
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical generation of acyl and carbamoyl radicals using a nucleophilic organic catalyst: applications and mechanism thereof†
Eduardo
de Pedro Beato
 a,
Daniele
Mazzarella
a,
Matteo
Balletti
a and
Paolo
Melchiorre‡
*ab
a,
Daniele
Mazzarella
a,
Matteo
Balletti
a and
Paolo
Melchiorre‡
*ab
aICIQ – Institute of Chemical Research of Catalonia, The Barcelona Institute of Science and Technology, Avenida Països Catalans 16, 43007, Tarragona, Spain. E-mail: pmelchiorre@iciq.es; Web: http://www.iciq.org/research/research_group/prof-paolo-melchiorre/
bICREA, Passeig Lluís Companys 23, 08010, Barcelona, Spain
First published on 4th June 2020
Abstract
We detail a strategy that uses a commercially available nucleophilic organic catalyst to generate acyl and carbamoyl radicals upon activation of the corresponding chlorides and anhydrides via a nucleophilic acyl substitution path. The resulting nucleophilic radicals are then intercepted by a variety of electron-poor olefins in a Giese-type addition process. The chemistry requires low-energy photons (blue LEDs) to activate acyl and carbamoyl radical precursors, which, due to their high reduction potential, are not readily prone to redox-based activation mechanisms. To elucidate the key mechanistic aspects of this catalytic photochemical radical generation strategy, we used a combination of transient absorption spectroscopy investigations, electrochemical studies, quantum yield measurements, and the characterization of key intermediates. We identified a variety of off-the-cycle intermediates that engage in a light-regulated equilibrium with reactive radicals. These regulated equilibriums cooperate to control the overall concentrations of the radicals, contributing to the efficiency of the overall catalytic process and facilitating the turnover of the catalyst.
Introduction
Free radical chemistry is extensively used for the synthesis of agrochemicals, pharmaceuticals, and materials.1 This is because radicals offer ways of making molecules that are often complementary to strategies proceeding via ionic pathways.2 Traditional methods for generating radicals require unstable initiators, or stoichiometric oxidants or reductants.3 These relatively harsh conditions (high temperatures, UV light, toxic reagents, or strong redox-active reagents) significantly affect the applicability and generality of radical processes. Recently, photoredox catalysis4,5 has provided milder and more effective platforms for generating open-shell intermediates. Overall, these radical generation strategies mainly rely on either atom abstraction or electron transfer mechanisms. Therefore, they require precursors with suitable bond-dissociation energies (BDE) or redox properties6 (Fig. 1a). These intrinsic features pose a limit to the generality of the radical precursors that can be used.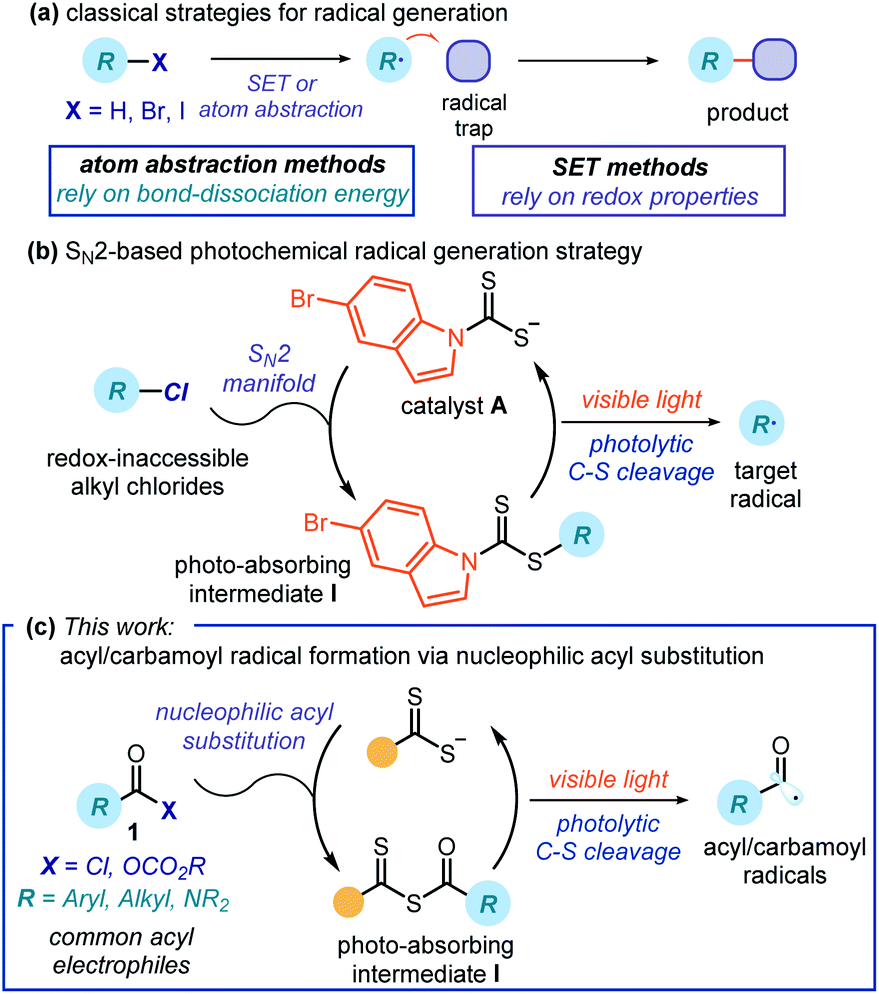 | ||
| Fig. 1 (a) Traditional strategies for generating radicals require precursors with suitable bond-dissociation energies (BDE) or redox properties. (b) Our recently developed SN2-based method uses a nucleophilic dithiocarbamate anion catalyst A to photochemically generate radicals based on the electrophilicity of substrates. (c) Presented photochemical generation of acyl and carbamoyl radicals based on a nucleophilic acyl substitution mechanism. | ||
Our laboratory recently reported a photochemical catalytic strategy that harnesses different physical properties of the substrates to generate radicals. Specifically, our method capitalizes solely on the electrophilic properties of a given precursor (Fig. 1b).7 Inspired by pioneering contributions on xanthate transfer chemistry by Barton8 and Zard,9 we designed the air- and moisture-stable dithiocarbamate anion catalyst A,7 which is adorned with an indole chromophoric unit. This nucleophilic10 organic catalyst can activate alkyl electrophiles by displacing a variety of leaving groups via an SN2 pathway. The ensuing photon-absorbing intermediates I afford radicals upon excitation by visible light and homolytic cleavage of the weak C–S bond. This catalytic SN2-based strategy grants access to stabilized radicals (including benzyl, allyl, and radicals bearing either a heteroatom or an electron-withdrawing moiety at the α-position) from substrates that would be inert to other radical-forming methodologies. These open-shell intermediates have been used to forge new C–C7,11 and C–B bonds.12
To date, our strategy has been limited by the need for substrates amenable to an SN2 displacement. Herein, we expand the potential of this photochemical catalytic approach to include nucleophilic acyl substitution as a suitable manifold for radical generation. Specifically, we generated acyl and carbamoyl radicals upon activation of the corresponding chlorides and anhydrides. We then intercepted the resulting nucleophilic radicals with electron-poor olefins in a Giese-type addition process. The chemistry uses commercially available nucleophilic organic catalysts and low-energy photons (blue LEDs) to activate acyl and carbamoyl chlorides that, due to their high reduction potential, are not readily prone to a SET-based activation mechanism. In addition, we detail how a combination of photophysical investigations, electrochemical studies, and quantum yield measurements along with the characterization of key intermediates provided a more comprehensive mechanistic picture of this photochemical catalytic radical generation strategy.
Design plan
Fig. 2 details the proposed mechanism for the photochemical catalytic generation of acyl and carbamoyl radicals from the corresponding chlorides. This initial mechanistic picture was based on our previous works dealing with the SN2 activation of alkyl electrophiles.7,11,12 By analogy, we envisioned a catalytic cycle where the dithiocarbamate anion catalyst A would activate acyl/carbamoyl chlorides 1 by means of a nucleophilic acyl substitution path. The resulting intermediate I possesses a weak C–S bond, which could be cleaved by low-energy photons to generate the target nucleophilic radical II and the dithiocarbonyl radical III. The feasibility of this crucial step found support in pioneering studies by Barton and Zard,13 who demonstrated the tendency of stoichiometric acyl xanthates to undergo photolysis affording acyl radicals. A few studies have also established the tendency of thiocarbonyl compounds of type I to generate radicals upon photolytic cleavage and promote polymerizations acting as photoiniferters.14 In addition, a report from Grainger highlighted how carbamoyl dithiocarbamates could generate carbamoyl radicals upon UV irradiation.15 Overall, these strategies required the use of stoichiometric pre-formed intermediates of type I and were all characterized by a group transfer mechanism.9,13–15 In addition, these approaches found limited application in processes involving acyl radicals, mainly because of their tendency to generate alkyl radicals upon decarbonylation.13b,c Our goal was to implement a general and effective catalytic process to generate acyl/carbamoyl radicals using blue light to trigger the photolysis of the catalytic intermediate I, which synthetically was not limited within the constraints of a group transfer mechanism. Along these lines, we surmised that the photogenerated acyl/carbamoyl radical II would be intercepted by an electron-poor olefin 2 to forge a new C–C bond. The emerging electrophilic radical IV would then abstract a hydrogen atom from γ-terpinene (a H donor), thus forming the final product 3 and the cyclohexadienyl radical V. Crucial for catalyst turnover would be an SET reduction of the dithiocarbonyl radical III from the cyclohexadienyl radical V, which would eventually close the catalytic cycle by returning catalyst A.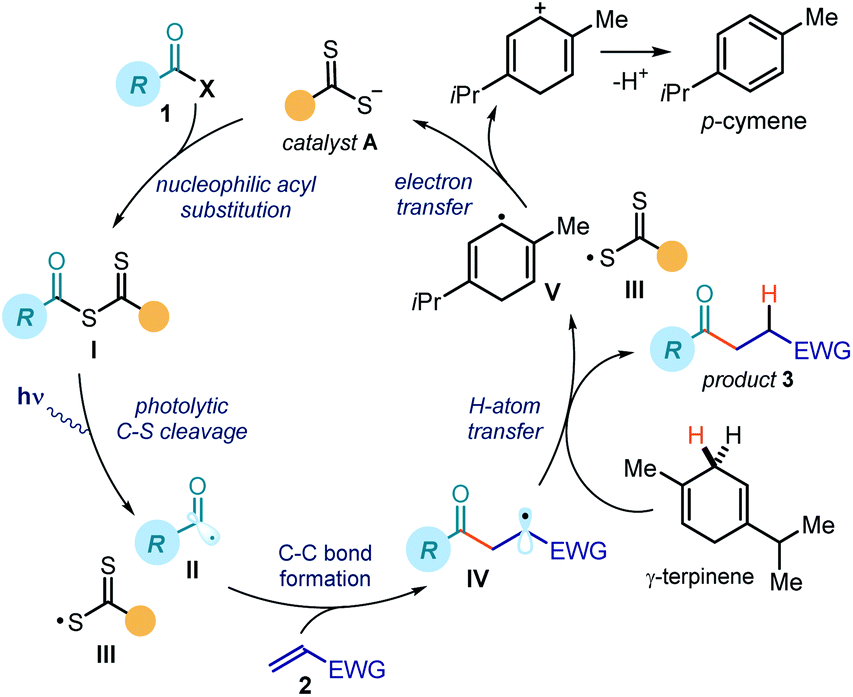 | ||
| Fig. 2 Initial design plan; the mechanism strictly resembles the machinery proposed in our previous studies (ref. 7). | ||
Here, we detail the successful realization of this proposal. We also report extensive mechanistic studies, which highlight how the underlying mechanism of this catalytic radical generation strategy is more complex than originally thought.
Results and discussion
Acyl radical formation and applications
Acyl radicals are nucleophilic intermediates that are useful for many organic transformations.16 Traditional strategies for their generation relied on the activation of stoichiometric acyl chalcogens by means of radical initiators or photolytic bond cleavage.13,16c–e Catalytic methods for acyl radical formation mainly rely on hydrogen atom transfer (HAT) processes17 or electron transfer mechanisms.18,19 While the HAT process from aldehydes provides an elegant and atom-economical entry to acyl radicals, this approach is biased by regioselectivity issues when it is applied to complex molecules.17c Photoredox catalytic methods20 require purposely designed substrates adorned with redox active auxiliaries. For example, acyl radicals could be achieved either by SET oxidation of α-keto acids,18a–d acyl silanes,18e or 4-acyl dihydropyridine derivatives,18f,g or upon SET reduction of hypervalent iodine reagent.19a,b Acyl chlorides21 and anhydrides22 are rarely used in acyl radical chemistry,23 since the radical formation can be further complicated by polar side reactions.24 In addition, these substrates are not easily prone to SET activation. For example, while the redox potentials of aromatic acid chlorides (e.g. benzoyl chloride, Ered = −1.6 V vs. SCE)25 fall within the range of commonly used photoredox catalysts, their alkyl counterparts cannot be easily reduced (e.g. cyclohexyl carbonylchloride, Ered = −2.1 V vs. SCE).26We therefore envisaged that the photochemical strategy depicted in Fig. 2 could be a suitable platform for generating acyl radicals from the chloride precursors. We anticipated that, when successfully developed, this approach would provide significant synthetic benefits. For example, the use of mild reaction conditions would preclude undesired decarbonylation processes, which are often a problem when acyl radical formation is achieved at high temperatures.27 In addition, since our strategy relies on the electrophilicity of the acyl radical precursors, this approach should be viable for a variety of readily available acyl electrophiles, including chlorides and anhydrides, independent of the nature of the substituent (aryl or alkyl).
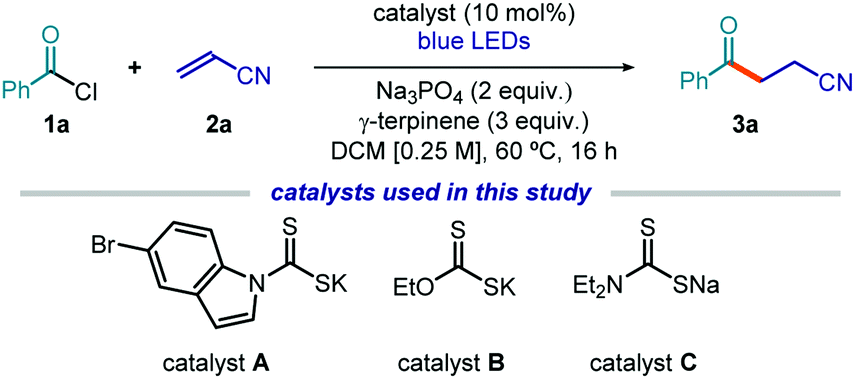
To test the feasibility of our strategy, we selected commercially available benzoyl chloride 1a as the acyl radical precursor and acrylonitrile 2a as the radical trap (Table 1). The experiments were conducted at 60 °C in dichloromethane (DCM) using a blue LED strip emitting at 465 nm, γ-terpinene as the H donor (3 equiv.), and 10 mol% of the nucleophilic catalyst. Na3PO4 (2 equiv.) was used to neutralize the hydrochloric acid generated during the process. Surprisingly, the indole-containing dithiocarbonyl anion catalyst A, which was successful in our previous studies,7,11,12 provided poor results and was not suitable for the generation of acyl radicals (entry 1). We then used the commercially available potassium ethyl xanthate catalyst B. This choice was informed by the notion that acyl xanthates of type I (see Fig. 2), resulting from the nucleophilic acyl substitution of 1, are known to undergo photolytic cleavage.13,14 We confirmed by UV/Vis spectroscopy that an authentic sample of the intermediate of type I, generated upon nucleophilic acyl substitution of B with 1a, could absorb light in the visible region, despite the lack of the indole chromophoric unit (see Fig. S10 in the ESI†). Importantly, the xanthate catalyst B provided the target product 3a with high chemical yield (entry 2). The commercially available sodium diethyldithiocarbamate C was not a suitable catalyst for this transformation (entry 3). However, this commercial salt is sold in its hydrate form (C·3H2O). Since the presence of water was deleterious for the stability of substrate 1a, we synthesized catalyst C as an anhydrous salt, which showed a fully recovered catalytic activity (entry 4). Based on these results, we selected the commercially available xanthate catalyst B for further investigations (see Section C1.2, Tables 1–6 in the ESI† for full details of the optimization phase). The reaction efficiency was affected by a lower temperature and by the presence of air, but synthetically useful yields were still achieved (entries 5 and 6, respectively). Control experiments showed the need for light and for the nucleophilic catalyst (entries 7 and 8).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Deviation | Yield 3ac (%) |
| a Reactions performed on a 0.5 mmol scale at 60 °C for 16 h using 2.0 mL of DCM under illumination by blue LED strip (λmax = 465 nm, 14 W) and using catalyst (10 mol%), 1.5 equiv. of 1a, and 2 equiv. of Na3PO4. b Yield determined by 1H NMR analysis of the crude mixture using trichloroethylene as the internal standard. c Yield of the isolated product 3a. LED: light-emitting diode. | |||
| 1 | A | None | 7 |
| 2 | B | None | 83 (82)b |
| 3 | C·3H2O | None | 34 |
| 4 | C | None | 86 |
| 5 | B | 40 °C | 40 |
| 6 | B | Under air | 60 |
| 7 | B | No light | 0 |
| 8 | None | None | 0 |
Using the optimized conditions detailed in entry 2 of Table 1, we then tested the possibility of using acyl electrophiles other than chlorides as the radical precursors. Benzoic anhydride 1b provided the acylation product 3a in moderate yield (Scheme 1a). The unsymmetrical anhydride 1c, generated by reaction of benzoic acid and ethyl chloroformate and directly used upon simple filtration, afforded 3a in a yield comparable to when using acyl chloride (Scheme 1b). Interestingly, this simple procedure avoids the need for an external base, since its role is played by the ethoxide anion generated during the process. Overall, this sequence allows the use of easily available carboxylic acids as acyl radical precursors.28
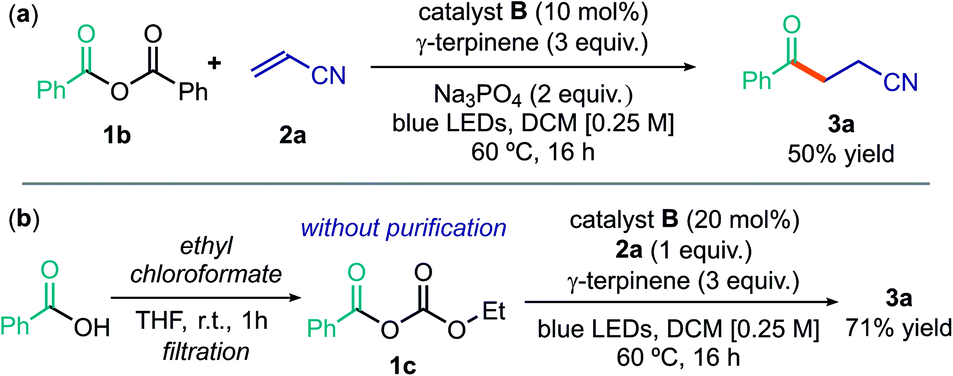 | ||
| Scheme 1 Use of other acyl radical precursors: (a) benzoic anhydride; (b) sequential procedure for the formation of anhydride 1c from benzoic acid and the ensuing acyl radical chemistry. | ||
The optimized conditions were then used to evaluate the scope of this catalytic acyl radical generation strategy (Fig. 3). In general, the reaction proceeded smoothly leading to the exclusive formation of the desired acylation products. Alkylated adducts, arising from a possible decarbonylation of the acyl radicals, were not observed. The main by-products formed during the process, which account for the moderate yields observed in some examples, arose from the dimerization of the acyl radicals and, sometimes, from the formation of the corresponding aldehydes via an HAT process, presumably from γ-terpinene.
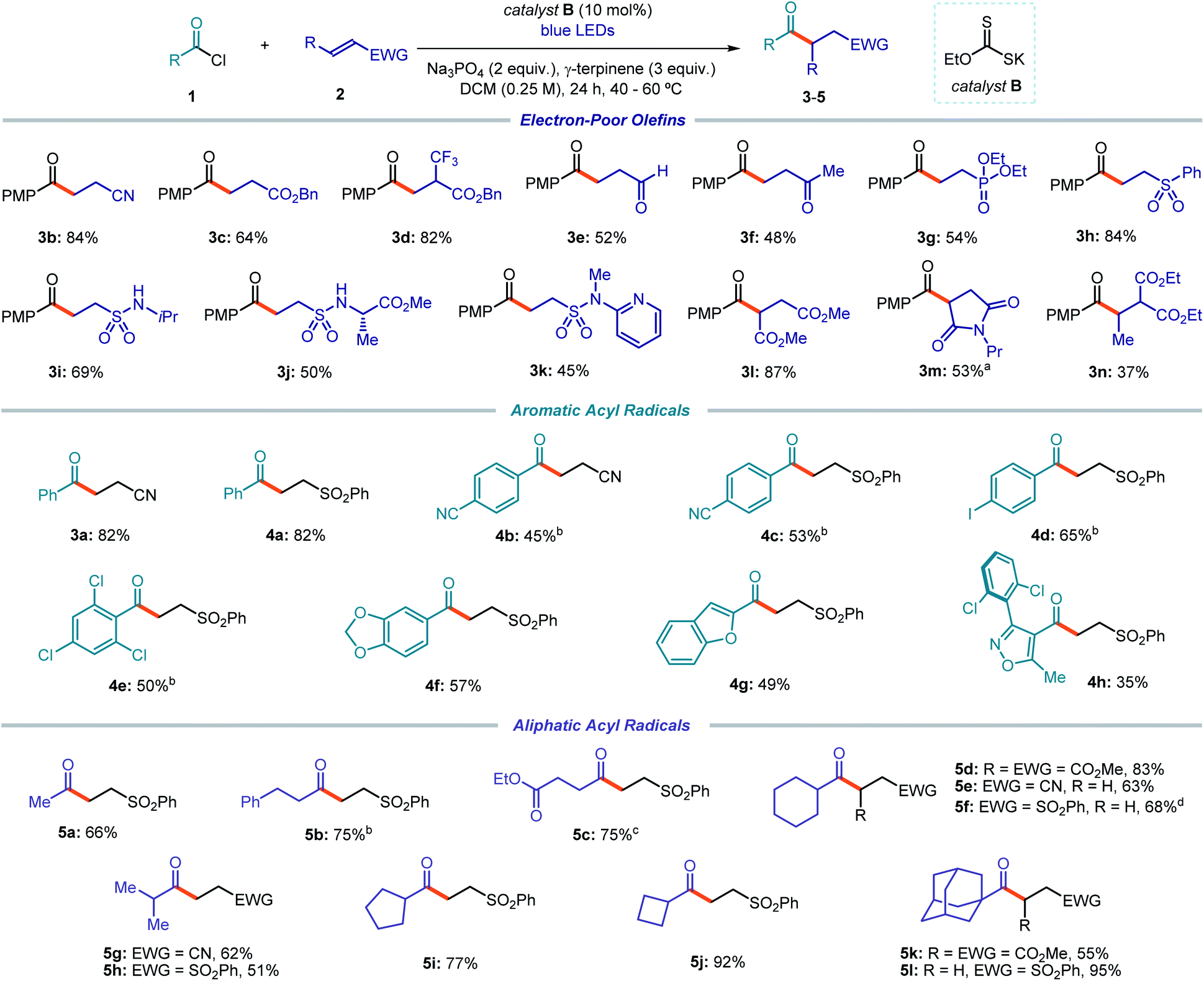 | ||
Fig. 3 Photochemical catalytic generation of acyl radicals from the chloride precursors and their use in Giese addition processes. Reactions performed on 0.5 mmol scale using 1.5 equiv. of acyl chloride 1 and 2.0 mL of dichloromethane (DCM); yields of products refer to isolated materials after purification; the bold orange bond denotes the newly formed C–C bond. Unless otherwise indicated, reactions with aromatic acyl radical precursors were performed at 60 °C, and with alkyl acyl radical precursors at 40 °C. a Yield measured by 1H NMR analysis using trichloroethylene as the internal standard. b Reaction time: 60 h. c Reaction temperature: 60 °C. d Product isolated as a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with the olefin substrate; the corrected yield is reported. PMP: p-methoxyphenyl; Pr: propyl; EWG: electron-withdrawing group. :1 mixture with the olefin substrate; the corrected yield is reported. PMP: p-methoxyphenyl; Pr: propyl; EWG: electron-withdrawing group. | ||
We first focused on the Giese addition using a variety of electron-poor olefins as the radical trap. Commercially available para-methoxy benzoyl chloride was used as the radical precursor, since it facilitated the purification of products 3 while providing comparable results to 1a (e.g. reaction with acrylonitrile 2a provided the corresponding products 3b and 3a in 84% and 82% yield, respectively). Terminal olefins with different electron withdrawing groups, including a cyano, an ester, an aldehyde, a ketone, a phosphonate, a sulfone, and a sulfonamide moiety, all delivered the acylation products 3b–k in synthetically useful yields. Internal olefins could also be used, as shown by the reaction of dimethyl fumarate (3l), propyl maleimide (3m), and diethyl ethylidene malonate (3n).
We then evaluated the acyl radical precursors suitable for this transformation. Aroyl chlorides bearing electron-poor, electron-rich, or hindered moieties on the aryl ring all provided the Giese addition products to phenyl vinyl sulfone in synthetically useful yields (4a–f). Aromatic acyl chlorides bearing heterocyclic moieties, which are common scaffolds in medicinal chemistry programs,29 including benzofuran (4g) and isoxazole (4h), were successfully used as radical precursors.
Because of the unique mechanism of activation of this radical generation strategy, which exploits the electrophilicity of the substrates, alkyl acyl chlorides could also be readily activated by the xanthate catalyst B to give the corresponding radicals. Alkyl acyl chlorides would be recalcitrant to SET activation strategies due to their high redox potential. Primary (5a–c), secondary (5d–j), and bridged tertiary (5k–l) acyl electrophiles reacted with Michael acceptors to afford products 5 in moderate to excellent yields. As a limitation of the system, benzylic and non-bridged tertiary acyl electrophiles were not suitable substrates, since the resulting radicals underwent decarbonylation. A complete list of moderately successful and unsuccessful substrates is reported in Section C7 of the ESI.† Notably, our strategy tolerates functionalities that would be incompatible with other approaches relying on HAT or SET mechanisms. For example, aldehydes (3e), iodides (4d), and dioxolanes (4f) smoothly reacted under the optimized conditions.
We then implemented a two-step telescoped sequence to form the acyl chlorides 1in situ and use it without further purification (Fig. 4a). This one-pot procedure allowed access to functionalized acyl radical precursors from readily available carboxylic acids upon simple treatment with oxalyl chloride. For example, highly functionalized heteroaromatic derivatives (6a) could be used as radical precursors. Alkyl derivatives bearing a chloride substituent selectively reacted at the acyl moiety under the optimized conditions (6b). Moreover, acyl radical precursors containing 5-, 4- and 6-membered rings, adorned with nitrogen (6c, g), fluorine (6d, e), or oxygen (6f) atoms or a tertiary bridged structure (6h), successfully delivered the corresponding products upon Giese reaction with electron-poor olefins.
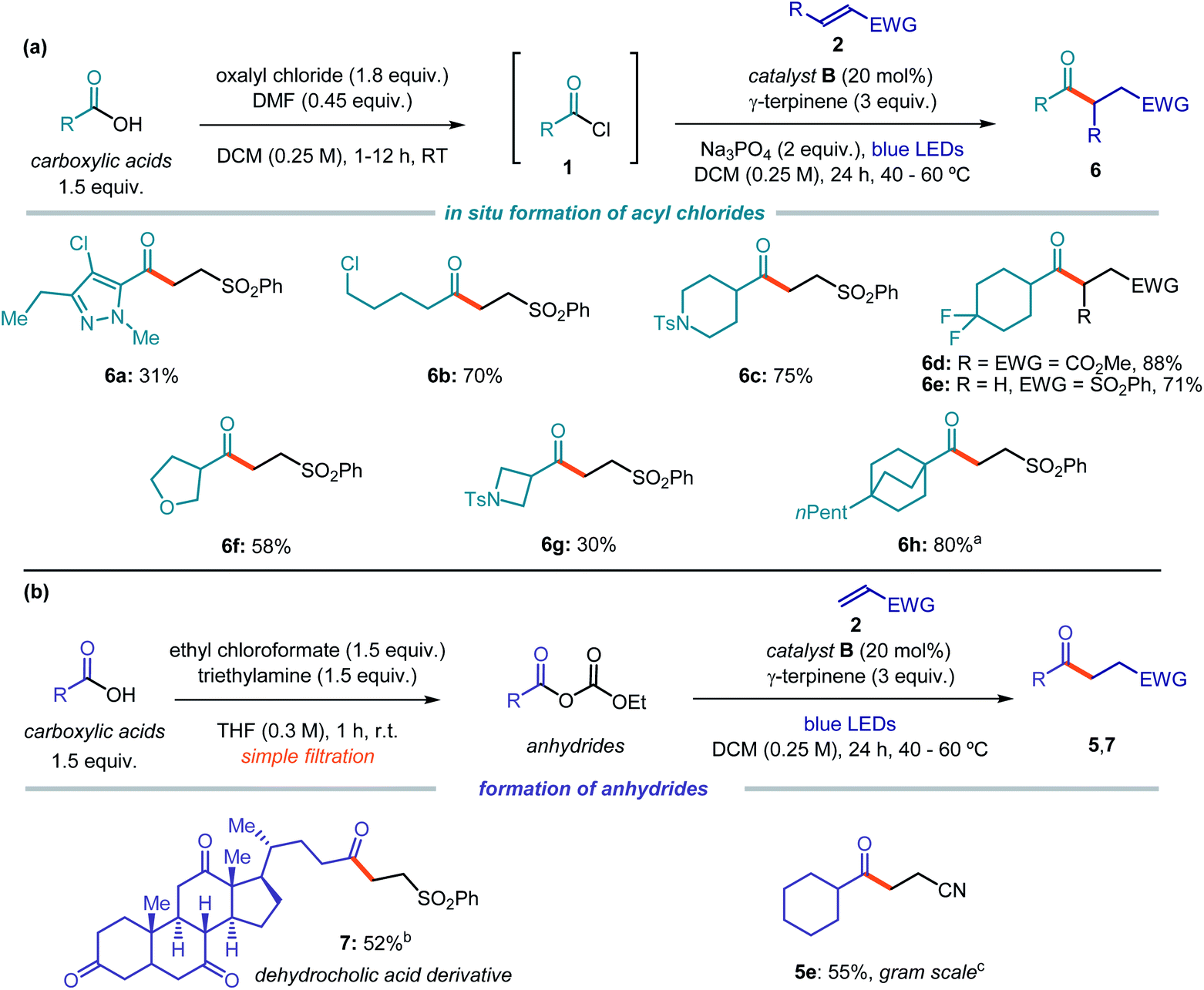 | ||
| Fig. 4 Photochemical catalytic generation of acyl radicals from carboxylic acids upon (a) in situ preparation of acyl chlorides and (b) sequential formation of anhydrides. Reactions performed on 0.5 mmol scale using 1.5 equiv. of 1 and 2.0 mL of dichloromethane; yields of products refer to isolated material after purification; the bold orange bond denotes the newly formed C–C bond. Unless otherwise indicated, all entries with aromatic acyl radical precursors were performed at 60 °C, and with alkyl acyl radical at 40 °C. a Yield calculated by 1H NMR using trichloroethylene as internal standard. b Reaction temperature: 60 °C. c 30 mol% of catalyst B. Ts: Tosyl, EWG: electron-withdrawing group. | ||
In a different procedure, already highlighted in Scheme 1b, we showed that unsymmetrical anhydrides could be generated by reaction of carboxylic acids and ethyl chloroformate. Upon simple filtration, and without further purification, they are then activated by catalyst B to form an acyl radical. This approach may offer a powerful alternative when using substrates bearing functionalities that are incompatible with the generation of acyl chlorides. For example, the natural product dehydrocholic acid, which did not react under the conditions reported in Fig. 4a, could be effectively functionalized to afford product 7 upon treatment with ethyl chloroformate and formation of its mixed anhydride (Fig. 4b). This procedure was also suitable for a gram scale synthesis of adduct 5e starting from the carboxylic acid substrate: using common laboratory glassware and a simple set-up, 1.13 grams of product 5e (55% yield) could be obtained.
Carbamoyl radical formation and applications
The general applicability of a chemical strategy is a crucial parameter for evaluating its usefulness. To further explore the potential of our photochemical radical generation approach, we investigated the activation of carbamoyl chlorides. The formation of carbamoyl radicals upon SET reduction of the progenitor chlorides is hampered by their exceedingly high reduction potential. Indeed, such an SET-based approach has never been reported to the best of our knowledge. The available strategies to generate carbamoyl radicals rely on a HAT process from the corresponding formamide17c,30 or on SET activation of substrates purposely adorned with suitable electron-auxiliary groups.18a,31 We reasoned that, because of the unique mechanism of substrate activation that relies on the electrophilicity of the substrates, our strategy could be successful for activating carbamoyl chlorides towards the formation of radicals. Our reasoning was supported by a previous report15 demonstrating that stoichiometric carbamoyl dithiocarbamates (which, in our approach, would be generated upon acyl substitution of the chloride substrates from the nucleophilic catalyst) can generate carbamoyl radicals32 upon light irradiation. Translating the catalytic system to the photochemical activation of carbamoyl chlorides required slightly different conditions. Because of the lower electrophilicity of carbamoyl chlorides, the more nucleophilic10 commercially available dithiocarbamate catalyst C·3H2O was used. Acetonitrile (CH3CN) emerged as the most appropriate solvent, while potassium phosphate was used as the base. Finally, irradiation at 405 nm secured an efficient photolysis of the carbamoyl dithiocarbamate intermediate of type I, whose absorption is blue-shifted with respect to the analogous acyl intermediate (see Fig. S14† for UV-VIS absorption studies). The progress toward the optimized conditions for the carbamoylation catalytic system is detailed in Table S8 in the ESI.†The results summarized in Fig. 5 illustrate the potential of this catalytic approach to generate carbamoyl radicals from the chloride precursors and their synthetic application in Giese addition processes. Different electron-poor olefins smoothly reacted with dimethylcarbamoyl chloride to furnish the corresponding amides (Fig. 5a, 9a–9c). A variety of carbamoyl chloride substrates could be successfully reacted with vinyl sulfone. Interestingly, this process offers a synthetic entry into Weinreb amide (9d) through an unusual disconnection compared to the classical one. Oxygen-, sulfur-, nitrogen-, chlorine- and fluorine-containing chlorides (9e–9i) reacted with good levels of efficiency. The carbamoyl chloride obtained from proline methyl ester was also successfully functionalized (9j).
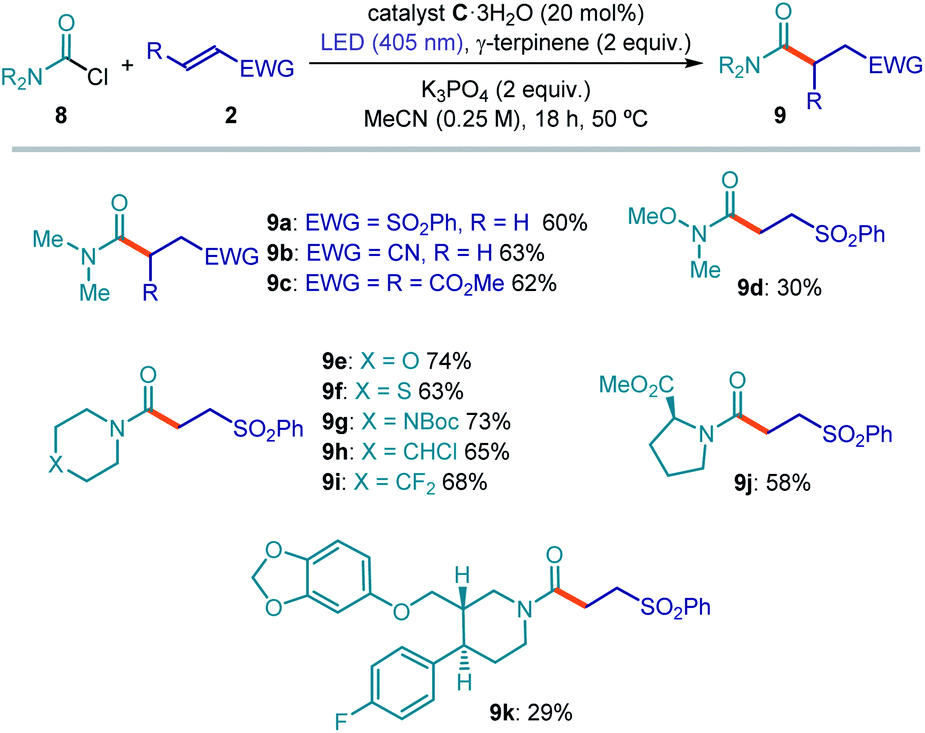 | ||
| Fig. 5 Photochemical catalytic generation of carbamoyl radicals from the chloride precursors and their use in intermolecular Giese addition processes. Reactions performed on 0.2 mmol scale using 2 equiv. of 8 and 0.8 mL of acetonitrile; yields of products refer to isolated material after purification; the bold orange bond denotes the newly formed C–C bond. | ||
Since substrates containing substituted piperidines proved suitable for this transformation, we tested the reactivity of piperidine-containing pharmaceutically active compounds. Indeed, the paroxetine-derived carbamoyl chloride delivered the corresponding Giese addition product (9k), although with a lower chemical yield than simpler substrates. In general, for low-yielding substrates we observed partial degradation of the catalyst and the formation of byproducts arising from dimerization or H-abstraction (HAT process) of the carbamoyl radicals, the latter path leading to the corresponding formamide derivatives.
We then tested the possibility of applying this carbamoyl radical generation strategy to drive an intramolecular process leading to cyclic products. We synthesized carbamoyl chlorides 10 with nitrogen atoms decorated with an alkyl chain bearing a double bond, primed to a radical cyclization (Fig. 6). By using the optimized conditions and simply lowering the amount of γ-terpinene (1.5 equivalents), substrates 10 could be efficiently converted to the corresponding β-lactams fused with a 6-membered ring (11a). Since this strategy of radical formation is not reliant on the redox potential of the substrate, both electron-withdrawing (11b) and electron-donating (11c) substituents at the nitrogen atom were tolerated well. Additionally, a nitrogen benzyl moiety on the substrate was not needed for the positive outcome of the reaction: a simple alkyl chain delivered the product, albeit with lower yields (11d). The method allows the straightforward synthesis of β-lactams33 fused with a 6-membered, a 5-membered (11e), or without any fused ring (11f). Here the efficiency of the cyclization was particularly plagued by the competitive HAT of the carbamoyl radical, leading to the formamide byproduct instead of the desired lactam. In addition, both 5-membered and 6-membered ring lactams could be obtained in good yields (11g, h).
 | ||
| Fig. 6 Carbamoyl radical formation and intramolecular processes. Reactions performed on 0.2 mmol scale using 0.8 mL of acetonitrile; yields of products refer to isolated material after purification; the bold orange bond denotes the newly formed C–C bond. | ||
Mechanistic studies
Our mechanistic understanding of the photochemical catalytic generation of acyl and carbamoyl radicals depicted in Fig. 2 was originally based on our related works7–12 dealing with the use of nucleophilic dithiocarbamate anion catalysts. The crucial steps of this mechanistic machinery are: (i) the ability of the nucleophilic organic catalyst to activate substrates upon nucleophilic attack; (ii) the photoactivity of the ensuing intermediate of type I, which can undergo photolytic cleavage upon visible light absorption; and (iii) the ability of the emerging dithiocarbonyl radical III to undergo SET reduction from the cyclohexadienyl radical V, which represents the turnover event of the proposed catalytic cycle. While evidence in support of points (i) and (ii) were collected in our previous studies, other important mechanistic questions remained unanswered. For example, the proposed catalyst turnover step (point iii) requires an SET event between two fleeting radical intermediates, which is a rather unlikely scenario. In addition, xanthates and thiocarbamates of type I have a great tendency to participate in group transfer manifolds,9 and it is not clear whether this mechanistic path could play an important role in our catalytic system too. We therefore performed extensive mechanistic investigations on the catalytic activation of acyl chlorides by the xanthate catalyst B and the ensuing Giese addition. Our reasoning was that this process might serve as a suitable probe for testing the mechanism and elucidating unclear aspects, thus providing general information on this radical generation strategy.We selected the reaction between benzoyl chloride 1a and acrylonitrile 2a catalyzed by catalyst B for mechanistic studies. Specifically, we deconstructed the general catalytic cycle depicted in Fig. 2 to focus on the most relevant individual steps, while characterizing the key intermediates responsible for the reactivity.
Nucleophilic acyl substitution and photochemical behavior of intermediate I
Potassium ethyl xanthate B is nucleophilic enough10 to readily react with 1a and afford the quantitative formation of the acylxanthate intermediate Ia (Fig. 7a). We wanted to verify if intermediate Ia, which absorbs light in the visible region (see Fig. S10 in the ESI†), could generate the benzoyl radical IIa upon illumination by a blue LED, the light source used in the synthetic applications. A stoichiometric amount of acylxanthate Ia, prepared in situ by reacting B and 1a, was irradiated in the presence of the radical trap 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO). This experiment afforded product 12, arising from the TEMPO trapping of the photochemically generated benzoyl radical IIa (Fig. 7b). When adding TEMPO to the catalytic model reaction (Fig. 7c), the reactivity was almost completely inhibited, leading to only traces of hydroacylation product 3a. Collectively, these experiments indicate the radical nature of the process and that the transiently generated acylxanthate Ia plays an active role in the chemistry. | ||
| Fig. 7 (a) The possible role of the acylxanthate Ia. Mechanistic experiments to probe that Ia is an active reaction intermediate: (b) the benzoyl radical IIa is generated during the process, and (c) the model reaction is inhibited by a radical scavenger. (d) Transient absorption spectroscopic studies: absorption at 620 nm of the transient xanthyl radical IIIa generated upon 355 nm laser excitation of acylxanthate Ia ([Ia]0 = 3 × 10−3 M in CH3CN); absorption decay (black line) processed through Savinsky Golay filter to facilitate lifetime measurement. Note logarithmic scale for time; ΔOD: optical density variation. (e) EPR spectrum generated from Ia ([Ia]0 = 0.1 M in toluene) at 298 K after 12.5 minutes (black line) of light irradiation by a 100 W mercury lamp. | ||
To better corroborate that the light-induced homolytic cleavage of Ia was the key acyl radical generation step (Fig. 7a), we used laser flash photolysis to detect the transient formation of the sulfur-centered radical IIIa. The xanthyl radical IIIa has been reported to display a characteristic absorption peak with a maximum at 620 nm.34 Accordingly, when a freshly prepared authentic sample of intermediate Ia was excited with a laser beam centered at 355 nm, we observed the formation of a transient species absorbing at 620 nm (half lifetime = 0.1 ± 0.01 ms), consistent with the characteristic line shape of xanthyl radical IIIa (Fig. 7d).
We also tried to detect that benzoyl radical IIa by electron paramagnetic resonance (EPR) spectroscopy. We conducted EPR analysis of a solution of Ia in toluene, performed at 298 K under light illumination by a mercury lamp. However, this provided a signal that was not consistent with that of benzoyl radical (singlet).35a The EPR spectrum (Fig. 7e) showed a pattern (sharp triplet centered at 3505 G, g-value = 2.00272, hyperfine splitting value = 2.6 G) that is consistent35b with a more stabilized carbon radical of type VI, which lies in proximity of two sulfur atoms and an ethoxy moiety. The formation of radical VI is consistent with the high tendency of xanthates of type I, and related thiocarbonylthio congeners, to trap radicals. This feature is the underlying reason for the xanthates' success as RAFT agents in polymerization.36 Indeed, xanthates of type I can readily react with acyl radicals of type II in a reversible addition–fragmentation equilibrium, leading to intermediate VI (Fig. 8).
 | ||
| Fig. 8 The reaction of the acyl radical IIa with its precursor Ia is reversible and imparts an increased lifetime to IIa. | ||
This equilibrium controls the overall concentration of highly reactive active radicals (such as II) by forming the more stabilized radical VI, which acts as a dynamic reservoir, while increasing their effective lifetime in the medium. This mechanism37 is at the basis of the success of the degenerative radical addition transfer of xanthates, and may also be operative in the present catalytic system. This possibility is further discussed below.
Fate and behavior of the xanthyl radical IIIa
One of the main mechanistic aspects to elucidate concerned the reactivity of the sulfur-centered radical of type III, generated upon photolytic cleavage of intermediate I. During a recent study11b on the use of the indole-based dithiocarbamate anion catalyst A, we detected the formation in small amounts of the dimer VII, which arises from the self-reaction of the sulfur-centered radical III. We wondered if the catalyst dimer could play an important role in the present process too. Dimer VIIa, readily synthesized in one step from the xanthate catalyst B, can absorb in the visible region (Fig. 9).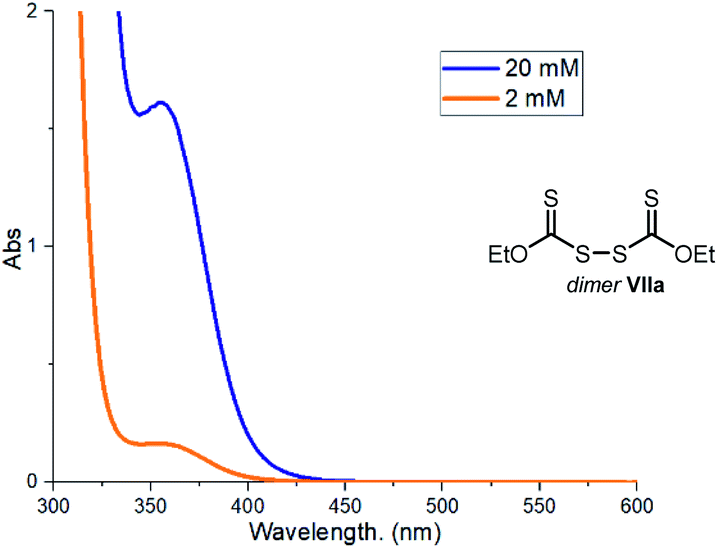 | ||
| Fig. 9 Absorption of dimer VIIa at different concentration in CH3CN. | ||
To verify that this intermediate was catalytically competent, we used an authentic sample of dimer VIIa (5 mol%) to catalyze the model reaction (Scheme 2). Irradiation with a blue LED afforded product 3a in high yield.38 Importantly, no reaction was observed when the same experiment was performed in the dark. These results are very informative of the reaction mechanism, since they imply that dimer VIIa is a photoactive species in equilibrium with the progenitor xanthyl radical IIIa. This dimerization manifold confers a longer lifetime to IIIa,39 preventing other reaction pathways. Thanks to the dimerization, the sulfur-centered radical IIIa gains a persistent radical character. On this basis, the proposed turnover of catalyst B, based on the SET reduction of radical IIIa from radical V (see Fig. 2) becomes feasible. Indeed, according to the persistent radical effect,39 processes between two fleeting radicals are feasible when one of them has a relatively longer lifetime.
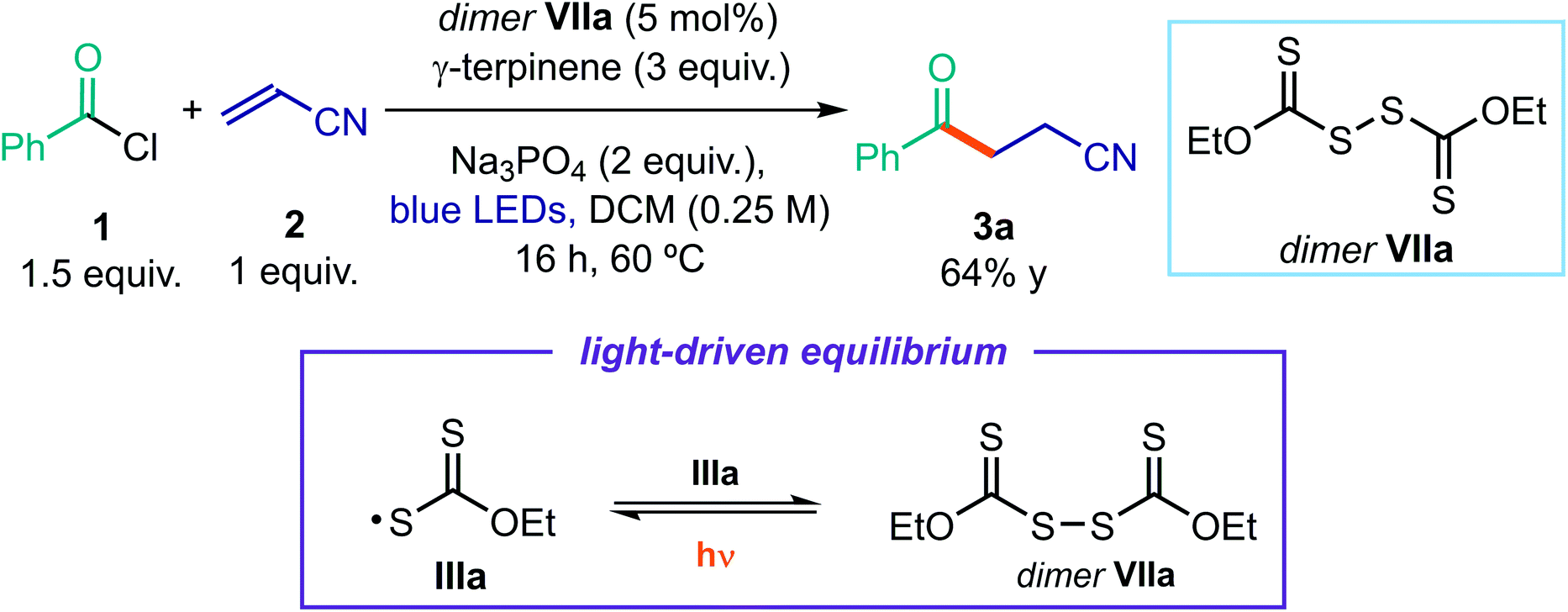 | ||
| Scheme 2 Experiment indicating that dimer VIIa is a catalytically (photo)active species and the light-driven equilibrium between the xanthyl radical IIIa and the dimer VIIa. | ||
To gain further insights into the light-driven equilibrium between the dimer and its radical progenitor IIIa shown in Scheme 2, we evaluated the behavior of dimer VIIa under the reaction conditions using 1 equivalent of γ-terpinene and under blue light illumination (Scheme 3). Dimer VIIa was completely consumed to produce 1.65 equivalents of catalyst B. This result provides compelling evidence to rationalize the mechanism of turnover of catalyst B. Importantly, considering the stoichiometry of the reaction in Scheme 3, it appears that γ-terpinene can provide two different pathways to reduce the xanthyl radical IIIa, namely an HAT process followed by an SET reduction from the ensuing cyclohexadienyl-type radical V.
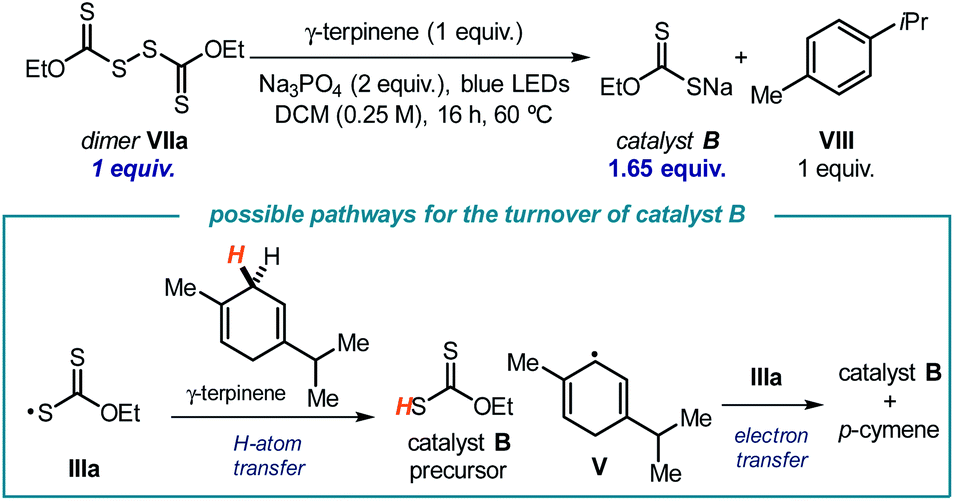 | ||
| Scheme 3 Experiment that probes the ability of γ-terpinene to reduce the xanthyl radical IIIavia both an HAT and an SET manifold. | ||
We also performed laser flash photolysis studies on dimer VIIa (Fig. 10a). When exposed to irradiation at 355 nm, the same transient species observed in the experiment with intermediate I was detected (Fig. 7d), which is consistent with the formation of the xanthyl radical IIIa. This transient was also detected when a laser beam centered at 420 and 460 nm was used (see Fig. S27 and S28 in the ESI†). This observation further confirms the light-driven equilibrium that links dimer VIIa with its radical progenitor IIIa (shown in Scheme 2). Moreover, the lifetime of the transient IIIa decreased upon addition of γ-terpinene (orange, green, and blue lines in Fig. 10b), which corroborates the notion, discussed in Scheme 3, that these two species can readily react.
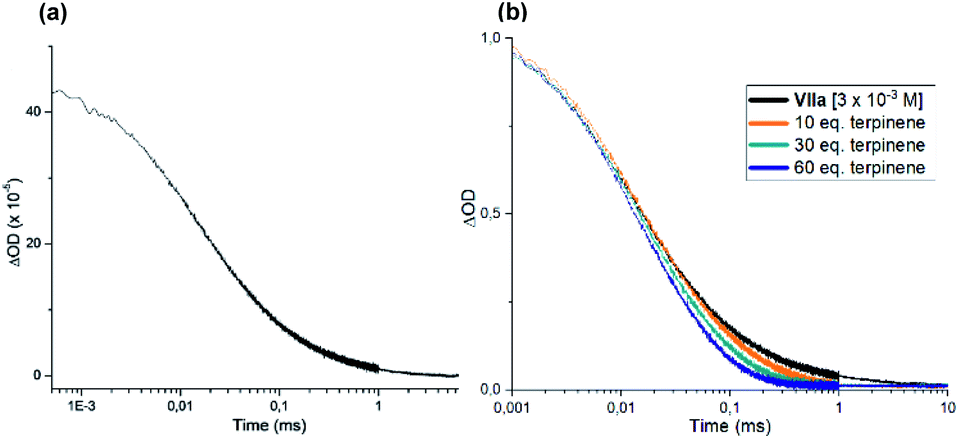 | ||
| Fig. 10 (a) Absorption at 620 nm of the transient xanthyl radical IIIa generated upon 355 nm laser excitation of dimer VIIa ([VIIa]0 = 3 × 10−3 M in acetonitrile). (b) Decrease of the lifetime of IIIa upon addition of γ-terpinene. Note logarithmic scale for time; ΔOD: optical density variation. Absorption decays (orange, green, and blue lines) observed in the presence of increasing amounts of γ-terpinene. Green line: ratio VIIa/γ-terpinene mimics the reaction conditions. ΔOD: optical density variation. | ||
Collectively, the experiments detailed in Schemes 2 and 3 along with the laser flash photolysis studies reported in Fig. 10 provide compelling evidence that dimer VIIa can form xanthyl radical IIIa upon photolytic cleavage. To collect direct evidence of the dimerization tendency of the sulphur-centered radical IIIa, we performed cyclic voltammetry analysis of both catalyst B and dimer VIIa (Fig. 11). We first focused on catalyst B, which was electrochemically analyzed during the initial forward scan applying an increasingly reducing potential (0 to −2.0 V, Fig. 11a). No reduction peaks were observed. During the reverse scan towards oxidation (from −2.0 V to +1.5 V), an irreversible peak appeared (EAp = Eox = 0.73 V in CH3CN vs. Ag/AgCl), which is ascribable to the oxidation of catalyst B to form the xanthyl radical IIIa (orange dot). We then electrochemically analyzed catalyst B in the opposite order (Fig. 11b), first applying an increasingly oxidizing potential (0 to +1.5 V). As expected, during oxidation, we observed the same signal as in the previous experiment, which is congruent with the formation of radical IIIa upon SET oxidation of B (orange dot). Interestingly, during the reverse scan towards reduction (from +1.5 V to −2.0 V), an additional peak was observed (ECp = Ered = −1.40 V in CH3CN vs. Ag/AgCl), which was not detected in the previous experiment (blue dot).
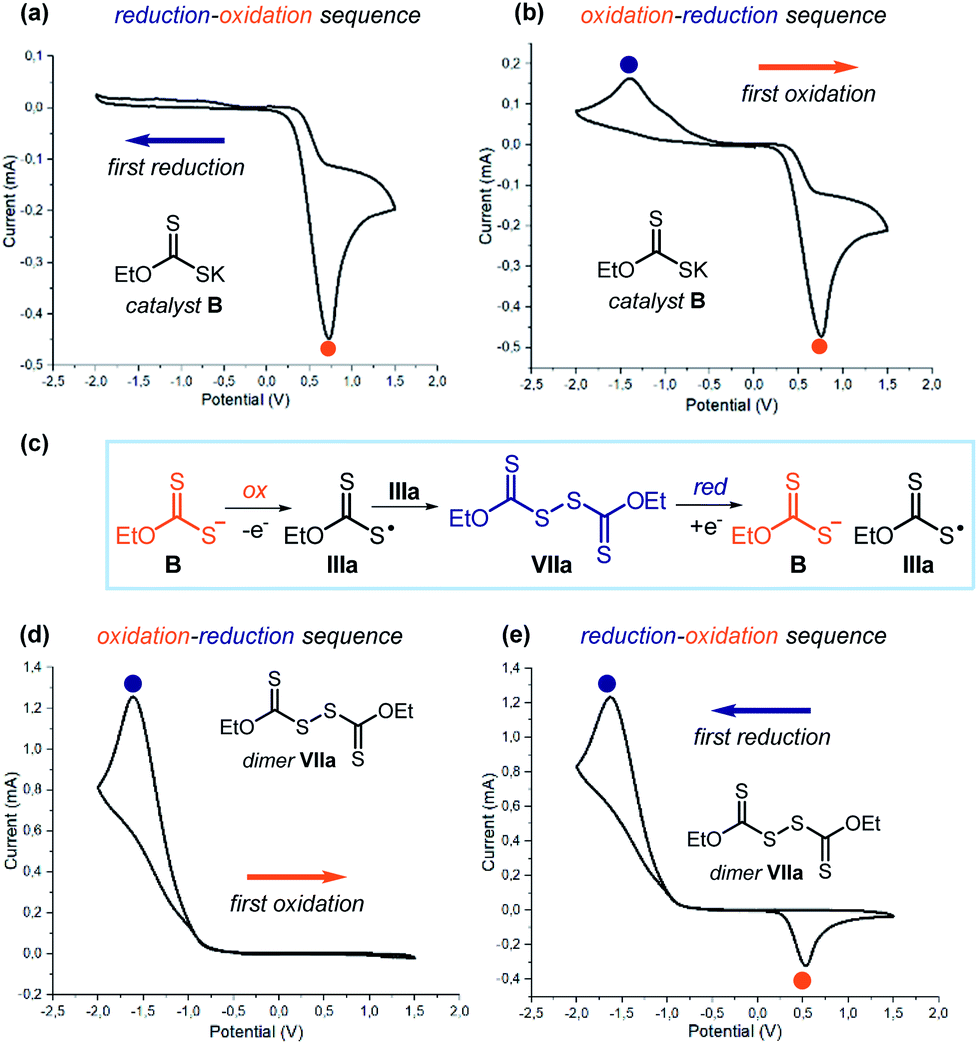 | ||
| Fig. 11 Cyclovoltammetric studies of catalyst B [0.02 M] in [0.1 M] TBAPF6 in CH3CN: (a) reduction–oxidation scan sequence; (b) oxidation–reduction scan sequence. (c) The xanthyl radical IIIa, which can be formed by SET reduction of VIIa and oxidation of B, is the link between catalyst B and dimer VIIa. (d and e) Cyclovoltammetric studies of dimer VIIa [0.02 M] in [0.1 M] TBAPF6 in CH3CN: (d) oxidation–reduction scan sequence; (e) reduction–oxidation scan sequence. Sweep rate: 500 mV s−1. Pt electrode working electrode, Ag/AgCl (KCl 3.5 M) reference electrode, Pt wire auxiliary electrode. | ||
These results can be rationalized based on a fast dimerization of radical IIIa, generated during the forward oxidation scan, that forms dimer VIIa, which is prone to SET reduction (Fig. 11c). This proposal is consonant with the following observation: (i) the formation of the stable dimer makes the oxidation of catalyst B irreversible, since IIIa is rapidly sequestered in the form of its dimer; (ii) it is only in the cyclic voltammetric analysis that starts with oxidation (Fig. 11b) that a reduction peak appears, which is ascribable to the reduction of dimer VIIa: this is because the dimer first requires the oxidation of catalyst B to be generated (Fig. 11c).
To corroborate this proposal, we repeated the same analysis using dimer VIIa. Applying an initial forward scan towards an increasingly oxidizing potential (0 to +1.5, Fig. 11d), no oxidation was observed. As expected, during the reverse scan (from +1.5 V to −2.0 V), the reduction peak of dimer VIIa (blue dot) was recorded (ECp = Ered = −1.61 V in CH3CN vs. Ag/AgCl). But when we started the electrochemical analysis towards reduction (0 to −2.0 V and then to +1.5 V, Fig. 11e), an additional oxidation peak could be observed (EAp = Eox = 0.53 V in CH3CN vs. Ag/AgCl). This is because the reduction of dimer VIIa furnishes the xanthyl radical IIIa along with the anionic catalyst B; the latter species is then oxidized during the reverse scan phase (orange dot), in agreement with the experiments depicted in Fig. 11a and b.
The comparison of the cyclic voltammograms indicates a clear correlation between the new reduction peak observed in the analysis of catalyst B in Fig. 11b with the peak observed upon reduction of dimer VIIa (Fig. 11e). A correlation exists also between the new oxidation peak in the analysis of dimer VIIa in Fig. 11e and the oxidation of catalyst B (Fig. 11a and b).40 Collectively, these studies indicate that the link between catalyst B and dimer VIIa is the xanthyl radical IIIa (Fig. 11c). They also show the strong tendency of the xanthyl radical IIIa to dimerize and form VIIa.
Trap of the acyl radical and ensuing processes
The mechanistic investigations conducted so far allowed us to elucidate the photochemical acyl radical generation mechanism while providing a rationale for the turnover of catalyst B. We then focused on the C–C bond formation, which is triggered by the trap of photogenerated acyl radical IIa by acrylonitrile 2a (Fig. 12a). The emerging electrophilic radical IVa would then abstract a hydrogen atom from γ-terpinene, affording the final product 3a and the cyclohexadienyl radical V. Since xanthates and thiocarbamates of type I have a strong tendency to participate in group transfer manifolds,9 we wondered whether this mechanistic path played an important role in the present catalytic system. We therefore conducted investigations to probe the possible intermediacy of the group transfer product of type IXa. Intermediate IXa might be generated via a xanthate transfer manifold between IVa and Ia, which would also generate a benzoyl radical IIa (Fig. 12a). IXa could also arise from a cross-coupling process between radicals IIIa and IVa.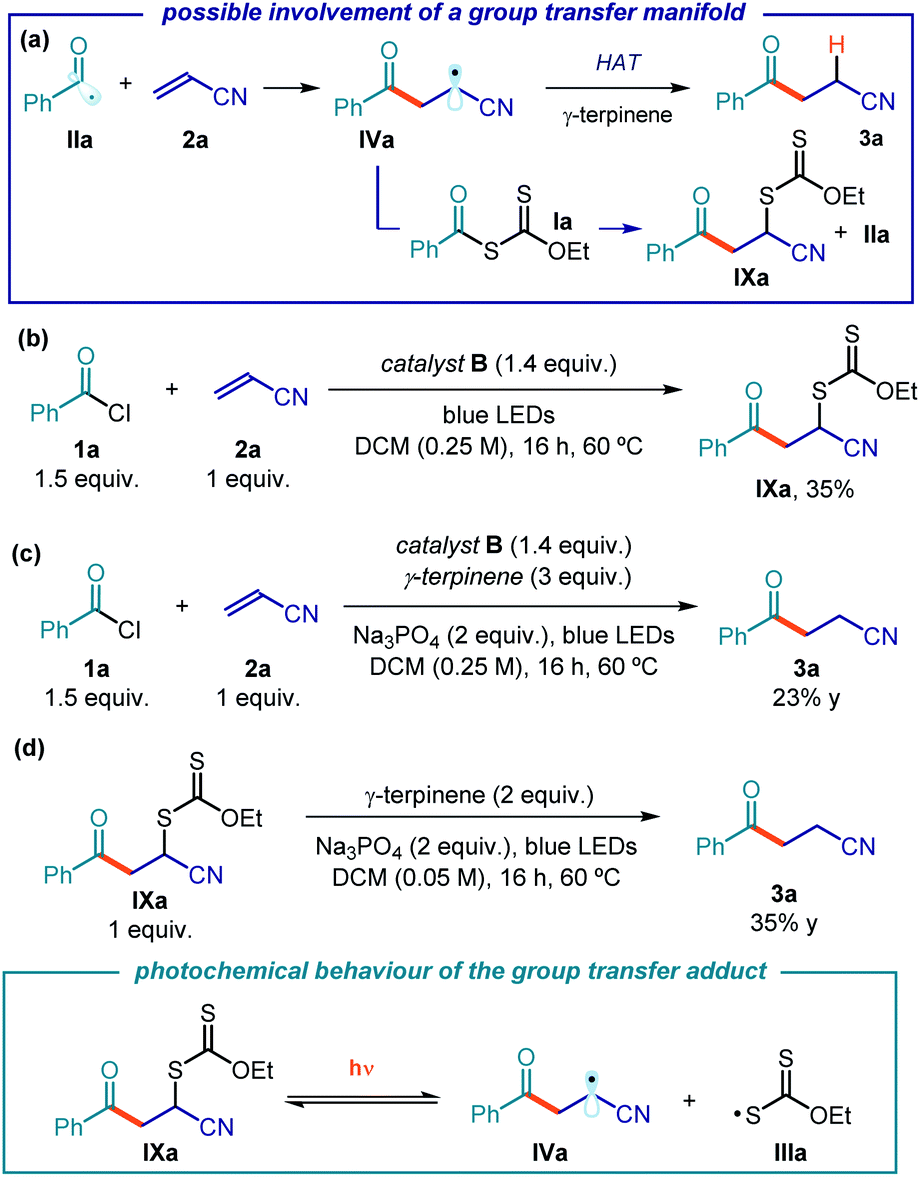 | ||
| Fig. 12 (a) Focusing on the acyl radical addition step. The model reaction in the presence of a stoichiometric amount of B and (b) in the absence or (c) in the presence of γ-terpinene (group transfer conditions). (d) The behavior of group transfer product IXa under the reaction conditions and upon blue light irradiation. | ||
When excluding γ-terpinene from the reaction, performed using an excess of xanthate B (1.4 equiv.), the group transfer product IXa was isolated in 35% yield (Fig. 12b). The Giese-type adduct 3a was not detected, which indicates the importance of γ-terpinene in enabling the radical conjugate addition manifold. By contrast, under identical conditions but in the presence of γ-terpinene, no traces of the group transfer product IXa could be detected, while the product 3a was exclusively formed (23% yield, Fig. 12c). Interestingly, the yield of 3a was much lower with respect to the experiment conducted using catalytic amounts of B (10 mol%, 82% yield). We suppose that the lower efficiency of the Giese addition path under stoichiometric conditions could be a consequence of the equilibrium showed in Fig. 8, i.e. a higher amount of intermediate I inhibits the radical addition to the olefin since the acyl radicals are sequestered by I.
We then prepared an authentic sample of the group transfer product IXa and subjected it to the optimized reaction conditions, but in the absence of catalyst B: the Giese-type adduct 3a was obtained in moderate yield (35% yield, Fig. 12d).41 This experiment is relevant to the reaction mechanism, since it implies the ability of the group transfer product, upon irradiation by blue LEDs and in the presence of γ-terpinene, to afford product 3a. This suggests that the group transfer product IXa, upon irradiation, may regenerate the progenitor radicals IIIa and IVa (inset in Fig. 12d). We also performed laser flash photolysis studies of intermediate IXa, observing a transient that is consonant with the xanthyl radical IIIa, generated upon photolysis (see Fig. S30 in the ESI†).
Although we never detected the group transfer adduct of type IX under catalytic conditions, we cannot exclude that this pathway is operative in our catalytic system. It is possible that intermediate IX may serve as another stabilizing, off-the-cycle intermediate, which can control the concentration of radical IIIa in solution through an additional equilibrium.
Overall catalytic cycle and full mechanistic picture
The mechanistic studies provided important clues to better delineate the mechanism of the photochemical Giese addition process. The full mechanistic picture is detailed in Fig. 13. We have identified that the overall catalytic cycle is regulated by different equilibriums that are essential to stabilizing the radical intermediates and controlling their overall concentration. For example, the highly reactive acyl radical II could be intercepted by xanthates of type I in a reversible addition–fragmentation equilibrium, leading to intermediate VI. At the same time, the xanthyl radical IIIa can both dimerize to form dimer VIIa and engage in a cross-coupling process with radical IVa to afford the xanthate transfer product IXa. These equilibriums, which are regulated by light, confer a persistent radical character to the xanthyl radical IIIa, facilitating its reduction and the turnover of the catalyst B. Importantly, we have demonstrated that γ-terpinene can use two different pathways to reduce the xanthyl radical IIIa, e.g. a direct HAT process and an SET reduction from the ensuing cyclohexadienyl-type radical V.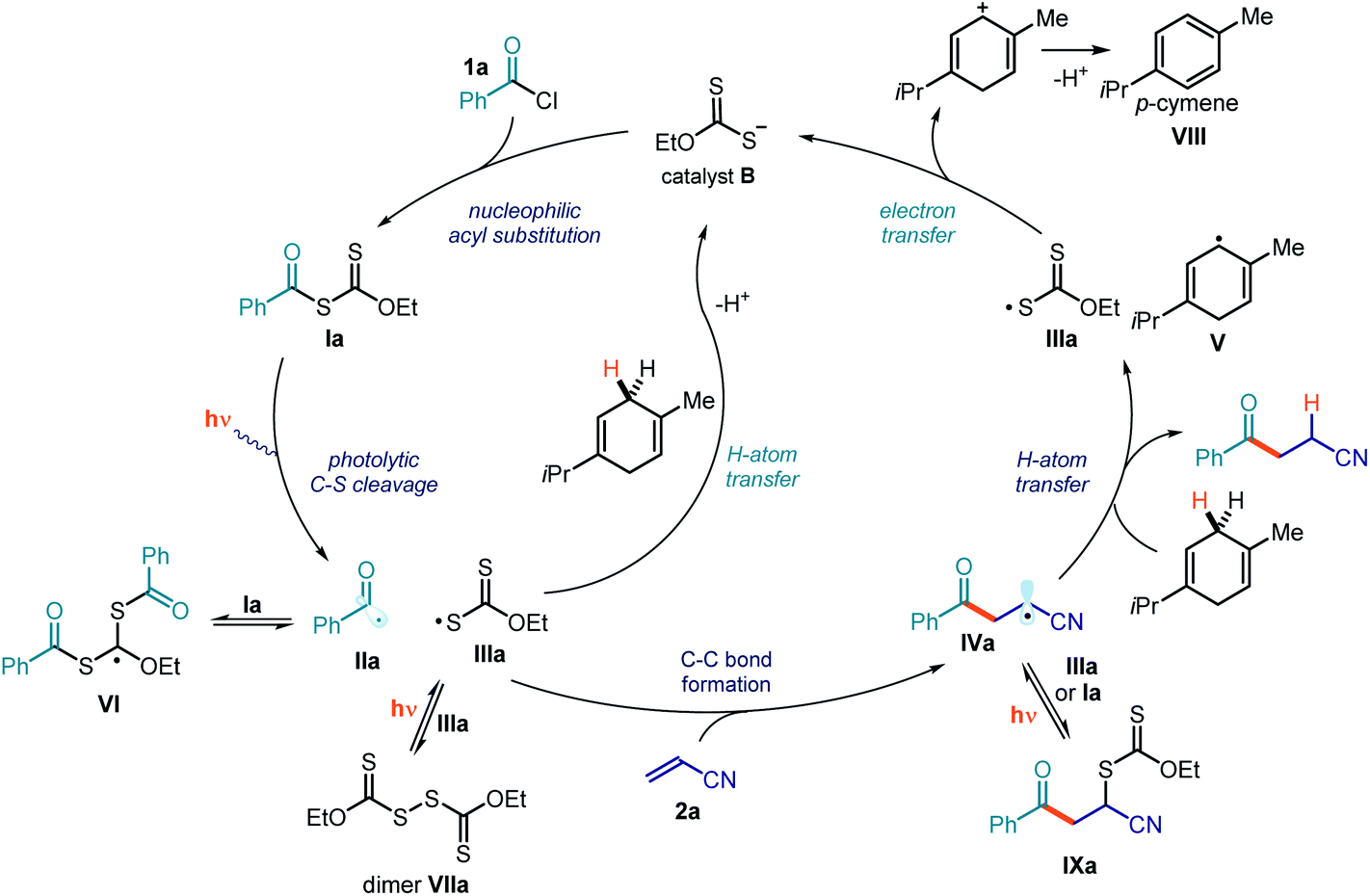 | ||
| Fig. 13 Detailed mechanistic picture, comprehensive of the off-the-cycle equilibriums, based on the mechanistic investigations conducted on the model reaction of acyl chloride 1a and acceptor 2a catalyzed by B. | ||
The main feature of the proposed mechanism in Fig. 13 is that it proceeds through a photocatalytic process, where the photoactivity of intermediate I dictates the formation of any molecule of the product. This scenario stands in contrast to a possible alternative radical chain propagation manifold, which would require the photoactivity of intermediate I to serve only as an initiation event. A tool to discriminate between these two mechanistic possibilities is to measure the quantum yield (Φ) of the overall reaction.42 We performed this measurement on the model reaction, and the quantum yield was found to be as low as 0.034 (λ = 460 nm, using potassium ferrioxalate as the actinometer). This result suggests that a radical chain process, based on either a xanthate group transfer manifold (Fig. 12a) or an SET from intermediate IV to intermediate I, is unlikely.43 Instead, the low quantum yield is congruent with the proposed photocatalytic mechanism depicted in Fig. 13.
Conclusions
In summary, we have developed a catalytic methodology that requires visible light and commercially available organic catalysts to access a wide range of acyl and carbamoyl radicals. These reactive intermediates are then trapped with a variety of electron-poor olefins in a Giese addition manifold. This methodology relies on the electrophilicity of the substrates rather than their reduction potential, allowing the use of readily available carboxylic acids/chlorides and carbamoyl chlorides (which would be recalcitrant to other radical generation strategies) as acyl and carbamoyl radical precursors. Extensive mechanistic investigations have been performed, including transient absorption spectroscopy investigations, electrochemical studies, quantum yield measurements, and the characterization of key intermediates. Collectively, these studies identified a variety of off-the-cycle intermediates that are in a light-regulated equilibrium with the reactive radicals. These regulated equilibriums cooperate to control the overall concentrations of the radicals, contributing to the efficiency of the process. The knowledge of the mechanistic details underlying this radical generation strategy may enable the rational development of new reactions. Our ongoing efforts are directed toward these aims.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank MINECO (CTQ2016-75520-P), and the European Research Council (ERC 681840 – CATA-LUX) for financial support. D. M. thanks H2020-MSCA-ITN-2016 (722591 – PHOTOTRAIN) for a predoctoral fellowship. We thank Dr Bertrand Schweitzer-Chaput (ICIQ), Dr Frank A. Arroyave (ICIQ), Mr Davide Spinnato (ICIQ), and Dr Robert Davidson (Dr Reddy's Laboratories) for useful discussions and precious mechanistic insights. We thank Dr Javier Pérez Hernández (ICIQ) for assistance with laser flash photolysis experiments.Notes and references
- M. Yan, J. C. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed.
- J. Lalevée and J. P. Fouassier, Overview of Radical Initiation in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, 2012, vol. 1, pp. 37–56 Search PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) N. Romero and D. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed.
- For radical formation using electrochemistry, see: C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed.
- (a) D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC; (b) D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC.
- Substrates bearing thio functions, including xanthates, have been extensively used in stoichiometric amounts to generate radicals, see: (a) D. H. R. Barton and S. Z. Zard, Pure Appl. Chem., 1986, 58, 675–684 CAS; (b) S. Z. Zard, Angew. Chem., Int. Ed., 1997, 36, 672–685 CrossRef.
- X.-H. Duan, B. Maji and H. Mayr, Org. Biomol. Chem., 2011, 9, 8046–8050 RSC.
- (a) S. Cuadros, M. A. Horwitz, B. Schweitzer-Chaput and P. Melchiorre, Chem. Sci., 2019, 10, 5484–5488 RSC; (b) D. Spinnato, B. Schweitzer-Chaput, G. Goti, M. Ošeka and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 9485–9490 CrossRef CAS PubMed.
- D. Mazzarella, G. Magagnano, B. Schweitzer-Chaput and P. Melchiorre, ACS Catal., 2019, 9, 5876–5880 CrossRef CAS.
- (a) D. H. R. Barton, M. V. George and M. Tomoeda, J. Chem. Soc., 1962, 1967–1974 RSC; (b) P. Delduc, C. Tailham and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1988, 308–310 RSC; (c) M. R. Heinrich and S. Z. Zard, Org. Lett., 2004, 6, 4969–4972 CrossRef CAS PubMed; (d) J. H. Penn and F. Liu, J. Org. Chem., 1994, 59, 2608–2612 CrossRef CAS; (e) F. Mestre, C. Tailham and S. Z. Zard, Heterocycles, 1989, 171–174 CAS.
- J. Lalevée, N. Blanchard, M. El-Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2347–2352 CrossRef.
- R. S. Grainger and P. Innocenti, Angew. Chem., Int. Ed., 2004, 43, 3445–3448 CrossRef CAS PubMed.
- (a) T. Caronna, G. P. Gardini and F. Minisci, J. Chem. Soc. D, 1969, 201 RSC; (b) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed; (c) D. L. Boger and R. J. Mathvink, J. Org. Chem., 1989, 54, 1777–1779 CrossRef CAS; (d) C. Chen, D. Crich and A. Papadatos, J. Am. Chem. Soc., 1992, 114, 8313–8314 CrossRef CAS; (e) D. Crich, C. Chen, J.-T. Hwang, H. Yuan, A. Papadatos and R. I. Walter, J. Am. Chem. Soc., 1994, 116, 8937–8951 CrossRef CAS.
- For selected recent examples based on a HAT mechanism, see: (a) X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353–11356 CrossRef CAS PubMed; (b) S. Mukherjee, R. A. Garza-Sanchez, A. Tlahuext-Aca and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 14723–14726 CrossRef CAS PubMed; (c) D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS; (d) M. Zhang, R. Ruzi, J. Xi, N. Li, Z. Wu, W. Li, S. Yu and C. Zhu, Org. Lett., 2017, 19, 3430–3433 CrossRef CAS PubMed; (e) M. D. Vu, M. Das and X.-W. Liu, Chem.–Eur. J., 2017, 23, 15899–15902 CrossRef CAS PubMed.
- For selected examples using α-keto acids as acyl radical precursors, see: (a) W.-M. Cheng, R. Shang, H.-Z. Yu and Y. Fu, Chem.–Eur. J., 2015, 21, 13191–13195 CrossRef CAS PubMed; (b) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4833 CrossRef CAS PubMed; (c) T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS PubMed; (d) J.-J. Zhao, H.-H. Zhang, X. Shen and S. Yu, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed. For an example using acyl silanes, see: (e) L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, ACS Catal., 2018, 8, 304–309 CrossRef CAS. For selected examples using 4-acyl-dihydropyridines, see: (f) G. Goti, B. Bieszczad, A. Vega-Peñaloza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS PubMed; (g) K. Zhang, L.-Q. Lu, Y. Jia, Y. Wang, F.-D. Lu, F. Pan and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 13375–13379 CrossRef CAS PubMed.
- For selected examples using hypervalent iodine reagents as acyl radical precursors, see: (a) H. Huang, G. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 7872–7876 CrossRef CAS PubMed; (b) H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374–8377 CrossRef CAS PubMed.
- C. Raviola, S. Protti, D. Ravelli and M. Fagnoni, Green Chem., 2019, 21, 748–764 RSC.
- S.-M. Xu, J.-Q. Chen, D. Liu, Y. Bao, Y.-M. Liang and P.-F. Xu, Org. Chem. Front., 2017, 4, 1331–1335 RSC.
- For selected examples using anhydride derivatives, see: (a) G. Bergonzini, C. Cassani and C.-J. Wallentin, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef CAS PubMed; (b) G. Bergonzini, C. Cassani, H. Lorimer-Olsson, J. Hörberg and C.-J. Wallentin, Chem.–Eur. J., 2016, 22, 3292–3295 CrossRef CAS PubMed; (c) S. Dong, G. Wu, X. Yuan, C. Zou and J. Ye, Org. Chem. Front., 2017, 4, 2230–2234 RSC.
- (a) N. C. Billingham, R. A. Jackson and F. Malek, J. Chem. Soc., Perkin Trans. 1, 1979, 1137–1141 RSC; (b) M. Ballestri, C. Chatgilialoglu, N. Cardi and A. Sommazzi, Tetrahedron Lett., 1992, 33, 1787–1790 CrossRef CAS.
- (a) J. Lusztyk, E. Lusztyk, B. Maillard, L. Lunazzi and K. U. Ingold, J. Am. Chem. Soc., 1983, 105, 4475–4477 CrossRef CAS; (b) J. Lusztyk, E. Lusztyk, B. Maillard and K. U. Ingold, J. Am. Chem. Soc., 1984, 106, 2923–2931 CrossRef CAS.
- D. Occhialini, K. Daasbjerg, H. Lund, K. Hult, T. Norin, P. Coppens and O. Buchardt, Acta Chem. Scand., 1993, 47, 1100–1106 CrossRef CAS.
- G. A. Urove and D. G. Peters, J. Electrochem. Soc., 1993, 140, 932–935 CrossRef CAS.
- (a) C. Chatgilialoglu and M. Lucarini, Tetrahedron Lett., 1995, 36, 1299–1302 CrossRef CAS; (b) H. Fischer and H. Paul, Acc. Chem. Res., 1987, 20, 200–206 CrossRef CAS.
- For a recent photoredox catalytic method of acyl radical formation from carboxylic acids, see: (a) E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed; (b) M. Zhang, J. Xie and C. Zhu, Nat. Commun., 2018, 9, 3517 CrossRef PubMed; (c) J. I. Martinez Alvarado, A. B. Ertel, A. Stegner, E. E. Stache and A. G. Doyle, Org. Lett., 2019, 21, 9940–9944 CrossRef CAS PubMed.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- (a) M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Commun., 2017, 53, 2335–2338 RSC; (b) I. Kim, G. Kang, K. Lee, B. Park, D. Kang, H. Jung, Y.-T. He, M.-H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 9239–9248 CrossRef PubMed.
- For examples on carbamoyl radicals generated through single electron oxidation: (a) M. Jouffroy and J. Kong, Chem.–Eur. J., 2019, 25, 2217–2221 CrossRef CAS PubMed; (b) Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172–2175 CrossRef CAS PubMed. For examples of radical formation through single electron reduction: (c) A. H. Jatoi, G. G. R. F. Pawar and Y. Landais, Chem. Commun., 2019, 55, 466–469 RSC; (d) W. F. Petersen, R. J. K. Taylor and J. R. Donald, Org. Lett., 2017, 19, 874–877 CrossRef CAS PubMed; (e) N. Alandini, L. Buzzetti, G. Favi, T. Schulte, L. Candish, K. D. Collins and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 5248–5253 CrossRef CAS PubMed.
- For the generation of carbamoyl radicals from carbamoylxanthates through the use of a thermal initiator, see: (a) G. López-Valdez, S. Olguín-Uribe and L. D. Miranda, Tetrahedron Lett., 2007, 48, 8285–8289 CrossRef; (b) A. Millán-Ortiz, G. López-Valdez, F. Cortez-Guzmán and L. D. Miranda, Chem. Commun., 2015, 51, 8345–8348 RSC.
- S. Hosseyni and A. Jarrahpour, Org. Biomol. Chem., 2018, 16, 6840–6852 RSC.
- A. Kaga, X. Wu, J. Y. J. Lim, H. Hayashi, Y. Lu, E. K. L. Yeow and S. Chiba, Beilstein J. Org. Chem., 2018, 14, 3047–3058 CrossRef CAS PubMed.
- (a) B. Bieszczad, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16878–16883 CrossRef CAS PubMed; (b) D. G. Hawthorne, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 5457–5459 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- S. Z. Zard, Helv. Chim. Acta, 2019, 102, e1900134 CrossRef.
- In analogy with the experiment depicted in Scheme 2, we also found that the dimer of catalyst C, which is commercially available, is a competent catalyst to promote the formation of carbamoyl radicals. Specifically, under the conditions reported in Fig. 5, 10 mol% of the dimer of C promotes the reaction of dimethylcarbamoyl chloride and vinyl sulfone to afford the Giese adduct 9a in 53% yield, see Section D1.5 in the ESI† for details.
- (a) D. Griller and U. K. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS; (b) K. S. Focsaneanu and J. C. Scaiano, Helv. Chim. Acta, 2006, 89, 2473–2482 CrossRef CAS; (c) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed.
- The small discrepancy in the values of the redox potentials are due to the difference in concentrations of the redox-active species. For example, in the case of the reduction peaks (Ered), which refer to the SET reduction of dimer VIIa, the concentration of VIIa is much higher in the analysis depicted in Fig. 11d and e than in Fig. 11b. This is because, in the latter case, dimer VIIa is in situ generated upon oxidation of catalyst B and dimerization of the ensuing xanthyl radical IIIa.
- We collected evidence that the group transfer intermediate of type IX may play a mechanistic relevant role also in the intramolecular process involving carbamoyl radicals detailed in Fig. 6. Specifically, when an authentic sample of a group transfer adduct, prepared according to ref. 15, was subjected to the optimized conditions for the intramolecular process, but in the absence of the dithiocarbamate catalyst C, the cyclization product 11a was obtained in moderate yield (48% yield). This experiment implies that, in analogy to the intermolecular process, the group transfer adduct may be a relevant intermediate also in the intramolecular variant. See Section D1.6, page S48 of the ESI† for details.
- (a) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed; (b) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- In general, a low quantum yield (Φ < 1) does not completely exclude a possible radical chain process, since it can be a consequence of a highly inefficient initiation step. In our case, the non-productive photoprocesses depicted in Fig. 13 (e.g., VIIa to IIIa, IXa to IVa), while inferring long lifetime to radical IIIa, can affect the quantum yield of the overall process.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and full compound characterization. See DOI: 10.1039/d0sc02313b |
| ‡ In memory of Kilian Muñiz (1970–2020). |
| This journal is © The Royal Society of Chemistry 2020 |