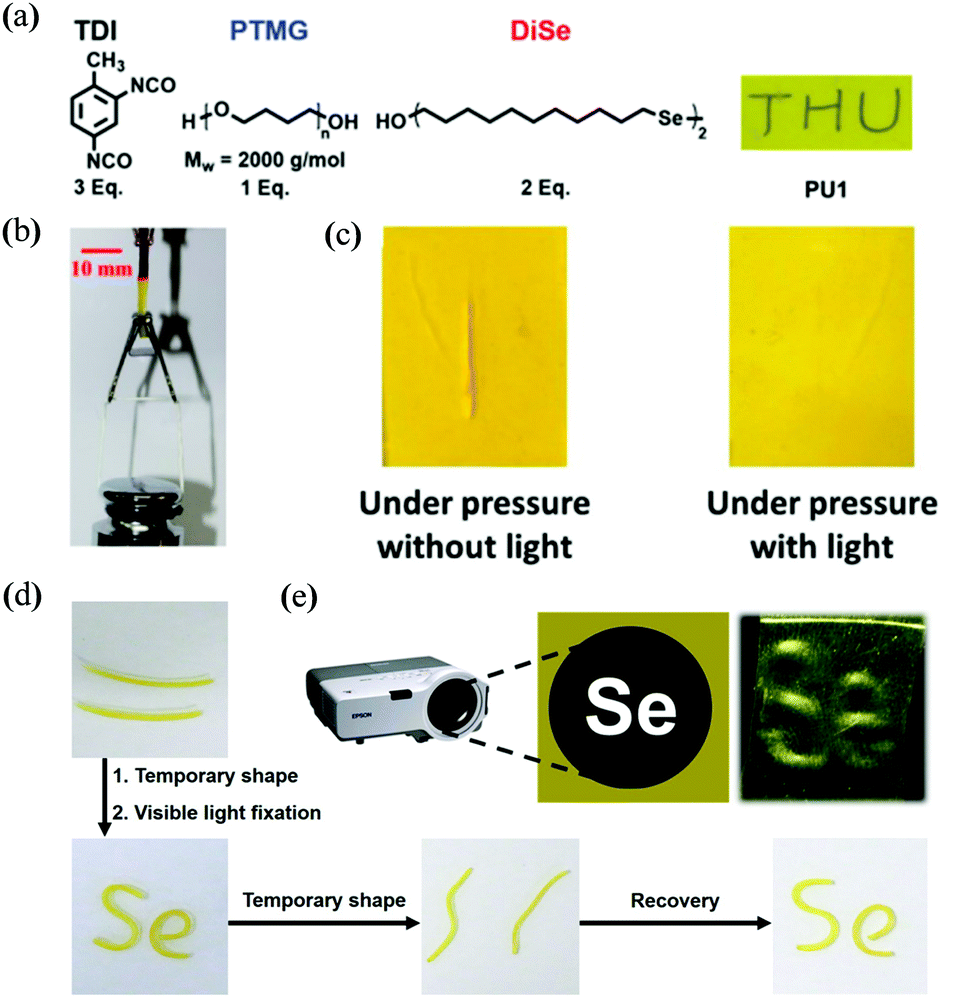 | ||
| Fig. 66 (a) Composition of the diselenide-bond-containing PU. (b) Material could bear 200 g weight after healing; the red part was stained with Nile red for clarity. (c) The crack disappeared after 24 h of light irradiation, demonstrating the healing property. (d) Typical shape-memory cycle of the material. (e) Material patterning achieved by a normal projector. Reprinted from ref. 503 and 504 with permission. Copyright (2015 and 2017) from Wiley-VCH and American Chemical Society. | ||
Apart from bulk materials, diselenide bonds could also be incorporated with surface/interface chemistry. Xu et al. developed the diselenide dynamic covalent bonds into a versatile surface modification method with fast response (within 30 s) and reversibility.506 The diselenide bonds could be modified onto different substrates such as PDMS, quartz, and ITO conductive film glass. Different diselenide molecules could then be immobilized onto the surface via the diselenide metathesis reaction to achieve a series of functions such as adjusting the wettability and photosurface patterning. Moreover, by using this technique, they successfully achieved liquid motion by visible-light irradiation in a tube under capillary force. Moreover, this technique could also provide an efficient modification method for surface bioconjugation, which has potential applications in clinical usage.
7.3 Self-assembly of hyperbranched-based SPs
One important component of the studies on the self-assembly of polymers is to figure out the correlation between the behaviors/properties of self-assemblies and the topological architecture of the involved polymers. Various behaviors and morphologies of the self-assembly of covalent polymers with different architectures have been formulated.507 Although studies on the same issue of SPs have started, the category and topological structure of SPs, however, are still limited when compared with the large number of covalent polymers. Theoretically, it will be easier to prepare SPs with different architectures, especially complex ones, because modules with different topologies can be separately synthesized and selected to associate via specific and strong supramolecular interactions according to the requirement. Moreover, dynamic supramolecular bonds may endow the self-assemblies with interesting properties. In this subsection, the design strategy, self-assembly, and properties of a series of SPs with different architectures are summarized.As simple architecture, the synthesis and self-assembly of supramolecular linear block copolymers (LBCs) were first reported.508–510 Two linear building blocks with a recognition unit at one end of each block was mixed, and complexation happened between them. For example, Yuan et al. synthesized two end-decorated homopolymers, namely, poly(styrene)–β-cyclodextrin (PS–β-CD) and poly(ethylene oxide)–ferrocene (PEO–Fc), which could orthogonally self-assemble into a supramolecular diblock copolymer (PS–β-CD/PEO–Fc) in aqueous solutions based on terminal host–guest interactions.508 Supramolecular PS-b-PEO further self-assembled into vesicles. The vesicles exhibited voltage-responsive behavior, in which the assembly and disassembly speed could be controlled by the applied voltage strength.
To increase structural complexity, one block of linear polymer can be replaced with dendritic polymers to yield a linear–dendritic block copolymers (LDBC). LDBC refers to polymers comprising a linear chain that is attached to a dendritic block. As a mimic of nature, LDBCs resemble trees in architecture and can be categorized into two types: LDBCs and linear–hyperbranched block copolymers (LHBCs).511,512 Although intriguing properties were exhibited, the preparation of LHBCs was usually based on covalent polymerization, which involved complicated multistep synthesis, and the covalent nature lacked dynamic controls over their self-assemblies.513,514 By introducing supramolecular interactions into the polymers, supramolecular LHBCs were successfully synthesized.515–517 For example, Zhou et al. reported a linear–hyperbranched supramolecular amphiphile and studied its self-assembly behaviors.515 A supramolecular LHBC was constructed via the noncovalent coupling of hyperbranched polyglycerol (HPG) grafted from β-cyclodextrin (CD-g-HPG) and adamantane-functionalized long alkyl chain (AD-Cn, n = 18), namely, AD-C18, by the specific AD/CD host–guest interactions (Fig. 67). The as-prepared supramolecular C18-b-HPG exhibited amphiphilic characteristics due to the hydrophilic HPG block and hydrophobic alkyl chain. The self-assembly of C18-b-HPG in water resulted in the formation of vesicles with considerable ductility and they were readily deformed by applying force with AFM tips. The addition of a competitive host of β-CD led to the disassembly of vesicles into unimers. The appealing mechanical properties and additive-responsive behaviors of the vesicles were supposed to arise from the supramolecular nature of C18-b-HPG.
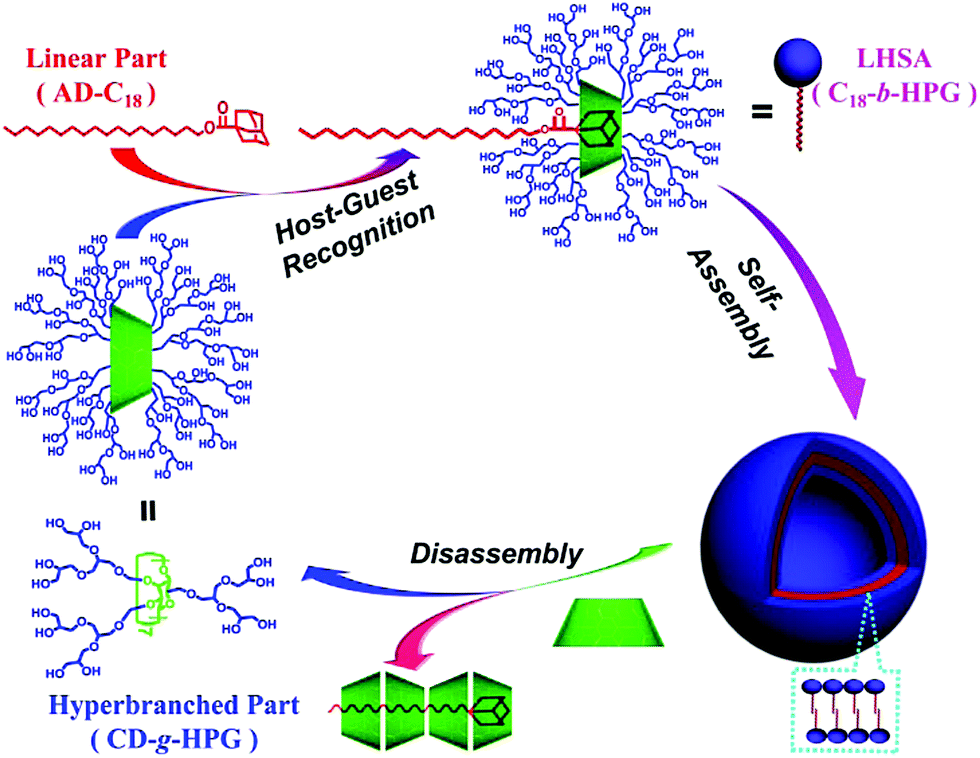 | ||
| Fig. 67 Preparation, self-assembly, and disassembly processes of C18-b-HPG. Reprinted from ref. 515 with permission. Copyright (2012) American Chemical Society. | ||
The dynamic control over the self-assembly behavior of supramolecular LHBCs could be smarter, more complicated, and hierarchical via an appropriate molecular design. Subsequently, Zhou et al. prepared a “latent double-amphiphilic” LHBC and studied its morphological transition under light stimulus.516 Two amphiphiles, namely, linear AZO–PEG (azobenzene-terminated PEG) and hyperbranched CD-g-HBPO (β-cyclodextrin-grafted HBPO), were coupled together to form a supramolecular LHBC of PEG-b-HBPO through “host–guest” interactions between AZO and CD under visible light. Amphiphilic PEG-b-HBPO self-assembled into vesicles in water. Under UV irradiation, however, PEG-b-HBPO was conversely dissociated into amphiphilic AZO–PEG and CD-g-HBPO. Meanwhile, the vesicles disappeared and turned into a mixture of nanofibers from AZO–PEGs and nanosheets from CD-g-HBPOs. This transition was totally reversible: the nanofibers and nanosheets could change back into vesicles again under visible light. The morphological transition, which represented a transition from one regular morphology (vesicle) into two regular ones (fiber and sheet), was reported for the first time and was considered to benefit from the dynamic supramolecular bonds in the polymer.
To further increase the structural complexity, one can replace both linear blocks with dendritic blocks to finally obtain a Janus macromolecule. Janus particles have attracted extensive attention owing to their unique and fascinating properties correlated to their asymmetric structure and functionalization as well as their promising potential to be used as new building blocks in self-assembly.518,519 Janus dendritic polymers, which possess a 3D structure, were regarded as molecular Janus particles. Although covalent Janus dendrimers were successfully obtained through complicated multiple synthesis steps, the preparation of Janus hyperbranched polymers (JHBPs) was difficult because the apex in HBP may be destroyed by intramolecular cyclization during the “one-pot” synthesis process. Zhou et al. overcame the obstacle by introducing supramolecular bonds into HBPs, and they reported the first example of JHBPs.520 The JHBP of HBPO-b-HPG was obtained by complexation between CD-g-HPG and hyperbranched HBPO with an azobenzene group (AZO-g-HBPO) at the apex via specific AZO/CD host–guest interactions (Fig. 68). HBPO-b-HPG self-assembled into narrowly distributed vesicles in water under visible light; on the contrary, the vesicles disassembled into unimers under UV-light irradiation. In addition, data from dissipative particle dynamics (DPD) simulations matched very well with the experimental data and revealed the dynamics of the self-assembly process from randomly distributed molecules into the final vesicles and detailed structures of the vesicles.
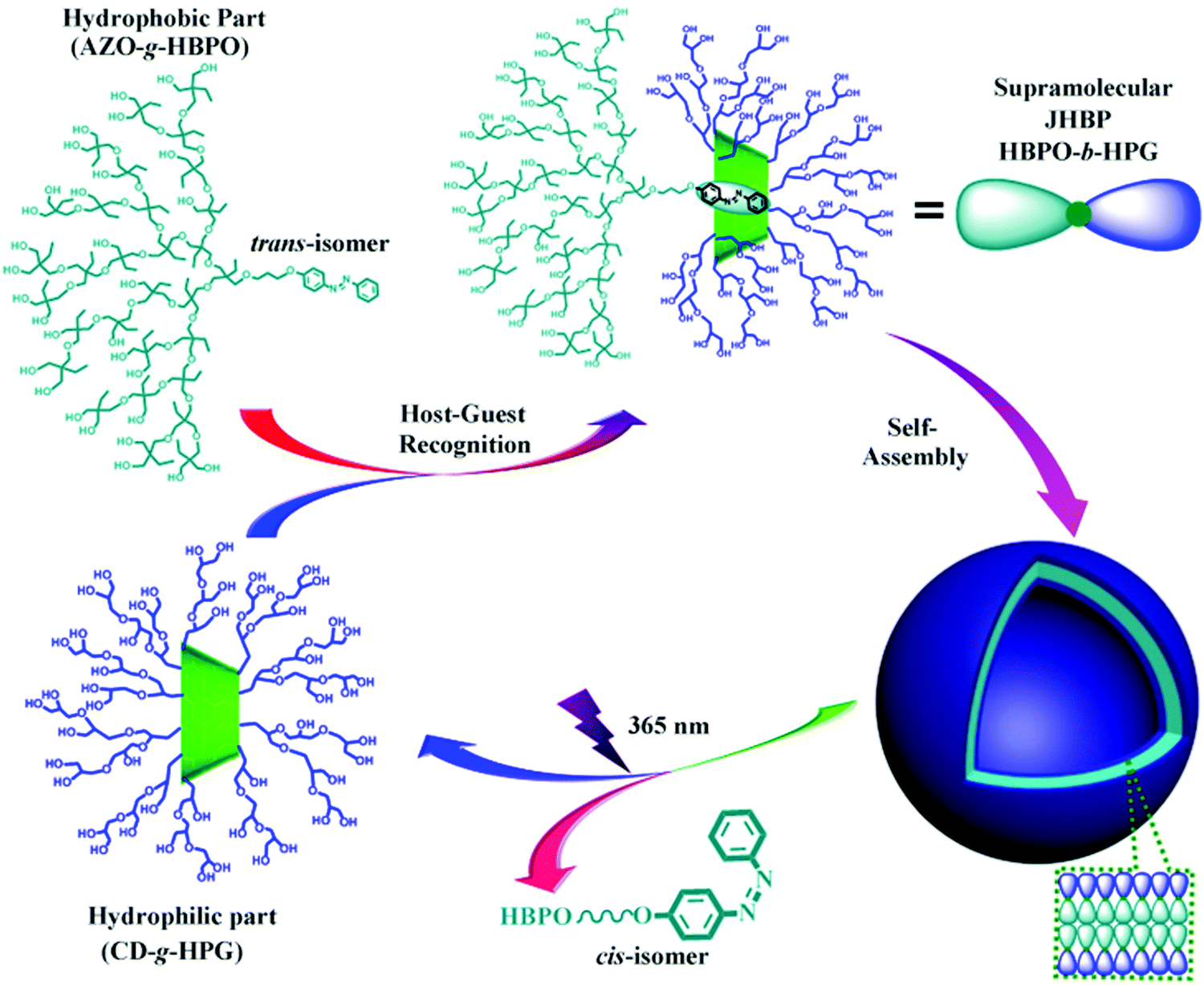 | ||
| Fig. 68 Preparation, self-assembly, and disassembly processes of the supramolecular JHBP of HBPO-b-HPG. Reprinted from ref. 520 with permission. Copyright (2013) American Chemical Society. | ||
By adding one more block, Zhou et al. further increased the structural complexity and obtained a “hyperbranched–linear–hyperbranched” triblock copolymer that resembled a dumbbell in shape.521 The dumbbell-like supramolecular triblock copolymer (DSTC) was constructed via noncovalent host–guest coupling between the monotelechelic HPG grafted from β-cyclodextrin (CD-g-HPG) and ditelechelic azobenzenes-endcapped linear polystyrene (AZO–PS–AZO). The resulting DSTC was amphiphilic and self-assembled into vesicles with a monolayer structure in water under visible light, and the vesicle size depended on the molecular weight of the PS block in the DSTC. In addition, the vesicles could disassemble into unimers under UV-light irradiation, revealing light-responsive self-assembly behavior.
Later on, the same group reported an even more complexed dandelion-like SP (DSP) with a “sphere–star–parachute” structure.522 The DSP was prepared by the noncovalent coupling of β-CD-endcapped HBPO-star-PEO and adamantane-1-3,4,5-tris(n-dodecan-1-yloxy)benzamide (AD-tC12) comprising an adamantane head and three dodecyl tails via specific AD/CD host–guest interactions (Fig. 69). The DSPs exhibited novel hierarchical self-assembly behavior from vesicles to nanotubes through a fusion process in water. As a proof of concept, the DSPs were used to build an artificial light-harvesting system in an aqueous environment, taking advantage of the supramolecular nature and the self-assembled nanotube structure. The system was constructed by incorporating hydrophobic donors in the hydrophobic hyperbranched cores inside the nanotubes and the hydrophilic acceptors on the nanotube surfaces. This system could effectively prevent π–π stacking between the donors and could provide a suitable distance between the donors and acceptors within the Förster radius, thereby yielding high energy transfer efficiency of over 90% in water. This work provides a solution to the low energy transfer efficiency in aqueous light-harvesting systems due to the poor solubility of fluorophores in water.
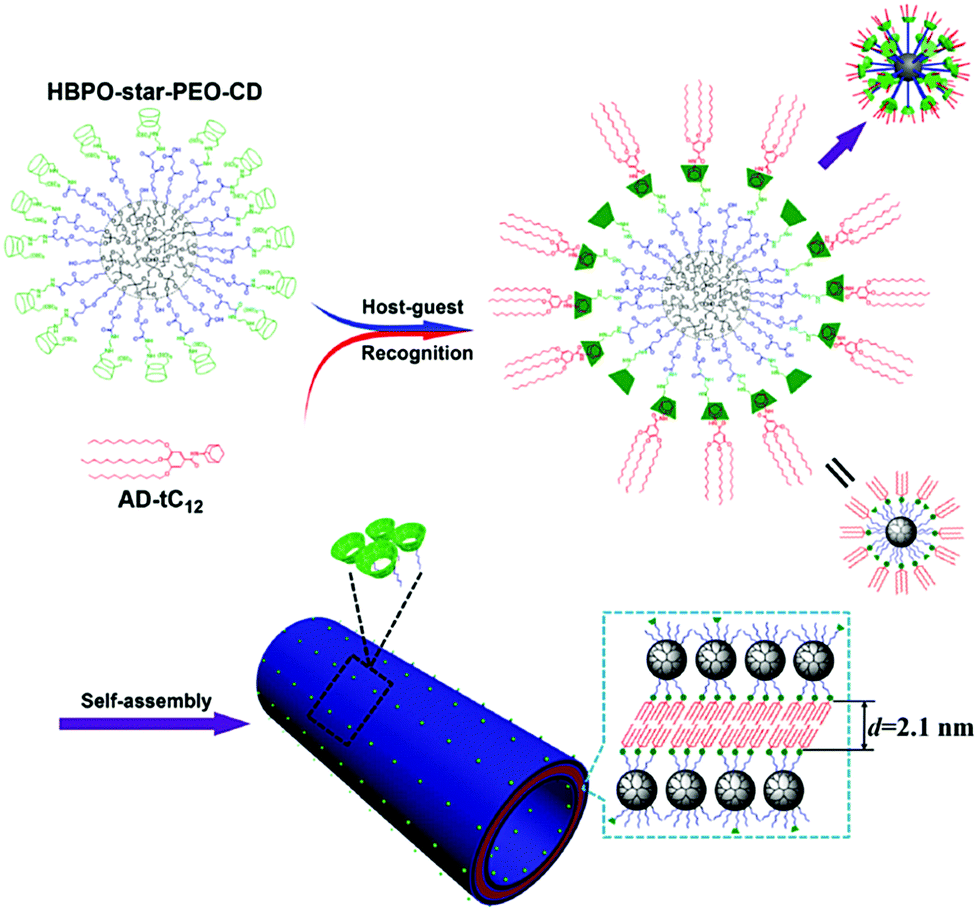 | ||
| Fig. 69 Preparation and self-assembly processes of DSPs. Reprinted from ref. 522 with permission. Copyright (2016) from Wiley-VCH. | ||
The introduction of supramolecular chemistry into polymers considerably simplified the preparation of polymers with complex architectures. Different “modules” can be separately synthesized and assembled together through supramolecular bonds to finally get SPs with various topological structures. Following this design strategy, more and more SPs with different architectures and components are expected to emerge in the future, which can certainly extend the properties of polymers as well as their applications. Moreover, the complex structure and dynamic nature of supramolecular bonds in SPs may reveal more intriguing behaviors and properties of their self-assemblies.
7.4 Self-assembly of BCPs under confinement
Self-assembly of BCPs has attracted considerable attention for many decades since it can generate well-ordered structures in a wide range of morphologies.523,524 The preparation and use of polymer materials usually suffers because of the external environment. For example, strong shear, elevated temperature, high pressure, and limited space are required in the process of injection molding. These extreme external environments have a significant impact on the microstructure and properties of polymer materials, endowing them with unique functionalities. When the self-assembly of BCPs takes place within confined spaces, the interfacial interactions, symmetry breaking, structural frustration, and confinement-induced entropy loss of the polymer chains play a dominant role in the final self-assembled structure, thereby resulting in some novel nanostructures that are not available in bulk and solution states.524–528 Moreover, functional inorganic NPs are usually introduced to coassemble with BCPs or directly assembled with polymer-tethered NPs, which can effectively combine the respective functions of the polymers and NPs.529,530Despite the rapidly increasing number of publications related to the self-assembly of BCPs or polymer-tethered NPs under confinement, there are still many open questions. For instance, the mechanisms of the confined assembly of BCPs are still under careful investigation. Control over the uniformity and stability of the assemblies and to get functional assemblies under confinement are still challenging. Due to their broad potential applications, the assembly behavior of BCPs and NPs under confinement has attracted considerable attention and has been the main research topic of Zhu et al. in the past decade. In this subsection, recent progresses made with regard to the assemblies of BCPs and inorganic NPs under confinement are addressed.
 | ||
| Fig. 70 Factors affecting the assembly behavior of BCPs in 3D confinement. | ||
Using the concept of confinement, Zhu et al. constructed stimuli-responsive photonic crystal microspheres of BCPs.527 Recently, the polystyrene-block-poly(2-vinyl pyridine) (PS-b-P2VP) particles with tunable periodic onion-like microstructures (Fig. 71) have been prepared via confined self-assembly in emulsion droplets, which exhibited angle-independent structural color after swelling in an ethanol/water mixed solution.528 Interestingly, the structural color can be effectively tuned by varying the ethanol concentration because of the selective swelling of P2VP domains. These photonic microspheres showed potential applications in bio-diagnosis, colorimetric sensors, encoding, and active displays. This concept of confinement could be extended to construct confined BCP shells (e.g., capsules). For example, hollow microcapsules with tunable shell structures have been fabricated under confinement, and the release behavior of encapsulated cargoes in these structured capsules was found to be strongly dependent on the shell structure.528
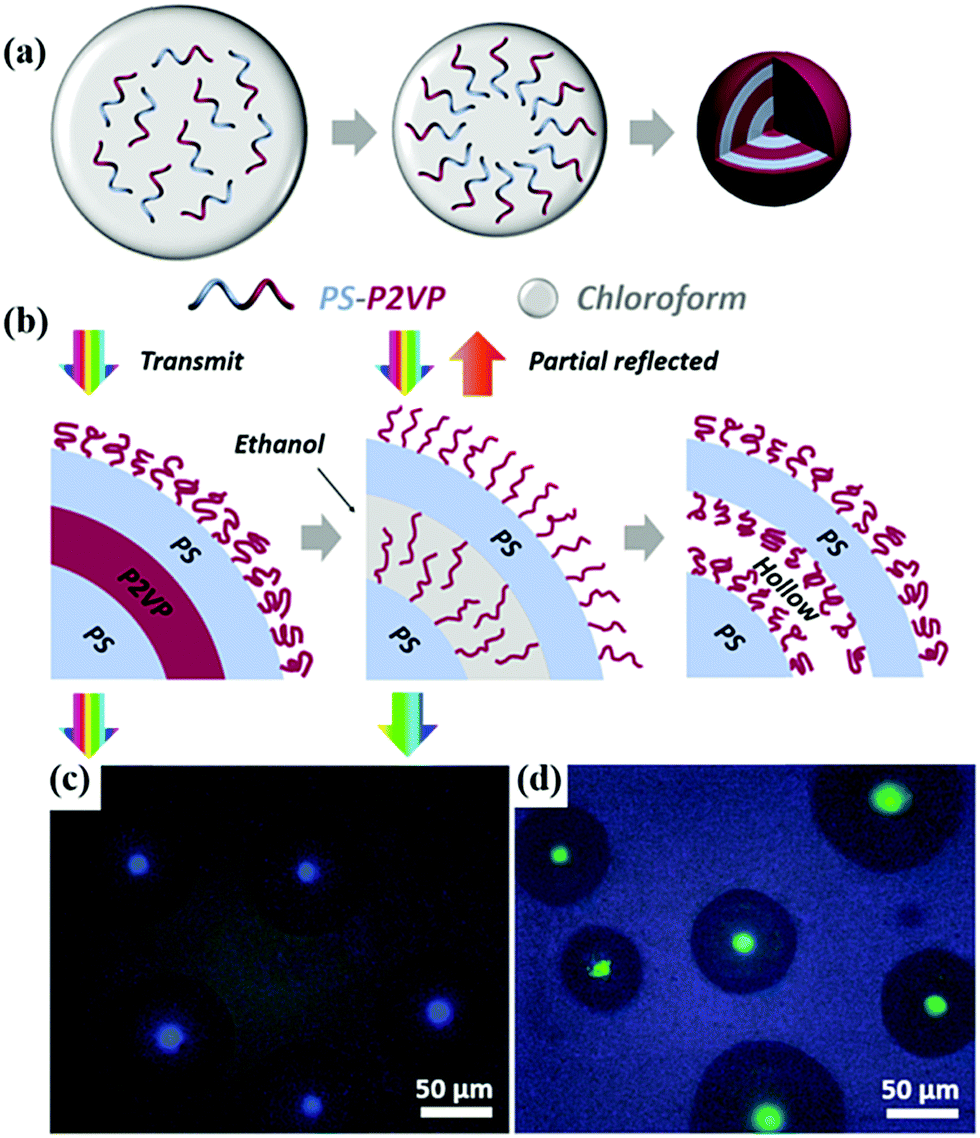 | ||
| Fig. 71 Schematic illustration showing the preparation of PS-b-P2VP photonic microspheres through confined self-assembly in emulsion droplets. Adapted from ref. 527 with permission. Copyright (2018) from Wiley-VCH. | ||
The above results indicate that confined assembly is a facile yet robust approach to control the shape and internal structure of BCP colloidal particles. Yet, despite the significant progress achieved in recent years, the assembly of BCPs in 3D confined space is still an emerging field. A better understanding of the control parameters that govern the confined assembly and nanostructures formation of BCPs should be explored in the future, which can certainly facilitate the design and fabrication of BCP materials with well-controlled structures and unique properties.
Furthermore, a “rapid liquid–liquid interface confined assembly” strategy was proposed, which has been used to generate a non-close-packed PS-tethered Au NPs superlattice monolayer (AuNPs@PS SM) on the centimeter scale.532 In this work, it was shown that a small amount (one drop, ∼1 μL) of PS-tethered Au NPs solution could spontaneously spread on the water surface to form a monolayer thin film. During this process, AuNPs@PS self-assembled into centimeter-sized free-standing thin film with a non-close-packing structure (Fig. 72). The generated film has been successfully used to fabricate high-performance pentacene-based organic nano-floating-gate memory (NFGM). The d space of NPs (e.g., interparticle distance) and the surface plasmon resonance spectra of the film can be tailored by adjusting the molecular weight of the tethered polymers. Precise control over the d value allows the successful fabrication of photosensitive NFGM devices with highly tunable performances from short-term memory to nonvolatile data storage. More interestingly, after incisively revealing the assembly mechanism, it was found that this strategy is general and can be applied to NPs with various sizes. This strategy is significantly meaningful to microelectronic, photovoltaic devices, and biochemical sensors. These results indicate that the confined assembly of a polymer–NP system is a promising approach for tuning the ordered structure and functionality of hybrid materials.
 | ||
| Fig. 72 Schematic illustration showing the strategy for fabricating AuNPs@PS SM through rapid liquid–liquid interface assembly on a convex water surface, and the design for NFGM devices based on a superlattice monomer. Adapted from ref. 532 with permission. Copyright (2018) from Wiley-VCH. | ||
The confined assembly of BCPs and inorganic NPs is a facile yet effective strategy to generate polymer and hybrid materials with well-ordered structures. Polymer particles with switchable shapes and internal structures can offer unique properties for this particular material. In addition, versatile hybrid nanomaterials can be designed on the basis of complementary advantages of functional inorganic NPs and polymeric materials via the confined assembly of polymer-tethered inorganic NPs.
However, despite the great progress made in recent years, the confined self-assemblies of BCPs and inorganic NPs still require more studies for a better understanding of the control parameters that govern the confined self-assembly and nanostructure formation. It still remains a challenge to utilize the well-defined BCPs or NP nanostructures for versatile applications (e.g., solar cells, sensors, targeted drug delivery, metal catalysis, and magnetic storage devices) by taking advantage of the integrated and unique properties of these hybrid materials.
We first discuss responsive fluorescent SP gels that have many potential applications as fluorescent sensors, biological probes, imaging agents, drug delivery, bioprobes, biological detection, etc. Polymer materials containing dynamic diselenide bonds are then introduced, because they can be endowed with many functionalities, such as self-healing properties, adjusting wettability of the surface, and photosurface patterning. The various topological nanostructures of hyperbranched SPs and BCPs can be self-assembled by synthesizing different hyperbranched BCPs or under different confinements, which is summarized in the last part of this section. In general, the recent developments of SPs have been briefly addressed, which may provide some clues to readers for designing more abundant and higher-level SPs to meet the needs of various fields. Many challenges still remain for SPs, such as the in situ monitoring of the assembly process, exposing the rules of electrons, energy and mass transfer, and realizing the construction and practical applications of smart devices made of SPs.
8. Stimuli-responsive polymer materials
Stimuli-responsive polymers are smart materials whose chemical or physical properties can undergo reversible or irreversible changes when they are exposed to external stimuli, such as temperature, pH, light, electricity, magnetic field, mechanical force, and biochemical molecules. They have drawn a great deal of research attention due to their applications in nanomedicine, diagnostics, flexible devices, smart actuation, optical storage, energy materials, and environmental governance. The existence, form, and mechanism of responsive polymer materials can be embodied in molecular chains, colloidal particles, self-assembled ordered aggregates, (hydro)gels, thin films, liquid crystal (LC) elastomers, and bulk materials. The response modes of such materials are different, including geometric dimension (shape) changes and recovery, changes in microenvironment polarity, cleavage or rebonding of covalent bonds, color and luminescence, surfaces infiltration, self-healing behavior, and so on. These different response modes are accompanied by their applications, such as shape changes that can mimic the action of muscles, cleavage or rebonding of covalent bonds that can control drug delivery and release, and self-healing behavior that can heal the polymers themselves. In this section, we only discuss electrochromic polymer materials and photoresponsive liquid crystal polymers (LCPs). SMPs, a type of stimuli-responsive polymers, will be addressed in the next section because of their unique mechanism and properties.8.1 Electrochromic polymer materials
Electrochromism is the phenomenon in which materials can reversibly change their optical properties such as transmittance, reflectance, or absorbance in response to external potential.533,534 Since the discovery of the electrochromic effect in transition-metal oxides by Deb et al. at the end of the 1960s, it has been applied in many fields such as smart windows, electronic displays, and mirror devices; therefore, state-of-the-art studies on electrochromism can have a significant impact on the environment, energy, and economy.535–537The main classes of electrochromic materials are CPs (polythiophene (PTH), polyaniline (PANI), polypyrrole (PPy), and their derivates), viologens and their derivates, and transition-metal oxides (WO3, NiO, and V2O5).538–540 All the abovementioned materials have gained popularity due to their ease of processability, high optical contrast, rapid switching times, and high coloration efficiency. In addition, many researchers have not only focused on improving the EC properties of the materials but also attempted to combine other special functions. Hence, we provide examples of instances in which such materials have been constructed.
In terms of CPs, PTH and their derivates are widely studied due to their modifiable structures to obtain multifunctional materials. In 2016, Xu's group designed a novel polymer with AIE and electrochromic properties, which was realized by introducing the TPE group into thiophene main chains (Fig. 73).541 The polymer exhibited weak photoluminescence in THF, but its corresponding film prepared by spray-coating exhibited intense yellow-green fluorescent light at 540 nm. The color of the polymer film could be switched from bright yellow to navy blue by applying a relatively low voltage. An electrochromic device (ECD) was fabricated by using this polymer, which differs from common ECDs because both its color and fluorescent state could be synchronously switched by applying different potential level; therefore, the polymer is a unique candidate for electrofluorochromic and electrochromic applications.
 | ||
| Fig. 73 Photographs of polymer-films-based ECDs and their corresponding transmittance and emission modulation. Reprinted from ref. 541 with permission. Copyright (2015) American Chemical Society. | ||
AIEgens are strongly emissive in their aggregate states but weakly emissive in dilute solutions. The dual-state emission (DSE) polymers have strong emissions in both solution and solid states, broadening the scope of fluorescent displays. Xu et al. reported two kinds of PTH derivatives containing trans-stilbene (P1) and fumaronitrile (P2) groups, respectively.542 These polymers emitted intense fluorescence in both solution and film states, attributable to the restriction of intramolecular rotation caused by multiple C–H⋯π bonds existing in the neighboring polymer backbones. Driven by different applied potential levels, the polymer films showed reversible electrochemical oxidation reduction reactions with colors changing from yellow-green to sky-blue and red to rufous. Furthermore, electrofluorochromism between the fluorescent state and nonfluorescent state happened synchronously (Fig. 74). This unique property makes the polymers a special candidate for optical applications.
 | ||
| Fig. 74 Electrofluorochromic and electrochromic bifunctional polymers with DSE by introducing multiple C–H⋯π bonds. Adapted from ref. 542 with permission. Copyright (2018) from Elsevier Ltd. | ||
With regard to composite electrochromic materials, Xu et al. investigated a quantum dots–polymer composite film, achieving the triple functions of electrochromism, electrofluorescence, and light-induced coloration effects (Fig. 75).543 Combining the advantages of both cadmium selenide (CdSe) quantum dots and conjugated electrochromic polymers, the composite film exhibited excellent electrochromic properties with low redox potential, high optical contrast, and fast response time. In addition, because of its enhanced performance, the composite film could switch between the nonfluorescent and fluorescent states, where the photoluminescence intensity was more than twice that of the pristine polymer film. Furthermore, the material could gradually become self-colored from the bleached state under sunlight irradiation.
 | ||
| Fig. 75 Trifunctional CdSe quantum dots–polymer composite film with electrochromic, electrofluorescent, and light-induced coloration effects. Adapted from ref. 543 with permission. Copyright (2018) from Elsevier Ltd. | ||
Insolubility is a major limitation of conducting electrochromic polymers. To allow materials to be incorporated into large-area device architectures of interest, considerable efforts have been devoted over the last few decades. Water processability offers a green alternative to conventional organic-solvent-based processing methods that can facilitate the utilization of electrochromic polymers in industrial-scale device applications. Xu et al. reported a highly regiosymmetric water-processable blue-to-transparent electrochromic film based on diethyl-malonate-derivatized PTH (Fig. 76).544 The polymer exhibited high electrochromic contrast (ΔTmax) of 56% at 580 nm and a relatively fast switching speed of 1.8 s; further, it exhibited contrast loss of only 11% (from 56% to 45%) for a squarewave potential step of 5 s over 11000 switching cycles, making it a desirable candidate for large-area electrochromic applications such as windows and displays, which have been long-lived aspirations, as reported by Xu et al.
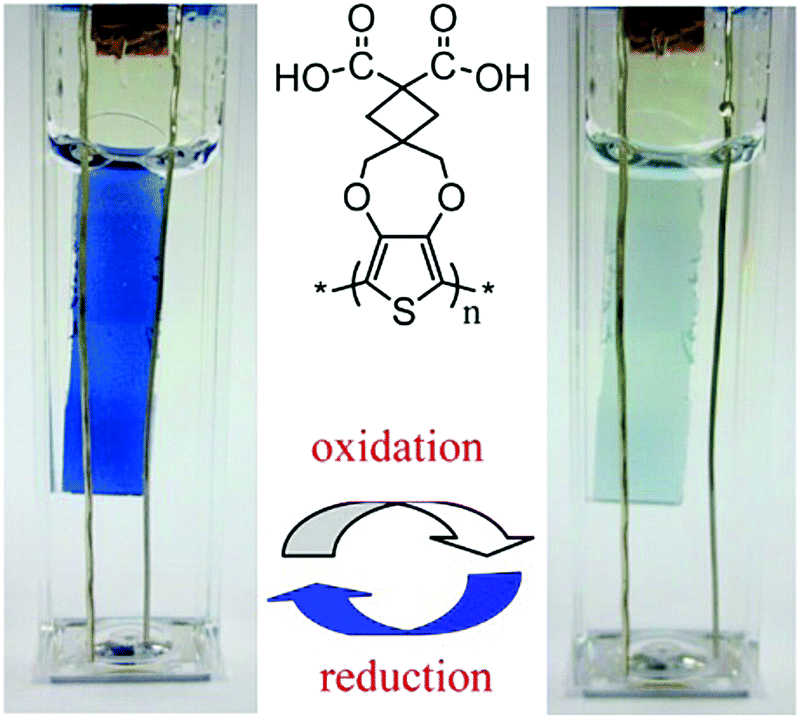 | ||
| Fig. 76 A highly regiosymmetric, water-processable blue-to-transparent PTH-derivatized EC film. Reprinted from ref. 544 with permission. Copyright (2015) from American Chemical Society. | ||
Viologen is another widely studied organic electrochromic material. In an electrochromic system, viologen and its derivates undergo reversible reduction under applied potential to form a viologen radical cation, which is considered to be one of the most stable radical molecules. The viologen radical cation exhibits a deep color because of its intense absorption of visible light. Xu et al. reported a novel electrofluorochromic dual-function devices based on aryl-substituted viologen derivatives.545 These devices comprise four viologen derivatives, namely, 4,4′-(1,4-phenylene)bis(1-butylpyridinium)hexafluorophosphate (PV), 4,4′-(naphthalene-1,4-diyl)bis(1-butylpyridinium)hexafluorophosphate (NV), 4,4′-(anthracene-9,10-diyl)bis(1-butylpyridinium)hexafluorophosphate (AV), and 4,4′-(benzo[c][1,2,5]thiaidazole-4,7-diyl)bis(1-butylpyridinium)hexafluorophosphate (BV). They were designed and functionally synthesized by introducing benzene, naphthalene, anthracene, and benzothiadiazole rings, respectively, between the two pyridine rings, which endowed these viologen derivatives with electrochromic and fluorescent switching properties. The devices comprising benzene viologen, naphthalene viologen, and benzothiadiazole viologen exhibited exceptional electrochromic properties with vivid color changes, high optical contrast, and fast response time under applied bias. In addition, owing to the increased conjugated p-framework degree of the designed compounds as compared to those of conventional viologens, all the fabricated devices could achieve intense fluorescence emissions (Fig. 77).
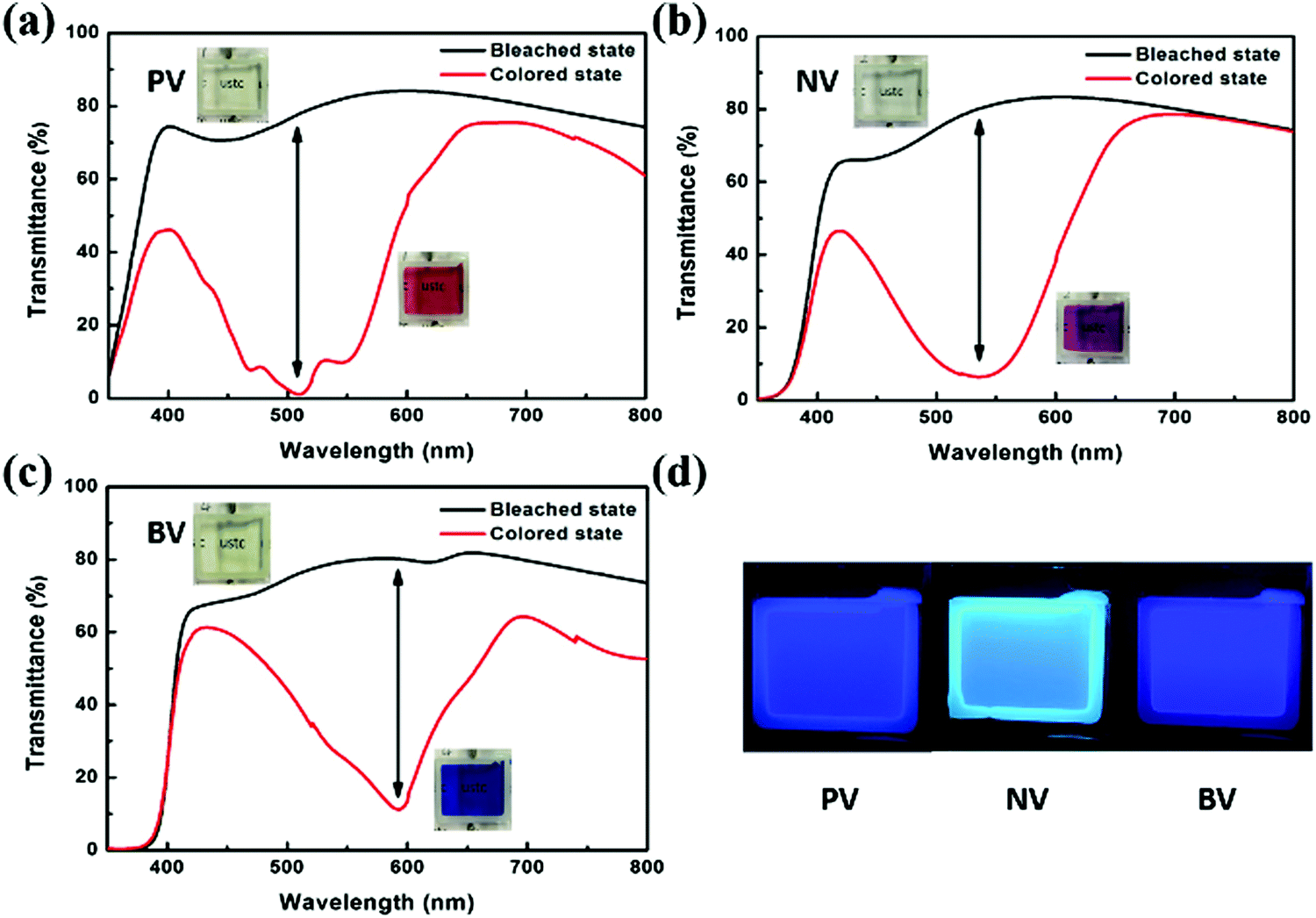 | ||
| Fig. 77 UV-vis transmittance spectra of ECDs: (a) PV, (b) NV, (c) BV, and (d) photographs of their emissive states. Adapted from ref. 545 with permission. Copyright (2018) from Elsevier Ltd. | ||
8.2 Photoresponsive LCPs
As a kind of intelligent material, stimuli-responsive crosslinked liquid crystal polymers (CLCPs) combine the advantages of polymer networks and liquid crystal molecules, such as elasticity, anisotropy, molecular cooperation effect, and stimuli responsiveness.546 Upon the application of appropriate external stimulation, the change in the mesogen arrangement in CLCPs could be transmitted to various macroscopic shape changes through the liquid crystal network. The incorporation of photochromic moieties, such as azobenzene, into CLCPs enables photoinduced deformation in a remote and precise manner. The reversible trans–cis isomerization of azobenzene upon specific light irradiation induces a change in the LC order and consequently leads to quick shape variations in the CLCP samples.547 The deformation process is based on the nanoscale molecular motion of the inner LC mesogens and requires neither complex architectural design nor precise assembly; therefore, photoresponsive CLCPs are appropriate materials to use for fabricating microscale actuators instead of traditional mechanical structures.548 This subsection mainly describes the recent research by Yu et al. regarding the fabrication of photoresponsive microstructured CLCP actuators for photonic crystals, superhydrophobic surfaces, and microfluidics.Photonic crystals contain periodic microstructures that reflect light of a particular wavelength. Adjustment of their microstructures can induce related reflection changes and have potential applications in display, communication, and anticounterfeiting technologies.549 Most of the existing phototunable photonic crystals comprise small LC molecules that can facilitate a change in their arrangement. Yu et al. firstly reported phototunable CLCP photonic crystals that exploited the advantage of photoinduced deformation (Fig. 78a).550 A two-dimensional (2D) periodic microarray was fabricated with azobenzene-containing CLCP via the replica molding technique. In accordance with geometry changes in the CLCP microarray induced by the photoisomerization of azobenzene groups, the ON/OFF switching behavior on the reflection spectra of the microarray was observed when it was alternately irradiated with UV-vis light (Fig. 78b and c). Subsequently, they prepared a photo and thermal dual-responsive CLCP inverse opal, which was the smallest CLCP photonic structure.551 Upon alternative irradiation of UV and visible light or temperature variations, the hole size of the inverse opal film changed (Fig. 78d), leading to rise and fall in the intensity of the reflection spectra (Fig. 78e), which could be ascribed to the deformation of CLCP induced by the photochemical reactions of azobenzene moieties or thermal-induced phase transition.
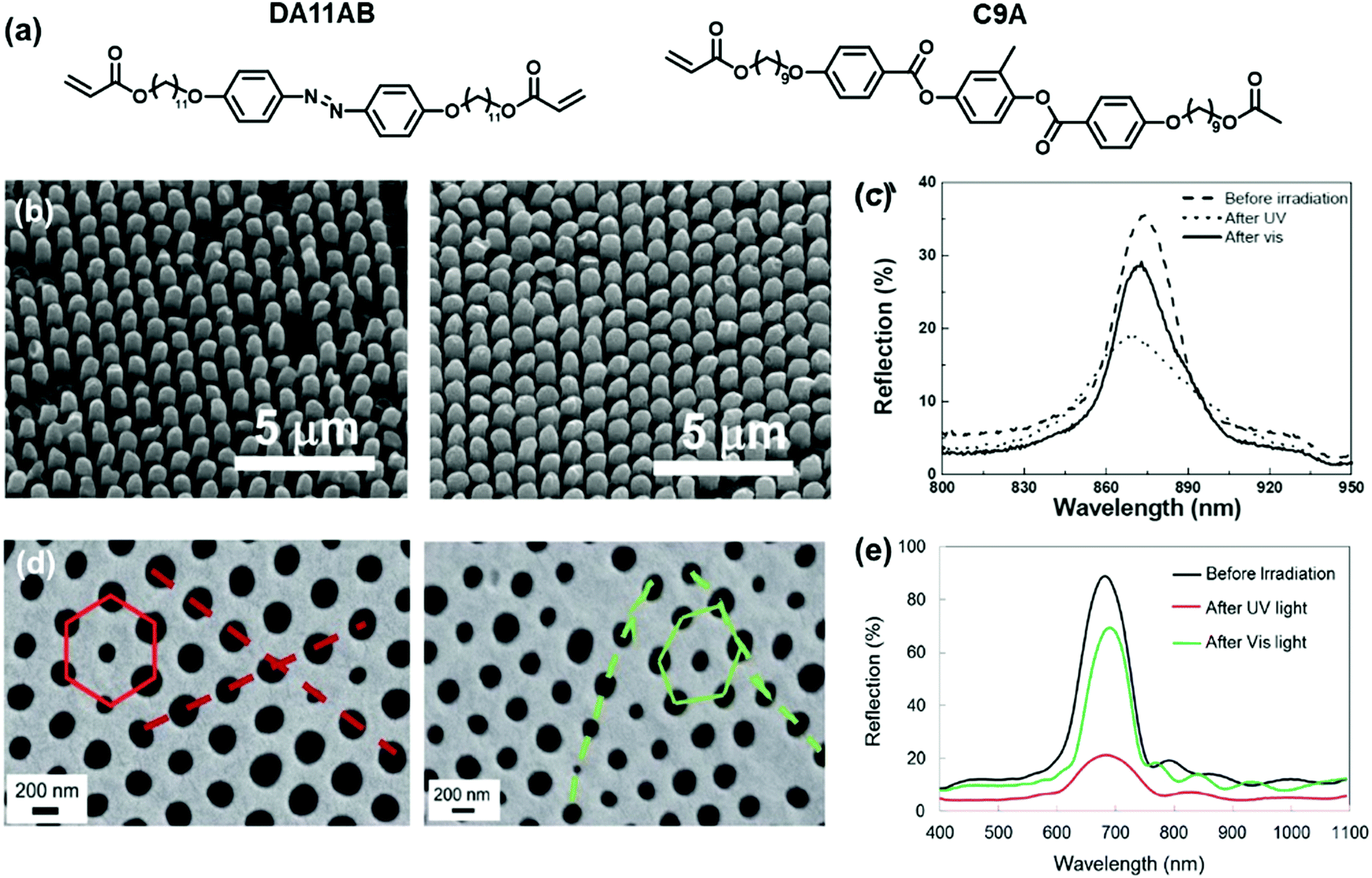 | ||
| Fig. 78 (a) Chemical structures of the LC monomers DA11AB and C9A. (b) SEM images of CLCP microarray before (left) and after (right) the irradiation of UV light. After UV-light irradiation, the mean diameter of the pillars increases from 0.587 to 0.695 μm, yielding a diameter increase of about 18%. (c) Reflection spectra of CLCP microarray under UV-light irradiation and subsequent visible-light irradiation. (d) SEM images of the inverse opal film (left) before and (right) after UV-light irradiation. The red and green regular hexagon and straight lines represent the arrangement of holes before and after UV-light irradiation. The distance between every two holes of the inverse opal film is 350 nm. (e) Reflection spectra of CLCP inverse opal film under UV-light irradiation and subsequent visible-light irradiation. Adapted from ref. 550 and 551 with permission. Copyright (2012 and 2014) from Wiley-VCH and Royal Society of Chemistry. | ||
Liu and Broer et al. designed a kind of novel azobenzene-containing CLCP coating with the ability to change its surface topology in the microscale upon UV-light irradiation (Fig. 79a).552 Due to the presence of chiral molecules in the LC mixture, LC mesogens spontaneously formed helical structures (the helix axis was parallel to the substrate), and the surface of the polymerized CLCP film was almost flat. Upon UV-light irradiation, protrusions were formed on the surface at planar positions, while wells were generated in homeotropic areas. As a result, the topological morphology of the CLCP surface was changed into a fingerprints-like structure (Fig. 79b). The mechanism of this remarkable photomechanical property is based on the reversible change in the order parameters by a photoinduced conformational change in the azobenzene units. Photoactivated generation of disorder in a LC network produces free volume that can lead to the controlled formation of dynamic corrugations at its surface. In addition to the use of dynamic friction in controlling the grip and release (Fig. 79c), this dynamic surface microstructure can yield versatile applications, such as tracking light in a solar energy system, liquid mixing, or flow regulation in microfluidic channels.553,554
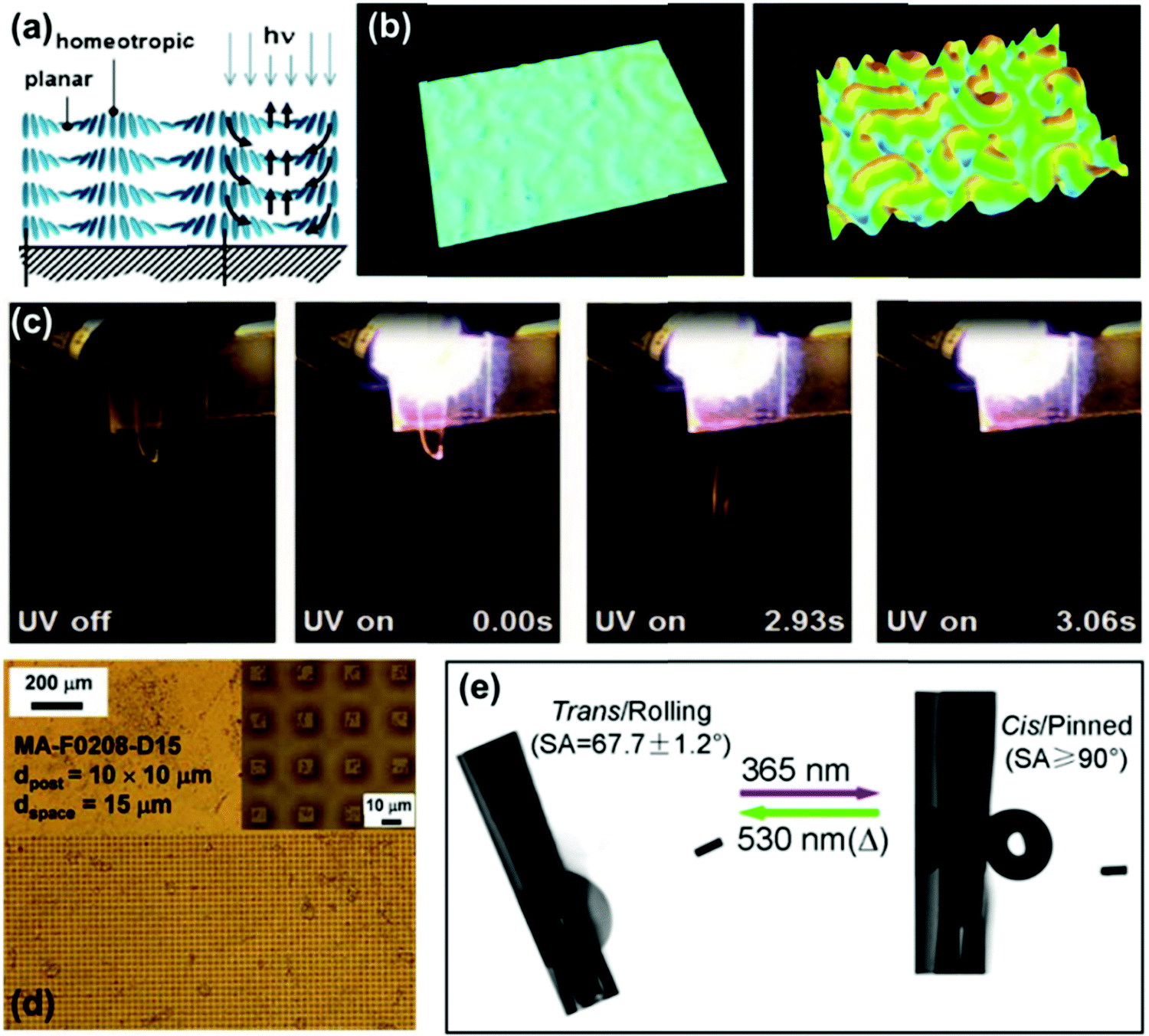 | ||
| Fig. 79 (a) Schematic representation of mesogen alignment in dynamic fingerprints. (b) 3D image of an initial flat state (left) and surface topographies under UV exposure (right). (c) Snapshots of a gripper that releases an object upon UV illumination. (d) Optical images of a microarrayed azobenzene CLCP film. (e) Light-controlled quick and reversible switching of superhydrophobic adhesion between rolling and pinning on microarrayed CLCP with a 2 μL water droplet. Adapted from ref. 552 and 555 with permission. Copyright (2014 and 2012) from Wiley-VCH and Royal Society of Chemistry. | ||
Superhydrophobic surfaces play wide-ranging roles in self-cleaning materials, lossless droplet transport, and open microreactors. When the adhesive force of a superhydrophobic surface is adjustable, it can work as a switch to control the movements of water droplets. In most of the existing works, the wettability and adhesive force of a superhydrophobic surface could be adjusted at the same time by changing the surface microstructures or chemical composition. It was challenging to regulate the adhesive force while maintaining the superhydrophobicity of a surface. In order to achieve the noncontact regulation of superhydrophobic adhesion, a CLCP microarrayed surface was fabricated by means of a secondary replicating method (Fig. 79d). The water sliding angle of the surface could be rapidly and reversibly adjusted by light irradiation while maintaining its large contact angle (Fig. 79e).555 Recently, Yu et al. fabricated two different submicroarrayed LCP surfaces using the high-throughput etching technique of colloidal lithography.556 These two surfaces were easily fabricated by modulating different types of etching masks, which were uniform with average diameters of sub-250 nm and sub-400 nm. Owing to the surface chemistry and surface morphology designed to induce substantial hydrophobicity, the two surfaces exhibited completely different wetting behaviors with regard to water adhesion, mimicking a rose petal and lotus leaf, respectively. Here, azotolane was used as the photoresponsive moiety instead of azobenzene to achieve all-visible-light control; therefore, the wettability of these films could be reversibly transferred when alternately irradiated by light at 470 and 530 nm. This kind of noncontact real-time photocontrollable switching of microdroplet adhesion might inspire and facilitate the designs and fabrication of novel microfluidic devices and lossless transportation.
In 2016, Yu et al. demonstrated a conceptually novel way to manipulate liquids in a micro-LCP channel by exploiting the asymmetric capillary force arising from photoinduced deformation (Fig. 80).557 Inspired by the lamellar structure of artery walls, they judiciously designed and synthesized a novel azobenzene-based linear liquid crystalline polymer (LLCP), where the flexible backbones and azobenzene mesogens self-assembled into ordered nanoscale lamellar structures due to the molecular cooperation effect. The non-crosslinked LLCP was firstly fabricated into 3D microtubes, which exhibited geometry changes from cylindrical to conical upon light irradiation with gradient intensity, inducing asymmetric capillary force to move the inner water slugs. When compared with existing photocontrollable water transportation technologies based on wettability gradients or the Marangoni effect, a tubular LLCP microactuator could transfer more types of liquids with faster speed. The transportation of multiphase liquids and even a different mix of liquids were achieved with remote photocontrol. Furthermore, the great advantage of water transportation in LLCP microtubes is that when compared with traditional microfluidics based on nonresponsive materials, air pumps and complex pipelines are not needed to drive the liquid movement. Hence, the technology developed by Yu's group has great potential to simplify the microfluidic system and create small-volume microreactors, laboratory-on-a-chip contexts, and microoptomechanical systems.
 | ||
| Fig. 80 (a) Schematics showing the motion of a slug of fully wetting liquid confined in a tubular microactuator (TMA) driven by photodeformation. (b) Lateral photographs of the light-induced motion of a silicone oil slug in a TMA fixed on a substrate that was taken through an optical filter to remove light with wavelengths below 530 nm. (c) Schematic illustration of the structure of artery walls. The middle coat of an artery, called the tunica media, comprising alternating muscle layers and elastic layers, which are responsible for stimuli-responsive deformation and mechanical robustness, respectively. (d) Molecular structure of LLCP. Reprinted from ref. 557 with permission. Copyright (2016) from Nature Publishing Group. | ||
In recent years, several burgeoning technologies, such as colloidal lithography, inkjet printing, molding, and electrospinning, have evolved to facilitate the fabrication of actuators at the micrometer and even nanometer scales, bringing photoresponsive LCP materials closer to real applications. It is anticipated that the preparation of microscale actuators will become the mainstream for the future development of photodeformable LCPs, which requires not only the guidance of new concepts and theories but also creative material design and application of novel processing methods.
Electrochromic polymers with other functions, such as electrofluorochromism, electrofluorescence, light-induced coloration effects, water processability, and fluorescent switching property, are briefly reviewed. The design and fabrication of photoresponsive LCP actuators for photonic crystals, superhydrophobic surfaces, and microfluidics are also addressed. However, further effort is needed for integrating stimuli-responsive polymers with other functions to fully demonstrate their broad potential applications. The future development of stimuli-responsive polymer materials is to further improve their specificity, sensitivity, and selectivity, as well as to promote their biomimic construction and functional integration, broadening the potential applications.
9. SMP materials
Shape-memory effect (SME) for polymers16,17 has been known for over 60 years. Products based on SMPs have also received commercial success since the early days, well-known examples of which are shrinkable tubing and shrinkable labels. However, this research field has only started to gain significant attention in this century, spurred by the realization of its enormous potential for use as deployable medical devices and aerospace structures.17 In general, SMPs fall into two categories: one-way and two-way SMPs (2W-SMPs). SMPs that can recover their permanent shape from a temporary shape when exposed to an external stimulus but cannot change back to its temporary shape unless they are reprogrammed by applying an external force are called one-way SMPs. SMPs that can reversibly change between two independent shapes under two various external stimuli are called 2W-SMPs. For re-imparting the SME to the former structure after recovery, a new programming step has to be applied, which is not suitable for advanced smart applications. In addition, new applications beyond the widely pursued medical devices (particularly in advanced manufacturing) have emerged. Other advances16,17 have also been made in extending shape recovery triggering mechanisms beyond direct heating. In this section, we briefly highlight the most notable recent developments in extending fundamental behaviors/mechanisms for one-way and 2W-SMPs.9.1 One-way SMPs
Depending on the number of memorized shapes, one-way SMPs can also be divided into two types: dual and multiple SMPs, according to their capability to memorize two or more shapes, respectively. Recently emergent thermadapt SMPs comprising dynamic covalent bonds are discussed in this subsection, because most examples of such SMPs are irreversible.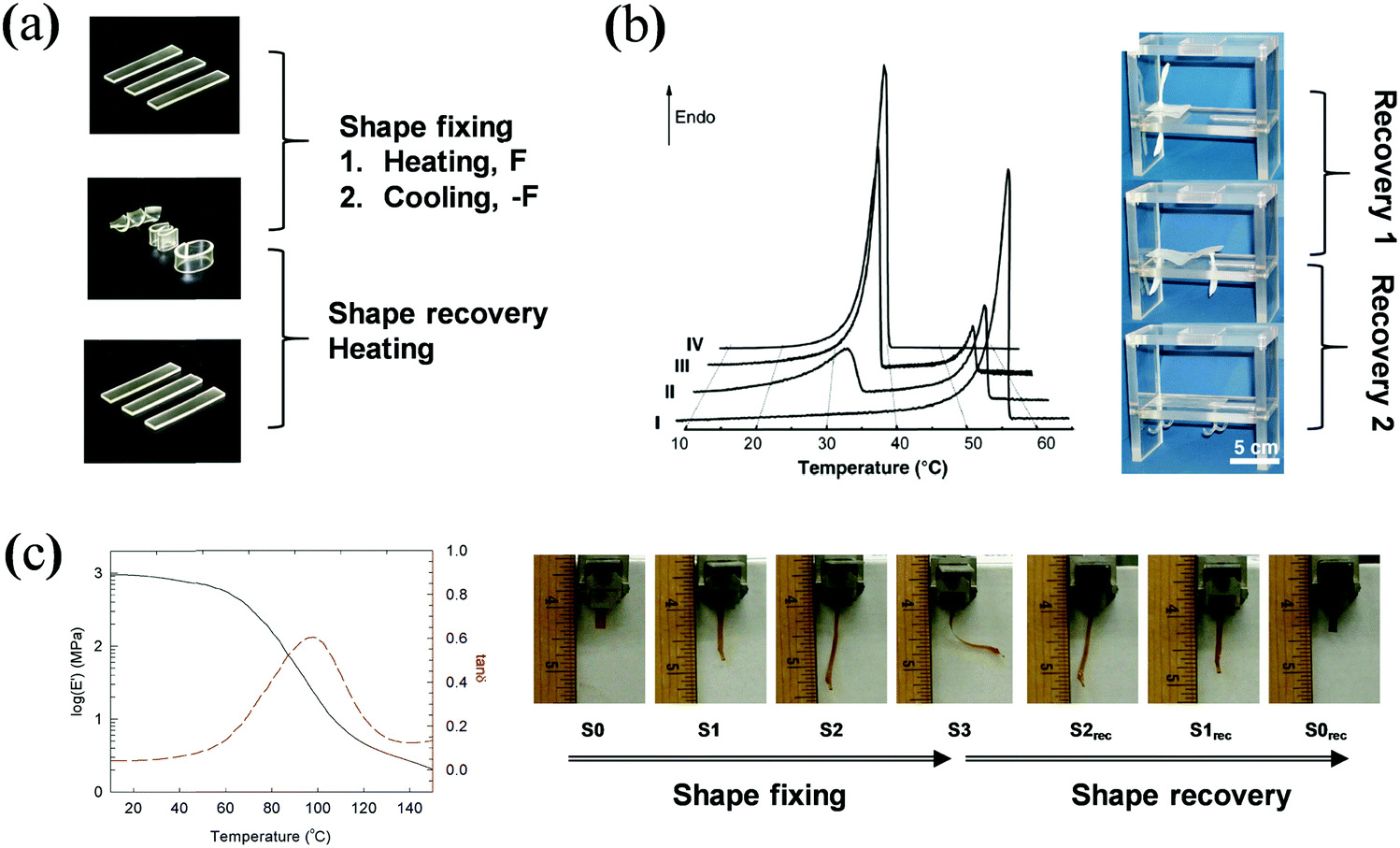 | ||
| Fig. 81 One-way SMEs. (a) dual-shape-memory cycle; (b) DSC curves for polymers with two distinct phase transitions and the associated triple-shape-memory recovery; (c) DMA curve for a polymer with a single broad transition and the associated multishape-memory cycle. Adapted from ref. 17, 558 and 559 with permission. Copyright (2015, 2006, and 2010) from Elsevier Ltd., National Academy of Sciences, U.S.A., and Nature Publishing Group. | ||
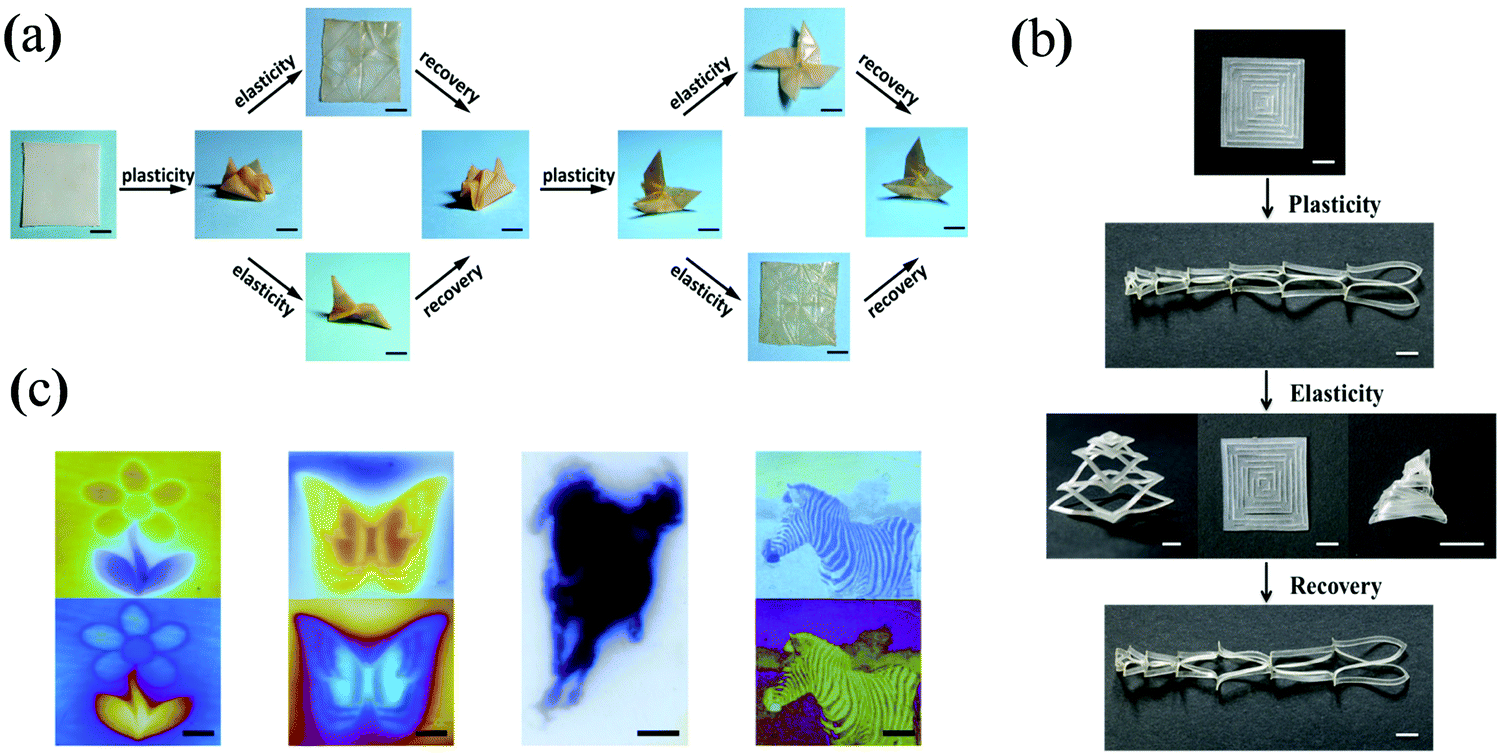 | ||
| Fig. 82 Thermadapt SMP. (a) Origami-enabled shape memory; (b) kirigami-enabled shape memory; (c) digitally defined stress map visualized as the birefringent color under polarized light. Adapted from ref. 561, 562 and 563 with permission. Copyright (2016, 2016, and 2018) from Wiley-VCH, AAAS, and Nature Publishing Group. | ||
9.2 2W-SMPs
Although the one-way effect is sufficient for numerous applications, it is highly desirable to formulate two-way SME (2W-SME). Until now, 2W-SMPs have been further classified into quasi and true 2W-SMPs, depending on whether they need applied force to take effect or not. The feasibility of quasi 2W-SME was demonstrated by Mather et al. in 2008.564 Their study demonstrated that a crystalline SMP, under a constant external load (Fig. 83a), could undergo reversible shape-shifting with temperature variations (below its crystallization and above its melting transition). Mechanistically, network anisotropy due to an external load causes directional crystallization/melting in the polymer, resulting in heating-induced contraction and cooling-induced elongation. Nevertheless, the versatility of this two-way shape-shifting behavior is severely constrained by the need to maintain an external load in the process. This limitation was elegantly addressed by Lendlein et al. in 2013 and Sheiko et al. in 2014.565,566 In a network system with two crystalline phases, the phase with higher melting transition was utilized to lock in internal stress, which permitted reversible shape-shifting via the other phase with the lower melting transition. Forgoing the external load, i.e., true 2W-SME, versatile reversible shape-shifting in a programmable manner can be realized (Fig. 83b).565 | ||
| Fig. 83 2W-SME. (a) Quasi two-way shape-memory cycles under various constant stresses; (b) stress-free programmable two-way shape memory; (c) soft robot based on light-programmable two-way shape memory. Adapted from ref. 564, 565 and 587 with permission. Copyright (2008, 2013, and 2018) from American Chemical Society, Wiley-VCH, and AAAS. | ||
In view of practical usability, true 2W-SMPs are obviously superior to the quasi versions. The available 2W-SMPs of this type include the following: (i) polymer laminates,567–570 which are made by stacking layers of crosslinked semicrystalline polymers or crosslinked semicrystalline polymers/elastomers; (ii) liquid crystalline elastomers (LCEs),571–573 and (iii) crosslinked semicrystalline polymers.574–582 Laminated SMPs are difficult to be fabricated because of macroscopic heterogeneity. Moreover, they can only implement two-way bending deformation. With respect to LCEs, which operate because of the reversible anisotropic phase transition of mesogens, the synthesis routes are complicated and the experimental conditions are rigorous. Comparatively, 2W-SMPs derived from crosslinked semicrystalline polymers are rather promising in consideration of the accessibility of raw materials as well as manufacturing and usage of the end products.
Lendlein et al. made use of a specific semicrystalline polymer with a broad melting temperature range, which was divided into an upper melting temperature (Tm) range (as the geometry-determining domain) and a lower Tm range (as the actuator domain).583 The material realized 2W-SME by repeatedly melting the lower Tm portion. Similarly, the method was also adopted by other researchers to enable 2W-SME via the partial melting of crystalline regions.566,584 Moreover, Wu et al. synthesized a two-way shape-memory IPN consisting of crosslinked elastomeric and crystalline components.585 Meng et al. reported a two-way shape actuator with double chemical networks.586 Xie et al. demonstrated much more complex 2W-SME in a polymer network with a single crystalline phase.587 In particular, the network comprised both thermo- and photoreversible bonds. Thermoreversible bonds were utilized to fabricate a complex 3D shape via solid-state plasticity. More importantly, photoreversible bonds allowed locking the network anisotropy in a spatioselective manner. Consequently, a flat sheet of polymer could be programmed into a 3D robotic crane that could undergo reversible wing-flapping (Fig. 83c). The rapid development in 2W-SME technology can open up future opportunities in unique device design.
By carefully analyzing the principles of documented systems, it is evident that an internal stress provider is imperative for building up a free-standing semicrystalline 2W-SMP regardless of its appearance and the way of interaction with the crystalline phases (Fig. 84), besides the chemical or physical crosslinkages that define the permanent shape.588 The internal stress provider (e.g., the higher Tm portion) plays the same role as external stress used for semicrystalline quasi 2W-SMPs and closely cooperates with the crystalline phase when temperature changes, inducing oriented recrystallization of the melted phase during cooling and restoring mechanical energy during heating.
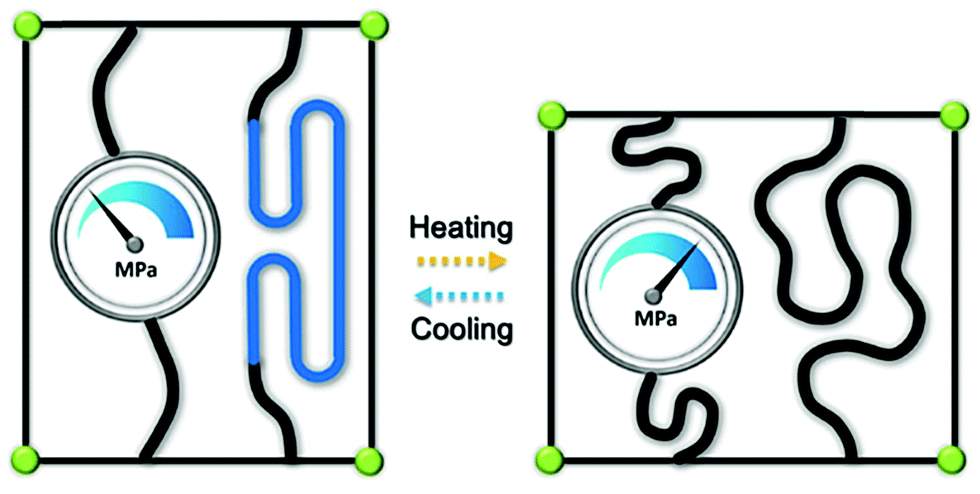 | ||
| Fig. 84 Design principle of a semicrystalline 2W-SMP with an internal stress provider. | ||
Inspired by this finding, a series of facile methods are proposed by Zhang et al. to prepare 2W-SMPs by introducing new internal stress providers. Crosslinked elastomeric styrene-butadiene-styrene block copolymer (SBS),589 for example, was successfully converted into an internal expansion stress provider in the crosslinked PU/SBS blend after appropriate programming. The temperature-induced reversible opposite directional deformability of the crystalline phase of PU, compressed SBS accompanying melting, and orientated recrystallization of PU led to the formation of the desired 2W-SME. Moreover, alkoxyamine moieties were introduced into crystalline polycaprolactone (PCL)-based crosslinked PU.590 Owing to the synchronous bond fission/radical recombination of C–ON bonds in alkoxyamine, the stretched PU network underwent uneven partial stress relaxation depending on the triggering status of C–ON bonds. Accordingly, the considerably relaxed portions tended to maintain their extended status, while the nearly unrelaxed portions tended to recover their original undeformed shape. These two opposite tendencies generated an internal tensional force, which worked with the bidirectional deformation driven by the melting/recrystallization of the crystalline phase such that 2W-SME could be detected.
In fact, PCL-based PU can be coupled with 2W-SME in a simpler way. As revealed by the recent exploration by Zhang et al.,588 proof-of-concept crosslinked crystalline PU was trained only by stretching and thermal treatment without using any additional chemicals. The reversible melting/recrystallization of the crystalline phases elaborately collaborated with the tensed hydrogen bond network, because the inherent hydrogen bond network could function as an internal stress provider. The programmed crosslinked PU exhibited reversible shrinkage and extension with changing temperatures.
It is worth noting that SME-aided self-healing has been extensively investigated with the rapid development of self-healing polymers.591 Contraction strain during the operation of SME can initially close cracks, facilitating intimate interfacial contact for the subsequent reestablishment of bonding. The method is particularly suited to the restoration of wider cracks; however, the existing solution mostly lies in one-way SME coupled with repeated self-healability. It is believed that an ideal case should be the cooperation between reversible SME and reversible self-healability. When reversible intrinsic self-healing ability is elaborately combined with 2W-SME in a single polymer, a larger crack can be repeatedly closed autonomously and then rebonded whenever it appears. In this context, no additional programming is needed to regain the SME after each healing event, which would give full play to multiple intrinsic self-healing events.
To verify this design, the abovementioned external stress-free two-way shape memory crosslinked PU/SBS589 was modified by including alkoxyamine into the skeleton (Fig. 85).592 The homolysis temperature of the included C–ON bond, i.e., self-healing temperature of the material, was tuned to be higher than the triggering temperature of the 2W-SME. The thermal retractability offered by the SME coupled with the reversible C–ON bonds proved to be able to indeed successively close and repair a wider crack (width: ∼250 μm). These works broaden the scope of SMP and intrinsic self-healing strategy.
 | ||
| Fig. 85 Schematic of a crosslinked PU containing reversible C–ON bond for the self-healing of wider cracks. Reprinted from ref. 592 with permission. Copyright (2018) American Chemical Society. | ||
In summary, significant progress has been recently made in the field of SMPs. Expansion of the shape-memory behavior beyond classical dual SMPs is particularly noteworthy, which includes multi-SMPs, thermadapt SMPs, and 2W-SMPs. These developments demonstrate the rich designability of SMPs, a key advantage over its metallic counterpart, namely, a shape-memory alloy (SMA). These advances at the fundamental material level can lead to practical benefits at the device level in the long run.
10. Polymer membranes for separation
In recent years, polymer membranes have become emerging materials for separation due to their obvious merits when compared with conventional separation techniques, such as distillation, extraction, crystallization, and filtration. In particular, polymer membranes possess low carbon footprint, low operating costs, energy efficiency, high product quality, small spatial requirements, and so on. The increasing concentration of CO2, oily wastewater, shortage of freshwater, and water pollution have promoted the development of separation membranes, particularly polymer membranes.593 Polymer membrane science is also an interdisciplinary subject, which involves polymer chemistry, polymer physics, materials science, material processing, and modification.594 The increasing demand for water resources, comprehensive treatment of drinking water, desalination of seawater and brackish water, and recycling of wastewater have become more serious problems than the energy issue. Therefore, the fabrication of separation polymer membranes with excellent properties has become imperative, yielding high efficiency, flux, and selectivity; good antifouling performance; and high chemical stability. Generally, the applications of polymer membranes include four main subgroups: reverse osmosis, pervaporation, gas separation, and electrodialysis. Each subgroup has its own applications. For example, the main industrial application of reverse osmosis is the desalination of brackish water and seawater. The principal application of pervaporation is the dehydration of organic solvents. In this section, we present the recent advances in polymer membranes for oil/water separation, blood purification and water treatment, and water desalination.10.1 Polymer membranes for oil/water separation
Polymer membranes have been widely used in diverse separation applications and have advantages in energy consumption, operation process, and secondary pollution problems over other separation technologies such as distillation, sublimation, crystallization, extraction, and ion exchange.595–598 However, besides these advantages, surface wettability is a major drawback of traditional polymer membranes. Most polymer membranes such as polysulfone (PS), polypropylene (PP), and polyvinylidene fluoride (PVDF), are not superwetting (superlyophilic or superlyophobic) and suffer from serious fouling, particularly with oil/water separation.599–605 The membrane flux and separation efficiency quickly decline due to fouling, which impedes the long-term use of such membranes and causes high cost for treating large amounts of oil/water mixtures. In contrast, superwetting polymer membranes have distinctly different wetting and dewetting behaviors with respect to oil and water. For instance, a superhydrophilic membrane is superoleophobic in water, and a superoleophilic membrane is superhydrophobic in oil. Therefore, superwetting polymer membranes are capable of more effective separation of oil from water or water from oil by the selective permeance and repellence and simultaneously control the fouling issue to the minimum.In order to improve the antifouling performance of polymer membranes, blending modifications that involve directly blending lyophilic or lyophobic additives in a polymer matrix during the membrane-manufacturing process have been widely adopted because of their ease of operation and scaling-up feasibility for industrialization.601–603 However, only a few blending additives can endow polymer membranes with superwetting property, particularly superhydrophilicity. An effective method for fabricating superwetting polymer membranes is surface modification that involves grafting or coating superlyophilic or superlyophobic modifiers onto the membrane surface. Another brand-new strategy for fabricating superwetting polymer membranes is to construct artificial hierarchical micro-/nanostructures on the membrane surface and accomplish superwettability via a combination of surface chemical composition and surface roughness. Very recently, nanomaterial-constructed membranes with the advantages of ultrathin thickness and tunable pore size at the nanometer scale have been comprehensively investigated. Polymer-decorated superwetting nanocomposite membranes were also tentatively applied for oil/water separation and exhibited both high efficiency and ultrahigh flux far beyond traditional polymer membranes.
Researchers usually adopt the surface grafting method in which superlyophilic polymer chains are immobilized onto membrane surfaces and function like “nanobrushes” to endow polymer membranes with superlyophilicity and antifouling property. In general, reactive groups need to be introduced onto the membrane surface in advance by introducing initiator sites or exposing the membrane to low-temperature plasma, ultraviolet, γ-ray, electron beam radiation, etc.612–614 The grafted polymers are usually grown via “grafting to” or “grafting from” approaches.615 Jin et al. synthesized the zwitterionic polyelectrolyte, poly(3-(N-2-methacryloxyethyl-N,N-dimethyl) ammonatopropanesultone), and grafted it onto a commercially available PVDF microfiltration membrane via a surface-initiated ATRP process.616 After grafting the zwitterionic “nanobrushes,” the wettability of the PVDF membrane transferred from hydrophobic to superhydrophilic and superoleophobic in the presence of water. Because of the superwetting property, this membrane could thoroughly separate a series of oils from oil-in-water emulsions with extremely high separation efficiency of above 99.999%, simultaneously with excellent anti-oil-fouling and cyclic performances. However, it has intrinsic drawbacks of time consumption and harsh chemical environment requirement during membrane fabrication, which considerably impedes the commercialization of this method to manufacture superwetting polymeric membranes.
Surface coating is another widely adopted method to fabricate superlyophilic polymer membranes and has promising potential for industrial mass production of membranes. PDA and polyethyleneimine (PEI) are a common coating combination for fabricating superhydrophilic polymer membranes, since PDA can be easily and robustly deposited on various solid substrates and superhydrophilic PEI is easy to be grafted on PDA via a Michael addition reaction. Xu et al. reported the fabrication of a superhydrophilic and underwater superoleophilic nanosilica-decorated PP microfiltration membrane with the assistance of a PDA/PEI intermediate layer.607,609 In this case, PEI played an important role in the polycondensation of silicic acid via the so-called biomineralization process. Benefiting from the robust superwetting property, this nanosilica-decorated PP membrane exhibited very high permeation flux and very high oil rejection for surfactant-stabilized oil-in-water emulsions, as well as outstanding flux recovery after simply rinsing with water. It is worth noting that PDA, a well-known “bio-glue,” is widely used to coat not only polymer membranes but also inorganic membranes, and it can act as an intermediate layer before the formation of the desired separation layer. Other coating materials, such as metallic oxide NPs, metallic carbonate NPs, and even 10 nm-thick metallic oxide nanolayers, have been adopted to fabricate superwetting and antifouling polymer membranes.610,611,617 Meanwhile, a collaborative mechanism exhibiting concomitant fouling resistance and fouling release during oil-in-water emulsion separation was proposed by Jiang et al. to explain the outstanding anti-oil-fouling performance of these membranes with both inorganic hydrophilic microdomains and organic hydrophobic microdomains. A drawback of the superwetting membranes obtained via surface coating is the instability of the coating layer, which is problematic for long-term use, particularly in acidic and alkaline environments.
A challenge in oil/water separation is separating highly viscous crude oil from oily water, where crude oil is easy to adhere onto the membrane surface and cause the fouling of membranes, even for those polymer membranes with underwater superoleophobicity toward light oil. A widely recognized theory is that the superoleophobicity and anti-oil-fouling property of a membrane is directly affected by its surface hydratability that can result in the formation of a sufficient hydrated layer to protect the membrane surface from oil contact and adhesion.618,619 Therefore, hydrogels with strong hydratability are ideal materials for modifying polymer membranes and achieve low-adhesive superoleophobicity for the antifouling separation of crude oil. The key point in modifying polymeric membranes by hydrogels is to form sufficiently small versions on the membrane surface such that the micro- and nanosized membrane pores are unblocked. Jin et al. used a LbL self-assembly method with molecular-scale controllability to coat an ultrathin alginate hydrogel layer on a poly(acrylic acid)-grafted polyvinylidene fluoride (PAA-g-PVDF) ultrafiltration membrane (Fig. 86).620 The thickness of the hydrogel layer was highly controllable from 3 to 17 nm, and the nanosized membrane pores were effectively maintained. An ionic bond was designed between the hydrogel layer and membrane substrate to ensure membrane stability. The hydrogel-modified membrane was superhydrophilic and exhibited underwater superoleophobic properties with nearly zero oil adhesion for crude oil and was capable of separating crude-oil-in-water emulsions with ultrahigh efficiency and outstanding antifouling ability. Another approach is to synthesize nanohydrogels and then decorate the nanohydrogel on polymer membranes. For instance, a sulfobetaine zwitterionic nanohydrogel (diameter: 50 nm) was synthesized via an inverse microemulsion polymerization process and then grafted onto a polymer microfiltration membrane.621 The resulting membrane was also endowed with superwetting and anti-oil-fouling properties. It should be noted that the size of the nanohydrogels should match with the surface pore size of the polymeric membrane.
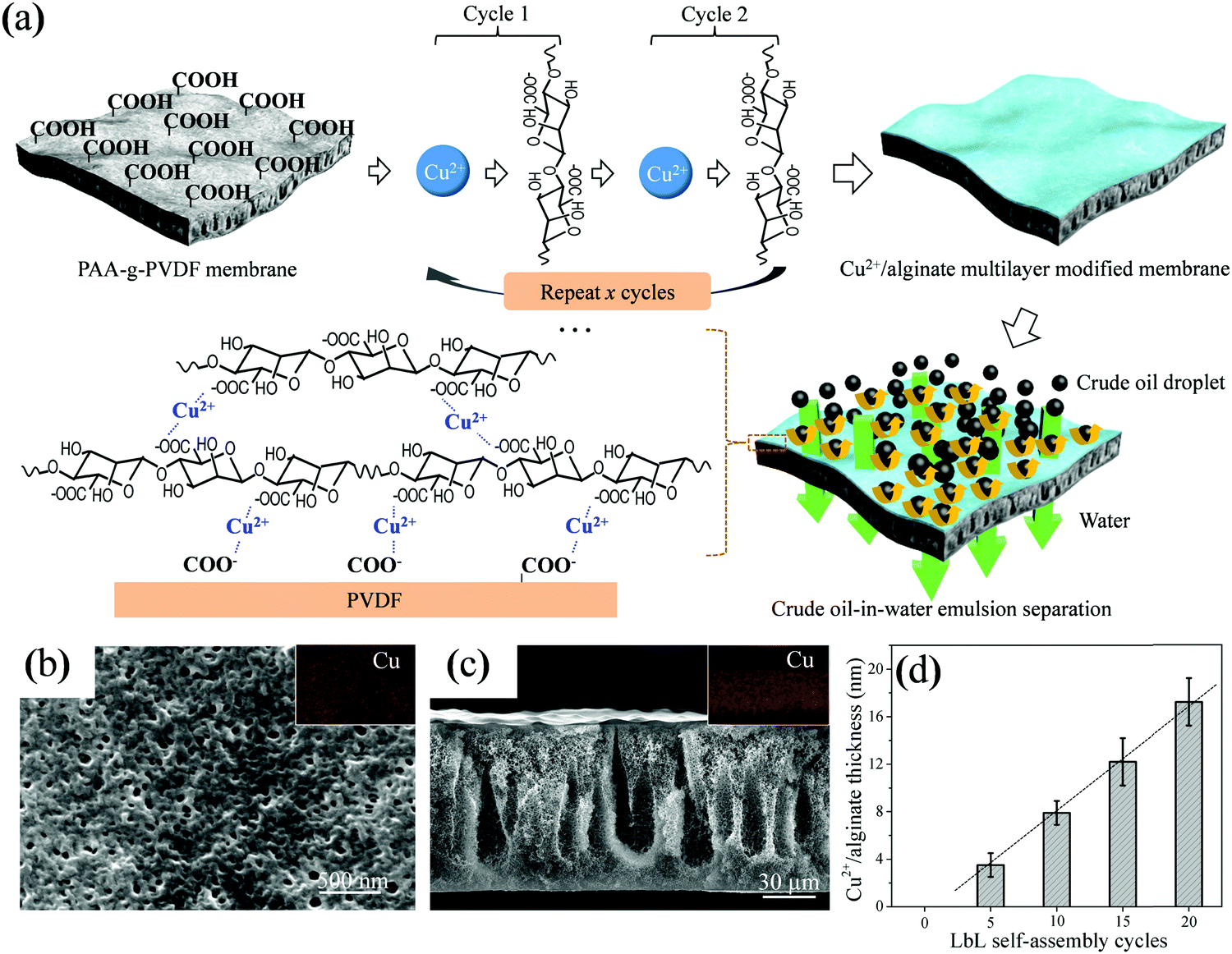 | ||
| Fig. 86 Ultrathin alginate-hydrogel-modified PAA-g-PVDF membrane. (a) Schematic showing the formation of alginate hydrogel on the membrane surface via LbL self-assembly and the antifouling separation of crude-oil-in-water emulsion. (b) Top-view and (c) cross-sectional SEM images of the membrane after 10 LbL cycles. Insets show the corresponding EDX images of Cu element distribution. (d) Thickness of the alginate hydrogel as a function of LbL cycles. Reproduced with permission from ref. 620. Copyright (2018) Wiley-VCH. | ||
The artificial construction of micro-/nanohierarchical surface structure with sufficient roughness during the membrane-forming process is crucial to obtain superwetting polymer membranes without subsequent surface modification. Jin et al. reported the construction of a spherical structure with the size of several hundreds of nanometers on a PAA-g-PVDF membrane via a salt-induced phase-inversion method (Fig. 87).624 Distinguished from the general approach of adding salt as a pore-forming additive to change the pore structure of the membrane, the salt that was added into the coagulation bath at a nearly saturated concentration can function as nucleates to induce the aggregation of polymer micelles. These micelle aggregates on the membrane surface grow additionally in the subsequent phase-inversion process into the desired spherical particles. Synergistically combined with the artificial micro-/nanohierarchical surface structure and the hydrophobicity of PAA-g-PVDF, the resulting PAA-g-PVDF membrane has intrinsic superhydrophilicity and underwater low-adhesive superoleophobicity. Meanwhile, this membrane could effectively separate both surfactant-free and surfactant-stabilized oil-in-water emulsions, either under low applied pressure or solely driven by gravity. An inert solvent-induced phase-inversion method was also used to generate a spherical structure on PVDF membranes.625 The added ammonia water was prone to induce localized microphase separation and caused the formation of “incipient precipitation” in the form of PVDF clusters, which functioned as a growing point to generate spherical microparticles during the subsequent phase-inversion process. As a result, the obtained PVDF membrane was uniformly skinless and comprised spherical microparticles with hierarchical micro-/nanostructures, which resulted in the superhydrophobicity and superoleophilicity of the membrane. Based on the oil-favoring and water-repellent properties, this PVDF membrane can also be applied for separating emulsified water from oil and organic solvents.
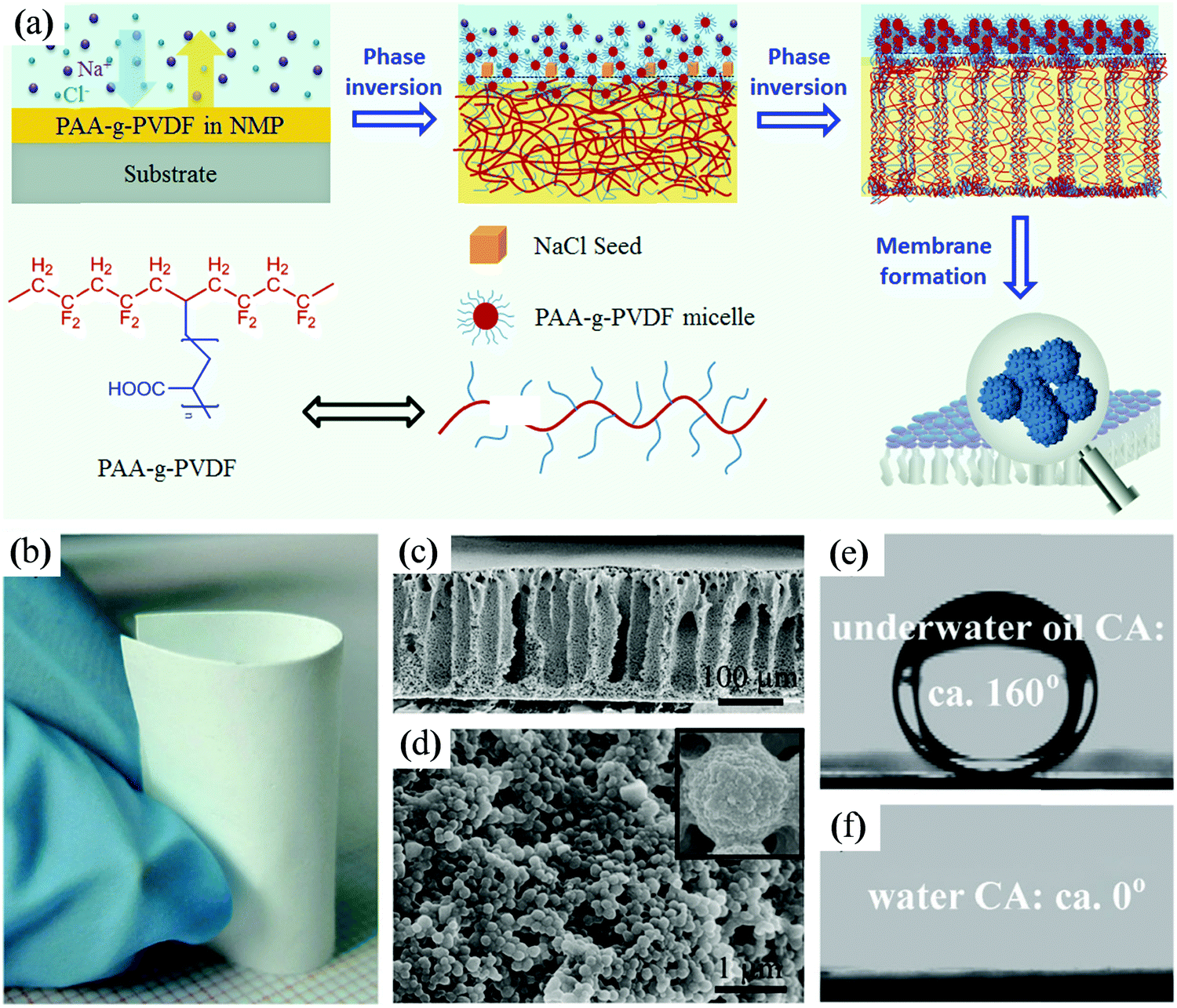 | ||
| Fig. 87 PAA-g-PVDF membrane with a micro-/nanohierarchical surface structure. (a) Schematic showing the fabrication of the membrane by a salt-induced phase-inversion process. (b) Photograph of the membrane. (c) Cross-section and (d) top-view SEM images of the membrane. Photographs of (e) an oil droplet on the membrane in water and (f) a water droplet on the membrane in air. Reproduced from ref. 624 with permission. Copyright (2014) Wiley-VCH. | ||
Generally, oil has much lower surface energy than water and wets a membrane more easily, so that the most hydrophilic or superhydrophilic membranes with high surface energy are simultaneously superoleophilic. Liu et al. reported this phenomenon with a PVDF-blend membrane. This membrane is superhydrophilic and underwater superoleophobic because of the inner hydrophilic additive and its spherulites–petaloid surface structure.626 On the other hand, the membrane is superoleophilic and underoil superhydrophobic. Therefore, such a type of superhydrophilic polymeric membranes can achieve both oil separation from water and water separation from oil. When compared with surface modification, the artificial construction of a micro-/nanohierarchical surface structure yields membranes with intrinsic surface components and omits the component-releasing problem for surface-modified membranes. However, it is still difficult to construct controllable micro-/nanohierarchical structures on the polymeric membrane surface by traditional methods. Innovative methodologies on this strategy are highly desired.
Single-walled carbon nanotube (SWCNT), a chemically stable and high-strength 1D nanomaterial, is a good choice to construct ultrathin network membranes with tunable thickness and pore size at the nanometer scale. Jin et al. constructed SWCNT membranes with nanopores and intrinsic superoleophilic–hydrophobic property, and they applied this membrane for the separation of water from water-in-oil emulsions for the first time.627 However, it was impossible for a pristine SWCNT membrane to achieve oil separation from oily water because of its wettability. Therefore, surface modification of the SWCNT membrane becomes imperative. The challenge in modifying an inert SWCNT membrane is to guarantee the uniform distribution of the modifier. In order to overcome this problem, a PDA layer with a thickness of several nanometers needs to be uniformly coated on every SWCNT in advance, which functions as an intermediate layer for the further grafting of PEI (Fig. 88).628 The fabricated SWCNT/PDA/PEI composite membrane exhibited superhydrophilicity and underwater superoleophobic property with ultralow oil adhesion, and it could effectively separate nanosized oil from oil-in-water nanoemulsions. According to the classic Hagen–Poiseuille fluid dynamic theory, membrane flux is inversely proportional to the total distance of the liquid running through the membrane.629 Hence, a thinner membrane corresponds to higher membrane flux. This nanomaterial-constructed composite membrane exhibited extremely high flux far beyond traditional polymeric and ceramic ultrafiltration membranes benefiting from its ultrathin thickness. It is believed that this new type of nanomaterial-constructed membrane is a promising alternative for traditional membranes in large-scale applications in the near future. A critical problem for the application of nanomaterial-constructed membranes is to achieve scalable and controllable membrane fabrication.
 | ||
| Fig. 88 SWCNT/PD/PEI composite membrane. (a) Schematic showing the fabrication of a membrane and the separation of oil-in-water nanoemulsion. (b) Digital photograph of a free-standing membrane. (c) AFM image and height profile of the membrane. (d) TEM image of the membrane. Photographs of (e) a water droplet on the membrane in air and (f) an oil droplet on the membrane underwater. Reproduced from ref. 628 with permission. Copyright (2015) Royal Society of Chemistry. | ||
10.2 Polyethersulfone (PES) membranes for blood purification and water treatment
Over the past several decades, polymeric membranes have been widely used for separation and purification requirements, such as molecular separation, water purification, plasma collection, and blood purification. Among the polymeric membranes used for blood purification and water treatment, PES membranes have shown good thermal and chemical resistance. However, some drawbacks associated with PES (such as its hydrophobic nature and adhesion of proteins and organisms) exist, which may yield undesirable results.Zhao et al. also developed a new method to modify PES membranes by “in situ crosslinking polymerization.” That is, functional monomers are introduced into a PES casting solution followed by a thermally induced crosslinking reaction, and the obtained solution is then directly converted into the membrane by a phase-inversion technique. For instance, PES was selected as the membrane matrix, and sodium acrylate (AANa) and sodium p-styrene sulfonate (SSNa) were selected as the functional monomers.631 After the in situ crosslinking polymerization of AANa and/or SSNa, a series of functional membranes were obtained, as shown in Fig. 89a. These functional membranes showed lower protein adsorption, suppressed platelet adhesion by 93%, and prolonged clotting times by 60 s for activated partial thromboplastin time (APTT) when compared with those obtainable from pristine PES membranes. Moreover, the membranes showed good antifouling property. These results confirmed that the highly efficient and convenient in situ crosslinking polymerization endowed PES membranes with excellent biocompatibility, thereby having great potential for use in blood purification.
 | ||
| Fig. 89 (a) In situ crosslinking polymerization of monomers; (b) in situ crosslinking polymerization combined with post-crosslinking of monomers; (c) surface coating followed by in situ crosslinking of polymers; (d) direct blending of nanofibers and nanotubes. Adapted from ref. 631–634 with permission. Copyright (2016, 2017, 2018, and 2017) Elsevier Ltd and American Chemical Society. | ||
By a combination of in situ crosslinking polymerization and post-functionalization, Zhao et al. could also integrate thin hydrogel films on the surface of PES membranes. For instance, hydroxyl groups were introduced onto the PES membrane by the use of in situ crosslinking polymerization of HEMA; then, double bonds were also introduced via the reaction between hydroxyl groups and acryloyl chloride. Sulfobetaine methacrylate (SBMA) and AANa were selected as the functional monomers to fabricate thin hydrogel layers onto the surfaces of PES membranes by UV-light-initiated polymerization.632 Ag ions were absorbed into the hydrogel layers by the carboxylate groups from AANa, as shown in Fig. 89b. The excellent hemocompatibility of the modified membranes was confirmed by undetectable plasma protein adsorption, suppressed platelet adhesion, prolonged clotting times, low hemolysis ratio, and suppressed blood-related complement activation. In addition, the strong antibacterial ability could last for over 5 weeks.
The in situ crosslinking reaction could also be performed between different polymers. Zhao et al. straightforwardly prepared a thin layer within a few minutes via the coordination reaction between tannic acid (TA) and FeIII. Thereafter, a reactive zwitterionic polymer (PEI-S) was post-crosslinked with the matrix layer via a dip-coating process at room temperature, as shown in Fig. 89c. Combining TA with an active zwitterionic polymer was an effective approach for constructing a universal, stable, and low-cost antifouling material surface.633
In spite of the addition of organic polymers, organic nanomaterials could also enhance the filtration and biological performances of polymeric membranes. For instance, Zhao et al. reported the usage of CNTs and aramid nanofibers (ANFs) to enhance the ultrafiltration and biological performances of PES membranes.634 The flux recovery ratio, protein adsorption, and bacterial adhesion tests validated that the modified membranes exhibited improved antifouling, blood compatibility, and efficient adsorption of small molecular toxins during dialysis applications of the composite membranes (Fig. 89d).
By simply blending stimuli-responsive materials into PES, a membrane with redox-responsive hydraulic permeability was fabricated for the first time by introducing a reversible redox-responsive system based on the host–guest complex between ferrocene and β-cyclodextrin.635 As a result, the water flux of the membrane with 18.3 wt% redox-responsive materials was 270 mL m−2 mmHg−1 h−1 in the original state, which increased up to 1022 mL m−2 mmHg−1 h−1 in the oxidation state and decreased to 538 mL m−2 mmHg−1 h−1 in the reduction state. Changes in the pore size and porosity of the modified membrane has outstanding potential for application in chemical detection and biological treatments, as shown in Fig. 90a.
 | ||
| Fig. 90 (a) Direct blending of redox-responsive materials; (b) in situ crosslinking copolymerization of ion-exchange monomers; (c) in situ crosslinking copolymerization of salt-responsive monomers; (d) direct blending of amphiphilic polymers coupled with electrospinning technology. Adapted from ref. 635, 636, 638, and 639 with permission. Copyright (2015, 2016, 2017, and 2018) Elsevier Ltd, American Chemical Society, and Royal Society of Chemistry. | ||
In situ crosslinking copolymerization method is also a good choice to fabricate functional PES membranes for water treatment. To develop PES membranes with high ion-exchange capacities (IECs) but low swelling degree, the universal mold casting approach was performed by in situ crosslinking negatively/positively charged monomers in the presence of PES, as shown in Fig. 90b. As a result, crosslinking monomers provided high IECs along with restricting the swelling of these hydrophilic monomers with high crosslinking density and hydrophobic polymers. For instance, Zhao et al. chose AA for fabricating cationic-exchange membranes (CEM) and 2-vinylpyridine (2VP) for fabricating anionic-exchange membranes (AEM).636 The new method yielded ultrahigh IECs of 7.88 mequiv g−1 for CEM and 6.27 mequiv g−1 for AEM, which are 8.8 and 7.0 times higher than that of Nafion 117 (0.89 mequiv g−1), respectively.
Salt-responsive PES membranes were prepared by the use of in situ crosslinking copolymerization method with methacrylatoethyl trimethyl ammonium chloride ([MTA][Cl]) as the functional monomer.637 The anion strength response of the membranes was positive, and the responsive coefficient of the membranes could be 26 times in response to an aqueous solution of NaCl (as shown in Fig. 90c). The cation species and concentrations had a minimal influence on the responsive behavior. Meanwhile, the membranes responded to both anion species (PF6−, BF4−, and SCN−) and their strengths. After slightly modifying the in situ crosslinking copolymerization method for a liquid system, novel anion-responsive smart PES membranes could be fabricated by filling PES microporous membranes (solid system) with poly(ionic liquid)s (PILs) gels.638 The wetting properties of these PILs could be controlled by changing their counteranions (CAs), and therefore, the filled PIL gel gates in the membrane pores could spontaneously switch from the “closed” state to the “open” state by recognizing the hydrophilic CAs in the environment and vice versa. As a result, the flux values of the smart membranes could be adjusted from a very low level (0 mL m−2 mmHg−1 for Cl−, Br−, and BF4−) to a relatively high one (430 mL m−2 mmHg−1 for TFSI). The anion-responsive gating behavior of PILs-filled membranes is fast, reversible, and reproducible. The proposed smart membranes are highly attractive for ion-recognizable chemical/biomedical purifications.
To rapidly remove toxins from wastewater, the simple blending of amphiphilic polymers coupled with the electrospinning method was developed. For instance, a novel ultrafast adsorption nanofibrous membrane (UFAM) for organic dyes was prepared via simple functional group surface migration.639 An amphiphilic copolymer of MMA and SSNa (mass ratio of 3:2) was synthesized by free radical polymerization and further blended into PES followed by electrospinning. Only by soaking in water and lyophilization, the hydrophilic and negatively charged groups migrated on the PES nanofibrous membrane surface, and the obtained UFAM was found to be exceptionally efficient in the removal of methylene blue (MB) from an aqueous solution, as shown in Fig. 90d. In particular, an ultrafast adsorption rate for MB with an adsorption equilibrium time of less than 5 min was reported. In addition, the UFAM showed excellent performance with respect to recyclability, dynamic filtration–purification, and selective adsorption of cationic dyes from mixed dye solutions.
As a short summary, for either blood purification or water treatment, the in situ crosslinking copolymerization method can avoid multistep purification, while the crosslinking degree should be controlled at a certain level. Otherwise, homogenous gels are formed in the case of a high crosslinking degree of monomers. In contrast to in situ crosslinking polymerization, the direct blending of amphiphilic polymers or small hydrophobic molecules may necessitate more steps for synthesis and/or purification, while the surface/interface migration of amphiphilic polymers may induce some dramatical merits, such as the fast adsorption of toxins. Different from in situ crosslinking polymerization and direct blending, the post-crosslinking of thin hydrogel films (even by several nanometers) could result in significant surface modification. However, precise control over the thickness of thin hydrogel films is desirable, because a thick hydrogel film can result in sacrificing the permeability of PES membranes. Notably, the abovementioned strategies for modifying PES membranes are also applicable to improve the performance of other polymeric matrixes.
10.3 Nanofiltration membranes (NFMs) for seawater desalination
Nanofiltration is a widely used pressure-driven membrane forming technique with low cost and high efficiency; it is used in separating organic dyes and multivalent ions, and it offers a promising solution to alleviate the formidable issue of freshwater scarcity.640 Diverse manipulation strategies have been developed to meet various expectations of NFMs, such as interfacial polymerization, mussel-inspired codeposition, and LbL assembly, for constructing selective layers on the top surfaces of porous substrates. Although these NFMs have made tremendous progress in the past few decades, they still suffer from a tradeoff between the water permeation flux and salt rejection. Therefore, efficient strategies are required to break the tradeoff effect and to simultaneously achieve high water permeation flux and high salt rejection. In recent years, a series of appealing approaches have been developed to improve the performances of NFMs, such as substrate optimization, sacrificial-layer application, interlayer introduction, and nanomaterials incorporation.Livingston et al. reported the formation of sub-10 nm polyamide nanofilms by controlling interfacial polymerization on a sacrificial layer of cadmium hydroxide nanostrands.641 However, these ultrathin nanofilms were only suggested as separating layers in NFMs that were used in processing organic solvents. Metal–organic framework NPs (ZIF-8) were also used as a sacrificial layer by Jin et al. to facilitate the formation of a rough polyamide selective layer with crumpled nanostructures.642 These crumpled nanostructures were believed to increase the effective area of the selective layer for water permeation. The NFMs thus obtained exhibited high water permeability (up to 53.5 L m−2 h−1 bar−1) with salt rejection of above 95% for Na2SO4. Nevertheless, it should be noted that heavy metal compounds such as cadmium hydroxide and ZIF-8 were used in the fabrication processes of NFMs, which are harmful to human health if their ions are left in the produced water. Therefore, Livingston et al. tried to fabricate ultrathin selective layers at the free aqueous–organic interface.643 Smooth sub-8 nm polyamide nanofilms were synthesized to provide fast water transport through the nanofilm composite membranes. Manipulating the intrinsic nanofilm thickness from ∼15 down to 8 nm revealed that water permeance proportionally increased with a decrease in the thickness, after which it increased nonlinearly to 2.7 L m−2 h−1 bar−1 as the thickness was further reduced to ∼6 nm. However, this method is limited for practical applications because of the difficulty in fabricating ultrathin films in a large area at the free aqueous–organic interface. On the other hand, Tan et al. modulated the interfacial polymerization by “controlling” monomer diffusion with water-soluble polymers to facilitate the formation of crumpled nanostructures in the polyamide selective layer.644 The thickness of the crumpled polyamide films (the authors called them nanoscale Turing structures) was about 20 nm or less, which was much thinner than those fabricated by traditional methods. Therefore, the as-prepared NFMs showed both high water permeability and high salt rejection, which could break the tradeoff effect.
Apart from polyamide ultrathin films obtained by interfacial polymerization, Xu et al. developed mussel-inspired chemistry to fabricate selective layers of NFMs. For example, as schematically shown in Fig. 91, CuSO4/H2O2 was found to trigger the rapid deposition of PDA-based ultrathin coatings with high uniformity and enhanced stability.645–648 These mussel-inspired coatings exhibit two kinds of nanopores (around 0.6 and 1.0 nm) for versatile molecular separation. The corresponding NFMs exhibited promising potential for water desalination as well as organic solvent separation.649 Polyamines such as PEI were used to promote the formation of mussel-inspired ultrathin selective layers via Michael addition and Schiff base reactions between a series of polyphenols and polyamines.650–655 At the same time, the surface potential and surface hydrophilicity were remarkably modulated because the used PEI was a type of positively charged polyelectrolyte.650,651 Therefore, the as-prepared NFMs showed different salt rejection properties via the Donner effect as compared to those exhibited by negatively charged polyamide-based membranes. Their surface properties could be further tuned by the addition of inorganic NPs or the biomineralization of inorganic nanofilms and nanorods.656–659 The resulting organic–inorganic composite NFMs could be applied for long-term salt rejection or dye removal from water due to the enhanced structural stability or the self-cleaning property endowed via photocatalysis. It should be pointed out that the mussel-inspired selective layers can be formed on the porous substrates via direct deposition649–659 or through the contradiffusion approach.660–662 In the latter case, the fabricated mussel-inspired films usually showed lower thickness with narrow pore size distribution for achieving precise separation.
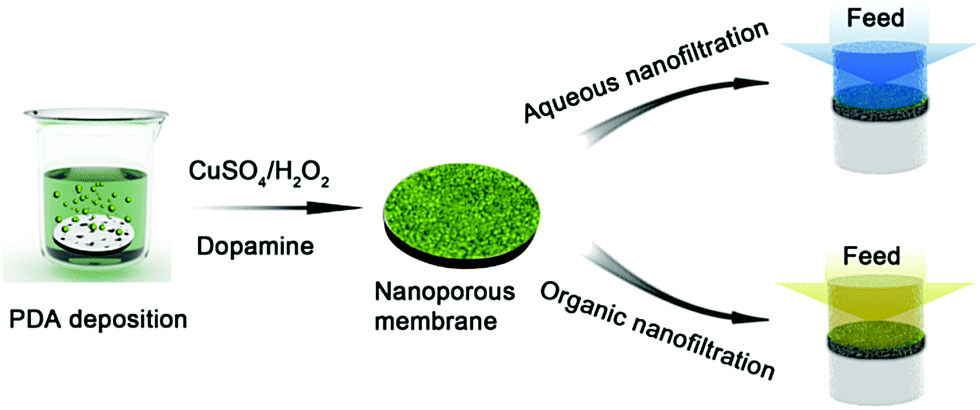 | ||
| Fig. 91 CuSO4/H2O2 triggered the rapid formation of PDA-based ultrathin selective layers with high uniformity and enhanced stability for both aqueous nanofiltration and organic nanofiltration. Reprinted from ref. 649 with permission. Copyright (2017) American Chemical Society. | ||
On the other hand, nanostructured interlayers were suggested by Xu et al. to optimize the substrate surfaces for modulating interfacial polymerization. Nanomaterials, such as CNTs, cellulose nanocrystals, and graphene oxide nanosheets or mussel-inspired coatings, were used to fabricate these interlayers.663–667 Taking cellulose nanocrystals as an example, the nanomaterials could be vacuum-filtrated on the porous substrate to form a uniform and hydrophilic thin coating. This coating adsorbed and distributed the aqueous solution of diamine on the substrate surface for interfacial polymerization. Therefore, the polymerization process could be controlled to a certain extent for facilitating the formation of ultrathin polyamide films with homogeneous characteristics. The thickness of the selective layer was normally lower than 50 nm and the corresponding NFMs exhibited overall desalination performance superior to that of state-of-the-art materials reported so far (Fig. 92). This work proposes a simple way to fabricate advanced NFMs with outstanding performance for practical applications.
 | ||
| Fig. 92 (a) Schematic illustration of the formation of a nanostructured interlayer from cellulose nanocrystals (CNC) and ultrathin polyamide selective layer by interfacial polymerization; (b) comparison of typical performances of the as-fabricated NFMs by mussel-inspired chemistry (●) and interlayer strategy (★) with those reported in the literature. Reprinted from ref. 664 with permission. Copyright (2017) Royal Society of Chemistry. | ||
Recent progresses made in the fabrication of polymer membranes for use in various applications, such as oil/water separation, blood purification, water treatment, and seawater desalination, are introduced. Moreover, design strategies and preparation methods of polymer membranes for obtaining good performances, such as good antifouling, high efficiency, ultrahigh flux, high mechanical strength, low protein adsorption, platelet adhesion abilities, good blood compatibility, high IECs, high salt rejection, etc., in the above applications are also presented. Although great advances in polymer membrane technology has been achieved, and a few of them have been industrialized, they still have their respective problems that need to be resolved. As an example, fabricating polymer membranes capable of good antifouling and chemical stability while maintaining high permeation flux and very high oil/salt rejection is still a challenge for researchers and engineers.
11. Energy storage polymers
The two worst crises for humans in the 21st century are energy shortage and environmental pollution. This is because fossil fuels—the dominant energy supplier—will eventually be depleted in the near future and they are also the cause of environmental pollution. Therefore, it is highly desirable to develop green and renewable sources of energy to replace fossil fuels as the main energy source. Energy storage is one of these solutions. Hence, dramatic progress in the field of energy storage materials and devices has been witnessed during the last decade. In the quest for high energy and power densities, fast electrode kinetics, long life cycle, and stable energy storage systems, various kinds of materials and different device configurations have been explored. The two main energy storage systems are supercapacitors and batteries. Although energy storage in both these systems depends on electrochemical processes, different mechanisms lead to different energy and power densities in such systems. Supercapacitors have been previously considered to have low energy density and high power density as well as long cycle life as compared to those obtainable from batteries, while the recent progresses made in both these fields have narrowed down this gap.668,669 Even though much progress has been made in the field of electrode materials, including various forms of carbons, nanostructured materials, transition metals, and metal oxides, the energy density of supercapacitors and the power density of batteries are still limited. The charge storage mechanism in such energy storage systems is mainly responsible for these limitations.670 This problem can be overcome either by inventing a new device with a different and effective charge storage mechanism or by selectively designing materials with superior properties. However, most energy storage devices, such as LIBs and supercapacitors, have some shortcomings, such as being heavy, rigid, and bulky, which cannot meet the demands of flexible and portable devices. In recent years, stretchable/portable devices have attracted considerable attention, such as wearable devices, artificial skin sensors, and bendable mobile phones.671,672 Organic molecules are considered to be promising materials for flexible energy storage devices due to their flexibility, diversity, relative abundance, low cost, etc. However, they still face challenges with regard to stability and low cycle life due to solubility in liquid electrolytes. This problem can be considerably resolved by the use of small organic molecules to build functional polymers. Therefore, we present the advances in energy storage devices in this section, such as LIBs, supercapacitors, and fiber-shaped energy devices made of polymers.11.1 Supercapacitors
Supercapacitors offer several advantages over batteries, such as high power density and long cycle life. It is always advantageous to have a device that can be charged at an ultrafast rate to its full capacity with a longer lifetime. In contrast to the ion diffusion mechanism in batteries that can reduce the overall kinetics, the ion adsorption mechanism on the surface of electrodes in supercapacitors can facilitate the fabrication of devices that can be charged within minutes.673 Supercapacitors are also expected to bridge the gap between batteries and capacitors. A typical example of the cyclic voltammetry (CV) curves of a supercapacitor and battery is shown in Fig. 93a and b, respectively. Based on the charge storage mechanisms, supercapacitors can be divided into two categories: electrostatic double-layer supercapacitors (EDLCs) and pseudocapacitors. Conventional materials with a high surface area have been explored in the field of capacitors, but interest regarding them has been rising in pseudocapacitors. Several new materials, such as polymers, are emerging as potential candidates for next-generation supercapacitors. Redox-active polymers and conducting polymers capable of pseudocapacitive energy storage are directly used as electrode materials. Porous polymers and heteroatom polymers are used to produce porous carbon and heteroatom-doped carbon, respectively. In this subsection, the applications of polymers in both types of supercapacitors are briefly discussed.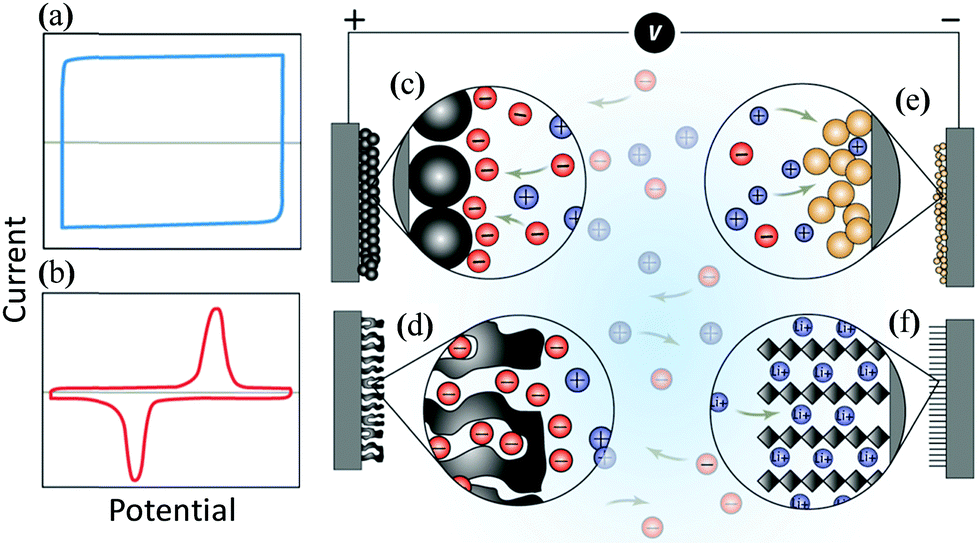 | ||
| Fig. 93 Typical CV curves of a (a) supercapacitor and (b) battery, and the charge storage mechanism for different materials: (c) surface charge accumulation in carbon and (d) porous carbon; pseudocapacitive behavior for (e) redox reaction and (f) ion intercalation. Reprinted from ref. 669 with permission. Copyright (2014) from AAAS. | ||
EDLCs primarily work through the interfacial accumulation of charges at the electrode–electrolyte surface.674 When a charged object is placed in a liquid, the countercharge from the liquid starts to accumulate near the surface, giving rise to an electric double layer.675 Therefore, surface-dependent charge storage mechanism in EDLCs needs materials with high specific surface, large porosity, and uniform pore distribution.676 Different types of carbons are the material of choice in this regard (Fig. 93c–f). A comprehensive report in this regard is out of the scope of this review and can be found elsewhere.677,678 For the sake of general understanding, we would like to introduce a few examples where polymers have been employed either to get heteroatom-doped porous carbon or composites with carbon for superior properties.
Heteroatom-doped carbon-based materials show superior properties in energy storage applications, particularly supercapacitors. There are various strategies to obtain heteroatom-doped carbon-based materials. Nitrogen- and oxygen-doped hierarchical porous carbon structures were fabricated via the carbonization of a quinone amine polymer precursor on the surface of MgO as a template.679 A high content of heteroatoms along with a large surface area were obtained, which ensures high pseudocapacitance as well as pathways for faster ion diffusion, leading to excellent charge storage properties. In another report, a template-free three-step carbonization process was developed to achieve nitrogen-doped 3D porous carbon-based materials with interconnected pores.680 The nitrogen content was found to be 9.71%, and the capacitor based on two-electrode systems delivered capacity of 221 F g−1 and showed excellent cycle life.
Although the stability of EDLCs is very high, they suffer from low gravimetric and volumetric energy densities.670 Research is being conducted for resolving this issue. One of the emerging trends is to investigate materials with pseudocapacitance.
Based on the mechanism of faradaic redox reaction, pseudo-supercapacitors usually offer higher theoretical capacity than EDLCs. Reversible redox reactions, similar to that in batteries, take place when potential is applied across the electrodes. Redox-active conducting polymers are one of the most promising candidates because of their low cost, structural diversity, high capacitance, and reasonable electronic conductivity. Various conducting polymers, such as PANI, PPy, PEDOT, and their derivatives, have been extensively studied.676,681,682 A few examples in this field will be summarized in the subsequent subsection.
Wei et al. synthesized large arrays of vertically aligned PANI through a one-step template-free approach on various conducting substrates (Fig. 94a–c).683 The vertical growth of nanowires on a conducting substrate with a narrow diameter was beneficial to the ion diffusion process. The highest specific capacitance of 950 F g−1 was obtained, and specific capacitance of 780 F g−1 was maintained at a high current density of 40 A g−1. The capacitance behaviors in the different electrolytes, such as 1 M HClO4, 1 M lithium bis(trifluoromethanesulfonyl), and nonsolvent electrolyte ionic liquid (IL), were also investigated. This study revealed that the oriented nanostructures could dramatically improve the electrochemical properties. Subsequently, the same group investigated the synergistic effect of PANI and graphene oxide by growing vertically aligned PANI nanowire array on the surface of graphene oxide sheets.684 Electrochemical capacitance and stability of the obtained nanocomposite were higher than those of both the individual components. Different dimensional materials can be composited to obtain functional nanomaterials with superior properties.
 | ||
| Fig. 94 Application of conducting polymers in supercapacitors: (a) structure of PANI, (b) schematic mechanism of faster ion diffusion in nanowire arrays of PANI, (c) morphology of PANI nanowire array, (d) interconnected porous network of 3D graphene-based flexible film, and (e) schematic of the growth of PANI nanowires on CNT yarn and SEM image of actual yarn. Reproduced from ref. 683, 685 and 686 with permission. Copyright (2010 and 2013) American Chemical Society and Wiley-VCH. | ||
Wei et al. synthesized a free-standing porous 3D reduced graphene oxide (3D-RGO) film using a template method for application in flexible supercapacitors.685 High charge–discharge rate performance was observed due to the presence of an interconnected porous structure, ensuring the diffusion of electrolyte ions into the inner surface electrodes (Fig. 94d). In addition, the growth of PANI nanowires on 3D-RGO resulted in excellent charge storage properties. Supercapacitors based on the 3D-RGO PANI nanocomposite could retain 90% of its initial capacity after 5000 charge–discharge cycles. In another study, PANI nanowires were grown on CNT yarn through an in situ process to assemble a wire-like supercapacitor (Fig. 94e).686 CNT yarn acted as both active material and current collector because of its worthwhile conductive properties. A thread-like supercapacitor exhibited excellent energy storage properties both in terms of rate performance and cycling stability. Capacity was retained under different bending states and angles, showing remarkable flexible properties. The free-standing flexible film composed of 3D-RGO PANI was also employed to assemble a flexible supercapacitor, which exhibited remarkable energy storage properties even under applied stress (Fig. 95a). The wire or fiber-shaped devices can be further woven into textiles to fabricate smart clothes (Fig. 95b). In a rather interesting study, a multifunctional flexible energy storage smart (ESS) window was fabricated by combining PANI nanowires with a transparent substrate (Fig. 95c).687 Energy storage and electrochromic functions were, therefore, integrated into a single device, leading to its potential application as a smart window. The device showed reasonable charge storage properties, which could be further improved by adjusting the structure and designing new materials. This ESS window can be further integrated with solar cells to combine energy production together with the storage function. Schon et al. joined fullerene monomers by a cyclobutene ring to synthesize a branched chain fullerene polymer.688 Based on the negative-charge-accepting pseudocapacitive mechanism, an n-type supercapacitor was fabricated, which exhibits good capacity and cycling stability.
 | ||
| Fig. 95 Different flexible energy storage devices: (a) flexible supercapacitor and schematic of bending at different angles, (b) photograph of a single flexible and transparent electrode, (c) yarn supercapacitor integrated into conventional cotton yarn, (d) wave-like flexible battery bent into different shapes, and (e) illumination of LEDs by a bent flexible battery. Reproduced from ref. 685, 686, 687 and 689 with permission. Copyright (2012, 2013, and 2018) from Royal Society of Chemistry and Wiley-VCH. | ||
These results suggest that polymers are potential candidates for next-generation commercial supercapacitors (either EDLCs or pseudocapacitors). Although significant progress has been made in the recent decade, there are still several challenges that need to be addressed. For example, energy density is still not sufficient. This may be achieved by designing new materials with high capacity or developing new electrolytes and processes. For example, lithium-ion-based supercapacitors can operate at higher voltages, leading to higher energy densities.668,670
11.2 LIBs
Batteries are another major area where polymers are being explored either as active material for energy storage or as additive for different purposes (Fig. 95d and e). In this subsection, we will summarize the recent examples elaborating the applications of functional polymers as active electrode materials. Electroactive small molecules are usually polymerized to improve stability in liquid electrolytes. For example, a novel hypercrosslinked polypillar[5]quinone (poly-P5Q) was used as the electrode material in LIBs.690 It offers very low practical capacity, but it provides new insights into the reduction of solubility of electroactive small molecules by crosslinking them. Solubility in the electrolyte is one of the major problems for organic materials. The stability of poly-P5Q in the liquid electrolyte was improved by this method. Two isomeric pairs of poly(anthraquinonyl imide)s were prepared, and their electrochemical properties as LIB cathodes were studied.691 It was shown that isomerism played an important role in electrochemical performance. An all-solid-state Li–organic battery with quinone-based polymer cathode and composite polymer electrolyte was reported.692 The ionic conductivity of the solid electrolyte was excellent when the temperature was 343 K. Although this performance was good, there is still a need to develop effective systems capable of the best performance at room temperature. An anode material for LIBs derived from aromatic imides with high capacity was reported due to its low potential.693 Based on the density functional theory calculation and natural bond orbital charge analysis for electron transfer processes and reaction free energies, they proposed a 12-electron transfer process to accommodate the high experimental capacity of aromatic amides. A quinone-based polymer accommodating 4 lithium ions was reported with high charge storage capacity.694 Lithiated poly(dihydroxyanthraquinonyl sulfide) (LiDHAQS) was selected as the material of choice because of its high degree of carbonyl groups. The experimental capacity of 3.5 Li+ per monomer was obtained, which is less than the theoretical value but higher than all the other quinone-based electrodes reported so far.An in situ polymerization method was adopted to synthesize 3,4,9,10-perylenetetracarboxylic dianhydride (PTCDA)-based polyimide/carbon nanotube (polyimide/CNT) nanocomposite electrode, which could be used in LIBs.695 Polymerization on the surface of CNTs was found to increase the capacity as well as cycling stability. Here, 93% of the initial capacity was found to be retained when the polyimide/CNT nanocomposite electrode was used after 300 cycles, while only 74% of the initial capacity was retained for PTCDA/CNTs. Pyromellitic dianhydride (PMDA) was polymerized with ethylene diamine in the presence of a 3D-RGO film to fabricate a flexible electrode for LIBs.250 The composition of the electrode was found to contain 80% of the weight of the active materials and 20% of 3D-RGO without any other additive or binder. 3D-RGO/polyimide flexible cathode exhibited initial capacity of 175 mA h g−1 as compared to 120 mA h g−1 when polyimide was bound with conductive carbon in the presence of a binder and a conventional electrode structure. Similarly, significant enhancement in the cycling performance was also reported. Through electrochemical impedance spectroscopy (EIS) studies, it was concluded that an effective 3D-RGO network could lead to excellent conductivity, which would eventually yield better rate and cycling performances. Polyimide suffers from poor conductivity, leading to poor performance and low material utilization. To overcome this issue, PMDA was in situ polymerized in the presence of CNTs to obtain composite materials with much better electronic properties.696 The vacuum filtration technique was used to fabricate a binder and metal-current-collector-free electrode for LIBs. In this case, the interconnective network of CNTs acted as the current collector, which is much lighter than the commercially available aluminum current collector, thereby effectively reducing the dead mass. Significant enhancement in electrochemical properties was reported in this case, where the electrode was still able to deliver high capacity of 120 mA h g−1 even at a very high rate of 20 C. The influence of monomer concentration on morphology was also studied in this research: it was observed that large aggregates start to grow instead of flakes on the CNT surface at a certain amount of monomer concentration. Therefore, the optimum monomer concentration is also important to obtain good results. Subsequently, the same group reported another interesting strategy to design large-area flexible cathodes for LIBs.697 A simple rolling method was used to fabricate the electrode film, which was mounted on a free-standing CNTs film by applying pressure. The CNT film acted as a mechanical support and current collector in this system (Fig. 96a). The polyimide obtained from the polymerization of PMDA and triethyl amine was used as the active material. The large-area flexible cathode showed superior electrochemical properties over a conventional electrode and retained 80% of its initial capacity after bending for 1000 times.
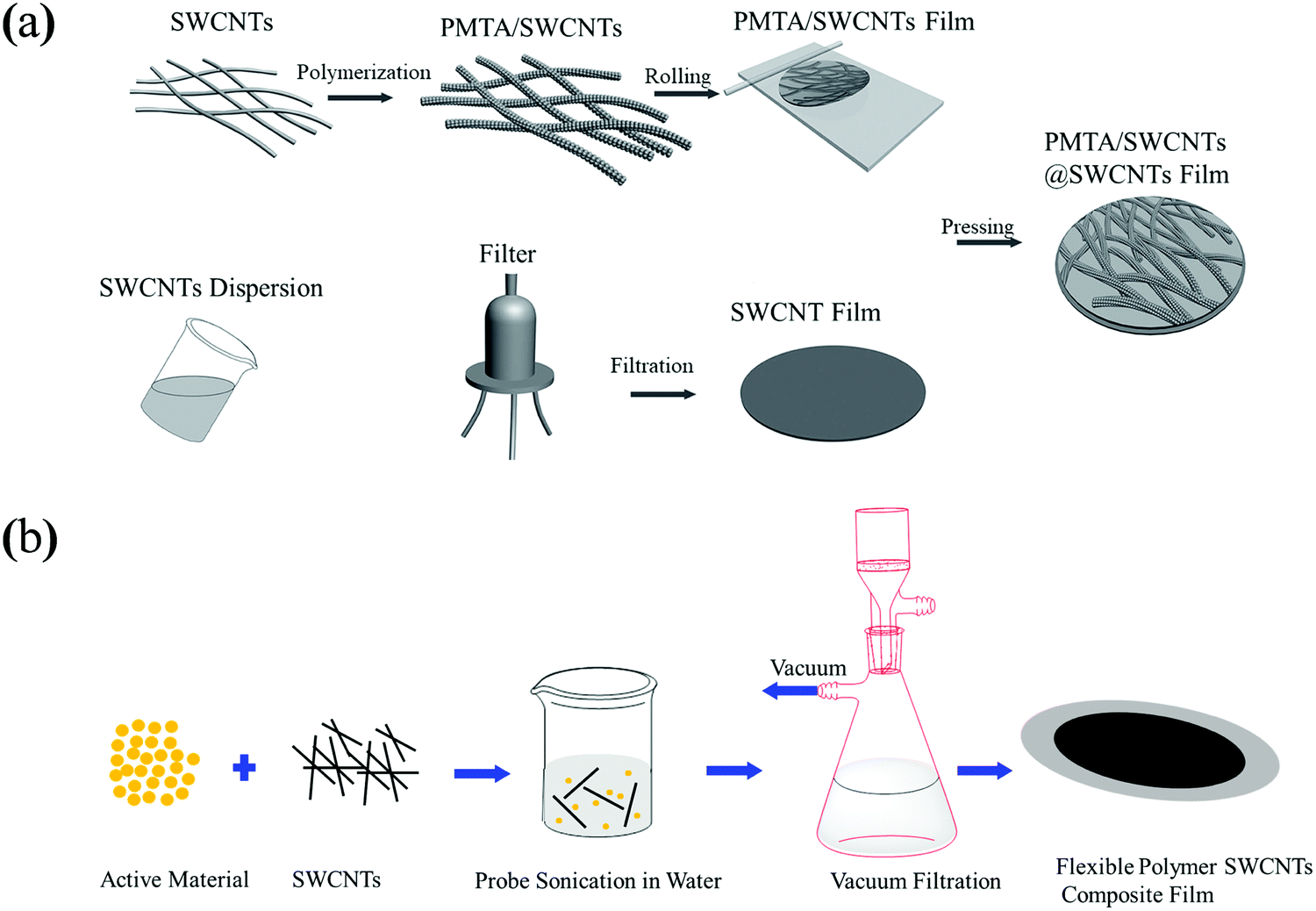 | ||
| Fig. 96 Schematic of fabricating a flexible electrode: (a) embedding a conventional cathode film into a CNT film acting as a flexible current collector and (b) fabrication of a free-standing film. Reprinted from ref. 689 and 697 with permission. Copyright (2015 and 2018) Wiley-VCH. | ||
In another example, PMDA and anthraquinone were polymerized together to obtain high-capacity polyimides.698 A simple vacuum filtration method together with CNTs was employed to obtain a flexible, free-standing, binder-free electrode for LIBs (Fig. 96b). High capacity of 190 mA h g−1 at 0.1 C was obtained due to the three-electron transfer system. Capacity of 120 mA h g−1 (∼63% of that at 0.1 C) was maintained at a high current density of 20 C. The cathode retained more than 95% of its initial capacity after 300 charge–discharge cycles. Excellent rate performance was assumed due to the interconnective network of CNTs in the cathode architecture. EIS results also showed that the charge transfer resistance decreased significantly with an increase in the concentration of CNTs, which has an influence on the electrochemical performance. The composite cathode showed optimal performance when the CNT content was 30%. A flexible cathode film exhibited good mechanical properties under stress. The tensile strength of the composite was comparable to that of CNTs. A sulfur-linked quinone-based polymer was reported for long-cycle-life LIBs.689 A free-standing electrode was assembled together with CNTs acting as a binder and current collector. When applied as the cathode, it was able to retain about 89% of its initial capacity after 500 charge–discharge cycles. Excellent rate performance was also observed. Furthermore, a flexible battery was assembled with a lithium metal anode, which could retain almost 88% of its initial capacity after 2000 bending and unbending cycles (Fig. 95d and e). In short, the CNTs played a crucial role in supporting the free-standing film due to their excellent mechanical properties and enhancing the electronic conductivity of the free-standing film.
A PMDA-based polyimide was synthesized via in situ polymerization on the surface of few-layer exfoliated graphene (FLEG) sheets as the cathode for LIBs.699 The utilization of the active material was improved by employing this composite material in the cathode, and polyimide-FLEG cathode delivered initial capacity of 177 mA h g−1 at 0.1 C. The cathode was able to retain almost 80% of its initial capacity after 200 charge–discharge cycles. The charge transfer resistance was significantly reduced by employing this cathode with a composite structure.
So far, polymer composites with CNTs or graphene are discussed. However, these two types of materials are expensive for commercial applications. A recent work reported an in situ polymerization strategy for synthesizing a polyimide composite with carbon black (CB).700 In this case, insulating polyimide and conductive additive can be tightly contacted, and the crystallization of polyimides can also be inhibited. By optimizing the synthesis conditions, a composite that could deliver extraordinary capacity at high current densities could be obtained, which yielded excellent rate performance. In comparison to the in situ polymerized composite, mechanically mixed composite electrodes exhibited much poorer performance. Consequently, CB can be an alternative to synthesize in situ polymerized composite electrode materials.
Based on the above discussion, it can be concluded that free-standing composite electrodes with CNTs or graphene offer various advantages over the conventional ones. These conductive additives can significantly improve the electronic conductivity. Because of good conductivity, free-standing films not only can act as a current collector but also offer excellent electron transport kinetics. Getting rid of the metal current collector in these films can lead to a reduction in the dead mass of the electrode, thereby effectively increasing the volumetric energy density. The microporous structure in these films is beneficial for the penetration of the electrolyte as well as fast diffusion of lithium ions in the electrode structure. These films offer reasonable mechanical properties with inherent flexibility, which is very beneficial for flexible LIBs.
Researchers are looking forward to some options other than LIBs. As alternatives, sodium-ion batteries (SIBs) are getting increasingly popular owing to their advantages over LIBs.701 In principle, polymers are promising candidates for next-generation SIBs since they follow the conversion-reaction-based energy storage mechanism, making them electroactive toward most metal-ion batteries. A PMDA-based polyimide with micro-flower-like morphology was tested as the anode in SIBs.702 A two-step enolization reaction with redox potential below 1.5 was observed during the electrochemical performance, suggesting its suitability as the anode for SIBs. Sodium terephthalate is one of the most promising anode materials for SIBs due to its low redox potential, while the performance is not stable due to its undesirable dissolution in the electrolyte. To overcome this problem, a polymer was reported bearing sodium terephthalate as the side group acting as the redox-active unit; here, the polymer backbone ensured stability.703 By suitably adjusting the structure with an electron-donating group, researchers were able to obtain lower potential and much better cycling performance with almost 100% coulombic efficiency for over 100 cycles at 30 mA g−1. Metal-ion batteries are going to become increasingly popular, and polymer materials can be the material of choice in the future.704,705
11.3 Fiber-shaped energy devices
Due to the high flexibility and easy fabrication typically derived from the solution process, a variety of functional polymers are explored for soft electronic devices. The use of polymers for thin-film electronic devices has been extensively discussed by many review articles, while it is rare to summarize their applications in a type of new soft electronic devices, i.e., fiber-shaped electronic devices that attract increasing attention with the rapid advance of information technology and artificial intelligence.706–711 When compared with the generally bulky or thin-film counterparts, fiber-shaped electronic devices demonstrate some unique and promising properties. For instance, they can accommodate complex deformations such as bending in various directions, twisting, and stretching. Furthermore, they can also be woven into soft, breathable, and comfortable electronic fabrics to satisfy the requirements of portable and wearable electronics.Functional polymers are the key components for the realization and high performance of fiber-shaped electronics devices. In this subsection, a fiber electrode is first presented; then, we summarize the different types of fiber-shaped electronic devices, and the future direction is highlighted at the end. Different from earlier review articles, in this paper, we focus more on discussing them from a viewpoint of practical applications.
Currently available fiber-shaped electronic devices include solar cells, triboelectric and piezoelectric generators, supercapacitors, LIBs, lithium–sulfur batteries, and metal–air batteries.712 With the help of a timeline, different types of fiber-shaped electronic devices are summarized in Fig. 97. It starts from sensors, solar cells, and light-emitting devices, and then extended to supercapacitors and batteries, almost according to the degree of difficulty in making them into a fiber shape in the lab. With further increasing interest from the industry, the main efforts are focused on the continuous fabrication of fiber-shaped energy-harvesting (e.g., solar cells) and storage (e.g., LIBs) devices, which may soon find large-scale applications in many fields.
 | ||
| Fig. 97 Advances in fiber electronic devices with four main functions: energy harvesting, energy storing, sensing, and lighting according to the timeline. | ||
The electrode has always played a key role in electronics, and it is even more important for fiber-shaped electronic devices. It is necessary to carefully control the composition and microstructure of fiber electrodes because, when compared with traditional plates or film substrates, the curved surface of a fiber electrode makes it much more difficult to deposit thin active layers, particularly under a continuous fabrication process. In addition, the fiber electrodes should be simultaneously flexible, strong, and conducting. This flexibility guarantees to meet the production process necessary in the industry, such as roll-to-roll fabrication. The fiber electrodes also serve as substrates; therefore, they must be sufficiently strong to bear various deformations without breaking during use. Of course, charges would transport along the fiber electrode, and the pathways are much longer in fiber-shaped electronic devices as compared to their planar counterparts. Therefore, they should have higher electrical conductivities to provide satisfactory performances at lengths ranging into meters or even kilometers for real-world applications.
Polymers, metals, and carbon nanomaterials represent the three main categories of candidates for fiber electrodes. Polymers have been mostly explored as they are efficient for both moiety control and surface modification. A spectrum of conducting polymers such as PANI, PPy, and PEDOT have been mainly explored. The resultant fibers were flexible but typically with relatively low conductivities (<102 S cm−1). A second phase with much higher conductivity, e.g., CNTs, is therefore added to increase the conductivities up to 103 S cm−1. However, it still needs to be further enhanced to meet the industrial production requirements of the resulting fiber-shaped electronic devices. They are expected to show conductivities on the level of 104 S cm−1 at room temperature. Recently, bare CNTs have also been assembled or coassembled with polymers into flexible and continuous fiber electrodes based on chemical vapor deposition processes, and the CNTs are highly aligned to provide high conductivities of 104 S cm−1.713–715 However, the use of chemical vapor deposition techniques makes the synthesis much more expensive when compared with those for polymers, and large-scale production remains difficult.
Generally, positive and negative fiber electrodes are twisted into fiber-shaped electronic devices.712 For instance, a fiber-shaped dye-sensitized solar cell was wound from a Ti wire coated with active materials and a CNT fiber (Fig. 98a), followed by the introduction of liquid electrolytes. However, the use of liquid electrolytes necessitated very strict sealing treatment that remains challenging, while the replacement with polymer gel electrolytes largely decreased the PCE. To this end, a fiber-shaped polymer solar cell that was completely solid had been invented by the use of CPs as active materials (Fig. 98b). Besides the above twisted structure, a lot of effort has also been devoted toward fabricating solar cells with a coaxial structure. In other words, a thin and transparent conducting layer was coated or wound on an active-layer-modified fiber electrode. For instance, a fiber-shaped PVSC was firstly produced by depositing active perovskite materials on the fiber electrode and then winding the aligned CNT sheet as the second electrode (Fig. 98c).711 The twisted structure favors continuous production, but the counter-electrode fiber may shield light with low PCE. In contrast, it is not easy to continuously prepare a thin-film electrode for a coaxial structure.
 | ||
| Fig. 98 Fiber-shaped solar cells with different active materials and architectures. (a) Fiber-shaped dye-sensitized solar cell twisted from Ti wire and CNT fiber. (b) Fiber-shaped polymer solar cell with a twisted structure. (c) Fiber-shaped PVSC with a coaxial structure. Reproduced from ref. 712 with permission. Copyright (2016) Wiley-VCH. | ||
It should be emphasized that the interface in a fiber device exhibits several unique features as compared to its 3D counterparts. For instance, for a fiber-shaped polymer solar cell with a twisted structure, the interface between the highly curved charge and hole transport layer are different from conventional flat interfaces. They are concentrated at the contact points of the two twisted fiber electrodes while uniformly distributed through the entire interface of the planar structure. In the case of coaxial structures, external electrodes are required to be stably and uniformly attached to the outer surface; therefore, high mechanical and electrical properties are desirable for achieving high PCEs.
When photoactive materials are replaced by electrochemically active materials, fiber-shaped supercapacitors, batteries, and other energy storage devices could be obtained.709–711 Like solar cells, the fiber-shaped energy storage devices are also made with both twisted and coaxial structures. Currently, fiber-shaped supercapacitors have been mostly explored possibly due to their easy fabrication, although they have found few applications. Recently, fiber-shaped batteries have started to attract increasing attention; now, they have become the primary candidate for real applications. Studies involving fiber-shaped LIBs are being conducted by a few companies for enabling their industrial production.
Fiber-shaped energy-harvesting and storage functionalities have also been integrated into a single fiber device. They may be further woven into flexible and even stretchable power textiles toward portable and wearable applications. Although it seems easy to assemble the above fiber devices into textiles based on the weaving process, it remains challenging to effectively connect so many fiber electrodes in the power textile. There exist many reports that have discussed solar cells or battery textiles, but they are also limited to relatively small sizes (in the range of centimeters). There is still a long way to go for fiber-shaped energy-harvesting and storage devices from the lab to the market. Scientifically speaking, further investigations into a distinctive interface are necessary to guide performance enhancement. Technically speaking, the emphasis will be made on generic and effective strategies for the continuous preparation of fiber electrodes and reliable connection of fiber-shaped devices.
The two main energy storage systems (supercapacitors and batteries) made of polymers are first introduced. However, such energy storage devices are usually rigid, heavy, and bulky. Subsequently, flexible and portable fiber-shaped energy devices are presented from the viewpoint of practical applications. From the above contents, it is evident that polymers have achieved considerable progress in basic research and practical applications in the field of energy storage. The main challenges needed to be addressed in the future are as follows: (1) fabricating supercapacitors with high energy density and batteries with high power density; (2) designing fiber-shaped energy devices with excellent electrochemical performance and mechanical stretchability.
12. COFs
COFs are a class of crystalline porous polymers assembled from organic building blocks by strong covalent bonds.716–722 Different from traditional polymers, they allow the precise and atomically controllable integration of struts into long-ranged ordered networks. COFs feature extended porous structures with high surface area, large cavity, good thermal stability, and low density. Moreover, the tunable pore size, shape, and environment, as well as the backbone functionalization of the network realized by pre- and/or post-synthesis modification, further expands their applications in gas storage and separation, optoelectronics, catalysis, energy storage, and electronic devices.723–730In COF chemistry, the molecular units can be accurately arranged into extended frameworks under the guidance of reticular chemistry,731 where the functionality and pore metrics can be further tuned. Since the first report on COF-1 and COF-5 by Yaghi et al.,732 COFs with various structures and characteristics have been designed and synthesized. The structures of COFs are largely determined by the geometry and dimensions of the building units and linkages, which can be classified into 2D and 3D frameworks. In 2D COFs, the structural units are covalently aligned in 2D atomic layers that further stack via noncovalent forces to form eclipsed or staggered configurations.733,734 For 3D COFs, the building blocks are covalently and periodically oriented to form 3D architectures, and the entire crystalline structure gets retained by covalent bonds.
The syntheses of most organic polymeric materials are kinetically controlled reactions, where the covalent bonds are irreversibly formed, resulting in amorphous products. To afford extended crystalline structures, the chemical reactions should be reversible and allow for “error-checking” and “proofreading” to give thermodynamically stable and highly ordered networks. In this way, the structural defects can be repaired by a reverse reaction; consequently, the bonds can be reformed to achieve crystalline architectures. Several reversible reactions have been employed to prepare COFs, and they can be grouped into B–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, C–O, Si–O, C–N, N–N, C–C, etc. (Fig. 99) based on the linkage type. The first two COFs (COF-1 and COF-5) were synthesized by the self-condensation of boronic acid and the cocondensation of boronic acid and catechol to give boroxine- and boronate-ester-linked structures, respectively.732 Borosilicate and spiroborate were incorporated to prepare boron-based COFs, and most of them exhibited good thermal stability but poor stability when exposed to water or acid, because water promoted the reverse reaction and led to the decomposition of COFs.735,736 To improve the chemical stability of COFs in water and even strong acid or basic aqueous solutions, more chemical reactions, e.g., phenazine- and β-ketoenamine-based COFs, were further explored to fabricate COFs.737–739 Post-synthesis modification was also utilized to stabilize the COF structures. Further, it was possible to employ reversible–irreversible cascade reactions or even irreversible nucleophilic aromatic substitution reactions to synthesize COFs, and the irreversibility of the reaction imparts the resulting COFs with good chemical stability.737,740,741
N, C–O, Si–O, C–N, N–N, C–C, etc. (Fig. 99) based on the linkage type. The first two COFs (COF-1 and COF-5) were synthesized by the self-condensation of boronic acid and the cocondensation of boronic acid and catechol to give boroxine- and boronate-ester-linked structures, respectively.732 Borosilicate and spiroborate were incorporated to prepare boron-based COFs, and most of them exhibited good thermal stability but poor stability when exposed to water or acid, because water promoted the reverse reaction and led to the decomposition of COFs.735,736 To improve the chemical stability of COFs in water and even strong acid or basic aqueous solutions, more chemical reactions, e.g., phenazine- and β-ketoenamine-based COFs, were further explored to fabricate COFs.737–739 Post-synthesis modification was also utilized to stabilize the COF structures. Further, it was possible to employ reversible–irreversible cascade reactions or even irreversible nucleophilic aromatic substitution reactions to synthesize COFs, and the irreversibility of the reaction imparts the resulting COFs with good chemical stability.737,740,741
 | ||
| Fig. 99 Chemical reactions for the preparation of COFs. | ||
Under the topology direction, COFs with targeted structures could be fabricated by an appropriate selection of the building units. In the target topology net, the structure can be deconstructed into basic geometric units, and further substitution with building molecules of certain connectivity and geometry would yield a COF possessing the same underlying net. To ensure that the conformation of the building blocks remains unchanged during the assembly process, rigid units with discrete directionality are preferred to construct well-defined networks. A wide range of molecules, such as simple arenes to extended π aromatics with heteroatoms, coordinated strings, and large macrocycles, have been employed to construct COFs. In 2D COFs, topologies such as hcb, sql, kgm, and hxl have been realized in COF structures, where there is only one type of edge linking the vertices.732,742–744 To improve the structural complexity, Zhao et al. reported the heterostructural mixed linker strategy to construct COFs with three types of pores.745 Employing the condensation reaction of a tetraamine (D2h symmetry) and two dialdehydes (C2 symmetry) of different lengths, they successfully synthesized a COF with hierarchical pores of inequilateral hexagon, as well as small and big triangular shapes. Other strategies such as desymmetrized vertex design and utilizing macrocycles as building blocks have also proven to be capable of giving COFs with periodically heterogeneous pore structures.746,747 With regard to 3D COFs, due to the limited choices of building units and synthesis difficulties, the reported topologies of 3D structures in COFs are only bor, ctn, pts, dia, rra, srs, and lon (Fig. 100).748–753 Feng et al. introduced the flexible building block of γ-cyclodextrin (γ-CD) into 3D COFs for the first time.751 Utilizing the transesterification reaction between trimethyl borate and hydroxyl groups, they could construct CD-COFs possessing an anionic framework with spiroborate linkage and tunable counter ions identified as the rra topology.
 | ||
| Fig. 100 3D topological diagrams of COFs. Adapted from ref. 716, 748, 750, 751 and 752 with permission. Copyright (2017, 2007, 2009, 2016, 2017, and 2018) AAAS, American Chemical Society, and Wiley-VCH. | ||
Once the organic building units and chemical reactions are judiciously determined, finding the appropriate synthesis conditions is also important to obtain crystalline porous frameworks. COFs are generally prepared by solvothermal synthesis, and the reaction vessels charged with precursor solutions are usually vacuumed to low pressure before being sealed. After heating for several days, polycrystalline products with high surface areas can be obtained. The preparation of single crystals has always been a challenge in COF chemistry, and the products always end up as polycrystalline or even amorphous solids. Recently, Yaghi et al. reported the first examples of COF single crystals suitable for single-crystal X-ray diffraction data collection.753 The strategy here is to add excess aniline as a nucleation inhibitor and competitive modulator during the synthesis of imine-based COFs (COF-300, COF-303, LZU-79, and LZU-111) at ambient temperature, which improves the reversibility during the course of bond formation. Dichtel and coworkers developed a seeded-growth method to prepare single-crystal 2D boronate ester-linked COFs.754
However, solvothermal synthesis is not applicable for industrial production processes because of the long reaction time, involvement of a large number of solvents, and harsh reaction conditions. To shorten the reaction time, microwave heating is also employed to assist the synthesis of COFs with higher production yields and purity.755 Solid-state reaction is a good choice for the fast and scale-up production of COFs. Banerjee et al. successfully synthesized β-ketoenamine-linked COFs by using a solvent-free mechanochemical method and further improved the crystallinity and porosity of COFs by adding the external coagent of p-toluene sulfonic acid that helps to control the covalent bond formation during solid-state preparation.756,757 This solid-state synthesis was further applied with a twin-screw extruder for large-scale COF synthesis, showing promising potential for industrial production.758 Another solvent-free synthesis method is ionothermal synthesis, where molten salts or ILs were used as solvents.759 Fang et al. reported the successful preparation of a series of 3D COFs utilizing ILs as the green solvent at ambient temperature and pressure in a short reaction time, which opens up a new way for green and massive production of COFs.760 Other methods such as continuous flow synthesis, sonochemical method, and photochemical synthesis have also been reported for synthesizing COF powders with high production rates.761–763
For practical applications such as catalysis, electronic devices, and sensing, these powder-form materials need to be processed into certain shapes, particularly thin films. The most direct and common way to synthesize COF films is solvothermal synthesis. Here, the substrates are immersed in the precursor solutions during the reaction, resulting in the growth of COF coating layers on the substrate surface. Various substrates have been used to prepare COF films such as graphene, indium-doped tin oxide, gold, and silicon dioxide, and the type of substrates influences the quality and orientation of the films.764–767 However, it is difficult to control the morphology and achieve large-area films by using this method. Interfacial synthesis is also employed to prepare COF films with good control to be cast and evaporated giving free-standing membranes with preferential orientation.768 For the large-scale synthesis of COF membranes, the continuous flow method and molecular baking strategy show promise. The former technique developed by Dichteland and coworkers enables the growth of COFs under the continuous flow condition, achieving good control over the film thickness and avoiding contamination from the powdered precipitates.769 Standalone and defect-free COF membranes with large areas can be obtained by the molecular baking method, and the concept was proven in the cases of β-ketoenamine and imine COF membranes.770 Interfacial synthesis is also employed to prepare COF films with good control over the nucleation process and film thickness, and the growth of COFs can be on either the air–liquid interface or the liquid–liquid interface.726 Using this method, Feng et al. prepared large-area crystalline monolayers and multilayer 2D polymers with controlled thickness by the Schiff-base condensation reaction at the air–water and water–chloroform interfaces, respectively.771 The obtained monolayer film (∼0.7 nm) on a wafer (lateral size: 4 inch) showed high crystallinity and exceptional mechanical robustness. Another mild way to prepare COF films is room-temperature vapor-assisted conversion synthesis, which was first reported by Medina et al.772 Basically, the precursors were first dropped onto the substrate and then exposed to organic solvent vapor. The thickness of the resulting film could be precisely controlled by tuning the volume and concentration of the precursor droplet. This strategy was effective for preparing BDT-COF and COF-5. Later, colloidal suspension of COF was directly used.
Bulk 2D COFs are layered structures whose atomic layers are stacked via weak interactions such as van der Waals force and/or hydrogen bonding, and the breakage of the interlayer π–π interactions can lead to single- or multilayered COF thin films, namely, COF nanosheets (CONs). The force to exfoliate the bulk materials can be either internal forces (which is the case for the self-exfoliation method) or external forces (used in solvent-assisted exfoliation, mechanical delamination, and chemical exfoliation strategies).773–776 The solvent-assisted method is the most commonly used method due to its simplicity and low cost. Further, the mechanical delamination method usually involves grinding or ball milling, which is an effective and powerful way to exfoliate. The self-exfoliation and chemical exfoliation methods often require a special chemical structure designed by the building block. Gu et al. employed ionic building blocks to construct COFs with electrostatic repulsion that can resist interlayer interactions. As a result, the afforded framework can self-exfoliate into monolayers or multilayers in solutions, and the electrophoretic deposition was further utilized to prepare COF films with controllable thickness.777 Wang et al. reported a self-limiting solid–vapor interface reaction strategy to directly prepare large-scale crystalline COF sheets with one-atom thickness via a Schiff base reaction, avoiding the need for further exfoliation processes.778
COFs have been widely studied for gas storage and separation due to their high porosity and low density. Furthermore, their designable structure and diverse functionality make them highly promising for many other applications. In the field of energy storage and electronic devices, Wang et al. mechanically delaminated redox-active COFs into few-layer nanosheets and applied them as a battery cathode that yielded considerably improved performance because of the shortened Li+ transportation path in the exfoliated COFs (Fig. 101a).725 Furthermore, they prepared a series of COF films by interfacial polymerization, and these films showed ultralow dielectric constants under high humidity due to the porous and symmetrical structure as well as the incorporated alkoxy chains (Fig. 101b).726 In the field of photoluminescence, Wang et al. designed and synthesized an AIE-luminogens-based 3D COFs, which could emit yellow fluorescence upon excitation; their further coating on a blue LED afforded a white LED with long working life.779 As demonstrated by Feng et al., COFs are a good platform for phosphorescence study, and the phosphorescence behavior can be tuned by adjusting the interlayer distances in a crystallization-induced phosphorescence-phosphor-based COF.780 In the field of catalysis, the first example was reported by Wang et al., and the COF-based catalyst was prepared by the metalation of an imine-based 2D COF with Pd(II), showing excellent yields, high stability, and facile recyclability for the Suzuki–Miyaura coupling reaction. This can be attributed to the highly exposed active sites and the rapid diffusion of products in a porous scaffold.723 Cui et al. successfully prepared a series of multivariate chiral COFs with organocatalyst-modified channels, serving as efficient asymmetric catalysts with stereoselectivity and diastereoselectivity.724
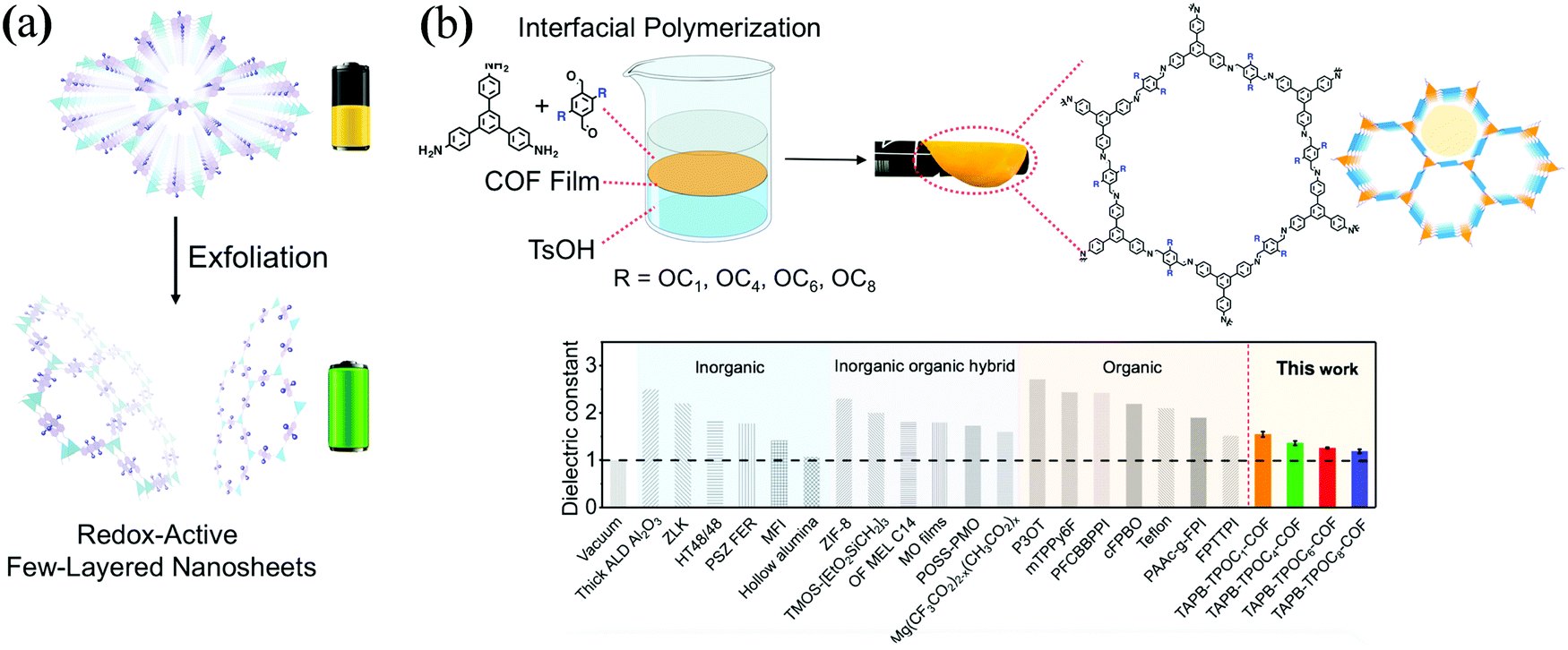 | ||
| Fig. 101 (a) Schematic illustration for the exfoliation of 2D redox-active COFs into exfoliated COFs as cathodes for LIBs. (b) COF films prepared by interfacial polymerization, and their dielectric constants compared with those of typical commercial and representative ultralow-k materials. Adapted from ref. 725 and 726 with permission. Copyright (2017 and 2018) American Chemical Society and Wiley-VCH. | ||
In summary, COFs have shown promise in diverse applications due to their unique characteristics. They are ideal platforms for various fundamental studies because of their highly ordered and porous structures. To further expand their structural complexity and diversity, multicomponent networks with hierarchical pores should be developed. The crystallinity of COFs still remains a challenge, and new reactions should be explored to construct highly crystalline materials with controllable stability and functionality. For functional exploration, COF-based composites are desirable. For example, well-aligned channels with tunable size and environment in COFs could be utilized to accommodate functional guest molecules. The final goal is to apply these porous materials in real-life applications. Therefore, low processability and scalability issues should be resolved. Researchers have proposed many promising strategies to deal with these problems and have made significant progress in this field. It is believed that the continued study on this emerging direction can yield practical applications of COFs in the future.
13. Conclusion and outlook
Advanced functional polymers are very interesting materials for broad applications requiring specific functions in their corresponding fields, such as biomedicine, aerospace, artificial intelligence, bionics, robotics, energy, electronics, separation, etc. During the past few decades, tremendous efforts have been devoted toward the advancement and development of this field. In this review, we addressed the notable recent progresses made in functional polymers, including their concepts, classifications, design strategies, preparation methodologies, performances, structure–property relationships, potential applications, and challenges. From these discussions, we can conclude that functional polymers play a critical role in the progress and development of our society and will continue to do the same in the future.Although great advances in functional polymeric materials have been achieved, numerous challenges still persist and should be seriously considered. A few examples are considered for each type of functional polymer as follows:
(1) Although ATRPs with organic photocatalysts have been rapidly developed in recent years, the synthesis is still limited to the polymerization of methyl acrylate in the presence of metal catalysts. Therefore, developing ATRP in the presence of a metal-free catalyst still needs to be determined.
(2) For luminescent polymers, pure and stable white-light emissions and highly efficient deep-blue-light emissions from phosphorescent materials have been rarely reported, which remain to be further developed.
(3) PCEs of OSCs are still much lower than those of PVSCs. Researchers should focus on the following aspects: (1) designing more efficient donors/acceptors, (2) controlling the aggregate structures of materials and the morphology of blend films, and (3) realizing the application of multifunctional organic photovoltaic devices.
(4) An ultralow-k polyimide still needs to be designed and prepared without sacrificing other properties. A simple synthesis method of NLO materials should be formulated.
(5) Many challenges in the drug delivery field need to be tackled and more efforts should be focused before translating them into clinical applications. For example, overcoming biological barriers and realizing highly efficient drug delivery systems need to be addressed. Because of the complexity of the ocean environment, marine antibiofouling polymer coatings with environmental adaption need to be developed in the future along with maintaining high efficiency and long service life.
(6) The main challenge of SPs is to design and fabricate materials that possess both dynamic capabilities and strong mechanical properties. The self-healing property can be facilitated in the soft state, while the mechanical strength of SPs is usually low. Materials with good mechanical properties usually have poor dynamic capabilities due to the presence of rigid polymer chains and restricted molecular mobility. This problem may be addressed by incorporating dynamic covalent bonds into SPs that can be cleaved and recombined when triggered by external stimuli, such as light or temperature.
(7) Electrochromic polymers have problems of trivial film processing, uneven electrochromism, ultraviolet degradation, etc., which can seriously limit large-scale preparation and cycling stability of the ECD.
(8) Integrating multifunctions, e.g., shape-memory and color-changing abilities, into a single SMP should be investigated for extending its potential applications. That is to say, shape and color may be synergistically changed when the SMP is triggered by an external stimulus. Moreover, SMPs are still investigated in the basic research phase; it is highly demanded to promote their development in practical applications.
(9) In the process of oil/water separation, seawater desalination, and blood purification, impurities, salts, sticky molecules, and organic compounds can be easily deposited on the surface of a membrane, promoting membrane fouling and therefore reducing membrane life. Antifouling polymer materials are highly desirable for membrane separation industries.
(10) Design of energy storage devices with high performances, such as high energy density, high power density, and long cycle life, is still challenging. Although flexible, bendable, twistable, and stretchable energy storage devices, i.e., fiber-shaped energy devices, have been investigated for several years, they are still in the early stages of lab-scale research. Therefore, the development of novel strategies for the fabrication of such devices in both academia and industry has become necessary.
(11) Although many strategies have been developed to improve the crystallinity of COFs, obtaining larger single crystals is fraught with difficulties.
Contributions
K. W. wrote sections of Abstract, Introduction, Conclusion and outlook, and transition sections. Z. A., D. H., A. Q., R. H., Y. H., J. Z., W. F., J. S., Z. C., B. T., Y. D., and Z. L. wrote the section of polymerization methodologies. J. G., A. Q., B. Y., and L. W. wrote the section of luminescent polymer materials. T. L., X. Z., H. C., J. L., Y. H., and F. H. wrote the section of photovoltaic polymer materials. Y. Z., Q. L., and Z. L. wrote the section of other electronical and optical polymer materials. X. J., X. Z., S. W., D. H., J. L., Z. Z., Y. S., X. Z., X. Z., S. L., H. C., and G. Z. wrote the section of bio-related polymer materials. H. W., F. H., H. X., Y. Z., and J. Z. wrote the section of supramolecular polymer materials. C. X., X. Z., X. Q., and Y. Y. wrote the section of stimuli-responsive polymer materials. T. X. and M. Q. Z. wrote the section of shape memory polymer materials. J. J., W. Z., C. Z., and Z. K. X. wrote the section of polymer membranes for separation. K. A., Z. W., X. S., and H. P. wrote the section of Energy storage polymers. Y. Z., X. F., and B. W. wrote the section of covalent organic frameworks. B. Z. T. proposed and corrected the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21788102, 51933010, 21922502, 51833008, 21890733, 51773229, 21774041, 21774114, 21734008, 21734003, 51690150, 51690151, 51625306, 21625102, 21634003, 21625402, 51525302, 51573061, 21525417, 21534003, 21490571, 21434005, 21490574), the State Key Research Development Program of China (2017YFA0403403, 2016YFA0203302, 2016YFC1103000, and 2016YFC1103001), the Natural Science Foundation of Guangdong Province (2016A030312002, 2019B030301003, and 2019A1515011585), the Innovation and Technology Commission of Hong Kong (ITC-CNERC14S01), the China Postdoctoral Science Foundation (2019M662902), and the Fundamental Research Funds for the Central Universities (2019MS007).References
- F. Zhao, Y. Shi, L. Pan and G. Yu, Multifunctional nanostructured conductive polymer gels: synthesis, properties, and applications, Acc. Chem. Res., 2017, 50, 1734–1743 CrossRef CAS PubMed
.
- W. Zhang, B. Aguila and S. Ma, Potential applications of functional porous organic polymer materials, J. Mater. Chem. A, 2017, 5, 8795–8824 RSC
- B. C. Anderson, G. D. Andrews, P. Arthur, H. W. Jacobson, L. R. Melby, A. J. Playtis and W. H. Sharkey, Anionic polymerization of methacrylates. Novel functional polymers and copolymers, Macromolecules, 1981, 14, 1599–1601 CrossRef CAS
- D. Kumar and R. C. Sharma, Advances in conductive polymers, Eur. Polym. J., 1998, 34, 1053–1060 CrossRef CAS
- J. Du and R. K. O'Reilly, Advances and challenges in smart and functional polymer vesicles, Soft Matter, 2009, 5, 3544–3561 RSC
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Multicompartment block polymer micelles, Macromolecules, 2011, 45, 2–19 CrossRef
- J. Jin, L. Cai, Y.-G. Jia, S. Liu, Y. Chen and L. Ren, Progress in self-healing hydrogels assembled by host–guest interactions: preparation and biomedical applications, J. Mater. Chem. B, 2019, 7, 1637–1651 RSC
- S. Strandman and X. X. Zhu, Self-healing supramolecular hydrogels based on reversible physical interactions, Gels, 2016, 2, 16 CrossRef PubMed
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Non-fullerene acceptors for organic solar cells, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS
- C. B. Nielsen, S. Holliday, H. Y. Chen, S. J. Cryer and I. McCulloch, Non-fullerene electron acceptors for use in organic solar cells, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed
- N. Liang, W. Jiang, J. Hou and Z. Wang, New developments in non-fullerene small molecule acceptors for polymer solar cells, Mater. Chem. Front., 2017, 1, 1291–1303 RSC
- A. Facchetti, Polymer donor–polymer acceptor (all-polymer) solar cells, Mater. Today, 2013, 16, 123–132 CrossRef CAS
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Functional supramolecular polymers for biomedical applications, Adv. Mater., 2015, 27, 498–526 CrossRef CAS PubMed
- W. Wu, W. Wang and J. Li, Star polymers: Advances in biomedical applications, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS
- K. Wang, S. Strandman and X. X. Zhu, A mini review: Shape memory polymers for biomedical applications, Front. Chem. Sci. Eng., 2017, 11, 143–153 CrossRef
- K. J. Wang, Y. G. Jia, C. Z. Zhao and X. X. Zhu, Multiple and two-way reversible shape memory polymers: Design strategies and applications, Prog. Mater. Sci., 2019, 105, 100572 CrossRef CAS
- Q. Zhao, H. J. Qi and T. Xie, Recent progress in shape memory polymer: New behavior, enabling materials, and mechanistic understanding, Prog. Polym. Sci., 2015, 49–50, 79–120 CrossRef CAS
- C. de Las Heras Alarcon, S. Pennadam and C. Alexander, Stimuli responsive polymers for biomedical applications, Chem. Soc. Rev., 2005, 34, 276–285 RSC
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Emerging applications of stimuli-responsive polymer materials, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed
- Q. Ma, H. Cheng, A. G. Fane, R. Wang and H. Zhang, Recent development of advanced materials with special wettability for selective oil/water separation, Small, 2016, 12, 2186–2202 CrossRef CAS PubMed
- Y. Zhang, X. Feng, S. Yuan, J. Zhou and B. Wang, Challenges and recent advances in MOF–polymer composite membranes for gas separation, Inorg. Chem. Front., 2016, 3, 896–909 RSC
- J. M. Stanley and B. J. Holliday, Luminescent lanthanide-containing metallopolymers, Coord. Chem. Rev., 2012, 256, 1520–1530 CrossRef CAS
- S. Dalapati, C. Gu and D. Jiang, Luminescent porous polymers based on aggregation-induced mechanism: design, synthesis and functions, Small, 2016, 12, 6513–6527 CrossRef CAS PubMed
- P. J. Waller, F. Gandara and O. M. Yaghi, Chemistry of covalent organic frameworks, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Supramolecular polymers, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, Metal-free atom transfer radical polymerization, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Electrochemically mediated atom transfer radical polymerization, Science, 2011, 332, 81–84 CrossRef CAS PubMed
- B. P. Fors and C. J. Hawker, Control of a living radical polymerization of methacrylates by light, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Organocatalyzed atom transfer radical polymerization driven by visible light, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed
- C.-H. Lim, M. D. Ryan, B. G. McCarthy, J. C. Theriot, S. M. Sartor, N. H. Damrauer, C. B. Musgrave and G. M. Miyake, Intramolecular charge transfer and ion pairing in n,n-diaryl dihydrophenazine photoredox catalysts for efficient organocatalyzed atom transfer radical polymerization, J. Am. Chem. Soc., 2017, 139, 348–355 CrossRef CAS PubMed
- R. M. Pearson, C.-H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, Organocatalyzed atom transfer radical polymerization using n-aryl phenoxazines as photoredox catalysts, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef CAS PubMed
- H. Gong, Y. Zhao, X. Shen, J. Lin and M. Chen, Organocatalyzed photocontrolled radical polymerization of semifluorinated (meth)acrylates driven by visible light, Angew. Chem., Int. Ed., 2018, 57, 333–337 CrossRef CAS PubMed
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, A robust and versatile photoinduced living polymerization of conjugated and unconjugated monomers and its oxygen tolerance, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed
- S. Shanmugam and C. Boyer, Stereo-, temporal and chemical control through photoactivation of living radical polymerization: synthesis of block and gradient copolymers, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed
- S. Shanmugam, J. Xu and C. Boyer, Exploiting metalloporphyrins for selective living radical polymerization tunable over visible wavelengths, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed
- J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, Selective photoactivation: From a single unit monomer insertion reaction to controlled polymer architectures, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS PubMed
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Aqueous photoinduced living/controlled
polymerization: tailoring for bioconjugation, Chem. Sci., 2014, 5, 3568–3575 RSC
- S. Shanmugam, J. Xu and C. Boyer, Utilizing the electron transfer mechanism of chlorophyll a under light for controlled radical polymerization, Chem. Sci., 2015, 6, 1341–1349 RSC
- S. Shanmugam, J. Xu and C. Boyer, Light-regulated polymerization under near-infrared/far-red irradiation catalyzed by bacteriochlorophyll a, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Organo-photocatalysts for photoinduced electron transfer-reversible addition–fragmentation chain transfer (PET-RAFT) polymerization, Polym. Chem., 2015, 6, 5615–5624 RSC
- S. Oliver, L. Zhao, A. J. Gormley, R. Chapman and C. Boyer, Living in the fast lane—high throughput controlled/living radical polymerization, Macromolecules, 2019, 52, 3–23 CrossRef CAS
- L. Shen, Q. Lu, A. Zhu, X. Lv and Z. An, Photocontrolled RAFT polymerization mediated by a supramolecular catalyst, ACS Macro Lett., 2017, 6, 625–631 CrossRef CAS
- J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O'Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Engineering live cell surfaces with functional polymers via cytocompatible controlled radical polymerization, Nat. Chem., 2017, 9, 537–545 CrossRef CAS PubMed
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Mechanically controlled radical polymerization initiated by ultrasound, Nat. Chem., 2017, 9, 135–139 CrossRef CAS
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Sono-RAFT polymerization in aqueous medium, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 CrossRef CAS PubMed
- Z. Liu, Y. Lv and Z. An, Enzymatic cascade catalysis for the synthesis of multiblock and ultrahigh-molecular-weight polymers with oxygen tolerance, Angew. Chem., Int. Ed., 2017, 56, 13852–13856 CrossRef CAS PubMed
- H. Z. Guo and Z. S. An, Enzyme catalysis for RAFT polymerization and functionalization: a sustainable strategy for synthesis of precision polymers, Acta Polym. Sin., 2018, 0, 1253–1261 CAS
- K. J. Rodriguez, B. Gajewska, J. Pollard, M. M. Pellizzoni, C. Fodor and N. Bruns, Repurposing biocatalysts to control radical polymerizations, ACS Macro Lett., 2018, 7, 1111–1119 CrossRef CAS
- C. Fodor, B. Gajewska, O. Rifaie-Graham, E. A. Apebende, J. Pollard and N. Bruns, Laccase-catalyzed controlled radical polymerization of N-vinylimidazole, Polym. Chem., 2016, 7, 6617–6625 RSC
- B. Zhang, X. Wang, A. Zhu, K. Ma, Y. Lv, X. Wang and Z. An, Enzyme-initiated reversible addition–fragmentation chain transfer polymerization, Macromolecules, 2015, 48, 7792–7802 CrossRef CAS
- A. Reyhani, M. D. Nothling, H. Ranji-Burachaloo, T. G. McKenzie, Q. Fu, S. Tan, G. Bryant and G. G. Qiao, Blood-catalyzed raft polymerization, Angew. Chem., Int. Ed., 2018, 57, 10288–10292 CrossRef CAS PubMed
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Highly controlled open vessel raft polymerizations by enzyme degassing, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Combinatorial low-volume synthesis of well-defined polymers by enzyme degassing, Angew. Chem., Int. Ed., 2016, 55, 4500–4503 CrossRef CAS PubMed
- Y. Lv, Z. Liu, A. Zhu and Z. An, Glucose oxidase deoxygenation-redox initiation for RAFT polymerization in air, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 164–174 CrossRef CAS
- Y. Wang, L. Fu and K. Matyjaszewski, Enzyme-deoxygenated low parts per million atom transfer radical polymerization in miniemulsion and ab initio emulsion, ACS Macro Lett., 2018, 7, 1317–1321 CrossRef CAS PubMed
- A. E. Enciso, L. Fu, A. J. Russell and K. Matyjaszewski, A breathing atom-transfer radical polymerization: fully oxygen-tolerant polymerization inspired by aerobic respiration of cells, Angew. Chem., Int. Ed., 2018, 57, 933–936 CrossRef CAS PubMed
- A. E. Enciso, L. Fu, S. Lathwal, M. Olszewski, Z. Wang, S. R. Das, A. J. Russell and K. Matyjaszewski, Biocatalytic “oxygen-fueled” atom transfer radical polymerization, Angew. Chem., Int. Ed., 2018, 57, 16157–16161 CrossRef CAS PubMed
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Click chemistry: diverse chemical function from a few good reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed
- A. Qin, J. W. Lam and B. Z. Tang, Click polymerization, Chem. Soc. Rev., 2010, 39, 2522–2544 RSC
-
A. Qin and B. Z. Tang, Click Polymerization, The Royal Society of Chemistry, 2018 Search PubMed
- D. Huang, Y. Liu, A. Qin and B. Tang, Recent advances in alkyne-based click polymerizations, Polym. Chem., 2018, 9, 2853–2867 RSC
-
D. Huang, A. Qin and B. Z. Tang, Click Polymerization, The Royal Society of Chemistry, 2018, ch. 2, pp. 36–85 Search PubMed
- Y. Shi, X. S. Cao, D. Q. Hu and H. F. Gao, Highly branched polymers with layered structures that mimic light-harvesting processes, Angew. Chem., Int. Ed., 2018, 57, 516–520 CrossRef CAS PubMed
- X. S. Cao, Y. Shi, X. F. Wang, R. W. Graff and H. F. Gao, Design a highly reactive trifunctional core molecule to obtain hyperbranched polymers with over a million molecular weight in one-pot click polymerization, Macromolecules, 2016, 49, 760–766 CrossRef CAS
- X. S. Cao, Y. Shi, W. P. Gan, H. Naguib, X. F. Wang, R. W. Graff and H. F. Gao, Effect of monomer structure on the CuAAC polymerization to produce hyperbranched polymers, Macromolecules, 2016, 49, 5342–5349 CrossRef CAS
- Y. Shi, R. W. Graff, X. S. Cao, X. F. Wang and H. F. Gao, Chain-growth click polymerization of ab(2) monomers for the formation of hyperbranched polymers with low polydispersities in a one-pot process, Angew. Chem., Int. Ed., 2015, 54, 7631–7635 CrossRef CAS PubMed
- A. J. Qin, J. W. Y. Lam, C. K. W. Jim, L. Zhang, J. J. Yan, M. Haussler, J. Z. Liu, Y. Q. Dong, D. H. Liang, E. Q. Chen, G. C. Jia and B. Z. Tang, Hyperbranched polytriazoles: Click polymerization, regioisomeric structure, light emission, and fluorescent patterning, Macromolecules, 2008, 41, 3808–3822 CrossRef CAS
- D. Huang, Y. Liu, A. Qin and B. Z. Tang, Structure–property relationship of regioregular polytriazoles produced by ligand-controlled regiodivergent Ru(II)-catalyzed azide–alkyne click polymerization, Macromolecules, 2019, 52, 1985–1992 CrossRef CAS
- A. J. Qin, Y. Liu and B. Z. Tang, Regioselective metal-free click polymerization of azides and alkynes, Macromol. Chem. Phys., 2015, 216, 818–828 CrossRef CAS
- H. Q. Wu, W. H. Dong, Z. T. Wang, B. C. Yao, M. Chen, J. Z. Sun, A. J. Qin and B. Z. Tang, An air-stable supported Cu(D dagger) catalyst for azide–alkyne click polymerization, Sci. China Chem., 2015, 58, 1748–1752 CrossRef CAS
- H. Wu, H. Li, R. T. Kwok, E. Zhao, J. Z. Sun, A. Qin and B. Z. Tang, A recyclable and reusable supported Cu(I) catalyzed azide–alkyne click polymerization, Sci. Rep., 2014, 4, 5107 CrossRef PubMed
- Q. Wei, J. Wang, X. Y. Shen, X. A. Zhang, J. Z. Sun, A. J. Qin and B. Z. Tang, Self-healing hyperbranched poly(aroyltriazole)s, Sci. Rep., 2013, 3, 1093 CrossRef
- Q. Wei, H. Deng, Y. Cai, J. W. Lam, J. Li, J. Sun, M. Gao, A. Qin and B. Z. Tang, Efficient polymerization of azide and active internal alkynes, Macromol. Rapid Commun., 2012, 33, 1356–1361 CrossRef CAS PubMed
- A. J. Qin, L. Tang, J. W. Y. Lam, C. K. W. Jim, Y. Yu, H. Zhao, J. Z. Sun and B. Z. Tang, Metal-free click polymerization: synthesis and photonic properties of poly(aroyltriazole)s, Adv. Funct. Mater., 2009, 19, 1891–1900 CrossRef CAS
- A. J. Qin, C. K. W. Jim, W. X. Lu, J. W. Y. Lam, M. Haussler, Y. Q. Dong, H. H. Y. Sung, I. D. Williams, G. K. L. Wong and B. Z. Tang, Click polymerization: Facile synthesis of functional poly(aroyltriazole)s by metal-free, regioselective 1,3-dipolar polycycloaddition, Macromolecules, 2007, 40, 2308–2317 CrossRef CAS
- X. Wang, R. R. Hu, Z. J. Zhao, A. J. Qin and B. Z. Tang, Self-healing hyperbranched polytriazoles prepared by metal-free click polymerization of propiolate and azide monomers, Sci. China: Chem., 2016, 59, 1554–1560 CrossRef CAS
- H. K. Li, H. Q. Wu, E. G. Zhao, J. Li, J. Z. Sun, A. J. Qin and B. Z. Tang, Hyperbranched poly(aroxycarbonyltriazole)s: metal-free click polymerization, light refraction, aggregation-induced emission, explosive detection, and fluorescent patterning, Macromolecules, 2013, 46, 3907–3914 CrossRef CAS
- H. K. Li, J. Mei, J. A. Wang, S. A. Zhang, Q. L. Zhao, Q. A. Wei, A. J. Qin, J. Z. Sun and B. Z. Tang, Facile synthesis of poly(aroxycarbonyltriazole)s with aggregation-induced emission characteristics by metal-free click polymerization, Sci. China: Chem., 2011, 54, 611–616 CrossRef CAS
- Q. Wang, H. K. Li, Q. Wei, J. Z. Sun, J. Wang, X. A. Zhang, A. J. Qin and B. Z. Tang, Metal-free click polymerizations of activated azide and alkynes, Polym. Chem., 2013, 4, 1396–1401 RSC
- Q. Wang, M. Chen, B. C. Yao, J. Wang, J. Mei, J. Z. Sun, A. J. Qin and B. Z. Tang, A polytriazole synthesized by 1,3-dipolar polycycloaddition showing aggregation-enhanced emission and utility in explosive detection, Macromol. Rapid Commun., 2013, 34, 796–802 CrossRef CAS PubMed
- Y. Liu, J. Wang, D. Huang, J. Zhang, S. Guo, R. R. Hu, Z. J. Zhao, A. J. Qin and B. Z. Tang, Synthesis of 1,5-regioregular polytriazoles by efficient NMe4OH-mediated azide–alkyne click polymerization, Polym. Chem., 2015, 6, 5545–5549 RSC
- B. X. Li, Y. Liu, H. Nie, A. J. Qin and B. Z. Tang, Phosphazene base-mediated azide–alkyne click polymerization toward 1,5-regioregular polytriazoles, Macromolecules, 2019, 52, 4713–4720 CrossRef CAS
- B. Yao, J. Sun, A. Qin and B. Z. Tang, Thiol–yne click polymerization, Chin. Sci. Bull., 2013, 58, 2711–2718 CrossRef CAS
- D. Konkolewicz, A. Gray-Weale and S. Perrier, Hyperbranched polymers by thiol–yne chemistry: from small molecules to functional polymers, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS PubMed
- R. Potzsch, H. Komber and B. C. Stahl, C. J. Hawker and B. I. Voit, Radical thiol–yne chemistry on diphenylacetylene: selective and quantitative addition enabling the synthesis of hyperbranched
poly(vinyl sulfide)s, Macromol. Rapid Commun., 2013, 34, 1772–1778 CrossRef PubMed
- Q. Wei, R. Potzsch, X. L. Liu, H. Komber, A. Kiriy, B. Voit, P. A. Will, S. Lenk and S. Reineke, Hyperbranched polymers with high transparency and inherent high refractive index for application in organic light-emitting diodes, Adv. Funct. Mater., 2016, 26, 2545–2553 CrossRef CAS
- S. Gazzo, G. Manfredi, R. Potzsch, Q. Wei, M. Alloisio, B. Voit and D. Comoretto, High refractive index hyperbranched polyvinylsulfides for planar one-dimensional all-polymer photonic crystals, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 73–80 CrossRef CAS
- J. Z. Liu, J. W. Y. Lam, C. K. W. Jim, J. C. Y. Ng, J. B. Shi, H. M. Su, K. F. Yeung, Y. N. Hong, M. Faisal, Y. Yu, K. S. Wong and B. Z. Tang, Thiol–yne click polymerization: regio- and stereoselective synthesis of sulfur-rich acetylenic polymers with controllable chain conformations and tunable optical properties, Macromolecules, 2011, 44, 68–79 CrossRef CAS
- C. K. W. Jim, A. Qin, J. W. Y. Lam, F. Mahtab, Y. Yu and B. Z. Tang, Metal-free alkyne polyhydrothiolation: synthesis of functional poly(vinylenesulfide)s with high stereoregularity by regioselective thioclick polymerization, Adv. Funct. Mater., 2010, 20, 1319–1328 CrossRef CAS
- D. Huang, Y. Liu, S. Guo, B. X. Li, J. Wang, B. C. Yao, A. J. Qin and B. Z. Tang, Transition metal-free thiol–yne click polymerization toward Z-stereoregular poly(vinylene sulfide)s, Polym. Chem., 2019, 10, 3088–3096 RSC
- B. C. Yao, J. Mei, J. Li, J. Wang, H. Q. Wu, J. Z. Sun, A. J. Qin and B. Z. Tang, Catalyst-free thiol–yne click polymerization: a powerful and facile tool for preparation of functional poly(vinylene sulfide)s, Macromolecules, 2014, 47, 1325–1333 CrossRef CAS
- B. C. Yao, T. Hu, H. K. Zhang, J. Li, J. Z. Sun, A. J. Qin and B. Z. Tang, Multi-functional hyperbranched poly(vinylene sulfide)s constructed via spontaneous thiol–yne click polymerization, Macromolecules, 2015, 48, 7782–7791 CrossRef CAS
- B. Z. He, S. J. Zhen, Y. W. Wu, R. R. Hu, Z. J. Zhao, A. J. Qin and B. Z. Tang, Cu(I)-Catalyzed amino-yne click polymerization, Polym. Chem., 2016, 7, 7375–7382 RSC
- B. He, H. Su, T. Bai, Y. Wu, S. Li, M. Gao, R. Hu, Z. Zhao, A. Qin, J. Ling and B. Z. Tang, Spontaneous amino-yne click polymerization: a powerful tool toward regio- and stereospecific poly(beta-aminoacrylate)s, J. Am. Chem. Soc., 2017, 139, 5437–5443 CrossRef CAS PubMed
- R. Hu, X. Chen, T. T. Zhou, H. Si, B. Z. He, R. T. K. Kwok, A. J. Qin and B. Z. Tang, Lab-in-cell based on spontaneous amino-yne click polymerization, Sci. China: Chem., 2019, 62, 1198–1203 CrossRef CAS
- X. M. Chen, R. Hu, C. X. Qi, X. Y. Fu, J. Wang, B. Z. He, D. Huang, A. J. Qin and B. Z. Tang, Ethynylsulfone-based spontaneous amino-yne click polymerization: a facile tool toward regio- and stereoregular dynamic polymers, Macromolecules, 2019, 52, 4526–4533 CrossRef CAS
- Y. Shi, T. Bai, W. Bai, Z. Wang, M. Chen, B. Yao, J. Z. Sun, A. Qin, J. Ling and B. Z. Tang, Phenol-yne click polymerization: an efficient technique to facilely access regio- and stereoregular
poly(vinylene ether ketone)s, Chem. – Eur. J., 2017, 23, 10725–10731 CrossRef CAS
- J. Wang, B. X. Li, D. H. Xin, R. R. Hu, Z. J. Zhao, A. J. Qin and B. Z. Tang, Superbase catalyzed regio-selective polyhydroalkoxylation of alkynes: a facile route towards functional poly(vinyl ether)s, Polym. Chem., 2017, 8, 2713–2722 RSC
- I. H. Lee, H. Kim and T. L. Choi, Cu-catalyzed multicomponent polymerization to synthesize a library of poly(N-sulfonylamidines), J. Am. Chem. Soc., 2013, 135, 3760–3763 CrossRef CAS PubMed
- H. Kim and T.-L. Choi, Preparation of a library of poly(N-sulfonylimidates) by Cu-catalyzed multicomponent polymerization, ACS Macro Lett., 2014, 3, 791–794 CrossRef CAS
- H. Kim, K. T. Bang, I. Choi, J. K. Lee and T. L. Choi, Diversity-oriented polymerization: one-shot synthesis of library of graft and dendronized polymers by Cu-catalyzed multicomponent polymerization, J. Am. Chem. Soc., 2016, 138, 8612–8622 CrossRef CAS PubMed
- H. Deng, T. Han, E. Zhao, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Multicomponent click polymerization: a facile strategy toward fused heterocyclic polymers, Macromolecules, 2016, 49, 5475–5483 CrossRef CAS
- L. Xu, T. Zhou, M. Liao, R. Hu and B. Z. Tang, Multicomponent polymerizations of alkynes, sulfonyl azides, and 2-hydroxybenzonitrile/2-aminobenzonitrile toward multifunctional iminocoumarin/quinoline-containing poly(N-sulfonylimine)s, ACS Macro Lett., 2019, 8, 101–106 CrossRef CAS
- L. Xu, F. Zhou, M. Liao, R. Hu and B. Z. Tang, Room temperature multicomponent polymerizations of alkynes, sulfonyl azides, and N-protected isatins toward oxindole-containing poly(N-acylsulfonamide)s, Polym. Chem., 2018, 9, 1674–1683 RSC
- L. Xu, R. Hu and B. Z. Tang, Room temperature multicomponent polymerizations of alkynes, sulfonyl azides, and iminophosphorane toward heteroatom-rich multifunctional poly(phosphorus amidine)s, Macromolecules, 2017, 50, 6043–6053 CrossRef CAS
- L. G. Xu, K. Yang, R. R. Hu and B. Z. Tang, Multicomponent polymerization of alkynes, sulfonyl azide, and iminophosphorane at room temperature for the synthesis of hyperbranched poly(phosphorus amidine)s, Synlett, 2018, 2523–2528 CAS
- T. Han, H. Deng, Z. Qiu, Z. Zhao, H. Zhang, H. Zou, N. L. C. Leung, G. Shan, M. R. J. Elsegood, J. W. Y. Lam and B. Z. Tang, Facile multicomponent polymerizations toward unconventional luminescent polymers with readily openable small heterocycles, J. Am. Chem. Soc., 2018, 140, 5588–5598 CrossRef CAS PubMed
- Q. Gao, L. H. Xiong, T. Han, Z. Qiu, X. He, H. H. Y. Sung, R. T. K. Kwok, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Three-component regio- and stereoselective polymerizations toward functional chalcogen-rich polymers with aie-activities, J. Am. Chem. Soc., 2019, 141, 14712–14719 CrossRef CAS PubMed
- Y. Hu, T. Han, N. Yan, J. Liu, X. Liu, W. X. Wang, J. W. Y. Lam and B. Z. Tang, Visualization of biogenic amines and in vivo ratiometric mapping of intestinal ph by aie-active polyheterocycles synthesized by metal-free multicomponent polymerizations, Adv. Funct. Mater., 2019, 29, 1902240 CrossRef
- H. Deng, R. Hu, E. Zhao, C. Y. K. Chan, J. W. Y. Lam and B. Z. Tang, One-pot three-component tandem polymerization toward functional poly(arylene thiophenylene) with aggregation-enhanced emission characteristics, Macromolecules, 2014, 47, 4920–4929 CrossRef CAS
- C. Zheng, H. Deng, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Multicomponent tandem reactions and polymerizations of alkynes, carbonyl chlorides, and thiols, Macromolecules, 2015, 48, 1941–1951 CrossRef CAS
- H. Deng, R. Hu, A. C. S. Leung, E. Zhao, J. W. Y. Lam and B. Z. Tang, Construction of regio- and stereoregular poly(enaminone)s by multicomponent tandem polymerizations of diynes, diaroyl chloride and primary amines, Polym. Chem., 2015, 6, 4436–4446 RSC
- X. Tang, C. Zheng, Y. Chen, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Multicomponent tandem polymerizations of aromatic diynes, terephthaloyl chloride, and hydrazines toward functional conjugated polypyrazoles, Macromolecules, 2016, 49, 9291–9300 CrossRef CAS
- X. Tang, L. Zhang, R. Hu and B. Z. Tang, Multicomponent tandem polymerization of aromatic alkynes, carbonyl chloride, and Fischer's base toward poly(diene merocyanine)s, Chin. J. Chem., 2019, 37, 1264–1270 CrossRef CAS
- C. Qi, C. Zheng, R. Hu and B. Z. Tang, Direct construction of acid-responsive poly(indolone)s through multicomponent tandem polymerizations, ACS Macro Lett., 2019, 8, 569–575 CrossRef CAS
- W. Tian, R. Hu and B. Z. Tang, One-pot multicomponent tandem reactions and polymerizations for step-economic synthesis of structure-controlled pyrimidine derivatives and poly(pyrimidine)s, Macromolecules, 2018, 51, 9749–9757 CrossRef CAS
- B. Wei, W. Li, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Metal-free multicomponent tandem polymerizations of alkynes, amines, and formaldehyde toward structure- and sequence-controlled luminescent polyheterocycles, J. Am. Chem. Soc., 2017, 139, 5075–5084 CrossRef CAS PubMed
- Y. Huang, P. Chen, B. Wei, R. Hu and B. Z. Tang, Aggregation-induced emission-active hyperbranched poly(tetrahydropyrimidine) s synthesized from multicomponent tandem polymerization, Chin. J. Polym. Sci., 2019, 37, 428–436 CrossRef CAS
- W. Q. Fu, L. C. Dong, J. B. Shi, B. Tong, Z. X. Cai, J. G. Zhi and Y. P. Dong, Synthesis of polyquinolines via one-pot polymerization of alkyne, aldehyde, and aniline under metal-free catalysis and their properties, Macromolecules, 2018, 51, 3254–3263 CrossRef CAS
- W. Q. Fu, L. C. Dong, J. B. Shi, B. Tong, Z. X. Cai, J. G. Zhi and Y. P. Dong, Synthesis and characterization of poly(etheneketone-arylene-ketone) s containing pendant methylthio groups via metal- free catalyzed copolymerization of aryldiynes with DMSO, Polym. Chem., 2018, 9, 4404–4412 RSC
- W. Q. Fu, L. C. Dong, J. B. Shi, B. Tong, Z. X. Cai, J. G. Zhi and Y. P. Dong, Multicomponent spiropolymerization of diisocyanides, alkynes and carbon dioxide for constructing 1,6-dioxospiro[4,4]nonane-3,8-diene as structural units under one-pot catalyst-free conditions, Polym. Chem., 2018, 9, 5543–5550 RSC
- W. Q. Fu, L. W. Kong, J. B. Shi, B. Tong, Z. X. Cai, J. G. Zhi and Y. P. Dong, Synthesis of poly(amine-furan-arylene)s through a one-pot catalyst-free in situ cyclopolymerization of diisocyanide, dialkylacetylene dicarboxylates, and dialdehyde, Macromolecules, 2019, 52, 729–737 CrossRef CAS
- W.-Q. Fu, G.-N. Zhu, J.-B. Shi, B. Tong, Z.-X. Cai and Y.-P. Dong, Synthesis and properties of photodegradable poly(furan-amine)s by a catalyst-free multicomponent cyclopolymerization, Chin. J. Polym. Sci., 2019, 37, 981–989 CrossRef CAS
- W. Q. Fu, J. B. Shi, B. Tong, Z. X. Cai, J. G. Zhi and Y. P. Dong, Synthesis and characterization of poly(iminofuran-arylene) containing bromomethyl groups linked at the 5-position of a furan ring via the multicomponent polymerizations of diisocyanides, dialkylacetylene dicarboxylates, and bis(2-bromoacetyl)biphenyl, Macromolecules, 2019, 52, 3319–3326 CrossRef CAS
- W. Li, X. Wu, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Catalyst-free, atom-economic, multicomponent polymerizations of aromatic diynes, elemental sulfur, and aliphatic diamines toward luminescent polythioamides, Macromolecules, 2015, 48, 7747–7754 CrossRef CAS
- T. Tian, R. Hu and B. Z. Tang, Room temperature one-step conversion from elemental sulfur to functional polythioureas through catalyst-free multicomponent polymerizations, J. Am. Chem. Soc., 2018, 140, 6156–6163 CrossRef CAS PubMed
- S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Phosphazene bases as organocatalysts for ring-opening polymerization of cyclic esters, Macromol. Rapid Commun., 2018, 39, 1800485 CrossRef PubMed
- J. J. Bozell and G. R. Petersen, Technology development for the production of biobased products from biorefinery carbohydrates-the US Department of Energy's “Top 10” revisited, Green Chem., 2010, 12, 539–554 RSC
- P. J. Yuan and M. Hong, Ring-opening polymerizations of the “non-strained” gamma-butyrolactone and its derivatives: an overview and outlook, Acta Polym. Sin., 2019, 50, 327–337 Search PubMed
- M. Hong and E. Y. Chen, Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of gamma-butyrolactone, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed
- M. Hong and E. Y. Chen, Towards truly sustainable polymers: a metal-free recyclable polyester from biorenewable non-strained gamma-butyrolactone, Angew. Chem., Int. Ed., 2016, 55, 4188–4193 CrossRef CAS PubMed
- C.-J. Zhang, L.-F. Hu, H.-L. Wu, X.-H. Cao and X.-H. Zhang, Dual organocatalysts for highly active and selective synthesis of linear poly(γ-butyrolactone)s with high molecular weights, Macromolecules, 2018, 51, 8705–8711 CrossRef CAS
- M. Hong, X. Tang, B. S. Newell and E. Y. X. Chen, “Nonstrained” γ-butyrolactone-based copolyesters: copolymerization characteristics and composition-dependent (thermal, eutectic, cocrystallization, and degradation) properties, Macromolecules, 2017, 50, 8469–8479 CrossRef CAS
- N. Zhao, C. Ren, H. Li, Y. Li, S. Liu and Z. Li, Selective ring-opening polymerization of non-strained γ-butyrolactone catalyzed by cyclic trimeric phosphazene base, Angew. Chem., Int. Ed., 2017, 56, 12987–12990 CrossRef CAS
- Y. Shen, J. Zhang, N. Zhao, F. Liu and Z. Li, Preparation of biorenewable poly([gamma]-butyrolactone)-b-poly(l-lactide) diblock copolyesters via one-pot sequential metal-free ring-opening polymerization, Polym. Chem., 2018, 9, 2936–2941 RSC
- Y. Shen, J. Zhang, Z. Zhao, N. Zhao, F. Liu and Z. Li, Preparation of amphiphilic poly(ethylene glycol)-b-poly(gamma-butyrolactone) diblock copolymer via ring opening polymerization catalyzed by a cyclic trimeric phosphazene base or alkali alkoxide, Biomacromolecules, 2019, 20, 141–148 CrossRef CAS
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Recent progress on polymer-based fluorescent and colorimetric chemosensors, Chem. Soc. Rev., 2011, 40, 79–93 RSC
- X. Feng, L. Liu, S. Wang and D. Zhu, Water-soluble fluorescent conjugated polymers and their interactions with biomacromolecules for sensitive biosensors, Chem. Soc. Rev., 2010, 39, 2411–2419 RSC
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole, Chem. Commun., 2001, 1740–1741 RSC
- H. Tong, Y. Hong, Y. Dong, Y. Ren, M. Haussler, J. W. Lam, K. S. Wong and B. Z. Tang, Color-tunable, aggregation-induced emission of a butterfly-shaped molecule comprising a pyran skeleton and two cholesteryl wings, J. Phys. Chem. B, 2007, 111, 2000–2007 CrossRef CAS PubMed
- H. Tong, Y. Hong, Y. Dong, M. Haussler, Z. Li, J. W. Lam, Y. Dong, H. H. Sung, I. D. Williams and B. Z. Tang, Protein detection and quantitation by tetraphenylethene-based fluorescent probes with aggregation-induced emission characteristics, J. Phys. Chem. B, 2007, 111, 11817–11823 CrossRef CAS PubMed
- Y. Q. Dong, J. W. Y. Lam, A. J. Qin, J. Z. Liu, Z. Li and B. Z. Tang, Aggregation-induced emissions of tetraphenylethene derivatives and their utilities as chemical vapor sensors and in organic light-emitting diodes, Appl. Phys. Lett., 2007, 91, 011111 CrossRef
- M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Tetraphenylpyrazine-based AIEgens: facile preparation and tunable light emission, Chem. Sci., 2015, 6, 1932–1937 RSC
- J. Liu, J. W. Y. Lam and B. Z. Tang, Aggregation-induced Emission of Silole Molecules and Polymers: Fundamental and Applications, J. Inorg. Organomet. Polym. Mater., 2009, 19, 249–285 CrossRef CAS
- J. W. Y. Lam, J. W. Chen, C. C. W. Law, H. Peng, Z. L. Xie, K. K. L. Cheuk, H. S. Kwok and B. Z. Tang, Silole-containing linear and hyperbranched polymers: Synthesis, thermal stability, light emission, nano-dimensional aggregation, and optical power limiting, Macromol. Symp., 2003, 196, 289–300 CrossRef CAS
- A. Qin, J. W. Y. Lam and B. Z. Tang, Luminogenic polymers with aggregation-induced emission characteristics, Prog. Polym. Sci., 2012, 37, 182–209 CrossRef CAS
- R. Hu, N. L. Leung and B. Z. Tang, AIE macromolecules: syntheses, structures and functionalities, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC
- Y. Chen, H. Han, H. Tong, T. Chen, H. Wang, J. Ji and Q. Jin, Zwitterionic phosphorylcholine-TPE conjugate for pH-responsive drug delivery and AIE active imaging, ACS Appl. Mater. Interfaces, 2016, 8, 21185–21192 CrossRef CAS PubMed
- H. Si, K. Wang, B. Song, A. Qin and B. Z. Tang, Organobase-catalysed hydroxyl-yne click polymerization, Polym. Chem., 2020, 11, 2568–2575 RSC
- J. Wang, T. Bai, Y. Chen, C. Ye, T. Han, A. Qin, J. Ling and B. Z. Tang, Palladium/benzoic acid-catalyzed regio- and stereoselective polymerization of internal diynes and diols through c(sp3)–h activation, ACS Macro Lett., 2019, 8, 1068–1074 CrossRef CAS
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Aggregation-induced emission: Together we shine, united we soar!, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Stabilizing triplet excited states for ultralong organic phosphorescence, Nat. Mater., 2015, 14, 685–690 CrossRef CAS
- S. Tao, S. Lu, Y. Geng, S. Zhu, S. A. T. Redfern, Y. Song, T. Feng, W. Xu and B. Yang, Design of metal-free polymer carbon dots: a new class of room-temperature phosphorescent materials, Angew. Chem., Int. Ed., 2018, 57, 2393–2398 CrossRef CAS PubMed
- C. Xia, S. Tao, S. Zhu, Y. Song, T. Feng, Q. Zeng, J. Liu and B. Yang, Hydrothermal addition polymerization for ultrahigh-yield carbonized polymer dots with room temperature phosphorescence via nanocomposite, Chem. – Eur. J., 2018, 24, 11303–11308 CrossRef CAS
- K. Jiang, Y. Wang, X. Gao, C. Cai and H. Lin, Facile, quick, and gram-scale synthesis of ultralong-lifetime room-temperature-phosphorescent carbon dots by microwave irradiation, Angew. Chem., Int. Ed., 2018, 57, 6216–6220 CrossRef CAS PubMed
- K. Jiang, Y. Wang, C. Cai and H. Lin, Conversion of carbon dots from fluorescence to ultralong room-temperature phosphorescence by heating for security applications, Adv. Mater., 2018, 30, 1800783 CrossRef PubMed
- Z. Tian, D. Li, E. V. Ushakova, V. G. Maslov, D. Zhou, P. Jing, D. Shen, S. Qu and A. L. Rogach, Multilevel data encryption using thermal-treatment controlled room temperature phosphorescence of carbon dot/polyvinylalcohol composites, Adv. Sci., 2018, 5, 1800795 CrossRef PubMed
- P. Long, Y. Feng, C. Cao, Y. Li, J. Han, S. Li, C. Peng, Z. Li and W. Feng, Self-protective room-temperature phosphorescence of fluorine and nitrogen codoped carbon dots, Adv. Funct. Mater., 2018, 28, 1800791 CrossRef
- F. Xu, H. U. Kim, J.-H. Kim, B. J. Jung, A. C. Grimsdale and D.-H. Hwang, Progress and perspective of iridium-containing phosphorescent polymers for light-emitting diodes, Prog. Polym. Sci., 2015, 47, 92–121 CrossRef CAS
- S. Shao, J. Ding, T. Ye, Z. Xie, L. Wang, X. Jing and F. Wang, A novel, bipolar polymeric host for highly efficient blue electrophosphorescence: a non-conjugated poly(aryl ether) containing triphenylphosphine oxide units in the electron-transporting main chain and carbazole units in hole-transporting side chains, Adv. Mater., 2011, 23, 3570–3574 CrossRef CAS PubMed
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, Highly efficient blue electrophosphorescent polymers with fluorinated poly(arylene ether phosphine oxide) as backbone, J. Am. Chem. Soc., 2012, 134, 15189–15192 CrossRef CAS PubMed
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, Synthesis and characterization of yellow-emitting electrophosphorescent polymers based on a fluorinated poly(arylene ether phosphine oxide) scaffold, J. Mater. Chem., 2012, 22, 24848–24855 RSC
- Z. Ma, J. Ding, B. Zhang, C. Mei, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Red-Emitting Polyfluorenes Grafted with Quinoline-Based Iridium Complex: “Simple Polymeric Chain, Unexpected High Efficiency”, Adv. Funct. Mater., 2010, 20, 138–146 CrossRef CAS
- Z. Ma, J. Ding, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Synthesis and characterization of red light-emitting electrophosphorescent polymers with different triplet energy main chain, Polymer, 2011, 52, 2189–2197 CrossRef CAS
- Z. Ma, L. Chen, J. Ding, L. Wang, X. Jing and F. Wang, Green electrophosphorescent polymers with poly(3,6-carbazole) as the backbone: a linear structure does realize high efficiency, Adv. Mater., 2011, 23, 3726–3729 CrossRef CAS PubMed
- G. Tu, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, White electroluminescence from polyfluorene chemically doped with 1,8-napthalimide moieties, Appl. Phys. Lett., 2004, 85, 2172–2174 CrossRef CAS
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, White electroluminescence from all-phosphorescent single polymers on a fluorinated poly(arylene ether phosphine oxide) backbone simultaneously grafted with blue and yellow phosphors, J. Am. Chem. Soc., 2012, 134, 20290–20293 CrossRef CAS PubMed
- S. Shao, S. Wang, X. Xu, Y. Yang, J. Lv, J. Ding, L. Wang, X. Jing and F. Wang, Realization of high-power-efficiency white electroluminescence from a single polymer by energy-level engineering, Chem. Sci., 2018, 9, 8656–8664 RSC
- P. Cheng and X. Zhan, Stability of organic solar cells: challenges and strategies, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, A wide-bandgap donor polymer for highly efficient non-fullerene organic solar cells with a small voltage loss, J. Am. Chem. Soc., 2017, 139, 6298–6301 CrossRef CAS PubMed
- Y. Lin, F. Zhao, Y. Wu, K. Chen, Y. Xia, G. Li, S. K. Prasad, J. Zhu, L. Huo, H. Bin, Z. G. Zhang, X. Guo, M. Zhang, Y. Sun, F. Gao, Z. Wei, W. Ma, C. Wang, J. Hodgkiss, Z. Bo, O. Inganas, Y. Li and X. Zhan, Mapping polymer donors toward high-efficiency fullerene free organic solar cells, Adv. Mater., 2017, 29, 1604155 CrossRef PubMed
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Efficient organic solar cells processed from hydrocarbon solvents, Nat. Energy, 2016, 1, 15027 CrossRef CAS
- P. Cheng, G. Li, X. Zhan and Y. Yang, Next-generation organic photovoltaics based on non-fullerene acceptors, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Organic and solution-processed tandem solar cells with 17.3% efficiency, Science, 2018, 361, 1094 CrossRef CAS PubMed
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, An electron acceptor challenging fullerenes for efficient polymer solar cells, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed
- Y. Lin, T. Li, F. Zhao, L. Han, Z. Wang, Y. Wu, Q. He, J. Wang, L. Huo, Y. Sun, C. Wang, W. Ma and X. Zhan, Structure evolution of oligomer fused-ring electron acceptors toward high efficiency of as-cast polymer solar cells, Adv. Energy Mater., 2016, 6, 1600854 CrossRef
- W. Wang, C. Yan, T. K. Lau, J. Wang, K. Liu, Y. Fan, X. Lu and X. Zhan, Fused hexacyclic nonfullerene acceptor with strong near-infrared absorption for semitransparent organic solar cells with 9.77% efficiency, Adv. Mater., 2017, 29, 1701308 CrossRef PubMed
- T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Fused tris(thienothiophene)-based electron acceptor with strong near-infrared absorption for high-performance as-cast solar cells, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed
- J. Zhu, Z. Ke, Q. Zhang, J. Wang, S. Dai, Y. Wu, Y. Xu, Y. Lin, W. Ma, W. You and X. Zhan, Naphthodithiophene-based nonfullerene acceptor for high-performance organic photovoltaics: effect of extended conjugation, Adv. Mater., 2018, 30, 1704713 CrossRef PubMed
- D. He, F. Zhao, J. Xin, J. J. Rech, Z. Wei, W. Ma, W. You, B. Li, L. Jiang, Y. Li and C. Wang, A fused ring electron acceptor with decacyclic core enables over 13.5% efficiency for organic solar cells, Adv. Energy Mater., 2018, 8, 1802050 CrossRef
- S. Dai, Y. Xiao, P. Xue, J. J. Rech, K. Liu, Z. Li, X. Lu, W. You and X. Zhan, Effect of core size on performance of fused-ring electron acceptors, Chem. Mater., 2018, 30, 5390–5396 CrossRef CAS
- S. Dai, T. Li, W. Wang, Y. Xiao, T. K. Lau, Z. Li, K. Liu, X. Lu and X. Zhan, Enhancing the performance of polymer solar cells via core engineering of nir-absorbing electron acceptors, Adv. Mater., 2018, 30, 1706571 CrossRef PubMed
- C. Huang, X. Liao, K. Gao, L. Zuo, F. Lin, X. Shi, C. Z. Li, H. Liu, X. Li, F. Liu, Y. Chen, H. Chen and A. K. Y. Jen, Highly efficient organic solar cells based on S,N-heteroacene non-fullerene acceptors, Chem. Mater., 2018, 30, 5429–5434 CrossRef CAS
- J. Sun, X. Ma, Z. Zhang, J. Yu, J. Zhou, X. Yin, L. Yang, R. Geng, R. Zhu, F. Zhang and W. Tang, Dithieno[3,2-b:2′,3′-d]pyrrol Fused Nonfullerene Acceptors Enabling Over 13% Efficiency for Organic Solar Cells, Adv. Mater., 2018, 30, 1707150 CrossRef PubMed
- Z. Xiao, X. Jia, D. Li, S. Wang, X. Geng, F. Liu, J. Chen, S. Yang, T. P. Russell and L. Ding, 26 mA cm−2 Jsc from organic solar cells with a low-bandgap nonfullerene acceptor, Sci. Bull., 2017, 62, 1494–1496 CrossRef CAS
- Y. Wu, H. Bai, Z. Wang, P. Cheng, S. Zhu, Y. Wang, W. Ma and X. Zhan, A planar electron acceptor for efficient polymer solar cells, Energy Environ. Sci., 2015, 8, 3215–3221 RSC
- F. Liu, J. Zhang, Z. Zhou, J. Zhang, Z. Wei and X. Zhu, Poly(3-hexylthiophene)-based non-fullerene solar cells achieve high photovoltaic performance with small energy loss, J. Mater. Chem. A, 2017, 5, 16573–16579 RSC
- J. Wu, Y. Xu, Z. Yang, Y. Chen, X. Sui, L. Yang, P. Ye, T. Zhu, X. Wu, X. Liu, H. Cao, A. Peng and H. Huang, Simultaneous enhancement of three parameters of p3ht-based organic solar cells with one oxygen atom, Adv. Energy Mater., 2018, 9, 1803012 CrossRef
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, High-performance fullerene-free polymer solar cells with 6.31% efficiency, Energy Environ. Sci., 2015, 8, 610–616 RSC
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Design, Synthesis, and photovoltaic characterization of a small molecular acceptor with an ultra-narrow band gap, Angew. Chem., Int. Ed., 2017, 56, 3045–3049 CrossRef CAS PubMed
- Y. Liu, C. Zhang, D. Hao, Z. Zhang, L. Wu, M. Li, S. Feng, X. Xu, F. Liu, X. Chen and Z. Bo, Enhancing the performance of organic solar cells by hierarchically supramolecular self-assembly of fused-ring electron acceptors, Chem. Mater., 2018, 30, 4307–4312 CrossRef CAS
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Energy-level modulation of small-molecule electron acceptors to achieve over 12% efficiency in polymer solar cells, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed
- H. Yao, L. Ye, J. Hou, B. Jang, G. Han, Y. Cui, G. M. Su, C. Wang, B. Gao, R. Yu, H. Zhang, Y. Yi, H. Y. Woo, H. Ade and J. Hou, Achieving highly efficient nonfullerene organic solar cells with improved intermolecular interaction and open-circuit voltage, Adv. Mater., 2017, 29, 1700254 CrossRef PubMed
- D. Xie, T. Liu, W. Gao, C. Zhong, L. Huo, Z. Luo, K. Wu, W. Xiong, F. Liu, Y. Sun and C. Yang, A novel thiophene-fused ending group enabling an excellent small molecule acceptor for high-performance fullerene-free polymer solar cells with 11.8% efficiency, Sol. RRL, 2017, 1, 1700044 CrossRef
- Z. Luo, H. Bin, T. Liu, Z. G. Zhang, Y. Yang, C. Zhong, B. Qiu, G. Li, W. Gao, D. Xie, K. Wu, Y. Sun, F. Liu, Y. Li and C. Yang, Fine-tuning of molecular packing and energy level through methyl substitution enabling excellent small molecule acceptors for nonfullerene polymer solar cells with efficiency up to 12.54, Adv. Mater., 2018, 30, 1706124 CrossRef PubMed
- S. Dai, F. Zhao, Q. Zhang, T. K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, Fused nonacyclic electron acceptors for efficient polymer solar cells, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C. J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, A facile planar fused-ring electron acceptor for as-cast polymer solar cells with 8.71% efficiency, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C. H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, High-efficiency and air-stable P3HT-based polymer solar cells with a new non-fullerene acceptor, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, High-performance electron acceptor with thienyl side chains for organic photovoltaics, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Single-junction binary-blend nonfullerene polymer solar cells with 12.1% efficiency, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed
- Y. Yang, Z. G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, Side-chain isomerization on an n-type organic semiconductor itic acceptor makes 11.77% high efficiency polymer solar cells, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed
- J. Wang, W. Wang, X. Wang, Y. Wu, Q. Zhang, C. Yan, W. Ma, W. You and X. Zhan, Enhancing performance of nonfullerene acceptors via side-chain conjugation strategy, Adv. Mater., 2017, 29, 1702125 CrossRef PubMed
- J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu, S. R. Marder and X. Zhan, Effect of isomerization on high-performance nonfullerene electron acceptors, J. Am. Chem. Soc., 2018, 140, 9140–9147 CrossRef CAS PubMed
- B. Jia, J. Wang, Y. Wu, M. Zhang, Y. Jiang, Z. Tang, T. P. Russell and X. Zhan, Enhancing the performance of a fused-ring electron acceptor by unidirectional extension, J. Am. Chem. Soc., 2019, 141, 19023–19031 CrossRef CAS PubMed
- Y. Lin and X. Zhan, Oligomer molecules for efficient organic photovoltaics, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed
- S. Chandrabose, K. Chen, A. J. Barker, J. J. Sutton, S. K. K. Prasad, J. Zhu, J. Zhou, K. C. Gordon, Z. Xie, X. Zhan and J. M. Hodgkiss, High exciton diffusion coefficients in fused ring electron acceptor films, J. Am. Chem. Soc., 2019, 141, 6922–6929 CrossRef CAS PubMed
- Y. Lin, F. Zhao, S. K. K. Prasad, J. D. Chen, W. Cai, Q. Zhang, K. Chen, Y. Wu, W. Ma, F. Gao, J. X. Tang, C. Wang, W. You, J. M. Hodgkiss and X. Zhan, Balanced partnership between donor and acceptor components in nonfullerene organic solar cells with >12% efficiency, Adv. Mater., 2018, 30, e1706363 CrossRef PubMed
- S. Li, L. Zhan, F. Liu, J. Ren, M. Shi, C.-Z. Li, T. P. Russell and H. Chen, An unfused-core-based nonfullerene acceptor enables high-efficiency organic solar cells with excellent morphological stability at high temperatures, Adv. Mater., 2018, 30, 1705208 CrossRef PubMed
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, Exploiting noncovalently conformational locking as a design strategy for high performance fused-ring electron acceptor used in polymer solar cells, J. Am. Chem. Soc., 2017, 139, 3356–3359 CrossRef CAS PubMed
- S. Yu, Y. Chen, L. Yang, P. Ye, J. Wu, J. Yu, S. Zhang, Y. Gao and H. Huang, Significant enhancement of photovoltaic performance through introducing S[three dots, centered]N conformational locks, J. Mater. Chem. A, 2017, 5, 21674–21678 RSC
- L. Zhan, S. Li, H. Zhang, F. Gao, T.-K. Lau, X. Lu, D. Sun, P. Wang, M. Shi, C.-Z. Li and H. Chen, A near-infrared photoactive morphology modifier leads to significant current improvement and energy loss mitigation for ternary organic solar cells, Adv. Sci., 2018, 5, 1800755 CrossRef PubMed
- L. Zhan, S. Li, S. Zhang, X. Chen, T.-K. Lau, X. Lu, M. Shi, C.-Z. Li and H. Chen, Enhanced charge transfer between fullerene and non-fullerene acceptors enables highly efficient ternary organic solar cells, ACS Appl. Mater. Interfaces, 2018, 10, 42444–42452 CrossRef CAS PubMed
- S. Li, L. Zhan, W. Zhao, S. Zhang, B. Ali, Z. Fu, T.-K. Lau, X. Lu, M. Shi, C.-Z. Li, J. Hou and H. Chen, Revealing the effects of molecular packing on the performances of polymer solar cells based on A-D-C-D-A type non-fullerene acceptors, J. Mater. Chem. A, 2018, 6, 12132–12141 RSC
- N. Wang, L. Zhan, S. Li, M. Shi, T.-K. Lau, X. Lu, R. Shikler, C.-Z. Li and H. Chen, Enhancement of intra- and inter-molecular π-conjugated effects for a non-fullerene acceptor to achieve high-efficiency organic solar cells with an extended photoresponse range and optimized morphology, Mater. Chem. Front., 2018, 2, 2006–2012 RSC
- R. Qin, W. Yang, S. Li, T.-K. Lau, Z. Yu, Z. Liu, M. Shi, X. Lu, C.-Z. Li and H. Chen, Enhanced intramolecular charge transfer of unfused electron acceptors for efficient organic solar cells, Mater. Chem. Front., 2019, 3, 513–519 RSC
- Z. Zhang, S. Zhang, Z. Liu, Z.-G. Zhang, Y. Li, C.-Z. Li and H. Chen, A simple electron acceptor with unfused backbone for polymer solar cells, Acta Phys.-Chim. Sin., 2019, 35, 394–400 Search PubMed
- S. Li, L. Zhan, C. Sun, H. Zhu, G. Zhou, W. Yang, M. Shi, C.-Z. Li, J. Hou, Y. Li and H. Chen, Highly efficient fullerene-free organic solar cells operate at near zero highest occupied molecular orbital offsets, J. Am. Chem. Soc., 2019, 141, 3073–3082 CrossRef CAS PubMed
- K. Zhao, L. Xue, J. Liu, X. Gao, S. Wu, Y. Han and Y. Geng, A new method to improve poly(3-hexyl thiophene) (P3HT) crystalline behavior: decreasing chains entanglement to promote order–disorder transformation in solution, Langmuir, 2010, 26, 471–477 CrossRef CAS PubMed
- Y. Xu, J. Liu, H. Wang, X. Yu, R. Xing and Y. Han, Formation of parallel aligned nano-fibrils of a donor–acceptor conjugated copolymer via controlling J-aggregates and post treatment, Soft Matter, 2013, 9, 9849–9856 RSC
- H. Wang, L. Chen, R. Xing, J. Liu and Y. Han, Simultaneous control over both molecular order and long-range alignment in films of the donor–acceptor copolymer, Langmuir, 2015, 31, 469–479 CrossRef CAS PubMed
- J. Liu, Y. Sun, X. Gao, R. Xing, L. Zheng, S. Wu, Y. Geng and Y. Han, Oriented poly(3-hexylthiophene) nanofibril with the π - π stacking growth direction by solvent directional evaporation, Langmuir, 2011, 27, 4212–4219 CrossRef CAS PubMed
- Q. Liang, J. Han, C. Song, X. Yu, D.-M. Smilgies, K. Zhao, J. Liu and Y. Han, Reducing the confinement of PBDB-T to ITIC to improve the crystallinity of PBDB-T/ITIC blends, J. Mater. Chem. A, 2018, 6, 15610–15620 RSC
- Q. Liang, X. Jiao, Y. Yan, Z. Xie, G. Lu, J. Liu and Y. Han, separating crystallization process of P3HT and O-IDTBR to construct highly crystalline interpenetrating network with optimized vertical phase separation, Adv. Funct. Mater., 2019, 29, 1807591 CrossRef CAS
- R. Zhang, H. Yang, K. Zhou, J. Zhang, X. Yu, J. Liu and Y. Han, Molecular orientation and phase separation by controlling chain segment and molecule movement in P3HT/N2200 blends, Macromolecules, 2016, 49, 6987–6996 CrossRef CAS
- H. Yang, R. Zhang, L. Wang, J. D. Zhang, X. H. Yu, J. G. Liu, R. B. Xing, Y. H. Geng and Y. C. Han, Face-on and edge-on orientation transition and self-epitaxial crystallization of all-conjugated diblock copolymer, Macromolecules, 2015, 48, 7557–7566 CrossRef CAS
- C. Duan, K. Zhang, C. Zhong, F. Huang and Y. Cao, Recent advances in water/alcohol-soluble pi-conjugated materials: new materials and growing applications in solar cells, Chem. Soc. Rev., 2013, 42, 9071–9104 RSC
- Z. Hu, K. Zhang, F. Huang and Y. Cao, Water/alcohol soluble conjugated polymers for the interface engineering of highly efficient polymer light-emitting diodes and polymer solar cells, Chem. Commun., 2015, 51, 5572–5585 RSC
- Z. Hu, F. Huang and Y. Cao, Layer-by-layer assembly of multilayer thin films for organic optoelectronic devices, Small, Methods, 2017, 1, 1700264 Search PubMed
- K. Zhang, Z. Hu, C. Sun, Z. Wu, F. Huang and Y. Cao, Toward solution-processed high-performance polymer solar cells: from material design to device engineering, Chem. Mater., 2016, 29, 141–148 CrossRef
-
Z. C. Hu, L. Ying, F. Huang and Y. Cao, Towards a bright future: polymer solar cells with power conversion efficiencies over 10%, Sci. China: Chem., 2017, 60, 571–582 Search PubMed
- C.-C. Chueh, C.-Z. Li and A. K. Y. Jen, Recent progress and perspective in solution-processed Interfacial materials for efficient and stable polymer and organometal perovskite solar cells, Energy Environ. Sci., 2015, 8, 1160–1189 RSC
- F. Huang, H. Wu, D. Wang, W. Yang and Y. Cao, Novel electroluminescent conjugated polyelectrolytes based on polyfluorene, Chem. Mater., 2004, 16, 708–716 CrossRef CAS
- H. Zhou, Y. Zhang, C. K. Mai, S. D. Collins, T. Q. Nguyen, G. C. Bazan and A. J. Heeger, Conductive conjugated polyelectrolyte as hole-transporting layer for organic bulk heterojunction solar cells, Adv. Mater., 2014, 26, 780–785 CrossRef CAS PubMed
- B. Xu, Z. Zheng, K. Zhao and J. Hou, A bifunctional interlayer material for modifying both the anode and cathode in highly efficient polymer solar cells, Adv. Mater., 2016, 28, 434–439 CrossRef CAS PubMed
- Z. C. He, C. M. Zhong, S. J. Su, M. Xu, H. B. Wu and Y. Cao, Enhanced power-conversion efficiency in polymer solar cells using an inverted device structure, Nat. Photonics, 2012, 6, 591–595 CrossRef
- K. Zhang, C. Zhong, S. Liu, C. Mu, Z. Li, H. Yan, F. Huang and Y. Cao, Highly efficient inverted polymer solar cells based on a cross-linkable water-/alcohol-soluble conjugated polymer interlayer, ACS Appl. Mater. Interfaces, 2014, 6, 10429–10435 CrossRef CAS PubMed
- J. Wang, K. Lin, K. Zhang, X.-F. Jiang, K. Mahmood, L. Ying, F. Huang and Y. Cao, Crosslinkable amino-functionalized conjugated polymer as cathode interlayer for efficient inverted polymer solar cells, Adv. Energy Mater., 2016, 6, 1502563 CrossRef
- S. Liu, K. Zhang, J. Lu, J. Zhang, H. L. Yip, F. Huang and Y. Cao, High-efficiency polymer solar cells via the incorporation of an amino-functionalized conjugated metallopolymer as a cathode interlayer, J. Am. Chem. Soc., 2013, 135, 15326–15329 CrossRef CAS PubMed
- Z. Wu, C. Sun, S. Dong, X. F. Jiang, S. Wu, H. Wu, H. L. Yip, F. Huang and Y. Cao, n-Type water/alcohol-soluble naphthalene diimide-based conjugated polymers for high-performance polymer solar cells, J. Am. Chem. Soc., 2016, 138, 2004–2013 CrossRef CAS PubMed
- Z. Hu, R. Xu, S. Dong, K. Lin, J. Liu, F. Huang and Y. Cao, Quaternisation-polymerized N-type polyelectrolytes: synthesis, characterisation and application in high-performance polymer solar cells, Mater. Horiz., 2017, 4, 88–97 RSC
- Z. Wang, N. Zheng, W. Zhang, H. Yan, Z. Xie, Y. Ma, F. Huang and Y. Cao, Self-doped, n-type perylene diimide derivatives as electron transporting layers for high-efficiency polymer solar cells, Adv. Energy Mater., 2017, 7, 1700232 CrossRef
- Z. Chen, Z. Hu, Z. Wu, X. Liu, Y. Jin, M. Xiao, F. Huang and Y. Cao, Counterion-tunable n-type conjugated polyelectrolytes for the interface engineering of efficient polymer solar cells, J. Mater. Chem. A, 2017, 5, 19447–19455 RSC
- C. Sun, Z. Wu, Z. Hu, J. Xiao, W. Zhao, H.-W. Li, Q.-Y. Li, S.-W. Tsang, Y.-X. Xu, K. Zhang, H.-L. Yip, J. Hou, F. Huang and Y. Cao, Interface design for high-efficiency non-fullerene polymer solar cells, Energy Environ. Sci., 2017, 10, 1784–1791 RSC
- S. Dong, Z. Hu, K. Zhang, Q. Yin, X. Jiang, F. Huang and Y. Cao, Cross-linkable and dual functional hybrid polymeric electron transporting layer for high-performance inverted polymer solar cells, Adv. Mater., 2017, 29, 1701507 CrossRef PubMed
- K. Zhang, B. Fan, R. Xia, X. Liu, Z. Hu, H. Gu, S. Liu, H.-L. Yip, L. Ying, F. Huang and Y. Cao, Highly efficient tandem organic solar cell enabled by environmentally friendly solvent processed polymeric interconnecting layer, Adv. Energy Mater., 2018, 8, 1703180 CrossRef
- K. Zhang, K. Gao, R. Xia, Z. Wu, C. Sun, J. Cao, L. Qian, W. Li, S. Liu, F. Huang, X. Peng, L. Ding, H. L. Yip and Y. Cao, High-performance polymer tandem solar cells employing a new n-type conjugated polymer as an interconnecting layer, Adv. Mater., 2016, 28, 4817–4823 CrossRef CAS PubMed
- K. Zhang, R. Xia, B. Fan, X. Liu, Z. Wang, S. Dong, H. L. Yip, L. Ying, F. Huang and Y. Cao, 11.2% All-polymer tandem solar cells with simultaneously improved efficiency and stability, Adv. Mater., 2018, 30, 1803166 CrossRef PubMed
- C. Sun, Z. Wu, H.-L. Yip, H. Zhang, X.-F. Jiang, Q. Xue, Z. Hu, Z. Hu, Y. Shen, M. Wang, F. Huang and Y. Cao, Amino-functionalized conjugated polymer as an efficient electron transport layer for high-performance planar-heterojunction perovskite solar cells, Adv. Energy Mater., 2016, 6, 1501534 CrossRef
- L. Tian, Z. Hu, X. Liu, Z. Liu, P. Guo, B. Xu, Q. Xue, H. L. Yip, F. Huang and Y. Cao, Fluoro- and amino-functionalized conjugated polymers as electron transport materials for perovskite solar cells with improved efficiency and stability, ACS Appl. Mater. Interfaces, 2019, 11, 5289–5297 CrossRef CAS PubMed
- Y. Meng, H. Wu, Y. Zhang and Z. Wei, A flexible electrode based on a three-dimensional graphene network-supported polyimide for lithium-ion batteries, J. Mater. Chem. A, 2014, 2, 10842–10846 RSC
- J. Hecht, The bandwidth bottleneck that is throttling the Internet, Nature, 2016, 536, 139–142 CrossRef CAS PubMed
- V. N. Egorov, V. L. Masalov, Y. A. Nefyodov, A. F. Shevchun, M. R. Trunin, V. E. Zhitomirsky and M. McLean, Dielectric constant, loss tangent, and surface resistance of PCB materials at K-band frequencies, IEEE Trans. Microw. Theory Techn., 2005, 53, 627–635 CAS
- B. Lee, Y. H. Park, Y. T. Hwang, W. Oh, J. Yoon and M. Ree, Ultralow-k nanoporous organosilicate dielectric films imprinted with dendritic spheres, Nat. Mater., 2005, 4, 147–151 CrossRef CAS PubMed
- T. M. Long and T. M. Swager, Molecular design of free volume as a route to low-kappa dielectric materials, J. Am. Chem. Soc., 2003, 125, 14113–14119 CrossRef CAS PubMed
- Y. Liu, Y. Zhang, Q. Lan, S. Liu, Z. Qin, L. Chen, C. Zhao, Z. Chi, J. Xu and J. Economy, high-performance functional polyimides containing rigid nonplanar conjugated triphenylethylene moieties, Chem. Mater., 2012, 24, 1212–1222 CrossRef CAS
- Y. Liu, Y. Zhang, Q. Lan, Z. Qin, S. Liu, C. Zhao, Z. Chi and J. Xu, Synthesis and properties of high-performance functional polyimides containing rigid nonplanar conjugated tetraphenylethylene moieties, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1302–1314 CrossRef CAS
- W. Chen, Z. Zhou, T. Yang, R. Bei, Y. Zhang, S. Liu, Z. Chi, X. Chen and J. Xu, Synthesis and properties of highly organosoluble and low dielectric constant polyimides containing non-polar bulky triphenyl methane moiety, React. Funct. Polym., 2016, 108, 71–77 CrossRef CAS
- R. Bei, C. Qian, Y. Zhang, Z. Chi, S. Liu, X. Chen, J. Xu and M. P. Aldred, Intrinsic low dielectric constant polyimides: relationship between molecular structure and dielectric properties, J. Mater. Chem. C, 2017, 5, 12807–12815 RSC
- Y. Liu, Z. Zhou, L. Qu, B. Zou, Z. Chen, Y. Zhang, S. Liu, Z. Chi, X. Chen and J. Xu, Exceptionally thermostable and soluble aromatic polyimides with special characteristics: intrinsic ultralow dielectric constant, static random access memory behaviors, transparency and fluorescence, Mater. Chem. Front., 2017, 1, 326–337 RSC
- Y.-W. Liu, L.-S. Tang, L.-J. Qu, S.-W. Liu, Z.-G. Chi, Y. Zhang and J.-R. Xu, Synthesis and properties of high performance functional polyimides containing rigid nonplanar conjugated fluorene moieties, Chin. J. Polym. Sci., 2019, 37, 416–427 CrossRef CAS
- Y. Liu, C. Qian, L. Qu, Y. Wu, Y. Zhang, X. Wu, B. Zou, W. Chen, Z. Chen, Z. Chi, S. Liu, X. Chen and J. Xu, A bulk dielectric polymer film with intrinsic ultralow dielectric constant and outstanding comprehensive properties, Chem. Mater., 2015, 27, 6543–6549 CrossRef CAS
- Q. Zeng, Z. Li, Z. Li, C. Ye, J. Qin and B. Z. Tang, Convenient attachment of highly polar azo chromophore moieties to disubstituted polyacetylene through polymer reactions by using “Click” chemistry, Macromolecules, 2007, 40, 5634–5637 CrossRef CAS
- W. Wu, S. Xin, Z. Xu, C. Ye, J. Qin and Z. Li, Main-chain second-order nonlinear optical polyaryleneethynylenes containing isolation chromophores: enhanced nonlinear optical properties, improved optical transparency and stability, Polym. Chem., 2013, 4, 3196 RSC
- Z. Li, W. Wu, Q. Li, G. Yu, L. Xiao, Y. Liu, C. Ye, J. Qin and Z. Li, High-generation second-order nonlinear optical (NLO) dendrimers: convenient synthesis by click chemistry and the increasing trend of NLO effects, Angew. Chem., Int. Ed., 2010, 49, 2763–2767 CrossRef CAS PubMed
- R. Tang, S. Zhou, W. Xiang, Y. Xie, H. Chen, Q. Peng, G. Yu, B. Liu, H. Zeng, Q. Li and Z. Li, New “X-type” second-order nonlinear optical (NLO) dendrimers: fewer chromophore moieties and high NLO effects, J. Mater. Chem. C, 2015, 3, 4545–4552 RSC
- W. Wu, C. Ye, G. Yu, Y. Liu, J. Qin and Z. Li, New hyperbranched polytriazoles containing isolation chromophore moieties derived from ab4 monomers through click chemistry under copper(i) catalysis: improved optical transparency and enhanced NLO effects, Chem. – Eur. J., 2012, 18, 4426–4434 CrossRef CAS PubMed
- W. Wu, R. Tang, Q. Li and Z. Li, Functional hyperbranched polymers with advanced optical, electrical and magnetic properties, Chem. Soc. Rev., 2015, 44, 3997–4022 RSC
- R. Tang, S. Zhou, Z. Cheng, G. Yu, Q. Peng, H. Zeng, G. Guo, Q. Li and Z. Li, Janus second-order nonlinear optical dendrimers: their controllable molecular topology and corresponding largely enhanced performance, Chem. Sci., 2017, 8, 340–347 RSC
- W. B. Wu, R. Xiao, W. D. Xiang, Z. P. Wang and Z. Li, Main chain dendronized polymers: design, synthesis, and application in the second-order nonlinear optical (NLO) area, J. Phys. Chem. C, 2015, 119, 14281–14287 CAS
- W. Wu, Z. Xu and Z. Li, Using low generation dendrimers as monomers to construct dendronized hyperbranched polymers with high nonlinear optical performance, J. Mater. Chem. C, 2014, 2, 8122–8130 RSC
- R. Tang, H. Chen, S. Zhou, W. Xiang, X. Tang, B. Liu, Y. Dong, H. Zeng and Z. Li, Dendronized hyperbranched polymers containing isolation chromophores: design, synthesis and further enhancement of the comprehensive NLO performance, Polym. Chem., 2015, 6, 5580–5589 RSC
- J. Li, X. Jin, Y. Liu, F. Li, L. Zhang, X. Zhu and Y. Lu, Robust enzyme-silica composites made from enzyme nanocapsules, Chem. Commun., 2015, 51, 9628–9631 RSC
- J. Du, J. Jin, M. Yan and Y. Lu, Synthetic nanocarriers for intracellular protein delivery, Curr. Drug Metab., 2012, 13, 82–92 CrossRef CAS PubMed
- Y. Liu, J. Du, M. Yan, M. Y. Lau, J. Hu, H. Han, O. O. Yang, S. Liang, W. Wei, H. Wang, J. Li, X. Zhu, L. Shi, W. Chen, C. Ji and Y. Lu, Biomimetic enzyme nanocomplexes and their use as antidotes and preventive measures for alcohol intoxication, Nat. Nanotechnol., 2013, 8, 187–192 CrossRef CAS
- Z. Zhang, Y. Gu, Q. Liu, C. Zheng, L. Xu, Y. An, X. Jin, Y. Liu and L. Shi, Spatial confined synergistic enzymes with enhanced uricolytic performance and reduced toxicity for effective gout treatment, Small, 2018, 14, 1801865 CrossRef
- S. Liang, Y. Liu, X. Jin, G. Liu, J. Wen, L. Zhang, J. Li, X. Yuan, I. S. Y. Chen, W. Chen, H. Wang, L. Shi, X. Zhu and Y. Lu, Phosphorylcholine polymer nanocapsules prolong the circulation time and reduce the immunogenicity of therapeutic proteins, Nano Res., 2016, 9, 1022–1031 CrossRef CAS
- X. Zhang, W. Chen, X. Zhu and Y. Lu, Encapsulating therapeutic proteins with polyzwitterions for lower macrophage nonspecific uptake and longer circulation time, ACS Appl. Mater. Interfaces, 2017, 9, 7972–7978 CrossRef CAS PubMed
- X. Zhang, D. Xu, X. Jin, G. Liu, S. Liang, H. Wang, W. Chen, X. Zhu and Y. Lu, Nanocapsules of therapeutic proteins with enhanced stability and long blood circulation for hyperuricemia management, J. Controlled Release, 2017, 255, 54–61 CrossRef CAS PubMed
- S. Liang, X. Jin, Y. F. Ma, J. Guo and H. Wang, Folic acid-conjugated BSA nanocapsule (n-BSA-FA) for cancer targeted radiotherapy and imaging, RSC Adv., 2015, 5, 88560–88566 RSC
- N. Wang, X. Jin, D. Guo, G. Tong and X. Zhu, Iron chelation nanoparticles with delayed saturation as an effective therapy for parkinson disease, Biomacromolecules, 2016, 18, 461–474 CrossRef PubMed
- N. Wang, X. Jin and X. Zhu, Construction of biomimetic long-circulation delivery platform encapsulated by zwitterionic polymers for enhanced penetration of blood–brain barrier, RSC Adv., 2017, 7, 20766–20778 RSC
- N. Wang, P. Sun, M. Lv, G. Tong, X. Jin and X. Zhu, Mustard-inspired delivery shuttle for enhanced blood–brain barrier penetration and effective drug delivery in glioma therapy, Biomater. Sci., 2017, 5, 1041–1050 RSC
- D. Chen, Y. Huang, H. Jiang, W. Yasen, D. Guo, Y. Su, B. Xue, X. Jin and X. Zhu, Fabrication of activity-reporting glucose oxidase nanocapsules with oxygen-independent fluorescence variation, ACS Appl. Mater. Interfaces, 2018, 10, 26005–26015 CrossRef CAS PubMed
- X. Duan, L. Liu, F. Feng and S. Wang, Cationic conjugated polymers for optical detection of DNA methylation, lesions, and single nucleotide polymorphisms, Acc. Chem. Res., 2010, 43, 260–270 CrossRef CAS
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Water-soluble conjugated polymers for imaging, diagnosis, and therapy, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed
- J. Wang, F. Lv, L. Liu, Y. Ma and S. Wang, Strategies to design conjugated polymer based materials for biological sensing and imaging, Coord. Chem. Rev., 2018, 354, 135–154 CrossRef CAS
- J. Li, C. Xie, J. Huang, Y. Jiang, Q. Miao and K. Pu, Semiconducting polymer nanoenzymes with photothermic activity for enhanced cancer therapy, Angew. Chem., Int. Ed., 2018, 57, 3995–3998 CrossRef CAS
- Q. Yang, Y. Dong, W. Wu, C. Zhu, H. Chong, J. Lu, D. Yu, L. Liu, F. Lv and S. Wang, Detection and differential diagnosis of colon cancer by a cumulative analysis of promoter methylation, Nat. Commun., 2012, 3, 1206 CrossRef PubMed
- L. Feng, L. Liu, F. Lv, G. C. Bazan and S. Wang, Preparation and biofunctionalization of multicolor conjugated polymer nanoparticles for imaging and detection of tumor cells, Adv. Mater., 2014, 26, 3926–3930 CrossRef CAS PubMed
- S. Li, T. Chen, Y. Wang, L. Liu, F. Lv, Z. Li, Y. Huang, K. S. Schanze and S. Wang, Conjugated polymer with intrinsic alkyne units for synergistically enhanced Raman imaging in living cells, Angew. Chem., Int. Ed., 2017, 56, 13455–13458 CrossRef CAS
- H. Yuan, H. Chong, B. Wang, C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chemical molecule-induced light-activated system for anticancer and antifungal activities, J. Am. Chem. Soc., 2012, 134, 13184–13187 CrossRef CAS PubMed
- S. Liu, H. Yuan, H. Bai, P. Zhang, F. Lv, L. Liu, Z. Dai, J. Bao and S. Wang, Electrochemiluminescence for electric-driven antibacterial therapeutics, J. Am. Chem. Soc., 2018, 140, 2284–2291 CrossRef CAS PubMed
- H. Chen, Z. Gu, H. An, C. Chen, J. Chen, R. Cui, S. Chen, W. Chen, X. Chen, X. Chen, Z. Chen, B. Ding, Q. Dong, Q. Fan, T. Fu, D. Hou, Q. Jiang, H. Ke, X. Jiang, G. Liu, S. Li, T. Li, Z. Liu, G. Nie, M. Ovais, D. Pang, N. Qiu, Y. Shen, H. Tian, C. Wang, H. Wang, Z. Wang, H. Xu, J.-F. Xu, X. Yang, S. Zhu, X. Zheng, X. Zhang, Y. Zhao, W. Tan, X. Zhang and Y. Zhao, Precise nanomedicine for intelligent therapy of cancer, Sci. China: Chem., 2018, 61, 1503–1552 CrossRef CAS
- F. Danhier, To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine?, J. Controlled Release, 2016, 244, 108–121 CrossRef CAS
- Q. Sun, Z. Zhou, N. Qiu and Y. Shen, Rational design of cancer nanomedicine: nanoproperty integration and synchronization, Adv. Mater., 2017, 29, 1606628 CrossRef PubMed
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Cancer nanomedicine: progress, challenges and opportunities, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed
- Z. Zhou, X. Liu, D. Zhu, Y. Wang, Z. Zhang, X. Zhou, N. Qiu, X. Chen and Y. Shen, Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration, Adv. Drug Delivery Rev., 2017, 115, 115–154 CrossRef CAS PubMed
- L. He, J. Liu, S. Li, X. Feng, C. Wang, X. Zhuang, J. Ding and X. Chen, Polymer nanoplatforms at work in prostate cancer therapy, Adv. Ther., 2019, 2, 1800122 CrossRef
- R. Sun, N. Qiu and Y. Shen, Polymeric cancer nanomedicines: challenge and development, Acta Polym. Sin., 2019, 50, 288–601 Search PubMed
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed
- C. F. Xu, H. B. Zhang, C. Y. Sun, Y. Liu, S. Shen, X. Z. Yang, Y. H. Zhu and J. Wang, Tumor acidity-sensitive linkage-bridged block copolymer for therapeutic siRNA delivery, Biomaterials, 2016, 88, 48–59 CrossRef CAS PubMed
- Y. Y. Yuan, C. Q. Mao, X. J. Du, J. Z. Du, F. Wang and J. Wang, Surface charge switchable nanoparticles based on zwitterionic polymer for enhanced drug delivery to tumor, Adv. Mater., 2012, 24, 5476–5480 CrossRef CAS PubMed
- J. Wei, X. Shuai, R. Wang, X. He, Y. Li, M. Ding, J. Li, H. Tan and Q. Fu, Clickable and imageable multiblock polymer micelles with magnetically guided and PEG-switched targeting and release property for precise tumor theranosis, Biomaterials, 2017, 145, 138–153 CrossRef CAS PubMed
- N. Song, M. Ding, Z. Pan, J. Li, L. Zhou, H. Tan and Q. Fu, Construction of targeting-clickable and tumor-cleavable polyurethane nanomicelles for multifunctional intracellular drug delivery, Biomacromolecules, 2013, 14, 4407–4419 CrossRef CAS PubMed
- C. Zhao, H. Deng, J. Xu, S. Li, L. Zhong, L. Shao, Y. Wu and X.-J. Liang, “Sheddable” PEG-lipid to balance the contradiction of PEGylation between long circulation and poor uptake, Nanoscale, 2016, 8, 10832–10842 RSC
- S. Liu, J. Pan, J. Liu, Y. Ma, F. Qiu, L. Mei, X. Zeng and G. Pan, Dynamically PEGylated and borate-coordination-polymer-coated polydopamine nanoparticles for synergetic tumor-targeted, chemo-photothermal combination therapy, Small, 2018, 14, 1703968 CrossRef PubMed
- M. R. Gordon, B. Zhao, F. Anson, A. Fernandez, K. Singh, C. Homyak, M. Canakci, R. W. Vachet and S. Thayumanavan, Matrix metalloproteinase-9-responsive nanogels for proximal surface conversion and activated cellular uptake, Biomacromolecules, 2018, 19, 860–871 CrossRef CAS PubMed
- J. Li, C. Sun, W. Tao, Z. Cao, H. Qian, X. Yang and J. Wang, Photoinduced PEG deshielding from ROS-sensitive linkage-bridged block copolymer-based nanocarriers for on-demand drug delivery, Biomaterials, 2018, 170, 147–155 CrossRef CAS PubMed
- Q. Zhou, Y. Hou, L. Zhang, J. Wang, Y. Qiao, S. Guo, L. Fan, T. Yang, L. Zhu and H. Wu, Dual-pH sensitive charge-reversal nanocomplex for tumor-targeted drug delivery with enhanced anticancer activity, Theranostics, 2017, 7, 1806–1819 CrossRef CAS PubMed
- V. A. Sethuraman and Y. H. Bae, TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors, J. Controlled Release, 2007, 118, 216–224 CrossRef CAS PubMed
- W.-H. Chen, G.-F. Luo, W.-X. Qiu, Q. Lei, S. Hong, S.-B. Wang, D.-W. Zheng, C.-H. Zhu, X. Zeng, J. Feng, S.-X. Cheng and X.-Z. Zhang, Programmed nanococktail for intracellular cascade reaction regulating self-synergistic tumor targeting therapy, Small, 2016, 12, 733–744 CrossRef CAS PubMed
- C.-Y. Sun, S. Shen, C.-F. Xu, H.-J. Li, Y. Liu, Z.-T. Cao, X.-Z. Yang, J.-X. Xia and J. Wang, Tumor acidity-sensitive polymeric vector for active targeted siRNA delivery, J. Am. Chem. Soc., 2015, 137, 15217–15224 CrossRef CAS PubMed
- E. Jin, B. Zhang, X. Sun, Z. Zhou, X. Ma, Q. Sun, J. Tang, Y. Shen, E. Van Kirk, W. J. Murdoch and M. Radosz, Acid-Active cell-penetrating peptides for in vivo tumor-targeted drug delivery, J. Am. Chem. Soc., 2013, 135, 933–940 CrossRef CAS PubMed
- Y. Yang, X. Xie, Y. Yang, Z. Li, F. Yu, W. Gong, Y. Li, H. Zhang, Z. Wang and X. Mei, Polymer nanoparticles modified with photo- and pH-dual-responsive polypeptides for enhanced and targeted cancer therapy, Mol. Pharmaceutics, 2016, 13, 1508–1519 CrossRef CAS PubMed
- J. Wu, H. Han, Q. Jin, Z. Li, H. Li and J. Ji, Design and proof of programmed 5-aminolevulinic acid prodrug nanocarriers for targeted photodynamic cancer therapy, ACS Appl. Mater. Interfaces, 2017, 9, 14596–14605 CrossRef CAS PubMed
- L. Zhu, P. Kate and V. P. Torchilin, Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting, ACS Nano, 2012, 6, 3491–3498 CrossRef CAS PubMed
- Y. Yang, Y. Yang, X. Xie, Z. Wang, W. Gong, H. Zhang, Y. Li, F. Yu, Z. Li and X. Mei, Dual-modified liposomes with a two-photon-sensitive cell penetrating peptide and NGR ligand for siRNA targeting delivery, Biomaterials, 2015, 48, 84–96 CrossRef CAS PubMed
- H. Han, D. Valdeperez, Q. Jin, B. Yang, Z. Li, Y. Wu, B. Pelaz, W. J. Parak and J. Ji, Dual enzymatic reaction-assisted gemcitabine delivery systems for programmed pancreatic cancer therapy, ACS Nano, 2017, 11, 1281–1291 CrossRef CAS PubMed
- Z. Zou, X. He, D. He, K. Wang, Z. Qing, X. Yang, L. Wen, J. Xiong, L. Li and L. Cai, Programmed packaging of mesoporous silica nanocarriers for matrix metalloprotease 2-triggered tumor targeting and release, Biomaterials, 2015, 58, 35–45 CrossRef CAS PubMed
- Z. Zhou, Y. Shen, J. Tang, E. Jin, X. Ma, Q. Sun, B. Zhang, E. A. Van Kirk and W. J. Murdoch, Linear polyethyleneimine-based charge-reversal nanoparticles for nuclear-targeted drug delivery, J. Mater. Chem., 2011, 21, 19114–19123 RSC
- P. Xu, E. A. Van Kirk, Y. Zhan, W. J. Murdoch, M. Radosz and Y. Shen, Targeted charge-reversal nanoparticles for nuclear drug delivery, Angew. Chem., Int. Ed., 2007, 46, 4999–5002 CrossRef CAS PubMed
- J. Li, X. Yu, Y. Wang, Y. Yuan, H. Xiao, D. Cheng and X. Shuai, A reduction and ph dual-sensitive polymeric vector for long-circulating and tumor-targeted siRNA delivery, Adv. Mater., 2014, 26, 8217–8224 CrossRef CAS PubMed
- Z. Zhou, W. J. Murdoch and Y. Shen, A linear polyethylenimine (LPEI) drug conjugate with reversible charge to overcome multidrug resistance in cancer cells, Polymer, 2015, 76, 150–158 CrossRef CAS
- S.-S. Han, Z.-Y. Li, J.-Y. Zhu, K. Han, Z.-Y. Zeng, W. Hong, W.-X. Li, H.-Z. Jia, Y. Liu, R.-X. Zhuo and X.-Z. Zhang, Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery, Small, 2015, 11, 2543–2554 CrossRef CAS PubMed
- S. Chen, L. Rong, Q. Lei, P.-X. Cao, S.-Y. Qin, D.-W. Zheng, H.-Z. Jia, J.-Y. Zhu, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, A surface charge-switchable and folate modified system for co-delivery of proapoptosis peptide and p53 plasmid in cancer therapy, Biomaterials, 2016, 77, 149–163 CrossRef CAS PubMed
- J. J. Chen, J. X. Ding, Y. C. Wang, J. J. Cheng, S. X. Ji, X. L. Zhuang and X. S. Chen, Sequentially responsive shell-stacked nanoparticles for deep penetration into solid tumors, Adv. Mater., 2017, 29, 1701170 CrossRef PubMed
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS PubMed
- J.-Z. Du, T.-M. Sun, W.-J. Song, J. Wu and J. Wang, A tumor-acidity-activated charge-conversional nanogel as an intelligent vehicle for promoted tumoral-cell uptake and drug delivery, Angew. Chem., Int. Ed., 2010, 49, 3621–3626 CrossRef CAS PubMed
- Y. Li, J. Yang, B. Xu, F. Gao, W. Wang and W. Liu, Enhanced therapeutic siRNA to Tumor cells by a pH-sensitive agmatine-chitosan bioconjugate, ACS Appl. Mater. Interfaces, 2015, 7, 8114–8124 CrossRef CAS PubMed
- Z. X. Zhou, Y. Q. Shen, J. B. Tang, M. H. Fan, E. A. Van Kirk, W. J. Murdoch and M. Radosz, Charge-reversal drug conjugate for targeted cancer cell nuclear drug delivery, Adv. Funct. Mater., 2009, 19, 3580–3589 CrossRef CAS
- J. Z. Du, C. Q. Mao, Y. Y. Yuan, X. Z. Yang and J. Wang, Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy, Biotechnol. Adv., 2014, 32, 789–803 CrossRef CAS PubMed
- Y. Shen, Y. Zhan, J. Tang, P. Xu, P. A. Johnson, M. Radosz, E. A. Van Kirk and W. J. Murdoch, Multifunctioning pH-responsive nanoparticles from hierarchical self-assembly of polymer brush for cancer drug delivery, AIChE J., 2008, 54, 2979–2989 CrossRef CAS
- W. He, X. Zheng, Q. Zhao, L. Duan, Q. Lv, G. H. Gao and S. Yu, pH-triggered charge-reversal polyurethane micelles for controlled release of doxorubicin, Macromol. Biosci., 2016, 16, 925–935 CrossRef CAS PubMed
- D. Hu, Z. Xu, Z. Hu, B. Hu, M. Yang and L. Zhu, pH-triggered charge-reversal silk sericin-based nanoparticles for enhanced cellular uptake and doxorubicin delivery, ACS Sustainable Chem. Eng., 2017, 5, 1638–1647 CrossRef CAS
- S. Lv, W. Song, Z. Tang, M. Li, H. Yu, H. Hong and X. Chen, Charge-conversional PEG-polypeptide polyionic complex nanoparticles from simple blending of a pair of oppositely charged block copolymers as an intelligent vehicle for efficient antitumor drug delivery, Mol. Pharmaceutics, 2014, 11, 1562–1574 CrossRef CAS PubMed
- P. P. Pillai, S. Huda, B. Kowalczyk and B. A. Grzybowski, Controlled pH stability and adjustable cellular uptake of mixed-charge nanoparticles, J. Am. Chem. Soc., 2013, 135, 6392–6395 CrossRef CAS PubMed
- Y. Huang, Z. Tang, X. Zhang, H. Yu, H. Sun, X. Pang and X. Chen, pH-triggered charge-reversal polypeptide nanoparticles for cisplatin delivery: preparation and in vitro evaluation, Biomacromolecules, 2013, 14, 2023–2032 CrossRef CAS PubMed
- Z. Wang, G. Ma, J. Zhang, Z. Yuan, L. Wang, M. Bernards and S. Chen, Surface protonation/deprotonation controlled instant affinity switch of nano drug vehicle (NDV) for pH triggered tumor cell targeting, Biomaterials, 2015, 62, 116–127 CrossRef CAS PubMed
- S. J. Tseng, Z. X. Liao, S. H. Kao, Y. F. Zeng, K. Y. Huang, H. J. Li, C. L. Yang, Y. F. Deng, C. F. Huang, S. C. Yang, P. C. Yang and I. M. Kempson, Highly specific in vivo gene delivery for p53-mediated apoptosis and genetic photodynamic therapies of tumour, Nat. Commun., 2015, 6, 6456 CrossRef CAS PubMed
- Q. Jin, Y. Deng, X. Chen and J. Ji, Rational design of cancer nanomedicine for simultaneous stealth surface and enhanced cellular uptake, ACS Nano, 2019, 13, 954–977 CAS
- S. Mitragotri and J. Lahann, Physical approaches to biomaterial design, Nat. Mater., 2009, 8, 15–23 CrossRef CAS PubMed
- M. Gaumet, A. Vargas, R. Gurny and F. Delie, Nanoparticles for drug delivery: The need for precision in reporting particle size parameters, Eur. J. Pharm. Biopharm., 2008, 69, 1–9 CrossRef CAS PubMed
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, The role of micelle size in tumor accumulation, penetration, and treatment, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS PubMed
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed
- Q. Sun, X. Sun, X. Ma, Z. Zhou, E. Jin, B. Zhang, Y. Shen, E. A. Van Kirk, W. J. Murdoch, J. R. Lott, T. P. Lodge, M. Radosz and Y. Zhao, Integration of nanoassembly functions for an effective delivery cascade for cancer drugs, Adv. Mater., 2014, 26, 7615–7621 CrossRef CAS PubMed
- Q. H. Sun, X. P. Ma, B. Zhang, Z. X. Zhou, E. L. Jin, Y. Q. Shen, E. A. Van Kirk, W. J. Murdoch, M. Radosz and W. L. Sun, Fabrication of dendrimer-releasing lipidic nanoassembly for cancer drug delivery, Biomater. Sci., 2016, 4, 958–969 RSC
- H.-J. Li, J.-Z. Du, J. Liu, X.-J. Du, S. Shen, Y.-H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, Smart superstructures with ultrahigh pH-sensitivity for targeting acidic tumor microenvironment: instantaneous size switching and improved tumor penetration, ACS Nano, 2016, 10, 6753–6761 CrossRef CAS PubMed
- H.-J. Li, J.-Z. Du, X.-J. Du, C.-F. Xu, C.-Y. Sun, H.-X. Wang, Z.-T. Cao, X.-Z. Yang, Y.-H. Zhu, S. Nie and J. Wang, Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS PubMed
- Y. Shen, X. Ma, B. Zhang, Z. Zhou, Q. Sun, E. Jin, M. Sui, J. Tang, J. Wang and M. Fan, Degradable dual pH- and temperature-responsive photoluminescent dendrimers, Chem. – Eur. J., 2011, 17, 5319–5326 CrossRef CAS PubMed
- M. Han, M.-Y. Huang-Fu, W.-W. Guo, N.-N. Guo, J. Chen, H.-N. Liu, Z.-Q. Xie, M.-T. Lin, Q.-C. Wei and J.-Q. Gao, MMP-2-sensitive HA end-conjugated poly(amidoamine) dendrimers via click reaction to enhance drug penetration into solid tumor, ACS Appl. Mater. Interfaces, 2017, 9, 42459–42470 CrossRef CAS PubMed
- R. Chen, Q. Chen, H. Qin and D. Xing, A photoacoustic shockwave triggers the size shrinkage of nanoparticles to obviously improve tumor penetration and therapeutic efficacy, Nanoscale, 2019, 11, 1423–1436 RSC
- T. Lang, Y. Liu, Z. Zheng, W. Ran, Y. Zhai, Q. Yin, P. Zhang and Y. Li, Cocktail strategy based on spatio-temporally controlled nano device improves therapy of breast cancer, Adv. Mater., 2018, 31, 1806202 CrossRef PubMed
- L. Mo, Z. Zhao, X. Hu, X. Yu, Y. Peng, H. Liu, M. Xiong, T. Fu, Y. Jiang, X. Zhang and W. Tan, Smart nanodrug with nuclear localization sequences in the presence of mmp-2 to overcome biobarriers and drug resistance, Chem. – Eur. J., 2019, 25, 1895–1900 CrossRef CAS PubMed
- S. Ruan, X. Cao, X. Cun, G. Hu, Y. Zhou, Y. Zhang, L. Lu, Q. He and H. Gao, Matrix metalloproteinase-sensitive size-shrinkable nanoparticles for deep tumor penetration and pH triggered doxorubicin release, Biomaterials, 2015, 60, 100–110 CrossRef CAS PubMed
- C. Wong, T. Stylianopoulos, J. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popovic, R. K. Jain, M. G. Bawendi and D. Fukumura, Multistage nanoparticle delivery system for deep penetration into tumor tissue, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426–2431 CrossRef CAS PubMed
- Z. Cao, Y. Ma, C. Sun, Z. Lu, Z. Yao, J. Wang, D. Li, Y. Yuan and X. Yang, ROS-sensitive polymeric nanocarriers with red light-activated size shrinkage for remotely controlled drug release, Chem. Mater., 2018, 30, 517–525 CrossRef CAS
- Q. Y. Feng and R. Tong, Anticancer nanoparticulate polymer–drug conjugate, Bioeng. Transl. Med., 2016, 1, 277–296 CrossRef CAS PubMed
- J. Bhattacharyya, J. J. Bellucci, I. Weitzhandler, J. R. McDaniel, I. Spasojevic, X. Li, C.-C. Lin, J.-T. A. Chi and A. Chilkoti, A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer
models, Nat. Commun., 2015, 6, 7939 CrossRef CAS PubMed
- T. Etrych, L. Daumova, E. Pokorna, D. Tuskova, O. Lidicky, V. Kolarova, J. Pankrac, L. Sefc, P. Chytil and P. Klener, Effective doxorubicin-based nano-therapeutics for simultaneous malignant lymphoma treatment and lymphoma growth imaging, J. Controlled Release, 2018, 289, 44–55 CrossRef CAS PubMed
- K. D. Zhao, D. Li, W. G. Xu, J. X. Ding, W. Q. Jiang, M. Q. Li, C. X. Wang and X. S. Chen, Targeted hydroxyethyl starch prodrug for inhibiting the growth and metastasis of prostate cancer, Biomaterials, 2017, 116, 82–94 CrossRef CAS PubMed
- D. Sun, J. Ding, C. Xiao, J. Chen, X. Zhuang and X. Chen, pH-responsive reversible pegylation improves performance of antineoplastic agent, Adv. Healthcare Mater., 2015, 4, 844–855 CrossRef CAS PubMed
- Z. Zhou, X. Ma, C. J. Murphy, E. Jin, Q. Sun, Y. Shen, E. A. Van Kirk and W. J. Murdoch, Molecularly precise dendrimer-drug conjugates with tunable drug release for cancer therapy, Angew. Chem., Int. Ed., 2014, 53, 10949–10955 CrossRef CAS PubMed
- X. Ling, J. Tu, J. Wang, A. Shajii, N. Kong, C. Feng, Y. Zhang, M. Yu, T. Xie, Z. Bharwani, B. M. Aljaeid, B. Shi, W. Tao and O. C. Farokhzad, Glutathione-responsive prodrug nanoparticles for effective drug delivery and cancer therapy, ACS Nano, 2019, 13, 357–370 CrossRef CAS PubMed
- Y. Su, Y. Hu, Y. Du, X. Huang, J. He, J. You, H. Yuan and F. Hu, Redox-responsive polymer drug conjugates based on doxorubicin and chitosan oligosaccharide-g-stearic acid for cancer therapy, Mol. Pharmaceutics, 2015, 12, 1193–1202 CrossRef CAS PubMed
- Y. Zhang, Q. Guo, S. An, Y. Lu, J. Li, X. He, L. Liu, Y. Zhang, T. Sun and C. Jiang, ROS-switchable polymeric nanoplatform with stimuli-responsive release for active targeted drug delivery to breast cancer, ACS Appl. Mater. Interfaces, 2017, 9, 12227–12240 CrossRef CAS PubMed
- J. Wang, S. Hu, W. Mao, J. Xiang, Z. Zhou, X. Liu, J. Tang and Y. Shen, Assemblies of peptide-cytotoxin conjugates for tumor-homing chemotherapy, Adv. Funct. Mater., 2019, 29, 1807446 CrossRef
- Z. Zhou, J. Tang, Q. Sun, W. J. Murdoch and Y. Shen, A multifunctional PEG-PLL drug conjugate forming redox-responsive nanoparticles for intracellular drug delivery, J. Mater. Chem. B, 2015, 3, 7594–7603 RSC
- I. Rosenbaum, A. J. Harnoy, E. Tirosh, M. Buzhor, M. Segal, L. Frid, R. Shaharabani, R. Avinery, R. Beck and R. J. Amir, Encapsulation and covalent binding of molecular payload in enzymatically activated micellar nanocarriers, J. Am. Chem. Soc., 2015, 137, 2276–2284 CrossRef CAS PubMed
- D. Zhang, J. Zhang, Q. Li, H. L. Tian, N. Zhang, Z. H. Li and Y. X. Luan, pH- and enzyme-sensitive ir820-paclitaxel conjugate self-assembled nanovehicles for near-infrared fluorescence imaging-guided chemo-photothermal therapy, ACS Appl. Mater. Interfaces, 2018, 10, 30092–30102 CrossRef CAS PubMed
- Z. X. Zhou, W. J. Murdoch and Y. Q. Shen, Synthesis of an esterase-sensitive degradable polyester as facile drug carrier for cancer therapy, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 507–515 CrossRef CAS
- Z. Li, L. Qiu, Q. Chen, T. Hao, M. Qiao, H. Zhao, J. Zhang, H. Hu, X. Zhao, D. Chen and L. Mei, pH-sensitive nanoparticles of poly(L-histidine)-poly(lactide-co-glycolide)-tocopheryl polyethylene glycol succinate for anti-tumor drug delivery, Acta Biomater., 2015, 11, 137–150 CrossRef CAS PubMed
- Y. J. Cheng, G. F. Luo, J. Y. Zhu, X. D. Xu, X. Zeng, D. B. Cheng, Y. M. Li, Y. Wu, X. Z. Zhang, R. X. Zhuo and F. He, Enzyme-induced and tumor-targeted drug delivery system based on multifunctional mesoporous silica nanoparticles, ACS Appl. Mater. Interfaces, 2015, 7, 9078–9087 CrossRef CAS PubMed
- C. Wang, S. Chen, Y. Wang, X. Liu, F. Hu, J. Sun and H. Yuan, Lipase-triggered water-responsive “pandora's box” for cancer therapy: toward induced neighboring effect and enhanced drug penetration, Adv. Mater., 2018, 30, 1706407 CrossRef PubMed
- K. Cheng, Y. Ding, Y. Zhao, S. Ye, X. Zhao, Y. Zhang, T. Ji, H. Wu, B. Wang, G. J. Anderson, L. Ren and G. Nie, Sequentially responsive therapeutic peptide assembling nanoparticles for dual-targeted cancer immunotherapy, Nano Lett., 2018, 18, 3250–3258 CrossRef CAS PubMed
- J. Rao and A. Khan, Enzyme sensitive synthetic polymer micelles based on the azobenzene motif, J. Am. Chem. Soc., 2013, 135, 14056–14059 CrossRef CAS PubMed
- C. Z. Sun, Y. Liang, N. Hao, L. Xu, F. R. Cheng, T. Su, J. Cao, W. X. Gao, Y. J. Pu and B. He, A ROS-responsive polymeric micelle with a pi-conjugated thioketal moiety for enhanced drug loading and efficient drug delivery, Org. Biomol. Chem., 2017, 15, 9176–9185 RSC
- W. G. Xu, J. X. Ding and X. S. Chen, Reduction-responsive polypeptide micelles for intracellular delivery of antineoplastic agent, Biomacromolecules, 2017, 18, 3291–3301 CrossRef CAS PubMed
- R. Mo, T. Jiang, R. DiSanto, W. Tai and Z. Gu, ATP-triggered anticancer drug delivery, Nat. Commun., 2014, 5, 3364 CrossRef PubMed
- Y. Yuan, C. Du, C. Sun, J. Zhu, S. Wu, Y. Zhang, T. Ji, J. Lei, Y. Yang, N. Gao and G. Nie, Chaperonin-GroEL as a smart hydrophobic drug delivery and tumor targeting molecular machine for tumor therapy, Nano Lett., 2018, 18, 921–928 CrossRef CAS PubMed
- C. Jiang, Z. Qi, H. Jia, Y. Huang, Y. Wang, W. Zhang, Z. Wu, H. Yang and J. Liu, ATP-responsive low-molecular-weight polyethylenimine-based supramolecular assembly via host–guest interaction for gene delivery, Biomacromolecules, 2019, 20, 478–489 CrossRef CAS PubMed
- Q. Zhou, Y. Wang, J. Xiang, Y. Piao, Z. Zhou, J. Tang, X. Liu and Y. Shen, Stabilized calcium phosphate hybrid nanocomposite using a benzoxaborole-containing polymer for pH-responsive siRNA delivery, Biomater. Sci., 2018, 6, 3178–3188 RSC
- Z. Jiang, Y. Wang, L. Sun, B. Yuan, Y. Tian, L. Xiang, Y. Li, Y. Li, J. Li and A. Wu, Dual ATP and pH responsive ZIF-90 nanosystem with favorable biocompatibility and facile post-modification improves therapeutic outcomes of triple negative breast cancer in vivo, Biomaterials, 2019, 197, 41–50 CrossRef CAS PubMed
- M. Ye, Y. Han, J. Tang, Y. Piao, X. Liu, Z. Zhou, J. Gao, J. Rao and Y. Shen, A Tumor-specific cascade amplification drug release nanoparticle for overcoming multidrug resistance
in cancers, Adv. Mater., 2017, 29, 1702342 CrossRef PubMed
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, S. Bhasin, Y. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. Shi and O. C. Farokhzad, ROS-responsive polyprodrug nanoparticles for triggered drug delivery and effective cancer therapy, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed
- S. Wang, Z. Wang, G. Yu, Z. Zhou, O. Jacobson, Y. Liu, Y. Ma, F. Zhang, Z. Y. Chen and X. Chen, Tumor-specific drug release and reactive oxygen species generation for cancer chemo/chemodynamic combination therapy, Adv. Sci., 2019, 6, 1801986 CrossRef PubMed
- Y. L. Dai, S. Y. Cheng, Z. L. Wang, R. L. Zhang, Z. Yang, J. J. Wang, B. C. Yung, Z. T. Wang, O. Jacobson, C. Xu, Q. Q. Ni, G. C. Yu, Z. J. Zhou and X. Y. Chen, Hypochlorous acid promoted platinum drug chemotherapy by myeloperoxidase-encapsulated therapeutic metal phenolic nanoparticles, ACS Nano, 2018, 12, 455–463 CrossRef CAS PubMed
- J. Li, A. Dirisala, Z. Ge, Y. Wang, W. Yin, W. Ke, K. Toh, J. Xie, Y. Matsumoto, Y. Anraku, K. Osada and K. Kataoka, Therapeutic vesicular nanoreactors with tumor-specific activation and self-destruction for synergistic tumor ablation, Angew. Chem., Int. Ed., 2017, 56, 14025–14030 CrossRef CAS PubMed
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Non-viral vectors for gene-based therapy, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed
- M. L. Forrest, J. T. Koerber and D. W. Pack, A degradable polyethylenimine derivative with low toxicity for highly efficient gene delivery, Bioconjugate Chem., 2003, 14, 934–940 CrossRef CAS PubMed
- A. Akinc, D. M. Lynn, D. G. Anderson and R. Langer, Parallel synthesis and biophysical characterization of a degradable polymer library for gene delivery, J. Am. Chem. Soc., 2003, 125, 5316–5323 CrossRef CAS PubMed
- X. Deng, N. Zheng, Z. Song, L. Yin and J. Cheng, Trigger-responsive, fast-degradable poly(beta-amino ester)s for enhanced DNA unpackaging and reduced toxicity, Biomaterials, 2014, 35, 5006–5015 CrossRef CAS PubMed
- Y. Yuan, C.-J. Zhang and B. Liu, A Photoactivatable AIE polymer for light-controlled gene delivery: concurrent endo/lysosomal escape and DNA unpacking, Angew. Chem., Int. Ed., 2015, 54, 11419–11423 CrossRef CAS PubMed
- Y. Y. He, Y. Nie, G. Cheng, L. Xie, Y. Q. Shen and Z. W. Gu, Viral mimicking ternary polyplexes: a reduction-controlled hierarchical unpacking vector for gene delivery, Adv. Mater., 2014, 26, 1534–1540 CrossRef CAS PubMed
- D. C. Zhu, H. J. Yan, X. Liu, J. J. Xiang, Z. X. Zhou, J. B. Tang, X. R. Liu and Y. Q. Shen, Intracellularly disintegratable polysulfoniums for efficient gene delivery, Adv. Funct. Mater., 2017, 27, 1606826 CrossRef
- X. Liu, J. Xiang, D. Zhu, L. Jiang, Z. Zhou, J. Tang, X. Liu, Y. Huang and Y. Shen, Fusogenic reactive oxygen species triggered charge-reversal vector for effective gene delivery, Adv. Mater., 2016, 28, 1743–1752 CrossRef CAS PubMed
- N. S. Qiu, X. R. Liu, Y. Zhong, Z. X. Zhou, Y. Piao, L. Miao, Q. Z. Zhang, J. B. Tang, L. Huang and Y. Q. Shen, Esterase-activated charge-reversal polymer for fibroblast-exempt cancer gene
therapy, Adv. Mater., 2016, 28, 10613–10622 CrossRef CAS PubMed
- N. S. Qiu, J. Q. Gao, Q. Liu, J. Q. Wang and Y. Q. Shen, Enzyme-responsive charge-reversal polymer-mediated effective gene therapy for intraperitoneal tumors, Biomacromolecules, 2018, 19, 2308–2319 CrossRef CAS PubMed
- J. Xiang, X. Liu, Z. Zhou, D. Zhu, Q. Zhou, Y. Piao, L. Jiang, J. Tang, X. Liu and Y. Shen, Reactive oxygen species (ROS)-responsive charge-switchable nanocarriers for gene therapy of metastatic cancer, ACS Appl. Mater. Interfaces, 2018, 10, 43352–43362 CrossRef CAS PubMed
- D. W. Zheng, L. Xu, C. X. Li, X. Dong, P. Pan, Q. L. Zhang, B. Li, X. Zeng and X. Z. Zhang, Photo-powered artificial organelles for atp generation and life-sustainment, Adv. Mater., 2018, 30, 1805038 CrossRef PubMed
- D. Zheng, B. Li, L. Xu, Q. L. Zhang, J. X. Fan, C. X. Li and X. Z. Zhang, Normalizing tumor microenvironment based on photosynthetic abiotic/biotic nanoparticles, ACS Nano, 2018, 12, 6218–6227 CrossRef CAS PubMed
- X. Q. Wang, F. Gao and X. Z. Zhang, Initiator-loaded gold nanocages as a light-induced free-radical generator for cancer therapy, Angew. Chem., Int. Ed., 2017, 56, 9029–9033 CrossRef CAS PubMed
- D. W. Zheng, Y. Chen, Z. H. Li, L. Xu, C. X. Li, B. Li, J. X. Fan, S. X. Cheng and X. Z. Zhang, Optically-controlled bacterial metabolite for cancer therapy, Nat. Commun., 2018, 9, 1680 CrossRef PubMed
- J. X. Fan, Z. H. Li, X. H. Liu, D. W. Zheng, Y. Chen and X. Z. Zhang, Bacteria-mediated tumor therapy utilizing photothermally-controlled TNF-alpha expression via oral administration, Nano Lett., 2018, 18, 2373–2380 CrossRef CAS PubMed
- W. L. Liu, T. Liu, M. Z. Zou, W. Y. Yu, C. X. Li, Z. Y. He, M. K. Zhang, M. D. Liu, Z. H. Li, J. Feng and X. Z. Zhang, Aggressive man-made red blood cells for hypoxia-resistant photodynamic therapy, Adv. Mater., 2018, 30, 1802006 CrossRef PubMed
- N. Ma, M. K. Zhang, X. S. Wang, L. Zhang, J. Feng and X. Z. Zhang, NIR light-triggered degradable mote2 nanosheets for combined photothermal and chemotherapy of cancer, Adv. Funct. Mater., 2018, 28, 1801139 CrossRef
- W. Song, J. Kuang, C. X. Li, M. K. Zhang, D. W. Zheng, X. Zeng, C. J. Liu and X. Z. Zhang, Enhanced immunotherapy based on photodynamic therapy for both primary and lung metastasis tumor eradication, ACS Nano, 2018, 12, 1978–1989 CrossRef CAS PubMed
- J. Y. Zhu, X. Zeng, S. Y. Qin, S. S. Wan, H. Z. Jia, R. X. Zhuo, J. Feng and X. Z. Zhang, Acidity-responsive gene delivery for “superfast” nuclear translocation and transfection with high efficiency, Biomaterials, 2016, 83, 79–92 CrossRef CAS PubMed
- D. W. Zheng, S. Hong, L. Xu, C. X. Li, K. Li, S. X. Cheng and X. Z. Zhang, Hierarchical micro-/nanostructures from human hair for biomedical applications, Adv. Mater., 2018, 30, e1800836 CrossRef PubMed
- X. D. Xu, P. E. Saw, W. Tao, Y. J. Li, X. Y. Ji, S. Bhasin, Y. L. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. J. Shi and O. C. Farokhzad, ROS-responsive polyprodrug nanoparticles for triggered drug delivery and effective cancer therapy, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed
- X. L. Hu, J. M. Hu, J. Tian, Z. S. Ge, G. Y. Zhang, K. F. Luo and S. Y. Liu, Polyprodrug amphiphiles: hierarchical assemblies for shape-regulated cellular internalization, trafficking, and drug delivery, J. Am. Chem. Soc., 2013, 135, 17617–17629 CrossRef CAS PubMed
- U. Hasegawa, A. J. van der Vlies, C. Wandrey and J. A. Hubbell, Preparation of well-defined ibuprofen prodrug micelles by RAFT polymerization, Biomacromolecules, 2013, 14, 3314–3320 CrossRef CAS PubMed
- X. L. Hu, G. H. Liu, Y. Li, X. R. Wang and S. Y. Liu, Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals, J. Am. Chem. Soc., 2015, 137, 362–368 CrossRef CAS PubMed
- X. L. Hu, S. D. Zhai, G. H. Liu, D. Xing, H. J. Liang and S. Y. Liu, Concurrent drug unplugging and permeabilization of polyprodrug-gated crosslinked vesicles for cancer combination chemotherapy, Adv. Mater., 2018, 30, 1706307 CrossRef PubMed
- J. J. Tan, Z. Y. Deng, G. H. Liu, J. M. Hu and S. Y. Liu, Anti-inflammatory polymersomes of redox-responsive polyprodrug amphiphiles with inflammation-triggered indomethacin release characteristics, Biomaterials, 2018, 178, 608–619 CrossRef CAS PubMed
- K. N. Zhu, G. H. Liu, J. M. Hu and S. Y. Liu, Near-infrared light-activated photochemical internalization of reduction-responsive polyprodrug vesicles for synergistic photodynamic therapy and chemotherapy, Biomacromolecules, 2017, 18, 2571–2582 CrossRef CAS PubMed
- B. Louage, L. Nuhn, M. D. P. Risseeuw, N. Vanparijs, R. De Coen, I. Karalic, S. Van Calenbergh and B. G. De Geest, Well-defined polymer-paclitaxel prodrugs by a grafting-from-drug approach, Angew. Chem., Int. Ed., 2016, 55, 11791–11796 CrossRef CAS PubMed
- P. Huang, D. L. Wang, Y. Su, W. Huang, Y. F. Zhou, D. X. Cui, X. Y. Zhu and D. Y. Yan, Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug-drug conjugate for cancer therapy, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed
- A. E. Rodda, L. Meagher, D. R. Nisbet and J. S. Forsythe, Specific control of cell–material interactions: Targeting cell receptors using ligand-functionalized polymer substrates, Prog. Polym. Sci., 2014, 39, 1312–1347 CrossRef CAS
- T. Sun, G. Qing, B. Su and L. Jiang, Functional biointerface materials inspired from nature, Chem. Soc. Rev., 2011, 40, 2909–2921 RSC
- D. Li, Q. Zheng, Y. Wang and H. Chen, Combining surface topography with polymer chemistry: exploring new interfacial biological phenomena, Polym. Chem., 2013, 5, 14–24 RSC
- K. von der Mark and J. Park, Engineering biocompatible implant surfaces, Prog. Mater. Sci., 2013, 58, 327–381 CrossRef CAS
- B. S. Gomes, B. Simões and P. M. Mendes, The increasing dynamic, functional complexity of bio-interface materials, Nat. Rev. Chem., 2018, 2, 0120 CrossRef
- J. J. Richardson, J. Cui, M. Bjornmalm, J. A. Braunger, H. Ejima and F. Caruso, Innovation in layer-by-layer assembly, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H. A. Klok, Surface-initiated controlled radical polymerization: state-of-the-art, opportunities, and challenges in surface and interface engineering with polymer brushes, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed
- Q. Wei and R. Haag, Universal polymer coatings and their representative biomedical applications, Mater. Horiz., 2015, 2, 567–577 RSC
- J. H. Ryu, P. B. Messersmith and H. Lee, Polydopamine surface chemistry: a decade of discovery, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed
- H. Ejima, J. J. Richardson and F. Caruso, Metal-phenolic networks as a versatile platform to engineer nanomaterials and biointerfaces, Nano Today, 2017, 12, 136–148 CrossRef CAS
- L. Moroni, M. Klein Gunnewiek and E. M. Benetti, Polymer brush coatings regulating cell behavior: passive interfaces turn into active, Acta Biomater., 2014, 10, 2367–2378 CrossRef CAS PubMed
- X. Liu and S. Wang, Three-dimensional nano-biointerface as a new platform for guiding cell fate, Chem. Soc. Rev., 2014, 43, 2385–2401 RSC
- D. Radke, W. Jia, D. Sharma, K. Fena, G. Wang, J. Goldman and F. Zhao, Tissue engineering at the blood-contacting surface: a review of challenges and strategies in vascular graft development, Adv. Healthcare Mater., 2018, 7, 1701461 CrossRef PubMed
- I. Reviakine, F. Jung, S. Braune, J. L. Brash, R. Latour, M. Gorbet and W. van Oeveren, Stirred, shaken, or stagnant: what goes on at the blood-biomaterial interface, Blood Rev., 2017, 31, 11–21 CrossRef CAS PubMed
- T. Wei, Z. Tang, Q. Yu and H. Chen, Smart antibacterial surfaces with switchable bacteria-killing and bacteria-releasing capabilities, ACS Appl. Mater. Interfaces, 2017, 9, 37511–37523 CrossRef CAS PubMed
- X. Ding, S. Duan, X. Ding, R. Liu and F.-J. Xu, Versatile antibacterial materials: an emerging arsenal for combatting bacterial pathogens, Adv. Funct. Mater., 2018, 28, 1802140 CrossRef
- Y. Zhu, K. Kekalo, C. Ndong, Y.-Y. Huang, F. Shubitidze, K. E. Griswold, I. Baker and J. X. J. Zhang, Magnetic-nanoparticle-based immunoassays-on-chip: materials synthesis, surface functionalization, and Cancer Cell Screening, Adv. Funct. Mater., 2016, 26, 3953–3972 CrossRef CAS
- M. W. Tibbitt, J. E. Dahlman and R. Langer, Emerging frontiers in drug delivery, J. Am. Chem. Soc., 2016, 138, 704–717 CrossRef CAS PubMed
- Y. Wang and D. S. Kohane, External triggering and triggered targeting strategies for drug delivery, Nat. Rev. Mater., 2017, 2, 17020 CrossRef CAS
- H. Chen, L. Yuan, W. Song, Z. Wu and D. Li, Biocompatible polymer materials: Role of protein-surface interactions, Prog. Polym. Sci., 2008, 33, 1059–1087 CrossRef CAS
- M. Rabe, D. Verdes and S. Seeger, Understanding protein adsorption phenomena at solid surfaces, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed
- R. A. Latour, Molecular simulation of protein-surface interactions: Benefits, problems, solutions, and future directions, Biointerphases, 2008, 3, FC2–FC12 CrossRef CAS PubMed
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Protein interactions with polymer coatings and biomaterials, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed
- T. A. Horbett, The role of adsorbed proteins in animal-cell adhesion, Colloids Surf., B, 1994, 2, 225–240 CrossRef CAS
- C. J. Wilson, R. E. Clegg, D. I. Leavesley and M. J. Pearcy, Mediation of biomaterial-cell interactions by adsorbed proteins: A review, Tissue Eng., 2005, 11, 1–18 CrossRef CAS PubMed
- P. Jonkheijm, D. Weinrich, H. Schroder, C. M. Niemeyer and H. Waldmann, Chemical strategies for generating protein biochips, Angew. Chem., Int. Ed., 2008, 47, 9618–9647 CrossRef CAS PubMed
- C. D. Walkey and W. C. W. Chan, Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC
- S. Jiang and Z. Cao, Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed
- B. K. D. Ngo and M. A. Grunlan, Protein resistant polymeric biomaterials, ACS Macro Lett., 2017, 6, 992–1000 CrossRef CAS
- L. Yuan, Q. Yu, D. Li and H. Chen, Surface modification to control protein-surface interaction, Macromol. Biosci., 2011, 11, 1031–1040 CrossRef CAS PubMed
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Stimuli-responsive interfaces and systems for the control of protein–surface and cell–surface interactions, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS PubMed
- Q. Yu, Y. Zhang, H. Wang, J. Brash and H. Chen, Anti-fouling bioactive surfaces, Acta Biomater., 2011, 7, 1550–1557 CrossRef CAS PubMed
- Y. Luan, D. Li, Y. Wang, X. Liu, J. L. Brash and H. Chen,
125I-radiolabeling, surface plasmon resonance, and quartz crystal microbalance with dissipation: three tools to compare protein adsorption on surfaces of different wettability, Langmuir, 2014, 30, 1029–1035 CrossRef CAS PubMed
- Y. Luan, D. Li, T. Wei, M. Wang, Z. Tang, J. L. Brash and H. Chen, “Hearing loss” in qcm measurement of protein adsorption to protein resistant polymer brush layers, Anal. Chem., 2017, 89, 4184–4191 CrossRef CAS PubMed
- X. Liu, L. Yuan, D. Li, Z. Tang, Y. Wang, G. Chen, H. Chen and J. L. Brash, Blood compatible materials: state of the art, J. Mater. Chem. B, 2014, 2, 5178–5738 Search PubMed
- B. D. Ippel and P. Y. W. Dankers, Introduction of nature's complexity in engineered blood-compatible biomaterials, Adv. Healthcare Mater., 2018, 7, 1700505 CrossRef PubMed
- D. Li, H. Chen and J. L. Brash, Mimicking the fibrinolytic system on material surfaces, Colloids Surf., B, 2011, 86, 1–6 CrossRef CAS PubMed
- D. Li, H. Chen, S. Wang, Z. Wu and J. L. Brash, Lysine-poly(2-hydroxyethyl methacrylate) modified polyurethane surface with high lysine density and fibrinolytic activity, Acta Biomater., 2011, 7, 954–958 CrossRef CAS PubMed
- D. Li, H. Chen, W. G. McClung and J. L. Brash, Lysine-PEG-modified polyurethane as a fibrinolytic surface: effect of PEG chain length on protein interactions, platelet interactions and clot lysis, Acta Biomater., 2009, 5, 1864–1871 CrossRef CAS PubMed
- Z. Tang, X. Liu, Y. Luan, W. Liu, Z. Wu, D. Li and H. Chen, Regulation of fibrinolytic protein adsorption on polyurethane surfaces by modification with lysine-containing copolymers, Polym. Chem., 2013, 4, 5597–5602 RSC
- Z. Tang, D. Li, X. Liu, Z. Wu, W. Liu, J. L. Brash and H. Chen, Vinyl-monomer with lysine side chains for preparing copolymer surfaces with fibrinolytic activity, Polym. Chem., 2013, 4, 1583–1589 RSC
- X. Shi, W. Zhan, G. Chen, Q. Yu, Q. Liu, H. Du, L. Cao, X. Liu, L. Yuan and H. Chen, Regulation of protein-binding capability of surfaces via host–guest interactions: effects of localized and average ligand density, Langmuir, 2015, 31, 6172–6178 CrossRef CAS PubMed
- W. Zhan, X. Shi, Q. Yu, Z. Lyu, L. Cao, H. Du, Q. Liu, X. Wang, G. Chen, D. Li, J. L. Brash and H. Chen, Bioinspired blood compatible surface having combined fibrinolytic and vascular endothelium-like properties via a sequential coimmobilization strategy, Adv. Funct. Mater., 2015, 25, 5206–5213 CrossRef CAS
- D. Li, S. Wang, Z. Wu, H. Chen and J. L. Brash, A new t-PA releasing concept based on protein–protein displacement, Soft Matter, 2013, 9, 2321–2328 RSC
- Z. Tang, Y. Luan, D. Li, H. Du, D. Haddleton and H. Chen, Surface immobilization of protease through an inhibitor-derived affinity ligand: a bioactive surface with defensive properties against inhibitor, Chem. Commun., 2015, 51, 14263–14266 RSC
- Z. Tang, D. Li, Y. Luan, L. Zhu, H. Du, Y. Tao, Y. Wang, D. Haddleton and H. Chen, Conjugation of polymers to proteins through an inhibitor-derived peptide: taking up the inhibitor “berth”, Chem. Commun., 2015, 51, 10099–10102 RSC
- Q. Liu, D. Li, W. Zhan, Y. Luan, H. Du, X. Liu, J. L. Brash and H. Chen, Surface having dual affinity for plasminogen and tissue plasminogen activator: in situ plasmin generation and clot lysis, J. Mater. Chem. B, 2015, 3, 6939–6944 RSC
- C. Li, H. Du, A. Yang, S. Jiang, Z. Li, D. Li, J. L. Brash and H. Chen, Thrombosis-responsive thrombolytic coating based on thrombin-degradable tissue plasminogen activator (t-PA) nanocapsules, Adv. Funct. Mater., 2017, 27, 1703934 CrossRef
- H. Du, C. Li, Y. Luan, Q. Liu, W. Yang, Q. Yu, D. Li, J. L. Brash and H. Chen, An antithrombotic hydrogel with thrombin-responsive fibrinolytic activity: breaking down the clot as it forms, Mater. Horiz., 2016, 3, 556–562 RSC
- Q. Xie, J. Pan, C. Ma and G. Zhang, Dynamic surface antifouling: mechanism and systems, Soft Matter, 2019, 15, 1087–1107 RSC
- M. Qi, Q. Song, J. Zhao, C. Ma, G. Zhang and X. Gong, Three-dimensional bacterial behavior near dynamic surfaces formed by degradable polymers, Langmuir, 2017, 33, 13098–13104 CrossRef CAS PubMed
- C. Ma, L. Xu, W. Xu and G. Zhang, Degradable polyurethane for marine anti-biofouling, J. Mater. Chem. B, 2013, 1, 3099–3106 RSC
- W. Xu, C. Ma, J. Ma, T. Gan and G. Zhang, Marine biofouling resistance of polyurethane with biodegradation and hydrolyzation, ACS Appl. Mater. Interfaces, 2014, 6, 4017–4024 CrossRef CAS PubMed
- J. Ma, C. Ma and G. Zhang, Degradable polymer with protein resistance in a marine environment, Langmuir, 2015, 31, 6471–6478 CrossRef CAS PubMed
- S. Chen, C. Ma and G. Zhang, Biodegradable polymer as controlled release system of organic antifoulant to prevent marine biofouling, Prog. Org. Coat., 2017, 104, 58–63 CrossRef CAS
- C. Ma, W. Zhang, G. Zhang and P.-Y. Qian, Environmentally friendly antifouling coatings based on biodegradable polymer and natural antifoulant, ACS Sustainable Chem. Eng., 2017, 5, 6304–6309 CrossRef CAS
- G. Dai, Q. Xie, S. Chen, C. Ma and G. Zhang, Biodegradable poly(ester)-poly(methyl methacrylate) copolymer for marine anti-biofouling, Prog. Org. Coat., 2018, 124, 55–60 CrossRef CAS
- X. Zhou, Q. Xie, C. Ma, Z. Chen and G. Zhang, Inhibition of marine biofouling by use of degradable and hydrolyzable silyl acrylate copolymer, Ind. Eng. Chem. Res., 2015, 54, 9559–9565 CrossRef CAS
- Q. Xie, C. Ma, G. Zhang and C. Bressy, Poly(ester)–poly(silyl methacrylate) copolymers: synthesis and hydrolytic degradation kinetics, Polym. Chem., 2018, 9, 1448–1454 RSC
- Q. Xie, Q. Xie, J. Pan, C. Ma and G. Zhang, Biodegradable polymer with hydrolysis-induced
zwitterions for antibiofouling, ACS Appl. Mater. Interfaces, 2018, 10, 11213–11220 CrossRef CAS PubMed
- B. Zheng, F. Wang, S. Dong and F. Huang, Supramolecular polymers constructed by crown ether-based molecular recognition, Chem. Soc. Rev., 2012, 41, 1621–1636 RSC
- X. Yan, F. Wang, B. Zheng and F. Huang, Stimuli-responsive supramolecular polymeric materials, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC
- S. Dong, B. Zheng, F. Wang and F. Huang, Supramolecular polymers constructed from macrocycle-based host–guest molecular recognition motifs, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed
- P. Wei, X. Yan and F. Huang, Supramolecular polymers constructed by orthogonal self-assembly based on host–guest and metal–ligand interactions, Chem. Soc. Rev., 2015, 44, 815–832 RSC
- H. Wang, X. Ji, Z. Li and F. Huang, Fluorescent supramolecular polymeric materials, Adv. Mater., 2017, 29, 1606117 CrossRef PubMed
- X. F. Ji, D. Y. Xia, X. Z. Yan, H. Wang and F. H. Huang, Supramolecular polymer materials based on crown ether and pillararene host–guest recognition motifs, Acta Polym. Sin., 2017, 0, 9–18 CAS
- Y. Li, T. Park, J. K. Quansah and S. C. Zimmerman, Synthesis of a redox-responsive quadruple hydrogen-bonding unit for applications in supramolecular chemistry, J. Am. Chem. Soc., 2011, 133, 17118–17121 CrossRef CAS PubMed
- D. C. Gonzalez, E. N. Savariar and S. Thayumanavan, Fluorescence patterns from supramolecular polymer assembly and disassembly for sensing metallo- and nonmetalloproteins, J. Am. Chem. Soc., 2009, 131, 7708–7716 CrossRef CAS PubMed
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, A healable supramolecular polymer blend based on aromatic pi-pi stacking and hydrogen-bonding interactions, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed
- S. Hackelbusch, T. Rossow, H. Becker and S. Seiffert, Multiresponsive polymer hydrogels by orthogonal supramolecular chain cross-linking, Macromolecules, 2014, 47, 4028–4036 CrossRef CAS
- N. Zweep, A. Hopkinson, A. Meetsma, W. R. Browne, B. L. Feringa and J. H. van Esch, Balancing hydrogen bonding and van der Waals interactions in cyclohexane-based bisamide and bisurea organogelators, Langmuir, 2009, 25, 8802–8809 CrossRef CAS PubMed
- Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Water-soluble supramolecular polymerization driven by multiple host-stabilized charge-transfer interactions, Angew. Chem., Int. Ed., 2010, 49, 6576–6579 CrossRef CAS PubMed
- M. Ikeda, K. Fukuda, T. Tanida, T. Yoshii and I. Hamachi, A supramolecular hydrogel containing boronic acid-appended receptor for fluorocolorimetric sensing of polyols with a paper platform, Chem. Commun., 2012, 48, 2716–2718 RSC
- A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, Cancer cell death induced by the intracellular self-assembly of an enzyme-responsive supramolecular gelator, J. Am. Chem. Soc., 2015, 137, 770–775 CrossRef CAS PubMed
- S. Srinivasan, P. A. Babu, S. Mahesh and A. Ajayaghosh, Reversible self-assembly of entrapped fluorescent gelators in polymerized styrene gel matrix: erasable thermal imaging via recreation of supramolecular architectures, J. Am. Chem. Soc., 2009, 131, 15122–15123 CrossRef CAS PubMed
- H. Wang, X. Ji, Z. Li, C. N. Zhu, X. Yang, T. Li, Z. L. Wu and F. Huang, Preparation of a white-light-emitting fluorescent supramolecular polymer gel with a single chromophore and use of the gel to fabricate a protected quick response code, Mater. Chem. Front., 2017, 1, 167–171 RSC
- H. Wang, X. Ji, Y. Li, Z. Li, G. Tang and F. Huang, An ATP/ATPase responsive supramolecular fluorescent hydrogel constructed via electrostatic interactions between poly(sodium p-styrenesulfonate) and a tetraphenylethene derivative, J. Mater. Chem. B, 2018, 6, 2728–2733 RSC
- J. M. Lehn, Dynamic combinatorial chemistry and virtual combinatorial libraries, Chem. – Eur. J., 1999, 5, 2455–2463 CrossRef CAS
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic covalent chemistry, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Dynamic combinatorial chemistry, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed
- A. Wilson, G. Gasparini and S. Matile, Functional systems with orthogonal dynamic covalent bonds, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC
- S. B. Ji, W. Cao, Y. Yu and H. P. Xu, Dynamic diselenide bonds: exchange reaction induced by visible light without catalysis, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed
- S. B. Ji, J. H. Xia and H. P. Xu, Dynamic chemistry of selenium: Se-N and Se-Se dynamic covalent bonds in polymeric systems, ACS Macro Lett., 2016, 5, 9–13 Search PubMed
- J. H. Xia, T. Y. Li, C. J. Lu and H. P. Xu, Selenium-containing polymers: perspectives toward diverse applications in both adaptive and biomedical materials, Macromolecules, 2018, 51, 7435–7455 CrossRef CAS
- S. Ji, W. Cao, Y. Yu and H. Xu, Visible-light-induced self-healing diselenide-containing polyurethane elastomer, Adv. Mater., 2015, 27, 7740–7745 CrossRef CAS PubMed
- S. Ji, F. Fan, C. Sun, Y. Yu and H. Xu, Visible light-induced plasticity of shape memory polymers, ACS Appl. Mater. Interfaces, 2017, 9, 33169–33175 CrossRef CAS PubMed
- F. Q. Fan, S. B. Ji, C. X. Sun, C. Liu, Y. Yu, Y. Fu and H. P. Xu, Wavelength-controlled dynamic metathesis: a light-driven exchange reaction between disulfide and diselenide bonds, Angew. Chem., Int. Ed., 2018, 57, 16426–16430 CrossRef CAS PubMed
- J. Xia, P. Zhao, K. Zheng, C. Lu, S. Yin and H. Xu, Surface Modification based on diselenide dynamic chemistry: towards liquid motion and surface bioconjugation, Angew. Chem., Int. Ed., 2019, 58, 542–546 CrossRef CAS PubMed
- W. Zhang and A. H. E. Müller, Architecture, self-assembly and properties of well-defined hybrid polymers based on polyhedral oligomeric silsequioxane (POSS), Prog. Polym. Sci., 2013, 38, 1121–1162 CrossRef CAS
- Q. Yan, J. Yuan, Z. Cai, Y. Xin, Y. Kang and Y. Yin, Voltage-responsive vesicles based on orthogonal assembly of two homopolymers, J. Am. Chem. Soc., 2010, 132, 9268–9270 CrossRef CAS PubMed
- Q. Yan, Y. Xin, R. Zhou, Y. Yin and J. Yuan, Light-controlled smart nanotubes based on the orthogonal assembly of two homopolymers, Chem. Commun., 2011, 47, 9594–9596 RSC
- X. J. Loh, J. del Barrio, T. C. Lee and O. A. Scherman, Supramolecular polymeric peptide amphiphile vesicles for the encapsulation of basic fibroblast growth factor, Chem. Commun., 2014, 50, 3033–3035 RSC
- F. Wurm and H. Frey, Linear–dendritic block copolymers: The state of the art and exciting perspectives, Prog. Polym. Sci., 2011, 36, 1–52 CrossRef CAS
- I. Gitsov, Hybrid linear dendritic macromolecules: From synthesis to applications, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5295–5314 CrossRef CAS
- F. Wurm, J. Klos, H. J. Rader and H. Frey, Synthesis and noncovalent protein conjugation of linear-hyperbranched PEG-poly(glycerol) alpha,omega(n)-telechelics, J. Am. Chem. Soc., 2009, 131, 7954–7955 CrossRef CAS PubMed
- V. Istratov, H. Kautz, Y.-K. Kim, R. Schubert and H. Frey, Linear-dendritic nonionic poly(propylene oxide)–polyglycerol surfactants, Tetrahedron, 2003, 59, 4017–4024 CrossRef CAS
- W. Tao, Y. Liu, B. Jiang, S. Yu, W. Huang, Y. Zhou and D. Yan, A linear-hyperbranched supramolecular amphiphile and its self-assembly into vesicles with great ductility, J. Am. Chem. Soc., 2012, 134, 762–764 CrossRef CAS PubMed
- W. Jiang, Y. Liu, C. Yu, S. Li, Y. Li and Y. Zhou, Light-triggered reversible “one-to-two” morphological transition in a “latent double-amphiphilic” linear-hyperbranched supramolecular block copolymer, Chem. Commun., 2016, 52, 8223–8226 RSC
- H. M. Li, J. Wang, Y. Z. Ni, Y. F. Zhou and D. Y. Yan, Synthesis of a linear-hyperbranched supramolecular polymer and its light-responsive self-assembly behavior, Acta Chim. Sin., 2016, 74, 415–421 CrossRef CAS
- A. H. Groschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. Muller, Precise hierarchical self-assembly of multicompartment micelles, Nat. Commun., 2012, 3, 710 CrossRef PubMed
- J. Yan, M. Bloom, S. C. Bae, E. Luijten and S. Granick, Linking synchronization to self-assembly using magnetic Janus colloids, Nature, 2012, 491, 578–581 CrossRef CAS PubMed
- Y. Liu, C. Yu, H. Jin, B. Jiang, X. Zhu, Y. Zhou, Z. Lu and D. Yan, A supramolecular Janus hyperbranched polymer and its photoresponsive self-assembly of vesicles with narrow size distribution, J. Am. Chem. Soc., 2013, 135, 4765–4770 CrossRef CAS PubMed
- D. P. Zhang, Y. J. Fan, H. M. Li, K. Li, Y. Yao, Y. F. Zhou and D. Y. Yan, A dumbbell-like supramolecular triblock copolymer and its self-assembly of light-responsive vesicles, RSC Adv., 2015, 5, 47762–47765 RSC
- D. P. Zhang, Y. N. Liu, Y. J. Fan, C. Y. Yu, Y. L. Zheng, H. B. Jin, L. Fu, Y. F. Zhou and D. Y. Yan, Hierarchical self-assembly of a dandelion-like supramolecular polymer into nanotubes for use as highly efficient aqueous light-harvesting systems, Adv. Funct. Mater., 2016, 26, 7652–7661 CrossRef CAS
- B. Sarkar and P. Alexandridis, Block copolymer-nanoparticle composites: Structure, functional properties, and processing, Prog. Polym. Sci., 2015, 40, 33–62 CrossRef CAS
- R. Deng, S. Liu, J. Li, Y. Liao, J. Tao and J. Zhu, Mesoporous block copolymer nanoparticles with tailored structures by hydrogen-bonding-assisted self-assembly, Adv. Mater., 2012, 24, 1889–1893 CrossRef CAS PubMed
- R. H. Deng, F. X. Liang, W. K. Li, Z. Z. Yang and J. T. Zhu, Reversible transformation of nanostructured polymer particles, Macromolecules, 2013, 46, 7012–7017 CrossRef CAS
- R. Deng, F. Liang, P. Zhou, C. Zhang, X. Qu, Q. Wang, J. Li, J. Zhu and Z. Yang, Janus nanodisc of diblock copolymers, Adv. Mater., 2014, 26, 4469–4472 CrossRef CAS PubMed
- Y. Yang, H. Kim, J. Xu, M. S. Hwang, D. Tian, K. Wang, L. Zhang, Y. Liao, H. G. Park, G. R. Yi, X. Xie and J. Zhu, Responsive block copolymer photonic microspheres, Adv. Mater., 2018, 30, 1707344 CrossRef PubMed
- J. Xu, J. Li, Y. Yang, K. Wang, N. Xu, J. Li, R. Liang, L. Shen, X. Xie, J. Tao and J. Zhu, Block copolymer capsules with structure-dependent release behavior, Angew. Chem., Int. Ed., 2016, 55, 14633–14637 CrossRef CAS PubMed
- R. J. Liang, J. P. Xu, R. H. Deng, K. Wang, S. Q. Liu, J. Y. Li and J. T. Zhu, Assembly of polymer-tethered gold nanoparticles under cylindrical confinement, ACS Macro Lett., 2014, 3, 486–490 CrossRef CAS
- K. Wang, S. M. Jin, J. Xu, R. Liang, K. Shezad, Z. Xue, X. Xie, E. Lee and J. Zhu, Electric-field-assisted assembly of polymer-tethered gold nanorods in cylindrical nanopores, ACS Nano, 2016, 10, 4954–4960 CrossRef CAS PubMed
- H. Tan, Q. Lyu, Z. Xie, M. Li, K. Wang, K. Wang, B. Xiong, L. Zhang and J. Zhu, Metallosupramolecular photonic elastomers with self-healing capability and angle-independent color, Adv. Mater., 2019, 31, 1805496 Search PubMed
- K. Wang, H. Ling, Y. Bao, M. Yang, Y. Yang, M. Hussain, H. Wang, L. Zhang, L. Xie, M. Yi, W. Huang, X. Xie and J. Zhu, A centimeter-scale inorganic nanoparticle superlattice monolayer with non-close-packing and its high performance in memory devices, Adv. Mater., 2018, 30, 1800595 CrossRef
- R. J. Mortimer, Electrochromic materials, Chem. Soc. Rev., 1997, 26, 147–156 RSC
- P. R. Somani and S. Radhakrishnan, Electrochromic materials and devices: present and future, Mater. Chem. Phys., 2003, 77, 117–133 CrossRef CAS
- A. Tsuboi, K. Nakamura and N. Kobayashi, A localized surface plasmon resonance-based multicolor electrochromic device with electrochemically size-controlled silver nanoparticles, Adv. Mater., 2013, 25, 3197–3201 CrossRef CAS PubMed
- S. K. Deb, Opportunities and challenges in science and technology of WO3 for electrochromic and related applications, Sol. Energy Mater. Sol. Cells, 2008, 92, 245–258 CrossRef CAS
- C. G. Granqvist, A. Azens, A. Hjelm, L. Kullman, G. A. Niklasson, D. Ronnow, M. Stromme Mattsson, M. Veszelei and G. Vaivars, Recent advances in electrochromics for smart windowsapplication, Sol. Energy Mater. Sol. Cells, 1998, 63, 199–216 CAS
- A. A. Argun, P.-H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Multicolored electrochromism in polymers: structures and devices, Chem. Mater., 2004, 16, 4401–4412 CrossRef CAS
- C. Xu, L. Liu, S. E. Legenski, D. Ning and M. Taya, Switchable window based on electrochromic polymers, J. Mater. Res., 2011, 19, 2072–2080 CrossRef
- E. Hwang, S. Seo, S. Bak, H. Lee, M. Min and H. Lee, An electrolyte-free flexible electrochromic device using electrostatically strong graphene quantum dot-viologen nanocomposites, Adv. Mater., 2014, 26, 5129–5136 CrossRef CAS
- S. Mi, J. Wu, J. Liu, Z. Xu, X. Wu, G. Luo, J. Zheng and C. Xu, AIEe-active and electrochromic bifunctional polymer and a device composed thereof synchronously achieve electrochemical fluorescence switching and electrochromic switching, ACS Appl. Mater. Interfaces, 2015, 7, 27511–27517 CrossRef CAS PubMed
- J. Wu, Y. Han, J. Liu, Y. Shi, J. Zheng and C. Xu, Electrofluorochromic and electrochromic bifunctional polymers with dual-state emission via introducing multiple C–H⋯π bonds, Org. Electron., 2018, 62, 481–490 CrossRef CAS
- J. Liu, G. Luo, S. Mi, J. Zheng and C. Xu, Trifunctional CdSe quantum dots-polymer composite film with electrochromic, electrofluorescent and light-induced coloration effects, Sol. Energy Mater. Sol. Cells, 2018, 177, 82–88 CrossRef CAS
- X. Chen, H. Liu, Z. Xu, S. Mi, J. Zheng and C. Xu, Highly regiosymmetric homopolymer based on dioxythiophene for realizing water-processable blue-to-transmissive electrochrome, ACS Appl. Mater. Interfaces, 2015, 7, 11387–11392 CrossRef CAS PubMed
- Y. Shi, J. Liu, M. Li, J. Zheng and C. Xu, Novel electrochromic-fluorescent bi-functional devices based on aromatic viologen derivates, Electrochim. Acta, 2018, 285, 415–423 CrossRef CAS
- J. Wei and Y. Yu, Photodeformable polymer gels and crosslinked liquid-crystalline polymers, Soft Matter, 2012, 8, 8050–8059 RSC
- Y. Yu, M. Nakano and T. Ikeda, Photomechanics: directed bending of a polymer film by light, Nature, 2003, 425, 145 CrossRef CAS PubMed
- Y. Yu, T. Maeda, J.-I. Mamiya and T. Ikeda, Photomechanical effects of ferroelectric liquid-crystalline elastomers containing azobenzene chromophores, Angew. Chem., Int. Ed., 2007, 119, 899–901 CrossRef
- Y. Zhao, Z. Xie, H. Gu, C. Zhu and Z. Gu, Bio-inspired variable structural color materials, Chem. Soc. Rev., 2012, 41, 3297–3317 RSC
- Z. Yan, X. Ji, W. Wu, J. Wei and Y. Yu, Light-switchable behavior of a microarray of azobenzene liquid crystal polymer induced by photodeformation, Macromol. Rapid Commun., 2012, 33, 1362–1367 CrossRef CAS PubMed
- J. Zhao, Y. Liu and Y. Yu, Dual-responsive inverse opal films based on a crosslinked liquid crystal polymer containing azobenzene, J. Mater. Chem. C, 2014, 2, 10262–10267 RSC
- D. Liu and D. J. Broer, Self-assembled dynamic 3D fingerprints in liquid-crystal coatings towards controllable friction and adhesion, Angew. Chem., Int. Ed., 2014, 126, 4630–4634 CrossRef
- M. Ibn-Elhaj and M. Schadt, Optical polymer thin films with isotropic and anisotropic nano-corrugated surface topologies, Nature, 2001, 410, 796–799 CrossRef CAS
- A. D. Stroock, Chaotic mixer for microchannels, Science, 2002, 295, 647–651 CrossRef CAS PubMed
- C. Li, F. Cheng, J.-A. Lv, Y. Zhao, M. Liu, L. Jiang and Y. Yu, Light-controlled quick switch of adhesion on a micro-arrayed liquid crystal polymer superhydrophobic film, Soft Matter, 2012, 8, 3730–3733 RSC
- Y. Zhan, J. Zhao, W. Liu, B. Yang, J. Wei and Y. Yu, Biomimetic submicroarrayed cross-linked liquid crystal polymer films with different wettability via colloidal lithography, ACS Appl. Mater. Interfaces, 2015, 7, 25522–25528 CrossRef CAS PubMed
- J. A. Lv, Y. Liu, J. Wei, E. Chen, L. Qin and Y. Yu, Photocontrol of fluid slugs in liquid crystal polymer microactuators, Nature, 2016, 537, 179–184 CrossRef CAS PubMed
- I. Bellin, S. Kelch, R. Langer and A. Lendlein, Polymeric triple-shape materials, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18043–18047 CrossRef CAS PubMed
- T. Xie, Tunable polymer multi-shape memory effect, Nature, 2010, 464, 267–270 CrossRef CAS PubMed
- L. Huang, R. Jiang, J. Wu, J. Song, H. Bai, B. Li, Q. Zhao and T. Xie, Ultrafast digital printing toward 4D shape changing materials, Adv. Mater., 2017, 29, 1605390 CrossRef PubMed
- Q. Zhao, W. Zou, Y. Luo and T. Xie, Shape memory polymer network with thermally distinct elasticity and plasticity, Sci. Adv., 2016, 2, e1501297 CrossRef PubMed
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Thermoset shape-memory polyurethane with intrinsic plasticity enabled by transcarbamoylation, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed
- G. Zhang, W. Peng, J. Wu, Q. Zhao and T. Xie, Digital coding of mechanical stress in a dynamic covalent shape memory polymer network, Nat. Commun., 2018, 9, 4002 CrossRef PubMed
- T. Chung, A. Rorno-Uribe and P. T. Mather, Two-way reversible shape memory in a semicrystalline network, Macromolecules, 2008, 41, 184–192 CrossRef CAS
- M. Behl, K. Kratz, J. Zotzmann, U. Nöchel and A. Lendlein, Reversible bidirectional shape-memory polymers, Adv. Mater., 2013, 25, 4466–4469 CrossRef CAS PubMed
- J. Zhou, S. A. Turner, S. M. Brosnan, Q. Li, J.-M. Y. Carrillo, D. Nykypanchuk, O. Gang, V. S. Ashby, A. V. Dobrynin and S. S. Sheiko, Shapeshifting: reversible shape memory in semicrystalline elastomers, Macromolecules, 2014, 47, 1768–1776 CrossRef CAS
- S. Chen, J. Hu, H. Zhuo and Y. Zhu, Two-way shape memory effect in polymer laminates, Mater. Lett., 2008, 62, 4088–4090 CrossRef CAS
- S. Chen, J. Hu and H. Zhuo, Properties and mechanism of two-way shape memory polyurethane composites, Compos. Sci. Technol., 2010, 70, 1437–1443 CrossRef CAS
- S. Chen, S. Yang, Z. Li, S. Xu, H. Yuan, S. Chen and Z. Ge, Electroactive two-way shape memory polymer laminates, Polym. Compos., 2015, 36, 439–444 CrossRef CAS
- H. Tamagawa, Thermo-responsive two-way shape changeable polymeric laminate, Mater. Lett., 2010, 64, 749–751 CrossRef CAS
- D. L. Thomsen, P. Keller, J. Naciri, R. Pink, H. Jeon, D. Shenoy and B. R. Ratna, Liquid crystal elastomers with mechanical properties of a muscle, Macromolecules, 2001, 34, 5868–5875 CrossRef CAS
- T. Ikeda, J. Mamiya and Y. Yu, Photomechanics of liquid-crystalline elastomers and other polymers, Angew. Chem., Int. Ed., 2007, 46, 506–528 CrossRef CAS
- H. Qin and P. T. Mather, Combined one-way and two-way shape memory in a glass-forming nematic network, Macromolecules, 2009, 42, 273–280 CrossRef CAS
- J. Li, W. R. Rodgers and T. Xie, Semi-crystalline two-way shape memory elastomer, Polymer, 2011, 52, 5320–5325 CrossRef CAS
- J. M. Raquez, S. Vanderstappen, F. Meyer, P. Verge, M. Alexandre, J. M. Thomassin, C. Jerome and P. Dubois, Design of cross-linked semicrystalline poly(epsilon-caprolactone)-based networks with one-way and two-way shape-memory properties through Diels–Alder reactions, Chem. – Eur. J., 2011, 17, 10135–10143 CrossRef CAS PubMed
- S. Pandini, S. Passera, M. Messori, K. Paderni, M. Toselli, A. Gianoncelli, E. Bontempi and T. Riccò, Two-way reversible shape memory behaviour of crosslinked poly(ε-caprolactone), Polymer, 2012, 53, 1915–1924 CrossRef CAS
- S. Pandini, F. Baldi, K. Paderni, M. Messori, M. Toselli, F. Pilati, A. Gianoncelli, M. Brisotto, E. Bontempi and T. Riccò, One-way and two-way shape memory behaviour of semi-crystalline networks based on sol–gel cross-linked poly(ε-caprolactone), Polymer, 2013, 54, 4253–4265 CrossRef CAS
- K. M. Lee, P. T. Knight, T. Chung and P. T. Mather, Polycaprolactone−POSS chemical/physical double networks, Macromolecules, 2008, 41, 4730–4738 CrossRef CAS
- R. M. Baker, J. H. Henderson and P. T. Mather, Shape memory poly(ε-caprolactone)-co-poly(ethylene glycol) foams with body temperature triggering and two-way actuation, J. Mater. Chem. B, 2013, 1, 4916–4920 RSC
- O. Dolynchuk, I. Kolesov, R. Androsch and H.-J. Radusch, Kinetics and dynamics of two-way shape-memory behavior of crosslinked linear high-density and short-chain branched polyethylenes with regard to crystal orientation, Polymer, 2015, 79, 146–158 CrossRef CAS
- K. Wang and X. X. Zhu, Two-way reversible shape memory polymers containing polydopamine nanospheres: light actuation, robotic locomotion, and artificial muscles, ACS Biomater. Sci. Eng., 2018, 4, 3099–3106 CrossRef CAS
- K. Wang, Y.-G. Jia and X. X. Zhu, Two-way reversible shape memory polymers made of cross-linked cocrystallizable random copolymers with tunable actuation temperatures, Macromolecules, 2017, 50, 8570–8579 CrossRef CAS
- M. Behl, K. Kratz, U. Noechel, T. Sauter and A. Lendlein, Temperature-memory polymer actuators, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12555–12559 CrossRef CAS PubMed
- M. Bothe and T. Pretsch, Bidirectional actuation of a thermoplastic polyurethane elastomer, J. Mater. Chem. A, 2013, 1, 14491–14497 RSC
- Y. Wu, J. Hu, J. Han, Y. Zhu, H. Huang, J. Li and B. Tang, Two-way shape memory polymer with “switch–spring” composition by interpenetrating polymer network, J. Mater. Chem. A, 2014, 2, 18816–18822 RSC
- Y. Meng, J. Jiang and M. Anthamatten, Shape actuation via internal stress-induced crystallization of dual-cure networks, ACS Macro Lett., 2015, 4, 115–118 CrossRef CAS
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Programming a crystalline shape memory polymer network with thermo- and photo-reversible bonds toward a single-component soft robot, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed
- L. F. Fan, M. Z. Rong, M. Q. Zhang and X. D. Chen, A very simple strategy for preparing external stress-free two-way shape memory polymers by making use of hydrogen bonds, Macromol. Rapid Commun., 2018, 39, e1700714 CrossRef PubMed
- L. F. Fan, M. Z. Rong, M. Q. Zhang and X. D. Chen, A facile approach toward scalable fabrication of reversible shape-memory polymers with bonded elastomer microphases as internal stress provider, Macromol. Rapid Commun., 2017, 38, 1700124 CrossRef
- L. F. Fan, M. Z. Rong, M. Q. Zhang and X. D. Chen, Dynamic reversible bonds enable external stress-free two-way shape memory effect of a polymer network and the interrelated intrinsic self-healability of wider crack and recyclability, J. Mater. Chem. A, 2018, 6, 16053–16063 RSC
- P. Zhang and G. Li, Advances in healing-on-demand polymers and polymer composites, Prog. Polym. Sci., 2016, 57, 32–63 CrossRef CAS
- L. F. Fan, M. Z. Rong, M. Q. Zhang and X. D. Chen, Repeated intrinsic self-healing of wider cracks in polymer via dynamic reversible covalent bonding molecularly combined with a two-way shape memory effect, ACS Appl. Mater. Interfaces, 2018, 10, 38538–38546 CrossRef CAS PubMed
- X. Q. Cheng, Z. X. Wang, X. Jiang, T. Li, C. H. Lau, Z. Guo, J. Ma and L. Shao, Towards sustainable ultrafast molecular-separation membranes: From conventional polymers to emerging materials, Prog. Mater. Sci., 2018, 92, 258–283 CrossRef CAS
- M. Ulbricht, Advanced functional polymer membranes, Polymer, 2006, 47, 2217–2262 CrossRef CAS
- M. M. Pendergast and E. M. V. Hoek, A review of water treatment membrane nanotechnologies, Energy Environ. Sci., 2011, 4, 1946–1971 RSC
- R. V. Reis and A. Zydney, Membrane separations in biotechnology, Curr. Opin. Biotechnol., 2001, 12, 208–211 CrossRef PubMed
- B. V. D. Bruggen, C. Vecasteele, T. V. Gestel, W. Doyen and R. Leysen, A review of pressure-driven membrane processes in wastewater treatment and drinking water production, Environ. Prog., 2003, 22, 46–56 CrossRef
- D. Rana and T. Matsuura, Surface modifications for antifouling membranes, Chem. Rev., 2010, 110, 2448–2471 CrossRef
- I. Sadeghi, A. Aroujalian, A. Raisi, B. Dabir and M. Fathizadeh, Surface modification of polyethersulfone ultrafiltration membranes by corona air plasma for separation of oil/water emulsions, J. Membr. Sci., 2013, 430, 24–36 CrossRef CAS
- Y. Yang, H. Zhang, P. Wang, Q. Zheng and J. Li, The influence of nano-sized TiO2 fillers on the morphologies and properties of PSF UF membrane, J. Membr. Sci., 2007, 288, 231–238 CrossRef CAS
- Y. H. Zhao, B. K. Zhu, L. Kong and Y. Y. Xu, Improving hydrophilicity and protein resistance of poly(vinylidene fluoride) membranes by blending with amphiphilic hyperbranched-star polymer, Langmuir, 2007, 23, 5779–5786 CrossRef CAS PubMed
- Y. Chen, M. Wei and Y. Wang, Upgrading polysulfone ultrafiltration membranes by blending with amphiphilic block copolymers: Beyond surface segregation, J. Membr. Sci., 2016, 505, 53–60 CrossRef CAS
- G.-D. Kang and Y.-M. Cao, Application and modification of poly(vinylidene fluoride) (PVDF) membranes – A review, J. Membr. Sci., 2014, 463, 145–165 CrossRef CAS
- Y. Z. Zhu, D. Wang, L. Jiang and J. Jin, Recent progress in developing advanced membranes for emulsified oil/water separation, NPG Asia Mater., 2014, 6, e101 CrossRef CAS
- X. Zhao, Y. Su, W. Chen, J. Peng and Z. Jiang, Grafting perfluoroalkyl groups onto polyacrylonitrile membrane surface for improved fouling release property, J. Membr. Sci., 2012, 415–416, 824–834 CrossRef CAS
- A. Rahimpour, S. S. Madaeni, S. Zereshki and Y. Mansourpanah, Preparation and characterization of modified nano-porous PVDF membrane with high antifouling property using UV photo-grafting, Appl. Surf. Sci., 2009, 255, 7455–7461 CrossRef CAS
- H.-C. Yang, K.-J. Liao, H. Huang, Q.-Y. Wu, L.-S. Wan and Z.-K. Xu, Mussel-inspired modification of a polymer membrane for ultra-high water permeability and oil-in-water emulsion separation, J. Mater. Chem. A, 2014, 2, 10225–10230 RSC
- S. Kasemset, A. Lee, D. J. Miller, B. D. Freeman and M. M. Sharma, Effect of polydopamine deposition conditions on fouling resistance, physical properties, and permeation properties of reverse osmosis membranes in oil/water separation, J. Membr. Sci., 2013, 425–426, 208–216 CrossRef CAS
- H. C. Yang, J. K. Pi, K. J. Liao, H. Huang, Q. Y. Wu, X. J. Huang and Z. K. Xu, Silica-decorated polypropylene microfiltration membranes with a mussel-inspired intermediate layer for oil-in-water emulsion separation, ACS Appl. Mater. Interfaces, 2014, 6, 12566–12572 CrossRef CAS
- X. Zhao, Y. Su, J. Cao, Y. Li, R. Zhang, Y. Liu and Z. Jiang, Fabrication of antifouling polymer–inorganic hybrid membranes through the synergy of biomimetic mineralization and nonsolvent induced phase separation, J. Mater. Chem. A, 2015, 3, 7287–7295 RSC
- H. C. Yang, Y. Xie, H. Chan, B. Narayanan, L. Chen, R. Z. Waldman, S. Sankaranarayanan, J. W. Elam and S. B. Darling, Crude-oil-repellent membranes by atomic layer deposition: oxide interface engineering, ACS Nano, 2018, 12, 8678–8685 CrossRef CAS PubMed
- F. J. Xu, J. P. Zhao, E. T. Kang, K. G. Neoh and J. Li, Functionalization of nylon membranes via surface-initiated atom-transfer radical polymerization, Langmuir, 2007, 23, 8585–8592 CrossRef CAS PubMed
- L.-P. Zhu, H.-B. Dong, X.-Z. Wei, Z. Yi, B.-K. Zhu and Y.-Y. Xu, Tethering hydrophilic polymer brushes onto PPESK membranes via surface-initiated atom transfer radical polymerization, J. Membr. Sci., 2008, 320, 407–415 CrossRef CAS
- H. Yu, Y. Cao, G. Kang, J. Liu, M. Li and Q. Yuan, Enhancing antifouling property of polysulfone ultrafiltration membrane by grafting zwitterionic copolymer via UV-initiated polymerization, J. Membr. Sci., 2009, 342, 6–13 CrossRef CAS
- J. Mansouri, S. Harrisson and V. Chen, Strategies for controlling biofouling in membrane filtration systems: challenges and opportunities, J. Mater. Chem., 2010, 20, 4567–4586 RSC
- Y. Zhu, F. Zhang, D. Wang, X. F. Pei, W. Zhang and J. Jin, A novel zwitterionic polyelectrolyte grafted PVDF membrane for thoroughly separating oil from water with ultrahigh efficiency, J. Mater. Chem. A, 2013, 1, 5758 RSC
- P. C. Chen and Z. K. Xu, Mineral-coated polymer membranes with superhydrophilicity and underwater superoleophobicity for effective oil/water separation, Sci. Rep., 2013, 3, 2776 CrossRef
- S. Gao, J. Sun, P. Liu, F. Zhang, W. Zhang, S. Yuan, J. Li and J. Jin, A robust polyionized hydrogel with an unprecedented underwater anti-crude-oil-adhesion property, Adv. Mater., 2016, 28, 5307–5314 CrossRef CAS PubMed
- F. Zhang, W. B. Zhang, Z. Shi, D. Wang, J. Jin and L. Jiang, Nanowire-haired inorganic membranes with superhydrophilicity and underwater ultralow adhesive superoleophobicity for high-efficiency oil/water separation, Adv. Mater., 2013, 25, 4192–4198 CrossRef CAS PubMed
- S. Gao, Y. Zhu, J. Wang, F. Zhang, J. Li and J. Jin, Layer-by-layer construction of Cu2+/alginate multilayer modified ultrafiltration membrane with bioinspired superwetting property for high-efficient crude-oil-in-water emulsion separation, Adv. Funct. Mater., 2018, 28, 1801944 CrossRef
- Y. Zhu, J. Wang, F. Zhang, S. Gao, A. Wang, W. Fang and J. Jin, Zwitterionic nanohydrogel grafted PVDF membranes with comprehensive antifouling property and superior cycle stability for oil-in-water emulsion separation, Adv. Funct. Mater., 2018, 28, 1804121 CrossRef
- A. B. D. Cassie and S. Baxter, Wettability of porous surfaces, Trans. Faraday Soc., 1944, 40, 546–551 RSC
- R. N. Wenzel, Resistance of Solid Surfaces to Wetting by Water, Ind. Eng. Chem., 1936, 28, 988–994 CrossRef CAS
- W. Zhang, Y. Zhu, X. Liu, D. Wang, J. Li, L. Jiang and J. Jin, Salt-induced fabrication of superhydrophilic and underwater superoleophobic PAA-g-PVDF membranes for effective separation of oil-in-water emulsions, Angew. Chem., Int. Ed., 2014, 53, 856–860 CrossRef CAS
- W. Zhang, Z. Shi, F. Zhang, X. Liu, J. Jin and L. Jiang, Superhydrophobic and superoleophilic PVDF membranes for effective separation of water-in-oil emulsions with high flux, Adv. Mater., 2013, 25, 2071–2076 CrossRef CAS
- M. Tao, L. Xue, F. Liu and L. Jiang, An intelligent superwetting PVDF membrane showing switchable transport performance for oil/water separation, Adv. Mater., 2014, 26, 2943–2948 CrossRef CAS
- Z. Shi, W. Zhang, F. Zhang, X. Liu, D. Wang, J. Jin and L. Jiang, Ultrafast separation of emulsified oil/water mixtures by ultrathin free-standing single-walled carbon nanotube network films, Adv. Mater., 2013, 25, 2422–2427 CrossRef CAS
- S. J. Gao, Y. Z. Zhu, F. Zhang and J. Jin, Superwetting polymer-decorated SWCNT composite ultrathin films for ultrafast separation of oil-in-water nanoemulsions, J. Mater. Chem. A, 2015, 3, 2895–2902 RSC
- X. S. Peng, J. Jin, Y. Nakamura, T. Ohno and I. Ichinose, Ultrafast permeation of water through protein-based membranes, Nat. Nanotechnol., 2009, 4, 353–357 CrossRef CAS PubMed
- R. Wang, T. Xiang, W.-F. Zhao and C.-S. Zhao, A facile approach toward multi-functional polyurethane/polyethersulfone composite membranes for versatile applications, Mater. Sci. Eng., C, 2016, 59, 556–564 CrossRef CAS PubMed
- S.-S. Li, Y. Xie, T. Xiang, L. Ma, C. He, S.-D. Sun and C.-S. Zhao, Heparin-mimicking polyethersulfone membranes - hemocompatibility, cytocompatibility, antifouling and antibacterial properties, J. Membr. Sci., 2016, 498, 135–146 CrossRef CAS
- M. He, Q. Wang, R. Wang, Y. Xie, W. Zhao and C. Zhao, Design of antibacterial poly(ether sulfone) membranes via covalently attaching hydrogel thin layers loaded with ag nanoparticles, ACS Appl. Mater. Interfaces, 2017, 9, 15962–15974 CrossRef CAS PubMed
- S. Chen, Y. Xie, T. Xiao, W. Zhao, J. Li and C. Zhao, Tannic acid-inspiration and post-crosslinking of zwitterionic polymer as a universal approach towards antifouling surface, Chem. Eng. J., 2018, 337, 122–132 CrossRef CAS
- C. Nie, Y. Yang, Z. Peng, C. Cheng, L. Ma and C. Zhao, Aramid nanofiber as an emerging nanofibrous modifier to enhance ultrafiltration and biological performances of polymeric membranes, J. Membr. Sci., 2017, 528, 251–263 CrossRef CAS
- W. Shi, L. Zhang, J. Deng, D. Wang, S. Sun, W. Zhao and C. Zhao, Redox-responsive polymeric membranes via supermolecular host–guest interactions, J. Membr. Sci., 2015, 480, 139–152 CrossRef CAS
- W. Zhao, C. He, C. Nie, S. Sun and C. Zhao, Synthesis and characterization of ultrahigh ion-exchange capacity polymeric membranes, Ind. Eng. Chem. Res., 2016, 55, 9667–9675 CrossRef CAS
- X. Zhang, J. Zhou, R. Wei, W. Zhao, S. Sun and C. Zhao, Design of anion species/strength responsive membranes via in-situ cross-linked copolymerization of ionic liquids, J. Membr. Sci., 2017, 535, 158–167 CrossRef CAS
- X. Zhang, S. Xu, J. Zhou, W. Zhao, S. Sun and C. Zhao, Anion-responsive poly(ionic liquid)s gating membranes with tunable hydrodynamic permeability, ACS Appl. Mater. Interfaces, 2017, 9, 32237–32247 CrossRef CAS PubMed
- Y. Xu, D. Yuan, J. Bao, Y. Xie, M. He, Z. Shi, S. Chen, C. He, W. Zhao and C. Zhao, Nanofibrous membranes with surface migration of functional groups for ultrafast wastewater remediation, J. Mater. Chem. A, 2018, 6, 13359–13372 RSC
- M. Elimelech and W. A. Phillip, The future of seawater desalination: energy, technology, and the environment, Science, 2011, 333, 712–717 CrossRef CAS PubMed
- S. Karan, Z. Jiang and A. G. Livingston, Sub-10 nm polyamide nanofilms with ultrafast solvent transport for molecular separation, Science, 2015, 348, 1347–1351 CrossRef CAS
- Z. Wang, Z. Wang, S. Lin, H. Jin, S. Gao, Y. Zhu and J. Jin, Nanoparticle-templated nanofiltration membranes for ultrahigh performance desalination, Nat. Commun., 2018, 9, 2004 CrossRef PubMed
- Z. Jiang, S. Karan and A. G. Livingston, Water transport through ultrathin polyamide nanofilms used for reverse osmosis, Adv. Mater., 2018, 30, 1705973 CrossRef PubMed
- Z. Tan, S. Chen, X. Peng, L. Zhang and C. Gao, Polyamide membranes with nanoscale Turing structures for water purification, Science, 2018, 360, 518–521 CrossRef CAS PubMed
- C. Zhang, Y. Ou, W. X. Lei, L. S. Wan, J. Ji and Z. K. Xu, CuSO4/H2O2-induced rapid deposition of polydopamine coatings with high uniformity and enhanced stability, Angew. Chem., Int. Ed., 2016, 55, 3054–3057 CrossRef CAS
- C. Zhang, M. Q. Ma, T. T. Chen, H. Zhang, D. F. Hu, B. H. Wu, J. Ji and Z. K. Xu, Dopamine-triggered one-step polymerization and codeposition of acrylate monomers for functional coatings, ACS Appl. Mater. Interfaces, 2017, 9, 34356–34366 CrossRef CAS
- C. Zhang, L. Gong, L. Xiang, Y. Du, W. Hu, H. Zeng and Z. K. Xu, Deposition and adhesion of polydopamine on the surfaces of varying wettability, ACS Appl. Mater. Interfaces, 2017, 9, 30943–30950 CrossRef CAS PubMed
- C. Zhang, H. N. Li, Y. Du, M. Q. Ma and Z. K. Xu, CuSO4/H2O2-Triggered polydopamine/poly(sulfobetaine methacrylate) coatings for antifouling membrane surfaces, Langmuir, 2017, 33, 1210–1216 CrossRef CAS PubMed
- C. Zhang, Y. Lv, W. Z. Qin, A. He and Z. K. Xu, Polydopamine coatings with nanopores for versatile molecular separation, ACS Appl. Mater. Interfaces, 2017, 9, 14437–14444 CrossRef CAS PubMed
- Y. Lv, H. C. Yang, H. Q. Liang, L. S. Wan and Z. K. Xu, Nanofiltration membranes via co-deposition of polydopamine/polyethylenimine followed by cross-linking, J. Membr. Sci., 2015, 476, 50–58 CrossRef CAS
- H.-C. Yang, M.-B. Wu, J. Hou, S. B. Darling and Z.-K. Xu, Nanofilms directly formed on macro-porous substrates for molecular and ionic sieving, J. Mater. Chem. A, 2018, 6, 2908–2913 RSC
- W. Z. Qiu, G. P. Wu and Z. K. Xu, Robust coatings via catechol-amine codeposition: mechanism, kinetics, and application, ACS Appl. Mater. Interfaces, 2018, 10, 5902–5908 CrossRef CAS PubMed
- W.-Z. Qiu, Y. Du, Y. Lv, H.-C. Yang and Z.-K. Xu, Codeposition of catechol-polyethyleneimine followed by interfacial polymerization for nanofiltration membranes with enhanced stability, J. Appl. Polym. Sci., 2017, 134, 45422 CrossRef
- W. Z. Qiu, Y. Lv, Y. Du, H. C. Yang and Z. K. Xu, Composite nanofiltration membranes via the co-deposition and cross-linking of catechol/polyethylenimine, RSC Adv., 2016, 6, 34096–34102 RSC
- W.-Z. Qiu, Q.-Z. Zhong, Y. Du, Y. Lv and Z.-K. Xu, Enzyme-triggered coatings of tea catechins/chitosan for nanofiltration membranes with high performance, Green Chem., 2016, 18, 6205–6208 RSC
- Y. Lv, Y. Du, Z. X. Chen, W. Z. Qiu and Z. K. Xu, Nanocomposite membranes of polydopamine/electropositive nanoparticles/polyethyleneimine for nanofiltration, J. Membr. Sci., 2018, 545, 99–106 CrossRef CAS
- Y. Lv, Y. Du, W. Z. Qiu and Z. K. Xu, Nanocomposite membranes via the codeposition of polydopamine/polyethylenimine with silica nanoparticles for enhanced mechanical strength and high water permeability, ACS Appl. Mater. Interfaces, 2017, 9, 2966–2972 CrossRef CAS PubMed
- Y. Lv, H. C. Yang, H. Q. Liang, L. S. Wan and Z. K. Xu, Novel nanofiltration membrane with ultrathin zirconia film as selective layer, J. Membr. Sci., 2016, 500, 265–271 CrossRef CAS
- Y. Lv, C. Zhang, A. He, S. J. Yang, G. P. Wu, S. B. Darling and Z. K. Xu, Photocatalytic nanofiltration membranes with self-cleaning property for wastewater treatment, Adv. Funct. Mater., 2017, 27, 1700251 CrossRef
- Y. Du, W. Z. Qiu, Y. Lv, J. Wu and Z. K. Xu, Nanofiltration membranes with narrow pore size distribution via contra-diffusion-induced mussel-inspired chemistry, ACS Appl. Mater. Interfaces, 2016, 8, 29696–29704 CrossRef CAS PubMed
- Y. Du, Y. Lv, W. Z. Qiu, J. Wu and Z. K. Xu, Nanofiltration membranes with narrowed pore size distribution via pore wall modification, Chem. Commun., 2016, 52, 8589–8592 RSC
- Y. Du, C. Zhang, Q. Z. Zhong, X. Yang, J. Wu and Z. K. Xu, Ultrathin alginate coatings as selective layers for nanofiltration membranes with high performance, ChemSusChem, 2017, 10, 2788–2795 CrossRef CAS PubMed
- M.-B. Wu, Y. Lv, H.-C. Yang, L.-F. Liu, X. Zhang and Z.-K. Xu, Thin film composite membranes combining carbon nanotube intermediate layer and microfiltration support for high nanofiltration performances, J. Membr. Sci., 2016, 515, 238–244 CrossRef CAS
- J.-J. Wang, H.-C. Yang, M.-B. Wu, X. Zhang and Z.-K. Xu, Nanofiltration membranes with cellulose nanocrystals as an interlayer for unprecedented performance, J. Mater. Chem. A, 2017, 5, 16289–16295 RSC
- X. Yang, Y. Du, X. Zhang, A. He and Z. K. Xu, Nanofiltration membrane with a mussel-inspired interlayer for improved permeation performance, Langmuir, 2017, 33, 2318–2324 CrossRef CAS PubMed
- X. Zhang, Y. Lv, H. C. Yang, Y. Du and Z. K. Xu, Polyphenol coating as an interlayer for thin-film composite membranes with enhanced nanofiltration performance, ACS Appl. Mater. Interfaces, 2016, 8, 32512–32519 CrossRef CAS PubMed
- H. Q. Liang, W. S. Hung, H. H. Yu, C. C. Hu, K. R. Lee, J. Y. Lai and Z. K. Xu, Forward osmosis membranes with unprecedented water flux, J. Membr. Sci., 2017, 529, 47–54 CrossRef CAS
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gomez-Romero, Hybrid energy storage: the merging of battery and supercapacitor chemistries, Chem. Soc. Rev., 2015, 44, 1777–1790 RSC
- P. Simon, Y. Gogotsi and B. Dunn, Where do batteries end and supercapacitors begin?, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed
- P. Jezowski, O. Crosnier, E. Deunf, P. Poizot, F. Beguin and T. Brousse, Safe and recyclable lithium-ion capacitors using sacrificial organic lithium salt, Nat. Mater., 2018, 17, 167–173 CrossRef CAS PubMed
- X. Wang, X. Lu, B. Liu, D. Chen, Y. Tong and G. Shen, Flexible energy-storage devices: design consideration and recent progress, Adv. Mater., 2014, 26, 4763–4782 CrossRef CAS
- Z. Yang, J. Deng, X. Chen, J. Ren and H. Peng, A highly stretchable, fiber-shaped supercapacitor, Angew. Chem., Int. Ed., 2013, 52, 13453–13457 CrossRef CAS
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Efficient storage mechanisms for building better supercapacitors, Nat. Energy, 2016, 1, 16070 CrossRef CAS
- W. K. Chee, H. N. Lim, Z. Zainal, N. M. Huang, I. Harrison and Y. Andou, Flexible graphene-based supercapacitors: a review, J. Phys. Chem. B, 2016, 120, 4153–4172 CAS
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Review on supercapacitors: Technologies and materials, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef
- Q. Meng, K. Cai, Y. Chen and L. Chen, Research progress on conducting polymer based supercapacitor electrode materials, Nano Energy, 2017, 36, 268–285 CrossRef CAS
- Y. Han, Z. Lai, Z. Wang, M. Yu, Y. Tong and X. Lu, Designing carbon based supercapacitors with high energy density: a summary of recent progress, Chem. – Eur. J., 2018, 24, 7312–7329 CrossRef CAS PubMed
- F. Ran, X. Yang and L. Shao, Recent progress in carbon-based nanoarchitectures for advanced supercapacitors, Adv. Compos. Hybrid Mater., 2018, 1, 32–55 CrossRef CAS
- Y. Zhang, T. Qu, K. Xiang, Y. Shen, S. Chen, M. Xie and X. Guo,
In situ formation/carbonization of quinone-amine polymers towards hierarchical porous carbon foam with high faradaic activity for energy storage, J. Mater. Chem. A, 2018, 6, 2353–2359 RSC
- M. Cheng, Y. Meng, Q. Meng, L. Mao, M. Zhang, K. Amin, A. Ahmad, S. Wu and Z. Wei, A hierarchical porous N-doped carbon electrode with superior rate performance and cycling stability for flexible supercapacitors, Mater. Chem. Front., 2018, 2, 986–992 RSC
- K. Wang, H. Wu, Y. Meng and Z. Wei, Conducting polymer nanowire arrays for high performance supercapacitors, Small, 2014, 10, 14–31 CrossRef CAS
- L. Nyholm, G. Nyström, A. Mihranyan and M. Strømme, Toward flexible polymer and paper-based energy storage devices, Adv. Mater., 2011, 23, 3751–3769 CAS
- K. Wang, J. Huang and Z. Wei, Conducting polyaniline nanowire arrays for high performance supercapacitors, J. Phys. Chem. C, 2010, 114, 8062–8067 CrossRef CAS
- J. Xu, K. Wang, S.-Z. Zu, B.-H. Han and Z. Wei, Hierarchical nanocomposites of polyaniline nanowire arrays on graphene oxide sheets with synergistic effect for energy storage, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed
- Y. Meng, K. Wang, Y. Zhang and Z. Wei, Hierarchical porous graphene/polyaniline composite film with superior rate performance for flexible supercapacitors, Adv. Mater., 2013, 25, 6985–6990 CrossRef CAS PubMed
- K. Wang, Q. Meng, Y. Zhang, Z. Wei and M. Miao, High-performance two-ply yarn supercapacitors based on carbon nanotubes and polyaniline nanowire arrays, Adv. Mater., 2013, 25, 1494–1498 CrossRef CAS PubMed
- K. Wang, H. Wu, Y. Meng, Y. Zhang and Z. Wei, Integrated energy storage and electrochromic function in one flexible device: an energy storage smart window, Energy Environ. Sci., 2012, 5, 8384–8389 RSC
- T. B. Schon, P. M. DiCarmine and D. S. Seferos, Polyfullerene electrodes for high power supercapacitors, Adv. Energy Mater., 2014, 4, 1301509 CrossRef
- K. Amin, Q. Meng, A. Ahmad, M. Cheng, M. Zhang, L. Mao, K. Lu and Z. Wei, A carbonyl compound-based flexible cathode with superior rate performance and cyclic stability for flexible lithium-ion batteries, Adv. Mater., 2018, 30, 1703868 CrossRef PubMed
- A. Ahmad, Q. Meng, S. Melhi, L. Mao, M. Zhang, B.-H. Han, K. Lu and Z. Wei, A hierarchically porous hypercrosslinked and novel quinone based stable organic polymer electrode for lithium-ion batteries, Electrochim. Acta, 2017, 255, 145–152 CrossRef CAS
- F. Xu, H. Wang, M. Wu, J. Nan, T. Li and S.-A. Cao, Electrochemical properties of poly(anthraquinonyl imide)s as high-capacity organic cathode materials for Li-ion batteries, Mater. Chem. Phys., 2018, 214, 120–125 CrossRef CAS
- W. Wei, L. Li, L. Zhang, J. Hong and G. He, An all-solid-state Li-organic battery with quinone-based polymer cathode and composite polymer electrolyte, Electrochem. Commun., 2018, 90, 21–25 CrossRef CAS
- Y. Wang, Z. Liu, H. Liu, H. Liu, B. Li and S. Guan, A novel high-capacity anode material derived from aromatic imides for lithium-ion batteries, Small, 2018, 14, 1704094 CrossRef PubMed
- A. Petronico, K. L. Bassett, B. G. Nicolau, A. A. Gewirth and R. G. Nuzzo, Toward a four-electron redox quinone polymer for high capacity lithium ion storage, Adv. Energy Mater., 2018, 8, 1700960 CrossRef
- H. Wu, K. Wang, Y. Meng, K. Lu and Z. Wei, An organic cathode material based on a polyimide/CNT nanocomposite for lithium ion batteries, J. Mater. Chem. A, 2013, 1, 6366–6372 RSC
- H. Wu, S. A. Shevlin, Q. Meng, W. Guo, Y. Meng, K. Lu, Z. Wei and Z. Guo, Flexible and binder-free organic cathode for high-performance lithium-ion batteries, Adv. Mater., 2014, 26, 3338–3343 CrossRef CAS PubMed
- H. Wu, Q. Meng, Q. Yang, M. Zhang, K. Lu and Z. Wei, Large-area polyimide/swcnt nanocable cathode for flexible lithium-ion batteries, Adv. Mater., 2015, 27, 6504–6510 CrossRef CAS PubMed
- H. P. Wu, Q. Yang, Q. H. Meng, A. Ahmad, M. Zhang, L. Y. Zhu, Y. G. Liu and Z. X. Wei, A polyimide derivative containing different carbonyl groups for flexible lithium ion batteries, J. Mater. Chem. A, 2016, 4, 2115–2121 RSC
- A. Ahmad, H. Wu, Y. Guo, Q. Meng, Y. Meng, K. Lu, L. Liu and Z. Wei, A graphene supported polyimide nanocomposite as a high performance organic cathode material for lithium ion batteries, RSC Adv., 2016, 6, 33287–33294 RSC
- G. Zhang, Z. Xu, P. Liu, Y. Su, T. Huang, R. Liu, X. Xi and D. Wu, A facile in-situ polymerization strategy towards polyimide/carbon black composites as high performance lithium ion battery cathodes, Electrochim. Acta, 2018, 260, 598–605 CrossRef CAS
- K. Amin, L. Mao and Z. Wei, Recent progress in polymeric carbonyl-based electrode materials for lithium and sodium ion batteries, Macromol. Rapid Commun., 2019, 40, 1800565 CrossRef
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, The lithium-ion battery: State of the art and future perspectives, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef
- R. Yamamoto, N. Yabuuchi and M. Miyasaka, Synthesis of conjugated carbonyl containing polymer negative electrodes for sodium ion batteries, J. Electrochem. Soc., 2018, 165, A434–A438 CrossRef CAS
- T. Bančič, J. Bitenc, K. Pirnat, A. Kopač Lautar, J. Grdadolnik, A. Randon Vitanova and R. Dominko, Electrochemical performance and redox mechanism of naphthalene-hydrazine diimide polymer as a cathode in magnesium battery, J. Power Sources, 2018, 395, 25–30 CrossRef
- G. Dawut, Y. Lu, L. C. Miao and J. Chen, High-performance rechargeable aqueous Zn-ion batteries with a poly(benzoquinonyl sulfide) cathode, Inorg. Chem. Front., 2018, 5, 1391–1396 RSC
- T. Chen, L. Qiu, Z. Yang, Z. Cai, J. Ren, H. Li, H. Lin, X. Sun and H. Peng, An integrated “energy wire” for both photoelectric conversion and energy storage, Angew. Chem., Int. Ed., 2012, 51, 11977–11980 CrossRef CAS PubMed
- J. Ren, Y. Zhang, W. Bai, X. Chen, Z. Zhang, X. Fang, W. Weng, Y. Wang and H. Peng, Elastic and wearable wire-shaped lithium-ion battery with high electrochemical performance, Angew. Chem., Int. Ed., 2014, 53, 7864–7869 CrossRef CAS
- Y. Zhang, W. Bai, X. Cheng, J. Ren, W. Weng, P. Chen, X. Fang, Z. Zhang and H. Peng, Flexible and stretchable lithium-ion
batteries and supercapacitors based on electrically conducting carbon nanotube fiber springs, Angew. Chem., Int. Ed., 2014, 53, 14564–14568 CrossRef CAS
- H. Lin, W. Weng, J. Ren, L. Qiu, Z. Zhang, P. Chen, X. Chen, J. Deng, Y. Wang and H. Peng, Twisted aligned carbon nanotube/silicon composite fiber anode for flexible wire-shaped lithium-ion battery, Adv. Mater., 2014, 26, 1217–1222 CrossRef CAS PubMed
- J. Ren, W. Bai, G. Guan, Y. Zhang and H. Peng, Flexible and weaveable capacitor wire based on a carbon nanocomposite fiber, Adv. Mater., 2013, 25, 5965–5970 CrossRef CAS PubMed
- J. Ren, L. Li, C. Chen, X. Chen, Z. Cai, L. Qiu, Y. Wang, X. Zhu and H. Peng, Twisting carbon nanotube fibers for both wire-shaped micro-supercapacitor and micro-battery, Adv. Mater., 2013, 25, 1155–1159 CrossRef CAS PubMed
- W. Weng, P. Chen, S. He, X. Sun and H. Peng, Smart electronic textiles, Angew. Chem., Int. Ed., 2016, 55, 6140–6169 CrossRef CAS PubMed
- P. Chen, Y. Xu, S. He, X. Sun, S. Pan, J. Deng, D. Chen and H. Peng, Hierarchically arranged helical fibre actuators driven by solvents and vapours, Nat. Nanotechnol., 2015, 10, 1077–1083 CrossRef CAS PubMed
- H. Peng, X. Sun, F. Cai, X. Chen, Y. Zhu, G. Liao, D. Chen, Q. Li, Y. Lu, Y. Zhu and Q. Jia, Electrochromatic carbon nanotube/polydiacetylene nanocomposite fibres, Nat. Nanotechnol., 2009, 4, 738–741 CrossRef CAS PubMed
- Z. T. Zhang, K. P. Guo, Y. M. Li, X. Y. Li, G. Z. Guan, H. P. Li, Y. F. Luo, F. Y. Zhao, Q. Zhang, B. Wei, Q. B. Pei and H. S. Peng, A colour-tunable, weavable fibre-shaped polymer light-emitting electrochemical cell, Nat. Photonics, 2015, 9, 233–238 CrossRef CAS
- C. S. Diercks and O. M. Yaghi, The atom, the molecule, and the covalent organic framework, Science, 2017, 355, eaal1585 CrossRef PubMed
- X. Feng, X. Ding and D. Jiang, Covalent organic frameworks, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC
- S. Y. Ding and W. Wang, Covalent organic frameworks (COFs): from design to applications, Chem. Soc. Rev., 2013, 42, 548–568 RSC
- P. J. Waller, F. Gandara and O. M. Yaghi, Chemistry of covalent organic frameworks, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS
- N. Huang, P. Wang and D. Jiang, Covalent organic frameworks: a materials platform for structural and functional designs, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS
- S. Kandambeth, K. Dey and R. Banerjee, Covalent organic frameworks: chemistry beyond the structure, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed
- S. Wang, X. Feng and B. Wang, Design and synthesis of covalent organic frameworks, Chin. Sci. Bull., 2018, 63, 2229–2245 CrossRef
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, Construction of covalent organic framework for catalysis: Pd/COF-LZU1 in Suzuki–Miyaura coupling reaction, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, Multivariate chiral covalent organic frameworks with controlled crystallinity and stability for asymmetric catalysis, J. Am. Chem. Soc., 2017, 139, 8277–8285 CrossRef CAS PubMed
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, Exfoliation of covalent organic frameworks into few-layer redox-active nanosheets as cathode materials for lithium-ion batteries, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed
- P. Shao, J. Li, F. Chen, L. Ma, Q. Li, M. Zhang, J. Zhou, A. Yin, X. Feng and B. Wang, Flexible films of covalent organic frameworks with ultralow dielectric constants under high humidity, Angew. Chem., Int. Ed., 2018, 57, 16501–16505 CrossRef CAS PubMed
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, A belt-shaped, blue luminescent, and semiconducting covalent organic framework, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed
- Y. Zeng, R. Zou and Y. Zhao, Covalent organic frameworks for CO2 capture, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS
- L. Ma, S. Wang, X. Feng and B. Wang, Recent advances of covalent organic frameworks in electronic and optical applications, Chin. Chem. Lett., 2016, 27, 1383–1394 CrossRef CAS
- L. Ma, X. Feng, S. Wang and B. Wang, Recent advances in AIEgen-based luminescent metal–organic frameworks and covalent organic frameworks, Mater. Chem. Front., 2017, 1, 2474–2486 RSC
- M. O’Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, the reticular chemistry structure resource (RCSR) database of, and symbols for, crystal nets, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Porous, crystalline, covalent organic frameworks, Science, 2005, 310, 1166–1170 CrossRef PubMed
- J. W. Colson and W. R. Dichtel, Rationally synthesized two-dimensional polymers, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed
- Y. Jin, Y. Hu and W. Zhang, Tessellated multiporous two-dimensional covalent organic frameworks, Nat. Rev. Chem., 2017, 1, 56 CrossRef
- J. R. Hunt, C. J. Doonan, J. D. LeVangie, A. P. Côté and O. M. Yaghi, Reticular Synthesis of covalent organic borosilicate frameworks, J. Am. Chem. Soc., 2008, 130, 11872–11873 CrossRef CAS
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Ionic covalent organic frameworks with spiroborate linkage, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, 3D microporous base-functionalized covalent organic frameworks for size-selective catalysis, Angew. Chem., Int. Ed., 2014, 53, 2878–2882 CrossRef CAS PubMed
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Conjugated organic framework with three-dimensionally ordered stable structure and delocalized pi clouds, Nat. Commun., 2013, 4, 2736 CrossRef
- P. F. Wei, M. Z. Qi, Z. P. Wang, S. Y. Ding, W. Yu, Q. Liu, L. K. Wang, H. Z. Wang, W. K. An and W. Wang, Benzoxazole-linked ultrastable covalent organic frameworks for photocatalysis, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, Crystalline dioxin-linked covalent organic frameworks from irreversible reactions, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS PubMed
- X. Ding, J. Guo, X. Feng, Y. Honsho, J. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. Jiang, Synthesis of metallophthalocyanine covalent organic frameworks that exhibit high carrier mobility and photoconductivity, Angew. Chem., Int. Ed., 2011, 50, 1289–1293 CrossRef CAS PubMed
- T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, One-step construction of two different kinds of pores in a 2D covalent organic framework, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS PubMed
- S. Dalapati, M. Addicoat, S. Jin, T. Sakurai, J. Gao, H. Xu, S. Irle, S. Seki and D. Jiang, Rational design of crystalline supermicroporous covalent organic frameworks with triangular topologies, Nat. Commun., 2015, 6, 7786 CrossRef CAS PubMed
- Z.-F. Pang, S.-Q. Xu, T.-Y. Zhou, R.-R. Liang, T.-G. Zhan and X. Zhao, Construction of covalent organic frameworks bearing three different kinds of pores through the heterostructural mixed linker strategy, J. Am. Chem. Soc., 2016, 138, 4710–4713 CrossRef CAS PubMed
- Y. Zhu, S. Wan, Y. Jin and W. Zhang, Desymmetrized vertex design for the synthesis of covalent organic frameworks with periodically heterogeneous pore structures, J. Am. Chem. Soc., 2015, 137, 13772–13775 CrossRef CAS
- X. Feng, Y. Dong and D. Jiang, Star-shaped two-dimensional covalent organic frameworks, CrystEngComm, 2013, 15, 1508–1511 RSC
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O’Keeffe and O. M. Yaghi, Designed synthesis of 3d covalent organic frameworks, Science, 2007, 316, 268–272 CrossRef CAS PubMed
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O’Keeffe and O. M. Yaghi, A crystalline imine-linked 3-d porous covalent organic framework, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, A pyrene-based, fluorescent three-dimensional covalent organic framework, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Three-dimensional anionic cyclodextrin-based covalent organic frameworks, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed
- O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Fröba, A. Thomas and J. Roeser, 3D anionic silicate covalent organic framework with srs topology, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS PubMed
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Single-crystal X-ray diffraction structures of covalent organic frameworks, Science, 2018, 361, 48–52 CrossRef CAS PubMed
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Seeded growth of single-crystal two-dimensional covalent organic frameworks, Science, 2018, 361, 52–57 CrossRef CAS PubMed
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Rapid microwave synthesis and purification of porous covalent organic frameworks, Chem. Mater., 2009, 204–206 CrossRef CAS
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, Mechanochemical synthesis of chemically stable isoreticular covalent organic frameworks, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed
- S. Karak, S. Kumar, P. Pachfule and R. Banerjee, Porosity prediction through hydrogen bonding in covalent organic frameworks, J. Am. Chem. Soc., 2018, 140, 5138–5145 CrossRef CAS PubMed
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, Constructing ultraporous covalent organic frameworks in seconds via an organic terracotta process, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed
- P. Kuhn, M. Antonietti and A. Thomas, Porous, covalent triazine-based frameworks prepared by ionothermal synthesis, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed
- X. Guan, Y. Ma, H. Li, Y. Yusran, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Fast, Ambient temperature and pressure ionothermal synthesis of three-dimensional covalent organic frameworks, J. Am. Chem. Soc., 2018, 140, 4494–4498 CrossRef CAS
- S.-T. Yang, J. Kim, H.-Y. Cho, S. Kim and W.-S. Ahn, Facile synthesis of covalent organic frameworks COF-1 and COF-5 by sonochemical method, RSC Adv., 2012, 2, 10179–10181 RSC
- Y. Peng, W. K. Wong, Z. Hu, Y. Cheng, D. Yuan, S. A. Khan and D. Zhao, Room temperature batch and continuous flow synthesis of water-stable covalent organic frameworks (COFs), Chem. Mater., 2016, 28, 5095–5101 CrossRef CAS
- S. Kim, C. Park, M. Lee, I. Song, J. Kim, M. Lee, J. Jung, Y. Kim, H. Lim and H. C. Choi, Rapid photochemical synthesis of sea-urchin-shaped hierarchical porous cof-5 and its lithography-free patterned growth, Adv. Funct. Mater., 2017, 27, 1700925 CrossRef
- D. D. Medina, V. Werner, F. Auras, R. Tautz, M. Dogru, J. Schuster, S. Linke, M. Doblinger, J. Feldmann, P. Knochel and T. Bein, Oriented thin films of a benzodithiophene covalent organic framework, ACS Nano, 2014, 8, 4042–4052 CrossRef CAS PubMed
- C. R. DeBlase, K. Hernandez-Burgos, K. E. Silberstein, G. G. Rodriguez-Calero, R. P. Bisbey, H. D. Abruna and W. R. Dichtel, Rapid and efficient redox processes within 2d covalent organic framework thin films, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS PubMed
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Oriented 2D covalent organic framework thin films on single-layer graphene, Science, 2011, 332, 228–231 CrossRef CAS PubMed
- X. Chen, H. Wang, H. Yi, X. Wang, X. Yan and Z. Guo, Anthraquinone on porous carbon nanotubes with improved supercapacitor performance, J. Phys. Chem. C, 2014, 118, 8262–8270 CrossRef CAS
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, Colloidal covalent organic frameworks, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS PubMed
- R. P. Bisbey, C. R. DeBlase, B. J. Smith and W. R. Dichtel, Two-dimensional covalent organic framework thin films grown in flow, J. Am. Chem. Soc., 2016, 138, 11433–11436 CrossRef CAS PubMed
- S. Kandambeth, B. P. Biswal, H. D. Chaudhari, K. C. Rout, H. S. Kunjattu, S. Mitra, S. Karak, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, Selective molecular sieving in self-standing porous covalent-organic-framework membranes, Adv. Mater., 2017, 29, 1603945 CrossRef PubMed
- H. Sahabudeen, H. Qi, B. A. Glatz, D. Tranca, R. Dong, Y. Hou, T. Zhang, C. Kuttner, T. Lehnert, G. Seifert, U. Kaiser, A. Fery, Z. Zheng and X. Feng, Wafer-sized multifunctional polyimine-based two-dimensional conjugated polymers with high mechanical stiffness, Nat. Commun., 2016, 7, 13461 CrossRef CAS PubMed
- D. D. Medina, J. M. Rotter, Y. Hu, M. Dogru, V. Werner, F. Auras, J. T. Markiewicz, P. Knochel and T. Bein, Room temperature synthesis of covalent-organic framework films through vapor-assisted conversion, J. Am. Chem. Soc., 2015, 137, 1016–1019 CrossRef CAS PubMed
- I. Berlanga, M. L. Ruiz-González, J. M. González-Calbet, J. L. G. Fierro, R. Mas-Ballesté and F. Zamora, Delamination of layered covalent organic frameworks, Small, 2011, 7, 1207–1211 CrossRef CAS PubMed
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, Chemically stable multilayered covalent organic nanosheets from covalent organic frameworks via mechanical delamination, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed
- Z. Kahveci, T. Islamoglu, G. A. Shar, R. Ding and H. M. El-Kaderi, Targeted synthesis of a mesoporous triptycene-derived covalent organic framework, CrystEngComm, 2013, 15, 1524–1527 RSC
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Chemically delaminated free-standing ultrathin covalent organic nanosheets, Angew. Chem., Int. Ed., 2016, 55, 15604–15608 CrossRef CAS PubMed
- L. Wang, C. Zeng, H. Xu, P. Yin, D. Chen, J. Deng, M. Li, N. Zheng, C. Gu and Y. Ma, A highly soluble, crystalline covalent organic framework compatible with device implementation, Chem. Sci., 2019, 10, 1023–1028 RSC
- X. H. Liu, C. Z. Guan, S. Y. Ding, W. Wang, H. J. Yan, D. Wang and L. J. Wan, On-surface synthesis of single-layered two-dimensional covalent organic frameworks via solid-vapor interface reactions, J. Am. Chem. Soc., 2013, 135, 10470–10474 CrossRef CAS PubMed
- H. Ding, J. Li, G. Xie, G. Lin, R. Chen, Z. Peng, C. Yang, B. Wang, J. Sun and C. Wang, An AIEgen-based 3D covalent organic framework for white light-emitting diodes, Nat. Commun., 2018, 9, 5234 CrossRef CAS PubMed
- S. Wang, L. Ma, Q. Wang, P. Shao, D. Ma, S. Yuan, P. Lei, P. Li, X. Feng and B. Wang, Covalent organic frameworks: a platform for the experimental establishment of the influence of intermolecular distance on phosphorescence, J. Mater. Chem. C, 2018, 6, 5369–5374 RSC
| This journal is © the Partner Organisations 2020 |