
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncreased hydrogen partial pressure suppresses and reverses hydrogen evolution during Pd catalysed electrolysis of CO2†
Martijn J. W.
Blom
 ab,
Wim P. M.
van Swaaij
a,
Guido
Mul
b and
Sascha R. A.
Kersten
*a
ab,
Wim P. M.
van Swaaij
a,
Guido
Mul
b and
Sascha R. A.
Kersten
*a
aA Sustainable Process Technology Group, Faculty of Science and Technology, University of Twente, P. O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: s.r.a.kersten@utwente.nl
bPhotoCatalytic Synthesis Group, MESA+ Institute for Nanotechnology, University of Twente, P. O. Box 217, 7500 AE Enschede, The Netherlands
First published on 30th June 2020
Abstract
Electrochemical reduction of CO2 on a Pd/C cathode produces formate and hydrogen at low overpotentials. We report on an innovative, effective approach to prevent hydrogen formation. By applying 4 bar partial hydrogen pressure, hydrogen evolution can be fully avoided at −0.05 V vs. RHE and 1 bar partial CO2 pressure.
Electrochemical conversion of CO2 to commodity chemicals using renewable electricity could be a key enabling technology for decoupling the chemical industry from fossil resources.1–3 Formate salts (M+ HCO2−) and formic acid are relatively high value CO2 reduction products, which can attain the roles of energy storage medium or C1 building block.4–6 The current industrial practice for the production of formic acid and formate is based on the conversion of fossil-based CO at 45 bar and 80 °C.7 Direct electrochemical reduction of CO2 to formate with renewable electricity could be a sustainable single-reactor alternative to the traditional route. However, the economic feasibility of such a process is largely determined by the overpotential and faradaic efficiency to formate (FE).4,8
Electrochemical conversion of CO2 to formate at near zero overpotential is only observed on Pd based electrocatalysts and the enzyme formate dehydrogenase and its derivatives,9–11 making Pd based catalysts especially relevant for heterogeneous, energy efficient electrocatalysis. When operated at less than 0.25 V overpotential, Pd based electrocatalysts produce mainly HCOO− (reaction 1) with H2 as a byproduct (reaction 2).12 Since Pd is an excellent hydrogen evolution catalyst and H2 and HCOO− are both formed via palladium hydride as an intermediate, suppressing hydrogen evolution is challenging.10,13 Many recent publications try to minimize hydrogen evolution by changing the nature of the catalyst via alloying (88–100% FE),14,15 doping (70% FE)16 or (nano)structuring (50–97% FE).17,18 Here we report on an alternative approach, which utilizes the reversible Pd-catalysed hydrogenation of CO2 (ref. 19 and 20) (reaction 3).
| CO2 + H2O + 2e− ⇄ HCOO− + OH− | (1) |
| 2H2O + 2e− ⇄ H2 + 2OH− | (2) |
| CO2 + OH− + H2 ⇄ HCOO− + H2O | (3) |
Generally, electrochemical setups for the electrochemical reduction of CO2 continuously sparge fresh CO2 through the catholyte, as is good practice, to avoid mass transfer limitations by undersaturation of the bulk electrolyte.21–23 However, CO2 sparging also strips any formed H2 from solution and pulls reaction 2 towards more hydrogen production. In contrast, in the absence of applied potential, Pd/C catalyses hydrogenation of CO2 dissolved in aqueous solution at elevated hydrogen partial pressure (pH2).20 Moreover, at electrochemical CO2 reduction conditions, reactions 2 and 3 were observed to occur simultaneously on Pd/C.24,25 The rate of reaction 3 increases with pH2,20 whereas the rate of reaction 2 decreases with pH2, but the latter is mostly governed by the applied potential.26 If, at a certain potential and pH2, the rates of reaction 2 and 3 are equal, net hydrogen production is zero. In a continuous reactor (without gaseous CO2 reduction products), those conditions correspond to an operating point where no external hydrogen supply is required and net no hydrogen is produced. Here, we systematically investigate the kinetics of combined chemical hydrogenation of CO2 and electrochemical reduction of CO2 with the aim to control the undesired net production of hydrogen and thus selectivity to formate. (We refer to selectivity instead of faraday efficiency, as the latter only concerns electrochemical reactions and selectivity concerns chemical reactions as well.) We decided to communicate our observations as such, without a complete understanding of the underlying phenomena yet, to make the findings available to the community for further exploration of its opportunities.
To study the effect of pH2 on the hydrogen evolution rate accurately, the electrochemical potential should be kept constant. Therefore, a reactor that can be pressurized and includes a stable (Ag/AgCl) reference electrode (RE) (Fig. 1), was developed, inspired by a geometry designed by Cave et al.27 The reactor contains a cation exchange membrane (CMI-7000) to divide the catholyte zone from the anolyte zone, and prevent product oxidation at the dimensionally stable anode. Both catholyte and anolyte are 1 M KHCO3. A control system is in place to keep both compartments at equal pressure, but separated, thus eliminating trans-membrane pressure drop and gas mixing (ESI Section S1.3†). The reference electrode is kept at the same pressure as the reactor, to eliminate any convective transport between the reference electrode and reactor, thereby maintaining a stable reference potential. During experiments, excess hydrogen, CO2, and argon are continuously sparged through the catholyte, to have accurate control over the gas phase composition. The reactor facilitates measurements in the kinetically limited regime over a wide (partial) pressure range, whilst maintaining accurate potential control (ESI Section S1.4†).
 | ||
| Fig. 1 Schematic representation of electrochemical cell. (A) Expanded view of electrochemical cell. (B) Schematic of operating cell. | ||
Electrochemical reduction of CO2 was performed on Pd/C coated titanium plate electrodes (ESI Section S1.2†). Each experiment took 60 minutes and formate was quantified by HPLC afterwards. Consequently, the hydrogen production was calculated by the difference from the accumulative charge and production of formate. A more detailed description of the experimental methods including error analysis is provided in the ESI in Section S1.†
The effect of hydrogen partial pressure on the net production of hydrogen was studied under three different conditions: at −0.05 V vs. RHE and pCO2 = 1 bar, at −0.10 V vs. RHE and pCO2 = 1 bar, and at −0.05 V vs. RHE and pCO2 = 3 bar, respectively. Under all conditions, the total pressure was maintained at 7 bar and H2/Ar partial pressures were varied. Thereby, the effect of pH2 is studied, with minimal bias from changes in flow, mixing (induced by gas bubbles) and CO2 concentration.
The results, presented in Fig. 2, show that under all conditions the average overall hydrogen production rate is significantly decreased by increasing the partial pressure of hydrogen. The effect is most pronounced at −0.05 V vs. RHE and partial CO2 pressure (pCO2) of 1 bar. At high enough pH2, overall hydrogen production can be prevented and can even become negative, implying hydrogen consumption, which must be viareaction 3. A higher cathodic potential (−0.10 V vs. RHE) results in a higher average rate to H2. This is a result of the increased electrochemical driving force for hydrogen evolution. At higher CO2 partial pressure the rate to H2 also increases, presumably due to kinetic effects induced by an increased acidity of the electrolyte.26 When pCO2 is increased from 1 to 3 bar, the proton concentration also increases threefold (ESI Section S1.9†) and a first order dependence of hydrogen evolution rate on the concentration of protons26 agrees with the threefold increase of hydrogen production at pH2 = 0 bar (Fig. 2). Extrapolation of the data indicates that also at more cathodic potential and at higher CO2 partial pressure, an operating point exists where overall hydrogen production equals zero.
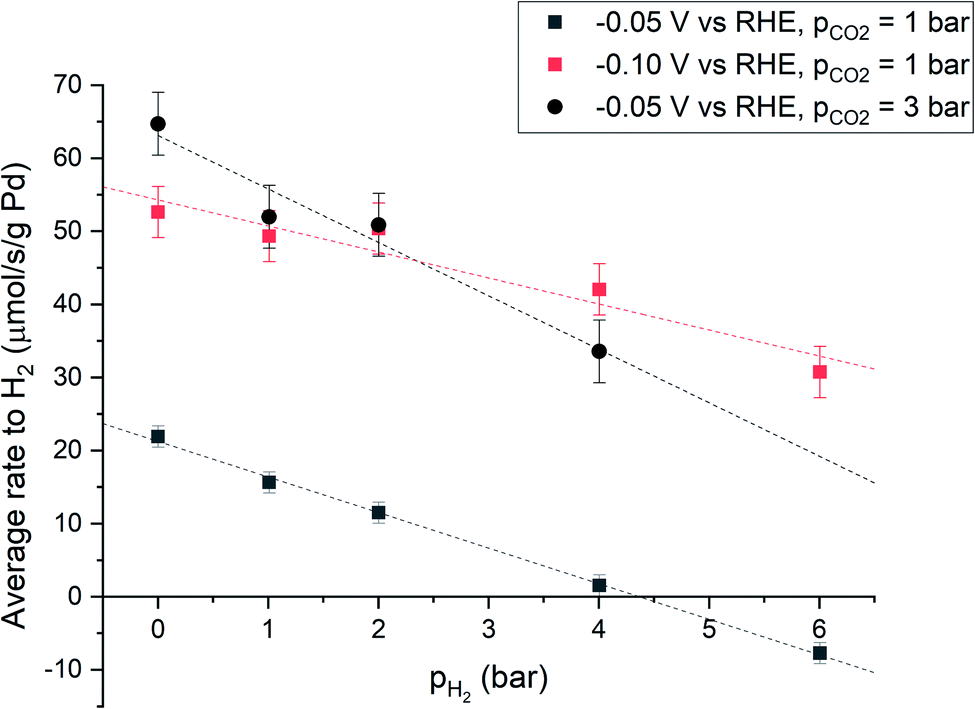 | ||
| Fig. 2 Hydrogen production during electrochemical CO2 reduction in 1 M KHCO3 sparged with a mixture of CO2, Ar and H2. ptotal = 7 bar, pCO2 is 1 bar or 3 bar and the applied potential is −0.05 V or −0.10 V vs. RHE. | ||
The hydrogen partial pressure does not significantly influence the average rate to formate (Fig. 3 and ESI Section S1.6†). This was observed for applied potentials of −0.05 V and −0.10 V vs. RHE, and at higher CO2 partial pressure of 3 bar. The average rate to formate increases at more negative cathodic potential and is similar to values reported in literature (6–52 μmole s−1 g−1 Pd) at comparable conditions.10,16 Furthermore, the rate to formate increases linearly with increased partial pressure of CO2, which is a continuation of the trend observed at partial pressures of CO2 below 1 bar, described by a first order dependence of the kinetics to HCOO− on the concentration of CO2.10
 | ||
| Fig. 3 Formate production during electrochemical CO2 reduction in 1 M KHCO3 sparged with a mixture of CO2, Ar and H2. ptotal = 7 bar, pCO2 is 1 bar or 3 bar and the applied potential is −0.05 V or −0.10 V vs. RHE. Average of data sets plotted as guide to the eye. | ||
Fig. 4 shows an overview of the reactions that are relevant for the overall conversion of CO2 and H2O into formate and OH−. Recent literature suggests an electro-hydrogenation mechanism for electrochemical formate production on Pd, wherein palladium hydride is the active catalyst phase.10,12 This is also the active catalyst phase for hydrogen evolution.13 In the absence of hydrogen in the feed, the hydride phase must be generated via electro-reduction of water (r1 in Fig. 4). Under the applied potential (<−0.05 V vs. RHE) hydrogen production is thermodynamically possible and occurs according to reaction r1 + r2. When pH2 in the reactor is raised, overall hydrogen production decreases. That is due to chemical hydrogenation (r−2 + r3 + r4), as the applied hydrogen pressure is far below the equilibrium pressure for electrochemical hydrogen evolution (49 bar at −0.05 V vs. RHE, based on Nernst's law).
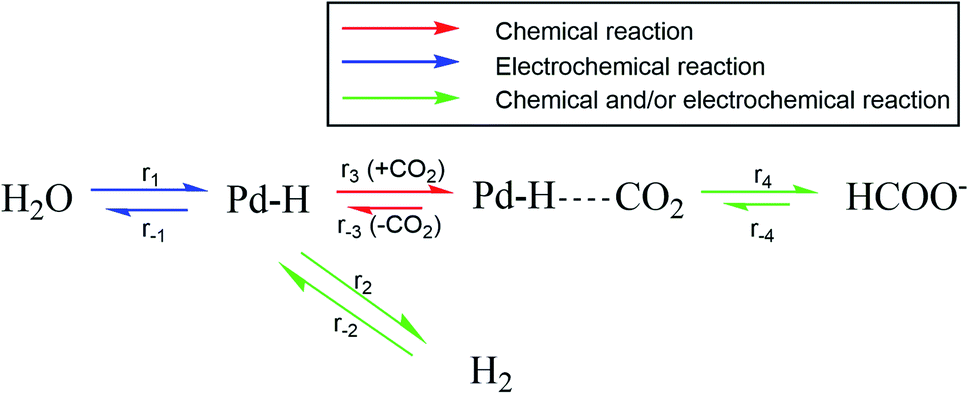 | ||
| Fig. 4 Reaction scheme for combined chemical and electrochemical reduction of CO2 to formate. | ||
Palladium hydride formation from molecular hydrogen (r−2) likely occurs via dissociation or electro-adsorption (Heyrovsky reaction) on Pd. Hydride formation from molecular hydrogen increases with increasing pH2 due to the increased H2 concentration in the aqueous phase. When r2 = r−2, hydrogen production is fully suppressed and the overall rate of formation (r2 − r−2) equals zero. Such a point is observed in Fig. 2 at −0.05 V vs. RHE, 1 bar CO2 partial pressure and at approximately 4 bar H2 partial pressure. Nearly all electrons added to the system are then used to produce formate, which would correspond to a faraday efficiency of approximately 100%. Since net production of H2 does not occur and the formate production is independent of pH2 (Fig. 3), the total current decreased as a function of pH2. When pH2 is increased further to 6 bar, r−2 exceeds r2, resulting in net hydrogen consumption. This indicates that overall, all electrons added to the system are used to make formate and even more formate is produced via chemical hydrogenation of CO2. Consequently, the hydrogen partial pressure can be used to control the net hydrogen production/consumption rate and thus the selectivity to formate.
At potentials less cathodic than −0.25 V vs. RHE, formate and hydrogen are the only significant products for electrochemical CO2 reduction using Pd. Palladium hydride formation (r1) is a relatively fast process,28,29 and no effect of the hydrogen partial pressure on the rate to formate was observed (Fig. 3). Therefore, the rate to formate seems not limited by hydride formation (r1 + r2) at the applied electrochemical conditions and r3 or r4 is likely the rate-limiting step for formate production, which is in agreement with recent literature.10 A yet unresolved question is the content of the Pd–H phase (Pd/H ratio) and the content of the Pd–HCO2 intermediate, and the relative size of r1 over r−2. We presently evaluate the mechanism and the rate limiting step by using isotopic labelling of H2 (feeding D2). Furthermore, we investigate why the rate to formate is unaffected by pH2 under electrochemical conditions and hypothesize this is due to a fully loaded Pd–H phase resulting from the cathodic potential. Finally, we assess the apparent activation energy for H2 and HCOO− formation under electrochemical and chemical conditions.
Conclusions
We developed an electrochemical CO2 reduction cell that operates at elevated pressure and can still employ a cation exchange membrane and reference electrode. Using this cell, we show that presence of H2 in the gas phase suppresses hydrogen evolution on a Pd/C catalyst, likely induced by chemical formation of a Pd–H phase and consecutive hydrogenation of CO2. This novel approach provides an alternative to the common practice of increasing the selectivity by changing the catalyst structure or composition. Under electrochemical CO2 reduction conditions of 1 bar CO2 partial pressure and −0.05 V vs. RHE, 4 bar H2 pressure is sufficient to completely eliminate net H2 evolution, and a further increase to 6 bar results in net H2 consumption. Therefore, by allowing a natural accumulation of electrochemically produced hydrogen, either via gas cap or recycle, the selectivity to formate can be controlled. This opens the possibility to use other parameters (pCO2, potential, temperature, etc.) to optimize the reaction rate, possibly creating the conditions for simultaneous high selectivity and conversion, as required for commercial implementation.Conflicts of interest
There are no conflicts to declare.References
- M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. technological use of CO2, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- H. R. Jhong, S. Ma and P. J. Kenis, Electrochemical conversion of CO2 to useful chemicals: current status, remaining challenges, and future opportunities, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- Y. Zheng, W. Zhang, Y. Li, J. Chen, B. Yu, J. Wang, L. Zhang and J. Zhang, Energy related CO2 conversion and utilization: advanced materials/nanomaterials, reaction mechanisms and technologies, Nano Energy, 2017, 40, 512–539 CrossRef CAS.
- S. Verma, B. Kim, H. R. M. Jhong, S. Ma and P. J. A. Kenis, A gross-margin model for defining technoeconomic benchmarks in the electroreduction of CO2, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS.
- T. Vo, K. Purohit, C. Nguyen, B. Biggs, S. Mayoral and J. L. Haan, Formate: An Energy Storage and Transport Bridge between Carbon Dioxide and a Formate Fuel Cell in a Single Device, ChemSusChem, 2015, 8, 3853–3858 CrossRef CAS PubMed.
- W. Supronowicz, I. A. Ignatyev, G. Lolli, A. Wolf, L. Zhao and L. Mleczko, Formic acid: a future bridge between the power and chemical industries, Green Chem., 2015, 17, 2904–2911 RSC.
- J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, in Ullmann's Encyclopedia of Industrial Chemistry, 2016 Search PubMed.
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Reversible interconversion of carbon dioxide and formate by an electroactive enzyme, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef CAS.
- X. Min and M. W. Kanan, Pd-Catalyzed Electrohydrogenation of Carbon Dioxide to Formate: High Mass Activity at Low Overpotential and Identification of the Deactivation Pathway, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- K. P. Sokol, W. E. Robinson, A. R. Oliveira, S. Zacarias, C. Y. Lee, C. Madden, A. Bassegoda, J. Hirst, I. A. C. Pereira and E. Reisner, Reversible and Selective Interconversion of Hydrogen and Carbon Dioxide into Formate by a Semiartificial Formate Hydrogenlyase Mimic, J. Am. Chem. Soc., 2019, 141, 17498–17502 CrossRef CAS PubMed.
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W. Bin Cai, X. Chen, R. Si, F. Yang, S. Miao, J. Wang, G. Wang and X. Bao, Switchable CO2 electroreduction via engineering active phases of Pd nanoparticles, Nano Res., 2017, 10, 2181–2191 CrossRef CAS.
- S. Sarkar and S. C. Peter, An overview on Pd-based electrocatalysts for the hydrogen evolution reaction, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC.
- R. Kortlever, I. Peters, S. Koper and M. T. M. Koper, Electrochemical CO2 Reduction to Formic Acid at Low Overpotential and with High Faradaic Efficiency on Carbon-Supported Bimetallic Pd–Pt Nanoparticles, ACS Catal., 2015, 5, 3916–3923 CrossRef CAS.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Exclusive Formation of Formic Acid from CO2 Electroreduction by a Tunable Pd–Sn Alloy, Angew. Chem., Int. Ed., 2017, 56, 12219–12223 CrossRef CAS PubMed.
- B. Jiang, X. G. Zhang, K. Jiang, D. Y. Wu and W. Bin Cai, Boosting Formate Production in Electrocatalytic CO2 Reduction over Wide Potential Window on Pd Surfaces, J. Am. Chem. Soc., 2018, 140, 2880–2889 CrossRef CAS.
- F. Zhou, H. Li, M. Fournier and D. R. MacFarlane, Electrocatalytic CO2 Reduction to Formate at Low Overpotentials on Electrodeposited Pd Films: Stabilized Performance by Suppression of CO Formation, ChemSusChem, 2017, 10, 1509–1516 CrossRef CAS PubMed.
- A. Klinkova, P. De Luna, C. T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, Rational Design of Efficient Palladium Catalysts for Electroreduction of Carbon Dioxide to Formate, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS.
- J. Su, L. Yang, M. Lu and H. Lin, Highly efficient hydrogen storage system based on ammonium bicarbonate/formate redox equilibrium over palladium nanocatalysts, ChemSusChem, 2015, 8, 813–816 CrossRef CAS PubMed.
- D. C. Engel, G. F. Versteeg and W. P. M. Van Swaaij, Chem. Eng. Res. Des., 1995, 73, 701–706 CAS.
- W. Yang, K. Dastafkan, C. Jia and C. Zhao, Design of Electrocatalysts and Electrochemical Cells for Carbon Dioxide Reduction Reactions, Adv. Mater. Technol., 2018, 3, 1–20 Search PubMed.
- S. Liang, N. Altaf, L. Huang, Y. Gao and Q. Wang, Electrolytic cell design for electrochemical CO2 reduction, J. CO2 Util., 2019, 35, 90–105 CrossRef.
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Effects of temperature and gas–liquid mass transfer on the operation of small electrochemical cells for the quantitative evaluation of CO2 reduction electrocatalysts, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC.
- F. Cai, D. Gao, H. Zhou, G. Wang, T. He, H. Gong, S. Miao, F. Yang, J. Wang and X. Bao, Electrochemical promotion of catalysis over Pd nanoparticles for CO2 reduction, Chem. Sci., 2017, 8, 2569 RSC.
- S. Y. Wu and H. T. Chen, CO2 Electrochemical Reduction Catalyzed by Graphene Supported Palladium Cluster: A Computational Guideline, ACS Appl. Energy Mater., 2019, 2, 1544–1552 CrossRef CAS.
- A. Lasia, Mechanism and kinetics of the hydrogen evolution reaction, Int. J. Hydrogen Energy, 2019, 44, 19484–19518 CrossRef CAS.
- E. R. Cave, J. H. Montoya, K. P. Kuhl, D. N. Abram, T. Hatsukade, C. Shi, C. Hahn, J. K. Nørskov and T. F. Jaramillo, Electrochemical CO2 reduction on Au surfaces: mechanistic aspects regarding the formation of major and minor products, Phys. Chem. Chem. Phys., 2017, 19, 15856–15863 RSC.
- Y. Li, S. Chen, R. Long, H. Ju, Z. Wang, X. Yu, F. Gao, Z. Cai, C. Wang, Q. Xu, J. Jiang, J. Zhu, L. Song and Y. Xiong, Near-surface dilution of trace Pd atoms to facilitate Pd–H bond cleavage for giant enhancement of electrocatalytic hydrogen evolution, Nano Energy, 2017, 34, 306–312 CrossRef CAS.
- J. Durst, C. Simon, F. Hasche and H. A. Gasteiger, Hydrogen oxidation and evolution reaction kinetics on carbon supported Pt, Ir, Rh, and Pd electrocatalysts in acidic media, J. Electrochem. Soc., 2015, 162, F190–F203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se00731e |
| This journal is © The Royal Society of Chemistry 2020 |