
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis[pyrrolyl Ru(II)] triads: a new class of photosensitizers for metal–organic photodynamic therapy†
Deborah A.
Smithen
 a,
Susan
Monro
b,
Mitch
Pinto
b,
John
Roque
III
cd,
Roberto M.
Diaz-Rodriguez
a,
Huimin
Yin
b,
Colin G.
Cameron
d,
Alison
Thompson
*a and
Sherri A.
McFarland
*d
a,
Susan
Monro
b,
Mitch
Pinto
b,
John
Roque
III
cd,
Roberto M.
Diaz-Rodriguez
a,
Huimin
Yin
b,
Colin G.
Cameron
d,
Alison
Thompson
*a and
Sherri A.
McFarland
*d
aDepartment of Chemistry, Dalhousie University, P. O. Box 15000, Halifax, NS B3H 4R2, Canada. E-mail: alison.thompson@dal.ca
bDepartment of Chemistry, Acadia University, Wolfville, NS B4P 2R6, Canada
cDepartment of Chemistry and Biochemistry, The University of North Carolina at Greensboro, PO Box 26170, Greensboro, NC 27402-6170, USA
dDepartment of Chemistry and Biochemistry, The University of Texas at Arlington, 700 Planetarium Pl, Arlington, TX 76019-0065, USA. E-mail: sherri.mcfarland@uta.edu
First published on 6th October 2020
Abstract
A new family of ten dinuclear Ru(II) complexes based on the bis[pyrrolyl Ru(II)] triad scaffold, where two Ru(bpy)2 centers are separated by a variety of organic linkers, was prepared to evaluate the influence of the organic chromophore on the spectroscopic and in vitro photodynamic therapy (PDT) properties of the compounds. The bis[pyrrolyl Ru(II)] triads absorbed strongly throughout the visible region, with several members having molar extinction coefficients (ε) ≥ 104 at 600–620 nm and longer. Phosphorescence quantum yields (Φp) were generally less than 0.1% and in some cases undetectable. The singlet oxygen quantum yields (ΦΔ) ranged from 5% to 77% and generally correlated with their photocytotoxicities toward human leukemia (HL-60) cells regardless of the wavelength of light used. Dark cytotoxicities varied ten-fold, with EC50 values in the range of 10–100 μM and phototherapeutic indices (PIs) as large as 5400 and 260 with broadband visible (28 J cm−2, 7.8 mW cm−2) and 625 nm red (100 J cm−2, 42 mW cm−2) light, respectively. The bis[pyrrolyl Ru(II)] triad with a pyrenyl linker (5h) was especially potent, with an EC50 value of 1 nM and PI > 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 with visible light and subnanomolar activity with 625 nm light (100 J cm−2, 28 mW cm−2). The lead compound 5h was also tested in a tumor spheroid assay using the HL60 cell line and exhibited greater photocytotoxicity in this more resistant model (EC50 = 60 nM and PI > 1200 with 625 nm light) despite a lower dark cytotoxicity. The in vitro PDT effects of 5h extended to bacteria, where submicromolar EC50 values and PIs >300 against S. mutans and S. aureus were obtained with visible light. This activity was attenuated with 625 nm red light, but PIs were still near 50. The ligand-localized 3ππ* state contributed by the pyrenyl linker of 5h likely plays a key role in its phototoxic effects toward cancer cells and bacteria.
000 with visible light and subnanomolar activity with 625 nm light (100 J cm−2, 28 mW cm−2). The lead compound 5h was also tested in a tumor spheroid assay using the HL60 cell line and exhibited greater photocytotoxicity in this more resistant model (EC50 = 60 nM and PI > 1200 with 625 nm light) despite a lower dark cytotoxicity. The in vitro PDT effects of 5h extended to bacteria, where submicromolar EC50 values and PIs >300 against S. mutans and S. aureus were obtained with visible light. This activity was attenuated with 625 nm red light, but PIs were still near 50. The ligand-localized 3ππ* state contributed by the pyrenyl linker of 5h likely plays a key role in its phototoxic effects toward cancer cells and bacteria.
1. Introduction
Light-responsive prodrugs are the basis for selectively targeting unwanted cells and tissue in photodynamic therapy (PDT). Activation of an otherwise nontoxic photosensitizer (PS) produces cytotoxic singlet oxygen (1O2) and other reactive oxygen species (ROS) in regions where the PS, light, and oxygen overlap spatiotemporally,1–3 thus confining toxicity to diseased tissue while sparing healthy tissue. The antitumor effects of PDT result from destruction of primary tumors and tumor vasculature, but can also include a systemic immunological response.4–12 Photofrin, a mixture of oligomeric tetrapyrroles, remains arguably the most utilized PS for PDT.12–15 However, a variety of second- and third-generation derivatives, including metallated tetrapyrroles, that seek to improve upon the properties of earlier PSs have gained attention and (in some cases) approval in certain countries.16,17Metal complexes that are not simply metallated tetrapyrroles are particularly intriguing as PSs for PDT,18,19 and there are numerous reports highlighting their rich photophysical and photochemical properties.20 Their modular architectures can be exploited to produce a variety of energetically accessible excited state configurations: metal-to-ligand charge transfer (MLCT),21 metal centered (MC),22–24 ligand centered (LC) or intraligand (IL),25–27 intraligand charge transfer (ILCT),28–30 ligand-to-ligand charge transfer (LLCT),31–33 ligand-to-metal charge transfer (LMCT),34 and metal-to-metal charge transfer (MMCT) in the case of multimetallic systems.35–38 Some of these excited states (and combinations thereof) may undergo the type I and II photoprocesses that define PDT or they may exert phototoxic effects via alternate mechanisms that do not involve oxygen. The oxygen-independent pathways, which includes stoichiometric photodissociation of ligands,22,24,39–45 have been collectively grouped as photochemotherapy (PCT) although no PCT agents have been approved for cancer therapy to date.16,46
Through our search for PSs that produce phototoxic effects in hypoxia via catalytic photosensitization pathways, we have found that the best features of both organic and inorganic PSs can be combined to produce hybrid systems, and the resulting metal–organic dyads exhibit unprecedented photocytotoxicities and phototherapeutic indices (PIs).26,47,48 Organic chromophores, either contiguously fused or tethered to coordinating diimine ligands, serve as excellent collection points for excitation energy from singlet excited states provided their localized 3IL states are energetically accessible through equilibration or relaxation. Organic triplets offer a unique means of slowing T → S intersystem crossing (ISC) in metal complexes, while the metal facilitates efficient formation of these triplet excited states and the possibility of oxygen-independent photoreactivity. Pure 3IL states that are lower in energy than the lowest lying 3MLCT state(s) tend to possess exceptionally long lifetimes (>20 μs) and proved very effective for in vitro PDT.26,47,49–52
From our extensive work in this area, we have found that organic triplets having charge transfer character (3ILCT) contributed by α-oligothienyl groups in certain systems are particularly photoreactive and make excellent PDT agents.16,48,53–61 Our TLD1433 is one example, which is a bis-heteroleptic Ru(II) complex based on the α-terthienyl-appended imidazo[4,5-f][1,10]phenanthroline (IP-3T) ligand that generates 1O2 with almost unity efficiency.16,17,48,62–66 TLD1433 is the first Ru(II) complex to enter a human clinical trial and is being evaluated in a Phase 2 clinical trial for treating non-muscle invasive bladder cancer with PDT (Clinicaltrials.gov identifier: NCT03945162).16,17,66
Our ongoing interest in exploring the photoreactivity of Ru(II) metal–organic systems, including TLD1433, inspired the present study. Herein, we explore the bis[Ru(II)-pyrrolide] scaffold, a metal–organic–metal triad, to push the envelope for achieving unprecedented in vitro PDT potency with 3IL excited states. This construct simultaneously satisfies three criteria: (i) low energy singlet and triplet MLCT states, (ii) utilization of two metal centers to funnel energy to an organic triplet, and (iii) incorporation of an organic chromophore with a triplet excited state of suitable energy and lifetime. Previously, we have shown that 2-formyl and 2-keto pyrroles can replace one of the 2,2′-bipyridyl (bpy) ligands in [Ru(bpy)3]2+ to form stable complexes under ambient conditions with MLCT states shifted as much as 1.52 eV relative to the parent complex,67 building on work involving 2-formylpyridyl ligands that convert to their hydrate form upon complexation.68 In our model mononuclear 2-formylpyrrolide complexes, continuous absorption out to 600 nm was achieved without the need for sterically-demanding diimines such as 2,2′-biquinoline (biq) that are known to lower the energies of both MLCT and MC states, leading to red-shifted absorption, but also photodissociation.
The small, bidentate pyrrolide ligand forms strong N-σ (η1) bonds to Ru(II), lowering the energy of MLCT states without promoting ligand loss from dissociative 3MC states. Conversion of this 2-formyl pyrrole ligand into its symmetric bis(formylpyrrole) counterpart with a central organic chromophore linker and coordination of the termini to Ru(II) diimine units was expected to result in complexes with a larger percentage of accessible 3IL triplets. Herein we report the synthesis and characterization of a family of bis[Ru(II)-pyrrolide] triads that differ in the identity of the organic chromophore used as the central linker. The influence of this unit on the photobiological activities within this class of compounds is examined in detail, and the potent in vitro PDT effects discussed.
2. Experimental procedures
2.1 Materials
All chemicals and reagents were purchased from commercial sources and were used as received, unless otherwise noted. Ethyl acetate, hexanes and dichloromethane were obtained crude and purified via distillation, under air and at 1 atm pressure, before use. Reagent-grade tetrahydrofuran (THF), ethylene glycol, isopropanol (IPA) and acetone were employed where stated. Anhydrous dichloromethane and dimethylformamide (DMF) were purchased from EMD Chemicals and Sigma Aldrich, respectively. All glassware was oven dried and purged with inert gas before use. Gravity column chromatography was performed using 230–400 mesh Silicycle ultra-pure silica gel or 150-mesh Brockman III activated neutral aluminum oxide. TLC was performed on silica gel or aluminum oxide plates and visualized using UV light (254 and/or 365 nm) and/or developed with vanillin stain.Characterized fetal bovine serum (FBS) and Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 4 mM L-glutamine were purchased from Fisher Scientific. Human promyelocytic leukemia cells (HL-60), Streptococcus mutans, and Streptococcus aureus were purchased from American Type Culture Collection (ATCC) through Cedarlane (Burlington, ON). Prior to use, FBS was aliquoted in 40 mL volumes, heat inactivated for 30 min at 55 °C, and stored at −20 °C. Water for biological experiments was deionized to a resistivity of 18 MΩ cm using a Barnstead filtration system.
2.2 Instrumentation
NMR spectra were recorded using a 500 MHz spectrometer. All 1H and 13C NMR chemical shifts are expressed in parts per million (ppm) using the solvent signal [CDCl3 (1H 7.26 ppm; 13C 77.16 ppm); DMSO-d6 (1H 2.50 ppm; 13C 39.52 ppm); THF-d8 (1H 1.73, 3.58 ppm; 13C 25.4, 67.6 ppm); CD2Cl2 (1H 5.32 ppm; 13C 53.8 ppm)] as the internal reference. Splitting patterns are indicated as follows: br, broad; s, singlet; d, doublet; t, triplet; at, apparent triplet; q, quartet; m, multiplet; sep, septet. All coupling constants (J) are reported in Hertz (Hz). Ultraviolet-visible spectra were recorded using a Varian Cary 100 Bio spectrophotometer. Mass spectra were recorded using ion trap (ESI or APCI) instruments. Microwave-promoted reactions were carried out using a Biotage Initiator 8 microwave with 0–400 W power at 2.45 GHz. Melting points are uncorrected.2.3 Synthesis and characterization
General procedure for the synthesis of bis(pyrrole)s (2) by Heck reaction (GP1). Palladium(II) acetate (1 mol%) and 2,4-pentanedione (2 mol%) were added to a solution of aryl dibromide (0.35 mmol, 1 equiv.) in anhydrous DMF (2.0 mL) at room temperature under argon, and stirred for 10 minutes. 2-Vinyl-N-Boc pyrrole (1a) (0.88 mmol, 2.5 equiv.) was then added as an oil, followed by potassium carbonate (0.7 mmol, 2 equiv.) as a solid in one portion, and the flask was sealed with a glass stopper before heating to 130 °C (Caution: always use a blast shield when heating a sealed system), using a sand bath covered with aluminum foil, with stirring for 6 hours. After cooling slightly, the reaction mixture was poured into ice-water (40 mL), neutralized with a few drops of 1 M HCl and refrigerated (4 °C) overnight. The resulting precipitate was collected using a Millipore filtration apparatus and then dried in a vacuum oven to give the crude product, which was subsequently washed with 0–30% diethyl ether/hexanes on a Millipore filter to give the desired bis(pyrrole) without the need for further purification, unless otherwise stated.
General procedure for the synthesis of bis(pyrrole)s (2) by Suzuki reaction (GP2). A solution of aryl dibromide (0.15 mmol, 1 equiv.) and 1-Boc-pyrrole-2-boronic acid (1b) (0.45 mmol, 3 equiv.) in anhydrous DMF (3 mL) was sparged with nitrogen gas for 10 minutes. Tetrakis(triphenylphosphine)palladium(0) (0.015 mmol, 0.1 equiv.) and potassium carbonate (0.60 mmol, 4 equiv.) were then added with stirring, and the solution was sparged with nitrogen for a further 5 minutes before the flask was sealed and heated to 110 °C for 24 hours. The reaction mixture was then cooled to room temperature and separated between dichloromethane (50 mL) and water (50 mL). The aqueous phase was extracted with dichloromethane (2 × 50 mL) and the combined organic extracts were washed with water (4 × 100 mL) and brine (100 mL), dried over anhydrous sodium sulfate, and concentrated to give the crude product which was purified using column chromatography on silica gel.
General procedure for the synthesis of bis(formylpyrrole)s (3) using Vilsmeier–Haack reaction (GP3). The desired bis(pyrrole) (2) (0.2 mmol, 1 equiv.) was dissolved in anhydrous DMF (4 mL) with stirring under nitrogen, and the solution was cooled to 0 °C in an ice bath. Phosphorous oxychloride (0.44 mmol, 2.2 equiv.), was then added drop-wise and the reaction mixture was warmed to 60 °C with stirring for 1.5 hours. After cooling to room temperature, 5% (w/v) aqueous potassium carbonate solution (∼3 mL) was added slowly until the solution became basic (∼pH 8, pH paper). The reaction mixture was then heated to 80 °C with stirring for 2 hours, before being poured into ice-water to precipitate the product which was collected using a Millipore filtration apparatus. The product was then dried in a vacuum oven and finally washed with 50–100% diethyl ether/hexanes.
General procedure for the synthesis of bis(ruthenium(II))hexafluorophosphate complex salts (4) (GP4). Triethylamine (0.24 mmol, 8 equiv.) was added to a suspension of dipyrrolic ligand (3) (0.031 mmol, 1.03 equiv.) and cis-bis-(2,2′-bipyridine)dichlororuthenium(II)dihydrate (0.06 mmol, 2 equiv.) in ethylene glycol (2.0 mL) in a Biotage microwave vial (2–5 mL capacity). The vial was then sealed using a manual cap crimper and placed in the microwave reactor, where it was heated at 125 °C for 80 minutes, at a maximum of 400 W power. After cooling, the reaction mixture was poured into a solution of ammonium hexafluorophosphate (0.45 mmol, 15 equiv.) in water (20 mL) and left to stand at room temperature overnight. The solution was then extracted thoroughly with dichloromethane (4 × 20 mL). The combined organic extracts were washed with brine (50 mL), dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified using column chromatography on silica gel (0–8% IPA/dichloromethane) and/or neutral alumina (0–8% methanol/dichloromethane).
General procedure for the conversion of bis(ruthenium(II))hexafluorophosphate complex salts to chloride salts (5) (GP5). Tetrabutylammonium chloride monohydrate (0.25 mmol, 20 equiv.) was added to a solution of the bis(ruthenium)hexafluorophosphate salt (4) (0.0125 mmol, 1.0 equiv.) in acetone (12 mL, 1 mM) with stirring at room temperature for 15 minutes. The desired chloride salt was generally observed to form as a precipitate during this time (unless otherwise stated), which was collected using Millipore filtration and washed with 30% acetone/hexanes before drying in a vacuum oven.
(E)-2-Styryl-1H-pyrrole (2a)69. Compound 2a was synthesized from 2-vinyl-N-Boc pyrrole (1a, 1.3 equiv.) and bromobenzene (a) using GP1 and a reaction time of 3 h. After cooling to room temperature, the reaction mixture was separated between diethyl ether (30 mL) and water (20 mL). The aqueous phase was extracted with diethyl ether (4 × 20 mL) and the combined organic extracts were washed with water (100 mL) and brine (100 mL), dried over anhydrous magnesium sulfate and concentrated in vacuo. The crude product was purified using column chromatography on silica gel eluting with 15% ethyl acetate/hexanes to give the title compound (34 mg, 64% yield) as a pale yellow solid. Mp 110–115 °C. 1H NMR (CDCl3, 500 MHz) δ: 7.43 (d, 2H, J = 7.5 Hz, ArH), 7.33 (at, 2H, J = 7.8 Hz, ArH), 7.21 (t, 1H, J = 7.3 Hz, ArH), 6.98 (d, 1H, J = 16.5 Hz, ArH), 6.83–6.82 (m, 1H), 6.67 (d, 2H, J = 16.5 Hz, ArH), 6.35–6.36 (m, 1H), 6.25 (aq, 1H, J = 3.0 Hz) ppm. 13C NMR (CDCl3, 125 MHz) δ: 137.6, 130.9, 128.8, 127.1, 126.0, 123.5, 119.2, 119.1, 110.2, 109.3 ppm. LRMS: 170.1 (M + H)+; HRMS calculated for C12H12N: 170.0964; found 170.0964.
1,4-Bis((E)-2-(1H-pyrrol-2-yl)vinyl)benzene (2b). Compound 2b was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 1,4-dibromobenzene (b) using GP1 to give the title compound (95 mg, 86% yield) as a dark yellow solid. Mp/dp > 280 °C. 1H NMR (THF-d8, 500 MHz) δ: 10.27 (br s, 2H, NH), 7.34 (s, 4H, ArH), 6.97 (d, 2H, J = 16.5 Hz, C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.72–6.71 (m, 2H, PyH), 6.68 (d, 2H, J = 16.5 Hz, CCH), 6.20–6.19 (m, 2H, PyH), 6.06–6.05 (m, 2H, PyH) ppm. 13C NMR (THF-d8, 125 MHz) δ: 137.5, 132.0, 126.6, 122.8, 120.0, 119.9, 109.9, 109.8 ppm. LRMS: 259.1 (M − H)−; HRMS calculated for C18H15N2: 259.1241; found 259.1238. ε386 nm = 48000 (THF).
CH), 6.72–6.71 (m, 2H, PyH), 6.68 (d, 2H, J = 16.5 Hz, CCH), 6.20–6.19 (m, 2H, PyH), 6.06–6.05 (m, 2H, PyH) ppm. 13C NMR (THF-d8, 125 MHz) δ: 137.5, 132.0, 126.6, 122.8, 120.0, 119.9, 109.9, 109.8 ppm. LRMS: 259.1 (M − H)−; HRMS calculated for C18H15N2: 259.1241; found 259.1238. ε386 nm = 48000 (THF).
4,4′-Bis((E)-2-(1H-pyrrol-2-yl)vinyl)-1,1′-biphenyl (2c). Compound 2c was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 4,4′-dibromobiphenyl (c) using GP1. The crude product was washed with 1
:1 diethyl ether:hexanes to give the title compound (95 mg, 86% yield) as a light brown solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 11.20 (br s, 2H, NH), 7.67 (d, 4H, J = 8.3 Hz, ArH), 7.52 (d, 4H, J = 8.3 Hz, ArH), 7.09 (d, 2H, J = 16.5 Hz, CCH), 6.88 (d, 2H, J = 16.5 Hz, CCH), 6.84 (br s, 2H, pyH), 6.28 (br s, 2H, PyH), 6.08 (br s, 2H, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 137.5, 136.8, 130.4, 126.5, 126.0, 121.6, 119.9, 119.8, 109.3, 109.0 ppm. LRMS: 337.2 (M + H)+; HRMS calculated for C24H21N2: 337.1699; found 337.1688. ε380 nm = 74000 (THF).
2,6-Bis((E)-2-(1H-pyrrol-2-yl)vinyl)naphthalene (2d). Compound 2d was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 2,6-dibromonaphthalene (d) using GP1 to give the title compound (95 mg, 97% yield) as a light brown solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 11.23 (br s, 2H, NH), 7.82 (d, 2H, J = 8.5 Hz, ArH), 7.76 (s, 2H, ArH), 7.69 (d, 2H, J = 8.5 Hz, ArH), 7.17 (d, 2H, J = 16.5 Hz, C
CH), 6.99 (d, 2H, J = 16.5 Hz, CCH), 6.86 (dd, 2H, J = 2.5, 4.0 Hz, PyH), 6.30 (br s, 2H, PyH), 6.09 (dd, 2H, J = 2.5, 5.5 Hz, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 135.0, 132.5, 130.5, 128.1, 124.5, 123.6, 122.2, 120.0, 119.9, 109.4, 109.0 ppm. LRMS: 311.2 (M + H)+; HRMS (APCI) calculated for C24H21N2: 311.1543; found 311.1528. ε384 nm = 59000 (THF).
4,7-Bis((E)-2-(1H-pyrrol-2-yl)vinyl)benzo[c][1,2,5]thiadiazole (2e). Compound 2e was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 4,7-dibromobenzo[c]-1,2,5-thiadiazole (e) using GP1. After cooling to room temperature, the reaction mixture was separated between 1
:2 THF:diethyl ether (30 mL) and water (20 mL). The aqueous phase was extracted with 1:2 THF:diethyl ether (4 × 20 mL) and the combined organic extracts were concentrated in vacuo. The crude product was purified using column chromatography on silica eluting with 30% ethyl acetate/hexanes to give the title compound (99 mg, 91% yield) as a red solid. Mp 200–205 °C. 1H NMR (CDCl3, 500 MHz) δ: 8.51 (br s, 2H, NH), 7.84 (d, 2H, J = 16.5 Hz, CCH), 7.52 (s, 2H, ArH), 7.17 (d, 2H, J = 16.5 Hz, CCH), 6.88 (dd, 2H, J = 2.5, 4.0 Hz, PyH), 6.51 (br s, 2H, PyH), 6.30 (dd, 2H, J = 2.5, 6.0 Hz, PyH) ppm. 13C NMR (CDCl3, 125 MHz) δ: 153.9, 131.5, 128.8, 125.9, 122.8, 120.1, 119.3, 110.6, 110.4 ppm. LRMS: 319.1 (M + H)+; HRMS (APCI) calculated for C18H15N4S: 319.1012; found 319.1000. ε520 nm = 18000; ε360 nm = 27000; ε266 nm = 14000 (THF).
9,10-Bis((E)-2-(1H-pyrrol-2-yl)vinyl)anthracene (2f). Compound 2f was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 9,10′-dibromoanthracene (f) using GP1. The crude product was washed with 10% diethyl ether/hexanes to give the title compound (114 mg, 97% yield) as a light brown solid. Mp 215–220 °C. 1H NMR (THF-d8, 500 MHz) δ: 10.60 (br s, 2H, NH), 8.44–8.42 (m, 4H, ArH), 7.56 (d, 2H, J = 16.5 Hz, C
CH), 7.43–7.41 (m, 4H, ArH), 6.84 (br s, 2H, PyH), 6.76 (d, 2H, J = 16.5 Hz, CCH), 6.33 (br s, 2H, PyH), 6.15 (br s, 2H, PyH) ppm. 13C NMR (THF-d8, 125 MHz) δ: 133.4, 131.8, 130.6, 129.4, 127.3, 125.6, 120.2, 118.8, 110.1, 109.9 ppm. LRMS: 361.2 (M + H)+; HRMS (APCI) calculated for C26H21N2: 361.1699; found 361.1688. ε424 nm = 12600; ε259 nm = 60000 (THF).
2,7-Bis((E)-2-(1H-pyrrol-2-yl)vinyl)-9H-fluorene (2g). Compound 2g was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 2,7-dibromofluorene (g) using GP1 to give the title compound (108 mg, quantitative) as a yellow/brown solid. Mp/dp > 250 °C. 1H NMR (THF-d8, 500 MHz) δ: 10.30 (br s, 2H, NH), 7.67 (d, 2H, J = 8.0 Hz, ArH), 7.61 (s, 2H, ArH), 7.39 (d, 2H, J = 8.0 Hz, ArH), 7.05 (d, 2H, J = 16.5 Hz, C
CH), 6.78 (d, 2H, J = 16.5 Hz, CCH), 6.73–6.72 (m, 2H, PyH), 6.22 (br s, 2H, PyH), 6.07–6.06 (m, 2H, PyH), 3.87 (s, 2H, CH2) ppm. 13C NMR (THF-d8, 125 MHz) δ: 144.8, 141.2, 137.8, 132.0, 125.7, 123.4, 122.5, 120.4, 120.0, 110.0, 109.8, 37.3 ppm (one signal missing). LRMS: 349.2 (M + H)+; HRMS (APCI) calculated for C25H21N2: 349.1699; found 349.1694. ε390 nm = 55000 (THF).
1,6-Bis((E)-2-(1H-pyrrol-2-yl)vinyl)pyrene (2h). Compound 2h was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 1,6-dibromopyrene (h) using GP1 to give the title compound (48 mg, quantitative) as a dark yellow/brown solid. Mp/dp > 250 °C. 1H NMR (THF-d8, 500 MHz) δ: 10.56 (br s, 2H, NH), 8.48 (d, 2H, J = 8.5 Hz, ArH), 8.30 (d, 2H, J = 8.5 Hz, ArH), 8.10 (d, 2H, J = 9.0 Hz, ArH), 8.05 (d, 2H, J = 9.0 Hz, ArH), 7.87 (d, 2H, J = 16.0 Hz, C
CH), 7.29 (d, 2H, J = 16.0 Hz, CCH), 6.83 (br s, 2H, PyH), 6.37 (br s, 2H, PyH), 6.14 (br s, 2H, PyH) ppm. 13C NMR (THF-d8, 125 MHz) δ: 133.7, 132.6, 131.0, 129.2, 127.9, 126.7, 125.7, 123.4, 123.2, 123.1, 120.4, 119.6, 110.8, 110.1 ppm (one signal missing). LRMS: 385.2 (M + H)+; HRMS calculated for C28H21N2: 385.1699; found 385.1686. ε433 nm = 37000; ε299 nm = 24000 (THF).
4,7-Bis(4-((E)-2-(1H-pyrrol-2-yl)vinyl)phenyl)benzo[c][1,2,5]thiadiazole (2i). Compound 2i was synthesized from 2-vinyl-N-Boc pyrrole (1a) and 4,7-bis(4-bromophenyl)benzo[c][1,2,5]thiadiazole (i)70 using GP1 to give the title compound (127 mg, quantitative) as a dark yellow/brown solid. Mp/dp > 250 °C. 1H NMR (THF-d8, 500 MHz) δ: 10.37 (br s, 2H, NH), 8.07 (d, 4H, J = 8.5 Hz, ArH), 7.91 (s, 2H, ArH), 7.58 (d, 4H, J = 8.5 Hz, ArH), 7.13 (d, 2H, J = 16.5 Hz, C
CH), 6.81 (d, 2H, J = 16.5 Hz, CCH), 6.77 (br s, 2H, PyH), 6.28 (br s, 2H, PyH), 6.09 (br s, 2H, PyH) ppm. 13C NMR (THF-d8, 125 MHz) δ: 155.1, 139.3, 136.3, 133.3, 131.9, 130.3, 128.4, 126.4, 122.4, 121.2, 120.3, 110.5, 110.0 ppm. LRMS: 471.2 (M + H)+; HRMS calculated for C30H23N4S: 471.1638; found 471.1624. ε447 nm = 31000; ε354 nm = 52000 (THF).
4,7-Bis(1-methyl-1H,1′H-[2,2′-bipyrrol]-5-yl)benzo[c][1,2,5]thiadiazole (2j). Compound 2j was synthesized from N-Boc-pyrrole-2-boronic acid (1b) and 4,7-bis(5-bromo-1-methyl-1H-pyrrol-2-yl)benzo[c][1,2,5]thiadiazole (j)71 using GP2. After cooling to room temperature the reaction mixture was separated between 2
:1 diethyl ether:THF (100 mL) and water (100 mL). The aqueous phase was extracted with 2:1 diethyl ether:THF (2 × 100 mL) and the combined organic extracts were washed with water (200 mL) and brine (200 mL), dried over anhydrous magnesium sulfate, and concentrated in vacuo. The crude product was washed with 0–20% diethyl ether/hexanes and then further purified using column chromatography on silica eluting with 50% diethyl ether/hexanes to give the title compound (140 mg, 85% yield) as a dark red/purple solid. Mp 184–187 °C. 1H NMR (THF-d8, 500 MHz) δ: 10.26 (br s, 2H, NH), 7.64 (s, 2H, ArH), 6.78 (br s, 2H, PyH), 6.54 (d, 2H, J = 3.5 Hz, PyH), 6.29 (d, 2H, J = 3.5 Hz, PyH), 6.26 (br s, 2H, PyH), 6.16 (d, 2H, J = 2.5 Hz, PyH), 3.68 (s, 6H, 2 × NCH3) ppm. 13C NMR (THF-d8, 125 MHz) δ: 155.2, 132.3, 132.1, 128.9, 126.1, 125.4, 118.8, 112.6, 109.3, 108.2, 107.7, 35.1 ppm. LRMS: 425.2 (M + H)+; HRMS calculated for C24H21N6S: 425.1543; found 425.1556. ε519 nm = 11300; ε311 nm = 29000 (THF).
N,N′-Bis(2-ethylhexyl)-6,6′-bis(1H-pyrrol-2-yl)isoindigo (2k). Compound 2k was synthesized from N-Boc-pyrrole-2-boronic acid (1b) and N,N′-bis(2-ethylhexyl)-6,6′-dibromoisoindigo (k)72 using GP2 with stirring at 115 °C for 18 h, then 125 °C for an additional 5 h. The crude product was purified using column chromatography on silica eluting with 30–60% diethyl ether in hexanes to give the title compound (203 mg, 53% yield) as a dark blue/black solid. Mp 232–234 °C. 1H NMR (THF-d8, 500 MHz) δ: 10.63 (br s, 2H, NH), 9.30 (d, 2H, J = 8.5 Hz, ArH), 7.16 (dd, 2H, J = 8.5, 1.5 Hz, ArH), 7.04 (d, 2H, J = 1.5 Hz, ArH), 6.85 (br s, 2H, PyH), 6.63 (br s, 2H, PyH), 6.18 (dd, 2H, J = 5.5, 2.5 Hz, PyH), 3.79–3.71 (m, 4H, 2× NCH2), 2.00–1.95 (m, 2H, 2× CH), 1.48–1.29 (m, 16H, 8× CH2), 0.97 (t, 6H, J = 7.5 Hz, 2× CH3), 0.91 (t, 6H, J = 7.0 Hz, 2× CH3) ppm. 13C NMR (THF-d8, 125 MHz) δ: 169.5, 146.6, 137.3, 132.9, 131.4, 131.2, 121.2, 120.4, 116.7, 110.6, 108.5, 103.5, 44.4, 38.6, 31.5, 29.5, 24.8, 24.0, 14.4, 11.0 ppm. LRMS: 617.4 (M + H)+; HRMS calculated for C40H49N4O2: 617.3850; found 617.3849. ε578 nm = 32
800; ε470 nm = 19200; ε310 nm = 31700 (THF).
(E)-5-Styryl-1H-pyrrole-2-carbaldehyde (3a)73. 2-Styryl pyrrole (2a, 53 mg, 0.31 mmol) was dissolved in anhydrous DMF (1.0 mL) with stirring under nitrogen, and the solution was cooled to 0 °C in an ice bath. Phosphorous oxychloride (30 μL, 0.33 mmol), was then added dropwise with continued stirring at 0 °C for 2 hours. 10% (w/v) aqueous potassium carbonate solution (2 mL) was then added, and the reaction mixture was separated between dichloromethane and water. The aqueous phase was extracted with dichloromethane (3 × 10 mL) and the combined organic extracts were washed with water (2 × 40 mL) and brine (30 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product was purified using column chromatography on silica eluting with 20–30% ethyl acetate in hexanes to give the title compound (26 mg, 42% yield) as a light yellow solid. Mp 141–144 °C. 1H NMR (CDCl3, 500 MHz) δ: 9.65 (brs, 1H, NH), 9.49 (s, 1H, CHO), 7.49 (d, 2H, J = 7.5 Hz, ArH), 7.38 (t, 2H, J = 7.5 Hz, ArH), 7.30 (t, 1H, J = 7.5 Hz, ArH), 7.07 (d, 1H, J = 16.5 Hz, CH
C), 6.99–6.97 (m, 1H, PyH), 6.97 (d, 1H, J = 16.5 Hz, CHC), 6.49 (dd, 1H, J = 3.5, 2.5 Hz, PyH) ppm. 13C NMR (CDCl3, 125 MHz) δ: 178.7, 139.1, 136.4, 133.0, 131.0, 129.0, 128.5, 126.7, 123.0, 117.4, 110.9 ppm. LRMS: 220.1 (M + Na)+; HRMS calculated for C13H11NONa: 220.0733; found 220.0734.
5,5′-((1E,1′E)-1,4-Phenylenebis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3b). Compound 3b was synthesized from 2b (130 mg, 0.50 mmol) using GP3 to give the title compound (135 mg, 85% yield) as a dark yellow solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.24 (br s, 2H, NH), 9.44 (s, 2H, CHO), 7.52 (s, 4H, ArH), 7.37 (d, 2H, J = 16.5 Hz, C
CH), 7.13 (d, 2H, J = 16.5 Hz, CCH), 7.03 (dd, 2H, J = 2.0, 3.5 Hz, PyH), 6.58 (dd, 2H, J = 2.0, 3.5 Hz, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.5, 138.8, 136.3, 133.3, 129.4, 126.8, 118.0, 110.5 ppm (one signal missing). LRMS: 315.1 (M − H)−; HRMS calculated for C20H15N2O2: 315.1139; found 315.1131. ε437 nm = 46000; ε413 nm = 59000 (DMSO).
5,5′-((1E,1′E)-[1,1′-Biphenyl]-4,4′-diylbis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3c). Compound 3c was synthesized from 2c (50 mg, 0.15 mmol) using GP3 to give the title compound (50 mg, 85% yield) as a dark yellow solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.25 (br s, 2H, NH), 9.44 (s, 2H, CHO), 7.76 (d, 4H, J = 8.0 Hz, ArH), 7.60 (d, 4H, J = 8.0 Hz, ArH), 7.42 (d, 2H, J = 16.5 Hz, C
CH), 7.17 (d, 2H, J = 16.5 Hz, CCH), 7.04 (d, 2H, J = 3.3 Hz, PyH), 6.60 (d, 2H, J = 3.3 Hz, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.5, 138.7, 135.9, 133.2, 129.3, 127.1, 126.9, 126.8, 118.1, 110.4 ppm (one signal missing). LRMS: 393.2 (M + H)+; HRMS (APCI) calculated for C26H21N2O2: 393.1598; found 393.1596. ε401 nm = 81000 (DMSO).
5,5′-((1E,1′E)-Naphthalene-2,6-diylbis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3d). Compound 3d was synthesized from 2d (60 mg, 0.19 mmol) using GP3 to give the title compound (54 mg, 76% yield) as a dark yellow solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.29 (br s, 2H, NH), 9.46 (s, 2H, CHO), 7.93 (d, 2H, J = 9.3 Hz, ArH), 7.90 (s, 2H, ArH), 7.75 (d, 2H, J = 9.3 Hz, ArH), 7.54 (d, 2H, J = 16.5 Hz, C
CH), 7.25 (d, 2H, J = 16.5 Hz, CCH), 7.06 (d, 2H, J = 3.9 Hz, PyH), 6.62 (d, 2H, J = 3.9 Hz, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.6, 138.8, 134.5, 133.4, 132.9, 129.8, 128.6, 126.2, 123.8, 118.6, 110.6 ppm (one signal missing). LRMS: 367.2 (M + H)+; HRMS (APCI) calculated for C24H18N2O2: 367.1441; found 367.1431. ε433 nm = 26000; ε408 nm = 31000 (DMSO).
5,5′-((1E,1′E)-Benzo[c][1,2,5]thiadiazole-4,7-diylbis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3e). Compound 3e was synthesized from 2e (50 mg, 0.16 mmol) using GP3 to give the title compound (57 mg, 97% yield) as a dark red solid. Mp/dp > 250 °C. 1H NMR (THF-d8, 500 MHz) δ: 11.57 (br s, 2H, NH), 9.48 (s, 2H, CHO), 8.13 (d, 2H, J = 16.3 Hz, C
CH), 7.68 (s, 2H, ArH), 7.64 (d, 2H, J = 16.3 Hz, CCH), 6.94 (br s, 2H, PyH), 6.62 (br s, 2H, PyH) ppm. 13C NMR (THF-d8, 125 MHz) δ: 178.5, 154.7, 139.6, 135.4, 130.1, 129.0, 126.5, 124.0, 122.0, 111.5 ppm. LRMS: 375.1 (M + H)+; HRMS (APCI) calculated for C20H15N4SO2: 375.0910; found 375.0892. ε500 nm = 28000; ε377 nm = 29000 (DMSO).
5,5′-((1E,1′E)-Anthracene-9,10-diylbis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3f). Compound 3f was synthesized from 2f (84 mg, 0.23 mmol) using GP3 to give the title compound (84 mg, 87% yield) as a dark yellow solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.51 (br s, 2H, NH), 9.53 (s, 2H, CHO), 8.44–8.42 (m, 4H, ArH), 8.31 (d, 2H, J = 16.5 Hz, C
CH), 7.60–7.58 (m, 4H, ArH), 7.13–7.12 (m, 2H, PyH), 6.89 (d, 2H, J = 16.5 Hz, CCH), 6.78–6.77 (m, 2H, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.9, 138.3, 133.5, 131.9, 128.8, 126.9, 126.1, 126.0, 125.8, 111.1 ppm (one signal missing). LRMS: 417.2 (M + H)+; HRMS (APCI) calculated for C28H21N2O2: 417.1598; found 417.1581. ε431 nm = 19000; ε334 nm = 22000; ε306 nm = 20000; ε264 nm = 62000 (DMSO).
5,5′-((1E,1′E)-(9H-Fluorene-2,7-diyl)bis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3g). Compound 3g was synthesized from 2g (97 mg, 0.28 mmol) using GP3 to give the title compound (106 mg, 94% yield) as a brown solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.25 (br s, 2H, NH), 9.44 (s, 2H, CHO), 7.90 (d, 2H, J = 8.0 Hz, ArH), 7.74 (s, 2H, ArH), 7.53 (d, 2H, J = 8.0 Hz, ArH), 7.46 (d, 2H, J = 16.5 Hz, C
CH), 7.17 (d, 2H, J = 16.5 Hz, CCH), 7.04 (d, 2H, J = 3.5 Hz, PyH), 6.59 (d, 2H, J = 3.5 Hz, PyH), 4.00 (s, 2H, CH2) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.4, 144.2, 140.8, 139.0, 135.5, 133.2, 130.3, 125.8, 122.6, 120.5, 117.5, 110.3, 36.3 ppm (one signal missing). LRMS: 405.2 (M + H)+; HRMS (APCI) calculated for C27H21N2O2: 405.1598; found 405.1580. ε436 nm = 55000; ε412 nm = 70000 (DMSO).
5,5′-((1E,1′E)-Pyrene-1,6-diylbis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3h). Compound 3h was synthesized from 2h (60 mg, 0.16 mmol) using GP3 to give the title compound (65 mg, 95% yield) as a brown solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.56 (br s, 2H, NH), 9.51 (s, 2H, CHO), 8.80 (d, 2H, J = 9.3 Hz, ArH), 8.60 (d, 2H, J = 16.5 Hz, C
CH), 8.48 (d, 2H, J = 8.0 Hz, ArH), 8.32 (d, 2H, J = 8.0 Hz, ArH), 8.27 (d, 2H, J = 9.3 Hz, ArH), 7.48 (d, 2H, J = 16.5 Hz, CCH), 7.11 (br s, 2H, PyH), 6.70 (br s, 2H, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.8, 139.1, 133.8, 131.3, 130.3, 128.5, 127.6, 125.6, 125.5, 124.7, 123.2, 122.9, 120.5, 112.5 ppm (one signal missing). LRMS: 441.2 (M + H)+; HRMS (APCI) calculated for C30H21N2O2: 441.1598; found 441.1588. ε453 nm = 55000; ε332 nm = 38000; ε257 nm = 31000 (DMSO).
5,5′-((1E,1′E)-(Benzo[c][1,2,5]thiadiazole-4,7-diylbis(4,1-phenylene))bis(ethene-2,1-diyl))bis(1H-pyrrole-2-carbaldehyde) (3i). Compound 3i was synthesized from 2i (44 mg, 0.11 mmol) using GP3 to give the title compound (113 mg, 92% yield) as a brown solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.30 (br s, 2H, NH), 9.46 (s, 2H, CHO), 8.11 (d, 2H, J = 7.5 Hz, ArH), 8.03 (s, 2H, ArH), 7.70 (d, 2H, J = 7.5 Hz, ArH), 7.48 (d, 2H, J = 16.0 Hz, C
CH), 7.23 (d, 2H, J = 16.0 Hz, CCH), 7.06 (br s, 2H, PyH), 6.63 (br s, 2H, PyH) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.6, 153.4, 138.7, 136.7, 136.1, 133.4, 131.7, 129.6, 129.3, 128.1, 126.5, 118.7, 110.6 ppm (one signal missing). LRMS: 527.1 (M + H)+; HRMS (APCI) calculated for C32H23N4SO2: 527.1536; found 527.1512. ε431 nm = 42000; ε373 nm = 53000 (DMSO).
5,5′-(Benzo[c][1,2,5]thiadiazole-4,7-diyl)bis(1′-methyl-1H,1′H-[2,2′-bipyrrole]-5-carbaldehyde) (3j). Compound 3j was synthesized from 2j (50 mg, 0.12 mmol) using GP3 and purified using column chromatography over silica eluting with 2
:1:2 diethyl ether:THF:hexane to give the title compound (47 mg, 84% yield) as a dark red solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.26 (br s, 2H, NH), 9.49 (s, 2H, CHO), 7.78 (s, 2H, ArH), 7.14 (d, 2H, J = 3.5 Hz, PyH), 6.82 (d, 2H, J = 3.5 Hz, PyH), 6.62 (d, 2H, J = 3.5 Hz, PyH), 6.60 (d, 2H, J = 3.5 Hz, PyH), 3.71 (s, 6H, NMe) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 178.4, 153.5, 133.0, 132.8, 132.5, 128.9, 128.1, 124.2, 112.3, 112.2, 110.1, 109.7, 35.1 ppm. LRMS: 481.1 (M + H)+; HRMS calculated for C26H21N6SO2: 481.1441; found 481.1422. ε496 nm = 32700; ε365 nm = 72400 (DMSO).
(E)-5,5′-(1,1′-Bis(2-ethylhexyl)-2,2′-dioxo-[3,3′-biindolinylidene]-6,6′-diyl)bis(1H-pyrrole-2-carbaldehyde) (3k). Compound 3k was synthesized from 2k (60 mg, 0.10 mmol) using GP3 to give the title compound (59 mg, 90% yield) as a dark purple/black solid. Mp/dp > 250 °C. 1H NMR (DMSO-d6, 500 MHz) δ: 12.54 (br s, 2H, NH), 9.56 (s, 2H, CHO), 9.04 (d, 2H, J = 8.0 Hz, ArH), 7.56 (d, 2H, J = 8.0 Hz, ArH), 7.48 (s, 2H, ArH), 7.16 (br s, 2H, PyH), 6.99 (br s, 2H, PyH), 3.62–3.55 (m, 4H, NCH2), 1.91–1.83 (m, 2H, CH), 1.35–1.25 (m, 10H, CH2), 1.25–1.17 (m, 6H, CH2), 0.85 (t, 6H, J = 6.8 Hz, CH3), 0.84–0.78 (m, 6H, CH3) ppm. 13C NMR (DMSO-d6, 125 MHz) δ: 179.3, 167.6, 145.5, 139.0, 134.5, 134.4, 131.1, 129.4, 122.4, 120.4, 118.8, 110.7, 104.9, 43.5, 36.7, 29.8, 27.8, 23.3, 22.6, 13.9, 10.4 ppm. LRMS: 673.4 (M + H)+; HRMS calculated for C42H49N4O4: 673.3748; found 673.3737. ε579 nm = 26
800; ε466 nm = 24200; ε331 nm = 29200 (DMSO).
[Ru(3a)(bpy)2]PF6 complex salt (4a). Complex salt 4a was synthesized from ligand 3a using GP4 and 1 equiv. cis-bis-(2,2′-bipyridine)dichlororuthenium(II) dihydrate for 1 h to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4b (50 mg, 96% yield) as a black glittery solid following isolation by Millipore filtration. Mp 170–175 °C. 1H NMR (CDCl3, 500 MHz) δ: 8.55 (s, 1H, CHO), 8.53 (d, 1H, J = 6.0 Hz, ArH), 8.39 (d, 1H, J = 8.0 Hz, ArH), 8.36 (t, 2H, J = 7.0 Hz, ArH), 8.30 (d, 1H, J = 8.0 Hz, ArH), 7.99 (t, 1H, J = 8.5 Hz, ArH), 7.94 (t, 1H, J = 7.0 Hz, ArH), 7.91–7.87 (m, 2H, ArH), 7.85 (t, 1H, J = 7.5 Hz, ArH), 7.79 (t, 1H, J = 7.5 Hz, ArH), 7.53–7.50 (m, 2H, ArH), 7.40 (t, 1H, J = 6.5 Hz, ArH), 7.28–7.21 (m, 3H, ArH), 7.18–7.15 (m, 3H, ArH), 6.82 (d, 1H, J = 16.5 Hz, CH
C), 6.73 (ad, 2H, J = 7.5 Hz, ArH), 6.70 (d, 1H, J = 4.5 Hz, ArH), 5.50 (d, 1H, J = 16.5 Hz, CHC) ppm. 13C NMR (CDCl3, 125 MHz) δ: 179.6, 159.4, 158.3, 158.1, 157.2, 155.0, 153.0, 151.9, 151.7, 150.6, 144.8, 136.8, 136.6, 135.9, 135.1, 132.0, 128.6, 128.1, 127.0, 126.9, 126.8, 126.7, 126.3, 125.8, 123.9, 123.5, 123.4, 120.6, 114.6 ppm (two signals missing). LRMS: 610.1 (M)+; HRMS calculated for C33H26N5ORu: 610.1175; found 610.1156. ε473 nm = 10900; ε346 nm = 27300; ε295 nm = 57100 (CH2Cl2). The corresponding chloride salt 5a was obtained following GP5, after which the reaction mixture was concentrated in vacuo and the residue was purified over basic alumina eluting with 10–40% methanol in ethyl acetate to give 5a (13 mg, 83%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 610.1 (M)+; PF6− ion not observed in negative mode.
[Ru2(3b)(bpy)4](PF6)2 complex salt (4b). Complex salt 4b was synthesized from ligand 3b using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4b (56 mg, 86% yield) as a black glittery solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.56 (s, 2H, 2× CHO), 8.56–8.55 (m, 2H, ArH), 8.40–8.37 (m, 4H, ArH), 8.32 (d, 2H, J = 8.5 Hz, ArH), 8.24 (d, 2H, J = 8.0 Hz, ArH), 8.00–7.92 (m, 8H, ArH), 7.89–7.82 (m, 4H, ArH), 7.55–7.53 (m, 4H, ArH), 7.44 (t, 2H, J = 6.5 Hz, ArH), 7.28–7.20 (m, 6H, ArH), 6.79 (d, 2H, J = 16.0 Hz, ArH), 6.70 (d, 2H, J = 4.0 Hz, ArH), 6.61 (s, 4H, ArH), 5.45 (d, 2H, J = 16.0 Hz, ArH) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 180.2, 159.7, 158.8, 158.1, 157.6, 155.1, 153.4, 152.5, 152.0, 151.0, 145.3, 136.8, 136.5, 136.0, 135.3, 131.5, 127.4, 126.94, 126.86, 126.8, 126.6, 125.8, 124.0, 123.5, 123.4, 123.3, 120.94, 120.92, 114.9, 70.8 ppm. LRMS: 571.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C60H46N10O2Ru2: 571.0941; found 571.0917. ε489 nm = 41
000; ε377 nm = 54000; ε294 nm = 112000 (CH2Cl2). The corresponding dichloride salt 5b was obtained following GP5 and isolated via Millipore filtration (13 mg, 73%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 571.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3c)(bpy)4](PF6)2 complex salt (4c). Complex salt 4c was synthesized from ligand 3c using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4c (22 mg, 61% yield) as a deep red solid. Mp/dp > 250 °C; 1H NMR (CD2Cl2, 500 MHz) δ: 8.59–8.56 (m, 2H, ArH), 8.56 (s, 2H, CHO), 8.45 (t, 2H, J = 7.3 Hz, ArH), 8.38–8.36 (m, 2H, ArH), 8.32 (d, 2H, J = 8.0 Hz, ArH), 8.25 (d, 2H, J = 8.0 Hz, ArH), 8.10 (t, 2H, J = 7.5 Hz, ArH), 7.99–7.95 (m, 6H, ArH), 7.91 (d, 2H, J = 5.5 Hz, ArH), 7.86–7.83 (m, 2H, ArH), 7.57–7.53 (m, 4H, ArH), 7.49 (d, 4H, J = 8.3 Hz, ArH), 7.44 (t, 2H, J = 6.0 Hz, ArH), 7.40–7.36 (m, 2H, ArH), 7.26 (d, 2H, J = 4.5 Hz, PyH), 7.22 (t, 2H, J = 6.5 Hz, ArH), 6.87 (d, 2H, J = 16.0 Hz, C
CH), 6.81 (d, 4H, J = 8.3 Hz, ArH), 6.73 (d, 2H, J = 4.5 Hz, PyH), 5.55 (d, 2H, J = 16.0 Hz, CCH), 1.53 (br s, 8H, H2O) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 180.2, 159.6, 158.8, 158.2, 157.7, 155.1, 153.3, 152.6, 152.0, 151.1, 145.2, 139.7, 136.7, 136.5, 136.2, 136.1, 136.0, 131.4, 127.3 (2× C) 127.1 (2× C), 126.9 (2× C), 125.9, 124.4, 123.4 (2× C), 123.3, 121.0, 114.7 ppm. LRMS: 609.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C66H50N10O2Ru2: 609.1097; found 609.1101. ε472 nm = 36000; ε430 nm = 42000; ε374 nm = 66000; ε294 nm = 106000 (CH2Cl2). The corresponding dichloride salt 5c was obtained following GP5 and isolated via Millipore filtration (11 mg, 72%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 609.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3d)(bpy)4](PF6)2 complex salt (4d). Complex salt 4d was synthesized from ligand 3d using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4d (18 mg, 42% yield) as a deep red solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.57 (s, 2H, CHO), 8.57–8.56 (m, 2H, ArH), 8.51 (dd, 2H, J = 4.5, 8.0 Hz, ArH), 8.42 (dd, 2H, J = 4.0, 8.0 Hz, ArH), 8.31 (d, 2H, J = 8.5 Hz, ArH), 8.25 (d, 2H, J = 8.0 Hz, ArH), 8.03–7.93 (m, 10H, ArH), 7.86–7.83 (m, 2H, ArH), 7.57–7.52 (m, 6H, ArH), 7.45–7.42 (m, 2H, ArH), 7.37–7.33 (m, 2H, ArH), 7.27 (d, 2H, J = 4.0 Hz, ArH), 7.23–7.21 (m, 4H, ArH), 6.98 (dd, 2H, J = 2.0, 16.0 Hz, C
CH), 6.78–6.76 (m, 4H, ArH), 5.59 (dd, 2H, J = 5.5, 16.0 Hz, CCH), 1.53 (br s, 8H, H2O) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 180.3, 159.7, 158.8, 158.2, 157.7, 155.1, 153.4, 152.5, 152.0, 151.0, 145.3, 136.7, 136.5, 136.0, 135.8, 134.9, 133.3, 131.9, 128.2, 127.4, 127.2, 126.9 (2× C), 125.84, 125.79, 124.8, 124.4, 123.5, 123.4, 123.3, 121.5, 114.9 ppm. LRMS: 596.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C64H48N10O2Ru2: 596.1019; found 596.1005. ε481 nm = 42000; ε437 nm = 42000; ε380 nm = 58000; ε294 nm = 116000 (CH2Cl2). The corresponding dichloride salt 5d was obtained following GP5 and isolated via Millipore filtration (7 mg, 78%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 596.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3e)(bpy)4](PF6)2 complex salt (4e). Complex salt 4e was synthesized from ligand 3e using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4e (24 mg, 61% yield) as a deep purple solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.62 (s, 2H, CHO), 8.52–8.50 (m, 2H, ArH), 8.46–8.39 (m, 4H, ArH), 8.32–8.29 (m, 2H, ArH), 8.24–8.22 (m, 2H, ArH), 8.00–7.95 (m, 6H, ArH), 7.91–7.86 (m, 4H, ArH), 7.85–7.81 (m, 2H, ArH), 7.54–7.51 (m, 2H, ArH), 7.49 (d, 2H, J = 7.0 Hz, ArH), 7.45–7.41 (m, 2H, ArH), 7.34 (dd, 2H, J = 2.0, 16.0, C
CH), 7.30 (d, 2H, J = 5.0 Hz, ArH), 7.24–7.17 (m, 4H, ArH), 6.93 (s, 2H, ArH), 6.86 (d, 2H, J = 4.5 Hz, ArH), 6.39 (dd, 2H, J = 2.0, 16.0, CCH), 1.54 (br s, 8H, H2O) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 180.7, 159.5, 158.8, 158.2, 157.8, 155.4, 153.7, 153.3, 152.6, 151.8, 150.9, 145.9, 136.8, 136.5, 136.1, 135.6, 129.1, 128.0, 127.4, 127.3, 127.0, 126.9, 126.8, 125.8, 125.4, 124.2, 123.5, 123.31, 123.26, 115.4 ppm. LRMS: 600.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C60H44N12SO2Ru2: 600.0753; found 600.0733. ε525 nm = 42000; ε358 nm = 40000; ε295 nm = 114000 (CH2Cl2). The corresponding dichloride salt 5e was obtained following GP5 and isolated via Millipore filtration (11 mg, 71%) as a brown solid. Mp/dp > 250 °C. LRMS: 600.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3f)(bpy)4](PF6)2 complex salt (4f). Salt 4f was synthesized from ligand 3f using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4f (24 mg, 45% yield) as a deep red solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.64 (s, 2H, CHO), 8.59 (d, 2H, J = 5.5 Hz, ArH), 8.36 (d, 2H, J = 8.0 Hz, ArH), 8.31–8.25 (m, 4H, ArH), 8.19 (d, 2H, J = 8.0 Hz, ArH), 8.12–8.08 (m, 2H, ArH), 8.04–8.01 (m, 4H, ArH), 8.00–7.96 (m, 2H, ArH), 7.93–7.89 (m, 4H, ArH), 7.81–7.77 (m, 2H, ArH), 7.68 (d, 2H, J = 16.0 Hz, C
CH), 7.62–7.59 (m, 2H, ArH), 7.52–7.48 (m, 4H, ArH), 7.44–7.42 (m, 4H, ArH), 7.37–7.35 (m, 2H, ArH), 7.18–7.15 (m, 2H, ArH), 6.99–6.95 (m, 2H, ArH), 6.54–6.49 (m, 2H, ArH), 6.10–6.04 (m, 2H, ArH), 5.04 (d, 2H, J = 16.0 Hz, CCH), 1.54 (br s, 8H, H2O) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 181.0, 158.7, 158.2, 157.6, 154.4, 152.5, 152.4, 152.3, 152.00, 151.96, 151.0, 145.1, 136.7, 136.4, 136.3, 133.7, 132.4, 131.0, 129.2, 127.5, 127.3, 127.0, 126.8, 126.5, 125.8, 125.7, 125.6, 123.9, 123.5, 123.3, 114.5 ppm. LRMS: 621.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C68H50N10O2Ru2: 621.1097; found 621.1074. ε508 nm = 30000; ε345 nm = 34000; ε295 nm = 110000 (CH2Cl2). The corresponding dichloride salt 5f was obtained following GP5 and isolated via Millipore filtration (9 mg, 52%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 621.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3g)(bpy)4](PF6)2 complex salt (4g). Complex salt 4g was synthesized from ligand 3g using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4g (39 mg, 66% yield) as a deep red solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.57 (d, 2H, J = 5.5 Hz, ArH), 8.55 (s, 2H, CHO), 8.51 (d, 2H, J = 8.0 Hz, ArH), 8.41 (d, 2H, J = 8.0 Hz, ArH), 8.31 (d, 2H, J = 8.0 Hz, ArH), 8.25 (d, 2H, J = 8.0 Hz, ArH), 8.07 (t, 2H, J = 8.0 Hz, ArH), 7.99–7.94 (m, 8H, ArH), 7.86–7.83 (m, 2H, ArH), 7.59–7.53 (m, 6H, ArH), 7.44 (t, 2H, J = 7.0 Hz, ArH), 7.40–7.37 (m, 2H, ArH), 7.26 (d, 2H, J = 4.5 Hz, ArH), 7.22 (t, 2H, J = 6.0 Hz, ArH), 6.93 (d, 2H, J = 16.5 Hz, C
CH), 6.88 (s, 2H, ArH), 6.85 (d, 2H, J = 8.0 Hz, ArH), 6.74 (d, 2H, J = 4.5 Hz, ArH), 5.57 (dd, 2H, J = 6.0, 16.5 Hz, CCH), 3.81 (s, 2H, CH2), 1.54 (br s, 8H, H2O) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 179.9, 159.7, 158.8, 158.2, 157.7, 155.3, 153.4, 152.5, 152.0, 151.0, 145.2, 144.4, 141.5, 136.7, 136.4, 136.0, 135.9, 135.7, 132.3, 127.3, 127.2, 126.9, 126.7, 125.9, 124.4, 123.5, 123.34, 123.26, 122.1, 120.5, 120.4, 120.2, 114.7, 36.6 ppm. LRMS: 615.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C67H50N10O2Ru2: 615.1097; found 615.1084. ε477 nm = 49000; ε435 nm = 54000; ε381 nm = 72000; ε294 nm = 124000 (CH2Cl2). The corresponding dichloride salt 5g was obtained following GP5 and isolated via Millipore filtration (13 mg, 75%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 615.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3h)(bpy)4](PF6)2 complex salt (4h). Complex salt 4h was synthesized from ligand 3h using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4h (39 mg, 69% yield) as a deep red solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.62 (s, 2H, CHO), 8.60 (d, 2H, J = 5.5 Hz, ArH), 8.38–8.30 (m, 8H, ArH), 8.27–8.24 (m, 4H, ArH), 8.04–8.03 (m, 2H, ArH), 8.02–7.95 (m, 6H, ArH), 7.92–7.91 (m, 2H, ArH), 7.88–7.84 (m, 4H, ArH), 7.77 (t, 2H, J = 7.5 Hz, ArH), 7.59–7.56 (m, 4H, ArH), 7.46 (t, 2H, J = 6.8 Hz, ArH), 7.33 (d, 2H, J = 4.5 Hz, ArH), 7.23 (t, 2H, J = 6.3 Hz, ArH), 7.17–7.12 (m, 4H, ArH), 6.94 (d, 2H, J = 4.5, ArH), 5.75 (d, 2H, J = 16.0 Hz, C
CH), 1.53 (br s, 8H, H2O) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 180.6, 159.7, 158.8, 158.2, 157.6, 155.3, 153.3, 152.5, 152.0, 151.1, 145.4, 136.8, 136.6, 136.1, 135.3, 131.8, 131.0, 128.6, 128.3, 128.0, 127.4, 127.0, 126.9 (2× C), 126.0, 125.5, 125.2, 125.1, 124.2, 124.1, 123.8, 123.4, 123.3, 123.0, 114.9 ppm. LRMS: 633.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C70H50N10O2Ru2: 633.1097; found 633.1119. ε511 nm = 64000; ε401 nm = 40000; ε294 nm = 132000 (CH2Cl2). The corresponding dichloride salt 5h was obtained following GP5 and isolated via Millipore filtration (15 mg, 83%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 633.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3i)(bpy)4](PF6)2 complex salt (4i). Complex salt 4i was synthesized from ligand 3i using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4i (46 mg, 72% yield) as a deep red solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.59 (s, 2H, CHO), 8.60–8.57 (m, 2H, ArH), 8.40–8.34 (m, 6H, ArH), 8.28 (d, 2H, J = 8.0 Hz, ArH), 8.01–7.96 (m, 10H, ArH), 7.86 (at, 8H, J = 8.3 Hz, ArH), 7.58–7.55 (m, 4H, ArH), 7.45 (t, 2H, J = 6.5 Hz, ArH), 7.34 (t, 2H, J = 6.8 Hz, ArH), 7.28 (d, 2H, J = 4.5 Hz, ArH), 7.23 (t, 2H, J = 6.5 Hz, ArH), 6.96–6.91 (m, 6H, ArH), 6.77 (d, 2H, J = 4.0 Hz, ArH), 5.62 (d, 2H, J = 16.5 Hz, C
CH), 1.55 (br s, 4H, H2O) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 180.4, 159.8, 158.7, 158.2, 157.5, 155.0, 154.3, 153.4, 152.5, 152.0, 151.1, 145.3, 137.1, 137.0, 136.8, 136.6, 136.0, 135.4, 132.7, 131.3, 129.6 (2× C), 128.4, 127.4, 127.1, 126.9, 126.6 (2× C), 125.9, 124.1, 123.44, 123.37 (2× C), 121.6, 114.8 ppm (one signal missing). LRMS: 676.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C72H52N12SO2Ru2: 676.1066; found 676.1039. ε475 nm = 47000; ε358 nm = 57000; ε295 nm = 122000 (CH2Cl2). The corresponding dichloride salt 5i was obtained following GP5 and isolated via Millipore filtration (13 mg, 75%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 676.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3j)(bpy)4](PF6)2 complex salt (4j). Complex salt 4j was synthesized from ligand 3j using GP4 to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4j (30 mg, 62% yield) as a deep red/black solid. Mp/dp > 250 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.73 (s, 2H, CHO), 8.54 (d, 2H, J = 5.5 Hz, ArH), 8.36–8.32 (m, 4H, ArH), 8.25–8.22 (m, 4H, ArH), 8.10 (t, 2H, J = 7.5 Hz, ArH), 8.03 (t, 2H, J = 7.3 Hz, ArH), 7.97 (t, 2H, J = 7.8 Hz, ArH), 7.80 (t, 2H, J = 7.8 Hz, ArH), 7.63–7.59 (m, 2H, ArH), 7.59–7.56 (m, 2H, ArH), 7.55–7.53 (m, 4H, ArH), 7.50 (t, 2H, J = 6.5 Hz, ArH), 7.42 (s, 2H, ArH), 7.35 (d, 2H, J = 4.0 Hz, ArH), 7.16 (t, 2H, J = 6.5 Hz, ArH), 6.97–6.93 (m, 2H, ArH), 6.43 (dd, 2H, J = 4.5, 1.0 Hz, ArH), 5.88 (dd, 2H, J = 3.5, 5.5 Hz, ArH), 5.46 (t, 2H, J = 3.0 Hz, ArH), 2.95 (s, 6H, NMe) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 182.4, 158.9, 158.5, 158.4, 158.3, 154.4, 152.8, 152.6, 152.2, 150.9, 148.1, 144.7, 136.7, 136.13, 136.07, 135.4, 130.6, 130.4, 129.2, 127.2, 126.7, 126.5, 126.2, 125.0, 124.6, 123.30, 123.26, 123.2, 122.9, 119.8, 111.9, 110.1, 33.4 ppm. LRMS: 653.1 (M/2)+ and 145.0 (PF6)−; HRMS calculated for C66H50N14SO2Ru2: 653.1019; found 653.1011. ε509 nm = 35
000; ε356 nm = 35000; ε294 nm = 127000 (CH2Cl2). The corresponding dichloride salt 5j was obtained following GP5 with 10:1 acetone:hexanes, and isolated via Millipore filtration (8 mg, 93%) as a red/brown solid. Mp/dp > 250 °C. LRMS: 653.1 (M/2)+; PF6− ion not observed in negative mode.
[Ru2(3k)(bpy)4](PF6)2 complex salt (4k). Complex salt 4k was synthesized from ligand 3k using GP4 in 9
:1 methanol:water for 1 h to give the corresponding bis(ruthenium(II))hexafluorophosphate salt 4k (18 mg, 70% yield) as a dark brown/black solid. Mp/dp 208–213 °C. 1H NMR (CD2Cl2, 500 MHz) δ: 8.76 (s, 2H, CHO), 8.60–8.58 (m, 2H, ArH), 8.51–8.48 (m, 2H, ArH), 8.35–8.29 (m, 4H, ArH), 8.22 (d, 2H, J = 7.0 Hz, ArH), 8.06–7.98 (m, 8H, ArH), 7.80 (t, 2H, J = 7.8 Hz, ArH), 7.58–7.55 (m, 2H, ArH), 7.51 (t, 2H, J = 6.3 Hz, ArH), 7.45–7.39 (m, 4H, ArH), 7.35–7.32 (m, 4H, ArH), 7.17 (t, 2H, J = 6.8 Hz, ArH), 6.79–6.71 (m, 2H, ArH), 6.46 (d, 2H, J = 4.0 Hz, ArH), 6.29–6.18 (m, 4H, ArH), 3.70–3.58 (m, 2H, NCH2), 3.47–3.33 (m, 2H, NCH2), 1.69 (br s, 2H, CHEt), 1.35–1.17 (m, 16H, CH2), 0.92–0.81 (m, 12H, CH3) ppm. 13C NMR (CD2Cl2, 125 MHz) δ: 182.9, 168.3, 158.9, 158.8, 158.5, 158.2, 157.0, 152.7, 152.3, 152.1, 151.3, 145.7, 144.7, 140.2, 136.8, 136.5, 136.1, 135.2, 132.4, 129.2, 127.3, 127.0, 126.6, 126.4, 126.2, 125.6, 123.5, 123.4, 123.0, 121.6, 120.8, 118.0, 107.6, 44.3, 37.9, 30.9, 29.0, 24.2, 23.4, 14.2, 10.7 ppm (some peaks were observed in duplicate suggesting diastereomeric effects). LRMS: 749.2 (M/2)+ and 144.9 (PF6)−; HRMS calculated for C82H78N12O4Ru2: 749.2173; found 749.2190. ε516 nm = 30100; ε377 nm = 32400; ε295 nm = 118400 (CH2Cl2). The corresponding dichloride salt 5k was obtained following GP5 with 10:1 acetone:hexanes, stirring at room temperature for 30 min. The reaction mixture was then concentrated in vacuo and the residue purified over neutral alumina, eluting with 3–8% methanol in dichloromethane to give 5k (14 mg, 84%) as a dark brown/black solid. Mp/dp > 250 °C. LRMS: 749.2 (M/2)+; PF6− ion not observed in negative mode.
2.4 Methods
:1 v/v ethanol–methanol glass21):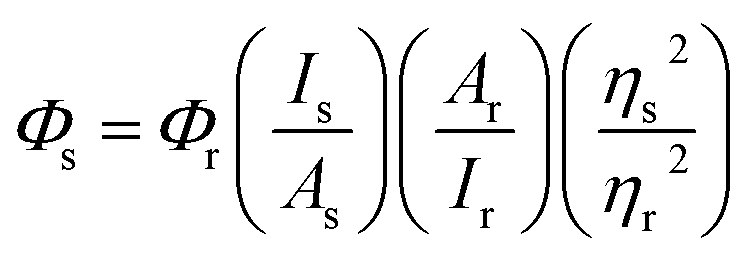 | (1) |
Singlet oxygen quantum yields (ΦΔ) were also estimated using eqn (1) with [Ru(bpy)3](PF6)2 as the standard (ΦΔ = 0.57 in aerated MeCN).75 Absorption spectra were recorded using a Jasco V-530 spectrophotometer, and luminescence spectra were collected using a PTI Quantamaster equipped with a standard photomultiplier tube (K170B) and a Hamamatsu R5509-42 photomultiplier tube for NIR detection (<1400 nm). Luminescence lifetimes were measured using a PTI LaserStrobe system incorporating a nitrogen-dye laser (GL-3300/GL-301) integrated with an R928 stroboscopic detector. Emission was also probed by gated methods using a pulsed xenon flash lamp and gated detector. Exponential curve fitting and corrections to the wavelength-dependence of lamp output and detector response were done with PTI Felix32 software.
000 cells per mL in 25 cm2 tissue culture flasks and were subcultured when growth reached approximately 1 × 106 cells per mL. Cytotoxicity and photocytotoxicity assays were performed on cells of mid-passage number (8–25 passages).
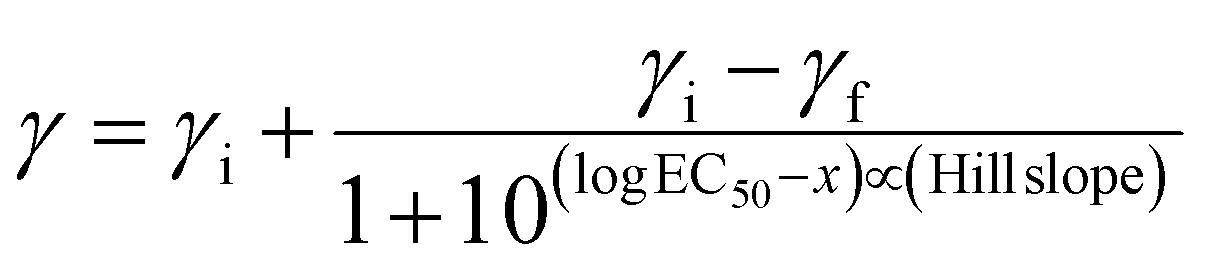 | (2) |
For cells growing in log phase and of similar passage number, EC50 values were reproducible to within ±25% in the submicromolar regime; ±10% below 10 μM; and ±5% above 10 μM. Photocytotoxicity indices (PIs), a measure of the therapeutic window, were calculated from the ratio of dark to light EC50 values obtained from the dose–response curves.
Serial dilutions of aqueous stock solutions of the Ru compounds were prepared in microcentrifuge tubes in TSB at 2× the concentration needed (final concentrations in the wells were 0.1 nM, 1 nM, 10 nM, 100 nM, 0.1 μM, 1 μM, 10 μM, and 50 μM). Prewarmed 100 μL aliquots of compounds were added to the sample wells (prewarmed TSB to the controls) and final assay volumes were 200 μL (final bacterial concentration ∼5 × 105 CFU mL−1). The PS-to-light interval was 1 h. Dark treatment microplates were wrapped in foil and placed in a dark drawer, while PDI-treated microplates were irradiated with visible light (400–700 nm, 40 ± 0.8 mW cm−2) using a 190 W BenQ MS510 overhead projector or with red light (625 nm, 35 ± 1.3 mW cm−2) from an LED array (Photodynamic Inc.). The irradiation time was 42 min and 48 min respectively, to yield light doses of approximately 100 J cm−2. Both dark and PDT-treated microplates were incubated overnight. The sample wells were carefully pipetted up and down to mix well and the absorbance at 562 nm was measured for all microplates with a BioTek EL800 plate reader. MIC50 values (the minimum inhibitory concentration at which ≥50% of the bacteria is inhibited) for antibiotic (dark) and antimicrobial PDI (light) activity were calculated from sigmoidal fits of the dose response curves using Graph Pad Prism 6.0 according to eqn (2) (above), where γi and γf are the initial and final absorbance intensities.
3. Results and discussion
3.1 Synthesis and characterization
We have previously reported the first synthesis of heteroleptic pyrrolyl/2,2′-bypyridyl complexes of ruthenium(II).67 Considering the high stability and unusual UV/vis properties of these mono-ruthenium complexes, we now explore the synthesis and properties of symmetric bis(ruthenium) complexes of this type, with the goal of determining the effect of varying the extent of conjugation in these bis[Ru(II)-pyrrolide] triads. Initial studies concerned the design and synthesis of a mono-pyrrolic ligand bearing extended conjugation, with intent to optimize the synthetic protocol.67,79 As such, N-Boc-2-vinyl pyrrole (1a)80 was synthesized in a two-step procedure from 2-formyl pyrrole and, following a modified procedure,81 was successfully employed as a Heck substrate with bromobenzene, providing the in situ-deprotected styryl-pyrrole 2a in good yield (64%, Scheme 1). Employing 1,4-dibromobenzene as the aryl halide along with 2 equivalents of vinyl-pyrrole 1a resulted in the conjugated, symmetric bis(pyrrole) 2b in high yield (86%).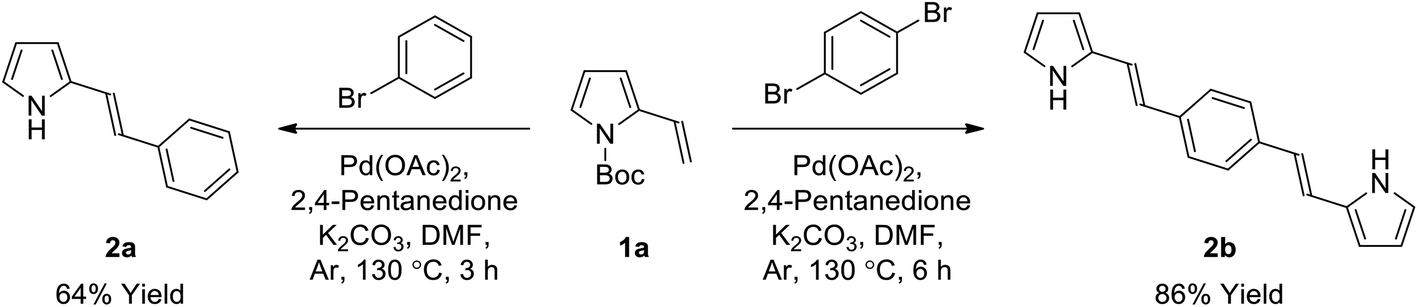 | ||
| Scheme 1 Synthesis of conjugated pyrrole 2a and bis(pyrrole) 2bvia Heck reaction. | ||
We then examined the scope of dibromoarene substrates in the double Heck reaction with vinyl pyrrole 1a (Table 1). A variety of linkers were selected for study, including bicyclic (entries 3 and 4), heterocyclic (entry 5), polycyclic compounds (entries 6–8), and linkers featuring extended conjugation (entries 9–11). The majority of substrates examined were well tolerated, giving bis(pyrrole)s 2b–i in excellent isolated yields (86–100%). Bithiophene, pyrazine and binaphthyl linkers were unsuccessful in this synthetic screen, as were extended linkers j and k. A double Suzuki reaction with N-Boc-pyrrole-2-boronic acid (1b) was subsequently investigated for linkers j and k, whereupon conditions were developed to generate the corresponding bis(pyrrole)s 2j and 2k in yields of 85 and 53%, respectively (entries 10 and 11).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pyrrole | Linker | n | Yield of 2b (%) | Yield of 3b (%) | |
| a Compounds 2a (see Scheme 1) and 3a are mono-pyrroles (pyrrole-CH = CHPh). b Isolated yield. c Heck reaction conditions: 1 equiv. 1a, Pd(OAc)2, 2,4-pentanedione, K2CO3, DMF, Ar, 130 °C, 3 h. d Heck reaction, 2 equiv. 1a, 6 h. e Suzuki reaction conditions: Pd(PPh3)4, K2CO3, DMF, 110 °C, 24 h. f Suzuki reaction, 115 °C for 18 h then 125 °C for 5 h. g Vilsmeier reaction, 1 equiv. POCl3. h Vilsmeier reaction, 2 equiv. POCl3. | ||||||
| 1 | 1a | a |
|
1 | 64 (2a)a,c | 86 (3a)a,g |
| 2 | 1a | b |
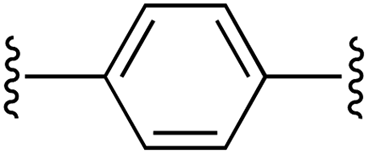
|
1 | 86 (2b)d | 85 (3b)h |
| 3 | 1a | c |
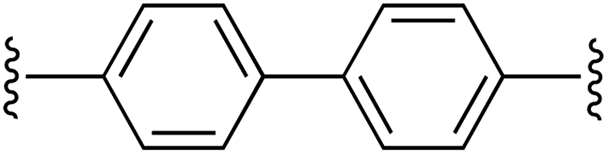
|
1 | 94 (2c)e | 85 (3c)h |
| 4 | 1a | d |
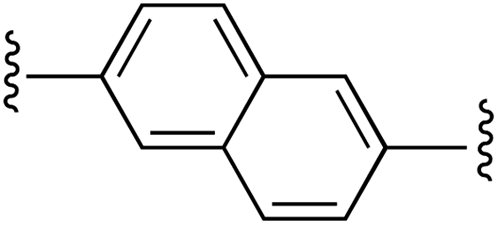
|
1 | 97 (2d)d | 76 (3d)h |
| 5 | 1a | e |
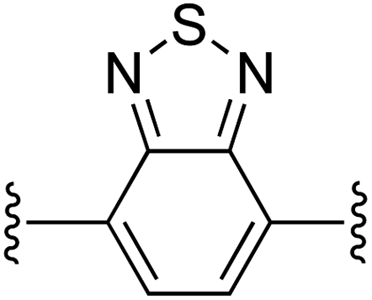
|
1 | 91 (2e)d | 97 (3e)h |
| 6 | 1a | f |
|
1 | 97 (2f)d | 87 (3f)h |
| 7 | 1a | g |
|
1 | 100 (2g)d | 94 (3g)h |
| 8 | 1a | h |
|
1 | 100 (2h)d | 95 (3h)h |
| 9 | 1a | i |
|
1 | 100 (2i)d | 92 (3i)h |
| 10 | 1b | j |
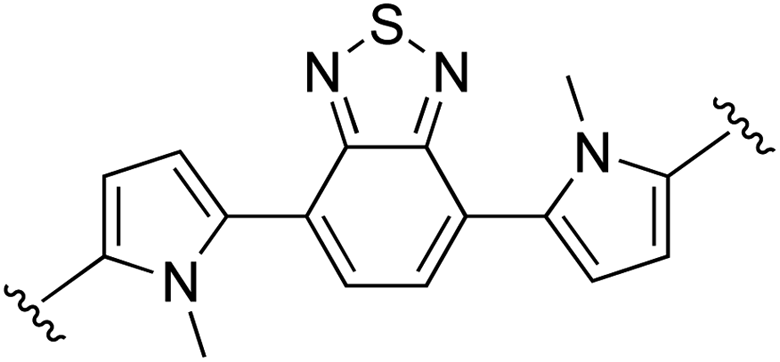
|
0 | 85 (2j)e | 84 (3j)h |
| 11 | 1b | k |
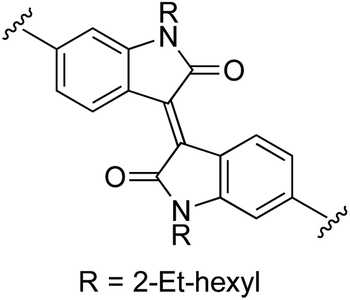
|
0 | 53 (2k)e,f | 90 (3k)h |
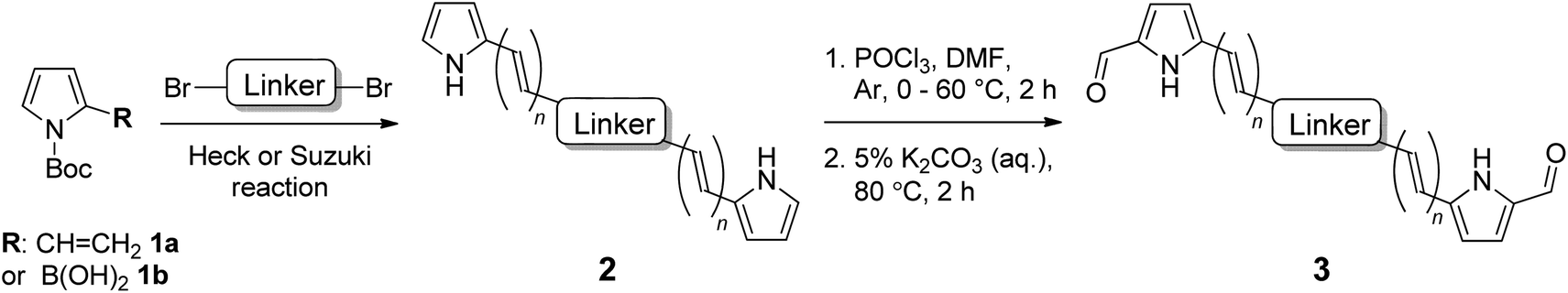
Using mono-pyrrole 2a as a model substrate, Vilsmeier–Haack formylation was found to be successful in installing an α-formyl group,82,83 providing bidentate ligand 3a in high yield (86%, Table 1, entry 1). Bis(pyrrole)s 2b–2k were subsequently subjected to Vilsmeier–Haack formylation conditions,84 employing 2 equivalents of phosphoryl chloride, whereby the corresponding bis(bidentate) ligands 3b–3k were isolated in good to excellent yields (76–97%, entries 2–11) following isolation by precipitation in water.
Mono-pyrrolic ligand 3a was again used as a model substrate for ruthenium complexation, using a previously reported microwave-promoted procedure,67,85 whereupon heteroleptic [Ru(3a)(bpy)2]PF6 complex salt 4a was isolated following treatment with aqueous ammonium hexafluorophosphate (96%, Table 2, entry 1). Complexation of bis(bidentate) ligands 3b–3j, using 2 equivalents of [Ru(bpy)2Cl2]·2H2O and slightly modified reaction conditions, was successful in generating the corresponding bis(ruthenium) complex salts 4b–4j, (42–86%, entries 2–10), which were purified using column chromatography on neutral alumina. Difficulties were encountered with ligand 3k, which underwent complexation and concomitant reduction of the central double bond of isoindigo linker k. This was thought to be an effect of the ethylene glycol solvent, which is known to oxidize during heating in air to generate the reductant glycolaldehyde.86 Altering the reaction solvent to 9:1 methanol:water overcame this problem and allowed for isolation of the desired complex salt 4k (70%, entry 11). For the purpose of assessing the photobiological activity of each bis[Ru(II)-pyrrolide] triad, salt conversion of the hexafluorophosphate salts (4a–4k) to the water-soluble chloride salts (5a–5k) was carried out by treatment with tetrabutylammonium chloride (TBAC) in acetone.87
|
|
||||
|---|---|---|---|---|
| Entry | Linker | n | Yield of 4b (%) | Yield of 5b (%) |
|
a Compounds 4a and 5a feature mono-pyrrolide ligands and one Ru centre (pyrrole-CH = CHPh).
b Isolated yield.
c 1 equiv. Ru(bpy)2Cl2·2H2O with reaction time of 60 min.
d 2 equiv. Ru(bpy)2Cl2·2H2O.
e Reaction solvent 9:1 methanol:water.
f Reaction solvent 10:1 acetone:hexanes.
|
||||
| 1 | a | 1 | 96 (4a)a,c | 83 (5a)a |
| 2 | b | 1 | 86 (4b)d | 73 (5b) |
| 3 | c | 1 | 61 (4c)d | 72 (5c) |
| 4 | d | 1 | 42 (4d)d | 78 (5d) |
| 5 | e | 1 | 61 (4e)d | 71 (5e) |
| 6 | f | 1 | 45 (4f)d | 52 (5f) |
| 7 | g | 1 | 66 (4g)d | 75 (5g) |
| 8 | h | 1 | 69 (4h)d | 83 (5h) |
| 9 | i | 1 | 72 (4i)d | 75 (5i) |
| 10 | j | 0 | 62 (4j)d | 93 (5j)f |
| 11 | k | 0 | 70 (4k)d,e | 84 (5k)f |
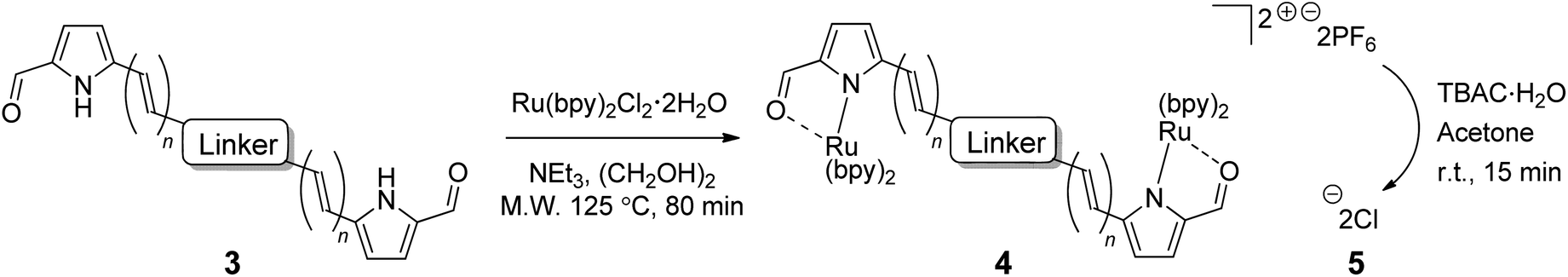
3.2 Spectroscopic properties
The MeCN-soluble PF6− salts of the complexes (4a–k) were used for all spectroscopic measurements, while the water-soluble Cl− salts of the complexes (5a–k) were used for biological studies. The reason MeCN was used as the solvent of choice for spectroscopy (instead of water or other aqueous solution) is that water quenches the 1O2 emission, precluding accurate determination of the upper limit for 1O2 quantum yields88 and because MeCN is the solvent used in many published spectroscopic studies.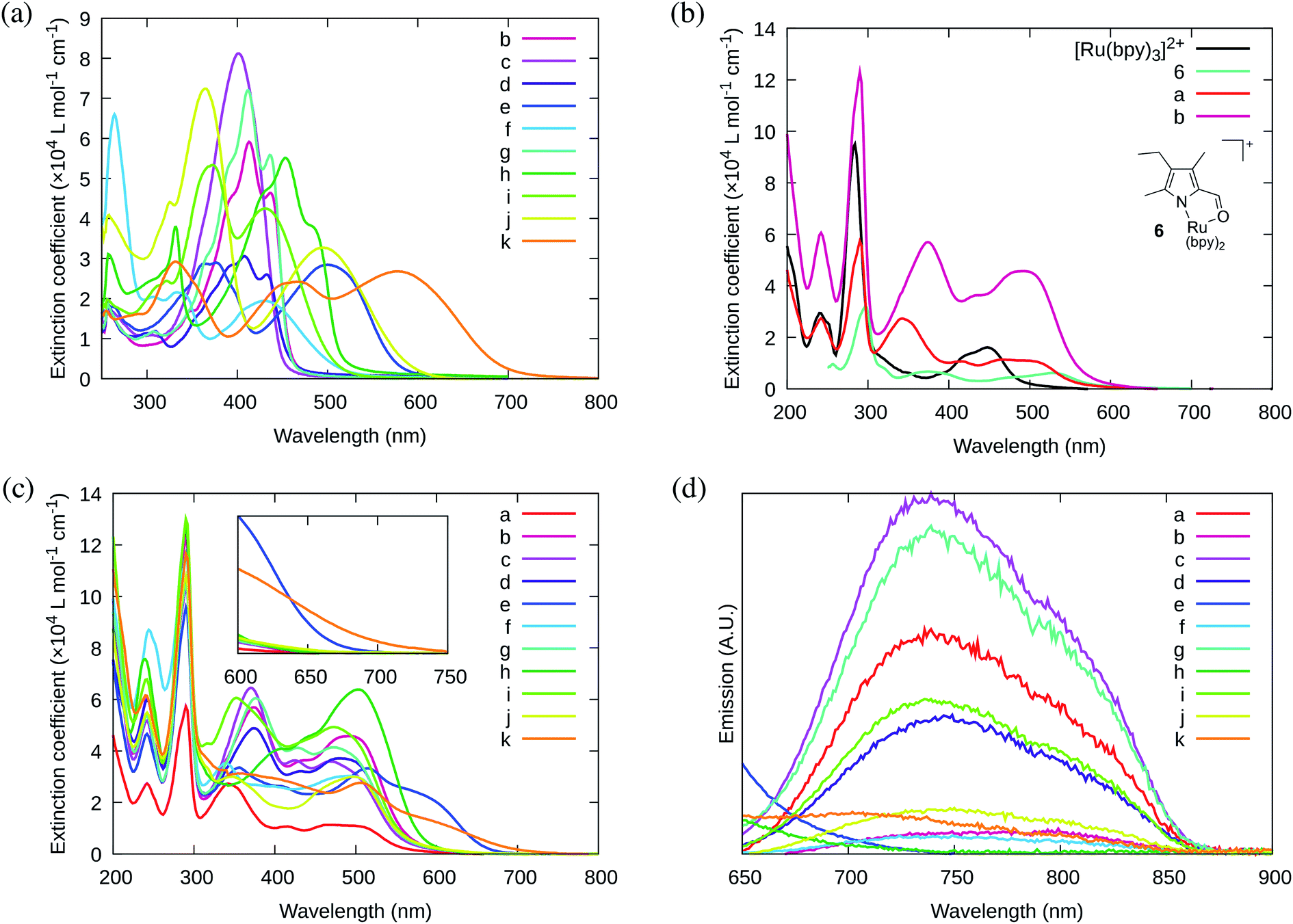 | ||
| Fig. 1 UV/Vis absorption spectra of (a) ligands 3b–k; (b) previously reported 6 and [Ru(bpy)3]2+ as reference complexes for mononuclear 4a and bis[Ru(II)-pyrrolide] triad 4b; and (c) mononuclear 4a and bis[Ru(II)-pyrrolide] triads 4b–k. (d) Phosphorescence emission spectra for mononuclear 4a and bis[Ru(II)-pyrrolide] triads 4b–k (collected in Ar using λex max). Absorption and emission spectra were collected on the PF6− salts of the complexes (5 μM) in MeCN. | ||
The absorption spectra of the free ligands are shown in Fig. 1a. For those ligands derived from (poly)cyclic aromatic hydrocarbon linkers (3b–d, 3f–h), the longest wavelength absorption maxima mirrored the 1ππ* transitions characteristic of the linker, but with bathochromic shifts and contributions arising from extended π-conjugation with the vinyl-appended 2-formyl pyrrolides. For example, free pyrene has a longest-wavelength absorption maximum just below 350 nm,89 whereas 3h, with pyrene as the linker, had its longest-wavelength absorption maximum near 448 nm, with a shoulder at 489 nm (≥100 nm red-shift relative to free pyrene). Notably, this significant bathochromic shift places the spectral window of the 1ππ* transition of ligand 3h in a similar position as the 1MLCT transition of [Ru(bpy)3]2+ (λmax = 448 nm). The longest-wavelength absorption maxima of 3e and 3j–k, with predicted ligand-localized contributions, are even more red-shifted, appearing at wavelengths ≥500 nm (λmax = 593 nm for 3k). It was anticipated that chelation of these unique π-expanded ligands to Ru(II) to form the bis[Ru(II)-pyrrolide] triads would further widen the visible spectral window and lead to enhanced molar extinction coefficients, especially at the longer wavelengths.
The UV/Vis absorption spectrum of our previously reported 2-formyl pyrrolide Ru(II) complex 6,67 representative of the core mononuclear N,O-coordinated system used in the triads but without extended conjugation, is compared to [Ru(bpy)3]2+, mononuclear 4a, and bis[Ru(II)-pyrrolide] 4b in Fig. 1b. Complex 6 was the first published example of a heteroleptic pyrrolide/2,2′-bipyridyl Ru(II) complex. This simple mononuclear construct displays continuous absorption between 200 and 600 nm, with a longest-wavelength absorption maximum near 528 nm for the 1MLCT transition, which is approximately 80 nm longer than that for [Ru(bpy)3]2+. Red-shifts of almost 100 nm for the lowest-energy 1MLCT transitions (relative to the corresponding Ru(II) systems containing neutral diimine ligands) agrees with what we have previously observed for Ru(II) complexes bearing anionic cyclometalating ligands, such as thionoester-substituted pyrrolides and deprotonated phenylpyridines.55,57,67,79 Presumably, this shift of the 1MLCT absorption band is a direct result of a concomitant increase in the energy of the Ru(dπ) orbitals arising from the strong N-σ (η1) donation of the pyrrolide nitrogen.
The styryl substituted pyrrolide complex (4a) led to significant absorption past 500 nm (ε510 = 1.1 × 104 M−1 cm−1) and doubled the extinction coefficients in this region compared to 6 (Fig. 1b). The slight blue-shift of about 13 nm for the longest-wavelength absorption maximum for 4a could reflect the enhanced conjugation of the pyrrolide ligand and weaker N-σ (η1) bonding to the Ru(II) center. Nevertheless, the extended conjugation provided by the styryl group in combination with the relatively strong N-σ donation of the N,O pyrrolide resulted in a Ru(II) complex that absorbs green light ten times more strongly than the related [Ru(bpy)3]2+ complex. In support of our hypothesis that these properties could be improved further, incorporation of two metal chromophores into a triad via two terminal 2-formylpyrrolyl ligands tethered to a central benzene linker through alkenyl groups (4b) resulted in a four-fold increase in the longest wavelength absorption maximum in comparison to its mononuclear counterpart 4a, and 40-fold relative to the parent [Ru(bpy)3]2+.
The absorption spectrum of the bis[Ru(II)-pyrrolide] complex 4b appeared to be more than a simple linear combination of two mononuclear fragments and the free organic ligand, thereby suggesting that the two metal centers are in conjugative communication mediated by the shared organic linker. This notion is supported by the observation that the longest-wavelength absorption maximum measured for the corresponding complex with a biphenyl linker (4c), which most likely adopts a nonplanar dihedral angle and decouples the two metal centers, is blue-shifted and of reduced intensity relative to both 4a and 4b. The other explored linkers can be structurally grouped as follows: polycyclic aromatics (4d, 4f–h), heterocycles based on benzothiadiazole (4e, 4i–j), or isoindigo (4k). Of all of the complexes, the pyrenyl linker (4h) exhibited the most intense transitions at its longest-wavelength absorption maximum, while the benzothiadiazole (4e) and isoindigo (4k) linkers yielded the longest-wavelength absorption maxima overall (albeit of reduced intensity relative to 4h). The absorption spectra of mononuclear 4a and the ten bis[Ru(II)-pyrrolide] complexes are compared in Fig. 1c.
Generally, complexation of the respective novel ligand 3 to two Ru(II) centers to produce the bis[Ru(II)-pyrrolide] triads 4 resulted in both a widening of the visible absorption window as well as a noticeable hyperchromic shift at these wavelengths for all bis[Ru(II)-pyrrolide] triads except for 4i and 4j. The longest-wavelength absorption bands in 4i were very similar to 3i, and in 4j, the free ligand was more absorptive at the longer wavelengths despite what appeared to be a longer wavelength absorption maximum for its complex. Notably, for the benzothiadiazoles (4e, 4i–j), the groups on either side of the benzothiadiazole had a marked impact on the longest wavelength transitions. For example, vinyl groups directly attached to the central benzothiadiazole group (4e) led to a longest wavelength absorption maximum near 615 nm, which was among the longest in the entire series. Adding phenyl groups between the benzothiadiazole and the vinyl groups (5i) or replacing the vinyl groups with N-methyl pyrrole groups shifted these bands hypsochromically by ≥100 nm. Clearly, there is much to be learned from these SARs and what they suggest in terms of the polarizabilities and CT characters of the ligands and their resulting bis[Ru(II)-pyrrolide] complexes, but the purpose of the present investigation was to provide a very general outline of these observations.
| Cmpd | Absmax/nm (logε) |
λ em max (λex)/nm | Φ p (1 × 10−3) | Φ Δ |
|---|---|---|---|---|
| a 298 K, Ar. b Emission from the 3MLCT state at 298 K was not observed (the tail of 1LC emission was observed). c Very weak 3MLCT emission that was continuous over the observation window. | ||||
| 5a | 244 (4.43), 284 (4.70), 290 (4.76), 340 (4.44), 416 (4.02), 464 (4.04), 514 (3.99) | 743 (466) | 1.07 | 0.13 |
| 5b | 242 (4.78), 284 (5.04), 290 (5.09), 378 (4.75), 434 (4.56), 494 (4.66), 515 (4.60) | 760 (500) | 0.10 | 0.69 |
| 5c | 244 (4.70), 284 (4.96), 290 (5.02), 372 (4.81), 428 (4.56), 470 (4.56), 504 (4.47) | 743 (470) | 1.20 | 0.57 |
| 5d | 244 (4.77), 284 (4.99), 288 (5.02), 376 (4.69), 436 (4.50), 484 (4.57) | 750 (484) | 0.52 | 0.61 |
| 5e | 244 (4.66), 282 (4.90), 290 (4.98), 360 (4.52), 414 (4.41), 518 (4.52), 602 (4.29) | —b | —b | 0.32 |
| 5f | 248 (4.93), 252 (4.91), 284 (5.02), 290 (5.07), 340 (4.55), 404 (4.42), 472 (4.47), 514 (4.44) | 765 (495) | 0.067 | 0.07 |
| 5g | 206 (4.91), 244 (4.72), 284 (4.97), 290 (5.02), 378 (4.78), 430 (4.61), 474 (4.62), 502 (4.56) | 743 (475) | 0.69 | 0.37 |
| 5h | 240 (4.88), 290 (5.11), 406 (4.61), 442 (4.66), 508 (4.80) | —b | —b | 0.68 |
| 5i | 242 (4.83), 292 (5.11), 318 (4.63), 354 (4.78), 476 (4.69), 510 (4.60) | 738 (474) | 0.68 | 0.77 |
| 5j | 244 (4.73), 290 (5.03), 316 (4.53), 352 (4.47), 438 (4.30), 504 (4.47) | 746 (500) | 0.28 | 0.33 |
| 5k | 242 (4.79), 292 (5.06), 398 (4.47), 510 (4.44), 618 (4.02) | 715 (507) | —c | 0.05 |
3.3 Photobiological activity
Cellular assays. The water-soluble Cl− salts (5a–k) were used for the biological experiments. The dark cytotoxicities of the mononuclear reference compound 5a and the bis[Ru(II)-pyrrolide] triads 5b–5k were determined using a human leukemia (HL-60) cell line. This cell line was chosen because it grows as a suspension rather than an adherent monolayer, thus eliminating some additional variability in the cellular assay that arises when treating differentially formed monolayers. Briefly, cells growing in log phase were dosed with the compounds at concentrations between 1 nM and 300 μM and assessed for viability after approximately 64 h using the Alamar Blue reagent. The photocytotoxicities were determined in an analogous manner except that a light treatment was delivered approximately 16 h after the cells were dosed with compound. The cell viability was quantified from dose–response curve fits to yield the effective concentration required to reduce cell viability by 50% (EC50) in the dark (dark EC50) and with the light treatment (light EC50). The phototherapeutic indices (PIs) were calculated as the ratios of the dark EC50 and light EC50 values, and represent the amplification of the cytotoxic effect with the light trigger. All cellular assays were carried out in triplicate under normoxic conditions, with representative data compiled in Table 4. For reference, the well-known cytotoxic chemotherapy agent cisplatin yields an EC50 value of approximately 25 μM with no difference between the dark and light condition (PI = 1).
| Complex | Dark EC50 (μM) | Vis lighta EC50 (μM) | Vis PIb | Red lightc EC50 (μM) | Red PIb |
|---|---|---|---|---|---|
| a Vis condition: 16 h DLI followed by broadband visible light irradiation (28 J cm−2, 7.8 mW cm−2). b PI = phototherapeutic index (ratio of dark EC50 to visible-light EC50). c Red condition: 16 h DLI followed by light irradiation with 625 nm LEDs (100 J cm−2, 42 mW cm−2). | |||||
| 5a | 1.69 ± 0.06 | 0.20 ± 0.01 | 8 | 0.29 ± 0.07 | 6 |
| 5b | 89.1 ± 0.8 | 0.55 ± 0.02 | 161 | 1.20 ± 0.03 | 74 |
| 5c | 31.6 ± 1.7 | 0.27 ± 0.04 | 115 | 0.79 ± 0.04 | 40 |
| 5d | 103 ± 0.6 | 0.19 ± 0.01 | 534 | 0.84 ± 0.02 | 123 |
| 5e | 173 ± 6 | 0.84 ± 0.01 | 206 | 0.73 ± 0.02 | 237 |
| 5f | 48.1 ± 0.4 | 3.05 ± 0.21 | 16 | 4.06 ± 0.09 | 12 |
| 5g | 54.4 ± 0.9 | 0.07 ± 0.01 | 734 | 0.35 ± 0.02 | 157 |
| 5h | 36.8 ± 2.9 | 0.01 ± 0.01 | 5439 | 0.14 ± 0.01 | 261 |
| 5i | 14.3 ± 0.4 | 0.15 ± 0.01 | 95 | 0.37 ± 0.05 | 39 |
| 5j | 39.8 ± 0.9 | 10.8 ± 0.3 | 4 | 10.2 ± 0.1 | 4 |
| 5k | 11.5 ± 0.3 | 6.36 ± 0.14 | 2 | 6.48 ± 0.16 | 2 |
Dark cytotoxicity. The dark cytotoxicities of the compounds investigated varied over two orders of magnitude from approximately 1.7 μM for the mononuclear 5a to just over 170 μM for the bis-Ru(II) triad 5e (Table 4, Fig. 2a). Notably, the mononuclear compound 5a was distinctly more cytotoxic than its triad counterparts, being seven-fold more cytotoxic than the most dark cytotoxic triad 5k (dark EC50 = 11.5 μM). There was a ten-fold variation among the Ru(II) triads that clustered into roughly three groups: least cytotoxic with dark EC50 values near 100 or more (5b, 5d–e), moderately cytotoxic with values near 30–50 μM (5c, 5f–h, 5j), and cytotoxic with values between 10–15 μM (5i, 5k).
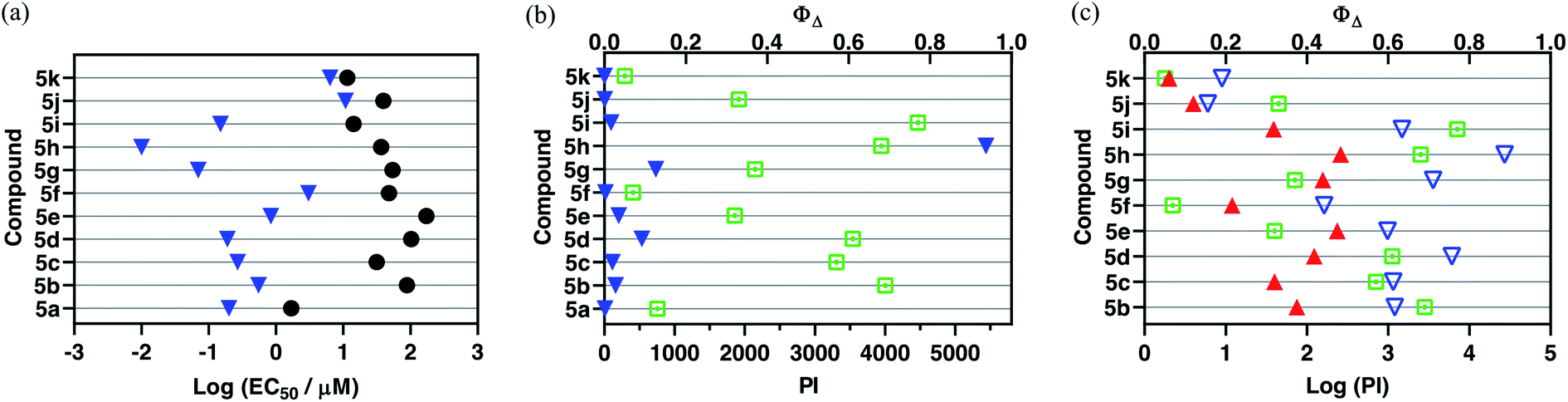 | ||
Fig. 2 (a) Activity plot for 5a–5k showing cytotoxicities in the dark (●) and with light activation using broadband visible light ( , 28 J cm−2, 7.8 mW cm−2); (b) activity plot for 5a–5k highlighting phototherapeutic indices (PIs) under the same light conditions as in (a), as well as 1O2 quantum yields ( , 28 J cm−2, 7.8 mW cm−2); (b) activity plot for 5a–5k highlighting phototherapeutic indices (PIs) under the same light conditions as in (a), as well as 1O2 quantum yields ( ); and (c) activity plot for the bis[Ru(II)-pyrrolide] triads 5b–5k showing their logPI values with visible ( ); and (c) activity plot for the bis[Ru(II)-pyrrolide] triads 5b–5k showing their logPI values with visible ( , 100 J cm−2, 28 mW cm−2) or 625 nm red ( , 100 J cm−2, 28 mW cm−2) or 625 nm red ( , 100 J cm−2, 42 mW cm−2) light. , 100 J cm−2, 42 mW cm−2) light. | ||
Structurally, the bis[Ru(II)-pyrrolide] systems can be divided into three classes: (i) those with aromatic hydrocarbon linkers that vary in the extent π-conjugation (5b–d, 5f–h), (ii) those with benzothiadiazole linkers with or without conjugated groups (5e, 5i–j), and (iii) one with an isoindigo linker (5k). The dark cytotoxicities of class (i) varied from 32 to 103 μM, while those for class (ii) varied from 14 to 173 μM. Complex 5k with the isoindigo linker was the most cytotoxic at 11.5 μM, and 5e with the benzothiadiazole linker was the least at 173 μM. Interestingly, incorporation of phenyl rings (5i) or N–Me pyrrole rings (5j) on either side of the benzothiadiazole group led to increased cytotoxicity relative to the parent 5e. Likewise, there was a substantial difference between incorporation of one phenyl ring (5b) as the linker and two (5c), with the latter resulting in elevated cytotoxicity. The incorporation of two fused rings, as in naphthalene (5d), resulted in a slightly reduced cytotoxicity relative to 5b.
Parameters such as lipophilicity and cellular uptake and distribution were not investigated as part of this study so it would be premature to speculate on reasons behind the observed differences in cytotoxicity. Rather, our intention here is to highlight the breadth of cytotoxic activity that can be obtained in a relatively small structural family of a new compound class and to also use the dark EC50 values as a reference point for assessing phototoxic effects and corresponding PIs. This significant variation within and between the classes underscores that the linker unit is an important point of variation for manipulating the inherent cytotoxicity of bis[Ru(II)-pyrrolide] triads, which could prove advantageous for optimization of PI values.
Photocytotoxicity. The photocytotoxicities of mononuclear 5a along with the bis[Ru(II)-pyrrolide] triads were determined with broadband visible light (28 J cm−2, 7.8 mW cm−2) and with 625 nm red light (100 J cm−2, 42 mW cm−2) (Fig. 2a and Table 4). Their visible light EC50 values under this condition varied by just over three orders of magnitude, ranging from approximately 3–11 μM for the least phototoxic systems (5f, 5j–k) to 10–70 nM for the most potent phototoxic compounds (5g, 5h). Other family members clustered near 150–270 nM (5a, 5c–d, 5i), with 5b and 5e much closer to 1 μM.
Because the light EC50 values contain contributions from the baseline dark cytotoxicity, the true phototoxic effects were assessed as PI values, or fold-amplification between the dark and light condition (Fig. 2b and Table 4). According to their PIs, the compounds could be grouped by having (i) very little phototherapeutic effect with PIs ≪100 (5a, 5f, 5j–k), (ii) marginal effects with PIs near 100–200 (5b–c, 5e, 5i), or (iii) very good effects with PIs >>200 (5d, 5g, 5h). Bis[Ru(II)-pyrrolide] 5h, exhibiting one of the larger 1O2 quantum yields, stood out from the rest with its visible PI exceeding 5000 using this relatively soft light dose. The PIs generally correlated with 1O2 quantum yields across the series (Fig. 3a), but the correlation was not strict when comparing individual compounds. For example, 5h had a much larger PI than the other family members (best emphasized in Fig. 3b), yet it did not have the largest 1O2 yield of the series. Certainly, other ROS and other phototoxic mechanisms could be at play, the cell-free 1O2 quantum yields may not reflect the cellular 1O2 quantum yields, and/or the subcellular targets may have a larger impact on the PI than the precise 1O2 quantum yield. Nevertheless, this compound class can be considered a new source of PSs for PDT.
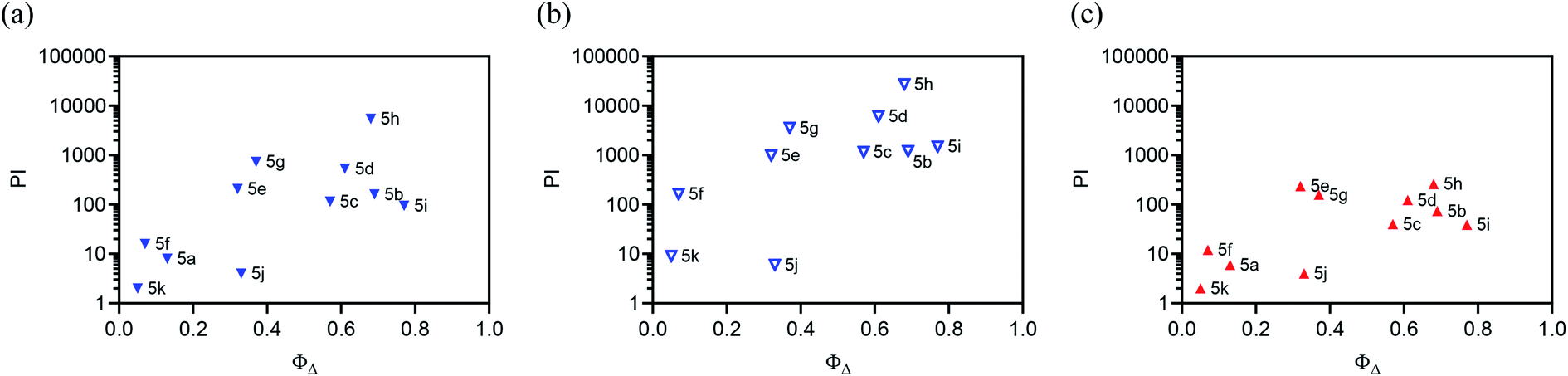 | ||
Fig. 3 Plots correlating PI value with the 1O2 quantum yield for each complex under three different light conditions: (a) broadband visible ( , 28 J cm−2, 7.8 mW cm−2) for 5a–k, (b) broadband visible ( , 28 J cm−2, 7.8 mW cm−2) for 5a–k, (b) broadband visible ( , 100 J cm−2, 28 mW cm−2) for 5b–k, and 625 nm red ( , 100 J cm−2, 28 mW cm−2) for 5b–k, and 625 nm red ( , 100 J cm−2, 42 mW cm−2) for 5a–k. , 100 J cm−2, 42 mW cm−2) for 5a–k. | ||
Structurally, the largest PIs were observed for the bis[Ru(II)-pyrrolide] systems with conjugated aromatic hydrocarbon linkers in the order: pyrene (5h) > fluorene (5g) > naphthalene (5d). The smallest PIs were obtained for the mononuclear 5a, which had very high dark cytotoxicity, and the bis[Ru(II)-pyrrolide] triads with anthracene (5f), isoindigo (5k), and bis(NMePy)benzothiadiazole (5j) as central linkers. The family members with intermediate and similar PIs contained phenyl and biphenyl linkers (5b), and (5c), respectively, as well as benzothiadiazole and diphenylbenzothiadiazole linkers (5e), and (5i), respectively. It is tempting to speculate that linkers with the requisite triplet state energies to act as excited state reservoirs might lead to increased sensitivity to oxygen (and other excited state quenchers) in these triads and thus larger PIs. However, as triplet state energies of the free ligands (and the corresponding 3IL or 3ILCT energies of the complexes) form part of a future extensive spectroscopic study we will not speculate at this time.
The photocytotoxicities and PIs for the bis[Ru(II)-pyrrolide] triads were also measured using a slightly stronger broadband visible light dose (100 J cm−2, 28 mW cm−2) from a different light source to cross-confirm the phototoxic effects across the series (Fig. 2c and 3b). The difference in light fluence or irradiance between the two experiments was almost four-fold, and the resulting PIs did not scale linearly with this change. However, the compounds clustered in the same groups based on their PIs and 1O2 quantum yields (Fig. 3b). The PI differences between the two visible light conditions were compound-dependent, ranging from two-fold (5j) to sixteen-fold (5i). Differences near ten-fold (5b–d and 5f) or five-fold (5e, 5g–h, 5k) were measured for the rest of the family. Notably, 5h had a visible EC50 value near 1 nM and PI > 27000, while the PI values for 5d and 5g were >6000 and >3500, respectively. 5h has one of the larger PI reported to date (Fig. 4).
 | ||
| Fig. 4 Molecular structures of the bis[Ru(II)-pyrrolides] with the largest PIs. | ||
Since mononuclear 5a and the bis[Ru(II)-pyrrolide] systems display longest-wavelength absorption maxima that are red-shifted compared to many well-studied Ru(II) polypyridyl complexes,21 their photocytotoxicities and PIs were also investigated using 625 nm red LEDs (100 J cm−2, 42 mW cm−2). As observed for the two different visible light treatments, the compounds clustered in the same groups based on their PIs and 1O2 quantum yields (Fig. 3c), but their PIs were attenuated. The red PIs ranged from 2 for the least photoactive compound (5k) to 260 for the most photoactive system (5h) (Table 4), with four of the triads maintaining PIs > 100 (5d–e, 5g–5h). The visible- and red-light treatments with a fluence of 100 J cm−2 (but different irradiances) are compared in Fig. 2c. The PIs for the bis[Ru(II)-pyrrolide] triads were attenuated to different extents using lower-energy red light, from 100-fold for 5h to two-fold with 5j. The order of attenuation appeared to parallel the magnitudes of the PIs with visible light rather than the molar extinction coefficients at 625 nm, with the more photoactive compounds being the most affected. Of the compounds considered most active under all three illumination conditions investigated, only 5e absorbs red light significantly (logε625 nm = 4.08) yet 5h (logε625 nm = 2.93) had a larger PI. The only other compound that absorbs light substantially at 625 nm is 5k (logε625 nm = 3.97), which was dark cytotoxic and considered relatively non-phototoxic under all light conditions explored. These variances present intriguing launch points for future investigation.
Wide concentration range photocytotoxicity assay. The visible light condition with a fluence of 100 J cm−2 described above yielded an EC50 value for 5h near 1 nM, which was the lowest concentration tested in that assay. To gain more insight regarding the visible light EC50 value with 100 J cm−2, we rescreened 5h starting at 100 pM and reduced the drug-to-light (DLI) interval from 16 h to 1 h (Fig. 5a). This new condition yielded a visible-light EC50 value for 5h of 1.33 nM (PI = 24
100). The PI was slightly reduced in this assay due to a higher dark cytotoxicity of 30.8 μM (versus 36.8 μM in the narrower range screen). In parallel, we also used red light (625 nm, 100 J cm−2, 29 mW cm−2) and obtained a red light EC50 value of 129 nM (PI = 239), which was similar to what was determined in the narrower concentration range assay.
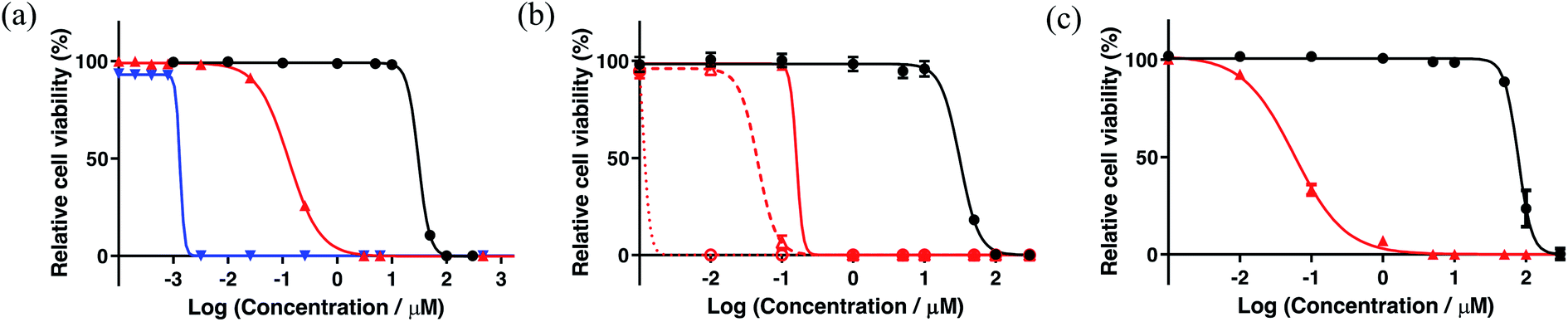 | ||
Fig. 5 (a) Wide concentration range dark/light cytotoxicity assay performed with 5h using the HL-60 cell line. Cells dosed with 5h received a dark (black) or light treatment with red (625 nm LEDs, red) or broadband visible (blue) light (100 J cm−2, 29 mW cm−2) with a DLI of 1 h. (b) Cytotoxicity (black) and photocytotoxicity (red) using the three 625 nm red light conditions: the red light dose used in (a) but with different concentrations of 5h ( ); 100 J cm−2 (29 mW cm−2) delivered in four 25 J cm−2 fractions separated by 15 min ( ); 100 J cm−2 (29 mW cm−2) delivered in four 25 J cm−2 fractions separated by 15 min ( ); and 200 J cm−2 delivered in two fractions of 100 J cm−2 separated by 1 h ( ); and 200 J cm−2 delivered in two fractions of 100 J cm−2 separated by 1 h ( ). (c) HL60 multicellular 3D spheroid cytotoxicity (black) and photocytotoxicity (red) assay with 5h using the red light condition described for (a). ). (c) HL60 multicellular 3D spheroid cytotoxicity (black) and photocytotoxicity (red) assay with 5h using the red light condition described for (a). | ||
Optimization of the red-light PI. Given that bis[Ru(II)-pyrrolide] 5h clearly emerged as a compound of interest for further investigation due to its unprecedented visible PI with both the high and low light fluences tested, we wondered whether the attenuated red-light PIs of ∼240–260 obtained with a fluence of 100 J cm−2 could be improved. The light parameter offers a unique opportunity to optimize the PI as the wavelength, fluence, irradiance, DLI, and dosing regimen can be manipulated. While the optimal light dosimetry parameters are not absolute and most certainly are compound-dependent, simple changes such as increasing the fluence and dosing interval are straightforward. We optimized the PI for 625 nm red light (100 J cm−2, 29 mW cm−2) with a 16 h DLI, where the red EC50 value in this assay was 161 nM and the PI was 195 (Fig. 5b). These unoptimized values differ slightly between assays16 so the reference condition was always run in parallel for comparison. Delivering the same total fluence but in four 25 J cm−2 intervals separated by 15 min increased the potency by almost four-fold (red EC50 = 45.7 nM, PI = 690). Increasing the light fluence to 200 J cm−2 delivered in two intervals of 100 J cm−2 separated by 1 h led to subnanomolar potency: red EC50 = 630 pM and PI = 50
000 (Fig. 5b). The superior potency with this light regimen exceeded even that of the visible light condition that yielded a PI > 27000. PIs of these magnitudes have not been reported. This very limited optimization study underscores how the light regimen can compensate for marginal extinction coefficients at the activation wavelength. In this preliminary investigation, we did not investigate the mechanism behind this improved response as part of this study, but it is known that fractionated dosing can (in some cases) improve response.12
Multicellular 3D tumor spheroid assay. The 3D multicellular tumor spheroid model can be exploited to mimic the highly plastic migratory/invasive tumor phenotypes that characterize some of the most aggressive conditions in vivo.90 For instance, they have hard-to-reach hypoxic regions that impart drug resistance. To test whether 5h could maintain potency against tumor spheroids of the same cell line used for the 2D suspension assays (HL-60), spheroids were grown to sizes of about 600 μm in diameter and treated with 5h in the concentration range of 1 nM to 300 μM. The spheroids were either kept in the dark or treated with 625 nm red light (100 J cm−2, 29 mW cm−2) with a 16 h DLI. As expected the HL-60 tumor spheroids were greater than two-fold more resistant to 5h in the dark (compared to 2D HL-60 cultures), with a dark EC50 of approximately 77 μM. Surprisingly, however, the photocytotoxicity was greater against the 3D tumor spheroids, with a red-light EC50 value of 60 nM and PI > 1200. We did not examine the source of this enhanced photocytotoxicity in the 3D tumor spheroid model, which should be scrutinized more closely across spheroids of different sizes and of different cell lines to assess whether this is a general property of 5h.
Bacterial survival assays. The ability of 5h to act as a photocytotoxic compound toward bacteria was briefly explored. Two bacterial species were grown as planktonic cultures and treated with 5h in the concentration range of 10 pM to 50 μM, where no dark cytotoxicity was apparent. Further treatment with either broadband visible or 625 nm red light (100 J cm−2, 28 mW cm−2) using a DLI of 1 h resulted in phototoxic effects toward both S. mutans and S. aureus (Fig. 6). There was no selectivity for either bacterial species, with visible EC50 values on the order of 130 to 160 nM and PIs >300 (PIs not determined because there was no dark cytotoxicity at the concentrations investigated). As observed with the HL-60 cells, the photocytotoxicity was attenuated upon moving to the use of red light, rather than visible light of the same fluence and irradiance. The reduction was approximately eight-fold, yielding PIs > 40–50. This result confirms that the phototoxic effect exhibited by 5h extends to other types of cells and that this new class of bis[Ru(II)-pyrrolide] triad shows potential for use as photoactive antimicrobials.
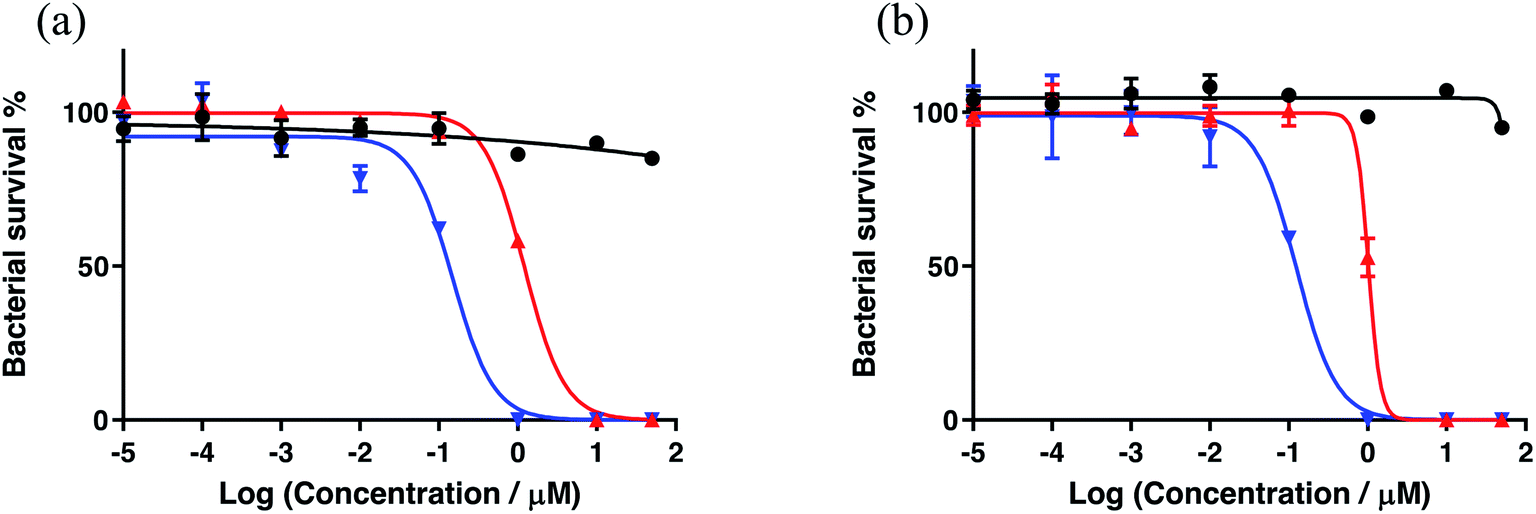 | ||
| Fig. 6 In vitro cytotoxic effects of 5h against S. mutans (a) and S. aureus (b) growing as planktonic cultures in the dark (black) or with a light treatment. The light treatments were broadband visible (blue) or 625 nm red (red) light (100 J cm−2, 28 mW cm−2) with a DLI of 1 h. | ||
4. Concluding remarks
In summary, the ten new bis[Ru(II)pyrrolide] triads reported herein demonstrated unequivocally that the central organic linker plays a pivotal role in determining the spectroscopic, biological, and photobiological properties of the metal–organic systems and that these properties in many cases are improved relative to the mononuclear counterpart 5a. The compounds demonstrated a large breadth of activity as exemplified by a very wide range for 1O2 quantum yields, dark cytotoxicities, and PIs. Simple variation of the central organic chromophore resulted in some compounds being excellent in vitro phototoxic agents, while others exhibited almost no photoactivity and could be considered traditional cytotoxic agents. The source of higher dark cytotoxicity for certain compounds is not known but could be related to their lipophilicities and resulting cellular uptake and/or localization.Although the generation of 1O2 under the cell-free condition does not establish ROS as the definitive mediator of photocytotoxicity, we presume PDT effects are responsible. As such, the excited state dynamics and redox characteristics of the complexes must be explored in order to propose a mechanism(s). However, the fact that 5h with the central pyrenyl group emerged as an extremely potent photosensitizer for in vitro PDT and that the triplet state energy of the isolated pyrenyl group is in energetic proximity to that of many well-studied 3MLCT states suggests at least a possible role for 3IL states in producing the larger 1O2 quantum yield and greater in vitro PDT potency toward cancer cells. At the time 5h was evaluated, PIs of such magnitude had not been reported and the opportunity to use interval dosing to achieve PIs >27000 had not been explored by groups developing new PSs. Compound 5h was also highly active toward the more resistant tumor spheroid model, which is characterized by multicellular resistance and regions of hypoxia, and also toward bacteria. The versatility of this new photosensitizer for both light-mediated anticancer and antimicrobial applications highlights the potential utility of the bis[Ru(II)-pyrrolide] scaffold for photobiological applications and introduces a new platform for further optimization of these important light-responsive agents.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
S. A. M. has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy.Acknowledgements
S. A. M. and A. T. thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support. S. A. M. and C. G. C. thank the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) for partial support of this work. The content in this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The TOC graphic was made using https://Biorender.com/.References
- R. Bonnett, Chemical Aspects of Photodynamic Therapy; Advanced chemistry texts, Gordon and Breach Science Publishers, Amsterdam, The Netherlands, 2000 Search PubMed.
- M. DeRosa, Photosensitized Singlet Oxygen and Its Applications, Coord. Chem. Rev., 2002, 233–234, 351–371, DOI:10.1016/S0010-8545(02)00034-6.
- T. Schlotthauer, R. Schroot, S. Glover, L. Hammarström, M. Jäger and U. S. Schubert, A Multidonor–Photosensitizer–Multiacceptor Triad for Long-Lived Directional Charge Separation, Phys. Chem. Chem. Phys., 2017, 19(42), 28572–28578, 10.1039/C7CP05593E.
- S. O. Gollnick, L. Vaughan and B. W. Henderson, Generation of Effective Antitumor Vaccines Using Photodynamic Therapy, Cancer Res., 2002, 62(6), 1604–1608 CAS.
- A. P. Castano, P. Mroz and M. R. Hamblin, Photodynamic Therapy and Anti-Tumour Immunity, Nat. Rev. Cancer, 2006, 6(7), 535–545, DOI:10.1038/nrc1894.
- P. Mroz, J. T. Hashmi, Y.-Y. Huang, N. Lange and M. R. Hamblin, Stimulation of Anti-Tumor Immunity by Photodynamic Therapy, Expert Rev. Clin. Immunol., 2011, 7(1), 75–91, DOI:10.1586/eci.10.81.
- S. O. Gollnick and C. M. Brackett, Enhancement of Anti-Tumor Immunity by Photodynamic Therapy, J. Immunol. Res., 2010, 46(1–3), 216–226, DOI:10.1007/s12026-009-8119-4.
- S. O. Gollnick, Photodynamic Therapy and Antitumor Immunity, J. Natl. Compr. Canc. Netw., 2012, 10(Suppl 2), S40–S43 CAS.
- M. Shams, B. Owczarczak, P. Manderscheid-Kern, D. A. Bellnier and S. O. Gollnick, Development of Photodynamic Therapy Regimens That Control Primary Tumor Growth and Inhibit Secondary Disease, Cancer Immunol. Immunother., 2015, 64(3), 287–297, DOI:10.1007/s00262-014-1633-9.
- F. Anzengruber, P. Avci, L. F. de Freitas and M. R. Hamblin, T-Cell Mediated Anti-Tumor Immunity after Photodynamic Therapy: Why Does It Not Always Work and How Can We Improve It?, Photochem. Photobiol. Sci., 2015, 14(8), 1492–1509, 10.1039/c4pp00455h.
- F. Vatansever and M. R. Hamblin, Photodynamic Therapy and Antitumor Immune Response, in Cancer Immunology, ed. N. Rezaei, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 383–399, DOI:10.1007/978-3-662-44946-2_21.
- D. van Straten, V. Mashayekhi, H. de Bruijn, S. Oliveira and D. Robinson, Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions, Cancers, 2017, 9(12), 19, DOI:10.3390/cancers9020019.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, Photodynamic Therapy, JNCI, J. Natl. Cancer Inst., 1998, 90(12), 889–905, DOI:10.1093/jnci/90.12.889.
- J. Levy and E. LevyPhotofrin-PDT from Bench to Bedside: Some Lessons Learned, in Handbook of photodynamic therapy: updates on recent applications of porphyrin-based compounds, ed. R. K. Pandey, D. Kessel and T. J. Dougherty, World Scientific, New Jersey, 2016 Search PubMed.
- D. Kessel and J. D. Thomas, An Appreciation, Photochem. Photobiol., 2020, 96(3), 454–457, DOI:10.1111/php.13144.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433, Chem. Rev., 2019, 119(2), 797–828, DOI:10.1021/acs.chemrev.8b00211.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Metal-Based Photosensitizers for Photodynamic Therapy: The Future of Multimodal Oncology?, Curr. Opin. Chem. Biol., 2020, 56, 23–27, DOI:10.1016/j.cbpa.2019.10.004.
- E. C. Glazer, Light-Activated Metal Complexes That Covalently Modify DNA, Isr. J. Chem., 2013, 53(6–7), 391–400, DOI:10.1002/ijch.201300019.
- N. A. Smith and P. J. Sadler, Photoactivatable Metal Complexes: From Theory to Applications in Biotechnology and Medicine, Philos. Trans. R. Soc., A, 2013, 371(1995), 20120519, DOI:10.1098/rsta.2012.0519.
- V. Balzani and A. Juris, Photochemistry and Photophysics of Ru(II)-polypyridine Complexes in the Bologna Group. From Early Studies to Recent Developments, Coord. Chem. Rev., 2001, 211(1), 97–115, DOI:10.1016/S0010-8545(00)00274-5.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence, Coord. Chem. Rev., 1988, 84, 85–277, DOI:10.1016/0010-8545(88)80032-8.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents, J. Am. Chem. Soc., 2012, 134(20), 8324–8327, DOI:10.1021/ja3009677.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Light-Activated Ruthenium Complexes Photobind DNA and Are Cytotoxic in the Photodynamic Therapy Window, Chem. Commun., 2012, 48(77), 9649, 10.1039/c2cc33359g.
- E. Wachter and E. C. Glazer, Mechanistic Study on the Photochemical “Light Switch” Behavior of [Ru(Bpy) 2 Dmdppz] 2+, J. Phys. Chem. A, 2014, 118(45), 10474–10486, DOI:10.1021/jp504249a.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Efficient DNA Photocleavage by [Ru(Bpy)2(Dppn)]2+ with Visible Light, Chem. Commun., 2010, 46(14), 2426, 10.1039/b925574e.
- R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, Exploitation of Long-Lived 3 IL Excited States for Metal–Organic Photodynamic Therapy: Verification in a Metastatic Melanoma Model, J. Am. Chem. Soc., 2013, 135(45), 17161–17175, DOI:10.1021/ja408426z.
- H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, S. Monro and S. A. McFarland, In Vitro Multiwavelength PDT with 3 IL States: Teaching Old Molecules New Tricks, Inorg. Chem., 2014, 53(9), 4548–4559, DOI:10.1021/ic5002368.
- C. Moorlag, B. Sarkar, C. N. Sanrame, P. Bäuerle, W. Kaim and M. O. Wolf, Conjugation Length Dependent Ground and Excited State Electronic Behavior in Oligothienyl Ru Complexes, Inorg. Chem., 2006, 45(18), 7044–7046, DOI:10.1021/ic060912n.
- M. B. Majewski, N. R. de Tacconi, F. M. MacDonnell and M. O. Wolf, Ligand-Triplet-Fueled Long-Lived Charge Separation in Ruthenium(II) Complexes with Bithienyl-Functionalized Ligands, Inorg. Chem., 2011, 50(20), 9939–9941, DOI:10.1021/ic201895y.
- M. B. Majewski, N. R. deTacconi, F. M. MacDonnell and M. O. Wolf, Long-Lived, Directional Photoinduced Charge Separation in Ru II Complexes Bearing Laminate Polypyridyl Ligands, Chem. – Eur. J., 2013, 19(25), 8331–8341, DOI:10.1002/chem.201203786.
- X. Han, L.-Z. Wu, G. Si, J. Pan, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, Switching between Ligand-to-Ligand Charge-Transfer, Intraligand Charge-Transfer, and Metal-to-Ligand Charge-Transfer Excited States in Platinum(II) Terpyridyl Acetylide Complexes Induced by PH Change and Metal Ions, Chem. - Eur. J., 2007, 13(4), 1231–1239, DOI:10.1002/chem.200600769.
- S. Verma, P. Kar, A. Das and H. N. Ghosh, Photophysical Properties of Ligand Localized Excited State in Ruthenium(Ii) Polypyridyl Complexes: A Combined Effect of Electron Donor–Acceptor Ligand, Dalton Trans., 2011, 40(38), 9765, 10.1039/c1dt10266d.
- L. K. Keniley, N. Dupont, L. Ray, J. Ding, K. Kovnir, J. M. Hoyt, A. Hauser and M. Shatruk, Complexes with Redox-Active Ligands: Synthesis, Structure, and Electrochemical and Photophysical Behavior of the Ru(II) Complex with TTF-Annulated Phenanthroline, Inorg. Chem., 2013, 52(14), 8040–8052, DOI:10.1021/ic4006949.
- Y. Zhao, J. A. Woods, N. J. Farrer, K. S. Robinson, J. Pracharova, J. Kasparkova, O. Novakova, H. Li, L. Salassa, A. M. Pizarro, G. J. Clarkson, L. Song, V. Brabec and P. J. Sadler, Diazido Mixed-Amine Platinum(IV) Anticancer Complexes Activatable by Visible-Light Form Novel DNA Adducts, Chem. – Eur. J., 2013, 19(29), 9578–9591, DOI:10.1002/chem.201300374.
- S. Swavey, Z. Fang and K. J. Brewer, Mixed-Metal Supramolecular Complexes Coupling Phosphine-Containing Ru(II) Light Absorbers to a Reactive Pt(II) through Polyazine Bridging Ligands, Inorg. Chem., 2002, 41(9), 2598–2607, DOI:10.1021/ic010806f.
- A. A. Holder, S. Swavey and K. J. Brewer, Design Aspects for the Development of Mixed-Metal Supramolecular Complexes Capable of Visible Light Induced Photocleavage of DNA, Inorg. Chem., 2004, 43(1), 303–308, DOI:10.1021/ic035029t.
- A. A. Holder, D. F. Zigler, M. T. Tarrago-Trani, B. Storrie and K. J. Brewer, Photobiological Impact of [{(Bpy)2Ru(Dpp)}2RhCl2]Cl5 and [{(Bpy)2Os(Dpp)}2RhCl2]Cl5 [Bpy = 2,2′-Bipyridine; Dpp = 2,3-Bis(2-Pyridyl)Pyrazine] on Vero Cells, Inorg. Chem., 2007, 46(12), 4760–4762, DOI:10.1021/ic0619916.
- D. F. Zigler, M. T. Mongelli, M. Jeletic and K. J. Brewer, A Trimetallic Supramolecular Complex of Osmium(II) and Rhodium(III) Displaying MLCT Transitions in the near-IR, Inorg. Chem. Commun., 2007, 10(3), 295–298, DOI:10.1016/j.inoche.2006.10.024.
- D. Havrylyuk, K. Stevens, S. Parkin and E. C. Glazer, Toward Optimal Ru(II) Photocages: Balancing Photochemistry, Stability, and Biocompatibility Through Fine Tuning of Steric, Electronic, and Physiochemical Features, Inorg. Chem., 2020, 59(2), 1006–1013, DOI:10.1021/acs.inorgchem.9b02065.
- B. A. Albani, C. B. Durr and C. Turro, Selective Photoinduced Ligand Exchange in a New Tris–Heteroleptic Ru(II) Complex, J. Phys. Chem. A, 2013, 117(50), 13885–13892, DOI:10.1021/jp4085684.
- J. D. Knoll, B. A. Albani, C. B. Durr and C. Turro, Unusually Efficient Pyridine Photodissociation from Ru(II) Complexes with Sterically Bulky Bidentate Ancillary Ligands, J. Phys. Chem. A, 2014, 118(45), 10603–10610, DOI:10.1021/jp5057732.
- A. Li, C. Turro and J. J. Kodanko, Ru(II) Polypyridyl Complexes as Photocages for Bioactive Compounds Containing Nitriles and Aromatic Heterocycles, Chem. Commun., 2018, 54(11), 1280–1290, 10.1039/C7CC09000E.
- A. J. Göttle, F. Alary, M. Boggio-Pasqua, I. M. Dixon, J.-L. Heully, A. Bahreman, S. H. C. Askes and S. Bonnet, Pivotal Role of a Pentacoordinate 3 MC State on the Photocleavage Efficiency of a Thioether Ligand in Ruthenium(II) Complexes: A Theoretical Mechanistic Study, Inorg. Chem., 2016, 55(9), 4448–4456, DOI:10.1021/acs.inorgchem.6b00268.
- L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. LeDévédec and S. Bonnet, A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells, Angew. Chem., Int. Ed., 2017, 56(38), 11549–11553, DOI:10.1002/anie.201703890.
- V. H. S. van Rixel, V. Ramu, A. B. Auyeung, N. Beztsinna, D. Y. Leger, L. N. Lameijer, S. T. Hilt, S. E. Le Dévédec, T. Yildiz, T. Betancourt, M. B. Gildner, T. W. Hudnall, V. Sol, B. Liagre, A. Kornienko and S. Bonnet, Photo-Uncaging of a Microtubule-Targeted Rigidin Analogue in Hypoxic Cancer Cells and in a Xenograft Mouse Model, J. Am. Chem. Soc., 2019, 141(46), 18444–18454, DOI:10.1021/jacs.9b07225.
- S. Bonnet, Why Develop Photoactivated Chemotherapy?, Dalton Trans., 2018, 47(31), 10330–10343, 10.1039/C8DT01585F.
- M. Stephenson, C. Reichardt, M. Pinto, M. Wächtler, T. Sainuddin, G. Shi, H. Yin, S. Monro, E. Sampson, B. Dietzek and S. A. McFarland, Ru(II) Dyads Derived from 2-(1-Pyrenyl)-1H-Imidazo[4,5-f][1,10]Phenanthroline: Versatile Photosensitizers for Photodynamic Applications, J. Phys. Chem. A, 2014, 118(45), 10507–10521, DOI:10.1021/jp504330s.
- G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Ru(II) Dyads Derived from α-Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT, Coord. Chem. Rev., 2015, 282–283, 127–138, DOI:10.1016/j.ccr.2014.04.012.
- C. Reichardt, M. Pinto, M. Wächtler, M. Stephenson, S. Kupfer, T. Sainuddin, J. Guthmuller, S. A. McFarland and B. Dietzek, Photophysics of Ru(II) Dyads Derived from Pyrenyl-Substitued Imidazo[4,5-f][1,10]Phenanthroline Ligands, J. Phys. Chem. A, 2015, 119(17), 3986–3994, DOI:10.1021/acs.jpca.5b01737.
- C. Reichardt, T. Sainuddin, M. Wächtler, S. Monro, S. Kupfer, J. Guthmuller, S. Gräfe, S. McFarland and B. Dietzek, Influence of Protonation State on the Excited State Dynamics of a Photobiologically Active Ru(II) Dyad, J. Phys. Chem. A, 2016, 120(32), 6379–6388, DOI:10.1021/acs.jpca.6b05957.
- C. Reichardt, K. R. A. Schneider, T. Sainuddin, M. Wächtler, S. A. McFarland and B. Dietzek, Excited State Dynamics of a Photobiologically Active Ru(II) Dyad Are Altered in Biologically Relevant Environments, J. Phys. Chem. A, 2017, 121(30), 5635–5644, DOI:10.1021/acs.jpca.7b04670.
- C. Reichardt, S. Monro, F. H. Sobotta, K. L. Colón, T. Sainuddin, M. Stephenson, E. Sampson, J. Roque, H. Yin, J. C. Brendel, C. G. Cameron, S. McFarland and B. Dietzek, Predictive Strength of Photophysical Measurements for in Vitro Photobiological Activity in a Series of Ru(II) Polypyridyl Complexes Derived from π-Extended Ligands, Inorg. Chem., 2019, 58(5), 3156–3166, DOI:10.1021/acs.inorgchem.8b03223.
- Y. Arenas, S. Monro, G. Shi, A. Mandel, S. McFarland and L. Lilge, Photodynamic Inactivation of Staphylococcus Aureus and Methicillin-Resistant Staphylococcus Aureus with Ru(II)-Based Type I/Type II Photosensitizers, Photodiagn. Photodyn. Ther., 2013, 10, 615–625, DOI:10.1016/j.pdpdt.2013.07.001.
- T. Sainuddin, M. Pinto, H. Yin, M. Hetu, J. Colpitts and S. A. McFarland, Strained Ruthenium Metal–Organic Dyads as Photocisplatin Agents with Dual Action, J. Inorg. Biochem., 2016, 158, 45–54, DOI:10.1016/j.jinorgbio.2016.01.009.
- G. Ghosh, K. L. Colón, A. Fuller, T. Sainuddin, E. Bradner, J. McCain, S. M. A. Monro, H. Yin, M. W. Hetu, C. G. Cameron and S. A. McFarland, Cyclometalated Ruthenium(II) Complexes Derived from á-Oligothiophenes as Highly Selective Cytotoxic or Photocytotoxic Agents, Inorg. Chem., 2018, 57(13), 7694–7712, DOI:10.1021/acs.inorgchem.8b00689.
- S. Monro, C. G. Cameron, X. Zhu, K. L. Colón, H. Yin, T. Sainuddin, M. Hetu, M. Pinto, A. Fuller, L. Bennett, J. Roque, W. Sun and S. A. McFarland, Synthesis, Characterization and Photobiological Studies of Ru(II) Dyads Derived from α-Oligothiophene Derivatives of 1,10-Phenanthroline, Photochem. Photobiol., 2019, 95(1), 267–279, DOI:10.1111/php.13012.
- J. McCain, K. L. Colón, P. C. Barrett, S. M. A. Monro, T. Sainuddin, J. Roque III, M. Pinto, H. Yin, C. G. Cameron and S. A. McFarland, Photophysical Properties and Photobiological Activities of Ruthenium(II) Complexes Bearing π-Expansive Cyclometalating Ligands with Thienyl Groups, Inorg. Chem., 2019, 58(16), 10778–10790, DOI:10.1021/acs.inorgchem.9b01044.
- J. A. Roque III, P. C. Barrett, H. D. Cole, L. M. Lifshits, G. Shi, S. Monro, D. von Dohlen, S. Kim, N. Russo, G. Deep, C. G. Cameron, M. E. Alberto and S. A. McFarland, Breaking the Barrier: An Osmium Photosensitizer with Unprecedented Hypoxic Phototoxicity for Real World Photodynamic Therapy, Chem. Sci., 2020, 11(36), 9784–9806 RSC.
- L. M. Lifshits, J. A. Roque III, H. D. Cole, R. P. Thummel, C. G. Cameron and S. A. McFarland, NIR-Absorbing Ru(II) Complexes Containing α-Oligothiophenes for Applications in Photodynamic Therapy, ChemBioChem, 2020 DOI:10.1002/cbic.202000419.
- J. A. Roque III, P. C. Barrett, H. D. Cole, L. M. Lifshits, E. Bradner, G. Shi, D. von Dohlen, S. Kim, G. Deep, C. G. Cameron, M. E. Alberto and S. A. McFarland, Os(II) Oligothienyl Complexes as a New Hypoxia-Active Photosensitizer Class for Photodynamic Therapy, Inorg. Chem., 2020 Search PubMed , accepted.
- Q. Chen, V. Ramu, Y. Aydar, A. Groenewoud, X.-Q. Zhou, M. J. Jager, H. Cole, C. G. Cameron, S. A. McFarland, S. Bonnet and B. E. Snaar-Jagalska, TLD1433 Photosensitizer Inhibits Conjunctival Melanoma Cells in Zebrafish Ectopic and Orthotopic Tumour Models, Cancers, 2020, 12(3), 587, DOI:10.3390/cancers12030587.
- J. Fong, K. Kasimova, Y. Arenas, P. Kaspler, S. Lazic, A. Mandel and L. Lilge, A Novel Class of Ruthenium-Based Photosensitizers Effectively Kills in Vitro Cancer Cells and in Vivo Tumors, Photochem. Photobiol. Sci., 2015, 14(11), 2014–2023, 10.1039/C4PP00438H.
- P. Kaspler, S. Lazic, S. Forward, Y. Arenas, A. Mandel and L. Lilge, A Ruthenium(II) Based Photosensitizer and Transferrin Complexes Enhance Photo-Physical Properties, Cell Uptake, and Photodynamic Therapy Safety and Efficacy, Photochem. Photobiol. Sci., 2016, 15(4), 481–495, 10.1039/C5PP00450K.
- M. A. Munegowda, C. Fisher, D. Molehuis, W. Foltz, M. Roufaiel, J. Bassan, M. Nitz, A. Mandel and L. Lilge, Efficacy of Ruthenium Coordination Complex–Based Rutherrin in a Preclinical Rat Glioblastoma Model, Neuro-Oncol. Adv., 2019, 1(1), vdz006, DOI:10.1093/noajnl/vdz006.
- L. Lilge, M. Roufaiel, S. Lazic, P. Kaspler, M. A. Munegowda, M. Nitz, J. Bassan and A. Mandel, Evaluation of a Ruthenium Coordination Complex as Photosensitizer for PDT of Bladder Cancer: Cellular Response, Tissue Selectivity and in Vivo Response, Transl. Biophotonics, 2020, 2(1–2) DOI:10.1002/tbio.201900032.
- L. Lilge, J. Wu, Y. Xu, A. Manalac, D. Molehuis, F. Schwiegelshohn, L. Vesselov, W. Embree, M. Nesbit, V. Betz, A. Mandel, M. A. S. Jewett and G. S. Kulkarni, Minimal Required PDT Light Dosimetry for Nonmuscle Invasive Bladder Cancer, J. Biomed. Optic., 2020, 25(6), 1–13, DOI:10.1117/1.JBO.25.6.068001.
- T. Lundrigan, C. L. M. Jackson, Md. I. Uddin, L. A. Tucker, A. A.-S. Ali, A. Linden, T. S. Cameron and A. Thompson, Synthesis of Heteroleptic Pyrrolide/Bipyridyl Complexes of Ruthenium(II), Can. J. Chem., 2012, 90(8), 693–700, DOI:10.1139/v2012-045.
- J. K. Blaho and K. A. Goldsby, Redox Regulation Based on the PH-Dependent Hydrolysis of 2-Pyridinecarboxaldehyde Coordinated to Ruthenium(II), J. Am. Chem. Soc., 1990, 112(16), 6132–6133, DOI:10.1021/ja00172a041.
- A. Spaggiari, D. Vaccari, P. Davoli and F. Prati, The Triphenyl Phosphite-Chlorine Reagent in the Synthesis of Pyrroles from N-Allylamides, Synthesis, 2006, No. 6, 995–998, DOI:10.1055/s-2006-926365.
- J. Liu, L. Bu, J. Dong, Q. Zhou, Y. Geng, D. Ma, L. Wang, X. Jing and F. Wang, Green Light-Emitting Polyfluorenes with Improved Color Purity Incorporated with 4,7-Diphenyl-2,1,3-Benzothiadiazole Moieties, J. Mater. Chem., 2007, 17(27), 2832, 10.1039/b700004a.
- M. Sun, L. Lan, L. Wang, J. Peng and Y. Cao, Synthesis of Novel Conjugated Polyelectrolytes for Organic Field-Effect Transistors Gate Dielectric Materials, Macromol. Chem. Phys., 2008, 209(24), 2504–2509, DOI:10.1002/macp.200800420.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Synthesis of Isoindigo-Based Oligothiophenes for Molecular Bulk Heterojunction Solar Cells, Org. Lett., 2010, 12(4), 660–663, DOI:10.1021/ol902512x.
- H. C. Kang and R. P. HauglandDibenzopyrrometheneboron Difluoride Dyes, US5433896, July 18, 1995.
- S. P. Foxon, C. Metcalfe, H. Adams, M. Webb and J. A. Thomas, Electrochemical and Photophysical Properties of DNA Metallo-Intercalators Containing the Ruthenium(II) Tris(1-Pyrazolyl)Methane Unit, Inorg. Chem., 2007, 46(2), 409–416, DOI:10.1021/ic0607134.
- A. A. Abdel-Shafi, P. D. Beer, R. J. Mortimer and F. Wilkinson, Photosensitized Generation of Singlet Oxygen from Ruthenium(II)-Substituted Benzoaza-Crown-Bipyridine Complexes, Phys. Chem. Chem. Phys., 2000, 2(14), 3137–3144, 10.1039/b002884n.
- W. Y. Ho, S. K. Yeap, C. L. Ho, R. A. Rahim and N. B. Alitheen, Development of Multicellular Tumor Spheroid (MCTS) Culture from Breast Cancer Cell and a High Throughput Screening Method Using the MTT Assay, PloS One, 2012, 7(9), e44640, DOI:10.1371/journal.pone.0044640.
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 7, ed. M. J. Ferraro, Clinical and Laboratory Standards Institute, Clinical and Laboratory Standards series; CLSI, Wayne, PA, 2006 Search PubMed.
- Clinical Microbiology Procedures Handbook, ed. H. D. Isenberg, American Society for Microbiology, ASM Press, Washington, D.C, 2nd edn, 2004 Search PubMed.
- D. A. Smithen, H. Yin, M. H. R. Beh, M. Hetu, T. S. Cameron, S. A. McFarland and A. Thompson, Synthesis and Photobiological Activity of Ru(II) Dyads Derived from Pyrrole-2-Carboxylate Thionoesters, Inorg. Chem., 2017, 56(7), 4121–4132, DOI:10.1021/acs.inorgchem.7b00072.
- J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins, J. Am. Chem. Soc., 2006, 128(35), 11693–11712, DOI:10.1021/ja062355+.
- X. Cui, J. Li, L. Liu and Q. X. Guo, 1,3-Dicarbonyl Compounds as Phosphine-Free Ligands for Pd-Catalyzed Heck and Suzuki Reactions, Chin. Chem. Lett., 2007, 18(6), 625–628, DOI:10.1016/j.cclet.2007.04.014.
- R. M. Silverstein, E. E. Ryskiewicz and C. Willard, 2-Pyrrolealdehyde, Org. Synth., 1956, 36, 74, DOI:10.15227/orgsyn.036.0074.
- R. P. Haugland and H. C. Kang, Chemically Reactive Dipyrrometheneboron Difluoride Dyes, US4774339A, September 27, 1988.
- J. Setsune and K. Watanabe, Cryptand-like Porphyrinoid Assembled with Three Dipyrrylpyridine Chains: Synthesis, Structure, and Homotropic Positive Allosteric Binding of Carboxylic Acids, J. Am. Chem. Soc., 2008, 130(8), 2404–2405, DOI:10.1021/ja710424n.
- S. J. Smalley, M. R. Waterland and S. G. Telfer, Heteroleptic Dipyrrin/Bipyridine Complexes of Ruthenium(II), Inorg. Chem., 2009, 48(1), 13–15, DOI:10.1021/ic8016497.
- S. E. Skrabalak, B. J. Wiley, M. Kim, E. V. Formo and Y. Xia, On the Polyol Synthesis of Silver Nanostructures: Glycolaldehyde as a Reducing Agent, Nano Lett., 2008, 8(7), 2077–2081, DOI:10.1021/nl800910d.
- M. Mariappan, M. Suenaga, A. Mukhopadhyay, P. Raghavaiah and B. G. Maiya, Synthesis, Structure, DNA Binding and Photocleavage Activity of a Ruthenium(II) Complex with 11-(9-Acridinyl)Dipyrido[3,2-a:2′,3′-c]Phenazine Ligand, Inorg. Chim. Acta., 2011, 376(1), 340–349, DOI:10.1016/j.ica.2011.06.042.
- P. R. Ogilby and C. S. Foote, Chemistry of Singlet Oxygen. 42. Effect of Solvent, Solvent Isotopic Substitution, and Temperature on the Lifetime of Singlet Molecular Oxygen (1.DELTA.g), J. Am. Chem. Soc., 1983, 105(11), 3423–3430, DOI:10.1021/ja00349a007.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley monographs in chemical physics; Wiley-Interscience, London, New York, 1970 Search PubMed.
- S. Sant and P. A. Johnston, The Production of 3D Tumor Spheroids for Cancer Drug Discovery, Drug Discov. Today Technol., 2017, 23, 27–36, DOI:10.1016/j.ddtec.2017.03.002.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic procedures for the synthesis of pyrrole 1a and aryl dibromides i, j and k. Figures giving 1H and 13C NMR spectra and UV/vis absorption spectra for all bis(pyrrole)s (2), ligands (3) and bis(ruthenium) complex salts (4). See DOI: 10.1039/d0sc04500d |
| This journal is © The Royal Society of Chemistry 2020 |