
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced catalytic reaction at an air–liquid–solid triphase interface
Liping
Chen
and
Xinjian
Feng
 *
*
College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China. E-mail: xjfeng@suda.edu.cn
First published on 2nd March 2020
Abstract
Gaseous reactant involved heterogeneous catalysis is critical to the development of clean energy, environmental management, health monitoring, and chemical synthesis. However, in traditional heterogeneous catalysis with liquid–solid diphase reaction interfaces, the low solubility and slow transport of gaseous reactants strongly restrict the reaction efficiency. In this minireview, we summarize recent advances in tackling these drawbacks by designing catalytic systems with an air–liquid–solid triphase joint interface. At the triphase interface, abundant gaseous reactants can directly transport from the air phase to the reaction centre to overcome the limitations of low solubility and slow transport of the dissolved gas in liquid–solid diphase reaction systems. By constructing a triphase interface, the efficiency and/or selectivity of photocatalytic reactions, enzymatic reactions, and (photo)electrochemical reactions with consumption of gaseous reactants oxygen, carbon dioxide, and nitrogen are significantly improved.
 Liping Chen | Liping Chen is a researcher at Soochow University. He obtained his B.S. degree (2009) from Jilin University, China and received his PhD (2014) from the College of Chemistry, Jilin University. He worked as a Postdoctoral Research Fellow at Soochow University (2015–2018) under the direction of Prof. Xinjian Feng. His current research focuses on design and fabrication of biomimetic interfacial electrodes for photoelectrochemical and biosensor applications. |
 Xinjian Feng | Xinjian Feng is a Professor in the Department of Chemistry at Soochow University. Prior to his appointment, he was a professor at the Suzhou Institute of Nanotech and Nanobionics, CAS. Before moving to Suzhou, he worked as a Humboldt Fellow at the Erlangen University of Germany and a postdoc at the Penn State University in America. He received his doctoral degree from the Institute of Chemistry, CAS. His current scientific interests mainly focus on the construction and application of triphase reaction interfaces for environmental remediation, health monitoring and energy conversion. |
1. Introduction
Heterogeneous catalysis is widely used in solar-driven water splitting, CO2/N2 reduction, fuel cells, pollutant degradation, fine chemical production and biomass detection, which are closely related to public issues of worldwide concern such as energy conversion, environmental remediation, health monitoring and chemical synthesis.1–4 Compared with homogeneous catalysis, heterogeneous catalysis possesses prominent advantages in reactant diversification and catalytic separation/regeneration. However, the kinetics and selectivity of catalytic reactions that take place at a liquid–solid diphase interface can be restricted by the low solubility and slow diffusion rate of gaseous reactants in the liquid phase when gaseous reactants are involved in aqueous heterogeneous reactions (such as catalytic O2 reduction reaction, CO2 reduction reaction, and N2 fixation). Although gas pre-saturation and/or continuous ventilation can address this issue, the essential problem remains unsolved: the low catalytic kinetics originating from the slow gas transport to the reaction centre cannot be increased. As a result, the performance of various systems based on conventional diphase catalytic reactions is generally limited.Inspired by natural surfaces with non-wetting features, artificial superhydrophobic substrates have been constructed based on the cooperative effect between micro-/nanocomposite surface structures and low surface energy materials.5 When these substrates are immersed in an aqueous solution, gas pockets will be trapped inside the rough surface structures leading to the formation of an air–liquid–solid triphase joint interface.6 In this triphase interface, gaseous reactants can be readily delivered to the catalytic centre from the air phase with continuous and sufficient supply—this eliminates the gas transport problem in traditional diphase systems and can boost the catalytic kinetics in heterogeneous reactions.
In this minireview, we focus on recent studies on the design and construction of air–liquid–solid triphase reaction interfaces and their effect on gas consumption related photocatalytic reactions, (photo)electrochemical reactions, and enzymatic reactions. By analysing the promoting effect of the triphase architecture in these reactions, we strive to help our readers construct a new concept of triphase reaction systems for improving the reaction efficiency and selectivity in gaseous reactant-based reactions with applications in environmental remediation, clean energy production, and health monitoring.
2. O2 consumption reactions with triphase interfaces
2.1 Photocatalytic reactions
Semiconductor-based photocatalysis with electron reduction or hole oxidation has attracted considerable interest. However, the separation of photogenerated electrons and holes is an essential factor that affects the reaction efficiency. When gaseous reactants are involved in photocatalytic reactions such as H2O2 production and organic pollutant degradation by O2 reduction, the solubility and diffusion rate of O2 are key issues that restrict the reaction rate and substrate–product conversion efficiency. Here, we describe the application of the air–liquid–solid triphase interface in photocatalysis and discuss the working principle of the triphase interface in improving the separation of photogenerated charges and enhancing the reaction efficiency.In the photocatalytic degradation reaction, the recombination of photogenerated electrons and holes is a major problem that hinders the degradation efficiency. Low-dimensional semiconductors and hybrid photocatalysts are commonly used to improve the separation of electrons and holes.7–10 However, this strategy can only be used for electrons and holes temporally separated within nanoscale photocatalysts. An effective approach is to remove them from the photocatalyst surface by using suitable and sufficient acceptors. O2 is an effective natural electron scavenger that can suppress the recombination of photogenerated electrons and holes. However, due to the low concentration and slow diffusion rate of O2 in aqueous environments, the removal of photogenerated electrons by coupling with O2 is strongly restricted: this leads to a low catalytic efficiency and low solar energy efficiency—especially at high pollutant concentrations or high light intensities.
To solve this problem, an air–liquid–solid triphase photocatalytic system is proposed.11Fig. 1a shows that TiO2 nanoparticles are immobilized on a hydrophobic carbon fibre mesh. Once immersed in an aqueous solution, air pockets will form in the empty space of the mesh substrate. Consequently, O2 can be directly and continuously delivered to the reaction zone, i.e., the aqueous-catalysis interface, from the air phase. This is an efficient route for removing photogenerated electrons from the photocatalyst surface to minimize the recombination of electron–hole pairs even under high light intensity. Fig. 1b shows that the degradation rate and apparent quantum yield of the triphase photocatalytic system are significantly increased versus a conventional liquid–solid diphase system—the disparity is more pronounced under high light intensity. It is worth noting that while decomposing the organic molecules in water, the photocatalytic reaction may also break carbon bonds of the superhydrophobic substrate. For practical application, to increase the long-term stability a thick photocatalyst layer or a stable inorganic film that can block the interaction between the reactive oxidizable species and the substrate can be adopted.
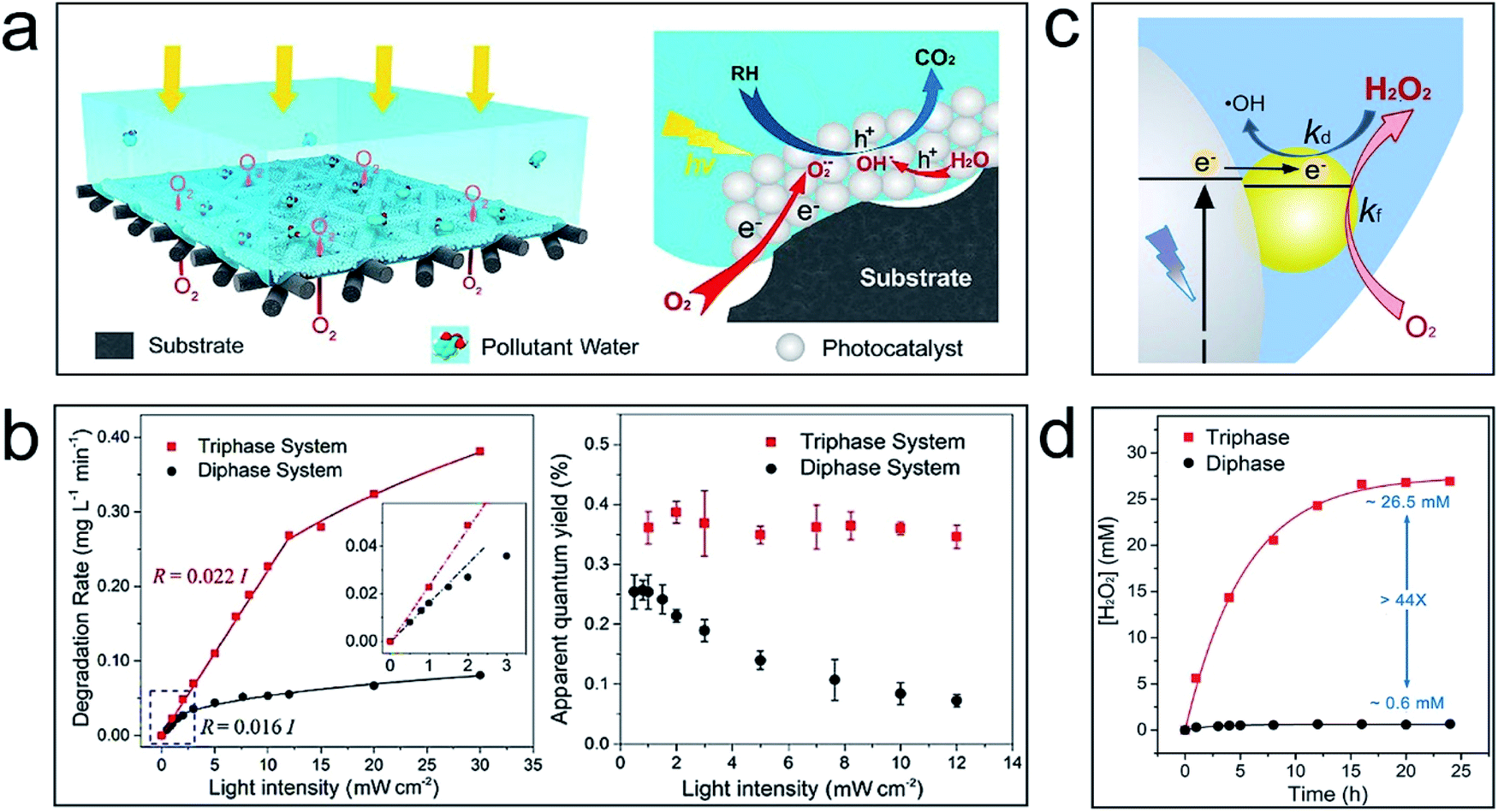 | ||
| Fig. 1 Application examples of the air–liquid–solid triphase system for photocatalytic reactions. (a) Schematic illustration of the triphase system for degradation of organic pollutants; (b) calibration plots of the degradation rate versus UV light intensity for triphase (red curve) and diphase (black curve) reaction systems (left), and the effect of the UV light intensity on the apparent quantum yield of the triphase and diphase systems (right). Reproduced with permission from ref. 11, Copyright 2017 American Chemical Society. (c) Schematic illustration of formation and degradation reactions of H2O2 at the triphase interface. (d) The steady-state concentration of H2O2 produced using the triphase system (red line) and diphase system (black line) under UV light illumination of 60 mW cm−2. Reproduced with permission from ref. 12, Copyright 2019 Elsevier. | ||
H2O2 is an energy carrier and an environmentally friendly oxidant useful in a wide variety of industrial and environmental areas. Photocatalytic H2O2 formation by O2 reduction has attracted extensive attention because it can proceed under mild conditions (ambient temperature and pressure) and requires only water, O2, and excitation light as the material and energy sources. However, due to the low concentration and slow diffusion rate of O2 in aqueous solution, there is often poor accessibility of O2 to the catalysts in a normal liquid–solid diphase system. The recombination of photogenerated charges can also impede the electron utilization efficiency. These problems are especially serious when the charge carrier concentrations become higher with increased light intensity; furthermore, the reverse reaction of H2O2 formation, i.e., the degradation of H2O2 by photogenerated electrons may also reduce the product yield.
The air–liquid–solid triphase photocatalytic reaction system can solve these issues and improve the H2O2 production rate.12Fig. 1c shows that reactant O2 can directly and continuously diffuse to the reaction interface from the air phase instead of slowly diffusing through the aqueous solution. This triphase architecture significantly increased the O2 concentration at the aqueous–catalysis reaction interface and enhanced the removal efficiency of photogenerated electrons by coupling with O2. This improves the utilization efficiency of photogenerated charges by suppressing the electron–hole recombination. Besides, the reverse reaction can also be reduced since photogenerated electrons are easily removed by accessible O2 from the air phase. As a result, a 44-fold enhancement in H2O2 yield can be achieved in the triphase photocatalytic system versus the diphase control (Fig. 1d).
Triphase interface photocatalytic reaction systems can also be used to boost plasmon-driven photocatalytic reactions, semiconductor-mediated photodegradation and other photo-energy conversion reactions as long as gaseous reactants are involved.13–16
2.2 Bio-(photo)electrochemical reactions
Oxidase-based bioassays that use natural O2 as an electron mediator have great potential in diagnosis of diabetes and other medical problems. Analyte levels can be measured by monitoring either O2 consumption or H2O2 formation (converted from O2 in the enzyme-regeneration reaction) that occurs in enzymatic reactions. However, conventional electrode-based biosensors usually operate in environments that contain a liquid–solid diphase interface where O2 is supplied from the liquid phase with inherent drawbacks of low solubility, slow diffusion rate, and fluctuating concentration in electrolyte. As a result, the O2 required for the enzymatic reaction at the electrolyte–enzyme layer interface is quickly depleted, leading to poor performance with low sensitivity, narrowed detection limits, and poor accuracy.To address these issues, oxidase-based enzymatic electrodes with an air–liquid–solid triphase interface have been developed.17 At the top of the sensor electrode (Fig. 2a), an enzyme layer (e.g., glucose oxidase, GOx) with H2O2 electrocatalysts (Pt nanoparticles) embedded beneath it maintained good contact with the analyte solution. At the bottom, a superhydrophobic substrate (carbon fibre mesh) traps air when immersed in an aqueous solution. The enzyme catalytic product (H2O2) is electrooxidized on the Pt surface to produce a rapid current response for analyte determination. At the air–liquid–solid triphase interface, the O2 required for enzyme regeneration can be readily provided from the air phase to ensure that a large amount of H2O2 is formed for accurate analyte determination. In a typical oxidase-based sensing experiment (Fig. 2b), a detection upper limit as high as 156 mM towards glucose can be obtained using the triphase biosensor—which is about 30-fold higher than that using the normal diphase one (inset in Fig. 2b). The authors have also studied the long-term stability of the superhydrophobic biosensor by testing its response to a high concentration glucose solution for more than eight months. The biosensor retains its response over the course of 250 days with a variation of less than 5%, indicating good stability of the superhydrophobic bioassay system. Using this triphase design, Mi and co-workers discussed the detailed kinetic parameters of the enzymatic reactions by regulating the constituents of the air phase or dissolved gas in the liquid phase.18 The results further confirmed the enhancement of the performance of the bioassay via the triphase structure.
 | ||
| Fig. 2 Application examples of the air–liquid–solid triphase system for bio-(photo)electrochemical reactions. (a) Schematic representation of the triphase biosensor with a thin layer of oxidase/chitosan composite film immobilized on the superhydrophobic substrate that is modified with the H2O2 catalyst. The enlarged view shows the air–liquid–solid triphase reaction zone where the oxidase catalytic bioreaction takes place. (b) Calibration plot of the triphase biosensor (red line) and normal diphase one (black curve in the inset). The linear detection upper limit is 156 mM for the triphase biosensor, which is about 30 times higher than that for the diphase one (∼5 mM). Reproduced with permission from ref. 17, Copyright 2015 Wiley-VCH. (c) Schematic illustration of the air–liquid–solid triphase bio-photoelectrode with a thin oxidase/chitosan composite layer immobilized atop a single-crystal TiO2 nanowire array film that is grown on a superhydrophobic substrate. The enlarged view shows the triphase reaction zone with rapid and continuous oxygen transport to the reaction interface. R denotes the reactant and P denotes the product. Reproduced with permission from ref. 23, Copyright 2018 Wiley-VCH. (d) Selectivity tests for glucose sensing. The presented curve shows the photocurrent responses for 1 mM glucose and successive addition of 0.1 mM different interferents using the triphase bio-photoelectrochemical assay system based on the reduction method. Reproduced with permission from ref. 26, Copyright 2018 Wiley-VCH. | ||
One lingering issue is selectivity. The photoelectrochemical (PEC) bioassay is an alternative that has attracted growing interest. Compared with electrochemical methods based on H2O2 oxidation with high oxidation potential, photo-generated holes can oxidize H2O2 at low overpotential to achieve the same advantages in sensitivity and the linear detection range but with improved selectivity. Nevertheless, similar to electrochemical methods, the O2 deficiency problem also hampers normal PEC biosensors with liquid–solid diphase interfaces. Besides, the electron transport in semiconductors may bring new issues for effective charge collection when semiconductor catalysts are used in photoelectrochemical reactions.
Single-crystal semiconductor nanowire arrays have excellent electron transport properties versus their nanoparticle counterparts.19–22 A triphase bio-photoelectrochemical assay system based on superhydrophobic substrate supported single-crystal nanowire arrays has been proposed to overcome both the O2 transport and electron transport limitations.23,24 As shown in Fig. 2c, a thin oxidase/chitosan composite layer is immobilized atop a single-crystal TiO2 nanowire array film, which was grown on superhydrophobic carbon cloth. In the presence of O2, the H2O2 enzymatic product will be generated when the analyte is introduced. Under excitation light irradiation, H2O2 can be subsequently oxidized by the photogenerated holes at the semiconductor surface resulting in a photocurrent response in proportion to the analyte level. Consequently, the combination of air–liquid–solid triphase electrode design and arrayed single-crystal nanowires can simultaneously lead to superior gas and charge transport pathways. This increases the upper detection limits and selectivity of the triphase bio-photoelectrochemical sensor at a relatively lower potential.
Based on the discussion above, we know that the (photo)electrochemical oxidation of H2O2 is an effective tool for bioassays. However, its inherent drawbacks, i.e., poor selectivity against easily oxidizable interferents, can only be weakened—they cannot be completely eliminated. The (photo)electrochemical reduction method for detecting H2O2 is an ideal solution for improving selectivity. Using this method, analytes are determined via H2O2 reduction at suitable potentials (e.g. −0.3 to 0 V vs. Ag/AgCl on the Pt catalyst) or with photogenerated electrons. Nevertheless, practical application of the H2O2 reduction-based method has been limited because O2 can also be reduced under the same conditions: this leads to interference currents. The interference current from O2 reduction will fluctuate because the O2 level at the traditional electrolyte–catalyst diphase interface is liquid phase dependent and varies widely. Thus, accurate detection of H2O2 by the reduction method is compromised.
The air–liquid–solid triphase joint interface is introduced for bioassays based on the H2O2 cathodic reaction.25,26 Unlike traditional liquid–solid diphase electrodes, the O2 level adjacent to the reaction sites is governed by the air phase. Due to the fast and constant O2 supply for the interfacial reaction centre from the air phase, the O2 level at the electrolyte–catalyst interface can remain constant and will not be affected by the concentration fluctuation of the dissolved O2 in the electrolyte. As a result, the interference current caused by the O2 reduction will stabilize and be treated as a constant signal background; thus, the accuracy of the bioassay based on H2O2 reduction can be guaranteed. This leads to a high selectivity of the triphase bioassay. In a bioassay with the air–liquid–solid triphase joint interface, the successive addition of different interferents did not result in any interference signal in the glucose assay (Fig. 2d), which confirmed the practicability of the triphase design for accurate analyte detection.
Other O2 consumption reactions like the O2 reduction reaction (ORR) are closely related to energy exploration or environmental governance. These reactions can also use triphase electrodes for performance enhancement. Through rational design, the air–liquid–solid triphase interface can be generated on electrodes assembled using Pt-loaded SiO2 pillar arrays, nickel foam, N-doped carbon nanotube arrays/nanosheets, or Au/NiFeOx modified with a low surface energy material or coated on porous hydrophobic polymer substrates.27–32 When used in the electrochemical ORR, triphase electrodes with balanced hydrophobicity/hydrophilicity ensured fast gas and ion transport to eventually improve ORR efficiency.
3. N2 and CO2 reduction reactions with the triphase interface
The N2 and CO2 reduction reactions (NRR and CRR) with photocatalytic or (photo)electrochemical methods can be used for sustainable energy, greenhouse gas elimination, and value-added chemical production.33,34 In conventional liquid–solid diphase reaction systems, the low dissolution and slow mass transfer of the N2 and CO2 in electrolytes seriously restrict the reaction kinetics. Besides, the un-wanted hydrogen evolution reaction (HER)35–38 will hinder the faradaic efficiency and/or the selectivity of the NRR and CRR. These limitations can be addressed using the air–liquid–solid triphase electrode.3.1 N2 reduction reactions
The use of the (photo)electrochemical NRR to produce NH3 is a particularly promising technique because the NH3 product is heavily used in chemical and pharmaceutical production, and is also a potential carbon-free energy carrier. However, in aqueous solutions, the (photo)electrochemical NRR is inhibited by the intense competition from the HER of water that results from the preferential adsorption of protons over nitrogen across all traditional NRR catalysts. Furthermore, once water exists in the reaction system (even the trace atmospheric water vapor), the HER will occur and generate dominant (photo)electrochemical current interference. Therefore, the intrinsic NRR (photo)electrochemical features can hardly be detected, let alone the further thermodynamic or kinetic studies.To achieve the (photo)electrochemical NRR with high selectivity and high efficiency under ambient conditions, an air–liquid–solid triphase photoelectrode structure is designed.39 As shown in Fig. 3a, a hydrophobic layer of the porous polytetrafluoroethylene (PTFE) framework was covered on the Ti–Si substrate with Au nanoparticles uniformly distributed on the framework. When the photoelectrode was immersed in electrolyte, an air phase will form by trapping gaseous N2 in the porous PTFE framework. Fig. 3b shows that the yield rate of NH3 (NRR product) and faradaic efficiency using the triphase electrode (Au–PTFE/TS) are higher than those using the diphase one (Au/TS) in a wide potential range. The excellent performance of the triphase reaction system can be ascribed to the direct transport of N2 molecules to the surface of the catalyst (Au nanoparticles) from the air phase. The hydrophobicity of the electrode ensured a low water contact area, which consequently restricts the contact between the proton and the catalysts. Eventually, the HER is suppressed, and the efficiency of the NRR is improved. Similar results for improving the NRR performance with electrochemical methods have been reported by constructing triphase interfaces with hydrophobic layers of the zeolitic imidazolate framework (ZIF) over the NRR electrocatalyst surfaces,40,41 indicating that the strategy of constructing electrodes with a triphase interface is feasible for the NRR under mild conditions.
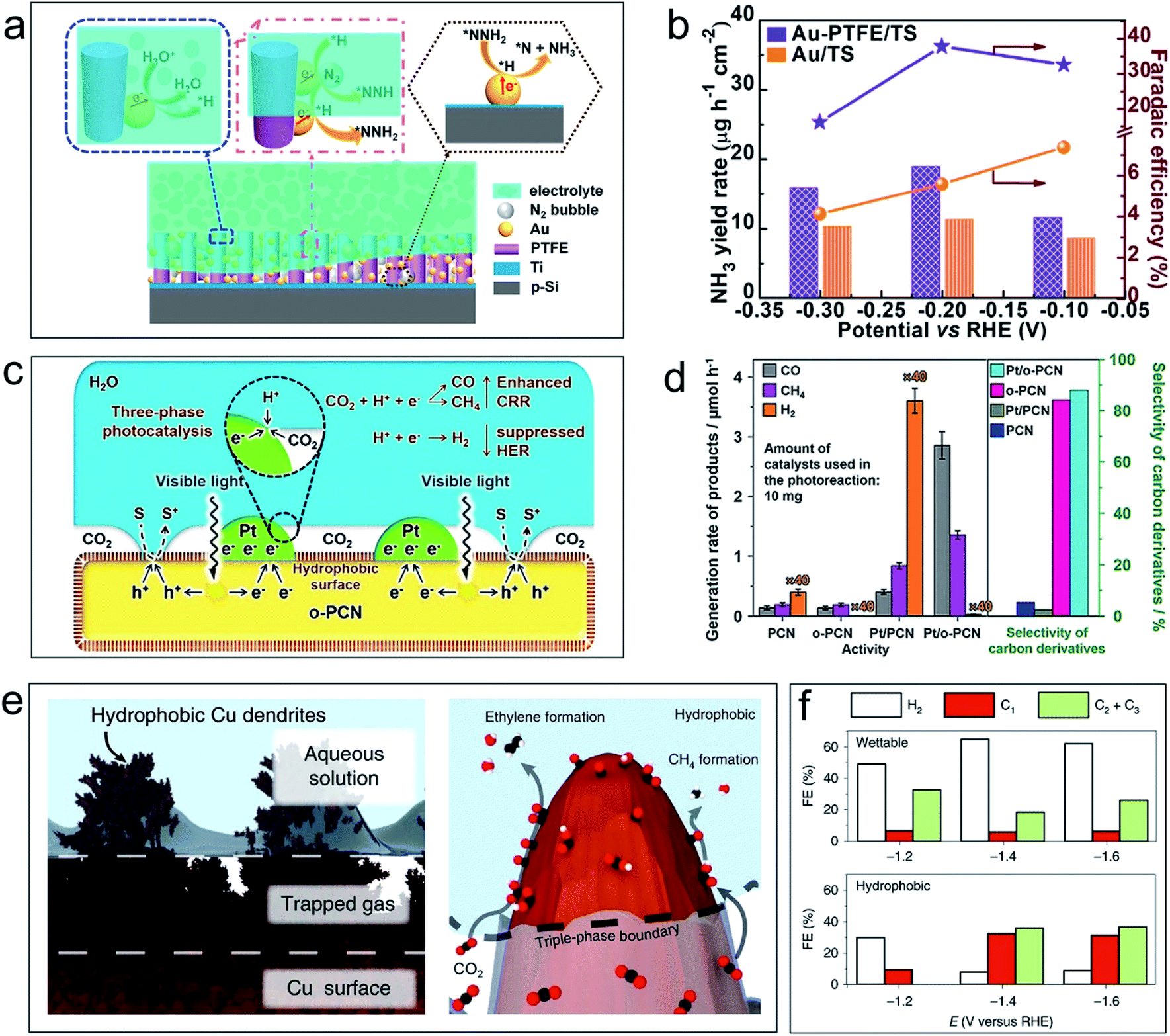 | ||
| Fig. 3 Application examples of the air–liquid–solid triphase system for the NRR and CRR. (a) Schematic illustration and the proposed reaction route of the triphase system for the electrochemical NRR on the Si-based photocathode. (b) Yield rate of NH3 (column diagrams) and faradaic efficiency (point plots) on Au/TS (orange) and Au–PTFE/TS (purple) at each given potential for 4 hours (TS denotes a thin Ti layer on the Si surface). Reproduced with permission from ref. 39, Copyright 2018 Elsevier. (c) Scheme of the triphase photocatalyst (hydrophobic polymeric carbon nitride nanosheets loaded with Pt nanoparticles) and the mechanism of the triphase photocatalytic CRR. S represents the hole sacrificial agent, and S+ represents the sacrificial agent which is oxidized by holes. (d) The activity of the photocatalytic CRR and selectivity towards carbon derivatives with the catalysts polymeric carbon nitride (PCN) and hydrophobic polymeric carbon nitride (o-PCN) with/without Pt partial loading. The note ×40 means the value of this column shown here is reduced by 40 times. Reproduced with permission from ref. 42, Copyright 2019 Wiley-VCH. (e) The illustration of the air–liquid–solid triphase interface when a hydrophobic Cu dendrite placed in water and gas trapped between dendrites (left), and the enhanced CO2 mass transport from the triple-phase boundary between the electrolyte, electrode and gaseous CO2 and the resultant formation of key products on the surface (right). (f) Controlled potential electrolysis product faradaic efficiencies from the wettable (diphase interface) and hydrophobic (triphase interface) dendrites at various potentials. Reproduced with permission from ref. 43, Copyright 2019 Springer Nature. | ||
3.2 CO2 reduction reactions
The photocatalytic or electrochemical CRR can eliminate greenhouse gases and harvest renewable resources. In the ideal case, both the hydrogen ion and CO2 source for the CRR can be supplied from the aqueous solution. However, in most cases, the dominant reaction in aqueous solution is the competing HER, i.e., direct water reduction by capturing photo-generated electrons or at cathodic polarized potentials. This leads to low selectivity and low activity of the CRR. Therefore, it is highly desirable to create an environment that simultaneously suppresses the HER and enhances the CRR.Among the different strategies for tackling this problem, a simple solution by constructing an air–liquid–solid triphase reaction interface has been proposed.42Fig. 3c shows that an efficient triphase contact of CO2 (air phase), H2O (liquid phase), and the catalyst (solid phase) is formed by modifying polymeric carbon nitride nanosheets with a hydrophobic polymer (with/without Pt nanoparticle loading). At the triphase joint interface, concentrated CO2 molecules in the air phase can directly transport to the surface of photocatalysts, which removes the mass-transfer limitations of CO2. The hydrophobic surface lowers the hydrogen ion contacts and suppresses the HER. In the photocatalytic CO2 reduction experiments (Fig. 3d), the hydrophobic catalysts (o-PCN) have high selectivity towards carbon-containing compounds, and a negligible amount of hydrogen is detected. This phenomenon becomes more pronounced even when Pt nanoparticles—one of the most efficient electron-collection agents and HER-promotion cocatalysts—are loaded for the photocatalytic reaction. These results proved the superior performance of the air–liquid–solid triphase interface and can improve the efficiency and selectivity of the photocatalytic CRR.
Electrochemical CO2 reduction is also a simple approach with great potential for the CRR. Similar to photocatalytic activities, the competitive HER predominates when cathodic polarization occurs in an aqueous environment. In addition, the reduction of extremely stable CO2 molecules is kinetically sluggish, and relatively high overpotentials are required. These features lead to low selectivity and low faradaic efficiency for CO2 reduction.
Wakerley and co-workers recently prepared an air–liquid–solid triphase electrode by modifying hierarchically structured Cu dendrites with 1-octadecanethiol—a low surface energy compound.43Fig. 3e shows that once immersed in electrolyte, the assembled hydrophobic electrode with the triphase joint interface can trap gaseous CO2 in the empty space of the Cu dendrites. During the electrochemical reduction process, gaseous CO2 in the air phase can be directly and continuously delivered to the reaction interface. This removes the transportation barriers of CO2 in aqueous phases and largely improves the CO2 concentration near the reaction centre. Meanwhile, the hydrophobic surface of the electrode minimized the water contact area and thus suppressed the HER. As a result, the electrochemical CRR with lowered HER efficiency and increased faradaic efficiency towards value-added carbon-containing chemicals is achieved (Fig. 3f). Using the same design principle, the triphase interface is constructed by modifying the catalyst (MoS2 nanosheets) with hydrophobic fluorosilane molecules or depositing gold nanoparticles on a hydrophobic nanoporous polyethylene membrane.44,45 This can also improve the CRR efficiency and enhance the electrochemical stability.
4. Conclusions and outlook
We briefly introduced the most recent progress in heterogeneous reactions involving gas consumption at air–liquid–solid triphase interfaces. By integrating hydrophobic materials with traditional hydrophilic catalysts, delicately designed triphase catalytic systems can be formed once the reactions occur in aqueous solution. At the triphase interface, gaseous reactants can be rapidly and constantly supplied to the reaction centre from the air phase. This removes barriers of low solubility and slow transport of gaseous reactants in traditional liquid–solid diphase systems. Furthermore, the hydrophobic surface of the catalysts can decrease the contact area for the hydrogen ion, which suppresses the competing hydrogen evolution reaction during CO2 or N2 reduction reactions.In this minireview, enhanced photocatalytic, bioelectronic and (photo)electrochemical reactions have been demonstrated using the rationally designed and constructed triphase system. However, only a few gas consumption reactions are covered in these reports of progress. Thus, it is intriguing to further explore the fundamental roles of the air–liquid–solid triphase interface in other interesting and valuable reactions. For example, the hydrogenation reaction is one of the most important reactions in the fine chemical and pharmaceutical industry. Applying the triphase reaction system to the liquid hydrogenation reaction could be an efficient way to increase H2 concentration on the catalyst surface, thus enhancing the hydrogenation reaction rate. Moreover, by rational design the triphase interface can also be used in gas evolution reactions like solar/electro-driven water splitting for H2 or O2 evolution. Given the wide variety of gaseous reactants involved in these reactions, the air–liquid–solid triphase reaction system could be used as a promising platform for environmental treatment, clean energy, value-added chemical production, and biological and clinical diagnosis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21988102 and 51772198) and the Jiangsu Province Science Foundation for Distinguished Young Scholars (BK20150032).Notes and references
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- B. Garlyyev, J. Fichtner, O. Piqué, O. Schneider, A. S. Bandarenka and F. Calle-Vallejo, Chem. Sci., 2019, 10, 8060–8075 RSC.
- W. Barthlott and C. Neinhuis, Planta, 1997, 202, 1–8 CrossRef CAS.
- Y. Wu, J. Feng, H. Gao, X. Feng and L. Jiang, Adv. Mater., 2019, 31, e1800718 CrossRef PubMed.
- L. Wei, C. Yu, Q. Zhang, H. Liu and Y. Wang, J. Mater. Chem. A, 2018, 6, 22411–22436 RSC.
- C. Liu, D. S. Kong, P. C. Hsu, H. T. Yuan, H. W. Lee, Y. Y. Liu, H. T. Wang, S. Wang, K. Yan, D. C. Lin, P. A. Maraccini, K. M. Parker, A. B. Boehm and Y. Cui, Nat. Nanotechnol., 2016, 11, 1098–1104 CrossRef CAS PubMed.
- S. Selim, L. Francàs, M. García-Tecedor, S. Corby, C. Blackman, S. Gimenez, J. R. Durrant and A. Kafizas, Chem. Sci., 2019, 10, 2643–2652 RSC.
- B. Song, Q. Wang, L. Wang, J. Lin, X. Wei, V. Murugadoss, S. Wu, Z. Guo, T. Ding and S. Wei, J. Colloid Interface Sci., 2020, 559, 124–133 CrossRef CAS PubMed.
- X. Sheng, Z. Liu, R. Zeng, L. Chen, X. Feng and L. Jiang, J. Am. Chem. Soc., 2017, 139, 12402–12405 CrossRef CAS PubMed.
- Z. Liu, X. Sheng, D. Wang and X. Feng, iScience, 2019, 17, 67–73 CrossRef CAS PubMed.
- H. Zhou, X. Sheng, J. Xiao, Z. Ding, D. Wang, X. Zhang, J. Liu, R. Wu, X. Feng and L. Jiang, J. Am. Chem. Soc., 2020, 142, 2738–2743 CrossRef CAS PubMed.
- Y. Gao, N. Yang, S. Lu, T. You and P. Yin, J. Mater. Chem. C, 2019, 7, 9926–9932 RSC.
- D. Aebisher, D. Bartusik, Y. Liu, Y. Zhao, M. Barahman, Q. Xu, A. M. Lyons and A. Greer, J. Am. Chem. Soc., 2013, 135, 18990–18998 CrossRef CAS PubMed.
- J. Liu, L. Ye, S. Wooh, M. Kappl, W. Steffen and H.-J. Butt, ACS Appl. Mater. Interfaces, 2019, 11, 27422–27425 CrossRef CAS PubMed.
- Y. Lei, R. Sun, X. Zhang, X. Feng and L. Jiang, Adv. Mater., 2016, 28, 1477–1481 CrossRef CAS PubMed.
- L. Mi, J. Yu, F. He, L. Jiang, Y. Wu, L. Yang, X. Han, Y. Li, A. Liu, W. Wei, Y. Zhang, Y. Tian, S. Liu and L. Jiang, J. Am. Chem. Soc., 2017, 139, 10441–10446 CrossRef CAS PubMed.
- J. Liu, X. Sheng, F. Guan, K. Li, D. Wang, L. Chen and X. Feng, Chem. Sci., 2018, 9, 7400–7404 RSC.
- X. Sheng, L. Chen, T. Xu, K. Zhu and X. Feng, Chem. Sci., 2016, 7, 1910–1913 RSC.
- X. Feng, K. Zhu, A. J. Frank, C. A. Grimes and T. E. Mallouk, Angew. Chem., Int. Ed., 2012, 51, 2727–2730 CrossRef CAS PubMed.
- X. Sheng, D. He, J. Yang, K. Zhu and X. Feng, Nano Lett., 2014, 14, 1848–1852 CrossRef CAS PubMed.
- L. Chen, X. Sheng, D. Wang, J. Liu, R. Sun, L. Jiang and X. Feng, Adv. Funct. Mater., 2018, 28, 1801483 CrossRef.
- D. Wang, L. Chen, Z. Ding and X. Feng, Sol. RRL, 2019 DOI:10.1002/solr.201900185.
- Z. Song, C. Xu, X. Sheng, X. Feng and L. Jiang, Adv. Mater., 2018, 30, 1701473 CrossRef PubMed.
- D. Wang, L. Chen, J. Liu, F. Guan, R. Sun, L. Jiang and X. Feng, Adv. Funct. Mater., 2018, 28, 1804410 CrossRef.
- P. Wang, T. Hayashi, Q. a. Meng, Q. Wang, H. Liu, K. Hashimoto and L. Jiang, Small, 2017, 13, 1601250 CrossRef PubMed.
- K. Xu, A. Loh, B. Wang and X. Li, J. Electrochem. Soc., 2018, 165, A809–A818 CrossRef CAS.
- Y. Li, H. Zhang, N. Han, Y. Kuang, J. Liu, W. Liu, H. Duan and X. Sun, Nano Res., 2019, 12, 177–182 CrossRef CAS.
- J. Li, Y. Zhu, W. Chen, Z. Lu, J. Xu, A. Pei, Y. Peng, X. Zheng, Z. Zhang, S. Chu and Y. Cui, Joule, 2019, 3, 557–569 CrossRef CAS.
- Y. Zhai, J. Wang, Q. Gao, Y. Fan, C. Hou, Y. Hou, H. Liu, Q. Shao, S. Wu, L. Zhao, T. Ding, F. Dang and Z. Guo, J. Catal., 2019, 377, 534–542 CrossRef CAS.
- W. Zhao, X. Li, R. Yin, L. Qian, X. Huang, H. Liu, J. Zhang, J. Wang, T. Ding and Z. Guo, Nanoscale, 2019, 11, 50–59 RSC.
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC.
- S. Fukuzumi, Y.-M. Lee, H. S. Ahn and W. Nam, Chem. Sci., 2018, 9, 6017–6034 RSC.
- G. Zheng, J. Wang, G. Zu, H. Che, C. Lai, H. Li, V. Murugadoss, C. Yan, J. Fan and Z. Guo, J. Mater. Chem. A, 2019, 7, 26077–26088 RSC.
- C. Wang, F. Lan, Z. He, X. Xie, Y. Zhao, H. Hou, L. Guo, V. Murugadoss, H. Liu, Q. Shao, Q. Gao, T. Ding, R. Wei and Z. Guo, ChemSusChem, 2019, 12, 1576–1590 CrossRef CAS PubMed.
- J. Desai, P. Baviskar, K. Hui and H. Pathan, ES Energy Environ., 2018, 2, 21–30 Search PubMed.
- S. Guo, J. Shang, T. Zhao, D. Hou, Z. Jin and G. Sun, ES Mater. Manuf., 2018, 2, 24–27 Search PubMed.
- J. Zheng, Y. Lyu, M. Qiao, R. Wang, Y. Zhou, H. Li, C. Chen, Y. Li, H. Zhou, S. P. Jiang and S. Wang, Chem, 2019, 5, 617–633 CAS.
- H. K. Lee, C. S. L. Koh, Y. H. Lee, C. Liu, I. Y. Phang, X. Han, C.-K. Tsung and X. Y. Ling, Sci. Adv., 2018, 4, eaar3208 CrossRef PubMed.
- J. Liu, R. Li, X. Zu, X. Zhang, Y. Wang, Y. Wang and C. Fan, Chem. Eng. J., 2019, 371, 796–803 CrossRef CAS.
- A. Li, Q. Cao, G. Zhou, B. Schmidt, W. Zhu, X. Yuan, H. Huo, J. Gong and M. Antonietti, Angew. Chem., Int. Ed., 2019, 58, 14549–14555 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- K. Lv, C. Teng, M. Shi, Y. Yuan, Y. Zhu, J. Wang, Z. Kong, X. Lu and Y. Zhu, Adv. Funct. Mater., 2018, 28, 1802339 CrossRef.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |