
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis, characterization, DNA/BSA/HSA interactions, molecular modeling, antibacterial properties, and in vitro cytotoxic activities of novel parent and niosome nano-encapsulated Ho(III) complexes†
Deng Yinhua ab,
Mohammad Mehdi Foroughic,
Zahra Aramesh-Boroujenid,
Shohreh Jahani*e,
Mohadesh Peydayesh*f,
Fariba Borhani*g,
Mehrdad Khatamieh,
Meysam Rohanii,
Michal Dusekj and
Vaclav Eignerj
ab,
Mohammad Mehdi Foroughic,
Zahra Aramesh-Boroujenid,
Shohreh Jahani*e,
Mohadesh Peydayesh*f,
Fariba Borhani*g,
Mehrdad Khatamieh,
Meysam Rohanii,
Michal Dusekj and
Vaclav Eignerj
aDepartment of Pharmacy, Hunan Provincial People's Hospital, Changsha 410005, P. R. China
bDepartment of Pharmacy, The First Hospital Affiliated to Hunan Normal University, Changsha 410005, P. R. China
cDepartment of Chemistry, Kerman Branch, Islamic Azad University, Kerman, Iran
dDepartment of Clinical Laboratory, AlZahra Hospital, Isfahan University of Medical Sciences, Isfahan, Iran
eNanobioelectrochemistry Research Center, Bam University of Medical Sciences, Bam, Iran. E-mail: shohre_jahani@yahoo.com; Tel: +98 35331321750
fClinical Research Center, Pastor Educational Hospital, Bam University of Medical Sciences, Bam, Iran. E-mail: drpedayesh57@gmail.com
gMedical Ethics and Law Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran. E-mail: faribaborhani@msn.com
hCell Therapy and Regenerative Medicine Comprehensive Center, Kerman University of Medical Sciences, Kerman, Iran
iDepartment of Medicine, Medical School, Bam University of Medical Sciences, Bam, Iran
jInstitute of Physics of the Czech Academy of Sciences, Na Slovance 2, 18221 Prague 8, Czech Republic
First published on 16th June 2020
Abstract
Based on the importance of metal-centered complexes that can interact with DNA, this research focused on the synthesis of a new Ho(III) complex. This complex was isolated and characterized via elemental analysis, and FT-IR, fluorescence, and UV-vis spectroscopy. Additional confirmation of the Ho(III) complex structure was obtained via single-crystal X-ray diffraction. DNA interaction studies were carried out via circular dichroism (CD) spectroscopy, UV-vis absorption spectroscopy, viscosity measurements and emission spectroscopy; it was proposed that the metal complex acts as an effective DNA binder based on studies in the presence of fish DNA (FS-DNA), showing high binding affinity to DNA in the presence of hydrophobic and electron donating substituents. Also, the interactions of this complex with human (HSA) and bovine serum albumin (BSA) proteins were studied via fluorescence spectroscopy techniques and the obtained results reveal an excellent propensity for binding in both cases. Furthermore, the interactions of the Ho(III) complex with DNA, BSA and HSA were confirmed via molecular docking analysis. The antimicrobial activities of the Ho(III) complex were tested against Gram-negative bacteria and Gram-positive bacteria. In addition, a niosome nano-encapsulated Ho(III) complex was synthesized, and the parent and encapsulated complexes were evaluated as potential antitumor candidates. The main structure of the Ho(III) complex is maintained after encapsulation using niosome nanoparticles. The MTT method was used to assess the anticancer properties of the Ho(III) complex and its encapsulated form toward human lung carcinoma and breast cancer cell lines. The anticancer activity in the encapsulated form was more than that of the parent Ho(III) complex. In conclusion, these compounds could be considered as new antitumor candidates.
1. Introduction
Today, cancer is a principal challenging human health issue and it is a primary target of therapeutic chemistry.1–3 Organic complexes and natural products are implemented in chemotherapy. However, numerous literature reports have noted that organic drugs show enhanced activity when coordinated with metal fragments.4 Therefore, metal complexes are the center of attention as possible chemotherapeutic agents.5The success of cisplatin and its analogues in treating numerous cancer cell lines has resulted in the development of inorganic metal-based drugs in the past few decades. However, the uses of cisplatin are limited because of its side effects and the human body's resistance to this drug.6 Studies have mostly considered the novel synthesis of specific extremely functional metal-based drugs with reduced toxicity and more developed therapeutic characteristics.7,8
Inner transition metal complexes are of interest in the coordination chemistry and bioinorganic fields. Numerous rare earth complexes have been implemented in the biomedical evaluation field as magnetic resonance imaging (MRI) contrast agents and as useful catalysts for the hydrolytic cleavage of phosphate ester bonds.9–14 Such rare earth complexes with organic ligands are of significant interest because of their structures and the possible usefulness of their luminescent characteristics. Due to specific biological and photophysical features, rare earth complexes have been implemented as biological probes in the fields of molecular biology and clinical chemistry. Various rare earth complexes can play a role in treating tumor cell lines. Due to their specific electron configurations, rare earth complexes are a source of inspiration for numerous efforts to design and synthesize possible antibacterial and anticancer agents.15 Rare earth complexes are able to directly interact with DNA or prevent complete DNA relaxation via topoisomerase inhibition.16,17
DNA is considered a substantial target for small molecule drugs, namely platinum drugs that irreversibly connect with DNA via covalent binding whilst preventing enzyme self-repair. Moreover, numerous metal complexes can bind with DNA via non-covalent binding modes, such as intercalative, electrostatic and groove binding modes.18,19 Such binding interactions, alongside metal complex catalytic hydrolysis or oxidation, may destroy the DNA structure and cause the demise of carcinoma cells.20
Human serum albumin (HSA) and bovine serum albumin (BSA) are the underlying elements of plasma proteins in humans and cows, correspondingly. HSA crystal formation analysis shows that drug binding sites are located in the IIA and IIIA subdomains.21 The geometry of the IIA pocket is rather different compared to that of IIIA, and a large hydrophobic cavity exists in IIA. BSA is homologous to HSA in terms of structure, with minor differences. HSA contains one tryptophan (Trp-214) within the IIA subdomain whilst BSA contains two tryptophan moieties (Trp-134 and Trp-213) in the IB and IIA subdomains, correspondingly.22,23
HSA makes up a 60% share of the proteins within blood plasma and adopts a vital role concerning drug delivery whilst typically accumulating in tumor tissues.24 Thus, a metal complex that binds to HSA may have enhanced anticancer and bioavailability activities.25
The implementation of rare earth complexes as in vitro biomarkers and, in particular, binding probes is of considerable interest, and substantial progress can be expected in the future. This paper addresses the characterization and synthesis of a novel Ho(III) complex coordinated by the 2,2′-bipyridine (bpy) ligand: [Ho(bpy)(H2O)6]Cl3. The crystal structure of the complex is ascertained via X-ray crystallography. The Ho(III) complex biological characteristics have been evaluated, including: (i) the study of the Ho(III) complex binding properties with FS-DNA via circular dichroism, emission titration and viscosity measurements; (ii) competitive binding evaluations with ethidium bromine, which was studied via fluorescence spectroscopy, for the purpose of examining the possible intercalation of the Ho(III) complex with DNA; (iii) the study of the Ho(III) complex binding characteristics with HSA and BSA via UV-vis and fluorescence spectroscopy at three various temperatures, as well as via circular dichroism; (iv) molecular docking studies of the interactions of the complex with HSA, BSA and DNA; (v) the evaluation of the Ho(III) complex antimicrobial characteristics against Gram-positive bacteria and Gram-negative bacteria; (vi) and studies of the in vitro cytotoxic activity of the Ho(III) complex and its encapsulated forms against two human carcinoma cell lines (MCF-7 and A-549) via MTT assays.
2. Experimental
2.1. Materials and instrumentation
In order to determine the FS-DNA solution concentration (ε260 = 6600 M−1 cm−1), as well as the purity (A260/A280 = 1.8–1.9), UV-vis spectrometry was utilized, which could indicate a lack of protein contamination. Solutions of FS-DNA and the complex with tris(hydroxymethyl-aminomethane)–HCl buffer, possessing 50 mM NaCl and 5 mM Tris–HCl (pH ≈ 7.2), were used to conduct tests relating to the interactions between FS-DNA and the complex. N, H and C content evaluations of the complex were conducted utilizing a PerkinElmer 2400 primary analyzer. The UV-vis absorption spectra were obtained via an Analytik Jena SPECORD S100 UV-vis spectrophotometer. At room temperature, fluorescence measurements were conducted using a PerkinElmer LS-3 spectrofluorophotometer that included 1 cm path length quartz cuvettes. An Aviv spectropolarimeter 215 model was used to record the CD spectra via a cylindrical cuvette with a 0.1 cm path length. The encapsulated version of the Ho(III) complex was analyzed using an inductively coupled plasma (ICP-Spectro ciros CCD instrument) spectrometer. By utilizing inductively coupled plasma measurements, the quantity of nano-encapsulated Ho(III) complex within the niosomes was ascertained. A LEO, LEO912-AB transmission electron microscope (TEM) and KYKY EM 3200 scanning electron microscope (SEM) were utilized to evaluate the surface morphology pertaining to the encapsulated version of the Ho(III) complex.2.2. Synthesis and characterization of [Ho(bpy)(H2O)6]Cl3
2.0 ml of ethanol solution was used to dissolve HoCl3·6H2O (1.0 mmol), and this was then added in a dropwise manner to 2,2′-bipyridine (1.0 mmol) ethanol solution. The resulting reaction mixture was refluxed for a period of 24 hours. After cooling down to room temperature, the resulting solution was filtered, and the white colored precipitate was gathered and washed three times using ethanol. Then, the resulting product was subjected to recrystallization from acetonitrile, taking the form of transparent crystals. Yield: 68.0%. Anal. calc. for C10H20N2O6HoCl3: C, 22.43%; H, 3.73%; N, 5.23%; found: C, 22.26%, H, 3.61%; N, 5.17%. 1H NMR (D2O) ppm: 8.08, 7.82, 7.70 (broad, H-bpy).2.3. X-ray data collection and crystallographic study
Ho(III) complex crystallographic data was gathered using a SuperNova diffractometer equipped with monochromated Mo-Kα radiation (0.71073 Å). Direct methods were used to solve the crystal structure with the Superflip program26 prior to refinement using the full matrix least squares F2 method via the Jana 2006 program.27 In various Fourier maps, hydrogen atoms were distinguished and refined to a rational geometry. Harmonic refinement was used to refine all non-hydrogen atoms. For further information on data collection and structure refinement, see Table 1.| Complex | 1 |
| Empirical formula | C10H20HoN2O6·3(Cl) |
| Formula weight | 535.6 |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Crystal size (mm) | 0.50 × 0.40 × 0.28 |
| a (Å) | 7.6541(6) |
| b (Å) | 14.2569(15) |
| c (Å) | 17.3435(11) |
| α (°) | 89.723(7) |
| β (°) | 87.557(6) |
| γ (°) | 87.201(8) |
| V (Å3) | 1888.6(3) |
| Z | 4 |
| Dcalc (g cm−3) | 1.884 |
| T (K) | 120 |
| θmax (°) | 29.6 |
| R[F2 > 3σ(F2)], wR(F2) | 0.026, 0.047 |
| Δρmax, Δρmin (e Å−3) | 1.29, −1.00 |
2.4. DNA-binding experiments
| [Q]/(εa − εf) = [Q]/(εb − εf) + 1/Kb(εb − εf) | (1) |
Eqn (2) is employed to derive the quantity of the binding constant and the number of binding sites from the emission spectra:
log(F0 − F/F) = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kb + nlog[Q] Kb + nlog[Q]
| (2) |
The Stern–Volmer eqn (3) was used to analyze the quenching data:
| F0/F = 1 + KSV[Q] | (3) |
Quantification of the Ho(III) complex binding capabilities with DNA was carried out, with F0 and F representing steady state fluorescence intensities in the existence and absence of a quencher, respectively. [Q] denotes the quenching agent concentration and KSV represents the Stern–Volmer constant. The F0/F ratio is mapped out vs. the quencher concentration in the form of a Stern–Volmer plot. The resulting slope depicts the Stern–Volmer constant in the case where there is a linear relationship.
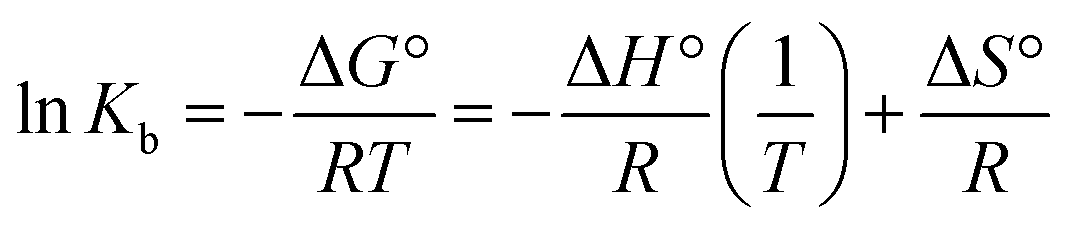 | (4) |
| ΔG° = ΔH° − TΔS° | (5) |
2.5. BSA- and HSA-binding experiments
BSA concentrated stock solution was obtained via dissolving a suitable quantity of BSA in Tris–HCl buffer (containing 5 mM Tris/50 mM NaCl at pH 7.2). The spectrophotometric determination of BSA concentration took place using the molar absorptivity relationship λ280 = 44300 M−1 cm−1.
Ho(III) complex and HAS stock solutions were assembled in Tris–HCl buffer (pH = 7.2). The HSA concentration was ascertained spectrophotometrically based on the molar absorptivity of 36500 M−1 cm−1 at 280 nm. The HAS solutions were preserved at 4 °C under dark conditions and used after no more than 4 days.
HSA fluorescence quenching spectra were obtained by maintaining the HSA concentration at a constant value (1.57 × 10−5 M) whilst enhancing the concentration of Ho(III) complex (1.25 × 10−6 M to 15.0 × 10−6 M) at room temperature. The fluorescence quenching spectra were recorded from 290–510 nm, and the excitation wavelength was 280 nm.
The transferred energy was deduced via FRET theory29 (implementing eqn (6)):
| E = 1 − (F0/F) = R06/(R06 + r6) | (6) |
|
R06 = 8.79 × 1025K2N−4ΦJ
| (7) |
 | (8) |
HSA and Ho(III) complex concentrations were maintained at 2 × 10−6 and 2 × 10−6 M, respectively, for CD spectra measurements. These spectra were documented within the 200–270 nm range with a response time of 4 s and a scan rate of 200 nm min−1. For every spectrum, three accumulated scans were used.
2.6. Docking protocol
To conduct docking calculations, the AutoDock 4.2.2 package was used. The Lamarckian genetic algorithm was used as a prominent search algorithm.30 The HSA (PDB ID: 1BMO) and BSA (PDB ID: 3V03) crystal structures and d(CGCGAATTCGCG)2 sequence dodecamer (PDB ID: 1BNA) DNA duplex information were downloaded from the Brookhaven Protein Data Bank. Hydrogen atoms and Gastiger charges were added to the protein structures and complex.31 During the docking procedure, BSA, DNA and HSA were kept stable whilst every torsional bond of the complex was freed. Spacing was induced via the AutoGrid program for a docking grip map of 80 × 80 × 80 points and a size of 0.375 Å. Thus, a maximum of 25000000 energy analysis calculations and 200 independent runs were implemented.
2.7. Antimicrobial assays
Antibacterial behavior was evaluated via the broth dilution technique against P. aeruginosa (ATCC 27853), E. coli (ATCC 25922), K. pneumonia (ATCC 10031), S. typhi (ATCC 1609), E. VRE, Acinetobacter, E. faecium, MRSA, and faecalis (ATCC 29212), which are causes of nosocomial infections. The bacterial growth intermediary used was Mueller–Hinton broth, including 2% glucose. The diameter of the inhibition zone was derived to acquire the Ho(III) complex antibacterial spectrum. A 107 colony-forming units (CFU) per ml standard inoculum for the purpose of studying stains on a Muller–Hinton agar plate was produced via a swab prior to filter paper discs infused with antibacterial agents (6 mg ml−1) being placed on the agar. Incubation was conducted at 37 °C overnight to calculate the inhibition zone diameter.The minimal bactericidal concentration (MBC) and minimal inhibitory concentration (MIC) values, obtained via the broth mixture technique, were used to quantitatively derive the Ho(III) complex antibacterial behavior. A bacterial suspension (107 CFU mL−1) was placed within test tubes of 5 ml of Mueller–Hinton broth containing complex mixtures within the 0.003–40 mg L−1 range. Incubation was conducted in an aerobic manner at 37 °C for a period of 24 hours. The non-shaken tubes were then evaluated to determine visible turbidity. The MIC denotes the lowest complex concentration without bacterial progression. Tests were conducted three times to validate the MIC results (μg ml−1) pertaining to each strain. Upon the determination of the MIC value, 0.1 ml of inoculum solution from the tubes with visible turbidity was subjected to subculturing on a nutrient agar plate surface prior to incubation at 37 °C for a period of 24 hours. The resulting colony count on the subculture was compared with the CFU ml−1 count from the initial inoculum. MBC denotes the minimum complex concentration that permits less than 0.1% of the initial inoculum to survive.
2.8. Cytotoxicity
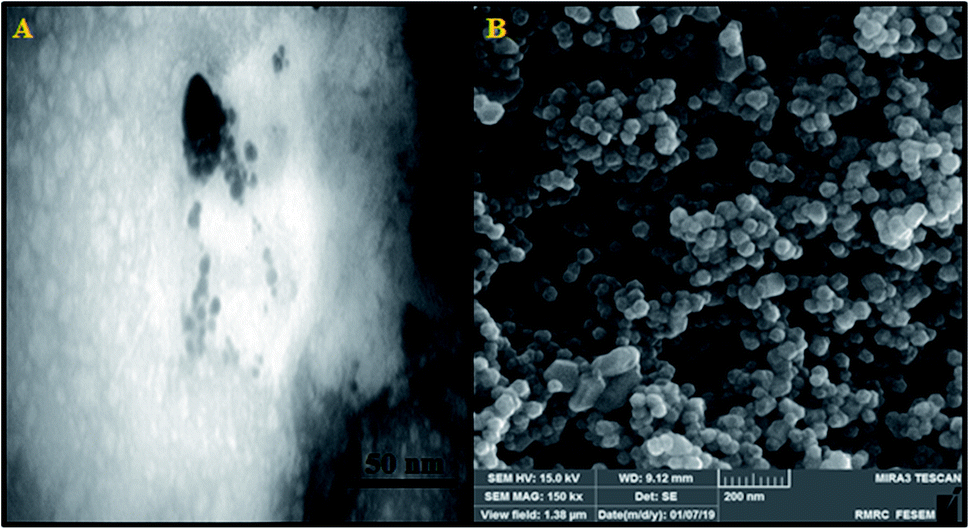 | ||
| Fig. 1 (A) TEM and (B) SEM images of the niosome nano-encapsulated Ho(III) complex (NN-En-Ho). | ||
| % cell cytotoxicity = [1 − Abs(drug)/Abs(control)] × 100. | (9) |
The IC50 values, denoting the concentration of drug leading to a 50% reduction in cellular viability, pertaining to the Ho(III) complex and NN-EN-Ho were derived.
The MCF-7 and A-549 cell lines in cell culture medium (100 μL) possessing 0.12 μg of Ho(III) complex (100 μM concentration) or 1.58 μg of NN-En-Ho (equivalent to 0.12 μg of Ho(III) complex) were subjected to incubation for 24 h in a 5% CO2 incubator. The resulting supernatant was dispensed, and the mixture, i.e. compound cells, was subjected to treatment via CHCl3, HNO3 and the Ho(III) complex; holmium content levels and the amount of niosome nano-encapsulated derivative (NN-En-Ho) were ascertained via ICP. Tests were performed with four repetitions and the number of wells tested/dose was two.
3. Results and discussion
3.1. Characterization of the complex
 | ||
| Fig. 2 An ORTEP view of the molecular structure of [Ho(bpy)(H2O)6]Cl3 with ellipsoids drawn at the 50% probability level. | ||
The hydrogen bonds are a driving force in the crystal structure formation, with a well-developed system of O–H⋯Cl hydrogen bonds forming plates in the structure and weaker C–H⋯Cl bonds connecting the plates together (see Fig. 3). For further information on the hydrogen bonds, see Table S2.†
 | ||
| Fig. 3 The packing structure of [Ho(bpy)(H2O)6]Cl3. | ||
In order to inspect the DNA, BSA and HSA interactions, the crystalline structure of the Ho(III) complex was ascertained, providing the Ho(III) complex structure for use in subsequent experiments. It is worth noting that the noncovalent interactions in the two complexes are identical, thus only one complex is focused on.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), (CC), and (C–H) bands, which appear at 1583–1579, 1453–1415, and 740 and 757 cm−1, respectively. In the complex, the stretching vibrations of these groups shift slightly to lower wavenumbers (1582–1545, 1437–1400, and 737 and 752 cm−1, respectively). Also, the spectrum of the Ho(III) complex show new absorptions in the regions of 3550–2600, 414, and 1622 cm−1, which are attributed to O–H and Ho–N stretching modes and the H–OH bending mode of H2O coordination.
N), (CC), and (C–H) bands, which appear at 1583–1579, 1453–1415, and 740 and 757 cm−1, respectively. In the complex, the stretching vibrations of these groups shift slightly to lower wavenumbers (1582–1545, 1437–1400, and 737 and 752 cm−1, respectively). Also, the spectrum of the Ho(III) complex show new absorptions in the regions of 3550–2600, 414, and 1622 cm−1, which are attributed to O–H and Ho–N stretching modes and the H–OH bending mode of H2O coordination.The f10 modification of Ho3+ involves 47 terms, creating 107 levels of different values of J due to spin orbit coupling. The Ho(III) ground state is 5I8. The Ho(III) absorption spectrum in methanol is provided in Fig. S4a.† There were five evident multiplet-to-multiplet conversions from the ground state to 5G3 and 3G5 (Ho-I); 5G6 (Ho-II); 5F2 (Ho-III); 5S2 and 5F4 (Ho-IV); and 5F5 (Ho-V) stimulated states in the case of the Ho(III) complex in the spectral range of 350 to 650 nm. Such conversions were assigned in the order of increasing conversion frequency. The most significant sensitivity towards the ligand and/or solvent atmosphere was evident in the case of the 5G6 ← 5I8 centered absorption close to 438 nm, out of all the Ho3+ conversions seen during the study. Fig. S4b† shows the Ho(III) complex emission spectrum when subjected to an excitation wavelength of 280 nm at room temperature in methanol, documented within the 350 to 650 nm range. The four emission peaks at 430, 479, 561, and 638 nm, can be allocated to the 5G3 + 3G5 → 5I8, 5G6 → 5I8, 5S2 + 5F4 → 5I8, and 5F5 → 5I8 transitions of the Ho3+ ion, respectively. In regard to the ligand centered emission band (495 nm), there were no efficient transfers of energy from bpy to the Ho3+ center; thus, energy back-transfer from the Ho3+ ion became prominent.35
3.2. DNA binding studies
 | ||
| Fig. 4 UV-vis spectra of the Ho(III) complex (Tris-buffer, pH 7.2) in the absence and presence of increasing amounts (1 is lowest and 6 is highest) of FS-DNA; DNA concentrations: 0, 1.1, 3.3, 5.5, 7.7 and 9.9 μM; inset: plot of [DNA]/(εa − εf) versus [DNA] during the titration of the complex with FS-DNA. | ||
The addition of FS-DNA to Ho(III) complex solutions at DNA/complex molar ratios within the range of 0 to 0.24 may lead to hypochromic shifts of between approximately 66.23% and 65.62% at 285 nm for the Ho(III) complex. Moreover, the Ho(III) complex absorption bands are reduced in intensity in the presence of increasing amounts of FS-DNA, showing the interactions among FS-DNA and the Ho(III) complex. The compound binding constant (Kb) was ascertained by utilizing eqn (1) to quantitatively acquire the Ho(III) complex binding strength with FS-DNA. A value of 0.6 ± 0.02 × 105 M−1 was determined for the DNA binding constant (Kb). This value is lower compared to the recorded values for conventional intercalators (ethidium bromide: Kb = 1.4 × 106 M−1; and [Ru(phen)DPPZ]2+: 106 to 107 M−1).36 Based on the results, the binding constant is higher when DNA groove binding is preserved.37
A fluorescence titration approach is implemented to study the interaction patterns and for a quantitative comparison of the relationship between the complex and DNA. The luminescence titration technique was used to acquire the binding constant of the Ho(III) complex to DNA. The reduction of the Ho(III) complex emission intensity upon the addition of FS-DNA is presented in Fig. 5. The binding site number (n) and Kb values are determined via plotting log(F0 − F/F) against log[DNA], obtaining a linear plot. The Kb and n values pertaining to the Ho(III) complex were derived to be 3.8 ± 0.03 × 105 M−1 and 1.05, respectively.
 | ||
| Fig. 5 The fluorescence spectra of complex–DNA; Ho(III) complex concentration: 2.0 × 10−5 M in Tris-buffer at pH 7.2; DNA concentrations (1 is lowest, 16 is highest): 0, 1.1, 2.2, 3.3, 4.4, 5.5, 6.6, 7.7, 8.8, 9.9, 11.0, 12.1, 13.2, 14.3, 15.4 and 16.5 μM; inset: plot of log[DNA] vs. log(F0 − F)/F. | ||
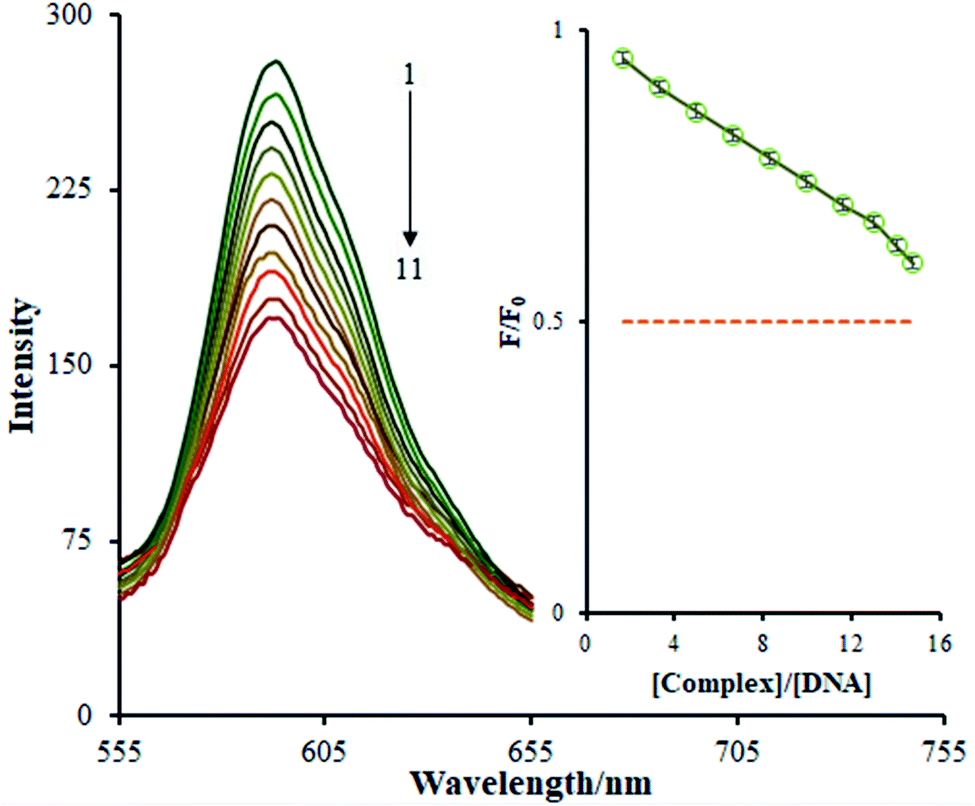 | ||
| Fig. 6 The emission spectra of the DNA–EB system in the presence of increasing amounts (1 is lowest, 11 is highest) of the Ho(III) complex; inset: a plot of F/F0 versus [complex]/[DNA]. | ||
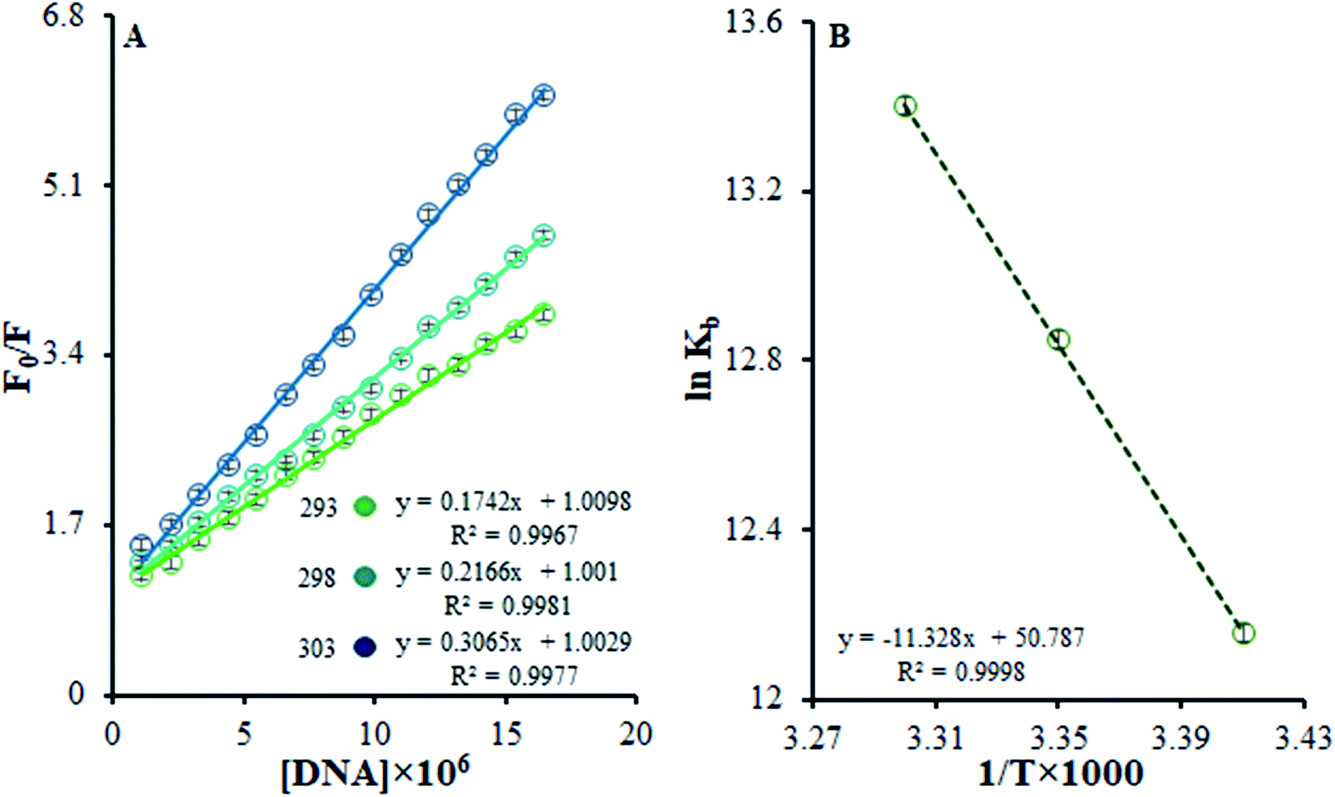 | ||
| Fig. 7 (A) Stern–Volmer quenching plots of F0/F versus [DNA] in the temperature range of 293–303 K. (B) The van't Hoff plot for the interaction of the Ho(III) complex and DNA. | ||
| Temperature (K) | KSV × 105 (M−1) | Kb × 105 (M−1) | ΔG° (kJ mol−1) | ΔH° (kJ mol−1) | ΔS° (J mol−1 K−1) | |
|---|---|---|---|---|---|---|
| DNA | 293 | 1.74 ± 0.02 | 1.9 ± 0.04 | −29.47 | 94.18 | 422.24 |
| 298 | 2.17 ± 0.04 | 3.8 ± 0.02 | −31.57 | |||
| 303 | 3.06 ± 0.03 | 6.6 ± 0.03 | −33.69 | |||
| BSA | 293 | 2.21 ± 0.02 | 6.81 ± 0.02 | −32.72 | −34.07 | −4.65 |
| 298 | 1.93 ± 0.03 | 5.52 ± 0.05 | −32.69 | |||
| 303 | 1.60 ± 0.02 | 4.29 ± 0.04 | −32.67 | |||
| HSA | 293 | 1.81 ± 0.03 | 8.35 ± 0.05 | −33.33 | −42.17 | −32.31 |
| 298 | 1.44 ± 0.03 | 6.42 ± 0.02 | −33.17 | |||
| 303 | 1.19 ± 0.04 | 4.68 ± 0.02 | −33.01 | |||
Eqn (3) was used to obtain KSV values by utilizing the linear relationships at different temperatures. It was shown that there is only one possible quenching procedure. According to Table 2, the KSV values are too large to occur because of dynamic quenching. Based on the outcomes, there is a specific interaction between FS-DNA and the Ho(III) complex, and the quenching technique is potentially a static procedure.
Table 2 shows that, based on the ΔG° negative value and the exothermic nature of the reaction coinciding with the positive entropy and enthalpy variations, there is spontaneous binding between DNA and the Ho(III) complex.
The most prominent force binding FS-DNA to the Ho(III) complex is the hydrophobic interactions resulting from the positive values of ΔS° and ΔH°. Evidently, the entropy of origin is the determining parameter influencing the stability of the DNA–complex. The discharge of counter ions or water molecules from the surface of the bound interacting partners may be determined during entropically driven DNA–complex interactions upon their binding, causing entropic gains. Adverse effects from enthalpy neutralize this impact due to the breaking of hydrogen bonds and non-covalent counter ion and water molecule interactions with non-complex partners. Furthermore, such interactions are enthalpically driven in the case of DNA intercalators and entropically driven in the case of groove binders.40 The thermodynamic information displayed the importance of entropy during DNA–complex creation, whilst the role of enthalpy was less significant. To conclude, the Ho(III) complex interacts with FS-DNA through hydrophobic groove binding.
The definition of intercalation is the insertion of a planar molecule among DNA base pairs, causing decreased DNA helical twisting and the extension of DNA. Thus, the lengthening and unwinding of the DNA helix in the presence of an intercalator may originate from base pairs dividing to adhere to the binding complex, hence enhancing DNA viscosity. To the contrary, there are no differences in the viscosity of DNA solution in the presence of agents that are bound to DNA through groove binding, and the twisting or bending of the DNA helix that may take place in the presence of agents that are electrostatically bound to DNA will decrease the viscosity and length concomitantly.28 Prior examinations exist in regard to the impact of Ho(III) complexes on the viscosity of FS-DNA. A plot of the relative specific viscosity (η/η0)1/3 versus the [complex]/[DNA] ratio for the current complex exhibits no substantial variations in terms of viscosity. Thus, it can be concluded that interactions between FS-DNA and the Ho(III) complex take place via a groove-binding mode. The results of the viscosity tests validate the results from spectroscopic investigations.
 | ||
| Fig. 8 CD spectra of (A) FS-DNA, (B) BSA and (C) HSA in the absence (solid line) and presence (dashed line) of the Ho(III) complex. | ||
High Ho(III) complex concentrations decreased the positive and negative band concentrations but there were no changes in the shape; this shows that the DNA binding of FS-DNA with the Ho(III) complex induces specific conformational variances whilst reducing base stacking. The changes in the FS-DNA conformation originate from a transition from a B-type to a C-type formation in the DNA molecule, as shown in Fig. 8a. The viscosity and spectral evaluations are validated by the CD spectra investigations.
3.3. BSA and HSA binding studies
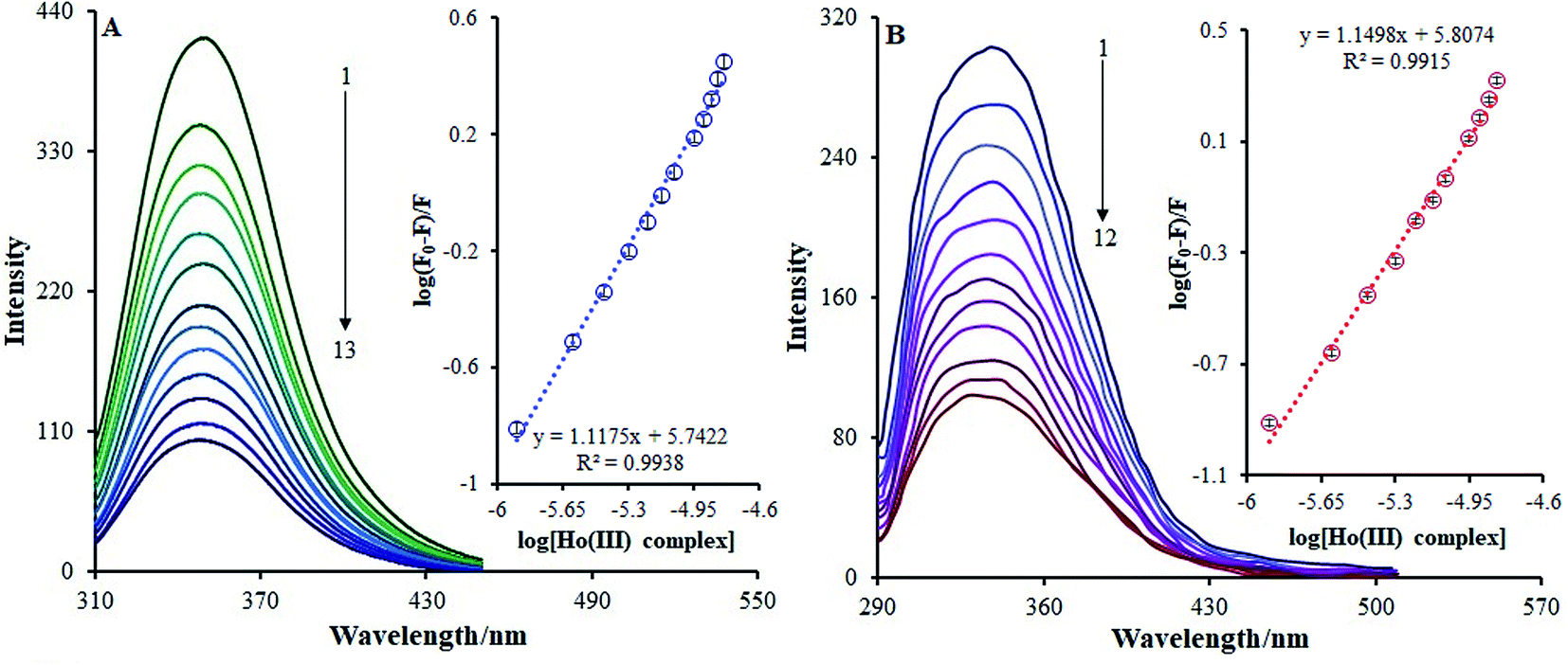 | ||
| Fig. 9 (A) The fluorescence spectra of BSA–Ho(III) complex; BSA concentration: 3.6 × 10−6 M in Tris-buffer at pH 7.2; Ho(III) complex (1 is lowest, 13 is highest): 0, 1.25, 2.51, 3.75, 5.0, 6.25, 7.5, 8.75, 11.25, 12.5, 13.75 and 16.25 μM; inset: plot of log[Ho(III) complex] vs. log(F0 − F)/F. (B) The fluorescent spectra of HSA–Ho(III) complex; HSA concentration: 1.57 μM in Tris-buffer at pH 7.2; Ho(III) complex (1 is lowest, 12 is highest): 0, 1.25, 2.51, 3.75, 5.0, 6.25, 7.5, 8.75, 11.25, 12.5, 13.75 and 15.0 μM; inset: plot of log[Ho(III) complex] vs. log(F0 − F)/F. | ||
In this case, F0 and F are the BSA fluorescence emission intensities in the presence and absence of the Ho(III) complex, respectively, n is the quantity of binding sites, Kb is the binding constant, and Q is the Ho(III) concentration acting as a quencher. The calculation of log(Kb) can be conducted based on the intercept on a plot of log[(F0/F)/F] versus log[Q] (inset Fig. 9a). Table 2 shows that the Kb value at 298 K is 5.52 × 105 M−1, and this is increased and decreased within the 293–303 K temperature range. It is noteworthy that a Kb value of 104 to 106 M−1 is suitable for drug–carrier complexes within the blood.43 Thus, such a Kb value indicates that BSA can be regarded as a favorable carrier for transferring the Ho(III) complex in vivo.
Via utilizing a fluorescence approach, useful information relating to the interactions between HSA and metal complexes can be acquired, e.g., binding modes, thermodynamic parameters, binding parameters, and the quenching mechanism.44
Fig. 9b shows the HSA fluorescence quenching spectra in the presence or absence of the Ho(III) complex. At 337 nm, the fluorescence emission of the HSA characteristic broad band was quenched as the concentration of the Ho(III) complex increased. Such quenching may be accredited to possible variances in the HSA secondary structures, which indirectly indicates the binding of Ho(III) to HSA.
The binding constant (Kb) and number of binding sites (n) can be acquired using the adjusted Stern–Volmer equation (eqn (2)). Upon plotting log(F0 − F)/F vs. log[Q] (Fig. 9b), the n and Kb values may be attained from the intercept and the slope of the double logarithm regression curve, and the relevant data are given in Table 2. The value of Kb at room temperature was 6.42 × 105 M−1, showing that the amino acid side chain within the Ho(III) complex did not have any impact on the Ho(III) complex association constant with respect to HSA. Moreover, the HSA-binding constant proves that HSA may be regarded as a favorable carrier for transferring the Ho(III) complex in vivo.44
F0 and F are the fluorescence intensities of BSA and HSA in the presence and absence of the quencher, respectively, [Q] is the Ho(III) complex quencher concentration, and KSV is the quenching constant.45,46 Fig. 10a and c depict F0/F vs. [Ho(III) complex] plots at various temperatures at λex = 295 nm. The KSV values are acquired from the slopes of the plots of F0/F vs. [Ho(III) complex] at various temperatures.
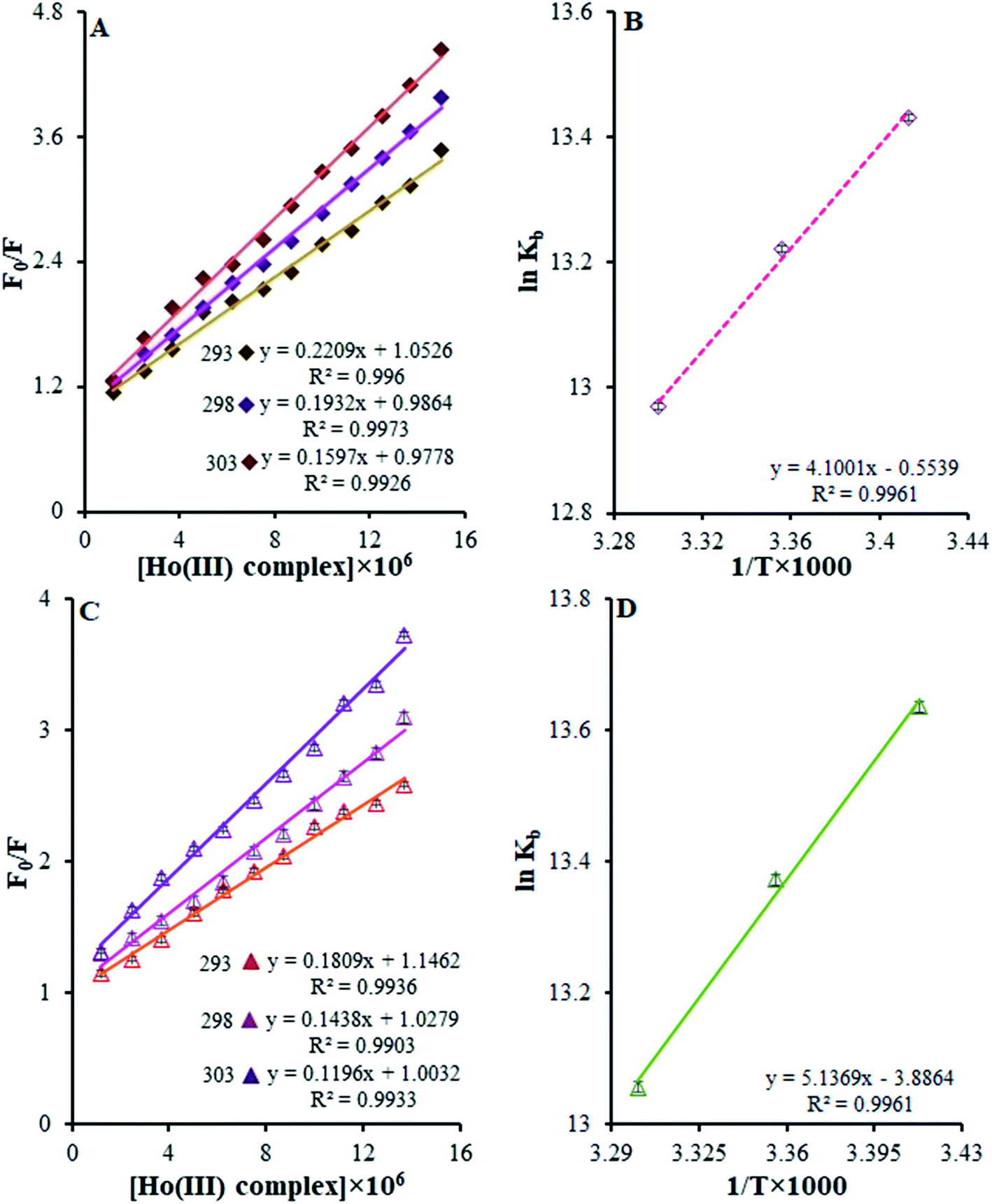 | ||
| Fig. 10 (A) Stern–Volmer quenching plots of F0/F versus [Ho(III) complex] in the temperature range of 293–303 K. (B) The van't Hoff plot for the interaction of the Ho(III) complex with BSA. (C) Stern–Volmer quenching plots of F0/F versus [Ho(III) complex] in the temperature range of 293–303 K. (D) The van't Hoff plot for the interaction of the Ho(III) complex and HSA. | ||
The curves exhibit linearity within the range of experimental concentrations. This implies that there is only one type of quenching during the complex–BSA and HSA interactions.45,46 The derived KSV values for the Ho(III) complex interactions with BSA and HSA at various temperatures are presented in Table 2. From this table, it is clear that the KSV values are reduced when the temperature rises. Such results indicate the possible static nature of the quenching mechanism.
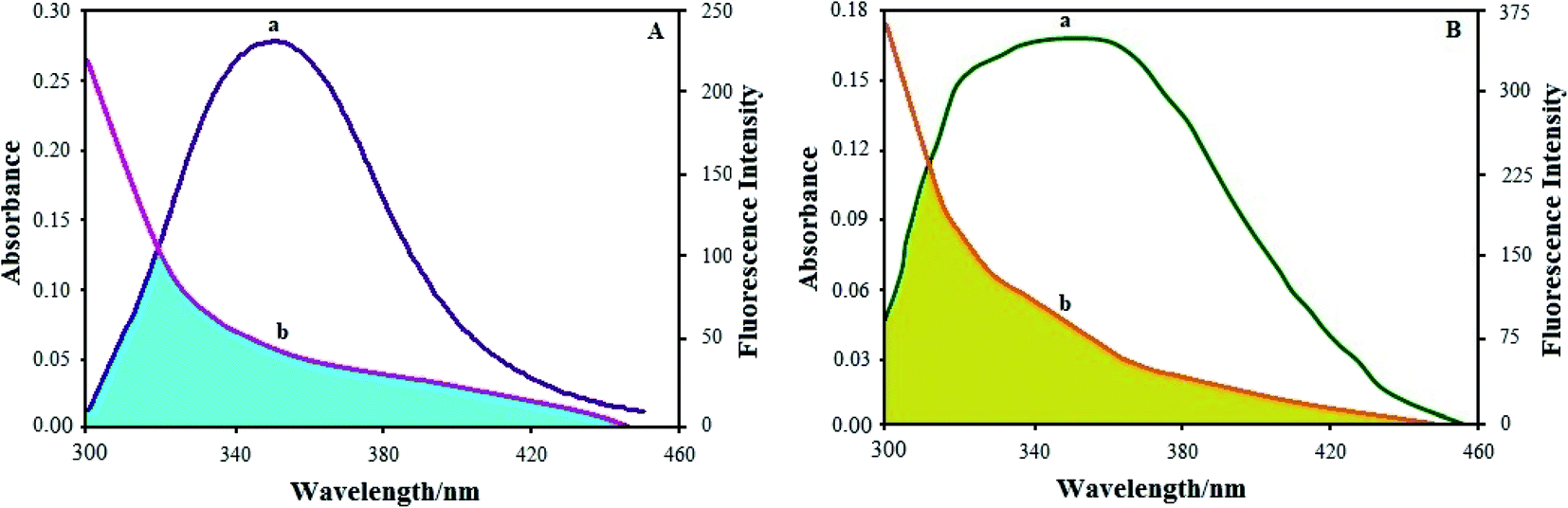 | ||
| Fig. 11 (A) Overlap of the fluorescence spectrum of BSA (T = 298 K) and the UV absorption spectrum of the Ho(III) complex: (a) fluorescence spectrum of BSA; (b) UV absorption spectrum of the Ho(III) complex. (B) Overlap of the fluorescence spectrum of HSA (T = 298 K) and the UV absorption spectrum of the Ho(III) complex: (a) fluorescence spectrum of HSA; (b) UV absorption spectrum of the Ho(III) complex. | ||
Based on Förster resonance energy transfer theory,29 the binding sites r must be within the vicinity of 2–8 nm in a state where 0.5R0 < r < 1.5R0. The current results from the BSA and HSA–Ho(III) complex interactions satisfy the aforementioned condition, thus displaying the high potential for energy transfer from BSA and HSA molecules in the excited state to the ground state of the quencher complex. The FRET studies complement our research on BSA and HSA fluorescence quenching by the Ho(III) complex via a static mechanism.
CD spectroscopy is considered an influential method for studying variances in protein secondary structures after interactions with metallodrugs. The HSA CD spectrum displays two negative bands at 221 nm and 209 nm, which represent the usual protein α-helical structure because of the involvement of n → π* transfer in the peptide bonds of the α-helix, as shown in Fig. 8c. To gain insight into the variance of the HSA secondary structures once it interacted with the surfactant–Ho(III) complex, HSA CD spectra were documented in the presence and absence of the surfactant–Ho(III) complex.46
3.4. Molecular docking results
 | ||
| Fig. 12 Detailed view of the interactions between the Ho(III) complex and (A) DNA, (B) BSA, and (C) HSA. Only the more important residues for binding are shown. | ||
The Ho(III) complex proved to have the best fit in the DNA minor groove, with a binding energy of −5.31 kcal mol−1. The energies acquired from docking research were in accordance with the obtained experimental results. The high negative energy suggests a formidable interaction between DNA and the Ho(III) complex, which was also shown through different spectral approaches. Thus, the docking results bolster the obtained experimental results and verify that a groove binding mode is operational between DNA and the Ho(III) complex.
3.5. Antibacterial effects
The antibacterial behavior of the Ho(III) complex is investigated based on inhibition region diameter measurements, and MBC and MIC methods. The inhibition region diameter range pertaining to the Ho(III) complex versus bacteria was within the 9.0 to 40.0 mm range, as shown in Table 3. The Ho(III) complex showed significant antibacterial behavior, namely versus S. typhi, VRE, and MRSA. Table 3 shows the adequate efficacy of the Ho(III) complex versus these bacteria. The MBC and MIC values of the Ho(III) complex versus bacteria were in the ranges of 0.14–4.0 mg ml−1 and 14.0–125.0 μg ml−1, respectively. Significant antibacterial behavior was shown by the Ho(III) complex with no consideration for Gram-class, especially toward MRSA, which is arduous to eliminate.49 No antibacterial activity was shown by the ligand whilst using the Ho(III) complex as a biomaterial. The antibacterial behavior of the Ho(III) complex may be explained according to the chelation concept.16 Metal ion polarity may be considerably condensed via chelation because the positive charge is partially shared with donor groups, and also due to the delocalization of electrons within the chelate ring. The condensation of metal ions may be intensified due to chelate lipophilic properties and interactions between the cell walls and metal ions. The charge dispersion and geometry of the metal complex molecules are consistent with bacterial cell walls, enhancing the penetration through the cell wall. Such unique structural adjustment can cause the collapse of the cell penetrability barrier and disrupt typical cell procedures. Interactions between cellular complexes and the metal ions are enhanced because of the diverse metal complex functional groups. The chelated complex may cause the deactivation of numerous cellular enzymes, modifying microorganism metabolic activity.50| Bacteria | Zone of inhibition (mm) | MIC (μg ml−1) | MBC (mg ml−1) | |
|---|---|---|---|---|
| Gram-positive | E. faecium | 15.0 | 125.0 | 3.64 |
| E. faecalis | 9.0 | 125.0 | 4.0 | |
| MRSA | 40.0 | 14.0 | 0.14 | |
| VRE | 37.0 | 16.0 | 0.91 | |
| Gram-negative | E. coli | 30.0 | 31.0 | 1.11 |
| P. aeruginosa | 18.0 | 114.0 | 2.22 | |
| K. pneumoniae | 29.0 | 7.0 | 1.11 | |
| A. baumannii | 28.0 | 31.0 | 1.82 | |
| S. typhi | 34.0 | 16.0 | 1.11 | |
3.6. In vitro cytotoxicity studies
The MTT technique was utilized to evaluate the Ho(III) complex and NN-En-Ho antitumor behavior, assaying the behaviors of MCF-7 and A-549 cancer cell lines in vitro, as presented in Fig. 13 and 14.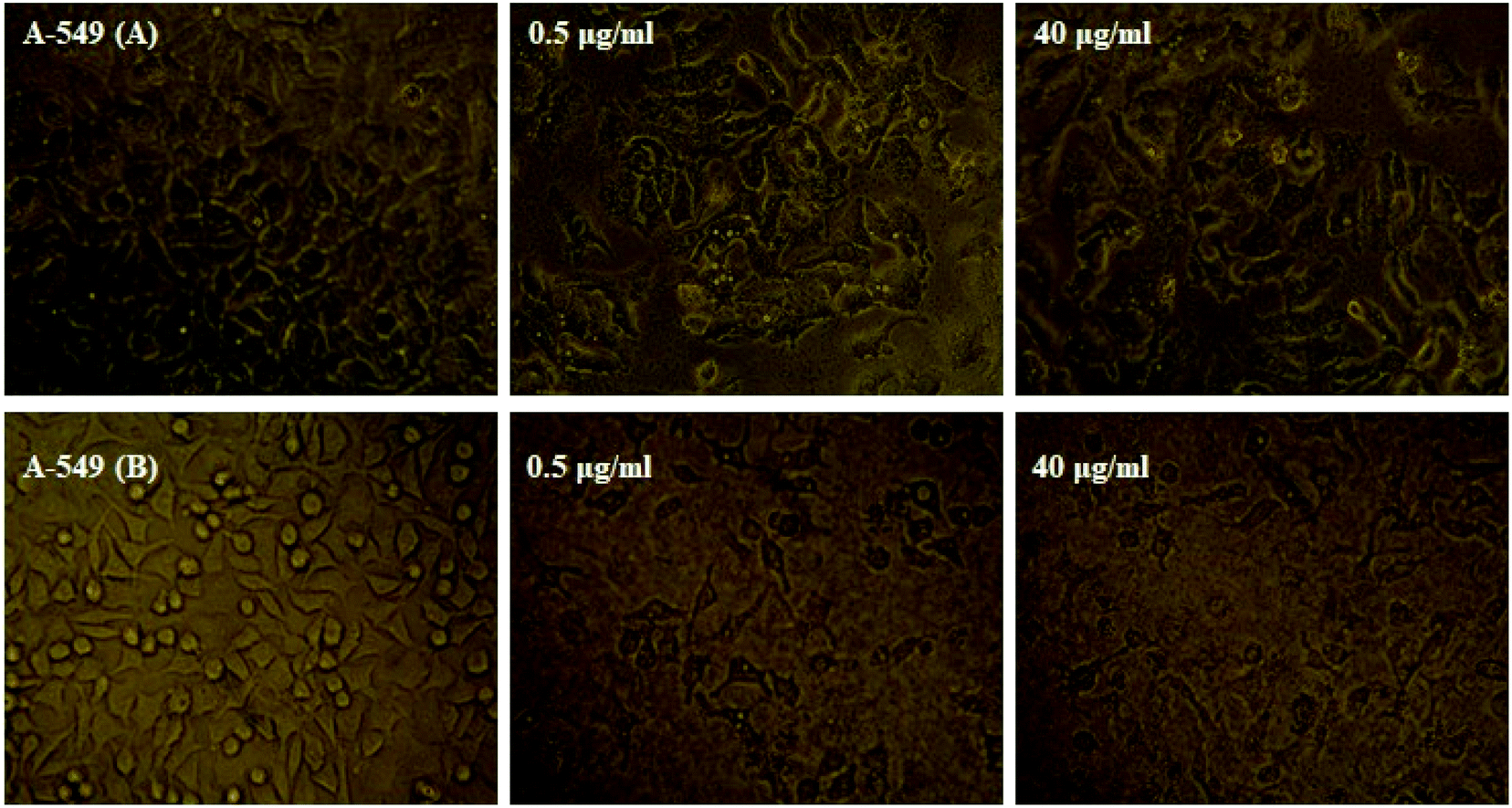 | ||
| Fig. 13 Microscopic photographs of A-549 cancer cells in the absence and presence of different concentrations of the (A) Ho(III) complex and (B) niosome nano-encapsulated Ho(III) complex (NN-En-Ho) (concentrations are indicated by the number inside each photo). | ||
 | ||
| Fig. 14 Microscopic photographs of MCF-7 cancer cells in the absence and presence of different concentrations of the (A) Ho(III) complex and (B) niosome nano-encapsulated Ho(III) complex (NN-En-Ho) (concentrations are indicated by the number inside each photo). | ||
Table S5† shows the inhibitory impact and IC50 values. The analysis results pertaining to variance indicate that the variations between the IC50 mean values of NN-EN-Ho and the Ho(III) complex with respect to both cell lines are statistically meaningful and not randomly created.
Furthermore, high levels of the compound groups produced identical results. The complex displays in vitro cytotoxicity versus the selected cell lines. Fig. 13 and 14 show that enhancing the concentrations of the complexes significantly reduces cancer cell proliferation, exhibiting a dose-reliant effect that hinders the growth of these cells.
Table S5† shows that the NN-En-Ho antitumor behavior is greater compared to the parent Ho(III) complex. The insertion of NN-En-Ho into the cell membrane occurs more effortlessly in comparison to the parent Ho(III) complex. Thus, it facilitates Ho(III) complex penetration into cancer cells whilst improving its antitumor behavior.
3.7. Comparison of the proposed complex with literature complexes
Table 4 presents a comparison of DNA/BSA/HSA binding in this research with other literature studies involving DNA/BSA/HSA binding.35,38,51,52 The proposed Ho(III) complex had larger Kb values in comparison to previously reported complexes in the literature, with the exception of two examples provided in ref. 35 and 54. The advantage of our research over ref. 35 is that we employ an inexpensive materials (bpy versus neo(2,9-dimethyl-1,10-phenanthroline)). In addition, in ref. 35, the interactions between BSA and HSA have not been investigated. Also, it is noteworthy that ref. 38 and 51–54 do not investigated the anticancer activities of the complexes or the construction of drug carriers. This proves that the Ho(III) complex in this study could be a favorable antitumor agent with more developed potential pharmaceutical functionality compared with the methods previously mentioned in the literature.| Complex | Kb × 105 (M−1) | (IC50) (μg ml−1) | Ref. | |||
|---|---|---|---|---|---|---|
| DNA | BSA | HSA | MCF-7 | A-549 | ||
| a Neo: 2,9-dimethyl-1,10-phenanthroline; Phen: 1,10-phanathroline; bpy: 2,2′-bipyridine. | ||||||
| [Ho(Neo)2Cl3·H2O] | 6.12 | — | — | 5.66 | 9.68 | 35 |
| [Ho(Phen)2Cl3]·H2O | 0.107 | — | — | — | — | 51 |
| [Eu(bpy)3Cl2(H2O)]Cl | 2.46 | — | — | — | — | 38 |
| [Tb(bpy)2Cl3(OH2)] | 1.01 | — | 3.63 | — | — | 52 and 53 |
| [Yb(bpy)2Cl3·OH2] | 4.85 | 5.07 | 2.57 | — | — | 54 and 53 |
| [Dy(bpy)2Cl3·OH2] | 2.30 | 1.18 | 2.95 | — | — | 55 and 53 |
| [Ho(bpy)(H2O)6]Cl3 | 3.80 | 5.52 | 6.42 | 7.23 | 11.45 | This work |
4. Conclusions
In the present study, independent [Ho(bpy)(H2O)6]3+ mononuclear complexes with six chloride anions were synthesized, exhibiting high binding affinity to FS-DNA. The interaction mechanism details were investigated for the binding of the Ho(III) complex to FS-DNA. Firstly, the interactions between the Ho(III) complex and FS-DNA were investigated via UV-vis and fluorescence methods under simulated physiological conditions. According to the presented findings, the intrinsic fluorescence of FS-DNA was quenched via a static quenching mechanism. Secondly, we studied the DNA-binding mode of the Ho(III) complex with FS-DNA. The results from EB–DNA competition and iodide quenching experiments, thermodynamic parameter studies, viscosity measurements, CD spectroscopy, and molecular docking studies suggested that the interaction mode between the Ho(III) complex and FS-DNA involves groove binding. The Ho(III) complex also shows good binding affinity with BSA and HSA. The fluorescence results show that the Ho(III) complex is a strong quencher and that it interacts with BSA and HSA through a dynamic quenching mechanism, as KSV decreases with increasing temperature. The signs and magnitudes of the thermodynamic parameters (ΔH°, ΔS° and ΔG°) indicate that the main forces acting between BSA, HSA and the Ho(III) complex are van der Waals and hydrogen bonding forces. According to the results, BSA and HSA can act as carrier proteins for this complex. Moreover, we prepared the parent and encapsulated forms of the Ho(III) complex. All of these complexes revealed significant inhibitory impacts on the A-549 and MCF-7 cell lines; higher in vitro antitumor activity was found in the case of NN-En-Ho because of its higher cellular penetration. Accordingly, it can be said that niosomes are potent drug carriers for this complex, which can act as an effective antitumor agent. We hope that our findings will be helpful for generating knowledge regarding the mechanisms of interaction between small molecule compounds and FS-DNA, BSA and HSA, as well as for developing the potential biological, pharmaceutical and physiological functions of these compounds in the future.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by Ren Shu Project of Hunan Provincial People's Hospital (RS201915). The crystallographic part was supported by Project 18-10504S of the Czech Science Foundation using instruments of the ASTRA lab established within the Operation program Pragu Competitiveness—Project CZ.2.16/3.1.00/24510.References
- G. Morris, N. Pahad, H. W. Dirr and S. Fanucchi, Arch. Biochem. Biophys., 2018, 657, 56 CrossRef CAS PubMed.
- M. Dickerson, B. Howerton, Y. Bae and E. C. Glazer, J. Mater. Chem. B, 2016, 4, 394 RSC.
- W. Huang, Y. Liu, J. Wang, X. Yuan, H. W. Jin, L. R. Zhang, J. T. Zhang, Z. M. Liu and J. R. Cui, Eur. J. Med. Chem., 2018, 157, 887 CrossRef CAS PubMed.
- S. Conoci, A. Mascali and F. Pappalardo, RSC Adv., 2014, 4, 2845 RSC.
- S. Komeda and A. Casini, Curr. Top. Med. Chem., 2012, 12, 219 CrossRef CAS PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481 CrossRef CAS PubMed.
- P. Adak, B. Ghosh, A. Bauzá, A. Frontera, A. J. Blake, M. Corbella, C. D. Mukhopadhyay and S. K. Chattopadhyay, RSC Adv., 2016, 6, 86851 RSC.
- K. J. Barnham, C. J. Bauer, M. I. Djuran, M. A. Mazid, T. Rau and P. J. Sadler, Inorg. Chem., 1995, 34, 2826 CrossRef CAS.
- T. Gyula, K. Zoltan and S. A. Dean, Inorg. Chem., 2006, 45, 9269 CrossRef PubMed.
- L. Luo, R. Xu, D. Zhao and S. Zhong, J. Rare Earths, 2010, 28, 106 CrossRef CAS.
- S. K. Agrawal and K. C. Gupta, Indian J. Chem., 1988, 27, 1008 Search PubMed.
- T. R. Li, Z. Y. Yang, B. D. Wang and D. D. Qin, Eur. J. Med. Chem., 2008, 43, 1688 CrossRef CAS PubMed.
- A. Zhu, Y. Zhou, Y. Wang, Q. Zhu, H. Liu, Z. Zhang and H. Lu, J. Rare Earths, 2018, 36, 1272 CrossRef CAS.
- S. Aime, S. G. Crich, E. Gianolio, G. B. Giovenzana, L. Tei and E. Terreno, Coord. Chem. Rev., 2006, 250, 1562 CrossRef CAS.
- S. V. Eliseeva and J. C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- T. F. Abbs Fen Reji, A. J. Pearl and B. A. Rosy, J. Rare Earths, 2013, 31, 1009 CrossRef.
- Y. Li and Z. Yang, J. Coord. Chem., 2010, 63, 1960 CrossRef CAS.
- P. M. Pithan, D. Decker, S. I. Druzhinin, H. Ihmels, H. Schönherr and Y. Voß, RSC Adv., 2017, 7, 10660 RSC.
- D. Inci, R. Aydn, Ö. Vatan, T. Sevgi, D. Y. lmaz, Y. Zorlu, B. Çosut, E. Demirkan and N. Çink, J. Biol. Inorg Chem., 2017, 22, 1 CrossRef PubMed.
- M. Raza, Y. Wei, Y. Jiang, A. Ahmad, S. Raza, S. Ullah, Y. Han, Q. Ullah Khan and Q. Yuan, New J. Chem., 2017, 41, 8203 RSC.
- G. Balakrishnan, T. Rajendran, K. Senthil Murugan, M. Ganesan, V. K. Sivasubramanian and S. Rajagopal, J. Lumin., 2019, 205, 51 CrossRef CAS.
- D. S. Reddy, M. Kongot, S. P. Netalkar, M. M. Kurjogi and A. Kumar, Eur. J. Med. Chem., 2018, 150, 864 CrossRef CAS PubMed.
- V. Sharma, P. Cantero-López, O. Yañez-Osses, C. Rojas-fuentes and A. Kumar, J. Mol. Liq., 2018, 271, 443 CrossRef CAS.
- F. Shen, Z. B. Ou, Y. J. Liu, W. Liu, B. F. Wang, Z. W. Mao and X. Y. Le, Inorg. Chim. Acta, 2017, 465, 1 CrossRef CAS.
- S. Dang, L. N. Sun, S. Y. Song, H. J. Zhang, G. L. Zheng, Y. F. Bi, H. D. Guo, Z. Y. Guo and J. Feng, Inorg. Chem. Commun., 2008, 11, 531 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345 CAS.
- M. Shahnawaz Khan, M. Khalid, M. Shahwaz Ahmad, M. Ahmad, M. Ashafaq, R. Arif and M. Shahid, J. Mol. Struct., 2019, 1175, 889 CrossRef.
- P. Singla, V. Luxami and K. Paul, Eur. J. Med. Chem., 2016, 117, 59 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639 CrossRef CAS.
- T. T. Zhao, X. Lu, X. H. Yang, L. M. Wang, X. Li, Z. C. Wang, H. B. Gong and H. L. Zhu, Bioorg. Med. Chem., 2012, 20, 3233 CrossRef CAS PubMed.
- L. Basiri, G. Rajabzadeh and A. Bostan, LWT–Food Sci. Technol., 2017, 84, 471 CrossRef CAS.
- S. Dang, L. N. Sun, S. Y. Song, H. J. Zhang, G. L. Zheng, Y. F. Bi, H. D. Guo, Z. Y. Guo and J. Feng, Inorg. Chem. Commun., 2008, 11, 531 CrossRef CAS.
- Y. C. Liu and Z. Y. Yang, J. Organomet. Chem., 2009, 694, 3091 CrossRef CAS.
- Sh. Jahani, M. Noroozifar, M. Khorasani-Motlagh, M. Torkzadeh-Mahani and M. Adeli-Sardou, J. Biomol. Struct. Dyn., 2019, 37, 4437 CrossRef CAS PubMed.
- S. Kashanian, M. B. Gholivand, F. Ahmadia, A. Taravati and A. Hosseinzadeh Colagar, Spectrochim. Acta, Part A, 2007, 67, 472 CrossRef CAS PubMed.
- F. Chen, Z. Xu, P. Xi, X. Liu and Z. Zeng, Anal. Sci., 2009, 25, 359 CrossRef CAS PubMed.
- Sh. Jahani, M. Khorasani-Motlagh and M. Noroozifar, J. Biomol. Struct. Dyn., 2016, 34, 612 CrossRef CAS PubMed.
- L. Guo, B. Qiu and G. Chen, Anal. Chim. Acta, 2007, 588, 123 CrossRef CAS PubMed.
- S. Niroomand, M. Khorasani-Motlagh, M. Noroozifar, Sh. Jahani and A. Moodi, J. Mol. Struct., 2017, 1130, 940 CrossRef CAS.
- F. Sama, M. Raizada, M. Ashafaq, M. Naqi Ahamad, I. Mantasha, I. Khushboo, M. Shahid, R. Arif, N. A. Shah and H. A. M. Saleh, J. Mol. Struct., 2019, 1176, 283 CrossRef CAS.
- B. Barut, U. Demirbaş, A. Şenocak, A. Özel and H. Kantekin, Synth. Met., 2017, 229, 22 CrossRef CAS.
- S. Nafisi and T. S. Vishkaee, J. Photochem. Photobiol., B, 2011, 105, 34 CrossRef CAS PubMed.
- S. Tabassum, M. Ahmad, M. Afzal, M. Zaki and P. K. Bharadwaj, J. Photochem. Photobiol., B, 2014, 140, 321 CrossRef CAS PubMed.
- M. Anjomshoa and M. Torkzadeh-Mahani, Spectrochim. Acta, Part A, 2015, 150, 390 CrossRef CAS PubMed.
- C. L. Zhang, Y. X. Liu, X. M. Zhang, S. Chen, F. Shen, Y. H. Xiong, W. Liu, Z. W. Mao and X. Y. Le, Mater. Sci. Eng. C, 2018, 91, 414 CrossRef CAS PubMed.
- V. Arumugam, R. Rajamanikandan, M. Ilanchelian, K. G. Moodley and G. G. Redhi, Spectrochim. Acta, Part A, 2019, 210, 299 CrossRef CAS PubMed.
- A. V. Protas, E. A. Popova, O. V. Mikolaichuk, Y. B. Porozov, A. R. Mehtive, I. Ott, G. V. Alekseev, N. A. Kasyanenko and R. E. Trifonov, Inorg. Chim. Acta, 2018, 473, 133 CrossRef CAS.
- F. C. Yang, K. H. Wu, W. P. Lin and M. K. Hu, Microporous Mesoporous Mater., 2009, 118, 467 CrossRef CAS PubMed.
- N. P. Priya, S. V. Arunachalam, N. Sathya, V. Chinnusamy and C. Jayabalakrishnan, Transition Met. Chem., 2009, 34, 437 CrossRef CAS.
- S. Niroomand, M. Khorasani-Motlagh and A. Moodi, J. Photochem. Photobiol., B, 2012, 117, 132 CrossRef CAS PubMed.
- Z. Aramesh-Boroujeni, M. Khorasani-Motlagh and M. Noroozifar, J. Biomol. Struct. Dyn., 2016, 34, 414 CrossRef CAS PubMed.
- Z. Aramesh-Boroujeni, A. K. Bordbar, M. Khorasani-Motlagh, E. Sattarinezhad, N. Fani and M. Noroozifar, J. Biomol. Struct. Dyn., 2019, 37, 1438 CrossRef CAS PubMed.
- Z. Aramesh-Boroujeni, Sh. Jahani, M. Khorasani-Motlagh, K. Kerman and M. Noroozifar, J. Biomol. Struct. Dyn., 2020, 38, 1711 CAS.
- Z. Aramesh-Boroujeni, S. Jahani, M. Khorasani-Motlagh, K. Kerman, N. Aramesh, S. Asadpour and M. Noroozifar, J. Biomol. Struct. Dyn., 2019, 1, DOI:10.1080/07391102.2019.1689170.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1882574. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03436c |
| This journal is © The Royal Society of Chemistry 2020 |