
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochromism in Ruddlesden–Popper copper-based perovskites: a light-induced change of coordination number at the surface†
B. G. H. M.
Groeneveld
,
H.
Duim
,
S.
Kahmann
 ,
O.
De Luca
,
E. K.
Tekelenburg
,
M. E.
Kamminga
,
L.
Protesescu
,
G.
Portale
,
G. R.
Blake
,
P.
Rudolf
and
M. A.
Loi
*
,
O.
De Luca
,
E. K.
Tekelenburg
,
M. E.
Kamminga
,
L.
Protesescu
,
G.
Portale
,
G. R.
Blake
,
P.
Rudolf
and
M. A.
Loi
*
Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, Groningen 9747 AG, The Netherlands. E-mail: m.a.loi@rug.nl
First published on 8th September 2020
Abstract
Ruddlesden–Popper organic–inorganic hybrid copper-based perovskites have been studied for decades owing to a variety of interesting properties, such as thermochromism and piezochromism, and the mechanisms behind these phenomena have been explained. Another possible property of these materials that has seldomly been investigated is photochromism. In this work, the photochromic properties of bis(phenethylammonium) tetrachlorocuprate (also known as phenethylammonium copper chloride) are reported for the first time. This material has attracted scientific interest owing to the fact that it shows both ferroelectric and ferromagnetic behavior. This work highlights the difference in stability between two Ruddlesden–Popper copper-based perovskites – with phenethylammonium (PEA) or methylammonium (MA) as the cations – during external stimuli. Various techniques, such as Raman and X-ray photoelectron spectroscopy, and grazing-incidence wide-angle X-ray scattering, combined with optical studies, were used to investigate the underlying photochemical processes at a molecular level. It is found that for the PEA compound, ultraviolet illumination causes a color change from yellow to brown. This is the result of two independent events, namely a Cu2+ reduction reaction and a transition from an octahedral copper-chloride structure to square-planar CuCl42−. After illumination, the material (brown color) is unstable in air, which is evident from a color change back to yellow. Interestingly, the similar compound bis(methylammonium) tetrachlorocuprate does not display photochromic behavior, which is attributed to the different nature of the two organic cations.
Introduction
The family of Ruddlesden–Popper organic–inorganic hybrid perovskites with the structural formula A2MX4 – where A is an organic cation with an ammonium group, M a metal anion (e.g., Cu2+, Pb2+, Sn2+, Ge2+, Mn2+, Eu2+) and X a halide anion – encompasses a large selection of materials, each with unique properties.1 The halides bind to the metal ions in the form of MX6 octahedra, which are connected by corner-sharing the four in-plane halide ions.2 The organic cations (e.g., methylammonium, butylammonium or phenethylammonium) are located in the voids between the octahedra and interact with them via Coulomb interaction and hydrogen bonding, forming an alternating layered two-dimensional structure.1 Depending on the structure of the organic cation, the inorganic octahedral framework might not form. An example is given by the difference in structure between bis(phenethylammonium) tetrabromocuprate, (C6H5C2H4NH3)2CuBr4, and bis[methyl(2-phenethyl)ammonium] tetrabromocuprate, (C6H5C2H4NH2CH3)2CuBr4. The former adopts the octahedral perovskite structure, but the latter has isolated CuBr42− anions; the steric hindrance of the methyl group prevents the formation of a perovskite structure because it hinders the formation of hydrogen bonds. The CuBr42− structure allows for more space for the organic cation, thereby enabling hydrogen bonding.2,3 Typical geometries for these CuX42− ions are square-planar (D4h symmetry) or distorted tetrahedral (D2d symmetry) (see Fig. S1 (ESI†) for a schematic overview of these geometries).4 The difference between these is caused by the strength of the hydrogen bonding of the cation with the halides, where stronger binding leads to a more square-planar character.5The variety in structures for copper halide phases, combined with the presence of Jahn–Teller effects in these d9 metal compounds, leads to interesting chemical effects. For instance, the distortion of the CuX42− tetrahedral structure and the elongation of certain Cu–X bonds in CuX6 octahedra can be attributed to the Jahn–Teller effect.6 Of particular interest are the copper chloride complexes. These were studied for their piezo- and thermochromism, i.e. the change of color upon pressure and temperature variation, respectively.5,7–15 Their piezochromism is explained by the tilting of the octahedra and reduction of the Jahn–Teller distortion by compressing the long Cu–Cl bond.7,8 Thermochromism often involves the change in coordination geometry from (nearly) square-planar CuCl42− at low temperature to a distorted tetrahedron at higher temperatures, due to the decreased strength of the hydrogen bonding.5,9,10,12,15 Riley et al. found that the color change in the thermochromic bis(piperazinium) tetrachlorocuprate could also be induced by laser illumination, but could not establish whether this was really caused by photoexcitation (photochromism) or by local heating due to the laser light.9
The work of Pan et al. demonstrated a photochromic effect in (C4H9NH3)2CuCl4, where a yellow film turned brown after illumination with UV light.16 They found little evidence for significant structural changes – infrared spectroscopy indicated a variation in the vibration of the N–H⋯Cl bond and the long Cu–Cl bond – and attributed the effect to a change in charge distribution of the dissymmetric Cl–Cu⋯Cl bond. In light of the importance of structural changes in the thermochromism described above, we find this conclusion not fully satisfying. In this work we investigated the photochromic properties of a similar material: bis(phenethylammonium) tetrachlorocuprate (also called phenetylammonium copper chloride), or (PEA)2CuCl4. This material, which has an octahedral CuCl6 structure with in-plane Jahn–Teller distortion, displays thermochromism and is a multiferroic due to its coexisting ferromagnetic and ferroelectric order, a property that is highly sought after.14,17
We found that (PEA)2CuCl4 changes color upon illumination depending on the atmosphere and the wavelength of the light. Grazing-incidence wide-angle X-ray scattering and Raman spectroscopy revealed that the color change is caused by a structural change in the inorganic framework, which takes place at the surface of the films. Moreover, using X-ray photoelectron spectroscopy we found that the Cu2+ at the surface reduces to Cu0/+, which might be explained by the formation of elemental copper in the form of clusters or nanoparticles. Air exposure reverses the color change, which is attributed to the absorption of water, leading to a different structure than the pristine material. The lack of photochromism in the similar compound bis(methylammonium) tetrachlorocuprate (or methylammonium copper chloride) – (MA)2CuCl4 – shows that the organic cation plays a decisive role in the process.
Results
Upon illuminating a yellow film of (PEA)2CuCl4 with ultraviolet (UV, λ = 254 nm) light in nitrogen atmosphere for 45 minutes, the color of the film changes to brown (see Fig. 1a). This color change is evident from the change in absorbance, which can be seen in Fig. 1b. The pristine material exhibits two absorption peaks in the ultraviolet region, namely the ligand-to-metal charge transfer transitions at 291 and 388 nm, which is characteristic of Jahn–Teller distorted CuCl42− compounds.18 After exposure to UV light, the intensity of these peaks decreases and both blueshift – to 266 and 385 nm, respectively. Simultaneously, an additional absorption feature forms between 450 and 800 nm. Pan et al. found very similar behavior when they studied the photochromism in the related compound bis(butylammonium) tetrachlorocuprate.16 We also studied (MA)2CuCl4 under UV illumination and found no color change after 45 minutes of exposure. The absorbance spectra of this film before and after UV-treatment can be seen in Fig. 1c. Apart from a small deviation in intensity, the two spectra are nearly identical. This result indicates that the organic cation plays an important role in the photochromism of (PEA)2CuCl4. | ||
| Fig. 1 (a) Photograph of (PEA)2CuCl4 (top row) and (MA)2CuCl4 (bottom row) thin films. These films are pristine (first column), UV-treated in N2 (second column) and UV-treated in air (third column). UV treatment was carried out with λ = 254 nm. Thin film absorbance spectra of (b) (PEA)2CuCl4 and (c) (MA)2CuCl4 in the case of a pristine film (black lines) and after 45 minutes of UV illumination in nitrogen environment (red lines). The narrow features between 850 and 900 nm are measurement artifacts. | ||
Raman spectroscopy has been used to characterize the vibrational modes of various copper chloride complexes.19–21 Our Raman spectrum collected from a single crystal of (PEA)2CuCl4 (Fig. 2) has clear peaks at 179, 247 and 287 cm−1, and is very similar to the low-frequency regime spectrum reported by Caretta et al.22 These peaks are all assigned to vibrational modes of Cu–Cl bonds: ν2 (A1g), ν4 (B2g) and ν1 (A1g), respectively.22,23 Notably, also prolonged and high intensity exposure to the laser (λ = 532 nm) of the Raman microscope converts the pristine material into the brown phase (Fig. S2, ESI†). We attribute this conversion to absorption taking place due to the small – but non-zero – absorption coefficient at this wavelength, combined with the high-power, focused laser light. This is accompanied by the appearance of a dominant peak at 271 cm−1. Increased laser power accelerates the conversion. This behavior is also observed in thin film samples, for which the Raman signal is naturally weaker (see Fig. S3, ESI†). Therefore, the color change is not only achieved with UV illumination, but also with focused visible light. Interestingly, the converted brown phase is stable in nitrogen, but not in air. In the latter case the color of the film changes back to yellow (see Fig. S4 (ESI†) for the absorbance after air exposure), which suggests that the brown phase might be sensitive to oxygen or water. We found that the conversion from the pristine to the brown phase depends on the wavelength and the atmosphere; this is described in the ESI† (see Fig. S5 and the accompanying section).
 | ||
| Fig. 2 Raman spectra of a pristine (PEA)2CuCl4 single crystal (black lines) and of the same crystal after 532 nm laser exposure (red lines). The inset displays a magnification of the low-energy part of the spectra. | ||
To investigate whether bulk structural changes occur during the conversion, we studied the pristine and converted films of both (PEA)2CuCl4 and (MA)2CuCl4 using X-ray powder diffraction (XRD). The corresponding diffraction patterns can be found in Fig. S6 (ESI†); they show the (00l) peaks (only even values of l for (PEA)2CuCl4),14,24 indicating that the inorganic plane is strongly oriented parallel to the substrate. No discernable new features were detected for the converted (PEA)2CuCl4 film compared to the pristine material. The same is true for (MA)2CuCl4, as expected since there was no change in the absorbance spectrum after UV treatment.
In our previous work we have demonstrated that XRD is not always the best choice for detailed structural characterization of metal halide perovskites, and we showed that grazing-incidence wide-angle X-ray scattering (GIWAXS) could provide more details.25 This is especially true if a texture is present in the perovskite layer.26 This advantage is combined with the ability to carry out depth-resolved measurements by varying the incident angle.27,28Fig. 3 shows the GIWAXS patterns of the pristine and UV-treated (PEA)2CuCl4 films at two different incident angles, αi = 0.2° and 0.8°, which correspond to roughly 25 and 2000 nm of X-ray penetration depth, respectively. The GIWAXS pattern of the pristine material (Fig. 3a and c) is composed of three strong signals focused along the quasi-vertical qz direction related to the (00l) reflections, one strong out-of-plane signal (113 reflection) and one weak signal along the qy direction, related to a (h00) signal. The pattern can be indexed using the known crystallographic structure for (PEA)2CuCl4, which is an orthorhombic unit cell of Pbca space group with axes of 7.31 Å, 7.34 Å and 38.64 Å (a × b × c).14 The anisotropic nature of the signals confirms the preferred orientation of the crystallites with the (00l) planes parallel to the substrate (see Fig. S7 (ESI†) for more details). The intense (00l) reflections with respect to the much weaker out-of-plane reflections can be explained by a higher degree of ordering of the stacking planes in the vertical z-direction rather than in the xy-plane. This is probably a result of the Jahn–Teller effect in these compounds.
 | ||
| Fig. 3 Wedge corrected GIWAXS patterns for a thin film of (PEA)2CuCl4 (a and c) before and (b and d) after UV irradiation measured at two different incident angles (αi = 0.2° (∼25 nm) for a and b and αi = 0.8° (∼2 μm) for c and d). | ||
Upon illumination with UV light, the (113) and (200) reflections practically disappear and the (00l) reflections becomes approximately five times weaker, although they do not vanish (see Fig. 3b and Fig. S8, ESI†). This indicates that UV treatment destroys the structural order in the xy-plane and reduces it in the z-direction. This effect is mostly limited to the top part of the film (top hundreds of nm), as the GIWAXS patterns before and after irradiation change negligibly when full X-ray penetration of the film is achieved (Fig. 3c and d). We also looked at the effect of the color change from brown back to yellow induced by air exposure. After several hours in air, the GIWAXS pattern characteristic of the pristine film is fully recovered (see Fig. 4).
 | ||
| Fig. 4 Evolution of the GIWAXS patterns for the (PEA)2CuCl4 film in air (a) just after irradiation with UV light, (b) 1 hour after irradiation and (c) 1 day after irradiation. The incident angle was 0.2°. Note that the sample was kept in the same place during this measurement, to ensure that a quantitative comparison between the different images can be made. | ||
Due to the conversion taking place mainly at the surface of the thin film, we resorted to X-ray photoelectron spectroscopy (XPS) to study the surface composition of the film. The full XPS spectra are displayed in Fig. S9 (ESI†). The Cu 2p XPS spectrum of the pristine (PEA)2CuCl4 film is shown in Fig. 5a. The fitted spectra of the other elements are included in the ESI† (Fig. S10). The film was UV-treated in the XPS setup under vacuum and measured afterwards. The UV-treatment was carried out for a cumulative duration of 45, 90 and 180 minutes. The satellite structures in Fig. 5a indicate the presence of Cu2+ species, as expected.29,30 As highlighted in Fig. 5b, there is an additional feature stemming from Cu0/+, for which the oxidation states cannot be discriminated precisely.30 However, it is clear that the ratio of Cu2+ to Cu0/+ decreases with UV exposure. Initially, Cu2+ and Cu0/+ make up about 75% and 25% of the Cu 2p3/2 signal, respectively, while after 180 minutes of exposure the Cu2+ feature represents 43% of the Cu 2p3/2 component (see Fig. S11, ESI†). Additionally, the Cl/Cu atomic ratio decreases from 5.0 in the initial measurement to 2.7 after 180 minutes of UV treatment (Fig. S12, ESI†).
 | ||
| Fig. 5 (a) XPS spectra of the Cu 2p core level region of a pristine (PEA)2CuCl4 film and after exposing it to UV light for various durations. The film was UV-converted in situ; XPS spectra were recorded after 45, 90 and 180 minutes. (b) Fits of the high-resolution Cu 2p3/2 peaks with Cu2+ (red curve) and Cu0/+ (blue curve) signals for the different data depicted in (a). | ||
We carried out photoluminescence measurements on both the pristine and UV-converted films to see whether the Cu0/+ led to new emission peaks. Laser excitation was performed at 267 nm. The photoluminescence (PL) experiment was carried out at 5 K, a temperature at which the conversion does not take place. Fig. 6 shows the PL intensity for both pristine and UV-converted (PEA)2CuCl4. It is clear that the pristine material does not emit. However, the UV-converted film has a strong signal centered around 550 nm. This is a very valuable finding for identification of the new species that form during the illumination of (PEA)2CuCl4.
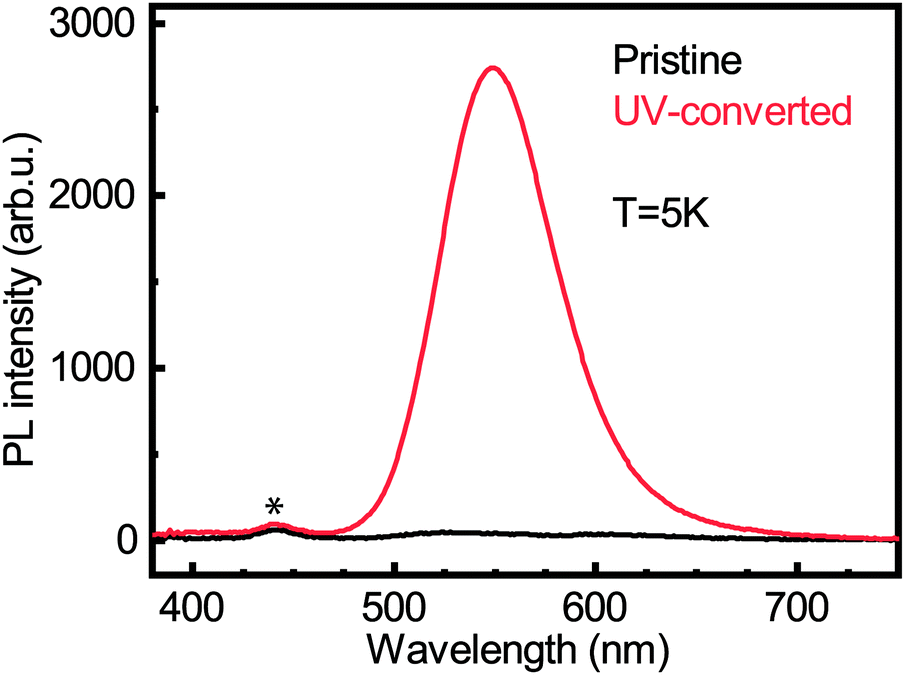 | ||
| Fig. 6 Photoluminescence spectra for pristine (black line) and UV-converted (red line) (PEA)2CuCl4 films acquired at 5 K. The peak that is marked with an asterisk is a measurement artifact due to stray background light. | ||
Discussion
From XPS we can conclude that UV-treatment of (PEA)2CuCl4 results in the reduction of Cu2+ to Cu0/+ (Fig. 5b). The copper reduction during UV irradiation is further confirmed by the decrease in satellite structures (Fig. 5a). The initial presence of the Cu0/+ component can be assigned to structural defects, partial reduction of Cu on the surface31,32 or traces of synthesis precursors.31 The decrease in Cl/Cu atomic ratio from 5.0 to 2.7 (Fig. S12, ESI†) is a clear indication that copper is in a lower oxidation state. Consequently, the formation of new compounds with Cu in a lower oxidation state has to be taken into consideration. Since the XPS measurement cannot distinguish between the two oxidation states of the Cu0/+ feature, both Cu+ and elemental copper could be formed in the process. Reports on the photoluminescence of Cu+ typically show emission with a peak around 515 nm,18,33 while copper(I) chloride (CuCl) emits around 390 nm.34 Both cannot explain what we observe in our measurements (550 nm, Fig. 6). Therefore, we have to look for a different origin. Considering elemental copper, we find that copper nanoparticles (NPs) embedded in a SiO2 matrix were reported to show photoluminescence around 550 nm, depending on their size.35 Such metallic copper NPs have been demonstrated to form through photochemical reduction of Cu+ and Cu2+, by illuminating precursors – such as CuCl2 – in solution with UV light.36–39 An example of photoinduced formation of metallic nanoparticles in solid state is the reduction of Au(III) to Au(0) nanoparticles, which occurs when AuCl4− is exposed to UV light.40 The presence of copper NPs could also explain the broad feature in the absorbance (Fig. 1b) between 450 and 800 nm.36 Another explanation for the broad absorption is the formation of Cu2O NPs.41 Considering that the conversion takes place in a nitrogen-filled glovebox, we deem the latter improbable.A partial contribution to the broad absorption feature could also be attributed to the presence of square-planar CuCl42− species. The CuCl42− ion has a centrosymmetric planar geometry with D4h symmetry;42 the inorganic plane of (PEA)2CuCl4 can be seen as square-planar CuCl42− ions linked to form infinite layers – a sort of polymerization – with Cu–Cl–Cu bonds linking the ions.4 The absorbance spectrum of square-planar CuCl42− displays ligand-field transitions between 500 and 850 nm.43,44 The formation of CuCl42− can also explain the shift in the UV-region of the absorption spectrum, because these ions have an absorption peak around 267–270 nm, matching perfectly with our spectrum.45,46 In addition, the new Raman peak can be attributed to square-planar CuCl42−: the totally symmetric stretch of the ion is found at 271 cm−1.43,47 Furthermore, there are reports in literature on a thermochromic effect in tetrachlorocuprates(II), where an increase in temperature leads to a (semi)reversible color change from green (square-planar CuCl42−) to yellow (CuCl42− in the form of a distorted tetrahedron).9,48 For bis(piperazinium) tetrachlorocuprate this effect also occurs upon illumination.9 In our case, the effect would involve a change from octahedral to square-planar CuCl42−, which would mean that the connected square-planar CuCl42− ions would become isolated. A similar process has been reported as a thermal effect in a hydrated nickel bromide compound.49 It is also important to underline that the transition from octahedral to square-planar CuCl42− could explain the disorder in the xy-plane, which was demonstrated with GIWAXS (see Fig. 3b).
The absorbance spectrum of UV-treated (PEA)2CuCl4 still shows features from the pristine layer in the form of a shoulder at 291 nm (see Fig. 1b). This can be explained by the results of the GIWAXS experiment, where we found that the conversion occurs in the first hundreds of nanometers from the surface. Below, in the bulk of the layer, no conversion takes place. This is the result of the limited penetration depth of UV light in our material. Therefore, the light can only convert (PEA)2CuCl4 in the first hundreds of nanometers.
Air exposure leads to the color change from brown to yellow. Bhattacharya et al. found that the thermochromic bis(benzimidazolium) tetrachlorocuprate can exist in two forms: the yellow non-hydrated phase and a green hydrated phase.11 The former consists of isolated distorted tetrahedral CuCl42− ions, the latter has alternating polymeric [CuCl42−]∞ and [CuCl2(H2O)2]∞ chains.50 Upon heating, the green hydrated phase loses the water molecules due to a thermochromic transition. The resulting yellow phase has isolated CuCl42− ions. The authors also demonstrated the reverse process, where the non-hydrated compound absorbs water to form the polymeric [CuCl2(H2O)2]∞ chains.50 Therefore, their system is a good example of a monomeric – polymeric transformation based on thermochromism and exposure to water molecules. We suppose that the mechanism behind the color change back to yellow in air for our system works in an analogous way, i.e. the square-planar CuCl42− ions in the brown phase could change to alternating polymeric [CuCl42−]∞ and [CuCl2(H2O)2]∞ chains after absorbing water molecules from the air. This would look more similar to the structure of pristine (PEA)2CuCl4 and could explain the recovery of the (113) and (200) reflections in the GIWAXS experiment due to the longer structural order of the polymeric chains.
At this point we are left to explain the lack of photochromic behavior in (MA)2CuCl4 (see Fig. 1c). We propose that this is due to a difference in the strength of the hydrogen bonding between the organic cation and the halide ions. Bloomquist et al. studied the thermochromic change of bis(isopropylammonium) tetrachlorocuprate and found that this involves a change from a linear structure of ribbon-like connected square-planar CuCl42− to isolated distorted tetrahedral CuCl42−.10 This is caused by a weakened hydrogen bond between the organic cations and the chloride ions due to an increase in temperature. Without the hydrogen bond there is an excess electrostatic charge on the chloride ions, which leads to a relaxation of the coordination of the copper complex to a distorted tetrahedral structure. The CH3-group in MA has an electron donating character, whereas the phenyl ring in PEA is electron withdrawing. This might lead to a difference in hydrogen bonding strength and stability of the octahedral structure. Additionally, methylamine does not absorb 254 nm light, whereas phenethylamine does.51,52 This means that only PEA will be in an excited state upon illumination. It has been shown that the most stable conformer of phenethylammonium has a gauche confirmation of the NH3+-group with respect to the alkyl chain, in which one hydrogen atom of the NH3+-group is oriented towards the benzene ring.53 In the excited state of this molecule, this hydrogen atom is situated closer to the benzene ring.54 Therefore, UV illumination could weaken the hydrogen bonding between PEA and the chloride ions.
Conclusions
This work is the first report on the photochromic properties of the hybrid organic–inorganic Ruddlesden–Popper perovskite (PEA)2CuCl4. Exposure to UV light (λ = 254 nm) in a nitrogen environment leads to a color change of the film from yellow to brown, but the color change can also occur in other conditions (312 nm light in air). This is accompanied by a loss in structural order in the xy-plane of the film's surface, as determined by GIWAXS. X-ray photoelectron spectroscopy shows that UV illumination causes the reduction of Cu2+ to Cu0/+. As a result of the illumination, new features were found in the absorbance and Raman spectra of UV-converted (PEA)2CuCl4, which can be explained by the presence of square-planar CuCl42− species. The instability of UV-converted (PEA)2CuCl4 in air is demonstrated by a color change back to yellow, which is accompanied by a recovery of the structural order, as seen in the GIWAXS pattern. The similar compound (MA)2CuCl4 does not show photochromic behavior, which we attribute to a difference in hydrogen bond strength between the organic cation and the chloride anions due to the different chemical nature of the two organic cations.Experimental section
Film processing
Preparing and spin coating the metal halide perovskite solutions took place in a glovebox with nitrogen environment. A solution of (PEA)2CuCl4 was made by dissolving phenethylammonium chloride (Sigma Aldrich) and copper(II) chloride (Sigma Aldrich) in N,N-dimethylformamide (DMF, Sigma Aldrich) in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. The final concentration of the solution was 1 M. For (MA)2CuCl4, methylammonium chloride (Sigma Aldrich) was used instead. Here, two solvents, DMF and dimethyl sulfoxide (DMSO, Alfa Aesar), were used in a 4:1 v/v ratio to improve the solubility. This solution had a 0.8 M concentration. Solutions were stirred at room temperature overnight before spin coating. The substrates were either made of glass or quartz and were cleaned with detergent solution, and subsequently ultrasonically cleaned in deionized water, acetone and isopropanol. After drying them in an oven at 130 °C for 10 min, they were treated with ultraviolet ozone (UV–O3) for 20 min. Spin coating involved a rotation of 5000 rpm during 45 seconds for (PEA)2CuCl4 and 3000 rpm for (MA)2CuCl4. To prevent charging effects in the XPS experiment we made a very thin film (50–150 nm) on a silicon substrate. This was done by blade coating because it offered us better control over the morphology of a very thin film compared to spin coating. For this experiment, the (PEA)2CuCl4 solution was diluted to 0.1 M and this was coated at 120 °C with a blade speed of 40 mm s−1.
:1 molar ratio. The final concentration of the solution was 1 M. For (MA)2CuCl4, methylammonium chloride (Sigma Aldrich) was used instead. Here, two solvents, DMF and dimethyl sulfoxide (DMSO, Alfa Aesar), were used in a 4:1 v/v ratio to improve the solubility. This solution had a 0.8 M concentration. Solutions were stirred at room temperature overnight before spin coating. The substrates were either made of glass or quartz and were cleaned with detergent solution, and subsequently ultrasonically cleaned in deionized water, acetone and isopropanol. After drying them in an oven at 130 °C for 10 min, they were treated with ultraviolet ozone (UV–O3) for 20 min. Spin coating involved a rotation of 5000 rpm during 45 seconds for (PEA)2CuCl4 and 3000 rpm for (MA)2CuCl4. To prevent charging effects in the XPS experiment we made a very thin film (50–150 nm) on a silicon substrate. This was done by blade coating because it offered us better control over the morphology of a very thin film compared to spin coating. For this experiment, the (PEA)2CuCl4 solution was diluted to 0.1 M and this was coated at 120 °C with a blade speed of 40 mm s−1.
Crystal synthesis
Single crystals of (PEA)2CuCl4 were grown by the slow evaporation of solvent at 60 °C. A 1:2 molar ratio of CuCl2·2H2O (Sigma Aldrich; ≥99.95%) and C6H5CH2CH2NH2·HCl (Sigma Aldrich; ≥98%) were dissolved in absolute ethanol (J.T. Baker) and placed in an oven at 60 °C. After approximately one week, transparent, yellow single crystals had formed. The crystals are shaped as platelets with sizes ranging from 0.1 to around 6 mm.
UV treatment
For the UV treatment the λ = 254 nm setting of a Spectroline EBF-280C/FE lamp was used. The UV treatment was carried out in a N2-filled glovebox for 45 minutes. The distance from the lamp to the sample was approximately 15 mm. For the experiment with the λ = 312 nm UV source, the longer wavelength setting on the same lamp was used.Absorbance
The thin film samples for the absorbance measurement were made on quartz, and characterized with a Shimadzu UV-3600 spectrophotometer with an integrating sphere attachment.X-Ray diffraction
Powder XRD data were collected under ambient conditions using a Bruker D8 Advance diffractometer in Bragg–Brentano geometry and operating with Cu Kα radiation (λ = 1.54 Å). A fixed slit width was used for the incident beam (1 mm) and diffracted beam (3 mm). Samples were rotated at 60 revolutions per minute. The angle 2θ was scanned between 5 and 40 degrees using a 0.015 degrees step size.Grazing-incidence wide-angle X-ray scattering
GIWAXS experiments were performed at the Multipurpose Instrument for Nanostructured Analysis (MINA) at RUG. The instrument is equipped with a rotating Cu anode source (λ = 0.15413 nm). GIWAXS patterns were acquired using a Pilatus 300 K solid state detector from Dectris with pixel size 0.172 mm × 0.172 mm. The sample alignment and the incident angle setting were achieved using motorized Huber positioning stages. The sample-to-detector distance (S-to-D) and the position of the direct beam on the detector where calibrated using known diffraction peaks from a silver behenate standard sample. The calibrated S-to-D value was 90 mm. The missing wedge correction and the intensity line cuts were obtained using the GIXGUI software.55 In the case of an ideally flat surface, the X-ray penetration depth (i.e. the depth into the material measured along the surface normal where the intensity of X-rays falls to 1/e of its value at the surface) depends on the wavelength λ of the X-rays, the critical angle of total reflection, αc, and the incident angle, αi, and can be estimated using the relation: , where β is the imaginary part of the complex refractive index of the compound.
, where β is the imaginary part of the complex refractive index of the compound.
X-ray photoelectron spectroscopy
XPS was performed using a Surface Science SSX-100 ESCA instrument with a monochromatic Al Kα X-ray source (hv = 1486.6 eV). The pressure in the measurement chamber was maintained below 1 × 10−9 mbar during data acquisition. The electron take-off angle with respect to the surface normal was 37°. The XPS data were acquired by using a spot size of 1000 μm diameter and the energy resolution was set to 1.3 eV for both the survey spectra and the detailed spectra of the C 1s, Cl 2p, Cu 2p, N 1s and O 1s core level regions. Furthermore, a gold mesh placed 1 mm above the sample was used during the XPS measurements in order to prevent the charging effect. Binding energies are reported ±0.1 eV and referenced to the C 1s photoemission peak centered at a binding energy of 284.8 eV.56 All XPS spectra were analyzed using the least-squares curve fitting program Winspec (LISE laboratory, University of Namur, Belgium). Deconvolution of the spectra included a Shirley57 baseline subtraction and fitting with a minimum number of peaks consistent with the structure of the components of the thin film, taking into account the experimental resolution. The profile of the peaks was taken as a convolution of Gaussian and Lorentzian functions. The uncertainty in the peak intensity determination is within 2% for all core levels reported. All measurements were carried out on freshly prepared samples. Exposure to UV (254 nm) light was conducted in situ through a special window of the vacuum chamber.Raman spectroscopy
Raman spectroscopy was carried out using a confocal inVia Qontor micro-Raman setup from Renishaw. Excitation occurred at 532 nm through a 50× objective, with a power between 39.5 and 73.4 μW. The light was collected in backscattering geometry, dispersed with a 2400 lines per millimeter grating and detected with a silicon CCD. Spectral calibration was assured through a silicon reference.Photoluminescence spectroscopy
For low-temperature photoluminescence spectroscopy, thin films were mounted into a cryostat (Oxford Instruments Optistat CF) in an inert atmosphere. The samples were excited at 4.6 eV (267 nm) using the third harmonic of a mode-locked Ti:sapphire laser (Mira 900, Coherent) operating at a repetition rate of 76 MHz. The excitation beam was spatially limited by an iris and focused onto the sample using a 150 mm focal length lens in a reflection geometry. The pump fluence was controlled using an adjustable neutral density filter. The photoluminescence was collected into a spectrometer and recorded by an ImagEM CCD camera (Hamamatsu, Japan).Author contribution statement
B. G., H. D. and M. A. L. designed the experiments. B. G., L. P. and M. A. L. conceived of the presented idea and crucial parts of the interpretation. H. D. carried out the photoluminescence experiment. S. K. was responsible for the Raman spectroscopy. The X-ray photoelectron spectroscopy experiment was carried out and analyzed by O. D. L. and P. R. X-ray diffraction characterization was conducted by E. T. and G. B. M. K. synthesized the single crystals. G. P. carried out the grazing-incidence wide-angle X-Ray scattering experiment and analysis. B. G. was responsible for all other experiments, made the thin film samples and wrote the manuscript. All authors provided critical feedback and helped writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to A. Kamp and T. Zaharia for technical support. This work is part of the research program of the Netherlands Organisation for Scientific Research (NWO). This is a publication of the FOM-focus Group ‘Next Generation Organic Photovoltaics’, participating in the Dutch Institute for Fundamental Energy Research (DIFFER). Financial support also came from the Advanced Materials research program of the Zernike National Research Centre under the Bonus Incentive Scheme (BIS) of the Dutch Ministry for Education, Culture and Science. S. K. acknowledges the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral research fellowship (grant no. 408012143).References
- D. B. Mitzi, Progress in Inorganic Chemistry, 1999, vol. 48, pp. 1–121 Search PubMed.
- D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1–12 RSC.
- H. Place and R. D. Willett, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 34–38 CrossRef.
- D. W. Smith, Coord. Chem. Rev., 1976, 21, 93–158 CrossRef CAS.
- R. D. Willett, J. A. Haugen, J. Lebsack and J. Morrey, Inorg. Chem., 1974, 13, 2510–2513 CrossRef CAS.
- L. L. Lohr, Proc. Natl. Acad. Sci. U. S. A., 1968, 59, 720–725 CrossRef CAS.
- A. Jaffe, Y. Lin, W. L. Mao and H. I. Karunadasa, J. Am. Chem. Soc., 2015, 137, 1673–1678 CrossRef CAS.
- P. Ghalsasi, N. Garg, M. N. Deo, A. Garg, H. Mande, P. Ghalsasi and S. M. Sharma, Phys. Chem. Chem. Phys., 2015, 17, 32204–32210 RSC.
- M. J. Riley, D. Neill, P. V. Bernhardt, K. A. Byriel and C. H. L. Kennard, Inorg. Chem., 1998, 37, 3635–3639 CrossRef CAS.
- D. R. Bloomquist, R. D. Willett and H. W. Dodgen, J. Am. Chem. Soc., 1981, 103, 2610–2615 CrossRef CAS.
- R. Bhattacharya, M. Sinha Ray, R. Dey, L. Righi, G. Bocelli and A. Ghosh, Polyhedron, 2002, 21, 2561–2565 CrossRef CAS.
- K. Sone and Y. Fukuda, Inorganic Thermochromism, 1987, pp. 104–131 Search PubMed.
- M. R. Bond, T. J. Johnson and R. D. Willett, Can. J. Chem., 1988, 66, 963–973 CrossRef CAS.
- A. Caretta, R. Miranti, A. H. Arkenbout, A. O. Polyakov, A. Meetsma, R. Hidayat, M. O. Tjia, T. T. M. Palstra and P. H. M. van Loosdrecht, J. Phys.: Condens. Matter, 2013, 25, 505901 CrossRef CAS.
- D. Xie, J. Xu, H. Cheng, N. Wang and Q. Zhou, J. Mol. Struct., 2018, 1161, 267–272 CrossRef CAS.
- X. Pan, G. Wu, M. Wang and H. Chen, J. Zhejiang Univ., Sci., A, 2009, 10, 710–715 CrossRef CAS.
- A. O. Polyakov, A. H. Arkenbout, J. Baas, G. R. Blake, A. Meetsma, A. Caretta, P. H. M. Van Loosdrecht and T. T. M. Palstra, Chem. Mater., 2012, 24, 133–139 CrossRef CAS.
- D. Cortecchia, H. A. Dewi, J. Yin, A. Bruno, S. Chen, T. Baikie, P. P. Boix, M. Grätzel, S. Mhaisalkar, C. Soci and N. Mathews, Inorg. Chem., 2016, 55, 1044–1052 CrossRef CAS.
- R. G. McDonald and M. A. Hitchman, Inorg. Chem., 1989, 28, 3996–4001 CrossRef CAS.
- A. Dick, H. Rahemi, E. R. Krausz, G. R. Hanson and M. J. Riley, J. Chem. Phys., 2008, 129, 214505 CrossRef.
- M. J. Riley, C. Boutchard, E. R. Krausz and M. A. Hitchman, Chem. Phys. Lett., 1996, 254, 403–409 CrossRef CAS.
- A. Caretta, R. Miranti, R. W. A. Havenith, E. Rampi, M. C. Donker, G. R. Blake, M. Montagnese, A. O. Polyakov, R. Broer, T. T. M. Palstra and P. H. M. van Loosdrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 024301 CrossRef.
- I. Pabst, J. Karolyi, H. Fuess and M. Couzi, Phys. Status Solidi, 1996, 155, 341–352 CrossRef CAS.
- I. Pabst, H. Fuess and J. W. Bats, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 413–416 CrossRef.
- B. G. H. M. Groeneveld, S. Adjokatse, O. Nazarenko, H.-H. Fang, G. R. Blake, G. Portale, H. Duim, G. H. ten Brink, M. V. Kovalenko and M. A. Loi, Energy Technol., 2019, 1901041 Search PubMed.
- S. Shao, J. Liu, G. Portale, H.-H. Fang, G. R. Blake, G. H. ten Brink, L. J. A. Koster and M. A. Loi, Adv. Energy Mater., 2018, 8, 1702019 CrossRef.
- J. Dong, S. Shao, S. Kahmann, A. J. Rommens, D. Hermida-Merino, G. H. ten Brink, M. A. Loi and G. Portale, Adv. Funct. Mater., 2020, 30, 2001294 CrossRef CAS.
- A. Vagias, Q. Chen, G. H. ten Brink, D. Hermida-Merino, J. Scheerder and G. Portale, ACS Appl. Polym. Mater., 2019, 1, 2482–2494 CrossRef CAS.
- F. Gao, Y. Wang, X. Wang and S. Wang, RSC Adv., 2016, 6, 34439–34446 RSC.
- W.-L. Dai, Q. Sun, J.-F. Deng, D. Wu and Y.-H. Sun, Appl. Surf. Sci., 2001, 177, 172–179 CrossRef CAS.
- S. K. Abdel-Aal, G. Kocher-Oberlehner, A. Ionov and R. N. Mozhchil, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 531 CrossRef.
- N. Akhtar, G. R. Blake, R. Felici, H. Amenitsch, T. T. M. Palstra and P. Rudolf, Nano Res., 2014, 7, 1832–1842 CrossRef CAS.
- S. Gomez, I. Urra, R. Valiente and F. Rodriguez, J. Phys.: Condens. Matter, 2010, 22, 295505 CrossRef.
- M. Nakayama, H. Ichida and H. Nishimura, J. Phys.: Condens. Matter, 1999, 11, 7653–7662 CrossRef CAS.
- O. A. Yeshchenko, I. M. Dmitruk, A. M. Dmytruk and A. A. Alexeenko, Mater. Sci. Eng., B, 2007, 137, 247–254 CrossRef CAS.
- X. Zhu, B. Wang, F. Shi and J. Nie, Langmuir, 2012, 28, 14461–14469 CrossRef CAS.
- G. G. Condorelli, L. L. Costanzo, I. L. Fragalà, S. Giuffrida and G. Ventimiglia, J. Mater. Chem., 2003, 13, 2409–2411 RSC.
- S. Giuffrida, G. G. Condorelli, L. L. Costanzo, I. L. Fragalà, G. Ventimiglia and G. Vecchio, Chem. Mater., 2004, 16, 1260–1266 CrossRef CAS.
- B. Wang, S. Chen, J. Nie and X. Zhu, RSC Adv., 2014, 4, 27381–27388 RSC.
- A. Taubert, I. Arbell, A. Mecke and P. Graf, Gold Bull., 2006, 39, 205–211 CrossRef CAS.
- S. Banerjee and D. Chakravorty, Europhys. Lett., 2000, 52, 468–473 CrossRef CAS.
- R. G. McDonald, M. J. Riley and M. A. Hitchman, Inorg. Chem., 1988, 27, 894–900 CrossRef CAS.
- A. Dick, H. Rahemi, E. R. Krausz, G. R. Hanson and M. J. Riley, J. Chem. Phys., 2008, 129, 214505 CrossRef.
- M. A. Hitchman and P. J. Cassidy, Inorg. Chem., 1979, 18, 1745–1754 CrossRef CAS.
- S. R. Desjardins, K. W. Penfield, S. L. Cohen, R. L. Musselman and E. I. Solomon, J. Am. Chem. Soc., 1983, 105, 4590–4603 CrossRef CAS.
- H.-B. Yi, F.-F. Xia, Q. Zhou and D. Zeng, J. Phys. Chem. A, 2011, 115, 4416–4426 CrossRef CAS.
- V. M. Masters, M. J. Riley and M. A. Hitchman, Chem. Phys. Lett., 1998, 288, 743–748 CrossRef CAS.
- R. L. Harlow, W. J. Wells, G. W. Watt and S. H. Simonsen, Inorg. Chem., 1974, 13, 2860–2864 CrossRef CAS.
- R. Tsuchiya, S. Joba, A. Uehara and E. Kyuno, Bull. Chem. Soc. Jpn., 1973, 46, 1454–1456 CrossRef CAS.
- M. Bukowska-Strzyżewska and J. Skoweranda, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2290–2293 CrossRef.
- L. Lagesson-Andrasko, V. Lagesson and J. Andrasko, Anal. Chem., 1998, 70, 819–826 CrossRef CAS.
- S. Sugasawa and R. Tachikawa, Tetrahedron, 1958, 4, 205–212 CrossRef CAS.
- J. J. Urban, C. W. Cronin, R. R. Roberts and G. R. Famini, J. Am. Chem. Soc., 1997, 119, 12292–12299 CrossRef CAS.
- G. Féraud, M. Broquier, C. Dedonder-Lardeux, G. Grégoire, S. Soorkia and C. Jouvet, Phys. Chem. Chem. Phys., 2014, 16, 5250 RSC.
- Z. Jiang, J. Appl. Crystallogr., 2015, 48, 917–926 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, Physical Electronics Division, PerkinElmer Corp., 1992 Search PubMed.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc03359f |
| This journal is © The Royal Society of Chemistry 2020 |