
New lead bromide chiral perovskites with ultra-broadband white-light emission†
Yalan
Liu‡
a,
Chao
Wang‡
a,
Yurong
Guo
a,
Linlin
Ma
a,
Chenyang
Zhou
a,
Ya
Liu
ab,
Lina
Zhu
a,
Xiaozeng
Li
a,
Mingxing
Zhang
ab and
Guangjiu
Zhao
 *a
*a
aTianjin Key Laboratory of Molecular Optoelectronic Sciences, National Demonstration Center for Experimental Chemistry & Chemical engineering Education, National Virtual Simulation Experimental Teaching Center for Chemistry & Chemical engineering Education, Department of Chemistry, School of Science, Tianjin University, Tianjin 300354, China. E-mail: gjzhao@tju.edu.cn
bState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 31st March 2020
Abstract
Herein, we present for the first time on the synthesis of two chiral perovskites with white-light emission and an achiral perovskite featuring a spectral blueshift. Moreover, the luminescence mechanism of these perovskites is investigated. The direct signals of the free excitons (FEs) and self-trapped excitons (STEs) were observed by performing time-resolved nanosecond transient absorption measurements for our perovskites. This directly verifies that the broad emission indeed originates from FE and STEs. The time-resolved transient emission curves manifest that the long-lived component is assigned to STEs transition and short-lived component is ascribed to FE transition. This study promotes the development of multifunctional applications of chiral white-light emissive perovskites.
Introduction
Organic–inorganic hybrid perovskites have drawn increasing attention for their promising applications in photovoltaic cells, light-emitting diodes (LEDs), photodetectors, and so on.1–9 Importantly, low-dimensional perovskites have a common broadband emission and large Stokes shifts, which is assigned to the existence of STEs and provides a probability of generating white-light emission.10–19 A single-component white-light emissive phosphor plays an important role in preparing high-quality white-light-emitting diodes (WLEDs), which is ascribed to resolving two problems of self-absorption and spectral shift.20–26 Therefore, significant efforts need to be made to obtain single-component white emissive low-dimensional hybrid perovskites.Meanwhile, the low-dimensional organic–inorganic hybrid perovskites feature the diversity of available ionic compositions and flexible structure, which makes it possible to achieve different optoelectronic properties via incorporating functional organic molecules into these perovskites.27–30 For example, Billing et al. synthesized one-dimensional (1D) chiral perovskites via the incorporation of chiral organic small molecules for the first time.31 Ma et al. reported the linear optical properties such as circularly polarized luminescence (CPL), and corresponding detection of chiral 2D perovskites.32 Xiong et al. reported the synthesis and ferroelectricity of a series of chiral perovskites.33 Current studies focus mostly on the linear optical properties,34,35 second harmonic generation (SHG),36–38 and electronic properties, including spintronics39 and ferroelectricity.40,41 However, to the best of our knowledge, there are no reports on the photoluminescence mechanism of low-dimensional hybrid perovskites with combined chirality and white-light emission.
Result and discussion
Here, we first reported the synthesis of chiral 2D perovskites (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 (β-MPEA = beta-methylphenethylamine, where S- and R-MPEA represent the molecules with opposite chirality) with ultra-broadband white-light emission. The schematic experimental operation of chiral perovskites is shown in Scheme 1a. Chiral perovskite crystals are synthesized via a solution method using chiral organic ligands, in which chiral cations are rationally introduced to achieve the spatial inversion asymmetry. We also synthesized achiral perovskite crystals ((rac)-MPEA)2PbBr4, (racemic beta-methylphenethylamine) using equal proportions of R-(+)- and S-(−)-β-MPEA as controls, further details are given in the ESI.†Scheme 1b displays the schematic structural illustration of chiral perovskite crystals. Two layers of chiral molecules sandwich a layer of corner-sharing [PbBr6]4− octahedral layer. Chiral organic cations in the perovskite crystal are held together by the van der Waals force and were stabilized here through π–π packing interactions. The inorganic layers and organic cations were connected by the hydrogen bonding of N–H⋯Br. (R-(+)-β-MPEA)2PbBr4 exhibits a white color and a needle-like shape (Fig. 1a), and emits white-light under the UV light of 365 nm (Fig. 1b). The scanning electron microscopy (SEM) image of (R-(+)-β-MPEA)2PbBr4 shows that the surface is smooth (Fig. 1c). To confirm the chemical composition of (R-(+)-β-MPEA)2PbBr4, we performed the energy-dispersive X-ray spectroscopy (EDS) measurement. As shown in Fig. S1 (ESI†), the C, N, Pb, and Br elements are distributed through the selected areas (marked by a square). The presence of the O element is assigned to the preparation in air. (S-(−)-β-MPEA)2PbBr4 and ((rac)-MPEA)2PbBr4 have similar properties (Fig. S2 and S3, ESI†). The X-ray diffraction (XRD) patterns of these as-synthesized perovskites exhibit sharp peaks with narrow full-width at half-maximum values, which further indicate that all of our perovskites have good crystallinity (Fig. 1d). We further investigated the thermal stability of these perovskite crystals. As shown in Fig. S4 (ESI†), the thermal gravimetric analysis (TGA) measurements reveal that our perovskite crystals exhibit thermal stability up to a decomposition temperature of approximately 220 °C.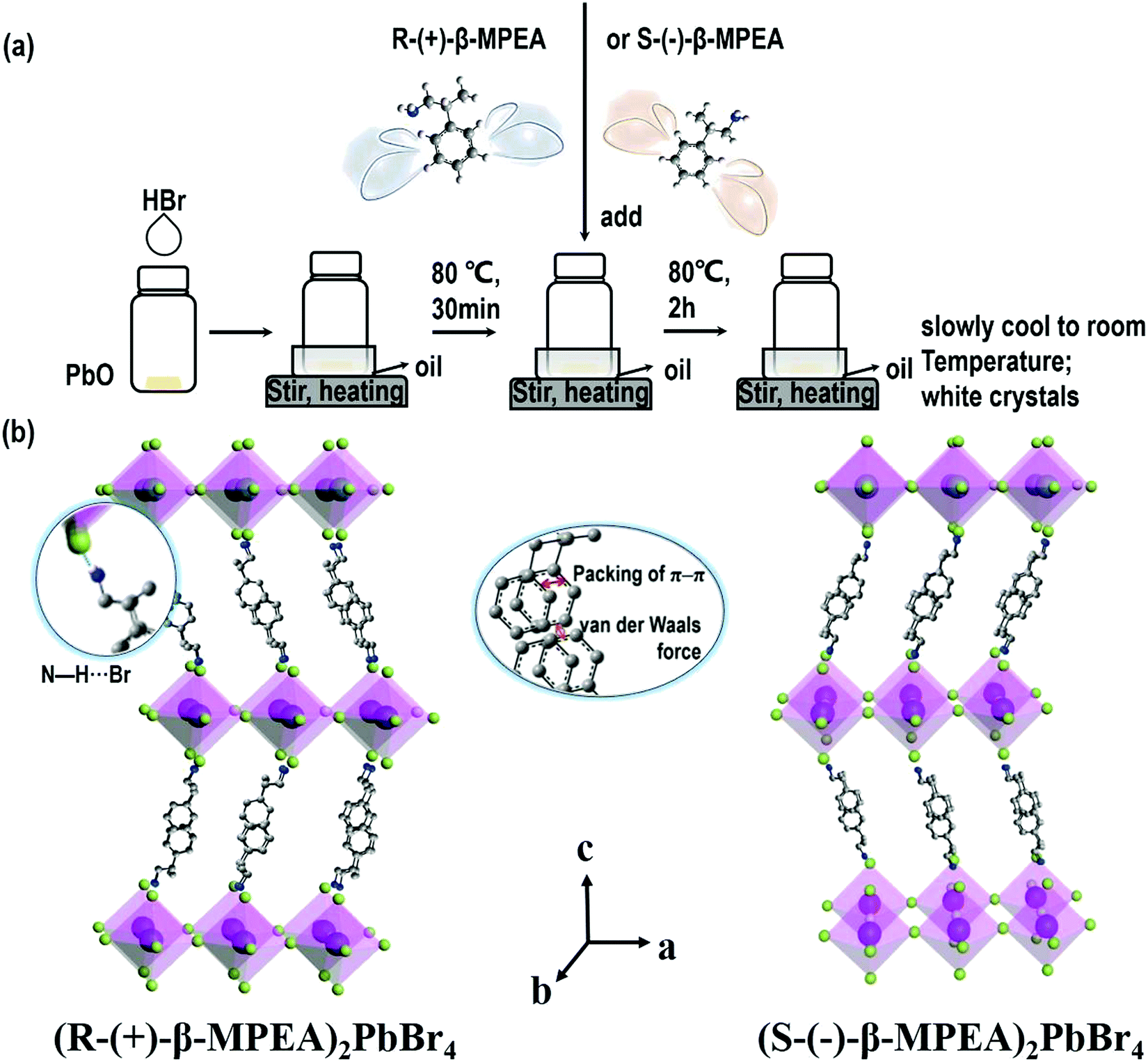 | ||
| Scheme 1 (a) The schematic experimental operation diagram. (b) The structural illustration of crystals of (R-(+)-β-and S-(−)-β-MPEA)2PbBr4 (yellow-green ball = bromide atom, sky-blue ball = lead atom, gray ball = carbon atom, deep-blue ball = nitrogen atom, and pink-white ball = hydrogen atom). | ||
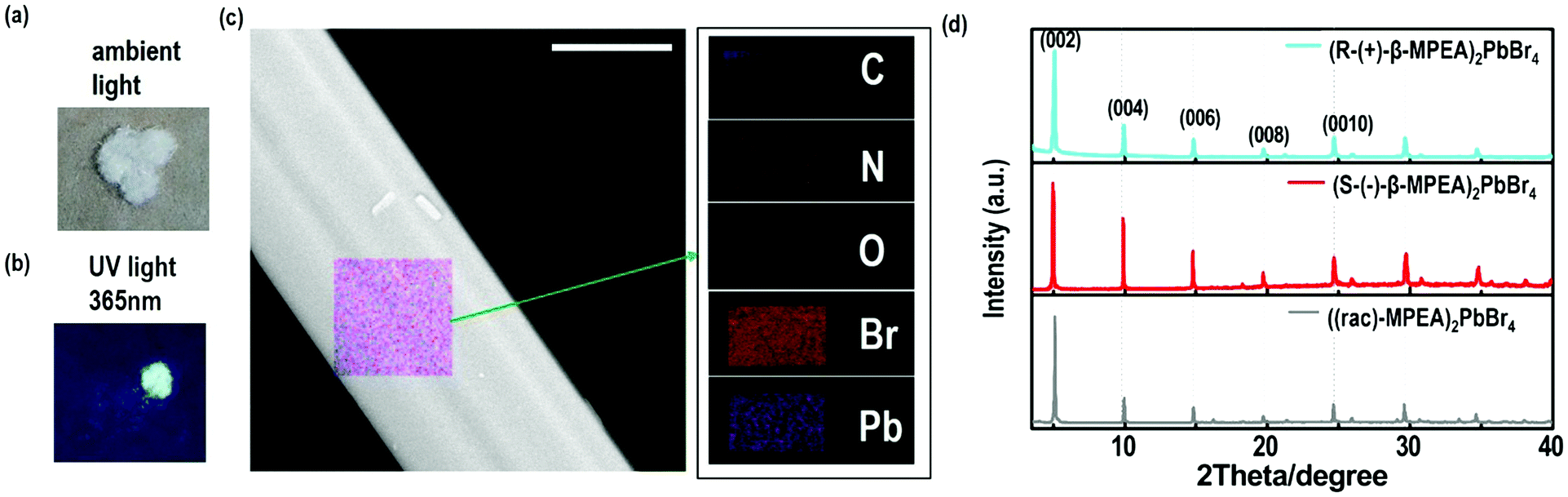 | ||
| Fig. 1 The optical image under (a) ambient light and (b) UV light (365 nm), (c) SEM image (scale bar is 5 μm) and element mapping images of (R-(+)-β-MPEA)2PbBr4. (d) The XRD patterns of perovskite crystals. | ||
To confirm the chirality of our perovskite crystals, we further carried out circular dichroism (CD). We measured CD using thin-films of these perovskites. Scheme S1 (ESI†) represents a spin-coating method to prepare thin-films. The as-formed perovskite crystals were spin-coated at 2000 rpm for 30 s on a clean quartz glass substrate with a 250 mg ml−1 precursor solution in dimethylformamide (DMF). After a heat treatment at 65 °C for 2 min, thin-films were formed (see the methods in the ESI†). The X-ray diffraction (XRD) patterns of the as-formed perovskite crystal thin-films are shown in Fig. 2a and Fig. S5 (ESI†). For comparison, the characteristic peak of the thin films appeared due to successful preparation. When the CD of the thin films was measured, (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 were found to have evident CD signals, while ((rac)-MPEA)2PbBr4 displayed a featureless flat signal (Fig. 3b). The peaks in the CD spectra of (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 are observed at the same position at 289 nm, 321 nm, 388 nm, and 398 nm. However, the peaks of (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 have opposite signs, as shown in Fig. 2b. The presence of the oppositely signed CD in (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 confirms that the incorporation of the chiral organic molecules can successfully endow perovskites with chirality. In detail, (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 do not show absolutely mirror-imaged CD signals with a weak dissymmetry factor. A comparison of the CD spectra and absorption spectra of the chiral films reveal that the CD peaks are located before the absorption band edge. The peak observed at 390 nm was extrapolated to 402 nm. It is found that the CD signal of (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 change the sign close to the absorption band edge. This phenomenon can be interpreted as the Cotton effect.42 Furthermore, the observed CD could be ascribed to the transition of electrons in the electronic bands, which in turn supported the observed Cotton effect. In addition, the changes in the CD signal in the wavelength region ranging from 250 to 325 nm can be attributed to additional excitonic transitions, whereas the CD signal in the longer wavelength region of the band edge actually originates from circular differential scattering according to the previous report.35 All of these findings support that chirality has been transferred from the solely organic cations to the overall (R-(+)-β-MPEA)2PbBr4 or (S-(−)-β-MPEA)2PbBr4 perovskite crystals, while chirality exists only in organic cations for ((rac)-MPEA)2PbBr4 perovskite crystals. These results contribute to further optical properties of chiral and achiral perovskites.
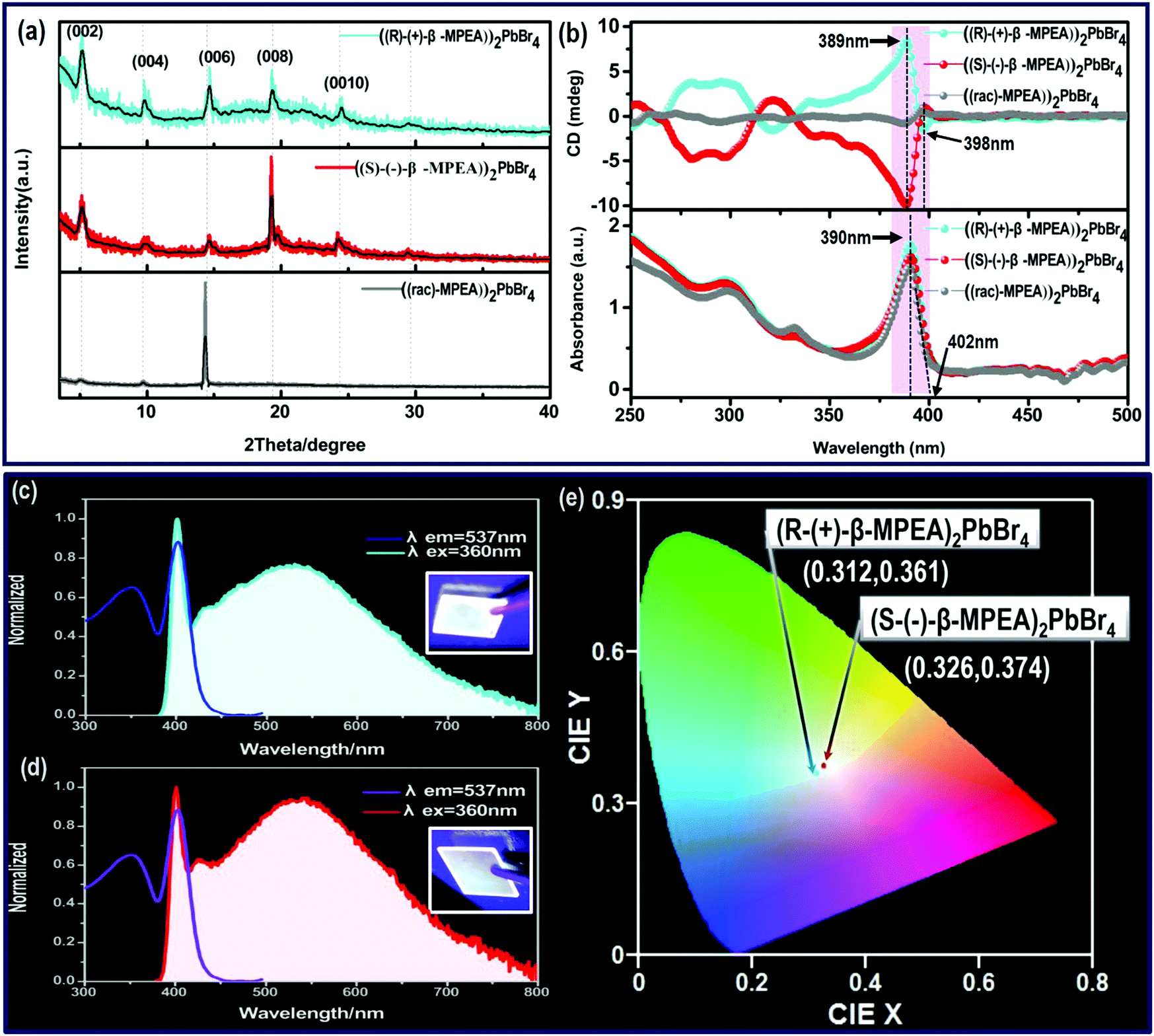 | ||
| Fig. 2 (a) The XRD patterns of chiral perovskite crystals (R-(+)-β-and S-(−)-β-MPEA)2PbBr4, (rac-MPEA)2PbBr4 film, (b) circular dichroism and steady-state absorption spectra of these perovskite crystals film. (c) PL and PLE spectra of (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 film (λex = 360 nm, λem = 537 nm), (d) PL and PLE spectra of (S-(−)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 film (λex = 360 nm, λem = 537 nm), (e) CIE coordinated of the emissions of (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 film. | ||
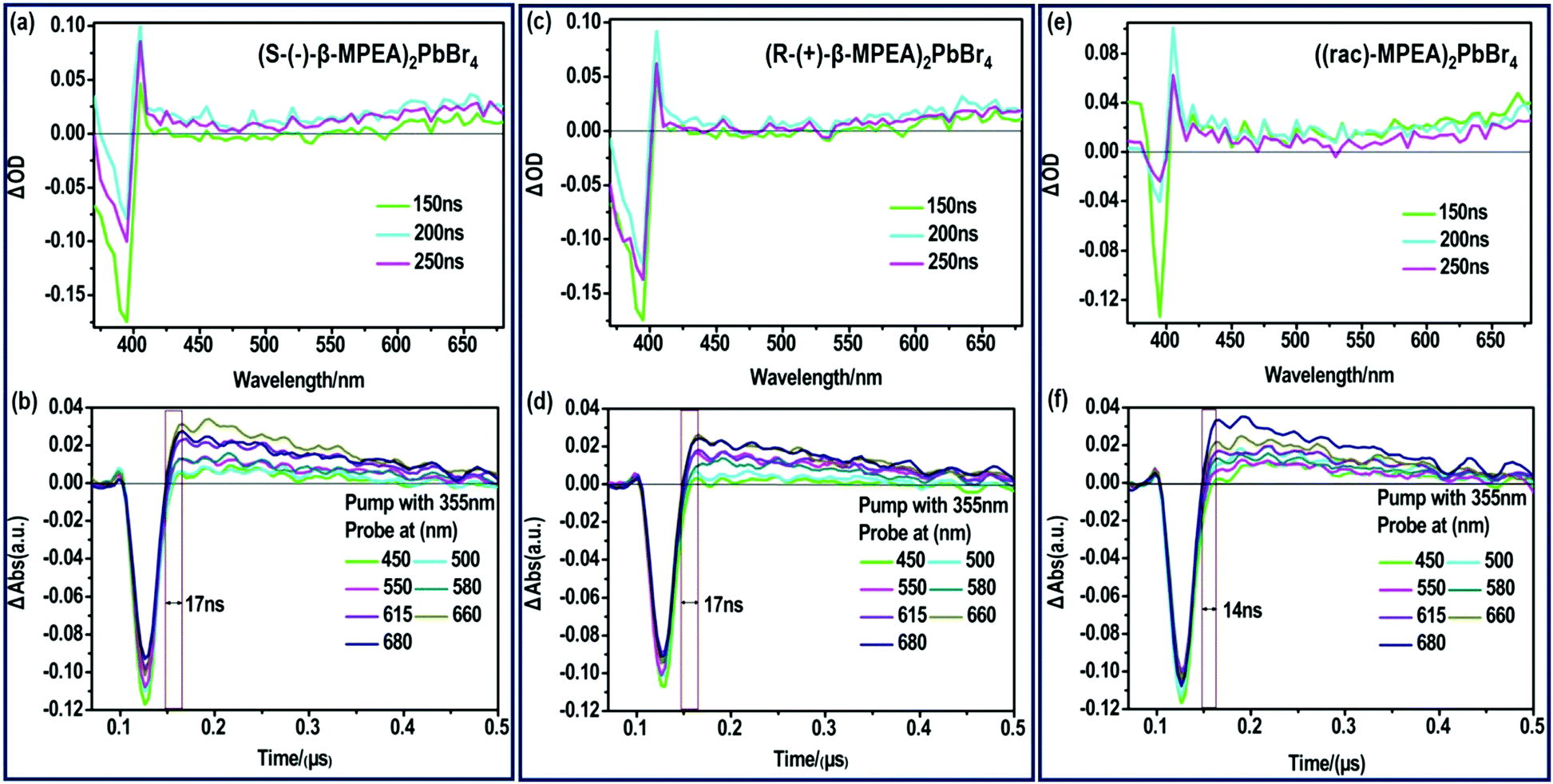 | ||
| Fig. 3 ns-TA spectra of (a) (S-β-MPEA)2PbBr4, (c) (R-β-MPEA)2PbBr4, (e) ((rac)-MPEA)2PbBr4 films upon excitation at 355 nm. TA onsets of (b) (S-β-MPEA)2PbBr4 (d) (R-β-MPEA)2PbBr4, (f) ((rac)-MPEA)2PbBr4 films probed at different wavelength. | ||
Next, the photophysical properties of the perovskite system were measured via photoluminescence excitation (PLE) and PL emission spectra. As shown in Fig. 2c and d, (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 both exhibit ultra-broadband emission spectrum ranging from 385 nm to 800 nm, and two emission peaks: a narrow emission located at 402 nm and a broad emission centered at 535 nm with an excitation wavelength of 360 nm. The origin of the unusual dual-peak emission is ascribed to free exciton (FE) and self-trapped exciton (STE) recombination, which will be discussed in detail in the later analysis of the ultra-fast spectra. Compared with (R-(+)-β-MPEA)2PbBr4, the intensity of the 402 nm emission of (S-(−)-β-MPEA)2PbBr4 remains unchanged, while the intensity of the 537 nm emission of (S-(−)-β-MPEA)2PbBr4 increased. Therefore, (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4 perovskite exhibit white-light emissions with the CIE chromaticity coordinates of (0.312, 0.361) and (0.326, 0.374), respectively, as shown in Fig. 2e. Meanwhile, both chiral perovskite films could emit white-light under UV light photoexcitation of 365 nm, as shown in Fig. 2a and b inset. Two chiral perovskites have a photoluminescence quantum yield (PLQY) of approximately 1%. According to the previous reports,43–45 the low-dimensional perovskites, particularly 1D or 2D perovskites emitting from both FE and STE excited state, rarely achieve high PLQY due to electronic band formation and aggregation-induced self-quenching in the solid-state. It is not difficult to understand that two chiral perovskites have a low PLQY. In addition, the PL spectrum of (rac-MPEA)2PbBr4 perovskite exhibits a non-Gaussian distribution emission located at 408 nm, as shown in Fig. S6 (ESI†). Compared with chiral perovskites, ((rac)-MPEA)2PbBr4 emits much stronger light with CIE chromaticity coordinates of (0.260, 0.290) and has a much higher PLQY of 5%. Therefore, the difference between R- and S-configure chiral cations may change the weak interactions existing in the perovskite crystal structure such as the hydrogen bonding and van der Waals force, which results in the optical difference, as shown in the CIE chromaticity coordinate and PLQY. Table S1 (ESI†) lists some reported PLQY and CIE chromaticity coordinates for white-light-emitting perovskites. Through comparison, the PLQY for our white-light-emitting perovskites is comparable and not particularly low. Besides, the presence of the same PbBr42− frameworks and cross-sectional aromatic area indicates a similar structure and is further attributed to optical similarity, as shown in the absorption spectra.
To further verify that the ultra-broadband emission of the perovskites originates from FE and STE, we subsequently performed nanosecond time-resolved transient absorption (ns-TA) spectroscopy to directly observe the signal of FE and STE, as presented in Fig. 3. The ns-TA spectra of (S-(−)-β-MPEA)2PbBr4, (R-(+)-β-MPEA)2PbBr4, and ((rac)-MPEA)2PbBr4 are shown in Fig. 3a, b, c, d, e and f, respectively. The ns-TA spectroscopy was carried out by conforming to the above-excitonic-peak excitation at 355 nm with pulse energy of 7 mJ. The characteristic bleach peak of 395 nm and a remarkable broad absorption with energy below that of excitons are observed in Fig. 4a–c, which support that the emission of our perovskites originates from FE and STE. The same position of the bleach peak exhibits similar exciton absorption for these perovskites, which is assigned to the presence of the same PbBr42− frameworks. In addition, it is reported that the formation time of STE demonstrates itself in the rise time of the observed induced absorption.10,12,46–50 The formation time of STE in (rac-MPEA)2PbBr4 is shorter than that of two chiral perovskites (Fig. 3b, d and f), which show a low potential barrier between the FE and STE states for ((rac)-MPEA)2PbBr4. (S-(−)-β-MPEA)2PbBr4 and (R-(+)-β-MPEA)2PbBr4 have the same formation time of STE, which indicates that there was no difference in the potential energy barrier between the FE and STE states for the two perovskites. To further comprehend the photoluminescence mechanism, we performed a Gaussian curve fitting and nanosecond time-resolved PL emission (ns-TE) spectroscopy.
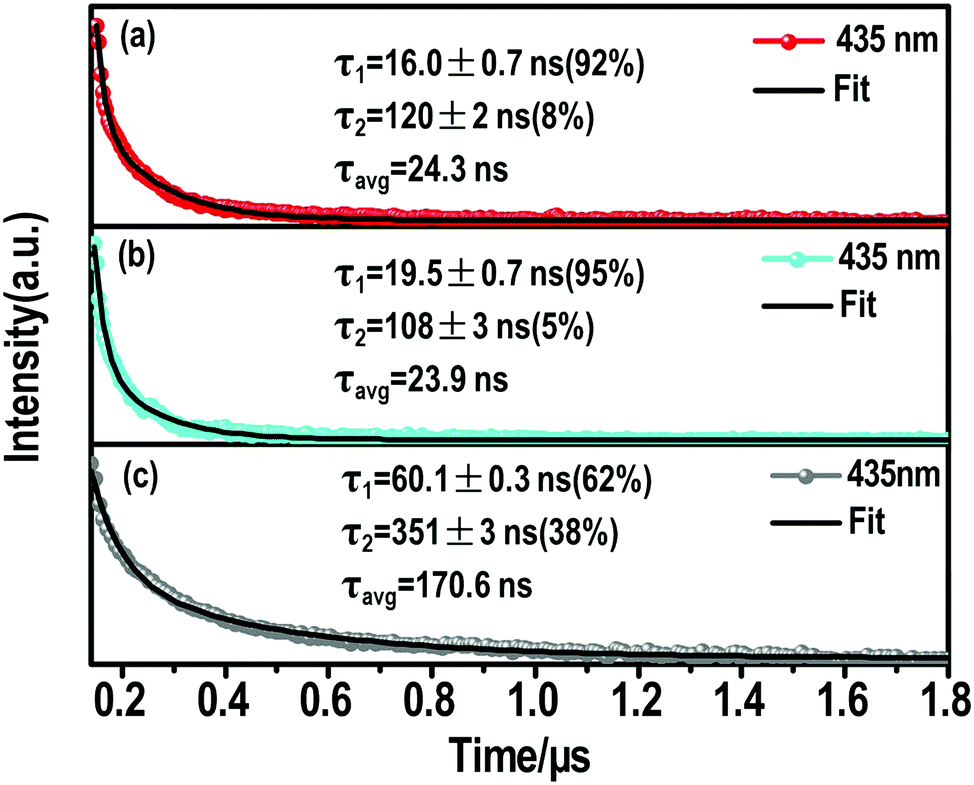 | ||
| Fig. 4 Nanosecond time-resolve emission (ns-TE) decays and fittings of the (a) (S-β-MPEA)2PbBr4, (b) (R-β-MPEA)2PbBr4, and (c) ((rac)-MPEA)2PbBr4 emission at 435 nm. | ||
The ns-TE spectra and Gaussian curve fitting of the PL spectra of these perovskites are shown in Fig. S6, ESI† and Fig. 4. These Gaussian peaks were found to give the optimal fit to the corresponding spectrum in Fig. S7a (ESI†). Compared with the chiral perovskites, the distribution of high-energy peaks increased for ((rac)-MPEA)2PbBr4. We assume that the self-depth of ((rac)-MPEA)2PbBr4 may be smaller than chiral perovskites. The comparison of ns-TE with the corresponding reverse-normalized PL spectra of three perovskites is shown in Fig. S7b–d (ESI†). The distribution of high-energy peaks in ((rac)-MPEA)2PbBr4 increases, while the distribution of low-energy peaks decreases, in accordance with the Gaussian curve fitting analysis. This further supports the previous assumption for the self-trapped depth. Another remarkable observation is that there is a common maximum emission peak at 435 nm. For all three perovskites, the PL decay monitored at 435 nm can be satisfactorily fitted with a biexponential decay (Fig. 4). The fast decay process (τ1) corresponds to the FE transition, while the slower decay process (τ2) is assigned to the STE transition. More explicitly, for (S-(−)-β-MPEA)2PbBr4, a biexponential decay has a 16 ns component (92%) and a 120 ns component (8%) in Fig. 4a. For (R-(+)-β-MPEA)2PbBr4, a biexponential decay has a 19.5 ns component (95%) and a 108 ns component (5%) in Fig. 4b. For ((rac)-MPEA)2PbBr4, a biexponential decay has a 60.1 ns component (62%) and a 351 ns component (38%) in Fig. 4c. The proportion of the STE component for (R-(+)-β-MPEA)2PbBr4 decrease in contrast to that of (S-(−)-β-MPEA)2PbBr4. The ns-TA data analysis shows the same potential barrier between FE and STE for (R-(+)-β-MPEA)2PbBr4 and (S-(−)-β-MPEA)2PbBr4. Both results manifest that some different defects tune radiative or nonradiative transitions and further change luminescence. Some defects that promote nonradiative transition between STE and ground state (GS) existed in (R-(+)-β-MPEA)2PbBr4, which is consistent with the decrease in the long-lived STE emission component. In addition, the significant increase in the long-lived component proportion for ((rac)-MPEA)2PbBr4 suggests that the transition from STEs to FEs has a low potential energy barrier, and more FEs were trapped to generate STEs, in accordance with the result of the ns-TA measurement. According to the previous reports,51,52 the inorganic and two kinds of chiral organic components couple to electronic subsystem and induce much rapid coherence loss for ((rac)-MPEA)2PbBr4. The rapid loss of coherence also explains the significant increase in the long-lived component proportions. Meanwhile, the PLQY of ((rac)-MPEA)2PbBr4 was much higher than that of chiral perovskites, which is ascribed to the removal of some defects that are in favor of nonradiative transition.
The photophysical mechanism of the ultra-broadband emission in the perovskite systems can be depicted, as shown in Fig. 5, based on the above discussions. Detail follows: upon photoexcitation, electrons in the GS were motivated to the FE state for the chiral perovskite systems. Some FEs can return to GS via radiative or nonradiative transition forms directly. Some FEs were trapped by the STE states to further produce the STE emission or non-radiative transition. Some FEs trapped by the STE states undergo back energy transfer to return to the FE state first, and then return to GS after emitting light. All the transitions happened in the chiral system simultaneously. The presence of chirality can generate some defects, which is in favor of nonradiative transitions (particularly STE nonradiative transition). It is not difficult to understand that the chiral perovskites have a PLQY of approximately 1% and lack a long-lived component. Compared with the chiral perovskites, the achiral perovskite has a low energy barrier between FE and STE and a higher proportion of the long-lived components, both manifesting that the transitions between the FE state and STE state are not restricted. The luminescence of achiral perovskite exhibits a blueshift. Moreover, the high energy emission peaks of the achiral perovskite increased. Both of them indicate that the self-trapped depth of the achiral perovskite was smaller than that of chiral perovskites, as shown in Fig. 5. In addition, the disappearance of chirality may remove some defects, which promotes the radiative transition. These all support that the PLQY of the achiral perovskite was much higher than that of two chiral perovskites. Finally, the ultra-broadband emission of our perovskites indeed originates from FE and STE.
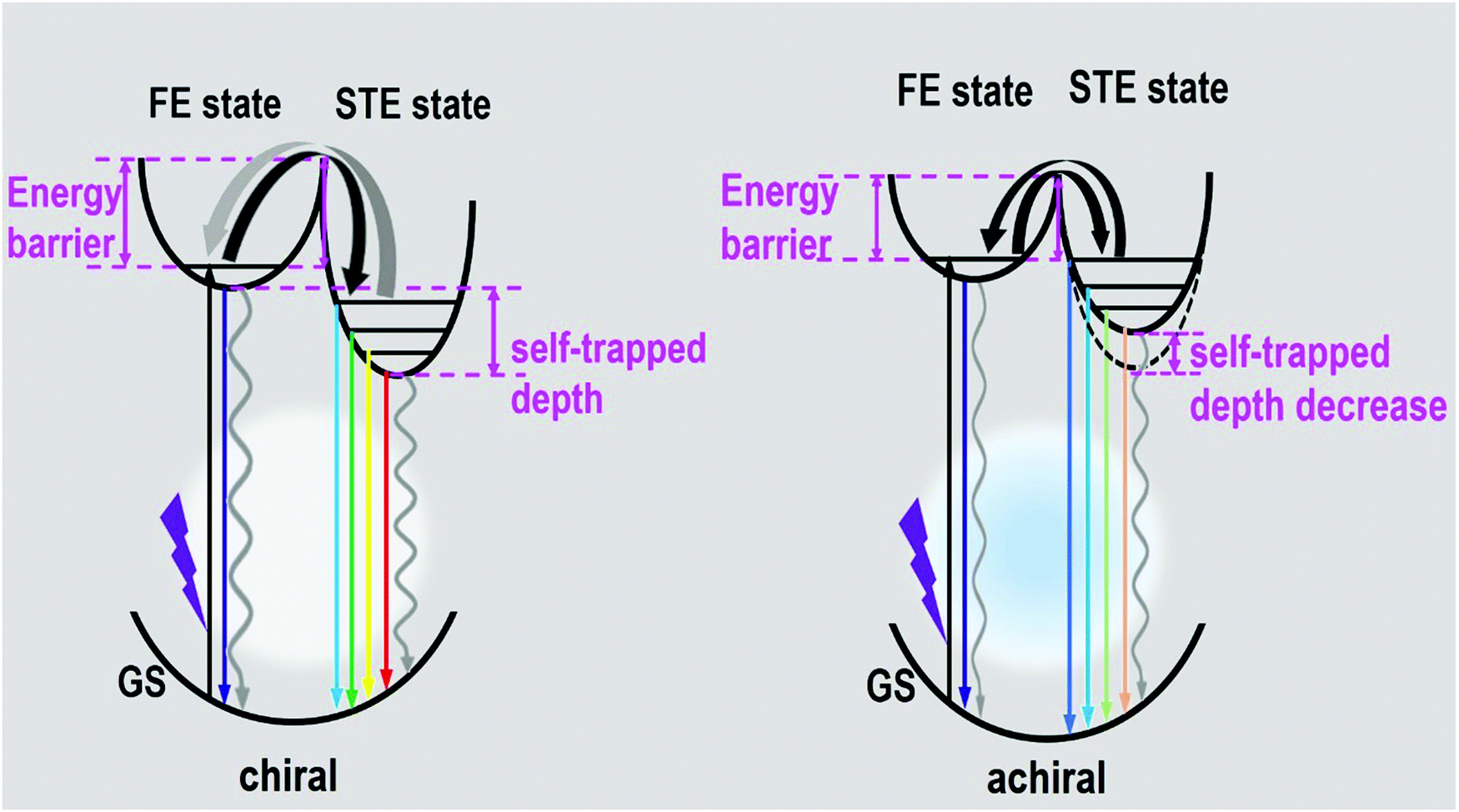 | ||
| Fig. 5 Diagram of luminescence processes in the as-formed perovskite crystals. (GS: ground state, FE state: free-exciton state, STE state: self-trapped exciton state.) | ||
Conclusions
In summary, we synthesized two chiral perovskites with broadband white-light emission and a remarkable CD signal, and an achiral perovskite ((rac)-MPEA)2PbBr4 with a featureless flat CD signal and spectral slight blue shift. The difference between R- and S-configure chiral cations may change the weak interaction existing in the perovskite crystal structure, which results in the optical difference, as shown in the luminescence and PLQY. In addition, the presence of the same PbBr42− frameworks and cross-sectional aromatic area suggests a similar structure and is further attributed to optical similarity, as shown in the absorption spectra. Moreover, the overall photoluminescence mechanism of these perovskites was formulated by a comprehensive analysis of the steady-state spectra and ultra-fast spectra. The direct signal of FE and STE was observed in the ns-TA spectra, which verifies that the ultra-broadband emission of our perovskites indeed originated from FE and STE. Meanwhile, the ns-TA spectra show that the formation time of STE in the achiral perovskite was shorter than that of chiral perovskites, which reveals a low potential barrier between the FE and STE states and the decrease in the self-trapped depth in the achiral perovskite. The ns-TE curves further manifest that the long-lived component is assigned to the STE transition, and the short-lived component is ascribed to the FE transition. These all support our luminescence mechanism. Thus, this study not only provides a new design by combining white-light emission and chirality but also promotes the development of the multifunctional application of white-light emissive perovskite materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 21573229, 21873068, and 21422309). G. J. Z. also thanks the financial support from the Frontier Science Project of the Knowledge Innovation Program of Chinese Academy of Science (CAS), Project for Excellent Member of CAS Youth Innovation Promotion Association, the Open Research Funds of State Key Laboratory of Bioelectronics (Southeast University) and Double First-Rate and Peiyang Scholar Projects (Tianjin University).Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed
- S. T. Ha, X. Liu, Q. Zhang, D. Giovanni, T. C. Sum and Q. Xiong, Adv. Opt. Mater., 2017, 2, 838–844 CrossRef
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed
- N. Wang, L. Cheng, R. Ge, S. Zhang, Y. Miao, W. Zou, C. Yi, Y. Sun, Y. Cao, R. Yang, Y. Q. Wei, Q. Guo, Y. Ke., M. T. Yu, Y. Z. Jin, Y. Liu, Q. Q. Ding, D. W. Di, L. Yang, G. C. Xing, H. Tian, C. H. Jin, F. Gao, R. H. Friend, J. P. Wang and W. Huang, Nat. Photonics, 2016, 10, 699–704 CrossRef CAS
- H. L. Wang, F. Yang, Y. R. Xiang, S. A. Ye, X. Peng, J. Song, J. L. Qu and W. Y. Wong, J. Mater. Chem. A, 2019, 7, 24191–24198 RSC
- B. Yang, J. S. Chen, Q. Shi, Z. J. Wang, M. Gerhard, A. Dobrovolsky, I. G. Scheblykin, K. J. Karki and K. L. Han, J. Phys. Chem. Lett., 2018, 8, 5017–5022 CrossRef PubMed
- Y. Cheng, Z. F. Shi, S. T. Yin, Y. li, S. Li, W. Q. Liang, D. Wu, Y. T. Tian and X. J. Li, Sol. Energy Mater. Sol. Cells, 2020, 204, 110230 CrossRef CAS
- A. Abate, M. Planells, D. J. Hollman, V. Barthi, S. Chand, H. J. Snaith and N. Robertson, Phys. Chem. Chem. Phys., 2015, 17, 2335–2338 RSC
- Y. Li, Z. F. Shi, L. Z. Lei, S. Li, D. W. Yang, T. T. Xu, Y. Z. Tian, Y. G. Lu, Y. Wang, L. J. Zhang, X. J. Li, Y. T. Zhang, G. T. Du and C. X. Shan, Adv. Mater. Interfaces, 2019, 6, 1900188 CrossRef
- Z. Z. Ma, Z. F. Shi, C. C. Qin, M. H. Cui, D. W. Yang, X. J. Wang, L. T. Wang, X. Z. Ji, J. L. Sun, D. Wu, Y. Zhang, X. J. Li, L. J. Zhang and C. X. Shan, ACS Nano, 2020 DOI:10.1021/acsnano.9b10148
- T. Hu, M. D. Smith, E. R. Dohner, M. J. Sher, X. Wu, M. T. Trinh, A. Fisher, J. Corbett, X. Y. Zhu, H. I. Karunadasa and A. M. Lindenberg, J. Phys. Chem. Lett., 2016, 7, 2258–2263 CrossRef CAS PubMed
- M. G. Ju, J. Dai, L. Ma, Y. Y. Zhou and X. C. Zeng, J. Am. Chem. Soc., 2018, 140, 10456–10463 CrossRef CAS PubMed
- M. D. Smith and H. I. Karunadasa, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS
- Z. Yuan, C. K. Zhou, Y. Tian, Y. Shu, J. Messier, J. C. Wang, L. J. van de Burgt, K. Kountouriotis, Y. Xin, E. Holt, K. Schanze, R. Clark, T. Siegrist and B. W. Ma, Nat. Commun., 2017, 8, 14051 CrossRef CAS PubMed
- R. T. Williams and K. S. Song, J. Phys. Chem. Solids, 1990, 51, 679–716 CrossRef CAS
- L. L. Mao, P. J. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, R. D. Schaller, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 13078–13088 CrossRef CAS PubMed
- K. M. Mccall, C. C. Stoumpos, S. S. Kostina, M. G. Kanatzidis and B. W. Wessels, Chem. Mater., 2017, 29, 4129–4145 CrossRef CAS
- Y. L. Liu, C. Wang, C. Y. Zhou, P. Li, L. N. Zhu, S. Q. Sun, X. Feng, Y. Sun and G. J. Zhao, J. Lumin., 2020, 221, 117045 CrossRef CAS
- J. S. Kim, P. E. Jeon, Y. H. Park, J. C. Choi, H. L. Park, G. C. Kim and T. W. Kim, Appl. Phys. Lett., 2004, 85, 3696–3698 CrossRef CAS
- C. C. Lin and R.-S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268–1277 CrossRef CAS PubMed
- Q.-Y. Yang and J.-M. Lehn, Angew. Chem., Int. Ed., 2014, 53, 4572–4577 CrossRef CAS PubMed
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed
- Z. Yuan, C. Zhou, J. Messier, Y. Tian, Y. Shu, J. Wang, Y. Xin and B. W. Ma, Adv. Opt. Mater., 2016, 4, 2009–2015 CrossRef CAS
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao and Q. Dong, Nature, 2018, 563, 541 CrossRef CAS
- S. Yang, Z. Lin, J. Wang, Y. Chen, Z. Liu, E. Yang, J. Zhang and Q. Ling, ACS Appl. Mater. Interfaces, 2018, 10, 15980–15987 CrossRef CAS PubMed
- D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1, 1–12 RSC
- Y. Zhou, Z. J. Yong, K. C. Zhang, B. M. Liu, Z. W. Wang, J. S. Hou, Y. Z. Fang, Y. Zhou, H. T. Sun and B. Song, J. Phys. Chem. Lett., 2016, 7, 2735–2741 CrossRef CAS PubMed
- C. M. Weidman, M. Seitz, S. D. Stranks and W. A. Tisdale, ACS Nano, 2016, 10, 7830–7839 CrossRef PubMed
- W. Li, Z. Wang, F. Deschler, S. Gao, R. H. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2, 16099 CrossRef
- D. G. Billing and A. Lemmerer, CrystEngComm, 2006, 8, 686–695 RSC
- J. Ma, C. Fang, C. Chen, L. Jin, J. Wang, S. Wang, J. Tang and D. H. Li, ACS Nano, 2019, 13, 3659–3665 CrossRef CAS PubMed
- H.-Y. Ye, Y.-Y. Tang, P.-F. Li, W.-Q. Liao, J.-X. Gao, X.-N. Hua, H. Cai, P.-P. Shi, Y.-M. You and R.-G. Xiong, Science, 2018, 361, 151–155 CrossRef CAS PubMed
- W. J. Chen, S. Zhang, M. H. Zhou, T. H. Zhao, X. J. Qin, X. F. Liu, M. H. Liu and P. F. Duan, J. Phys. Chem. Lett., 2019, 10, 3290–3295 CrossRef CAS PubMed
- J. Ahn, E. Lee, J. Tan, W. Yang, B. Kim and J. Moon, Mater. Horiz., 2017, 4, 851–856 RSC
- Y. Peng, Y. P. Yao, L. N. Li, Z. Y. Wu, S. S. Wang and J. H. Luo, J. Mater. Chem. C, 2018, 6, 6033–6037 RSC
- J. Wang, Y. Mi, X. Gao, J. Z. Li, S. G. Lan, C. Fang, H. Z. Shen, X. L. Wen, R. Chen, X. F. Liu, T. C. He and D. H. Li, Adv. Opt. Mater., 2019, 1900398 CrossRef
- C. Yuan, X. Li, S. Semin, Y. Feng, T. Rasing and J. Xu, Nano Lett., 2018, 18, 5411–5417 CrossRef CAS PubMed
- G. Long, C. Jiang, R. Sabatini, Z. Yang, M. Wei, L. N. Quan, Q. Liang, A. Rasmita, M. Askerka, G. Walters, X. Gong, J. Xing, X. Wen, R. Quintero-Bermudez, H. Yuan, G. Xing, X. R. Wang, D. Song, O. Voznyy, M. Zhang, S. Hoogland, W. Gao, Q. Xiong and E. H. Sargent, Nat. Photonics, 2018, 12, 528–533 CrossRef CAS
- C.-K. Yang, W.-N. Chen, Y.-T. Ding, J. Wang, W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Z.-X. Wang and R.-G. Xiong, Adv. Mater., 2019, 31, 1808088 CrossRef PubMed
- Y. Ai, X.-G. Chen, P.-P. Shi, Y.-Y. Tang, P.-F. Li, W.-Q. Liao and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 4474–4479 CrossRef CAS PubMed
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 21, 3885–3896 RSC
- W. W. Meng, X. M. Wang, Z. W. Xiao, J. B. Wang, D. B. Mitzi and Y. F. Yan, J. Phys. Chem. Lett., 2017, 8, 2999–3007 CrossRef CAS PubMed
- J. J. Luo, S. R. Li, H. D. Wu, Y. Zhou, Y. Li, J. Li, J. Liu, J. H. Li, K. H. Li, F. Yi, G. D. Niu and J. Tang, ACS Photonics, 2018, 5, 398–405 CrossRef CAS
- J. J. Luo, X. M. Wang, S. R. Li, J. Liu, Y. M. Guo, G. D. Niu, L. Yao, Y. H. Fu, L. Gao, Q. S. Dong, C. Y. Zhao, M. Y. Leng, F. S. Ma, W. X. Liang, L. D. Wang, S. Y. Jin, J. B. Han, L. J. Zhang, J. Etheridge, J. B. Wang, Y. F. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563 Search PubMed
- A. Ben-Moshe, A. Teitelboim, D. Oron and G. Markovich, Nano Lett., 2016, 16, 7467–7473 CrossRef CAS PubMed
- R. L. Zhang, X. Mao, Y. Yang, S. Q. Yang, W. Y. Zhao, T. Wumaier, D. H. Wei, W. Q. Deng and K. L. Han, Angew. Chem., Int. Ed., 2019, 58, 2725–2729 CrossRef CAS PubMed
- Y. Peng, L. N. Li, C. M. Ji, Z. Y. Wu, S. S. Wang, X. T. Liu, Y. P. Yao and J. H. Luo, J. Am. Chem. Soc., 2019, 141, 12197–12201 CrossRef CAS PubMed
- C. Wang, Y. Liu, X. Feng, C. Y. Zhou, Y. L. Liu and G. J. Zhao, Angew. Chem., Int. Ed., 2019, 58, 11642–11646 CrossRef CAS PubMed
- M. Z. Li, J. Zhou, G. J. Zhou, M. S. Molokeev, J. Zhao, V. Morad, M. V. Kovaleko and Z. G. Xia, Angew. Chem., Int. Ed., 2019, 58, 18670–18675 CrossRef CAS PubMed
- Z. S. Zhang, W. H. Fang, M. V. Tokina, R. Long and O. V. Prezhdo, Nano Lett., 2018, 18, 2459–2466 CrossRef CAS PubMed
- L. Liu, W.-H. Fang, R. Long and O. V. Prezhdo, J. Phys. Chem. Lett., 2018, 9, 1164–1171 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc00881h |
| ‡ These authors are equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |