
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent advances in tin cluster chemistry
Bertram
Peters†
,
Niels
Lichtenberger†
 ,
Eike
Dornsiepen†
and
Stefanie
Dehnen
*
,
Eike
Dornsiepen†
and
Stefanie
Dehnen
*
Fachbereich Chemie, Wissenschaftliches Zentrum für Materialwissenschaften (WZMW), Philipps-Universität Marburg, Hans-Meerwein-Straße 4, D-35043 Marburg, Germany. E-mail: dehnen@chemie.uni-marburg.de
First published on 21st October 2019
Abstract
This perspective summarizes highlights and most recent advances in tin cluster chemistry, thereby addressing the whole diversity of (mostly) discrete units containing tin atoms. Although being a (semi-)metallic element, tin is in the position to occur both in formally positive or negative oxidation states in these molecules, which causes a broad range of fundamentally different properties of the corresponding compounds. Tin(IV) compounds are not as oxophilic and not as prone to hydrolysis as related Si or Ge compounds, hence allowing for easier handling and potential application. Nevertheless, their reactivity is high due to an overall reduction of bond energies, which makes tin clusters interesting candidates for functional compounds. Beside aspects that point towards bioactivity or even medical applications, materials composed of naked or ligand-protected tin clusters, with or without bridging ligands, show interesting optical, and ion/molecule-trapping properties.
 Bertram Peters | Bertram Peters received his BSc in 2014 and his MSc in 2016 from Philipps University of Marburg, the latter with a thesis on the reactivity of sulfidometalates in ionic liquids under the supervision of Stefanie Dehnen. Currently, he is a doctoral research fellow in the Dehnen group, on a project dealing with ionothermal syntheses of chalcogenidometalates. |
 Niels Lichtenberger | Dr Niels Lichtenberger obtained his master's degree in Chemistry from the Philipps-Universität Marburg. He finished his doctoral studies in 2018 under the supervision of Stefanie Dehnen and currently works as a postdoctoral researcher in her group. His research concentrates on the formation, geometric and electronic structures of binary and ternary intermetalloid and heterometallic clusters of heavy group 13, group 15 (semi)metals in combination with d-block and f-block elements. During his graduate studies, he was supported by a PhD scholarship from the Marburg University Research Academy (MARA). In 2018, he received a scholarship from the Deutscher Akademischer Austauschdienst (DAAD) to join the group of Dr Stosh Kozimor at the Los Alamos National Laboratory (NM, USA) to further develop the field of intermetalloid clusters with actinide ions. |
 Eike Dornsiepen | Dr Eike Dornsiepen obtained his master's degree in Chemistry from the Philipps-Universität Marburg. He finished his doctoral studies in 2019 under the supervision of Stefanie Dehnen and currently works as a postdoctoral researcher in her group. He has been and is currently supported by the German Research Society (Deutsche Forschungsgemeinschaft, DFG) within the framework of the Graduate School GRK 1782 and the Collaborative Research Center SFB 1083. His research focuses on organotin chalcogenide clusters and their ternary derivatives, especially with particular emphasis on their nonlinear optical properties. |
 Stefanie Dehnen | Stefanie Dehnen obtained her diploma in 1993 and her doctoral degree in 1996 from the University of Karlsruhe (now KIT) under the supervision of Dieter Fenske. After a postdoctoral stay with Reinhart Ahlrichs (1997) she completed her Habilitation in Inorganic Chemistry in 2004. In the same year, she was awarded the Wöhler Young Scientists Award from the German Chemical Society (GDCh). As of 2006, she is Full Professor of Inorganic Chemistry and Director of the Scientific Center of Materials Science (Executive Director from 2012–2014) at Philipps-Universität Marburg. She is currently an elected member of the Board of GDCh, and the chairperson of the Division for Inorganic Chemistry (Wöhler-Vereinigung für Anorganische Chemie) at GDCh. She is spokesperson of the review board for molecular chemistry of the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG). In 2016, she has been elected full members of Göttingen Academy of Sciences and Humanity (Akademie der Wissenschaften zu Göttingen) and Academy of Sciences and Literature, Mainz (Akademie der Wissenschaften und der Literatur Mainz), and in 2019, she has been elected full member of the European Academy of Sciences (EurASc). In 2018, she was awarded the Philipps-Universität Marburg Award for Support of Women in Science. Her current research interests comprise synthesis, formation mechanisms, stability, reactivity, and physical properties of compounds and materials with binary and ternary chalcogenidometalate anions, organotetrel chalcogenide compounds, binary Zintl anions and ternary intermetalloid clusters. |
1. Introduction
According to the element tin's broad range of accessible oxidation states (+IV through −I), compounds of this main group element are multi-variant regarding structures, bonding modes, and reactivities.1 For organotin compounds, for instance, it is well-known that subtle changes of ligand types and numbers greatly affect bioactivity and toxicity, with mono-organotin species being relatively benign with respect to tin compounds with more than one organic ligand.2–5 Inorganic tin compounds, like tin chalcogenides, on the other hand, are semiconductors and significantly less harmful regarding their biological impact.6,7 Hence, investigations of tin compounds can be found in essentially all fields of chemical science.Currently, one of the most rapidly developing families of tin compounds to be obtained and analyzed in condensed phase are clusters, with their correspondingly wide range of properties. While some of the clusters give answers to fundamental aspects of chemical bonding and inorganic reaction pathways or even mechanisms, others point to new opportunities for nonlinear optics or semiconductor technologies. Clusters with organic ligands furthermore allow for fine-tuning of the biological activity of tin compounds through alterations of the organic moieties.4,8–11
This perspective article intends to highlight some of the most impressive results reported in this context in recent years and to point towards future developments. For addressing clusters with tin in its highest oxidation states as well as clusters with tin in its lowest oxidation states, the following sections will focus on (i) ligand-free chalcogenido stannate(-metalate) clusters, (ii) organotin(-metal) chalcogenide clusters, and (iii) tin-containing Zintl-type and related clusters. All three types of compounds are prepared from fundamentally different starting materials that typically differ in the oxidation state of the tin atoms. While this choice determines the general type of reaction product, an accurate prediction of reaction products is still almost impossible in cluster chemistry. Yet, upon surveying the field, one recognizes common patterns in structures and bonding, which allows to understand more about these compounds, their formation and their properties. In the meantime, there exist well-established synthesis protocols for reproducible cluster syntheses, indicating that these clearly developed away from purely black-box chemistry.
Without going too much into details of synthesis procedures that can be found elsewhere,12–17 structural, chemical, and physical properties of the selected examples of tin clusters will be named, illustrating the breadth and beauty of this area of research, and also pointing to possible implications of this field for inorganic, organoelement, and materials chemistry.
2. Chalcogenido stannate and metalate clusters
Chalcogenido stannate clusters can be understood as heavier homologues of molecular silicate anions, yet with notable structural differences from these as a consequence of larger ionic radii and longer bonds, and with smaller HOMO–LUMO gaps that cause different (photo-)physical properties. The expansion of the clusters' compositions towards ternary chalcogenido metalate clusters resembles the step from silicates to metalate-silicates, such as zeolites, with similar differences as indicated above, and the additional difference that zeolites never form discrete molecular clusters.Wet-chemical, solvothermal and flux processes belong to traditional cluster synthesis methods that have been known for several decades.12–17 Employed in chalcogenido stannate chemistry, they provide access to various cluster motifs in tetrahedral topologies, commonly known as Tn-type or Pn-type clusters.18 Tn (n = 1–6) denotes supertetrahedral fragments of the sphalerite-type structure with n “layers” of simple (T1-type) tetrahedra {XY4} being connected via corner-sharing. The smallest member of the penta-tetrahedral (Pn, n = 1, 2) family, P1, is based on an “inverse” T1 unit {YX4}, with its faces capped by four “normal” T1 units {XY4}. A vast number of open framework structures in different topologies have been reported, in which supertetrahedral clusters of the same or of different types are linked to form materials with photocatalytic or strong luminescent properties, or ion exchange capabilities.18–20 Isolated, ligand-free clusters, in contrast, have remained rare. Two T3-type clusters [M5Sn5S20]10− (M = Co, Zn; Fig. 1, top left), obtained by reactions in water,21 and a family of P1-type clusters [M4Sn4Ch17]10− (M = Fe, Zn, Co, Mn; Ch = S, Se, Te; Fig. 1, top center), obtained from wet-chemical approaches22–28 or from K2Sx flux reactions,29 show fine-tunable optical properties via alteration of their composition.
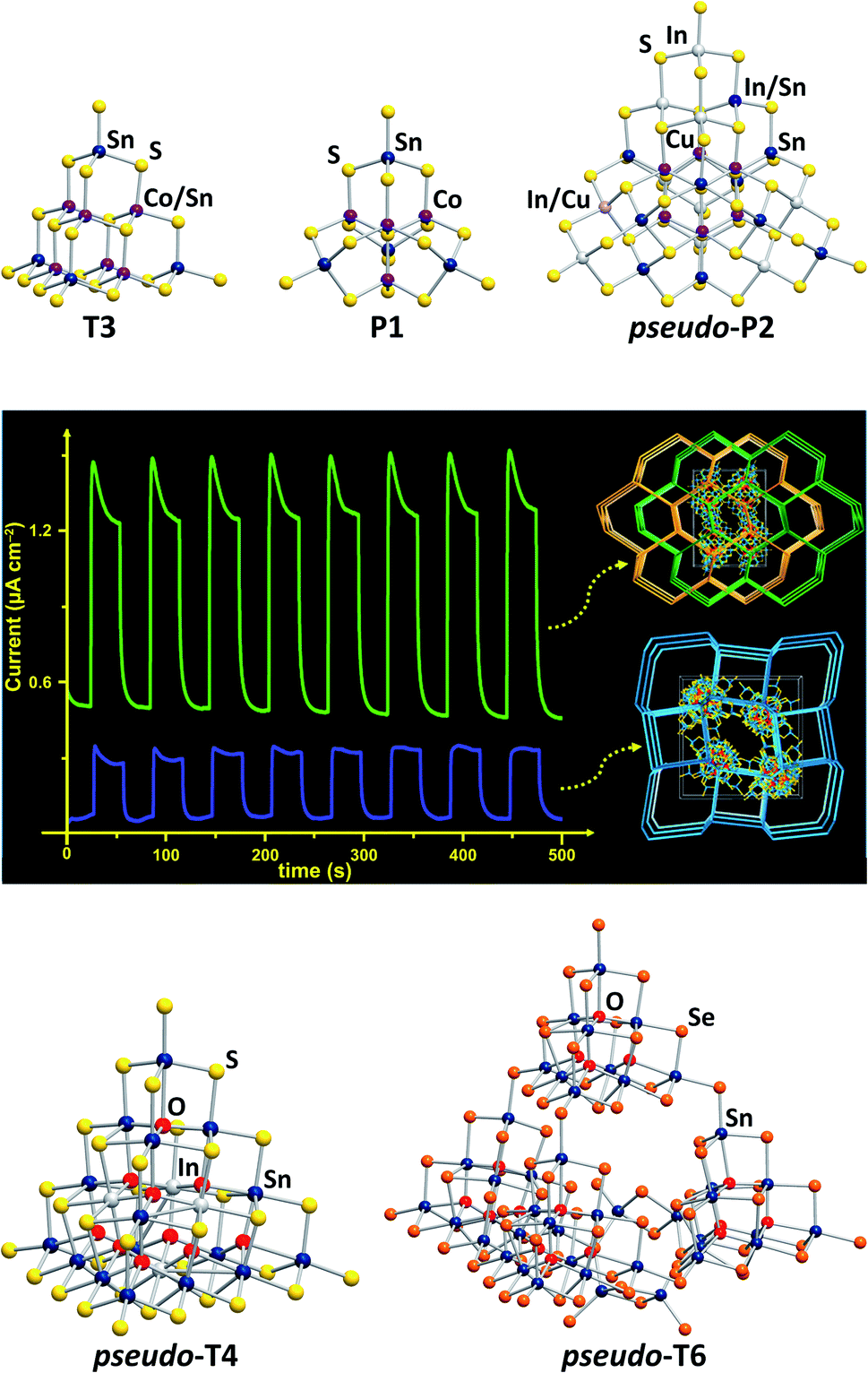 | ||
| Fig. 1 Molecular structures of various cluster anions adopting tetrahedral topologies. Top (from left): [Co5Sn5S20]10−,21 [Co4Sn4S17]10−,29 and [Cu6In12.5Sn7.5S42]10.5−.30 Center: photocurrent response curves of networks formed by corner-sharing of the pseudo-P2-type clusters [Cu6In12.5Sn7.5S42]10.5− or [Cu6.2In10.6Sn9.2S42.5]10.2− (reproduced from ref. 30 with permission from ACS). Bottom: [In4Sn16O10S34]12−,31 and [Sn42O16Se72]8−.32 | ||
Some open-framework compounds, obtained via solvothermal reactions, comprise pseudo-Tn-type clusters and feature a range of interesting (physico-)chemical properties. A network of the largest examples of (pseudo-)Pn-type clusters, which was also obtained by means of solvothermal reactions, comprises pseudo-P2-type clusters [Cu6.2In10.6Sn9.2S42.5]10.2− and [Cu6In12.5Sn7.5S42]10.5− (Fig. 1, top right).30 The compounds show a relatively rapid photocurrent response and high electrocatalytic oxygen reduction activity (Fig. 1, center). A network based on the pseudo-T4-type [In4Sn16O10S34]12− units (Fig. 1, bottom left), for instance, exhibits efficient heavy metal ion sequestration capabilities.31 The pseudo-T6-type cluster [Sn42O16Se72]8− (Fig. 1, bottom right), composed of four corner-linked T3-type {Sn10O4Se20} units and an {Sn2Se6} unit, forms an extremely robust framework with a narrow band gap.32
Both for isolated clusters and for networks of the latter, it was shown that the cluster size and composition, especially regarding the nature of involved transition metal atoms, has a major influence on the molecular electronic excitation energies and the band gaps of the solid materials, which in turn affect the photocatalytic activity.17–19
The most recent development for the synthesis of chalcogenido stannate and metalate clusters is the application of uncommon reaction media, like surfactants,33,34 hydrazine,35,36 or ionic liquids.17 Ionothermal reaction conditions, that is reaction in ionic liquids under slightly elevated temperatures, provide several advantages, the most important one being the large adjustability of the reaction media's properties. At the same time, the reaction conditions, including slightly elevated temperatures, provoke the formation of non-classical, highly exceptional structures. Products with extended anionic substructures featuring new network topologies as well as new molecular cluster motifs are obtained. One example for the latter is a giant spherical anion with 192 atoms, [Ge24Sn36Se132]24− (Fig. 2, top left).37 This cluster, called “Zeoball”, represents the largest main group element polyanion known to date. Salts comprising this spherical anion are capable of activating halogen–halogen bonds. A very uncommon, pseudo-P2-type cluster comprising an interstitial Cs+ cation, [Cs@Sn4(Ge4Se10)4]7− (Fig. 2, top right), was obtained under ionothermal conditions upon in situ reduction of Sn(IV) from SnCl4 to Sn(II) by a low-valent main group cluster in the presence of T2-type [Ge4Se10]4− anions.38 Very recently, a selective methylation of terminal chalcogenide ligands was observed to take place in imidazolium-based ionic liquids, with the (non-innocent) ionic liquid itself acting as a relatively benign methylation agent. The resulting clusters [Sn10O4S16(SMe)4]4− (Fig. 2, bottom left) and [Sn4Mn4Se13(SeMe4)]6− (Fig. 2, bottom right) consequently possess smaller anionic charges than the purely inorganic parent compounds.39
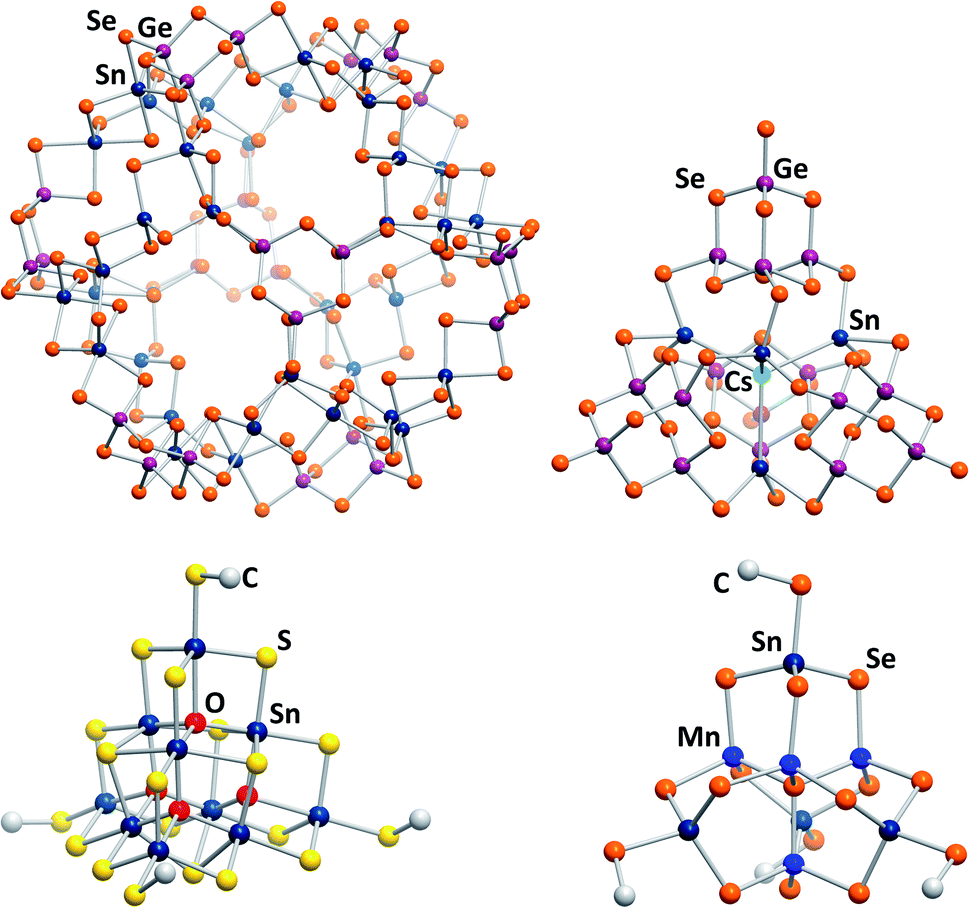 | ||
| Fig. 2 From top left: molecular structures of the “Zeoball” anion [Ge24Sn36Se132]24−,37,40 the pseudo-P2-type cluster [Cs@Sn4(Ge4Se10)4]7−,38 and the methylated cluster anions [Sn10O4S16(SMe)]4−, and [Mn4Sn4Se13(SeMe)4]6−.39 | ||
3. Organotin chalcogenide clusters and related compounds
In contrast to the chalcogenido stannate clusters described in the previous section, organotin chalcogenide clusters are usually neutral, hybrid inorganic/organic compounds that consist of an inorganic core comprising tin and chalcogen atoms, which is protected by organic substituents at the tin atoms. Organotin compounds have been used as biocidal agents, for antifouling treatment,8 fungicides, acaricides, and antimicrobial agents,9 which has been one of the motivations of extending the research in this field, besides the large variety of structures and the physical properties of such compounds.Historically, this chemistry was dominated by organotin oxide clusters.41–49 Such compounds were obtained by different procedures, including reactions of organotin chlorides with (tBu2SnO)3 as oxide source, which afforded compounds like the ladder-type cluster [{RSnCl(CH2)3SnCl(CH2)3SnClRO3}4] (R = CH2SiMe3; Fig. 3, left).46 Hydrolysis reactions led to a variety of organotin oxide clusters like [tBu2(OH)Sn(O)SnPh(CH2Me2SiO)-SntBu2O]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 47 or [RSn(OH)O]6 (R = 2,6-(Me2NCH2)2C6H3).48 A rather unusual way of preparing organotin oxide clusters is the reaction of organotin trichlorides with Na2O in liquid ammonia, yielding the adamantane-type cluster [(RSn)4O6] (R = C(SiMe3)3).49 Recent highlights in this field include [{(nBuSn)12O14(OH)6}(O3SC6H4–NH2)2], which exhibits significant cytostatic activity towards lung cancer cells,50 or the gold complex-decorated [({Me2Sn(dppba)(AuCl)}2O)2], (dppba = p-diphenylphosphino benzoate, Ph2P(4-(COO−)C6H4)) which exhibits multi-color photoluminescence upon incorporation of different aniline derivatives into the voids of its crystal structure.51
47 or [RSn(OH)O]6 (R = 2,6-(Me2NCH2)2C6H3).48 A rather unusual way of preparing organotin oxide clusters is the reaction of organotin trichlorides with Na2O in liquid ammonia, yielding the adamantane-type cluster [(RSn)4O6] (R = C(SiMe3)3).49 Recent highlights in this field include [{(nBuSn)12O14(OH)6}(O3SC6H4–NH2)2], which exhibits significant cytostatic activity towards lung cancer cells,50 or the gold complex-decorated [({Me2Sn(dppba)(AuCl)}2O)2], (dppba = p-diphenylphosphino benzoate, Ph2P(4-(COO−)C6H4)) which exhibits multi-color photoluminescence upon incorporation of different aniline derivatives into the voids of its crystal structure.51
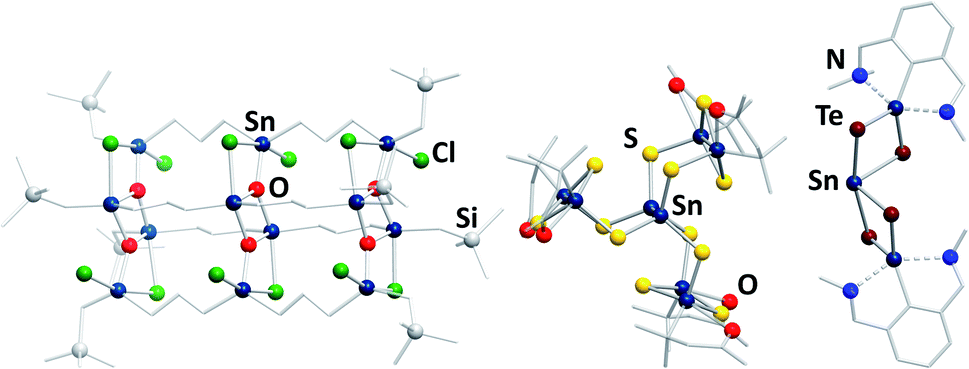 | ||
| Fig. 3 From left: molecular structures of [{RSnCl(CH2)3Sn(Cl)(CH2)3SnClRO3}4] (R = CH2SiMe3),46 [(RSnIV)6SnIII2S12] (R = CMe2CH2CO(Me)),60 and [(RSn)2SnTe4] (R = 2,6-(Me2NCH2)2C6H3).62 | ||
Organotin chalcogenide clusters, hence containing the heavier homologues of oxygen, are typically obtained by reactions of organotin trichlorides with a chalcogenide source (Ch = S, Se, Te). The first known cluster of this type, [(MeSn)4S6], was initially prepared in 1903,52 structurally characterized in 1968,53 and found to possess an adamantane-type inorganic scaffold. A second, “double-decker”-type isomer was found as late as in 2007 for [{o-(Me2NCH2)C6H4}Sn4S6]; the isomeric {Sn4S6} cluster core is stabilized by coordination of σ-donor ligands to the tin atoms, and a corresponding increase of the coordination number from four to five.54 Stepwise condensation reactions of RSnCl3 and (Me3Si)2Ch55,56 allowed for the isolation of several reaction intermediates, including semicube-type clusters [(RSn)3Ch4Cl] (R = CMe2CH2CO(Me)) that also feature five-coordinate tin atoms.56–58 In the aforementioned compounds, all tin atoms are in the formal +IV oxidation state. However, some mixed-valence clusters have also been reported, in which Sn(+IV) and Sn(+II) atoms coexist.59 Additionally, one example of a mixed-valence cluster, [(RSnIV)6SnIII2S12] (R = CMe2CH2CO(Me); Fig. 3, center), features tin atoms in the rare +III oxidation state.60,61
The treatment of low valent tin species, such as R2Sn (R = 2,6-(Me2NCH2)2C6H3), bearing sterically demanding organic substituents with elemental chalcogens yields compounds like [(RSn)2SnTe4] (R = 2,6-(Me2NCH2)2C6H3; Fig. 3, right),62 or the mixed-chalcogen compounds [(RSnCh)2Ch′] (R = 2,6-(Me2NCH2)2C6H3; Ch/Ch′ = S/Se, S/Te, Se/Te).63 Clusters of the composition [(RR′Sn)2SnTe4] (R = N{2,6-(iPr)2C6H3}SiMe3; R′ = 2,4,6-{CH(SiMe3)2}3C6H2) were also obtained by reaction of RR′SnLi2 with TeCl2.64 Another synthetic approach starts out from the organotin hydride RSnH3 (R = 2,6-{2,4,6-(iPr)C6H2}2C6H3), which is reacted with elemental chalcogens to yield clusters with trichalcogenide bridges.65
Organotin chalcogenide clusters can serve as starting ground for follow-up reactions, either by extension of their inorganic core, or by derivatization of their organic substituents. Modifications of the inorganic core are usually achieved by reactions with transition metal complexes.15 The first cluster with a respective ternary inorganic core, [(PhSn)2(CuPPhMe2)6S6], was obtained from [(PhSn)4S6], Na2S and [Cu(PPhMe2)3Cl].66
Some of the structurally most exceptional examples are mixed-valent clusters like [(RSnIV)4(SnIICl)2(MPPh3)2Ch8] (R = CMe2CH2CO(Me); M/Ch = Cu,Ag/S,55 and Cu,Ag/Se67,68), but also large clusters like the isomeric species [(RSn)12Ag14Se25] with R = CMe2CH2CO(Me) (Fig. 4, left) or R = CMe2CH2CNNH2(Me),67 or the cluster [(PhSn)18Cu10S31(PPh3)4Cl2] (Fig. 4, right).69
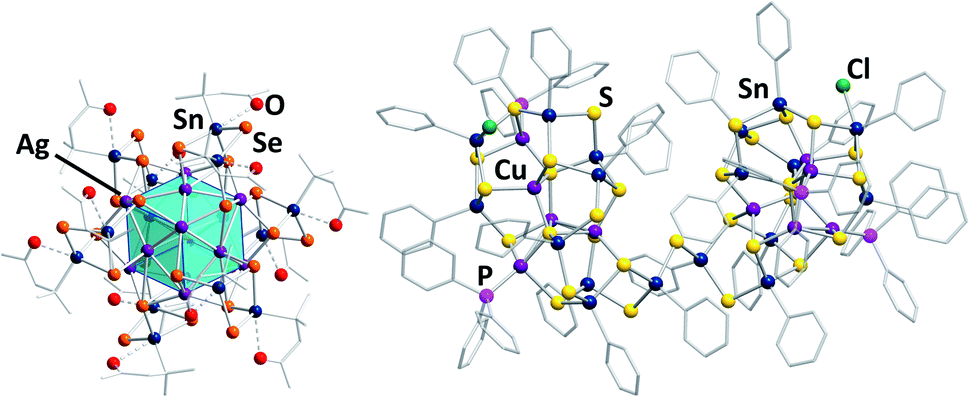 | ||
| Fig. 4 From left: molecular structures of [(RSn)12Ag14Se25] (R = CMe2CH2CO(Me))67 and [(PhSn)18Cu10S31(PPh3)4Cl2].69 | ||
Derivatization of the organic substituents require the presence of organic substituents with suitable functional groups. In most reported cases, condensation reactions between –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –NH2 groups were applied. A wide range of organic and organometallic molecules have been attached to organotin chalcogenide clusters this way, including poly- and heteroaromatic groups,70,71 metallocenes,72–74 or diamandoids.75
O and –NH2 groups were applied. A wide range of organic and organometallic molecules have been attached to organotin chalcogenide clusters this way, including poly- and heteroaromatic groups,70,71 metallocenes,72–74 or diamandoids.75
Reactions with bifunctional molecules cause intramolecular bridging of inorganic scaffolds by the organic substituents under formation of macrocyclic or cavitand-like molecules of the composition [(Sn6S10)2R4] (R = {CMe2CH2C(Me)NNH}2Naph or {CMe2CH2C(Me)NNCH}2C6H4),76,77 or [(Sn3Ch4)2R3] (R = {CMe2CH2C(Me)NNHCO}2(C2H4)4, {CMe2CH2C(Me)NNH}2Naph; Ch = S, Se).76–78 Here, two tin chalcogenide cages of either defect-heterocubane-type architecture {Sn3Ch4} or doubly μ-Ch-linked double-defect-heterocubane-type architecture {Sn6Ch10} are bridged by three or four organic linkers. With carbohydrazide as linker, an organotin telluride chain of the composition [(R)(HR′)(H2R′)2Sn7Te8Cl4] (R = CMe2CH2CO(Me), R′ = {CMe2CH2C(Me)NN}2CO) was obtained.56
Inspired by the biological activity of organotin compounds, a special emphasis was put on the attachment of biomolecules to organotin chalcogenide clusters in recent years, with the long-term goal of finding new enzyme inhibitors and cytostatic drugs. For this purpose, a wide range of amino acids and oligopeptides have successfully been attached either via reaction with amino acid hydrazides,79,80 or via click chemistry.81,82 Biological or medical use have not yet come into sight, but the groundwork for further extensions in this direction has been laid.
Another field of research that points towards potential applications of organotin chalcogenide clusters addresses extreme nonlinear optical properties of adamantane-type clusters of the general formula [(RSn)4Ch6]. An unprecedented form of white-light generation (WLG) by irradiation with a commercially available continuous-wave infrared laser diode, thus without the use of a pulsed laser, was demonstrated for the first time on [(StySn)4S6], an intrinsically amorphous compound (Fig. 5).83
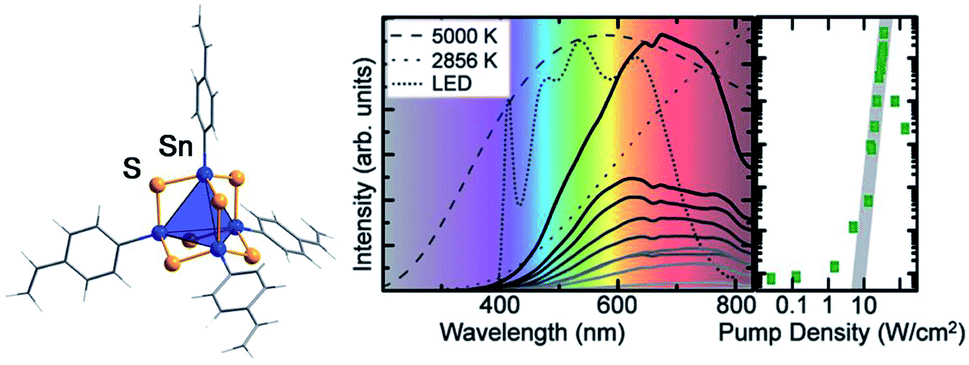 | ||
| Fig. 5 Left: molecular structure of [(StySn)4S6], as obtained from quantum chemical optimization procedures. Right: emission spectrum of [(StySn)4S6] for different excitation densities and an excitation wavelength of 980 nm compared with the emission spectra of black body emitters and a white LED (reproduced from ref. 83 with permission by AAAS). | ||
Further studies showed that this effect seems to be specific to adamantane-type organotetrel clusters and to organic adamantanes,84 given that the samples are amorphous and possess cyclic organic substituents.85,86 This represents the current state of knowledge, as the exact mechanism for this process is still unknown and under investigation. Crystalline samples of this class of compounds also possess nonlinear optical properties, but instead of WLG show efficient second harmonic generation (SHG).
Beside organotin clusters that are bridged by chalcogen atoms, several pnictogenide-bridged clusters were reported, most of which are based on heterocubane-type architectures like [Sn4(NR)4] (R = 2-Me-5-MeOC6H3, 2,5-(MeO)2C6H3, 3,5-(MeO)2C6H3),87 or corner-sharing heterocubanes like [Sn7(2-NR)8] (R = pyrimidinyl, 5-methylpyridinyl).88
In these clusters, however, the oxidation state of the tin atoms is exclusively +II, as the organic substituents are found at the pnictogen (Pn) atoms, which renders them as tin(II) imido clusters. The most recent examples include the organotin(IV) arsenide cluster [(TerSn)4As4] (Ter = Terphenyl).89 The ionic derivative of a tin(II) imide cluster, [Li(thf)Sn3(NtBu)4]−, was generated by aminolysis of [Me2Si(NtBu)2Sn] with H2NtBu and subsequent lithiation of the product [Sn3(NtBu)2(NtBuH)2].90 The lithiated compound can easily be oxidized by elemental selenium and tellurium to form [Li(thf)(SnCh)3(NtBu)4]− (Ch = Se, Te).91
4. Zintl and related low-valent clusters containing tin atoms
While all clusters that were discussed in the preceding sections contain tin atoms in positive oxidation states, this section will address clusters, in which tin atoms are (formally) negatively charged. Anionic main group element clusters were first mentioned in the literature in 1891 by Joannis,92 and famously investigated in a systematic fashion by Zintl in the 1930s.93 Milestone improvements to their synthesis and characterization followed with the introduction of ethylenediamine as a solvent and cation sequestering agents, like crypt-222.94–96Historically, the first stannide clusters were investigated alongside the corresponding lead and germanium clusters. Two structure types are dominant for the polytetrelide cluster anions, namely Tt44− tetrahedra and Tt94− cages.12,97 The latter are fluxional in shape, often adopting capped square antiprismatic or tricapped trigonal prismatic geometries.98,99 Investigations and follow-up chemistry of the Tt44− anions are restricted to liquid ammonia as a solvent due to the high charge per atom in these anions and Sn44− anions still have not been employed to date.100–103 Room temperature reactivity studies have therefore focused on the reactivity of the larger Tt94− anions, most commonly accessed through dissolution of the respective Zintl phases A4Tt9 (A: alkali metal, most often K; Tt = Ge, Sn, Pb).104–106 In recent times, new developments included oxidative coupling of such Zintl anions,107–109 the formation of organic derivatives,110–112 and the extension and transformation of the inorganic core by formation of intermetalloid or heterometallic clusters that combine main group (semi-)metal atoms with d- or f-block metal atoms M.12–14,113 Regarding clusters that comprise tin atoms, most results were obtained in the latter area of research, which will therefore be addressed herein.
Most syntheses of such mixed metallic M/Sn clusters from Zintl anions made use of redox-inert d10 transition metal compounds or such that provide stable complex fragments, prone to simple coordination chemistry. By reacting K4Sn9 in en with Cu(Mes) the simplest intermetalloid clusters possible, [Cu@Sn9]3−, can be obtained, in which the Cu+ ion is incorporated into the {Sn9} cage without affecting its structure.114 Similarly, reactions with [M(CO)3(Mes)] (M = Cr, Mo, W) afford the heterometallic cluster [(Sn9)M(CO)3]4− with an intact Sn94− cage acting as a 6 electron ligand to an {M(CO)3} fragment.115–117 However, Tt94− cages do not remain intact in all reactions; in fact, they are far more likely to rearrange and form new cluster ions that are only stable in the presence of the respective d- or f-block metal atoms. A process similar to the transformation of Pb94− into the first intermetalloid cluster, [Pt@Pb12]2−,118 was observed for Sn94−, affording the isoelectronic anion [Ir@Sn12]3−.119 All of these clusters can be interpreted in terms of Wade–Mingos' rules.120–123 Zintl chemists quickly explored this large reaction space, making use of a vast variety of available transition metal compounds and by reintroducing liquid ammonia as a solvent.
Despite the beauty and aesthetics of deltahedral Wade clusters, it should be noted that their formation is expected for group 14 metallide clusters, as Sn− (or its homologues) is isoelectronic to a BH group. Hence, group 14 (semi-)metal anions that deviate from these geometries are more interesting in terms of their electronic structure. Two of the first examples that featured drastically different geometries were the pentagonal prismatic clusters [M@Ge10]3− (M = Fe, Co).124,125 Later, the structural variety in 10, 12, and 14-vertex intermetalloid clusters of group 14 elements was investigated systematically in a series of quantum chemical studies.126–128 These studies were sparked by the synthesis and crystallization of the isoelectronic stannide cluster [Fe@Sn10]3−,126 which exhibits yet another cluster geometry that can be considered as an intermediate between the two extremes of a deltahedral and a pentagonal prismatic cage. A very flat potential energy hypersurface was found for this anion that led to severe disorder in the solid state and very poor single crystal data.
Another exceptional cluster geometry is observed in the anion [Ti@Sn15Ti3Cp5]4/5− (Fig. 6, top left) that was obtained from reactions of K4Sn9 with [TiCp2Cl2] in liquid ammonia.129 This study is interesting for a number of reasons. First, this cluster was the first polystannide to comprise hard, Lewis-acidic transition metal cations, which is rare in Zintl cluster chemistry in general; most intermetalloid and heterometallic clusters contain softer, electron rich metal atoms. Second, the cluster comprises a naked, endohedral Ti atom besides {TiCp} and {TiCp2} fragments, thereby blurring the boundaries between intermetalloid clusters (with the M atoms inside the stannide cage) and heterometallic clusters (with the M atoms being integrated in a joint cluster shell).
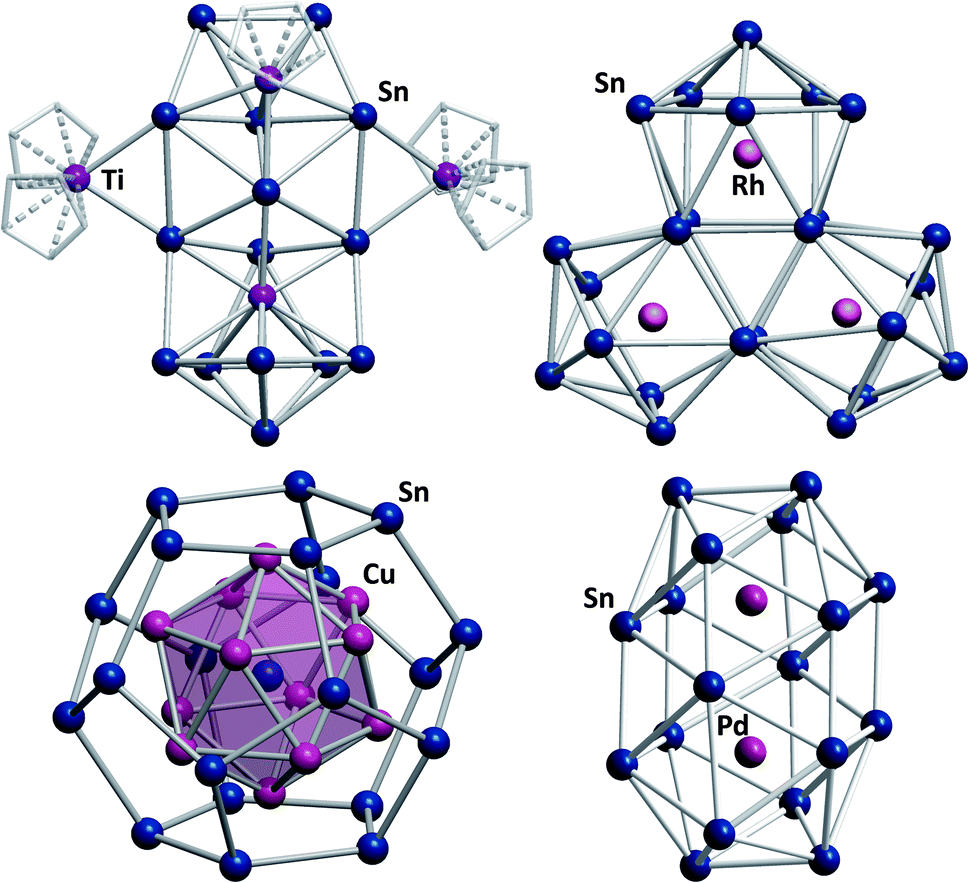 | ||
| Fig. 6 From top left: molecular structures of the cluster anions [Ti@Sn15Ti3Cp5]4/5−,129 [Rh3@Sn24]5−,131 [Sn@Cu12@Sn20]12−,132 and [Pd2@Sn18]4−.133,134 | ||
Lastly, some intermediates isolated alongside the formation of the product cluster provided real, experimental glimpses into potential cluster formation pathways. Indeed, the mechanisms of cluster growth remain a mostly speculative aspect in this chemistry due to short reaction times and a lack of spectroscopic handles.130 Very recently, another report was published that again provides some indirect evidence for a possible cluster formation pathway. Reactions of K4Sn9 with [(coe)2Rh(μ-Cl)]2 (coe = cyclooctene) afford a series of Rh-centered polystannide clusters: [Rh@Sn10]3−, [Rh@Sn12]3−, [Rh2@Sn17]6− and [Rh3@Sn24]5− (Fig. 6, top right).131 The first three of these anions represent well-known intermetalloid cluster types, even though (drastic) distortions away from idealized geometries are observed. This is most notable in [Rh@Sn10]3−, and these effects can be attributed to electron back-donation from the endohedral rhodium atom to the outer cage. [Rh3@Sn24]5− represents the largest polystannide cluster known to date. It was obtained via two different synthetic routes, both of which involve heating solutions in which [Rh@Sn10]3− anions are present. A closer look at the structure of [Rh3@Sn24]5− shows that it can be viewed as a fusion product of three [Rh@Sn10]3− clusters, suggesting that this is at least a plausible reaction pathway. Two further, remarkable structures that were reported some years ago, should also be mentioned: the onion type cluster [Sn@Cu12@Sn20]12− (Fig. 6, bottom left), which occurs in an intermetallic phase and was referred to as a molecular “bronze”,132 and the largest single-cage deltahedral cluster [Pd2@Sn18]4− (Fig. 6, bottom right).133,134
Another branch of Zintl chemistry makes use of small, binary anions as building blocks for larger intermetalloid and heterometallic clusters. The high charge of Sn44− tetrahedra can be reduced by formal replacement of Sn with group 15 elements, yielding isoelectronic (Tt2Pn2)2− anions. These are soluble in ethylenediamine and can be used in follow-up reactions, just like the homoatomic anions discussed previously. Such reactions afford ternary clusters, as first demonstrated with the synthesis of [Zn@Zn5Sn3Bi8]4−.135 The use of these binary precursors also allowed for the first incorporation of f-elements into polyanionic cages, as observed in [Eu@Sn6Bi8]4− (Fig. 7, left), which still has not been achieved with homoatomic tetrelide precursors.136 Since then, this chemistry has successfully been expanded to other elemental combinations, also including (TrBi3)2− anions (Tr = Ga, In, Tl).137–140 Reactions with transition metal complexes indicated drastic differences in reactivity and product formation with respect to homoatomic polystannides: reactions of binary (Sn2Sb2)2− with [K(thf)0.15][Co(cod)2] (cod = 1,5-cyclooctadiene) and [(nacnac)Cu(NCMe)] (nacnac = [(N(C6H3iPr2-2,6)C(Me))2CH]−) afforded the ternary clusters [Co@Sn6Sb6]4− (Fig. 7, right), and [(CuSn5Sb3)2]4−.141,142 As the electronic situation in such anions is often very complex, their characterization is bearing new challenges. The mentioned cluster, [(CuSn5Sb3)2]4− (Fig. 8, top), is a recent example for such a study.142 The anion is to be viewed as a dimer of two {CuSn5Sb3}2− 9-atom cages with weak contacts between them. Quantum chemical analyses suggested the absence of any significant bonding interactions between the copper atoms, despite a relatively short Cu–Cu distance. Detailed studies of the electronic situations addressed (i) the hypothetic halves of the cluster as well as (ii) the final product upon hypothetic dimerization of the cluster halves versus (iii) the respective combination of two homoatomic 9-atomic tin clusters (Fig. 8, center and bottom).
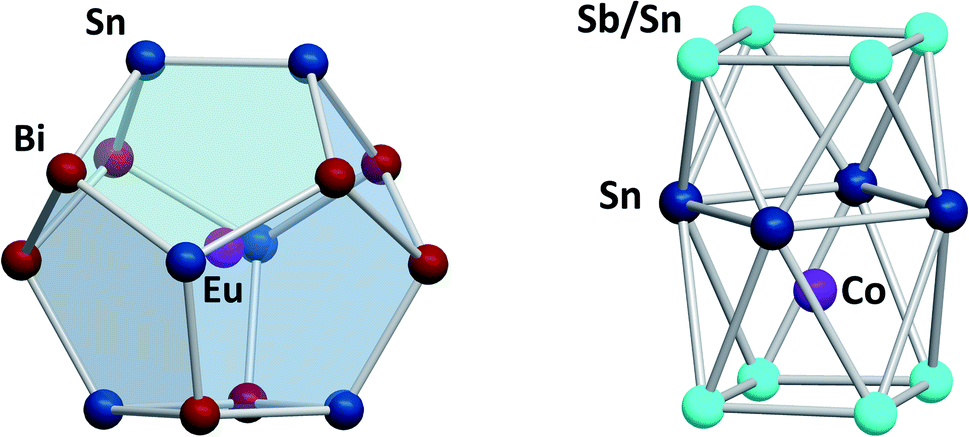 | ||
| Fig. 7 From left: molecular structures of the cluster anions [Eu@Sn6Bi8]4−,136 and [Co@Sn6Sb6]4−.141 The clusters show positional disorder of the main group elements in the cluster shell. The atom distributions depicted here are based on quantum chemical calculations and represent the energetically preferred distribution. | ||
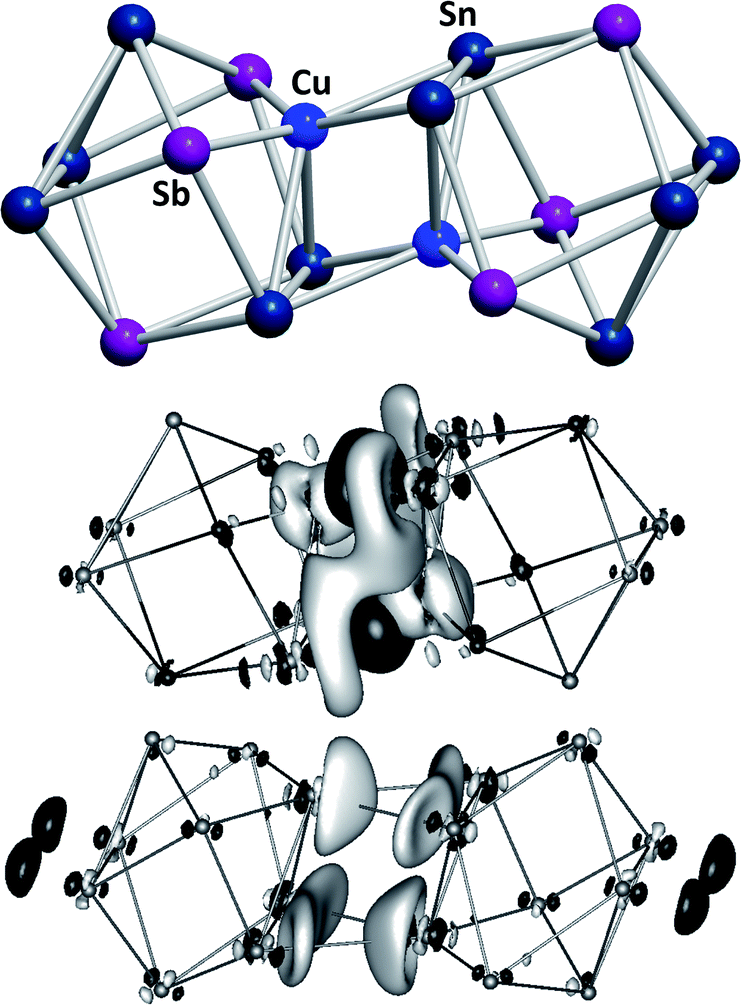 | ||
| Fig. 8 Top: molecular structure of the cluster anion [(CuSn5Sb3)2]4− .142 Center and bottom: difference of electron densities of dimer and monomers upon hypothetic dimerization reactions to form [(CuSn5Sb3)2]4− or the non-existent “(Sn92−)2” analogue. Contours are drawn at 0.0016 a.u. For black areas, electron density in the dimer is higher than in the monomers, for light areas it is lower (reproduced from ref. 142 with permission from Wiley-VCH). | ||
These indicated large differences between the homoatomic and the heteroatomic system, respectively, and thereby helped to understand the ternary cluster. Thus, studies of binary Zintl anions allow for a significant expansion of polyanionic main group element cluster chemistry and additionally provide further insights into aspects of cluster bonding and their formation.
In the past few years a novel direction of Zintl cluster chemistry, based on (element-)organic derivatives of Ge94− anions, most often (Ge9R3)− units, was developed.143 A rich coordination chemistry, based on these derivatives, has evolved since their first preparation.14,144–146 However, a transfer of this work to the less stable Sn94− cages has been done with limited success so far.112 The only reported compound that was obtained this way is [(Hyp)Au(Sn9Hyp3)Au(Sn9Hyp3)Au(Hyp)]− (Hyp = hypersilyl), bearing a cluster-based chain, which was most likely formed upon dissociation of [Sn10Hyp4]2− in the presence of [(PPh3)Au(SHyp)].147 However, several related compounds were synthesized starting out from low valent organotin compounds or low valent tin halides. These clusters typically contain tin atoms in oxidation states 0 and +I (besides a few atoms with oxidation states +II or −I). Consequently, the compounds feature drastically altered properties, including other solubilities and reactivities, as compared to the naked Sn94− anions. Examples are [Sn15R6] (R = N(2,6-iPr2C6H3)(SiMe3)),148 [tBu7MeSn4],149 [(tripSn)6] (trip = 2,4,6-triisopropylphenyl),150 [Sn10{Si(SiMe3)3}4]2−,151 (Fig. 9, left), [{(Me3Si)2CH}6Sn9(NHC)],152 (Fig. 9, right), and [Mes10Sn10].152
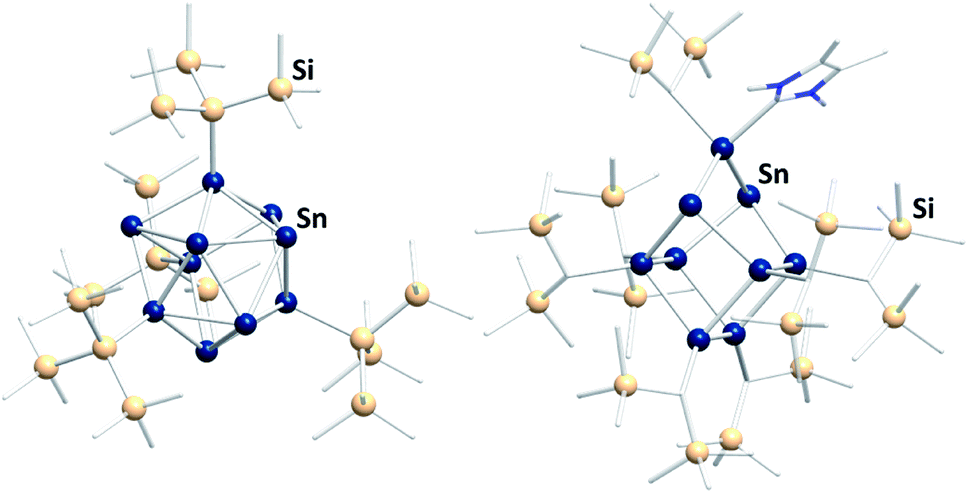 | ||
| Fig. 9 Fom left: molecular structures of the ligand-protected clusters [Sn10{Si(SiMe3)3}4]2−151 and [{(Me3Si)2CH}6Sn9(NHC)].152 | ||
Conclusions and outlook
Tin clusters belong to the most contemporary family of tin compounds and exhibit a high variability, owing to the large range of potential oxidation states to be adopted by this group 14 (semi-)metal, and due to its potential to bind to most other atoms.Very obviously, the oxidation state is correlated with a preference for certain coordination environments and consequently structural features. Tin atoms in higher/highest oxidation states prefer tetrahedral, trigonal bipyramidal, or even octahedral coordination, whereas (slightly) negatively charged or formally neutral tin atoms behave more like metals, and are thus are more flexible regarding coordination numbers and geometries.
In its extreme oxidation state, +IV, tin is not as oxophilic and not as sensitive to hydrolysis as the lighter homologues, which is due to the smaller Sn–O bond energy (414 kJ mol−1) as compared to Ge–O (473 kJ mol−1) and Si–O (598 kJ mol−1),153 which makes handling of its compounds under ambient conditions less problematic. At the same time, all other bond energies to its own substituents are also generally smaller than for the lighter group 14 elements, which allows for a significant reactivity in according reaction mixtures – especially in the less common oxidation states of tin. For this, tin clusters, with their finely tunable electronic properties, should be ideal candidates for bioactive compounds, for catalysts, or for precursors to innovative materials. Despite the high activity in the field, such expansions are yet in their infancy.
Hence, further developments are to be expected – in each of the discussed areas of research, and also in new directions. Maybe, future work will even end up in the co-existence of the extreme versions of Sn(+IV) and Sn(−I) clusters in the same compound, similar to the tetrelide–tetrelate compounds of the lighter homologues silicon and germanium, such as Cs10[Si4][Si3O9] and Rb14[Ge4][Si6O17].154
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) within individual projects and within the frameworks of GRK1782, SFB1082, SPP1415, SPP1708, and FOR2824, by the Alexander von Humboldt Foundation, the Friedrich-Ebert Foundation, the Marburg University Research Academy (MARA), the German Academic Exchange Service (Deutscher Akademischer Austauschdienst, DAAD), and the Fonds of Chemical Industries (FCI).References
- M. Gielen, A. Davies, K. Pannell and E. Tiekink, Tin Chemistry: Fundamentals, Frontiers, and Applications, John Wiley & Sons, 2008 Search PubMed.
- R. D. Kimbrough, Environ. Health Perspect., 1976, 14, 51–56 CrossRef CAS.
- G. Eng, C. Whitmyer, B. Sina and N. Ogwuru, Main Group Met. Chem., 1999, 22, 311 CAS.
- M. Gielen, Appl. Organomet. Chem., 2002, 16, 481–494 CrossRef CAS.
- A. G. Hadi, K. Jawad, D. S. Ahmend and E. Yousif, Syst. Rev. Pharm., 2019, 10, 26–31 CrossRef CAS.
- K. A. Winship, Adverse Drug React. Toxicol. Rev., 1988, 7, 19–38 CAS.
- J. P. Wise and Q. Qin, Hexavalent Chromium and DNA, Biological Implications of Interaction in Encyclopedia of Metalloproteins, ed. R. H. Kretsinger, V. N. Uversky and E. A. Permyakov, Springer, New York, NY, 2013, pp. 975–978 Search PubMed.
- I. Omae, Appl. Organomet. Chem., 2003, 17, 81–105 CrossRef CAS.
- M. Nath, Appl. Organomet. Chem., 2008, 22, 598–612 CrossRef CAS.
- M. Hong, H. Geng, M. Niu, F. Wang, D. Li, J. Liu and H. Yin, Eur. J. Med. Chem., 2014, 86, 550–561 CrossRef CAS.
- M. Martins, P. V. Baptista, A. S. Mendo, C. Correia, P. Videira, A. S. Rodrigues, J. Muthukumaran, T. Santos-Silva, A. Silva, M. F. C. Guedes da Silva, J. Gigante, A. Duarte, M. Gajewska and A. R. Fernandes, Mol. BioSyst., 2016, 12, 1015–1023 RSC.
- S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS.
- R. S. P. Turbervill and J. M. Goicoechea, Chem. Rev., 2014, 114, 10807–10828 CrossRef CAS.
- R. J. Wilson, N. Lichtenberger, B. Weinert and S. Dehnen, Chem. Rev., 2019, 119, 8506–8554 CrossRef CAS.
- E. Dornsiepen, E. Geringer, N. Rinn and S. Dehnen, Coord. Chem. Rev., 2019, 380, 136–169 CrossRef CAS.
- S. Dehnen and M. Melullis, Coord. Chem. Rev., 2007, 251, 1259–1280 CrossRef CAS.
- S. Santner, J. Heine and S. Dehnen, Angew. Chem., Int. Ed., 2016, 55, 876–893 CrossRef CAS.
- P. Feng, X. Bu and N. Zheng, Acc. Chem. Res., 2005, 38, 293–303 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS.
- M. J. Manos and M. G. Kanatzidis, Chem. Sci., 2016, 7, 4804–4824 RSC.
- C. Zimmermann, C. E. Anson, F. Weigend, R. Clérac and S. Dehnen, Inorg. Chem., 2005, 44, 5686–5695 CrossRef CAS.
- S. Dehnen and M. K. Brandmayer, J. Am. Chem. Soc., 2003, 125, 6618–6619 CrossRef CAS PubMed.
- E. Ruzin, S. Jakobi and S. Dehnen, Z. Anorg. Allg. Chem., 2008, 634, 995–1001 CrossRef CAS.
- E. Ruzin, A. Fuchs and S. Dehnen, Chem. Commun., 2006, 4796–4798 RSC.
- C. Zimmermann, M. Melullis and S. Dehnen, Angew. Chem., Int. Ed., 2002, 41, 4269–4272 CrossRef CAS.
- M. K. Brandmayer, R. Clérac, F. Weigend and S. Dehnen, Chem.–Eur. J., 2004, 10, 5147–5157 CrossRef CAS.
- F. Lips and S. Dehnen, Inorg. Chem., 2008, 47, 5561–5563 CrossRef CAS.
- E. Ruzin, E. Zent, E. Matern, W. Massa and S. Dehnen, Chem.–Eur. J., 2009, 15, 5230–5244 CrossRef CAS.
- O. Palchik, R. G. Iyer, J. H. Liao and M. G. Kanatzidis, Inorg. Chem., 2003, 42, 5052–5054 CrossRef CAS.
- J. Lv, J. Zhang, C. Xue, D. Hu, X. Wang, D.-S. Li and T. Wu, Inorg. Chem., 2019, 58, 3582–3585 CrossRef CAS.
- X.-M. Zhang, D. Sarma, Y.-Q. Wu, L. Wang, Z.-X. Ning, F.-Q. Zhang and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 5543–5546 CrossRef CAS PubMed.
- Q. Lin, X. Bu and P. Feng, Chem. Commun., 2014, 50, 4044–4046 RSC.
- F. Camerel, M. Antonietti and C. F. J. Faul, Chem.–Eur. J., 2003, 9, 2160–2166 CrossRef CAS.
- W. Zhu, X. Ou, Z. Lu, K. Chen, Y. Ling and H. Zhang, J. Mater. Sci.: Mater. Electron., 2019, 30, 5760–5770 CrossRef CAS.
- W.-W. Xiong, J. Miao, P.-Z. Li, Y. Zhao, B. Liu and Q. Zhang, J. Solid State Chem., 2014, 218, 146–150 CrossRef CAS.
- J. Han, S. Li, C. Tang, W. Zheng, W. Jiang and D. Jia, RSC Adv., 2018, 8, 34078–34087 RSC.
- Y. Lin, W. Massa and S. Dehnen, J. Am. Chem. Soc., 2012, 134, 4497–4500 CrossRef CAS.
- S. Santner, A. Wolff, M. Ruck and S. Dehnen, Chem.–Eur. J., 2018, 24, 11899–11903 CrossRef CAS.
- B. Peters, S. Santner, C. Donsbach, P. Vöpel, B. Smarsly and S. Dehnen, Chem. Sci., 2019, 10, 5211–5217 RSC.
- S. Santner, S. Yogendra, J. J. Weigand and S. Dehnen, Chem.–Eur. J., 2017, 23, 1999–2004 CrossRef CAS.
- R. O. Day, J. M. Holmes, V. Chandrasekhar and R. R. Holmes, J. Am. Chem. Soc., 1987, 109, 940–941 CrossRef CAS.
- H. Puff and H. Reuter, J. Organomet. Chem., 1989, 373, 173–184 CrossRef CAS.
- V. Chandrasekhar and R. Thirumoorthi, Organometallics, 2009, 28, 2096–2106 CrossRef CAS.
- Q. Li, F. Wang, R. Zhang, J. Cui and C. Ma, Polyhedron, 2015, 85, 361–368 CrossRef CAS.
- Y.-P. Xie, J.-F. Ma, J. Yang, Y.-Y. Liu, J.-C. Ma and M.-Z. Su, Eur. J. Inorg. Chem., 2009, 2144–2152 CrossRef CAS.
- M. Mehring, M. Schürmann, H. Reuter, D. Dakternieks and K. Jurkschat, Angew. Chem., Int. Ed. Engl., 1997, 36, 1112–1114 CrossRef CAS.
- S. Baba Haj, C. Dietz, M. Lutter and K. Jurkschat, Organometallics, 2015, 34, 5555–5565 CrossRef CAS.
- M. Bouška, L. Dostál, R. Jirásko, A. Růžička and R. Jambor, Organometallics, 2009, 28, 4258–4261 CrossRef.
- K. Wraage, T. Pape, R. Herbst-Irmer, M. Noltemeyer, H.-G. Schmidt and H. W. Roesky, Eur. J. Inorg. Chem., 1999, 869–872 CrossRef CAS.
- G.-H. Wen, R.-F. Zhang, Q.-L. Li, S.-L. Zhang, J. Ru, J.-Y. Du and C.-L. Ma, J. Organomet. Chem., 2018, 861, 151–158 CrossRef CAS.
- C. Jobbágy, P. Baranyai, Á. Gömöry and A. Deák, CrystEngComm, 2018, 20, 5935–5939 RSC.
- P. Pfeiffer and L. Rügheimer, Ber. Dtsch. Chem. Ges., 1903, 36, 3027–3030 CrossRef CAS.
- C. Dorfelt, A. Janeck, D. Kobelt, E. F. Paulus and H. Scherer, J. Organomet. Chem., 1968, 14, 22–24 CrossRef.
- R. A. Varga and C. Silvestru, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2789 CrossRef CAS.
- J. P. Eußner and S. Dehnen, Chem. Commun., 2014, 50, 11385–11388 RSC.
- J. P. Eußner, R. O. Kusche and S. Dehnen, Chem.–Eur. J., 2015, 21, 12376–12388 CrossRef.
- J. P. Eußner, B. E. K. Barth, E. Leusmann, Z. You, N. Rinn and S. Dehnen, Chem.–Eur. J., 2013, 19, 13792–13802 CrossRef.
- N. Rinn, J. P. Eußner, W. Kaschuba, X. Xie and S. Dehnen, Chem.–Eur. J., 2016, 22, 3094–3104 CrossRef CAS.
- Z. You, K. Harms and S. Dehnen, Eur. J. Inorg. Chem., 2015, 5322–5328 CrossRef CAS.
- Z. Hassanzadeh Fard, C. Müller, T. Harmening, R. Pöttgen and S. Dehnen, Angew. Chem., Int. Ed., 2009, 48, 4441–4444 CrossRef.
- J. P. Eußner, B. E. K. Barth, U. Justus, N. W. Rosemann, S. Chatterjee and S. Dehnen, Inorg. Chem., 2015, 54, 22–24 CrossRef PubMed.
- M. Bouška, L. Dostál, Z. Padělková, A. Lyčka, S. Herres-Pawlis, K. Jurkschat and R. Jambor, Angew. Chem., Int. Ed., 2012, 51, 3478–3482 CrossRef.
- M. Bouška, L. Střižík, L. Dostál, A. Růžička, A. Lyčka, L. Beneš, M. Vlček, J. Přikryl, P. Knotek, T. Wágner and R. Jambor, Chem.–Eur. J., 2013, 19, 1877–1881 CrossRef.
- T. Tajima, N. Takeda, T. Sasamori and N. Tokitoh, Eur. J. Inorg. Chem., 2005, 4291–4300 CrossRef CAS.
- M. Saito, H. Hashimoto, T. Tajima and M. Ikeda, J. Organomet. Chem., 2007, 692, 2729–2735 CrossRef CAS.
- R. Hauser and K. Merzweiler, Z. Anorg. Allg. Chem., 2002, 628, 905–906 CrossRef CAS.
- N. Rinn, L. Guggolz, K. Gries, K. Volz, J. Senker and S. Dehnen, Chem.–Eur. J., 2017, 23, 15607–15611 CrossRef CAS PubMed.
- N. Rinn, L. Guggolz, J. Lange, S. Chatterjee, T. Block, R. Pöttgen and S. Dehnen, Chem.–Eur. J., 2018, 24, 5840–5848 CrossRef CAS PubMed.
- E. Dornsiepen, F. Weigend and S. Dehnen, Chem.–Eur. J., 2019, 25, 2486–2490 CrossRef CAS PubMed.
- B. E. K. Barth, E. Leusmann, K. Harms and S. Dehnen, Chem. Commun., 2013, 49, 6590–6592 RSC.
- E. Leusmann, F. Schneck and S. Dehnen, Organometallics, 2015, 34, 3264–3271 CrossRef CAS.
- C. Pöhlker, I. Schellenberg, R. Pöttgen and S. Dehnen, Chem. Commun., 2010, 46, 2605–2607 RSC.
- Z. You, R. Möckel, J. Bergunde and S. Dehnen, Chem.–Eur. J., 2014, 20, 13491–13496 CrossRef CAS PubMed.
- E. Leusmann, M. Wagner, N. W. Rosemann, S. Chatterjee and S. Dehnen, Inorg. Chem., 2014, 53, 4228–4233 CrossRef CAS PubMed.
- B. E. K. Barth, B. A. Tkachenko, J. P. Eußner, P. R. Schreiner and S. Dehnen, Organometallics, 2014, 33, 1678–1688 CrossRef CAS.
- Z. H. Fard, M. R. Halvagar and S. Dehnen, J. Am. Chem. Soc., 2010, 132, 2848–2849 CrossRef CAS PubMed.
- M. R. Halvagar, Z. Hassanzadeh Fard and S. Dehnen, Chem.–Eur. J., 2011, 17, 4371–4374 CrossRef CAS PubMed.
- K. Hanau, N. Rinn, M. Argentari and S. Dehnen, Chem.–Eur. J., 2018, 24, 11711–11716 CrossRef CAS PubMed.
- N. Rinn, J.-P. Berndt, A. Kreher, R. Hrdina, M. Reinmuth, P. R. Schreiner and S. Dehnen, Organometallics, 2016, 35, 3215–3220 CrossRef CAS.
- A. Engel and S. Dehnen, Eur. J. Inorg. Chem., 2019 DOI:10.1002/ejic.201900528.
- J.-P. Berndt, A. Engel, R. Hrdina, S. Dehnen and P. R. Schreiner, Organometallics, 2019, 38, 329–335 CrossRef CAS.
- A. Engel, E. Dornsiepen and S. Dehnen, Inorg. Chem. Front., 2019, 6, 1973–1976 RSC.
- N. W. Rosemann, J. P. Eußner, A. Beyer, S. W. Koch, K. Volz, S. Dehnen and S. Chatterjee, Science, 2016, 352, 1301–1304 CrossRef CAS PubMed.
- N. W. Rosemann, H. Locke, P. R. Schreiner and S. Chatterjee, Adv. Opt. Mater., 2018, 6, 1701162 CrossRef.
- N. W. Rosemann, J. P. Eußner, E. Dornsiepen, S. Chatterjee and S. Dehnen, J. Am. Chem. Soc., 2016, 138, 16224–16227 CrossRef CAS PubMed.
- E. Dornsiepen, F. Dobener, N. Mengel, O. Lenchuk, C. Dues, S. Sanna, D. Mollenhauer, S. Chatterjee and S. Dehnen, Adv. Opt. Mater., 2019, 7, 1801793 CrossRef.
- A. Bashall, A. Ciulli, E. A. Harron, G. T. Lawson, M. McPartlin, M. E. G. Mosquera and D. S. Wright, J. Chem. Soc., Dalton Trans., 2002, 1046–1050 RSC.
- F. Benevelli, E. L. Doyle, E. A. Harron, N. Feeder, E. A. Quadrelli, D. Sáez and D. S. Wright, Angew. Chem., Int. Ed., 2000, 39, 1501–1503 CrossRef CAS.
- A. Hinz and J. M. Goicoechea, Dalton Trans., 2018, 47, 8879–8883 RSC.
- M. Veith, M.-L. Sommer and D. Jäger, Chem. Ber., 1979, 112, 2581–2587 CrossRef CAS.
- T. Chivers and D. J. Eisler, Angew. Chem., Int. Ed., 2004, 43, 6686–6689 CrossRef CAS.
- M. Joannis, C. R. Hebd. Seances Acad. Sci., 1891, 113, 795–798 Search PubMed.
- E. Zintl, J. Goubeau and W. Dullenkopf, Z. Phys. Chem., 1931, 154, 1–46 CAS.
- crypt-222: 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane.
- D. Kummer and L. Diehl, Angew. Chem., Int. Ed., 1970, 9, 895 CrossRef CAS.
- J. D. Corbett, D. G. Adolphson, D. J. Merryman, P. A. Edwards and F. J. Armatis, J. Am. Chem. Soc., 1975, 97, 6267–6268 CrossRef CAS.
- S. C. Sevov and J. M. Goicoechea, Organometallics, 2006, 25, 5678–5692 CrossRef CAS.
- T. F. Fässler, Angew. Chem., Int. Ed., 2001, 40, 4161–4165 CrossRef.
- L. L. Lohr, Inorg. Chem., 1981, 20, 4229–4235 CrossRef CAS.
- K. Wiesler, K. Brandl, A. Fleischmann and N. Korber, Z. Anorg. Allg. Chem., 2009, 635, 508–512 CrossRef CAS.
- M. Waibel, F. Kraus, S. Scharfe, B. Wahl and T. F. Fässler, Angew. Chem., Int. Ed., 2010, 49, 6611–6615 CrossRef CAS PubMed.
- F. Fendt, C. Koch, S. Gartner and N. Korber, Dalton Trans., 2013, 42, 15548–15550 RSC.
- M. Neumeier, F. Fendt, S. Gartner, C. Koch, T. Gartner, N. Korber and R. M. Gschwind, Angew. Chem., Int. Ed., 2013, 52, 4483–4486 CrossRef CAS PubMed.
- V. Queneau and S. C. Sevov, Inorg. Chem., 1998, 37, 1358–1360 CrossRef CAS PubMed.
- C. Hoch, M. Wendorff and C. Röhr, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, i45–i46 CrossRef PubMed.
- S. Ponou and T. F. Fässler, Z. Anorg. Allg. Chem., 2007, 633, 393–397 CrossRef CAS.
- A. Ugrinov and S. C. Sevov, J. Am. Chem. Soc., 2002, 124, 10990–10991 CrossRef CAS PubMed.
- R. Hauptmann and T. F. Fässler, Z. Anorg. Allg. Chem., 2003, 629, 2266–2273 CrossRef CAS.
- C. Downie, Z. Tang and A. M. Guloy, Angew. Chem., Int. Ed., 2000, 39, 337–340 CrossRef PubMed.
- C. B. Benda, H. He, W. Klein, M. Somer and T. F. Fässler, Z. Anorg. Allg. Chem., 2015, 641, 1080–1086 CrossRef CAS.
- M. M. Bentlohner, W. Klein, Z. H. Fard, L. A. Jantke and T. F. Fässler, Angew. Chem., Int. Ed., 2015, 54, 3748–3753 CrossRef CAS PubMed.
- D. J. Chapman and S. C. Sevov, Inorg. Chem., 2008, 47, 6009–6013 CrossRef CAS PubMed.
- R. J. Wilson, B. Weinert and S. Dehnen, Dalton Trans., 2018, 47, 14861–14869 RSC.
- S. Scharfe, T. F. Fässler, S. Stegmaier, S. D. Hoffmann and K. Ruhland, Chem.–Eur. J., 2008, 14, 4479–4483 CrossRef CAS PubMed.
- B. W. Eichhorn, R. C. Haushalter and W. T. Pennington, J. Am. Chem. Soc., 1988, 110, 8704–8706 CrossRef CAS.
- B. Kesanli, J. Fettinger and B. Eichhorn, Chem.–Eur. J., 2001, 7, 5277–5285 CrossRef CAS.
- J. Campbell, H. P. A. Mercier, H. Franke, D. P. Santry, D. A. Dixon and G. J. Schrobilgen, Inorg. Chem., 2002, 41, 86–107 CrossRef CAS PubMed.
- E. N. Esenturk, J. Fettinger, Y. F. Lam and B. Eichhorn, Angew. Chem., Int. Ed., 2004, 43, 2132–2134 CrossRef CAS PubMed.
- J. Q. Wang, S. Stegmaier, B. Wahl and T. F. Fässler, Chem.–Eur. J., 2010, 16, 1793–1798 CrossRef CAS PubMed.
- K. Wade, J. Chem. Soc. D, 1971, 792–793 RSC.
- K. Wade, Adv. Inorg. Chem. Radiochem., 1976, 18, 1–66 CrossRef CAS.
- D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- D. M. P. Mingos, T. Slee and L. Zhenyang, Chem. Rev., 1990, 90, 383–402 CrossRef CAS.
- J. Q. Wang, S. Stegmaier and T. F. Fässler, Angew. Chem., Int. Ed., 2009, 48, 1998–2002 CrossRef CAS PubMed.
- B. Zhou, M. S. Denning, D. L. Kays and J. M. Goicoechea, J. Am. Chem. Soc., 2009, 131, 2802–2803 CrossRef CAS PubMed.
- T. Kramer, J. C. Duckworth, M. D. Ingram, B. Zhou, J. E. McGrady and J. M. Goicoechea, Dalton Trans., 2013, 42, 12120–12129 RSC.
- J. M. Goicoechea and J. E. McGrady, Dalton Trans., 2015, 44, 6755–6766 RSC.
- X. Jin, V. Arcisauskaite and J. E. McGrady, Dalton Trans., 2017, 46, 11636–11644 RSC.
- C. B. Benda, M. Waibel and T. F. Fässler, Angew. Chem., Int. Ed., 2015, 54, 522–526 CAS.
- B. Weinert, S. Mitzinger and S. Dehnen, Chem.–Eur. J., 2018, 24, 8470–8490 CrossRef CAS PubMed.
- C. Liu, X. Jin, L.-J. Li, J. Xu, J. E. McGrady and Z.-M. Sun, Chem. Sci., 2019, 10, 4394–4401 RSC.
- S. Stegmaier and T. F. Fässler, J. Am. Chem. Soc., 2011, 133, 19758–19768 CrossRef CAS PubMed.
- Z. M. Sun, H. Xiao, J. Li and L. S. Wang, J. Am. Chem. Soc., 2007, 129, 9560–9561 CrossRef CAS PubMed.
- F. S. Kocak, P. Zavalij, Y. F. Lam and B. W. Eichhorn, Inorg. Chem., 2008, 47, 3515–3520 CrossRef CAS PubMed.
- F. Lips and S. Dehnen, Angew. Chem., Int. Ed., 2009, 48, 6435–6438 CrossRef CAS PubMed.
- F. Lips, R. Clérac and S. Dehnen, Angew. Chem., Int. Ed., 2011, 50, 960–964 CrossRef CAS PubMed.
- N. Lichtenberger, R. J. Wilson, A. R. Eulenstein, W. Massa, R. Clérac, F. Weigend and S. Dehnen, J. Am. Chem. Soc., 2016, 138, 9033–9036 CrossRef CAS PubMed.
- N. Lichtenberger, N. Spang, A. Eichhöfer and S. Dehnen, Angew. Chem., Int. Ed., 2017, 56, 13253–13258 CrossRef CAS PubMed.
- B. Weinert, F. Weigend and S. Dehnen, Chem.–Eur. J., 2012, 18, 13589–13595 CrossRef CAS PubMed.
- B. Weinert, F. Müller, K. Harms, R. Clérac and S. Dehnen, Angew. Chem., Int. Ed., 2014, 53, 11979–11983 CrossRef CAS PubMed.
- R. J. Wilson, F. Hastreiter, K. Reiter, P. Buschelberger, R. Wolf, R. M. Gschwind, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2018, 57, 15359–15363 CrossRef CAS PubMed.
- R. J. Wilson, L. Broeckaert, F. Spitzer, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2016, 55, 11775–11780 CrossRef CAS PubMed.
- A. Schnepf, Angew. Chem., Int. Ed., 2003, 42, 2624–2625 CrossRef CAS PubMed.
- F. Li and S. C. Sevov, Inorg. Chem., 2015, 54, 8121–8125 CrossRef CAS PubMed.
- F. Li, A. Muñoz-Castro and S. C. Sevov, Angew. Chem., Int. Ed., 2016, 55, 8630–8633 CrossRef CAS PubMed.
- O. Kysliak, D. D. Nguyen, A. Z. Clayborne and A. Schnepf, Inorg. Chem., 2018, 57, 12603–12609 CrossRef CAS PubMed.
- M. Binder, C. Schrenk, T. Block, R. Pöttgen and A. Schnepf, Chem. Commun., 2017, 53, 11314–11317 RSC.
- M. Brynda, R. Herber, P. B. Hitchcock, M. F. Lappert, I. Nowik, P. P. Power, A. V. Protchenko, A. Ruzicka and J. Steiner, Angew. Chem., Int. Ed., 2006, 45, 4333–4337 CrossRef CAS PubMed.
- M.-L. Lechner, K. Fürpaß, J. Sykora, R. C. Fischer, J. Albering and F. Uhlig, J. Organomet. Chem., 2009, 694, 4209–4215 CrossRef CAS.
- C. P. Sindlinger and L. Wesemann, Chem. Sci., 2014, 5, 2739–2746 RSC.
- C. Schrenk, F. Winter, R. Pöttgen and A. Schnepf, Chem.–Eur. J., 2015, 21, 2992–2997 CrossRef CAS PubMed.
- J.-J. Maudrich, C. P. Sindlinger, F. S. W. Aicher, K. Eichele, H. Schubert and L. Wesemann, Chem.–Eur. J., 2017, 23, 2192–2200 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Taylor & Francis Group, 90th edn, 2009–2010 Search PubMed.
- S. Hoffmann, T. F. Fässler, C. Hoch and C. Röhr, Angew. Chem., Int. Ed., 2001, 40, 4398–4400 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |