
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single-ion conducting covalent organic framework for aqueous rechargeable Zn-ion batteries†
Sodam
Park‡
 a,
Imanuel
Kristanto‡
b,
Gwan Yeong
Jung
b,
David B.
Ahn
a,
Kihun
Jeong
*a,
Sang Kyu
Kwak
*a and
Sang-Young
Lee
*c
a,
Imanuel
Kristanto‡
b,
Gwan Yeong
Jung
b,
David B.
Ahn
a,
Kihun
Jeong
*a,
Sang Kyu
Kwak
*a and
Sang-Young
Lee
*c
aDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: jkh1905@unist.ac.kr; skkwak@unist.ac.kr
bDepartment of Chemical Engineering, School of Energy and Chemical Engineering, UNIST, Ulsan 44919, Republic of Korea
cDepartment of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 120-749, Republic of Korea. E-mail: sangyounglee87@gmail.com
First published on 2nd October 2020
Abstract
Despite their potential as promising alternatives to current state-of-the-art lithium-ion batteries, aqueous rechargeable Zn-ion batteries are still far away from practical applications. Here, we present a new class of single-ion conducting electrolytes based on a zinc sulfonated covalent organic framework (TpPa-SO3Zn0.5) to address this challenging issue. TpPa-SO3Zn0.5 is synthesised to exhibit single Zn2+ conduction behaviour via its delocalised sulfonates that are covalently tethered to directional pores and achieve structural robustness by its β-ketoenamine linkages. Driven by these structural and physicochemical features, TpPa-SO3Zn0.5 improves the redox reliability of the Zn metal anode and acts as an ionomeric buffer layer for stabilising the MnO2 cathode. Such improvements in the TpPa-SO3Zn0.5–electrode interfaces, along with the ion transport phenomena, enable aqueous Zn–MnO2 batteries to exhibit long-term cyclability, demonstrating the viability of COF-mediated electrolytes for Zn-ion batteries.
Introduction
The forthcoming smart and ubiquitous energy era has inspired the relentless pursuit of advanced power sources with high electrochemical performances and safety that can outperform current state-of-the-art Li-ion batteries.1,2 Moreover, the rapid growth of power sources in emerging application fields has brought up new issues on their environmental benignity and cost competitiveness.3–5Among various power sources explored to date, aqueous Zn-ion batteries have garnered considerable attention as a promising candidate to achieve this challenging goal owing to the use of water-based electrolytes and multielectron redox (Zn0/2+)-driven high energy density.3–8 Notably, recent advances in electrochemical rechargeability enabled by introduction of mild acidic electrolytes have encouraged potential use of Zn-ion batteries.6–13 However, practical application of these batteries has still been staggering mainly due to the lack of suitable electrolytes ensuring interfacial stability with electrodes. In particular, poor redox reversibility of the Zn metal electrode poses a formidable challenge to the electrochemical performance sustainability.14–19
Single-ion conductors have been investigated as an appealing electrolyte platform beyond conventional liquid electrolytes since their high cation transference number (t+) can mitigate unwanted interfacial side reactions with electrodes.20–22 Despite these advantageous effects, a limited number of single Zn2+ conductors have been reported in comparison to monovalent cation (e.g., Li+ and Na+) conductors. An inorganic single-ion conductor based on ZnPS3 was reported as a solid-state electrolyte.23 However, strongly bound Zn2+ in the lattices resulted in poor ion transport (e.g., ionic conductivity (σ) = 10−8 to 10−6 S cm−1 at 60 °C). Other approaches include the use of anionic metal–organic frameworks (MOFs) and polymers.24–26 Unfortunately, the instability of electrolyte–electrode interfaces has not yet been resolved, making it difficult to enable practical Zn-ion batteries. Thus, an innovative concept of single Zn2+ conductors that can secure interfacial compatibility with electrodes as well as reliable ion transport behaviour is urgently needed.
Here, we demonstrate for the first time the use of a single-ion conducting covalent organic framework (COF) as a viable electrolyte strategy for aqueous rechargeable Zn-ion batteries. COFs have been regarded as appealing ion transport media owing to their ordered porous structure, functionalities and structural stability.27–33 A zinc sulfonated COF (TpPa-SO3Zn0.5; Fig. 1a) is synthesised to build well-defined directional channels in which covalently tethered and delocalised sulfonates play key roles in realising single Zn2+ transport. In addition, chemically stable β-ketoenamine linkages are introduced into the framework to achieve structural robustness against water. Benefiting from these structural and physicochemical uniqueness, TpPa-SO3Zn0.5 successfully exhibits single Zn2+ conduction characteristics (σ = 2.2 × 10−4 S cm−1 at room temperature and tZn2+ = 0.91), thereby allowing sustainable redox of the Zn metal anode. Moreover, TpPa-SO3Zn0.5 acts as an ionomeric buffer layer that can suppress structural disruption of the MnO2 cathode during repeated redox. These advantageous effects of TpPa-SO3Zn0.5 on the interfacial compatibility with the electrodes, in combination with its single-ion conduction properties, enable Zn–MnO2 cells to provide an excellent electrochemical performance with long-term cyclability.
 | ||
| Fig. 1 (a) Chemical structure, (b) SEM and EDS mapping (for S and Zn) images, (c) structural model (Zn2+: purple, TpPa-SO3–: grey), (d) XRD patterns and (e) N2 gas isotherms of TpPa-SO3Zn0.5. | ||
Results and discussion
TpPa-SO3Zn0.5 was synthesised via the solvothermal reaction of 1,3,5-triformylphloroglucinol (Tp) and 1,4-phenylenediamine-2-sulfonic acid (Pa-SO3H) to obtain a sulfonic acid COF (TpPa-SO3H; yield: 96%),28,34 followed by reaction with zinc acetate to exchange H+ with Zn2+ (yield: 91%; Scheme S1†). The CHN analysis and inductively coupled plasma optical emission spectrometry (ICP-OES) results show that the elemental composition of the synthesised TpPa-SO3Zn0.5 matches well with the theoretical composition (e.g., for the Zn content, 10.09 (calcd.) and 9.82 wt% (found); Table S1†). The formation of β-ketoenamine linkages in TpPa-SO3Zn0.5 was confirmed by the characteristic cross-polarisation magic angle spinning 13C nuclear magnetic resonance (CP-MAS 13C NMR) spectrum (C atoms assigned to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (184 ppm) and –HN–CC– groups (147 and 108 ppm); Fig. S1a†). This result was further verified by Fourier transform infrared (FT-IR) spectrometry (CC (1574 cm−1) and C–N stretches (1251 cm−1); Fig. S1b†).
O (184 ppm) and –HN–CC– groups (147 and 108 ppm); Fig. S1a†). This result was further verified by Fourier transform infrared (FT-IR) spectrometry (CC (1574 cm−1) and C–N stretches (1251 cm−1); Fig. S1b†).
Scanning electron microscopy (SEM), transmission electron microscopy (TEM) and energy dispersive spectrometry (EDS) results reveal the formation of TpPa-SO3Zn0.5 crystallites with a uniform distribution of S and Zn (Fig. 1b and S2†). The structural model of TpPa-SO3Zn0.5 (Fig. 1c) was constructed based on a triclinic crystal system (space group = P1; Table S2†)28,34 in which hexagonal pores are vertically stacked to form a stable eclipsed configuration with a slight offset (Fig. S3a†). The interplanar stacking distance along the c-axis was estimated to be ca. 3.4 Å (Fig. S3b†). In addition, the geometry of Zn2+ coordinated by three O atoms originating from two –CO and one –SO3– was suggested to be most thermodynamically stable (Fig. S3c†). The charge neutrality of the framework was secured by the localised charge distribution, in which Zn2+ and non-coordinating –SO3– produce the electron-deficient and -rich regions, respectively (Fig. S4†). The X-ray diffraction (XRD) pattern of TpPa-SO3Zn0.5 shows characteristic peaks at 2θ = 4.6 and 27.0° assigned to the (100) and (001) facets, respectively (Fig. 1d, black), which is similar to the simulated pattern obtained from the structural model (Fig. 1d, green). The difference in the peak width and intensity ratio between the experimental and simulated patterns might be due to the small crystallite size and deviation from perfect crystallinity.35,36 A series of TpPa-SO3X (X = H, Li, Zn0.5) afforded similar XRD patterns,28,34 showing that various positive charge carriers can be paired with the isostructural framework. In addition, the N2 gas sorption isotherms of TpPa-SO3Zn0.5 reveal a porous structure with a Brunauer–Emmett–Teller (BET) surface area of 472 m2 g−1 and a pore size of ca. 13 Å (Fig. 1e, black and S5†). This well-ordered porous structure of TpPa-SO3Zn0.5 could contribute to facilitating uniform and directional ion transport.27–33
The stability of TpPa-SO3Zn0.5 in water was evaluated as a requirement for use in aqueous batteries. After treatment in H2O at 100 °C for 7 days, TpPa-SO3Zn0.5 still showed the characteristic XRD pattern (Fig. 1d, purple) and porosity (BET surface area = 365 m2 g−1; Fig. 1e, purple). This good stability in water could be ascribed to the presence of chemically robust β-ketoenamine linkages.36–38
The ionic conductivity of TpPa-SO3Zn0.5 was examined by electrochemical impedance spectrometry (EIS) using Zn2+ blocking Ti||Ti cells. The self-standing pellet of TpPa-SO3Zn0.5 used in this analysis (the inset in Fig. 2a, left) was prepared by a cold-pressing method and showed a densely packed morphology (Fig. S6a†). The obtained pellet, in which H2O (100 wt% of the pellet weight) was incorporated to hydrate Zn2+,24,25 yielded σ = 2.2 × 10−4 S cm−1 at room temperature (Fig. 2a, black and S7†). The temperature-dependent ionic conductivity showed Arrhenius behaviour with an activation energy (Ea) of 0.19 eV, similar to that reported for the hydrated Zn2+ conducting MOF.24 To elucidate the role of Zn2+ in the COF media, a non-sulfonated COF comprising monophenyl building blocks (TpPa)38 was synthesised as a control sample without Zn2+ (Fig. S8†). TpPa showed a considerably low conductivity after hydration (σ = 4.3 × 10−6 S cm−1; Fig. S9†), verifying that the charge transport in the COF media is predominantly enabled by Zn2+.
 | ||
| Fig. 2 (a) Arrhenius plots for the ionic conductivity of hydrated TpPa-SO3Zn0.5. The inset shows optical images of a TpPa-SO3Zn0.5 pellet (left) and a TpPa-SO3Zn0.5–PTFE composite membrane (right). (b) EIS profiles and time-dependent current profile for a Zn|TpPa-SO3Zn0.5|Zn cell recorded at 20 mV polarisation. (c) tZn2+ values obtained for TpPa-SO3Zn0.5 (purple) and LE16 (2 M ZnSO4 in H2O; green). Representative snapshots obtained from the MD simulations showing time-dependent ion distributions in (d) TpPa-SO3Zn0.5 and (e) LE. Zn2+: coloured diversely for a clear representation of the movement, TpPa-SO3–: grey, SO42−: green, H2O: omitted for clarity. | ||
The mechanically flexible membrane of TpPa-SO3Zn0.5 was prepared by mixing a small amount of polytetrafluoroethylene (PTFE; 5 wt% of the TpPa-SO3Zn0.5 weight). The SEM image of the resultant composite membrane shows that the PTFE webs as a binder tightly entangle the TpPa-SO3Zn0.5 powders (Fig. S6b†), thereby resulting in good flexibility (the inset in Fig. 2a, right). Notably, the ion conduction characteristics of this membrane (σ = 1.8 × 10−4 S cm−1 at room temperature, Ea = 0.20 eV after hydration; Fig. 2a, purple) are comparable to those of the pellet, indicating that the PTFE binder minimally impedes the ion transport of TpPa-SO3Zn0.5. It should be noted that the electrochemical tests subsequently shown were performed using this practical composite membrane (identically denoted as TpPa-SO3Zn0.5).
To demonstrate the single-ion conduction behaviour of TpPa-SO3Zn0.5, its tZn2+ was examined using a potentiostatic polarisation method according to the following equation:39,40
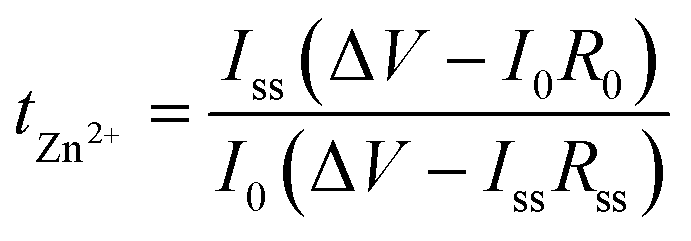
The ion conduction behaviour of TpPa-SO3Zn0.5 was further investigated via molecular dynamics (MD) simulations. For this analysis, a TpPa-SO3Zn0.5 model saturated with H2O and a control model of the LE (2 M ZnSO4 in H2O) were constructed (Table S4†). The application of an external electric field (1.0 V Å−1 in the –z-axis direction) to both model systems induced the ion movements (Fig. 2d and e). A notable finding is that a significantly uniform ion distribution was observed in TpPa-SO3Zn0.5 compared with the LE, which could result from the difference in the dynamic behaviour of the anion groups in both systems. In TpPa-SO3Zn0.5, the –SO3– group is covalently tethered along the directional pores, forming anionic channels that allowed the uniform Zn2+ flux (Fig. 2d). However, freely mobile SO42− in the LE formed randomly spread ion clusters with Zn2+ due to their strong electrostatic interactions (Fig. 2e). These random ion movements in the LE resulted in the velocity distribution to be nearly neutral against the direction of the applied electric field (Fig. S10a†, green). Meanwhile, a shift of the velocity distribution following the direction of the electric field was observed in TpPa-SO3Zn0.5 (Fig. S10a†, purple), correspondingly verifying the characteristic movement of Zn2+ in the anionic channels. Moreover, Zn2+ in TpPa-SO3Zn0.5 was found to be readily hydrated (i.e., a large fraction of O atoms coordinated to Zn2+ was derived from H2O; Fig. S10b†), indicating the promoted ionic dissociation between the anionic framework and Zn2+ that is highly advantageous for facile ion transport. These results verify that the immobilised and delocalised –SO3– group in TpPa-SO3Zn0.5 plays key roles in enabling fast single-ion conduction.
In addition to the ion conduction characteristics of TpPa-SO3Zn0.5, its thermal and electrochemical stabilities were investigated. The thermogravimetric analysis (TGA) curve shows no obvious weight loss up to 200 °C under a N2 atmosphere (Fig. S11†). The linear sweep voltammogram (LSV) shows that TpPa-SO3Zn0.5 is electrochemically stable up to ca. 2 V vs. Zn/Zn2+ (Fig. S12†), thus fulfilling the requirement for building practical aqueous batteries.
To examine the applicability of TpPa-SO3Zn0.5 to the Zn metal electrode, the galvanostatic cyclability of a Zn|TpPa-SO3Zn0.5|Zn cell was examined and compared with that of a Zn|LE|Zn cell (LE = 2 M ZnSO4 in H2O) at room temperature (current density = 0.1 mA cm−2, capacity = 0.1 mA h cm−2). The cell containing the LE, although it initially operated with a low overpotential (ca. 40 mV), showed fluctuating behaviour after 180 h and eventually failed (Fig. 3a, green), which might result from an electrical short-circuit. In sharp contrast, TpPa-SO3Zn0.5 allowed stable operation of the cell for 500 h, during which the overpotential gradually decreased from ca. 100 to 60 mV (Fig. 3a, purple). In addition, the structural ordering of TpPa-SO3Zn0.5 was not disrupted after the cycling test (Fig. S13†).
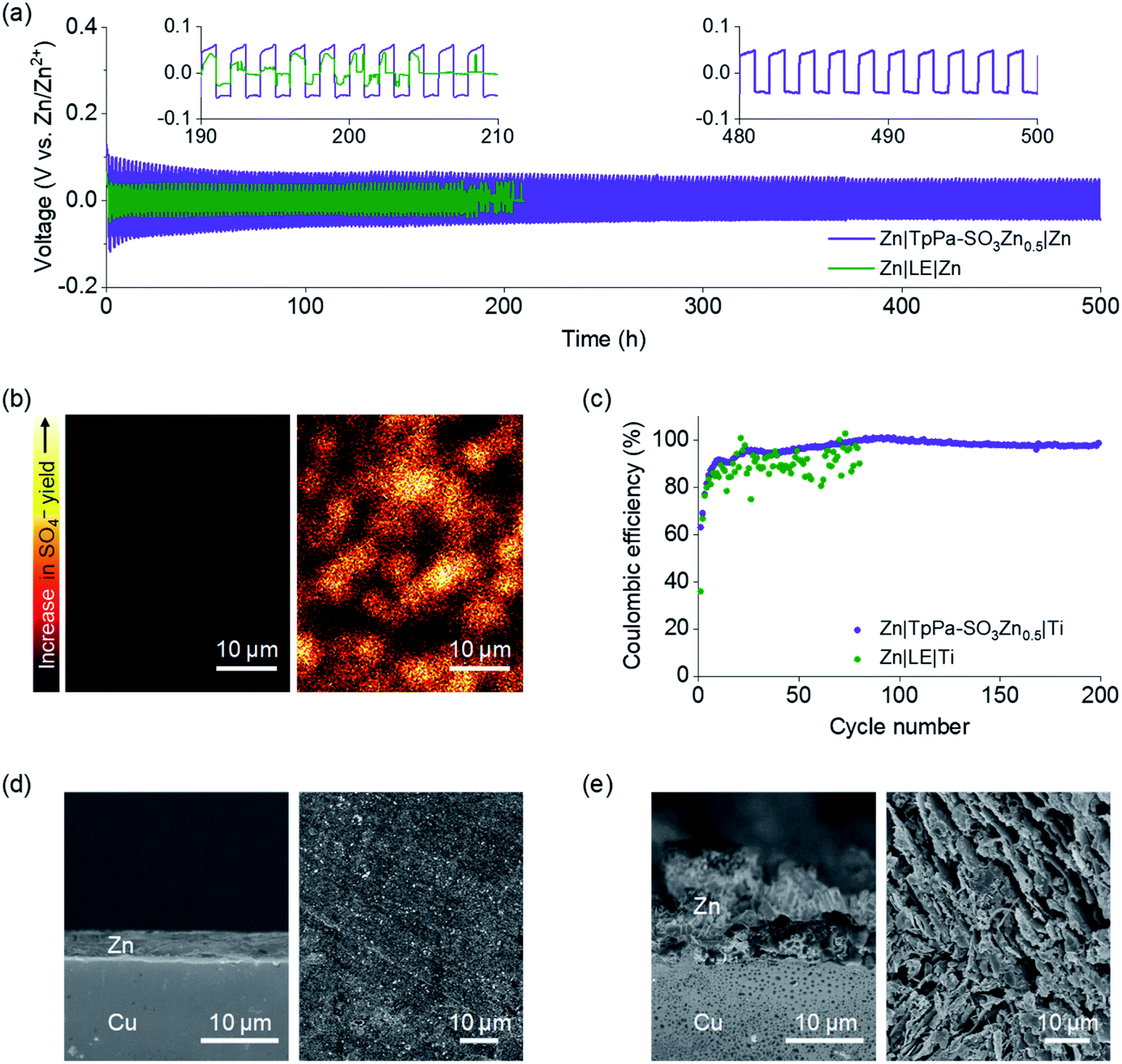 | ||
| Fig. 3 (a) Galvanostatic voltage profiles of Zn|TpPa-SO3Zn0.5|Zn (purple) and Zn|LE|Zn cells (LE = 2 M ZnSO4 in H2O; green). (b) TOF-SIMS mapping images for SO4– yielded on Zn metal electrodes in contact with TpPa-SO3Zn0.5 (left) or LE (right) after cycling tests. (c) Coulombic efficiency plots for Zn plating/stripping processes of Zn|TpPa-SO3Zn0.5|Ti (purple) and Zn|LE|Ti cells (green). Cross-sectional and surface SEM images for electrochemically plated Zn metals in contact with (d) TpPa-SO3Zn0.5 or (e) LE. | ||
Such an obvious difference in the stability of the cells was investigated by analysing the Zn metal electrodes after the cycling tests. Fig. 3b shows the mapping images of SO4– as a representative ion species yielded from time-of-flight secondary ion mass spectrometry (TOF-SIMS) conducted on the Zn metal electrodes of both cells. SO4– was minimally observed on the Zn metal electrode in contact with TpPa-SO3Zn0.5 (left), exhibiting good interfacial stability. In contrast, randomly proliferated SO4– was observed on the Zn metal surface in contact with the LE (right), indicating the formation of a deposition phase that could result from the nonuniform ion transport and adverse reactions between mobile SO42− and Zn metal. The X-ray photoelectron spectrometry (XPS) and XRD results correspondingly reveal that the unwanted corrosion products (e.g., Zn4SO4(OH)6·5H2O)41 were exclusively formed on the Zn metal electrode cycled in the LE (Fig. S14†).
The reversibility of Zn plating/stripping in the two electrolyte systems was compared using the Zn||Ti configurations. During a cycle, Zn metal was electrochemically plated on the Ti working electrode and subsequently stripped out (current density = 0.1 mA cm−2, capacity = 0.1 mA h cm−2). TpPa-SO3Zn0.5 allowed this process to persist over 200 cycles with high reversibility (Fig. 3c, purple), in contrast to the LE that resulted in cycling with severe fluctuation (Fig. 3c, green). The initially observed increase in the Coulombic efficiency of the Zn|TpPa-SO3Zn0.5|Ti cell might include gradual stabilisation of the TpPa-SO3Zn0.5–Ti metal interface.16
The advantages of TpPa-SO3Zn0.5 were further highlighted by monitoring the Zn electroplating behaviour. As shown in the SEM images, densely plated Zn metal with a smooth surface was observed on the Cu substrate in contact with TpPa-SO3Zn0.5 (Fig. 3d), whereas porous and rough Zn metal plating was formed in the LE (Fig. 3e). These results demonstrate the beneficial effects of single-ion transport in TpPa-SO3Zn0.5 on the stable and reversible Zn plating/stripping behaviour.
The applicability of TpPa-SO3Zn0.5 as a new electrolyte in aqueous rechargeable Zn-ion batteries was investigated using the Zn||MnO2 configurations. α-MnO2 used as a cathode material was hydrothermally synthesised according to a previous report,11 yielding brown powders that revealed a regular nanorod shape and a characteristic XRD pattern (Fig. S15a and b†). In addition, the normal redox behaviour of α-MnO2 (cathodic peaks at 1.2–1.4 and anodic peaks at ca. 1.6 V in the cyclic voltammograms (CVs); Fig. S15c†) consistent with the previous reports11,13 was observed.
The electrochemical performance of a Zn|TpPa-SO3Zn0.5|MnO2 cell was tested at a current density of 0.6 A g−1 at room temperature. Notably, cycling stability over 800 cycles was observed for this cell (Fig. 4a). The cell initially showed a specific capacity of 196.0 mA h g−1 and still delivered 144.0 mA h g−1 at the 800th cycle, representing 73% capacity retention. The discharge curves include two sloping plateaus at 1.2–1.4 V (Fig. 4b), similar to those observed in the CVs. In contrast, the cell containing a typical LE (2 M ZnSO4 + 0.2 M MnSO4 in H2O) showed rapid capacity decay during the cycling (40% capacity retention after the 400th cycle; Fig. S16a†).
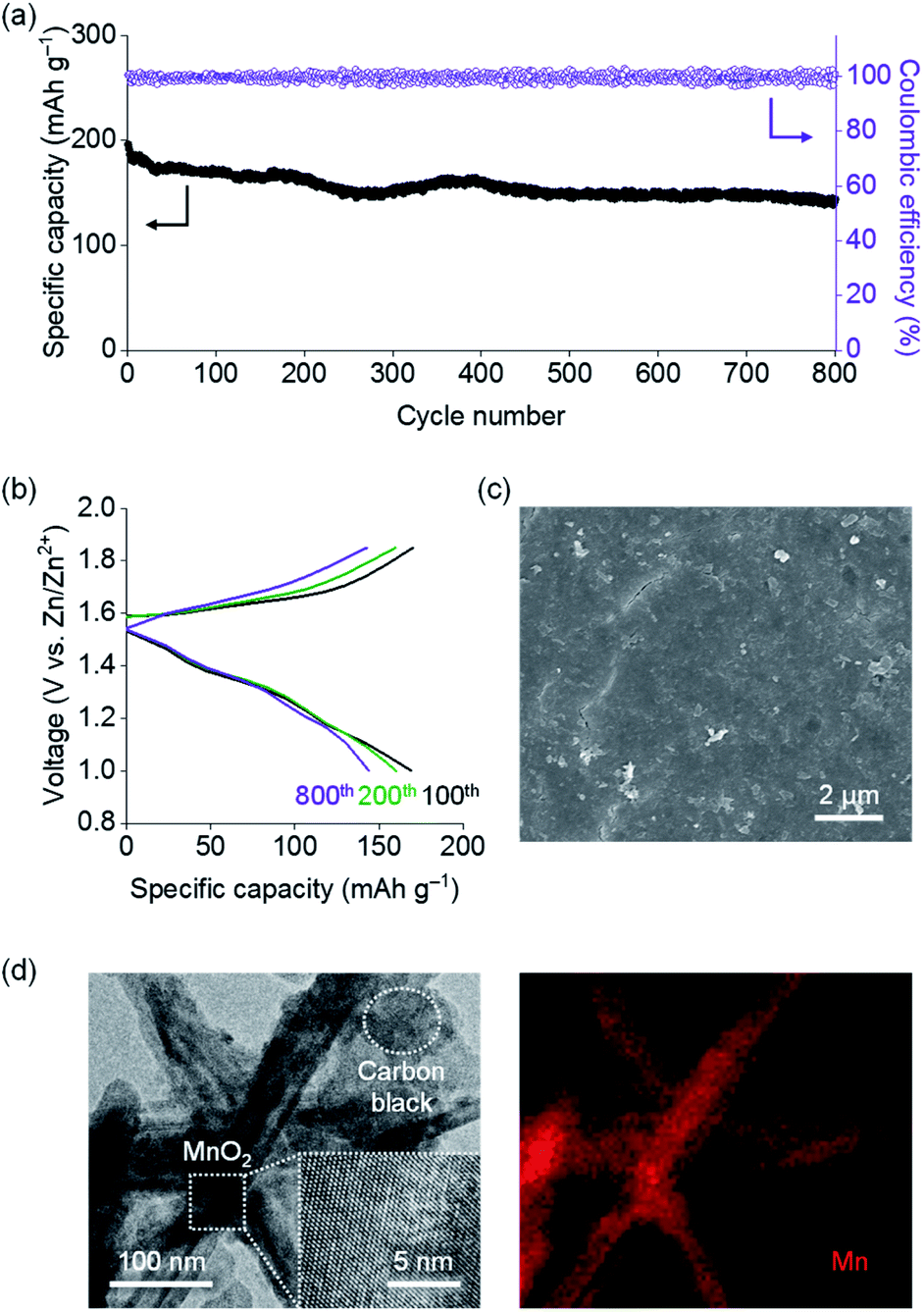 | ||
| Fig. 4 (a) Cycling performance of a Zn|TpPa-SO3Zn0.5|MnO2 cell. (b) Charge/discharge profiles at the 100th, 200th and 800th cycles. (c) SEM image of the Zn metal anode after the 800th cycle. (d) TEM (left) and EDS mapping (for Mn; right) images of the MnO2 cathode after the 800th cycle. | ||
Postmortem analyses were conducted on the decent cyclability of the Zn|TpPa-SO3Zn0.5|MnO2 cell. As shown in the SEM image (Fig. 4c), TpPa-SO3Zn0.5 allowed redox stability of the Zn metal anode in the cell, similar to that observed in the Zn plating/stripping tests (vide supra: Fig. 3). Moreover, the characteristic nanorod shape of MnO2 with structural ordering was well-maintained after the cycling (Fig. 4d), indicating that TpPa-SO3Zn0.5 might function as an ionomeric buffer layer to suppress the structural disruption of MnO2.42 The XPS results further show that the formation of decomposition products43 was retarded in the MnO2 cathode in contact with TpPa-SO3Zn0.5 (Fig. S17†). In contrast, the LE in the cell generated severely pulverised Zn metal and MnO2 particles (Fig. S16b and c†). These results verify that the good compatibility of TpPa-SO3Zn0.5 with the electrodes, which is ascribed to the unique structural features and single-ion conduction behaviour, significantly contributed to the cycling sustainability of the Zn|TpPa-SO3Zn0.5|MnO2 cell.
The performance of electrochemical cells containing TpPa-SO3Zn0.5 was compared with that of the cells containing other advanced electrolytes (Table S5†). Among the numerous previous approaches, the systems using single Zn2+ conductors, including an anionic MOF24 and sulfonated polymers,25,26 were exclusively chosen for fair comparison. TpPa-SO3Zn0.5 allowed superior cyclability of the electrochemical cells with a reliable operating voltage compared with the previously reported materials, underscoring that the single-ion conducting COF offers great promise as an attractive electrolyte for aqueous Zn-ion batteries.
Conclusions
We have presented the single-ion conducting COF, TpPa-SO3Zn0.5, as a viable electrolyte for aqueous rechargeable Zn-ion batteries. TpPa-SO3Zn0.5 featured immobilised and delocalised sulfonates in the directional pores (enabling single Zn2+ transport) and the stable β-ketoenamine linkages (allowing structural robustness against water). The unusual single-ion conduction behaviour of TpPa-SO3Zn0.5, which was elucidated by experimental and theoretical studies, allowed Zn plating/stripping reliability. In addition, TpPa-SO3Zn0.5 as an ionomeric buffer layer effectively prevented the structural disruption of the MnO2 cathode during repeated redox. Driven by these improvements in the TpPa-SO3Zn0.5–electrode interfaces and the ion transport phenomena, TpPa-SO3Zn0.5 enabled the Zn–MnO2 cells to provide long-term cycling performance (over 800 cycles at a current density of 0.6 A g−1 at an average operating voltage of 1.4 V). Future development of single Zn2+ conducting COFs will be directed to material optimisation and engineering with Zn-ion batteries. The COF-mediated electrolytes described herein move us a step closer toward practical use of rechargeable Zn-ion batteries and open a new route for the design of advanced aqueous electrolytes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program (2018R1A2A1A05019733 and 2018M3D1A1058624) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning. This work was also supported by the Samsung Research Funding Centre of Samsung Electronics under project no. SRFC-MA1702-04. KJ acknowledges the financial support from the Basic Science Research Program (2017R1D1A1B03033699 and 2020R1I1A1A01064798) through the NRF funded by the Ministry of Education. SKK acknowledges the financial support from ERC (NRF-2014R1A5A1009799) and computational support from UNIST-HPC.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- Z. Liu, Y. Huang, Y. Huang, Q. Yang, X. Li, Z. Huang and C. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 RSC.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS.
- H. Jia, Z. Wang, B. Tawiah, Y. Wang, C.-Y. Chan, B. Fei and F. Pan, Nano Energy, 2020, 70, 104523 CrossRef CAS.
- J. Shin, J. Lee, Y. Park and J. W. Choi, Chem. Sci., 2020, 11, 2028–2044 RSC.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS.
- S. Higashi, S. W. Lee, J. S. Lee, K. Takechi and Y. Cui, Nat. Commun., 2016, 7, 11801 CrossRef.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef.
- K. Jeong, S. Park and S.-Y. Lee, J. Mater. Chem. A, 2019, 7, 1917–1935 RSC.
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- A. J. Martinolich, C.-W. Lee, I.-T. Lu, S. C. Bevilacqua, M. B. Preefer, M. Bernardi, A. Schleife and K. A. See, Chem. Mater., 2019, 31, 3652–3661 CrossRef CAS.
- Z. Wang, J. Hu, L. Han, Z. Wang, H. Wang, Q. Zhao, J. Liu and F. Pan, Nano Energy, 2019, 56, 92–99 CrossRef CAS.
- C. Hänsel and D. Kundu, ACS Omega, 2019, 4, 2684–2692 CrossRef.
- B.-S. Lee, S. Cui, X. Xing, H. Liu, X. Yue, V. Petrova, H.-D. Lim, R. Chen and P. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 38928–38935 CrossRef CAS.
- X. Li and K. P. Loh, ACS Materials Lett., 2019, 1, 327–335 CrossRef CAS.
- K. Jeong, S. Park, G. Y. Jung, S.-H. Kim, Y.-H. Lee, S. K. Kwak and S.-Y. Lee, J. Am. Chem. Soc., 2019, 141, 5880–5885 CrossRef CAS.
- Z. Li, Z.-W. Liu, Z. Li, T.-X. Wang, F. Zhao, X. Ding, W. Feng and B.-H. Han, Adv. Funct. Mater., 2020, 30, 1909267 CrossRef CAS.
- Z. Guo, Y. Zhang, Y. Dong, J. Li, S. Li, P. Shao, X. Feng and B. Wang, J. Am. Chem. Soc., 2019, 141, 1923–1927 CrossRef CAS.
- S. Ashraf, Y. Zuo, S. Li, C. Liu, H. Wang, X. Feng, P. Li and B. Wang, Chem.–Eur. J., 2019, 25, 13479–13483 CrossRef CAS.
- Q. Xu, S. Tao, Q. Jiang and D. Jiang, J. Am. Chem. Soc., 2018, 140, 7429–7432 CrossRef CAS.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S.-H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS.
- S. Chandra, T. Kundu, K. Dey, M. Addicoat, T. Heine and R. Banerjee, Chem. Mater., 2016, 28, 1489–1494 CrossRef CAS.
- E. Vitaku and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 12911–12914 CrossRef CAS.
- S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- N. S. Schauser, R. Seshadri and R. A. Segalman, Mol. Syst. Des. Eng., 2019, 4, 263–279 RSC.
- H. Glatz, E. Tervoort and D. Kundu, ACS Appl. Mater. Interfaces, 2020, 12, 3522–3530 CrossRef CAS.
- Y. Zeng, X. Zhang, Y. Meng, M. Yu, J. Yi, Y. Wu, X. Lu and Y. Tong, Adv. Mater., 2017, 29, 1700274 CrossRef.
- Y. Li, S. Wang, J. R. Salvador, J. Wu, B. Liu, W. Yang, J. Yang, W. Zhang, J. Liu and J. Yang, Chem. Mater., 2019, 31, 2036–2047 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02785e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |